

Abstract
Cortical pyramidal neurons (PNs) containing non-phosphorylated neurofilaments (NNFs) localized with the SMI-32 monoclonal antibody have been shown to be especially vulnerable to degeneration in Alzheimer’s disease (AD). The present investigation is the first to study the expression of SMI-32+ NNFs in neurons of the basolateral nuclear complex of the amygdala (BNC), which contains cortex-like PNs and nonpyramidal neurons (NPNs). We observed that PNs in the rat basolateral nucleus (BL), but not in the lateral (LAT) or basomedial (BM) nuclei, have significant levels of SMI-32-ir in their somata with antibody diluents that did not contain Triton X-100, but staining in these cells was greatly attenuated when the antibody diluent contained 0.3% Triton. Using Triton-containing diluents we found that all SMI-32+ neurons in all three of the BNC nuclei were NPNs. Using a dual-labeling immunoperoxidase technique we demonstrated that most of these SMI-32+ NPNs were parvalbumin-positive (PV+) or somatostatin-positive NPNs, but not vasoactive intestinal peptide-positive or neuropeptide Y-positive NPNs. Using a technique that combines retrograde tracing with SMI-32 immunohistochemistry using intermediate levels of Triton in the diluent we found that all BNC neurons projecting to the mediodorsal thalamic nucleus (MD) were large NPNs, and most were SMI-32+. In contrast, BNC neurons projecting to the ventral striatum or cerebral cortex were PNs that expressed low levels of SMI-32 immunoreactivity (SMI-32-ir) in the BL, and no SMI-32-ir in the LAT or BM. These data suggest that the main neuronal subpopulations in the BNC that degenerate in AD may be PV+ and MD-projecting NPNs.
Keywords: basolateral amygdala, non-phosphorylated neurofilaments, immunohistochemistry, retrograde tract tracing, pyramidal neurons, nonpyramidal neurons
Graphical Abstract
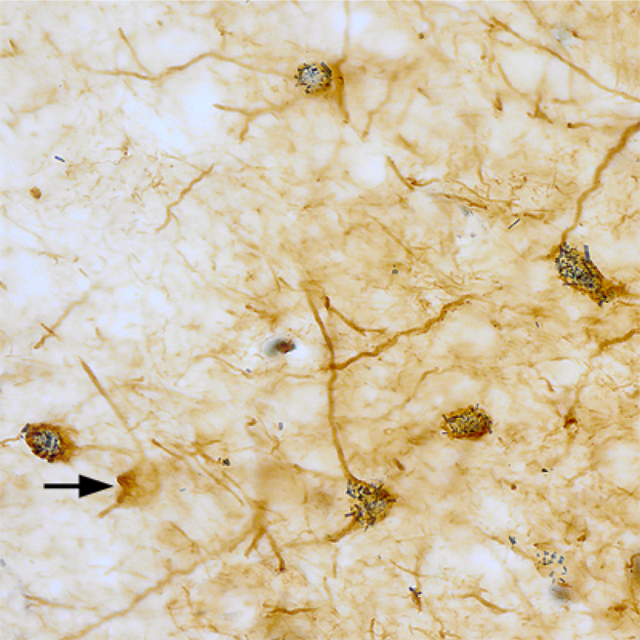
1. INTRODUCTION
Neurofilaments (NFs) are 10 nm-wide strands of neuron-specific intermediate filaments that are an important component of the neuronal cytoskeleton. There are several major NF proteins, including the structurally and genetically-related “triplet” proteins consisting of low (ca. 68 kD, NF-L) middle (ca. 150 kD, NF-M) and high (ca. 200 kD, NF-H) molecular weight subunits which copolymerize to form NFs (Yuan et al., 2017; Bott and Winckler, 2020). NFs are among the most highly phosphorylated proteins in the brain; posttranslational phosphorylation stabilizes NFs and increases their interaction with each other and with other cytoskeletal proteins (Matus 1988; Julien and Mushynski, 1982; Lewis and Nixon, 1988; Bott and Winckler, 2020). NF phosphorylation is usually confined to axons; little or no phosphorylated NFs are observed in most neuronal somata, except in neurological diseases or in axotomized neurons (Lee et al., 1988; Klosen et al., 1990; Yuan et al., 2017). Most NFs in somata and dendrites are nonphosphorylated, with phosphorylation occurring during transport of NFs down the axon (Sternberger and Sternberger, 1983). A monoclonal antibody developed by the Sternbergers (SMI-32) recognizes nonphosphorylated epitopes on NF-M and NF-H proteins (Sternberger and Sternberger, 1983; Lee et al., 1988), but only stains particular neuronal subpopulations. For example, early studies demonstrated that SMI-32 immunoreactive (SMI-32+) pyramidal cells in the human and monkey neocortices were located in particular layers in different areas, and that the staining of these neurons, which was confined to somata and dendrites, was significantly reduced in Alzheimer’s disease (Campbell and Morrison, 1989; Hof et al., 1990). Most of the SMI-32+ PNs in the cortex are involved in intracortical connections (Hof et al., 1990). Likewise, specific neuronal subpopulations are SMI-32+ in the rodent cortex (Budinger et al., 2000; Kirkcaldie et al., 2002; Boire et al., 2005; Voelker et al., 2004; Ouda et al., 2012).
There have been no immunohistochemical studies of SMI-32 non-phosphorylated NF epitope expression in neurons of the basolateral nuclear complex (BNC) of the amygdala. The BNC consists of three main nuclei: the lateral, basolateral, and basomedial nuclei. Like the cortex there are two main types of neurons in the BNC: (1) spiny glutamatergic pyramidal projection neurons (PNs), and (2) spine-sparse or spine-free nonpyramidal neurons (NPNs) (McDonald, 2020). Although these neurons do not exhibit a laminar or columnar organization like cortical neurons, their morphology, synaptology, electrophysiology and pharmacology are remarkably similar to their counterparts in the cortex (Carlsen and Heimer, 1988; Sah et al., 2003; Muller et al., 2005, 2006, 2007). As in the cortex, most NPNs are GABAergic interneurons (INs). Like the cortex, several distinct NPN subpopulations can be identified in the BNC on the basis of their expression of neuropeptides and calcium-binding proteins, including (1) parvalbumin+/calbindin+ (PV+/CB+) neurons, (2) somatostatin+/calbindin+ (SOM+/CB+) neurons, (3) large multipolar cholecystokinin+ (CCK+) neurons that are often CB+, and (4) small bipolar and bitufted interneurons that exhibit extensive colocalization of calretinin (CR), CCK, and vasoactive intestinal peptide (VIP) (Spampanato et al., 2011; Capogna 2014; McDonald, 2020). In addition, a subpopulation of SOM+ neurons, but not other NPN subpopulations, expresses neuropeptide Y (NPY) (McDonald, 1989; Urban, 2020). These separate NPN subpopulations in rodents play discrete roles in the intrinsic circuitry of the BNC by innervating distinct compartments of PNs and/or other INs, and by having different inputs/receptors and electrophysiological properties (Ehrlich et al., 2009; Spampanato et al., 2011; Bienvenu et al., 2012; Capogna 2014; Krabbe et al.,, 2018; Rovira-Esteban et al., 2017; Lucas and Clem, 2018; Beyeler and Dabrowska, 2020; McDonald, 2020). In addition to these NPNs there is a distinct NPN subpopulation in the basolateral and basomedial nuclei that projects to the mediodorsal thalamic nucleus (McDonald, 1987; 1996). There are also discrete PN subpopulations that can be identified on the basis of connections with specific cortical or subcortical regions, and/or specific functional roles (Herry et al., 2008; Senn et al., 2014; Namburi et al., 2015; Beyeler et al., 2016; Kim et al., 2016, 2017; McDonald, 2020).
Since the BNC is a cortex-like structure, it is of interest to determine if the laminar and areal specificity of SMI-32 expression in PNs of the cortex is mirrored by similar specificity of SMI-32 expression in PNs in discrete nuclear subdivisions of the BNC. Our working hypothesis was that SMI-32 staining in the BNC would be confined to PN subpopulations with specific projections, and that NPNs would exhibit little if any SMI-32 expression, similar to the cortex. This hypothesis was tested in the present study by combining immunohistochemistry for SMI-32 with retrograde tract tracing or immunohistochemistry for several NPN markers in the rat BNC.
2. MATERIALS AND METHODS
2.1. Animals
Twenty-four male Sprague-Dawley rats (250–350 g; Harlan, Indianapolis, Indiana) were used for these studies. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Use and Care committee (IACUC) of the University of South Carolina. All efforts were made to minimize animal suffering and to use the minimum number of animals necessary to produce reliable scientific data.
2.2. Single-labeling immunoperoxidase technique for SMI-32 localization
Localization of SMI-32 immunoreactivity (SMI-32-ir) was performed in six rats using the avidin–biotin immunoperoxidase (ABC) technique. Five rats were anesthetized with chloral hydrate (350 mg/kg, i.p.) and perfused intracardially with phosphate-buffered saline (PBS; pH 7.4) containing 1.0 % sodium nitrite (50 ml), followed by 4.0% paraformaldehyde in 0.1 M phosphate buffer (PB) at pH 7.4 (500 ml). One additional rat was perfused with a mixture of 4.0% paraformaldehyde and 0.2% glutaraldehyde; there was no obvious difference in SMI-32 staining in the brain of the latter rat compared with those that were perfused with just 4.0% paraformaldehyde. After perfusion, brains were removed and postfixed for 3–5 hours in the perfusate. Most brains (n = 4) were then sectioned on a vibratome at a thickness of 50 μm in the coronal plane. Two brains were bisected with a midline cut and then one hemisphere was sectioned at a thickness of 50 μm in the coronal plane and the other hemisphere was sectioned at a thickness of 70 μm in the sagittal (n = 1) or horizontal plane (n = 1).
Sections were processed for immunohistochemistry in wells of tissue culture plates. They were incubated overnight in the mouse monoclonal SMI-32 antibody for non-phosphorylated neurofilaments (1:25,000; Sternberger-Meyer Immunocytochemicals, Jarrettsville. MD; Table 1). All antibodies were diluted in 0.1 M PBS containing 1% normal goat serum. In three brains the antibody diluent also contained 0.3% Triton X-100, but in two brains the diluent was Triton X-100 free. In one brain half of the sections were processed using a diluent that contained 0.3% Triton X-100, and the other half of the sections were incubated in a diluent that was Triton X-100 free. Following primary antibody incubation, sections were processed for the ABC immunoperoxidase technique using a Vectastain mouse ABC kit (Vector Laboratories, Burlingame, CA). DAB (3,3’-diaminobenzidine-4HCl; Sigma Chemical Co., St. Louis, MO) was used as a chromogen to generate a brown reaction product (0.05% DAB, 0.01% hydrogen peroxide, in 0.01 M Tris buffer, pH 7.2). After the immunohistochemical procedures, sections were mounted on gelatinized slides, dried overnight, dehydrated in ethanols, cleared in xylene, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA). In this portion of the study, and all other portions, slides were analyzed with an Olympus BX51 microscope (Tokyo, Japan), and digital light micrographs were taken with an Olympus DP2-BSW camera system. Brightness and contrast were adjusted in Photoshop CS2 (Adobe Systems, San Jose, CA).
Table 1.
Antibodies and dilutions used in this study (see text for specificity data).
| Antibody | Source | Cat. # | Immunogen | Species | Dilution | RRID |
|---|---|---|---|---|---|---|
| SMI-32 | Sternberger-Meyer Immunocytochemicals | 32 | Nonphosphorylated neurofilaments from rat brain separated by SDS-PAGE | Mouse | 1:10,000 and 1:25,000 | NA |
| PV | Gift of K. Baimbridge | R-301 | Rat muscle PV | Rabbit | 1:10,000 | NA |
| SOM | ImmunoStar Inc. | 20067 | Synthetic SOM coupled to keyhole limpet hemocyanin | Rabbit | 1:4000 | AB_572264 |
| NPY | Peninsula Laboratories International. | T-4070 | Amino acids 30–65 of rat NPY | Rabbit | 1:10,000 | AB_518507 |
| VIP | ImmunoStar. | 20077 | Porcine VIP conjugated to bovine thyroglobulin | Rabbit | 1:2000 | AB_572270 |
2.3. Dual-labeling immunoperoxidase technique for SMI-32 colocalization with interneuronal markers
The results of the single-labeling immunoperoxidase studies indicated that many NPNs in the BNC were stained for SMI-32. To determine if separate subpopulations of NPNs express non-phosphorylated neurofilaments (NNFs) an avidin-biotin modification of a sequential two-color immunoperoxidase procedure (Levey et al., 1986) was used in ten rats to study possible colocalization of SMI-32 with markers for several NPN subtypes: (1) parvalbumin (PV); (2) somatostatin (SOM); (3) neuropeptide Y (NPY); or (4) vasoactive intestinal peptide (VIP).
Rats were anesthetized with chloral hydrate (350 mg/kg, i.p.) and perfused intracardially with PBS containing 1.0 % sodium nitrite (50 ml), followed by 4.0% paraformaldehyde in PB (500 ml). Four of the ten rats received bilateral injections of colchicine dissolved in PBS into the lateral ventricles (45 μg into each ventricle) 24 h before perfusion to enhance staining of neuropeptides. The three neuropeptides studied (VIP, NPY, and SOM) were stained in colchicine-injected brains; only non-colchicine-injected rats were used for SMI-32/PV dual-staining. SMI-32-ir appeared to be identical in colchicine-injected and non-colchicine-injected brains, but levels of neuropeptide staining were enhanced in colchicine-injected brains as in previous studies in this lab (McDonald, 1985, 1989; McDonald and Pearson, 1989). After perfusion, brains were removed and postfixed for 3–5 hours in the perfusate. Antibodies were diluted in 0.1 M PBS containing 1% normal goat serum and 0.3% Triton X-100. Sections (50 μm thick) were first incubated in the first sequence primary antibody (SMI-32) overnight at 4°C, and then processed for the avidin-biotin immunoperoxidase technique using a mouse Vectastain ABC kit. DAB was used as the first sequence chromogen to generate a diffuse, brown reaction product (0.05% DAB, 0.01% hydrogen peroxide, in 0.01 M Tris buffer, pH 7.2). Following extensive rinsing in Tris buffer followed by PBS, SMI-32-stained sections were incubated in one of the second sequence IN marker primary antibodies (to PV, SOM, NPY, or VIP; Table 1) overnight at 4°C and then processed for avidin-biotin immunohistochemistry using a Vectastain rabbit ABC kit. Benzidine dihydrochloride (BDHC; Sigma Chemical Co.) was used as the second sequence chromogen to generate a granular, dark blue reaction product. Sections were then rinsed thoroughly in 0.01 M phosphate buffer (pH 6.8), mounted on gelatinized slides, dried overnight, dehydrated in alcohols, cleared in xylene, and coverslipped with Permount.
Several controls were performed to test the method specificity of the colocalization procedure. In every animal used for SMI-32/marker colocalization some sections were processed with the marker antibodies omitted or replaced by diluted normal rabbit serum (1:5000). There was no BDHC reaction product in these controls, indicating that the BDHC reaction product was due to the presence of the marker antibody, and not the result of residual peroxidase activity remaining from the first sequence (DAB) procedure, or interactions of the biotinylated antibody or ABC complex used for the marker antibodies with the reagents used in the first sequence DAB procedure. In other controls the SMI-32 antibody was omitted; in these controls no DAB staining was observed, only BDHC marker staining.
The staining for SMI-32 appeared identical in all brains used for SMI-32/marker dual localization. The pattern of staining for markers was similar to that of previous studies in the rodent BNC (McDonald, 1985, 1989; Gustafson et al., 1986; McDonald and Betette, 2001; Rhomberg et al., 2018), but the intensity of staining for each marker varied among brains. To quantitate the extent of SMI-32/marker colocalization, the two brains with the most robust staining for each marker were selected for counts of single- and double-labeled neurons. Separate counts were performed in the lateral and basolateral nuclei; SMI-32+ neurons were sparse in the basomedial nucleus. Since there were no obvious differences in the extent of colocalization in the left and right amygdalas, or in different subdivisions of the nuclei, counts were pooled bilaterally from all three subdivisions of the lateral nucleus (dorsolateral, ventromedial, and ventrolateral subdivisions) for lateral nucleus (LAT) counts, and the two main subdivisions of the basolateral nucleus (anterior and posterior) for basolateral nucleus (BL) counts. In each brain counts were obtained from sections through each nucleus starting at the anterior pole of these nuclei (bregma −1.8) and proceeding caudally (see Tables 2–4 for the number of amygdalae analyzed in each brain). For SMI-32+/VIP+ and SMI-32+/NPY+ preparations in both nuclei, and SMI-32+/SOM+ preparations in the BL, all of which showed little or no colocalization, counts were made of single-labeled marker+ neurons and double-labeled marker+/SMI-32+ neurons until a total of 50 neurons were analyzed. For SMI-32+/SOM+ preparations in the LAT, which exhibited significant colocalization, counts were made of single-labeled SOM+ neurons, single-labeled SMI-32+ neurons, and double-labeled SOM+/SMI-32+ neurons until at least 125 neurons were counted in each brain. For SMI-32+/PV+ preparations in both the BL and LAT, which exhibited significant colocalization, counts were made of single-labeled PV+ neurons, single-labeled SMI-32+ neurons, and double-labeled PV+/SMI-32+ neurons until a total of at least 150 neurons were counted in each brain. Since stereological methods were not used, these analyses are considered semi-quantitative.
Table 2.
Cell counts of colocalization of SMI-32 with PV in the basolateral (BL) and lateral (LAT) nuclei.
| Nucleus | Brain | SMI-32 Single-labeled Neurons | PV Single-labeled Neurons | SMI-32/PV Double-labeled Neurons | % of SMI-32 Neurons Double-labeled | % of PV Neurons Double-labeled | Total Neurons Counted | Number of Amygdalae Analyzed |
|---|---|---|---|---|---|---|---|---|
| BL | S16 | 18 | 18 | 117 | 86.7% (117/135) | 86.7% (117/135) | 153 | 12 |
| S14 | 28 | 6 | 117 | 80.1% (117/145) | 95.1% (117/123) | 151 | 13 | |
| Total | 46 | 24 | 234 | 83.6% (234/280) | 90.7% (234/258) | 304 | 25 | |
| LAT | S16 | 56 | 10 | 85 | 60.3% (85/141) | 89.5% (85/95) | 151 | 11 |
| S14 | 62 | 12 | 78 | 55.8% (78/140) | 86.7% (78/90) | 152 | 14 | |
| Total | 118 | 22 | 163 | 58.0% (163/281) | 88.1% (163/185) | 303 | 25 |
Table 4.
Cell counts of colocalization of SMI-32 with neuropeptide markers in the basolateral (BL) and lateral (LAT) nucleus.
| IN Marker | Nucleus | Brain | SMI-32 Single-labeled Neurons | SMI-32/Marker Double-labeled Neurons | % of Marker Neurons Double-labeled | Number of Amygdalae Analyzed |
|---|---|---|---|---|---|---|
| VIP | BL | S13 | 50 | 0 | 0% | 16 |
| S18 | 50 | 0 | 0% | 19 | ||
| Total | 100 | 0 | 0% | 35 | ||
| LAT | S13 | 50 | 0 | 0% | 18 | |
| S18 | 50 | 0 | 0% | 18 | ||
| Total | 100 | 0 | 0% | 36 | ||
| NPY | BL | S13 | 50 | 0 | 0% | 17 |
| S15 | 50 | 0 | 0% | 19 | ||
| Total | 100 | 0 | 0% | 36 | ||
| LAT | S13 | 50 | 0 | 0% | 22 | |
| S18 | 50 | 0 | 0% | 20 | ||
| Total | 100 | 0 | 0% | 42 | ||
| SOM | BL | S13 | 49 | 1 | 2% | 15 |
| S17 | 49 | 1 | 2% | 15 | ||
| Total | 98 | 2 | 2% | 30 |
2.4. Retrograde tract tracing combined with SMI-32 immunohistochemistry
The main targets of BNC projections are the ventral striatum, mediodorsal thalamic nucleus (MD), and several cortical areas, including the medial and lateral prefrontal cortices (PFCs), insular cortex, temporal cortex, perirhinal cortex, entorhinal cortex and hippocampus (McDonald, 2020). Although there are a small number of SOM+ and NPY+ nonpyramidal neurons that project to the entorhinal cortex (McDonald and Zaric, 2015), the remaining cortical targets of the BNC, as well as the striatal targets, only receive inputs from BNC PNs (McDonald, 1991a, b, 1992, 2020). The BNC neurons projecting to MD are a special subpopulation of nonpyramidal neurons that are not immunoreactive for any of the calcium-binding proteins and neuropeptide markers expressed by INs (McDonald 1987, 1996, 2020); there is no evidence that these neurons are GABAergic, but some may be glutamatergic (Mátyás et al., 2014; Timbie and Barbas, 2015).
Retrograde tract tracing was combined with SMI-32 immunohistochemistry in a total of 8 male Sprague Dawley rats (250–350 g) to determine to what extent different subpopulations of BNC projection neurons were SMI-32+. Animals were anesthetized with chloral hydrate and placed into a stereotaxic apparatus (David Kopf Instruments, Tujunda, CA). Bilateral pressure injections of the retrograde tracer wheat germ agglutinin-conjugated horseradish peroxidase (WGA-HRP; 4%) were made into some of the major targets of the BNC using coordinates obtained from an atlas of the rat brain (Paxinos and Watson, 2007; all bregma coordinates mentioned in this paper are based on the Paxinos and Watson atlas). A 1.0-μl Hamilton microsyringe equipped with a 26-gauge needle was used to inject the medial and lateral PFC (2 rats), perirhinal/dorsolateral entorhinal cortices (2 rats), ventral striatum (2 rats), and MD thalamus (2 rats). Two injections, one rostral and one caudal, of 0.06 μl WGA-HRP were made into each target in each hemisphere. After a 20–24 h survival, animals were anesthetized with chloral hydrate and perfused intracardially with PBS containing 1.0 % sodium nitrite (50 ml), followed by a mixture of 4.0% paraformaldehyde/0.1% glutaraldehyde in PB (500 ml). Brains were removed and postfixed in the perfusate for 2–4 h..
Coronal sections (50 μm) were cut on a vibratome, processed for HRP histochemistry using tetramethylbenzidine (TMB) as a chromogen (deOlmos et al., 1978), and stabilized with diaminobenzidine-cobalt (Rye et al., 1984). The stabilization procedure changes the TMB reaction product from blue to black. Next, ABC immunohistochemistry, rinsing, and coverslipping was performed on free-floating amygdalar sections using the SMI-32 antibody as described above, but at a dilution of 1:10,000. The antibody diluent for these studies contained 0.2% Triton X-100 rather than the 0.3% used in the single-labeling studies. Sections containing injection sites in the cortex and striatum (i.e., at non-amygdalar levels) were not immunostained, but instead were Nissl-stained with cresyl violet. Sections containing the MD injection sites, which were at amygdalar levels, were SMI-32 immunostained, but not Nissl-stained. Slides were examined at 400X magnification to determine if SMI-32+ neurons were TMB+. The brown immunostaining of SMI-32+ neurons was easily distinguished from the black granular staining of retrogradely-labeled TMB+ neurons. Preliminary observations suggested that only MD injections resulted in retrograde-labeling of SMI-32+ NPNs in the BNC. To quantitate these observations, counts of single-labeled TMB+ neurons and double-labeled TMB+/SMI-32+ neurons were made in the two MD-injected brains that had the most accurate MD injections and the most TMB+ neurons in the BNC. Seventy-five TMB+ neurons were analyzed in each brain. Since stereological methods were not used, these analyses are considered semi-quantitative.
2.5. Antibody characterization and specificity
The antibodies used in this study are listed in Table 1. The monoclonal mouse SMI-32 antibody recognizes nonphosphorylated epitopes on NF-M and NF-H neurofilament proteins (Sternberger and Sternberger, 1983; Lee et al., 1988). All IN marker polyclonal antibodies were raised in rabbit. The PV antibody (1: 10,000; antiserum R-301) was generously donated by Dr. Kenneth Baimbridge (University of British Columbia). The PV antibody was raised in rabbit against rat muscle PV. Previous adsorption studies have shown that this antiserum recognizes PV, but not other calcium-binding proteins such as calbindin or calretinin (Condé et al. 1994). In our lab sections incubated in preimmune serum, normal rabbit serum, or PBS in place of the primary antiserum exhibited no immunostaining. The SOM antibody (1:4000, #20067, ImmunoStar Inc., Hudson, WI) was raised against synthetic somatostatin coupled to keyhole limpet hemocyanin with a carbodiimide linker. Somatostatin immunolabeling was completely abolished by preadsorption with somatostatin, somatostatin 25, and somatostatin 28, but preadsorption with numerous peptides, including NPY and VIP, resulted in no reduction of staining (manufacturer information). In our lab preadsorption of diluted antibody with SOM-14 (Sigma Chemical Co.) at a concentration of 100 μg/ml abolished all tissue staining, but preabsorption with NPY or VIP (Sigma Chemical Co.) resulted in no reduction of staining. The VIP antibody (1:2000, #20077, ImmunoStar Inc.) was raised against porcine VIP conjugated to bovine thyroglobulin with carbodiimide. VIP immunolabeling was completely abolished by preadsorption with VIP but preadsorption with numerous peptides, including somatostatin and NPY, resulted in no reduction of staining (manufacturer information). In our lab preadsorption of diluted antibody with VIP (Sigma Chemical Co.) at a concentration of 100 μg/ml abolished all tissue staining, but preabsorption with NPY or SOM-14 (Sigma Chemical Co.) resulted in no reduction of staining. The NPY antibody (1:10,000, #T-4070, Peninsula Laboratories International Inc., San Carlos, CA) was raised against amino acids 30–65 of rat NPY. In radioimmunoassays it reacts with human, rat, and porcine NPY but not unrelated peptides (manufacturer information). In our lab preadsorption of diluted antibody with NPY (Sigma Chemical Co.) at a concentration of 100 μg/ml abolished all tissue staining, but preadsorption with SOM-14 or VIP (Sigma Chemical Co.) resulted in no reduction of staining.
3. RESULTS
3.1. Single-labeling immunoperoxidase technique for SMI-32 localization
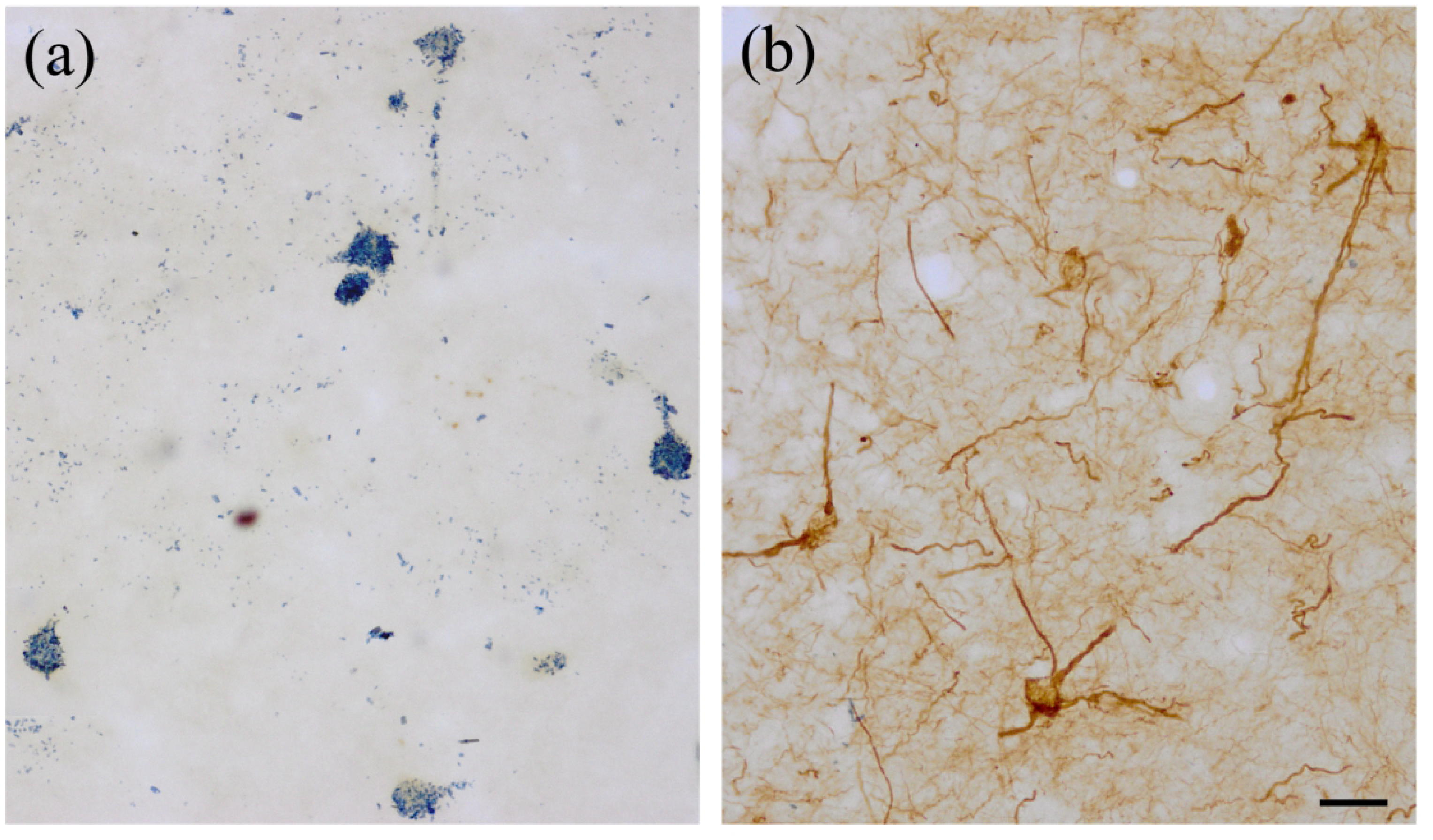
SMI-32-ir was widespread throughout the forebrain, but only particular neuronal subpopulations in each region appeared to be SMI-32+. Most amygdalar nuclei, including all of those in the BNC, contained a subpopulation of SMI-32+ NPNs (Figs. 1–4). Figures 1 and 2 show SMI-32-ir at three different anteroposterior levels of the amygdala (anterior [Fig. 1 (a)], middle [Fig. 1(b)], and posterior [Fig. 2(a)]) and allow a comparison with SMI-32-ir seen in a representative cortical area (somatosensory cortex; Fig. 2(b)). The basolateral and lateral nuclei contained many SMI-32+ NPNs, but relatively few were seen in the basomedial nucleus, except at caudal levels of the amygdala. These immunostained amygdalar NPNs exhibited moderate to intense SMI-32-ir in their somata and throughout the entire extent of their dendritic arborizations. In some SMI-32+ NPNs a stained axon was observed; it arose from a conical axon hillock and quickly assumed a uniform diameter of 0.5–0.8 μm. Axons could only be followed for 30–50 μm. In contrast to the amygdala, the most conspicuous subpopulation of SMI-32+ neurons in the neocortex were PNs (Fig. 2 (b)). Surprisingly, in sections stained for SMI-32 with antibodies whose diluent did not contain Triton X-100, many amygdalar PNs in the anterior and posterior subdivisions of the basolateral nucleus (BLa and BLp), but not in other amygdalar nuclei including other nuclei of the BNC, exhibited moderate SMI-32-ir that was restricted to their somata and proximal dendrites (Fig. 5). Some processes of these cells were thin and could be axons, but this identification was not definitive. The staining of NPNs in the BNC was not affected by the elimination of Triton X-100 in the diluent.
Fig. 1.
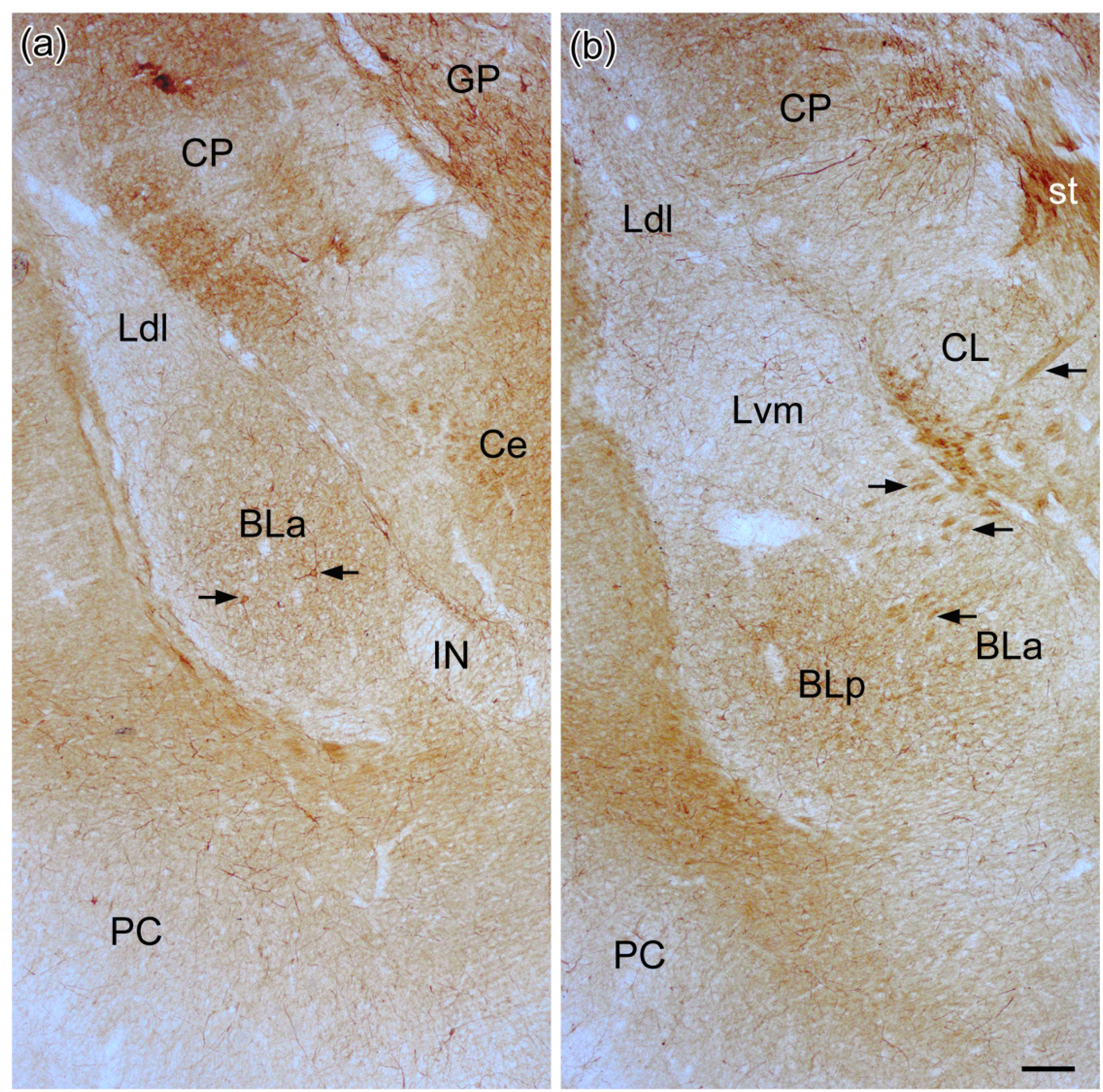
Photomicrographs showing SMI-32-ir in the BNC at the bregma −2.3 (a) and bregma −2.8 (b) levels. See Paxinos and Watson (2007) for depictions of the exact borders of these nuclei at these levels. See Figs. 3(a) and (b) for higher power photomicrographs of neurons in these sections or adjacent sections in the same brain. Arrows in (a) point to SMI-32+ neurons depicted in Fig. 3(a). Arrows in (b) point to bundles of SMI-32+ axons running between BL and the stria terminalis (st). Also note robust staining in the stria itself. Scale bar = 200 μm for (a) and (b). Other abbreviations: BLa, anterior subdivision of the basolateral nucleus; BLp, posterior subdivision of the basolateral nucleus; Ce, central nucleus; CL, lateral subdivision of the central nucleus; CP, caudatoputamen; GP, globus pallidus; IN, intercalated nucleus; Ldl, dorsolateral subdivision of the lateral nucleus; Lvm, ventromedial subdivision of the lateral nucleus; PC, piriform cortex.
Fig. 4.

Photomicrographs of SMI-32+ neurons in the lateral nucleus in a sagittal section. Scale bar = 50 μm.
Fig. 2.
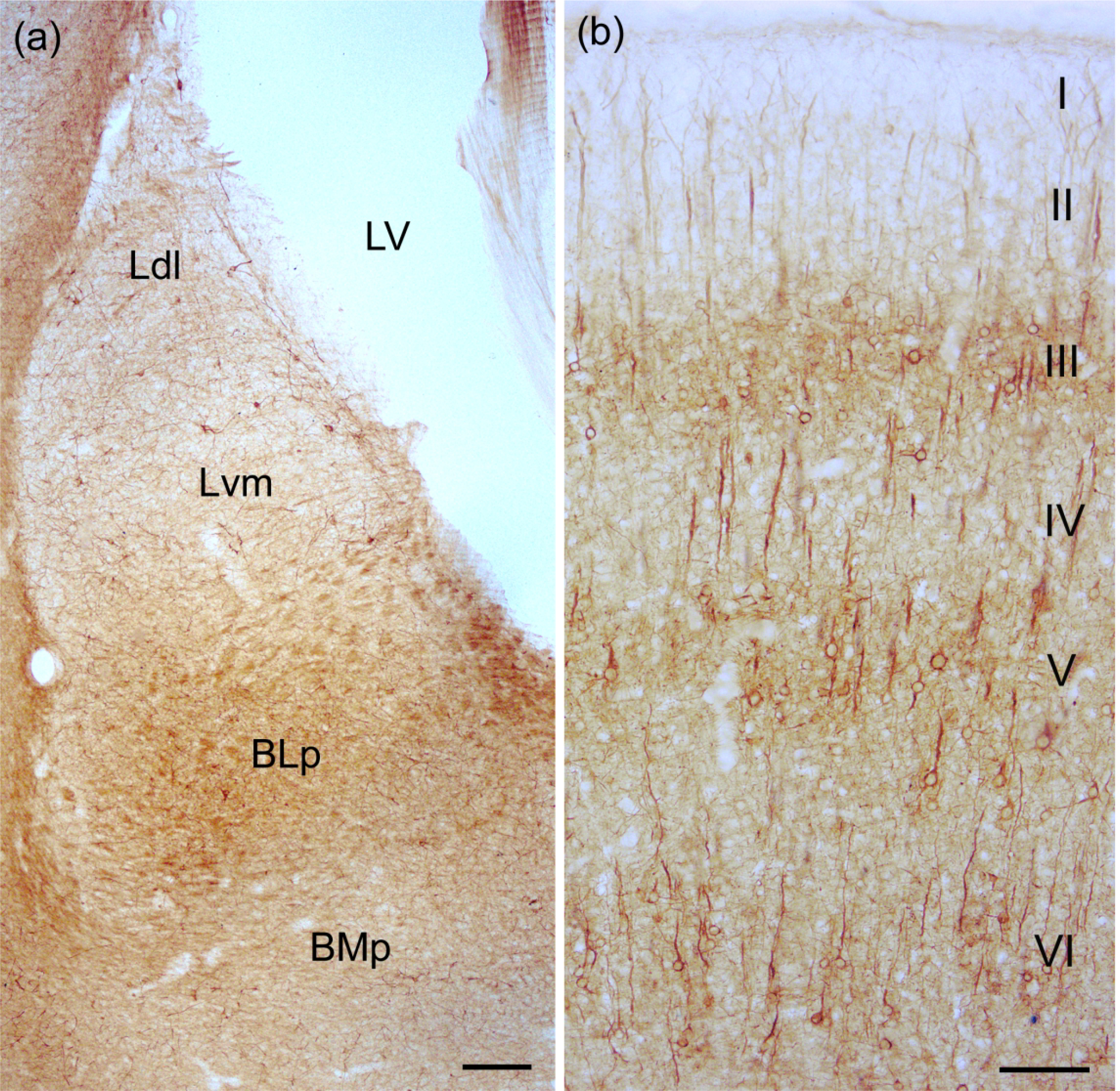
(a) Photomicrograph showing SMI-32-ir in the BNC at the bregma −3.8 level. See Paxinos and Watson (2007) for depictions of the exact borders of these nuclei at this level. See Figs. 3(c) and (d) for higher power photomicrographs of neurons in adjacent sections in the same brain. Scale bar = 200 μm. Additional abbreviations: BMp, posterior subdivision of the basomedial nucleus; LV, lateral ventricle. (b) SMI-32-ir in the somatosensory cortex. Note staining of PNs in three discrete layers (III, V, and VI). Scale bar = 100 μm.
Fig. 5.
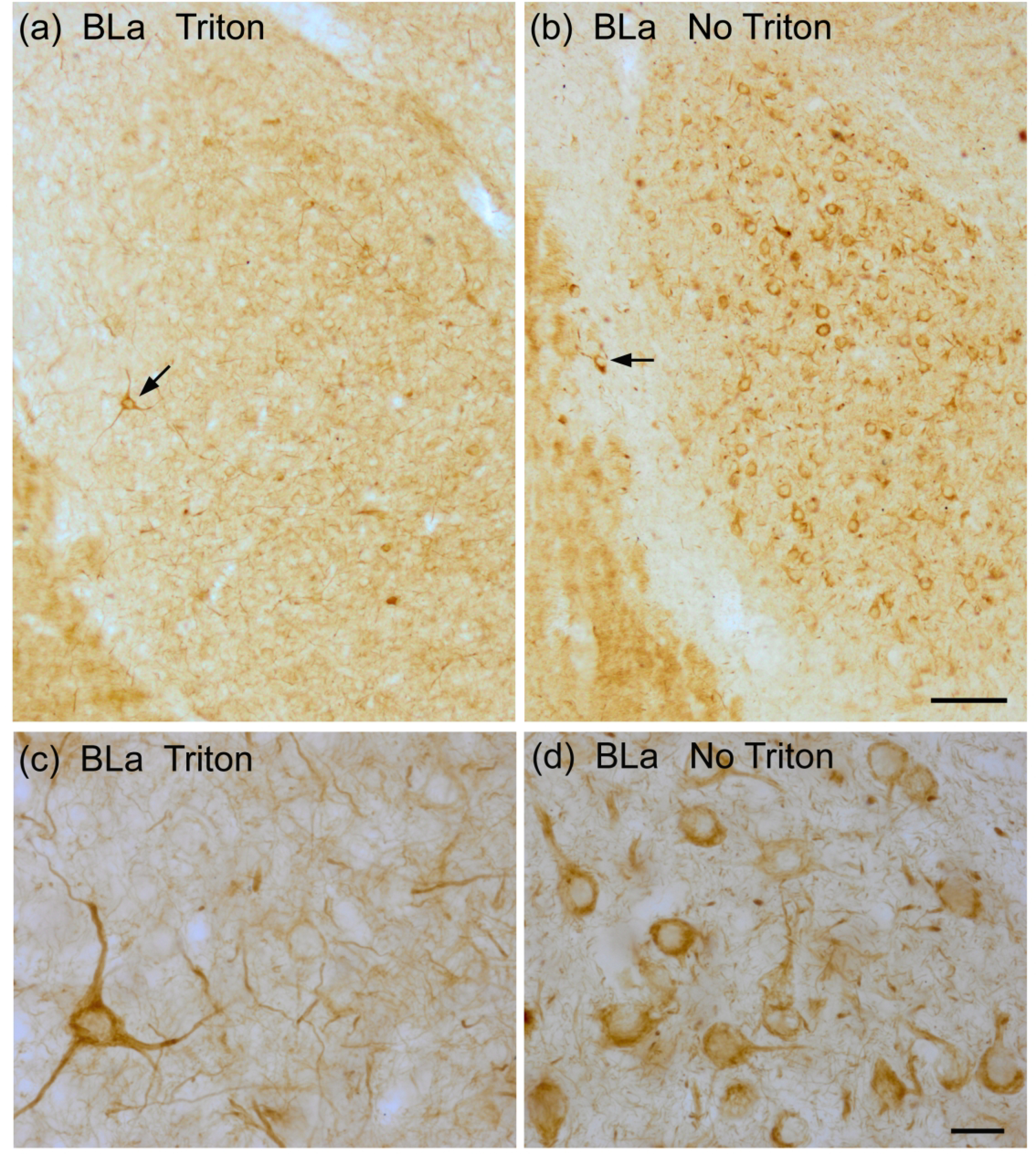
(a) and (b) Low power photomicrographs showing the differences in SMI-32-ir in the basolateral nucleus in sections incubated in antibodies whose diluents contained 0.3% Triton X-100 (a) versus no Triton X-100 (b). Both sections are from the same brain (bregma −1.8). Note that with no Triton X-100 it appears that virtually all PNs are stained whereas with 0.3% Triton X-100 PNs are barely visable. NPNs are stained with both levels of Triton X-100, but are much less noticeable in the no Triton X-100 preparation. Arrows indicate NPNs in the BLa (a) and the external capsule laterally-adjacent to BLa (b). Scale bar = 100 μm. (c) and (d) Higher power photomicrographs taken from the sections shown in (a) and (b). Scale bar in d = 20 μm. (c) is at the same magnification.
Nonpyramidal SMI-32+ neurons typically were multipolar neurons with 2–4 primary dendrites (Figs. 3, 4). Somatic size of SMI-32+ NPNs was fairly uniform in the basolateral nucleus (ca. 15–20 μm long and 13–17 μm wide) (Figs. 3(a), (b)). Somatic size of SMI-32+ NPNs in the lateral nucleus was more variable, with more smaller somata than in the basolateral nucleus; most somata were about 13–17 μm long and 10–13 μm wide (Figs. 3 (c), 4). Somatic size was quantitated by summing the lengths and widths of SMI-32+ somata using a 40X objective and a calibrated ocular reticule. The size of SMI-32+ NPN somata was quantitated in two subdivisions of the BNC in the same brain, the BLa and the Lvm (i.e., one representative subdivision in each of the two main BNC nuclei). The sum of the lengths and widths of SMI-32+ somata in the BLa was 31.30 ± 3.05 μm (mean ± SD; n = 25), while in the Lvm it was 27.65 ± 4.09 (mean ± SD; n = 25). These differences were statistically significant (unpaired T-test: P = 0.0008).
Fig. 3.
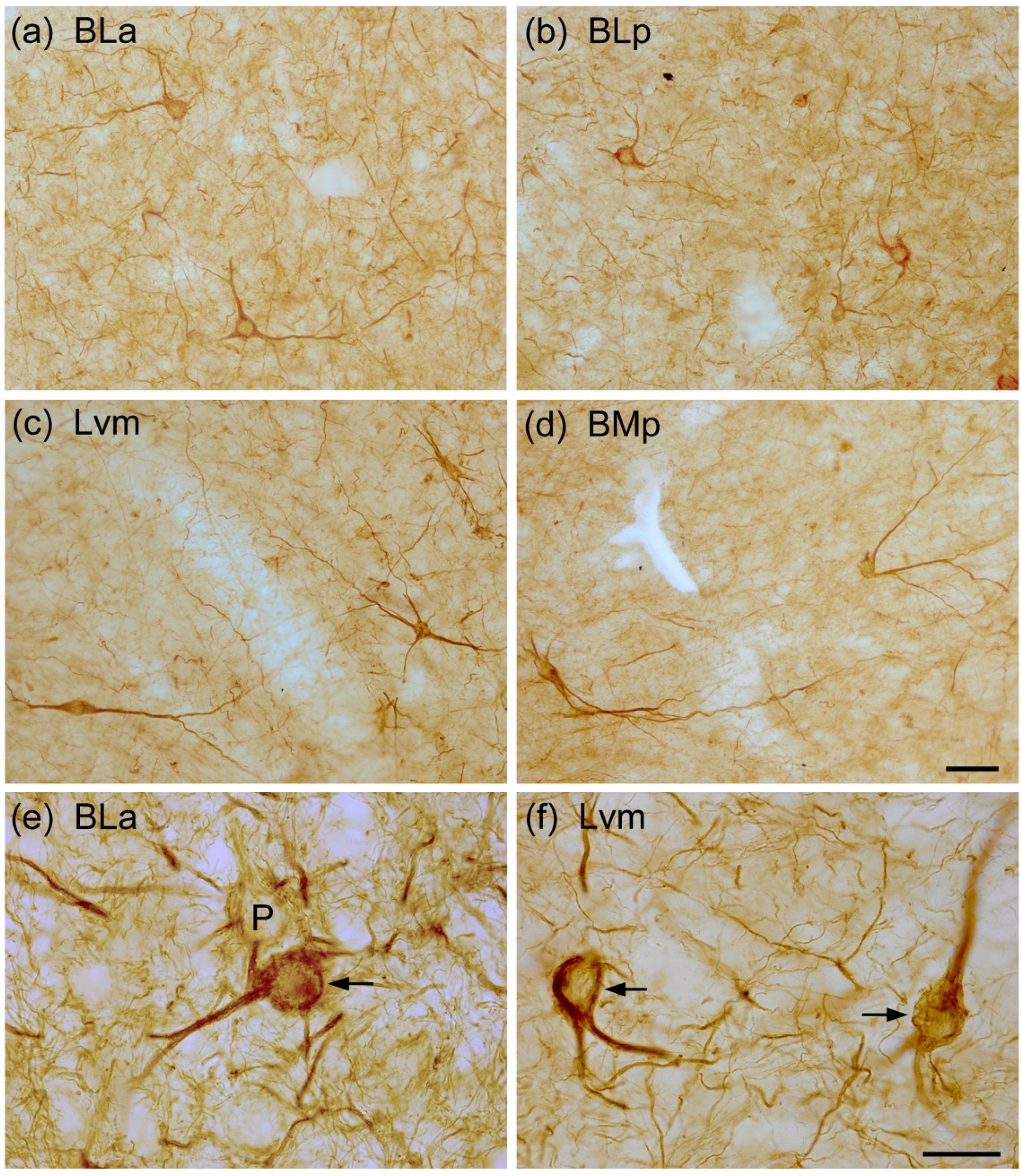
Photomicrographs of SMI-32+ NPNs in the BNC: (a) BLa (from the same section shown in Fig. 1(a)); (b) BLp (from a section adjacent to that shown in Fig. 1(b); (c) Lvm (from a section adjacent to that shown in Fig. 2(a)); (d) BMp (from a section adjacent to that shown in Fig. 2(a)). (e) and (f) Higher power photomicrographs showing SMI-32-ir in NPNs in the BLa (e) and Lvm (f). Arrows in (e) and (f) indicate SMI-32-ir NPNs. P in (e) indicates a lightly-labeled presumptive pyramidal neuron. Note many SMI-32+ processes in the neuropil. Scale bar = 50 μm in (d); (a)-(c) are at the same magnification. Scale bar = 20 μm in (f); (e) is at the same magnification.
The neuropil in the BNC contained many SMI-32+ processes (Fig. 3(e), (f)). Some processes were thicker and fairly rectilinear, suggesting that they were thin dendrites of SMI-32+ NPNs. Other processes were thinner (0.5–0.8 μm) and more tortuous, suggesting that they were axons. In addition, bundles of axons were observed extending between the BLa and BLp and the intra-amygdalar portion of the stria terminalis exiting the amygdala (Fig. 1(b)).
3.2. SMI-32 colocalization with NPN markers
Several distinct NPN subpopulations in the BNC can be identified using expression of calcium-binding proteins and neuropeptides as markers (McDonald, 2020). The present investigation performed dual immunolabeling of SMI-32 with four marker neurochemicals (PV, SOM, VIP, and NPY) to determine if SMI-32+ neurons belonged to one particular NPN subpopulation, using DAB to generate a diffuse brown reaction product for SMI-32, and BDHC to generate a blue particulate reaction product for different markers. These two reaction products were easily discriminated in all of these studies (Figs. 6–9).
Fig. 6.
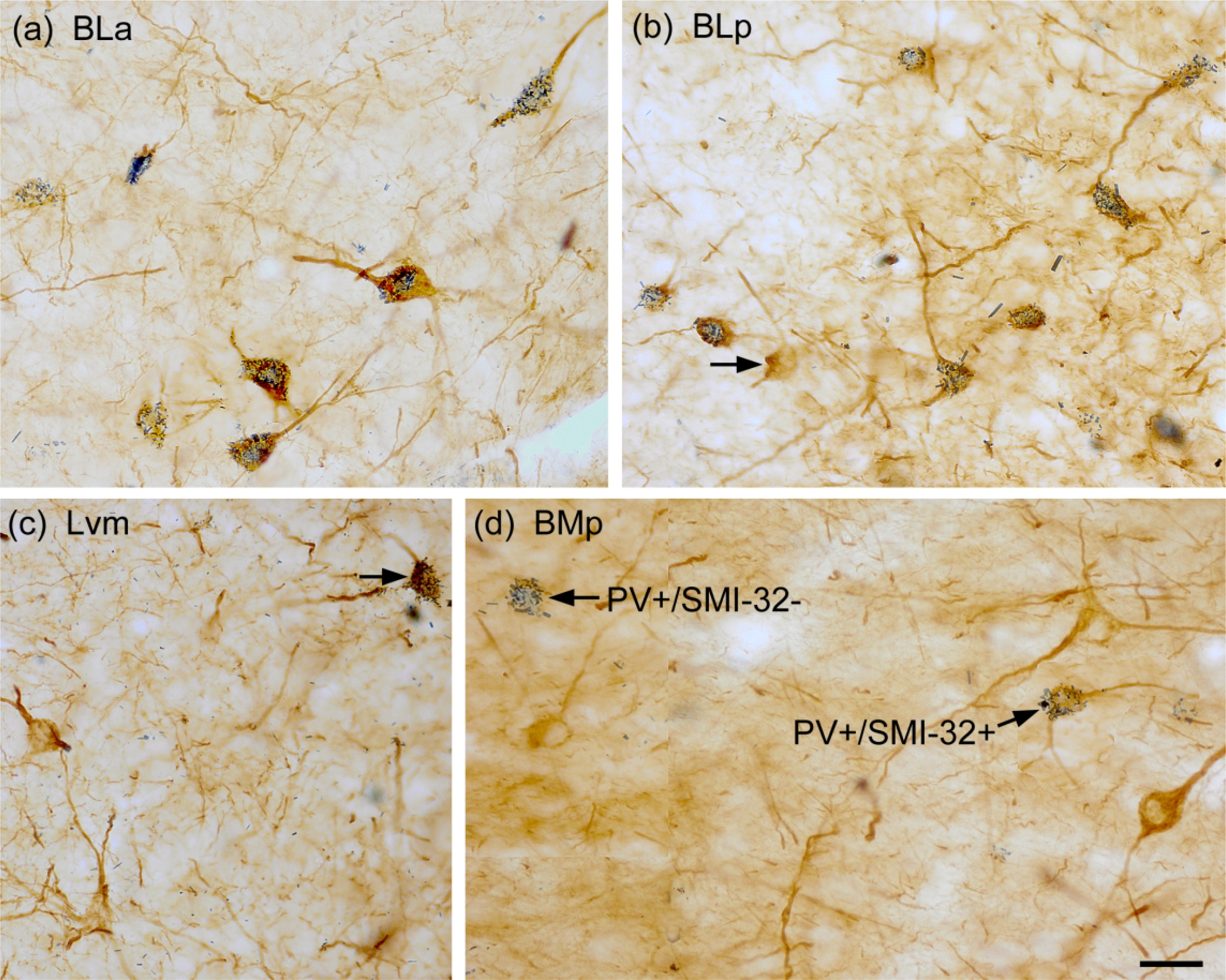
Photomicrographs showing dual-localization of SMI-32 (diffuse brown reaction product) and PV (blue particulate reaction product) in the BNC. (a) Extensive colocalization of SMI-32 (brown) and PV (blue) in NPNs in the BLa. (b) Extensive colocalization of SMI-32 (brown) and PV (blue) in NPNs in the BLp. Arrow points to one single-labeled SMI-32+ IN. (c) Two single-labeled SMI-32+ NPNs (brown) and one double-labeled SMI-32+/PV+ NPN (arrow) in the Lvm. (d) Three single-labeled SMI-32+ NPNs (brown), one single-labeled PV+/SMI-32− NPN, and one double-labeled SMI-32+/PV+ NPN in the BMp. Scale bar = 25 μm for (a)-(d).
Fig. 9.
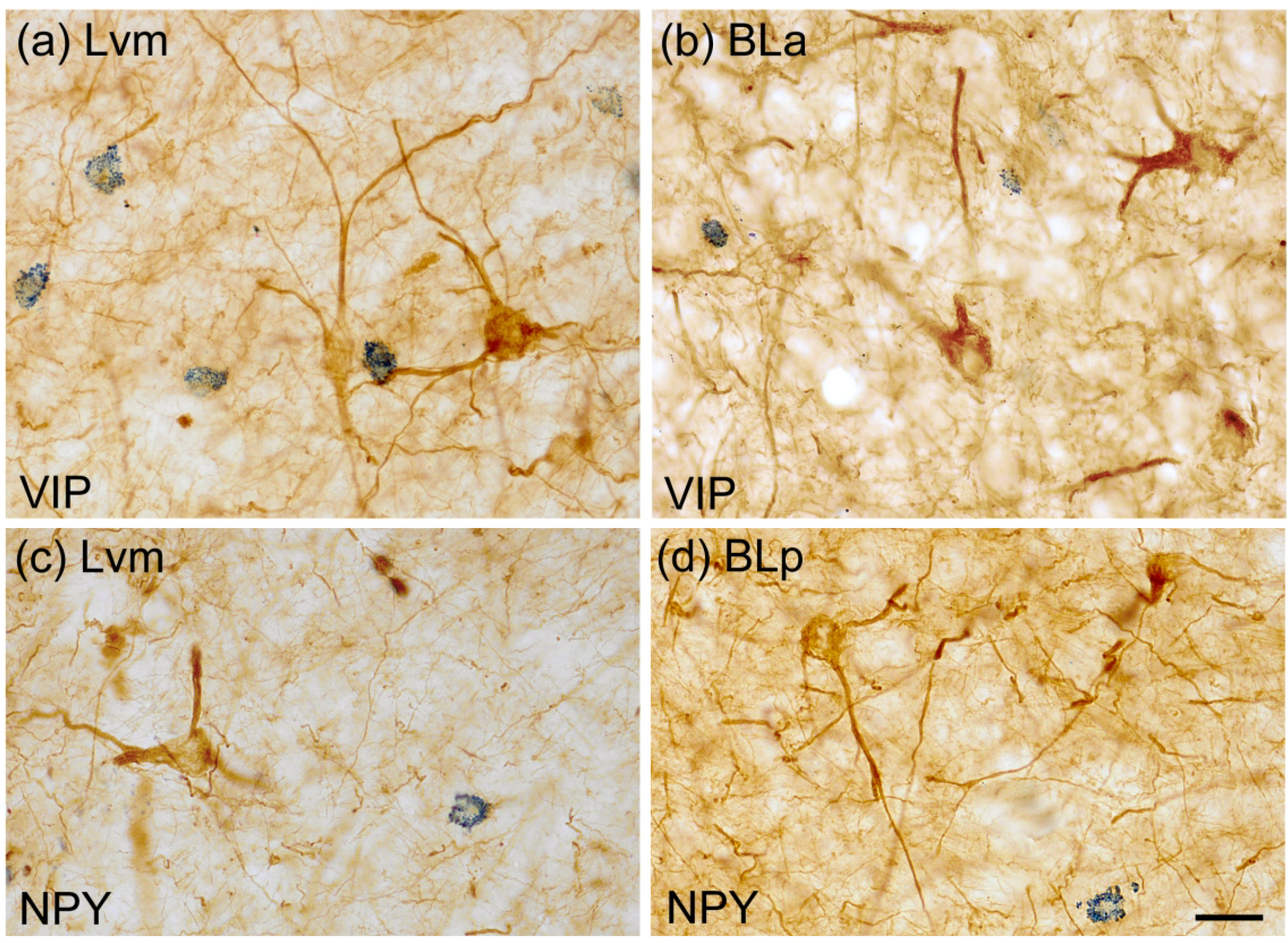
Photomicrographs of NPNs in the BNC in sections dual-labeled for SMI-32 (diffuse brown reaction product) and either VIP or NPY (blue particulate reaction product). (a) and (b) SMI-32 and VIP single-labeled NPNs in the Lvm (a) and BLa (b). (c) and (d) SMI-32 and NPY single-labeled NPNs in the Lvm (a) and BLp (b). No VIP+ or NPY+ NPNs in the BNC were SMI-32+. Scale bar = 20 μm in (a) – (d).
The majority of SMI-32+ NPNs in all BNC nuclei were also PV+ (Fig. 6). In the basolateral nucleus (BLa and BLp combined) 83.6% of SMI-32+ neurons were PV+, and these neurons constituted 90.7% of PV+ neurons (Table 2). In the lateral nucleus only 58.0% of SMI-32+ neurons were PV+, and these neurons constituted 88.1% of PV+ neurons (Table 2). Control sections processed for the DAB/BDHC dual immunohistochemical technique, but with the omission of either the SMI-32 or PV antibodies resulted in no staining for the omitted antibody (Fig. 7). Identical results were obtained in controls for the other SMI-32/marker studies.
Fig. 7.
Controls in a PV/SMI-32 preparation. (a) Section processed for dual-labeling but with the SMI-32 antibody omitted. Note only blue particulate BDHC labeling for PV is present (BLp). (b) Section processed for dual-labeling but with the PV antibody omitted. Note only diffuse brown DAB labeling for SMI-32 is present (Lvm). Scale bar = 25 μm for (a) and (b).
A significant number of SMI-32+ NPNs in the lateral nucleus were SOM+ (29.7%), and these neurons constituted 17.3% of SOM+ neurons (Fig. 8; Table 3). Only 2% of SOM+ neurons in the basolateral nuclei, and no VIP+ or NPY+ NPNs in the BNC, were SMI-32+ (Figs. 8 and 9; Table 4).
Fig. 8.
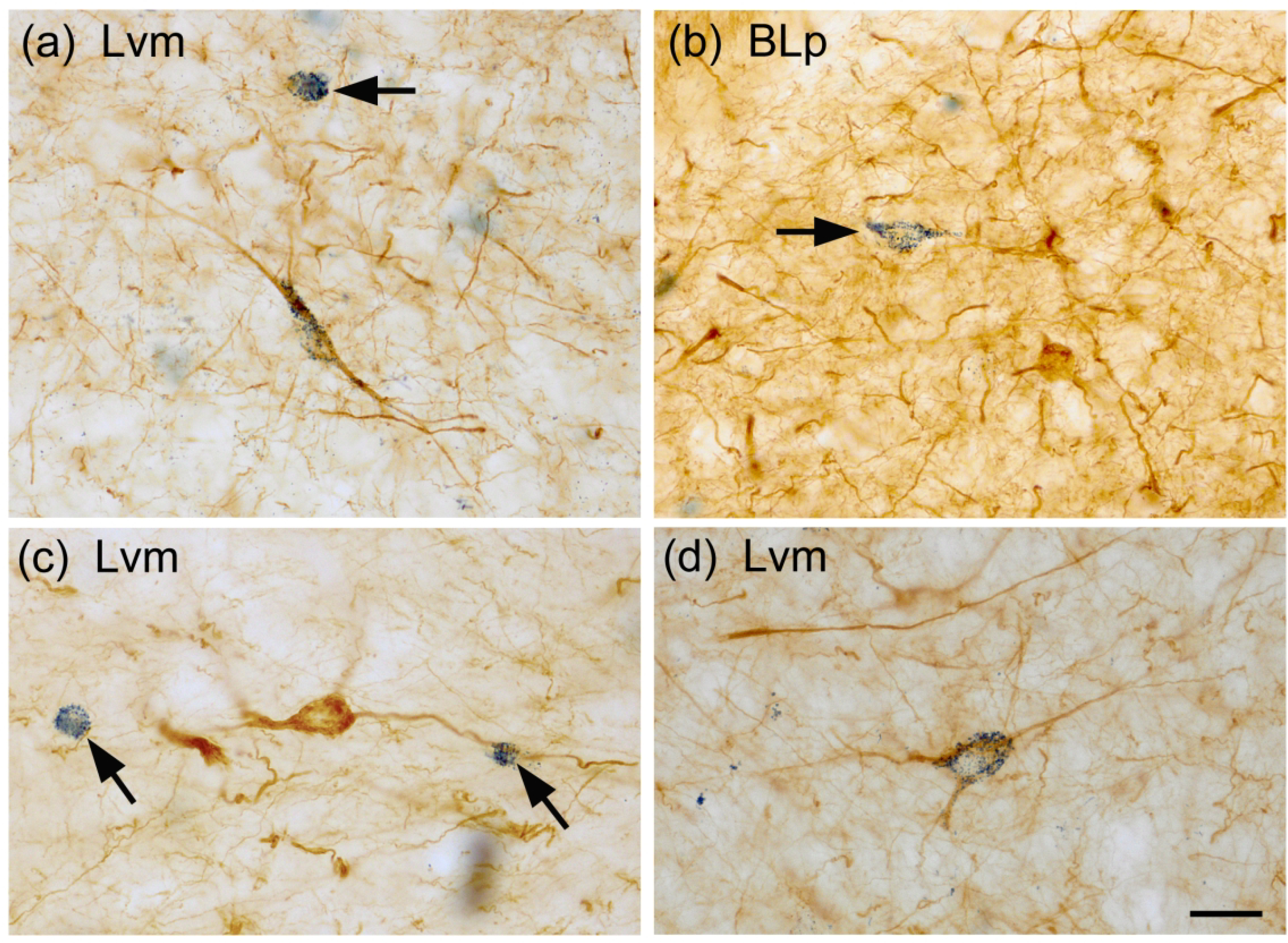
Photomicrographs of NPNs in the BNC in sections dual-labeled for SMI-32 (diffuse brown reaction product) and SOM (blue particulate reaction product). (a) A SMI-32+ single-labeled NPN (top, arrow) and a SMI-32+/SOM+ double-labeled NPN (center) in the Lvm. (b) A SOM+ single-labeled NPN (arrow) and several SMI-32+ single-labeled NPNs in the BLp. (c) Two small SOM+ single-labeled NPNs (arrows) and one larger SMI-32+ single-labeled NPN in the Lvm. (d) A SMI-32+/SOM+ double-labeled NPN in the Lvm. Scale bar = 20 μm for (a)-(d).
Table 3.
Cell counts of colocalization of SMI-32 with SOM in the lateral (LAT) nucleus.
| Nucleus | Brain | SMI-32 Single-labeled Neurons | SOM Single-labeled Neurons | SMI-32/SOM Double-labeled Neurons | % of SMI-32 Neurons Double-labeled | % of SOM Neurons Double-labeled | Total Neurons Counted | Number of Amygdalae Analyzed |
|---|---|---|---|---|---|---|---|---|
| LAT | S13 | 35 | 74 | 17 | 32.3% (17/52) | 18.8% (17/91) | 126 | 26 |
| S17 | 43 | 84 | 16 | 27.1% (16/59) | 16.0% (16/100) | 140 | 21 | |
| Total | 78 | 158 | 33 | 29.7% (33/111) | 17.3% (33/191) | 166 | 47 |
3.3. Retrograde tract tracing combined with SMI-32 immunohistochemistry
The antibody diluent for all studies combining retrograde tract tracing with SMI-32 immunohistochemistry contained 0.2% Triton X-100 rather than the 0.3% used in the single-labeleing studies; this resulted in strong staining of NPNs in all BNC nuclei and additional light staining of PNs in the BLa and BLp. Two animals received bilateral injections of WGA-HRP into the ventral striatum at two mediolateral and two anteroposterior sites for a total of 4 injections on each side. These injections filled the nucleus accumbens and fundus striati from bregma +2.7 to bregma +0.5 levels (Fig. 10 (a)), and produced numerous retrogradely-labeled neurons in the BLa and BLp from the rostral pole of BLa (bregma −1.6) to the middle of the amygdala (bregma −3.3). There were also some retrogradely-labeled neurons in the lateral nucleus and the ventral subdivision of the basolateral nucleus (BLv) at these levels. The black granular TMB reaction product was easily distinguished from the diffuse brown reaction product of SMI-32 immunohistochemistry. No SMI-32+ NPNs were retrogradely-labeled (Fig. 10 (b)). The TMB reaction product was located in the somata and proximal dendrites of presumptive PNs that were either SMI-32-negative or had very light SMI-32-ir. The somata of the retrogradely-labeled neurons in the BLa and BLp were roughly 15–18 μm in diameter; some of the SMI-32+ NPNs were larger than the retrogradely-labeled neurons, and some were smaller (Fig. 10 (b)).
Fig. 10.
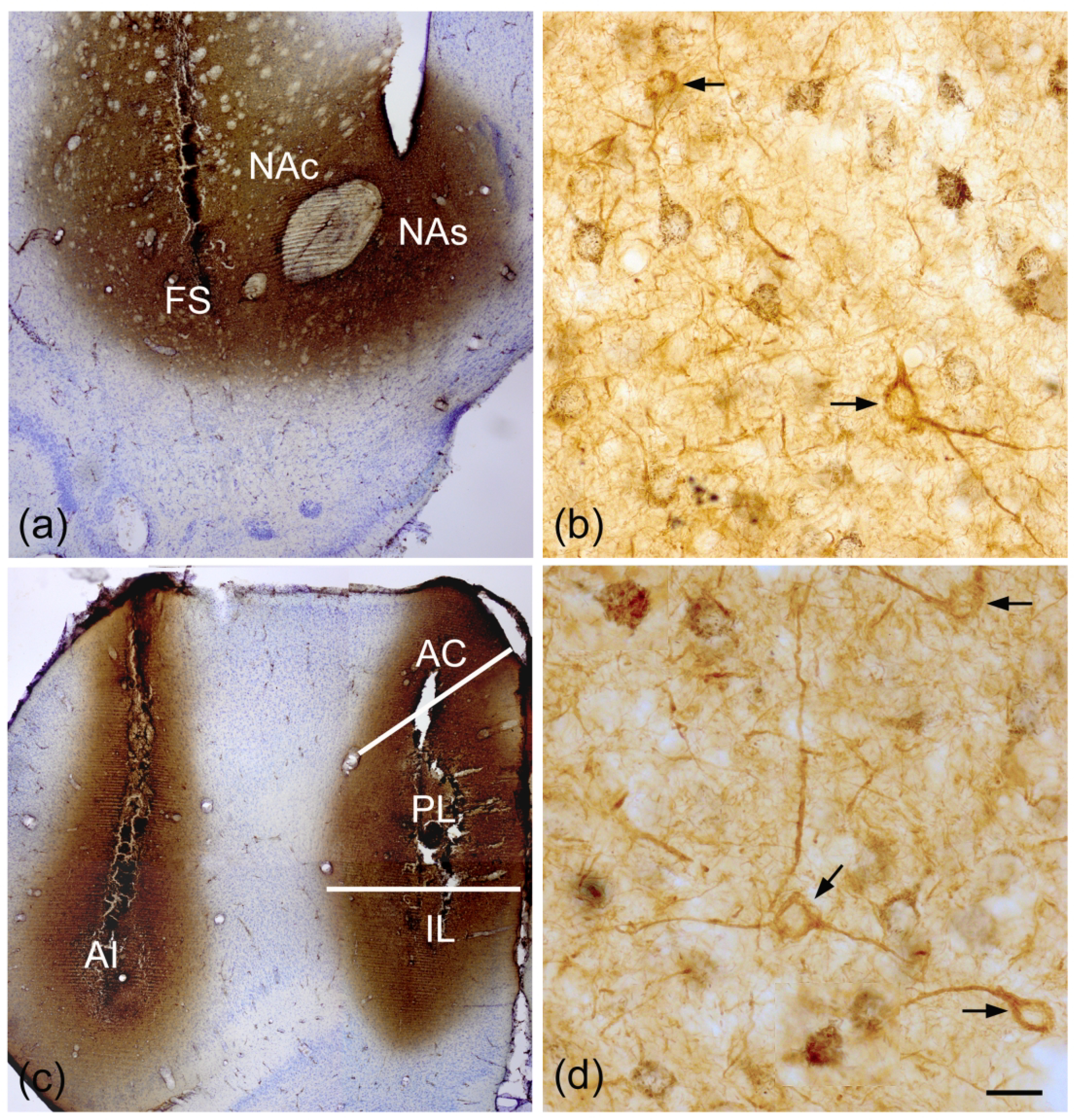
(a) Injections of WGA-HRP into the ventral striatum, including the nucleus accumbens shell and core (NAs, NAc), and the laterally-adjacent fundus striati (FS). (b) Presumptive PNs in the BLa retrogradely-labeled by ventral striatal injections contain black granular TMB reaction product. Two SMI-32+ NPNs (arrows) are not retrogradely-labeled. (c) Injections of WGA-HRP into the agranular insular area (AI) of the lateral prefrontal cortex, and the prelimbic (PL), infralimbic (IL), and anterior cingulate areas (AC) of the medial prefrontal cortex. (d) Presumptive PNs in the BLa retrogradely-labeled by prefrontal injections contain black granular TMB reaction product. Three SMI-32+ NPNs (arrows) are not retrogradely-labeled. Scale bar in (d) = 20 μm. (b) is at the same magnification.
Two animals received bilateral injections of WGA-HRP into the medial and lateral prefrontal cortex (PFC) at two anteroposterior sites for a total of 4 injections on each side. These injections filled the medial PFC (prelimbic, infralimbic and anterior cingulate cortices) from bregma +4.0 to bregma +2.7 levels, and the lateral PFC (rostral agranular insular cortices) from bregma +4.0 to bregma +1.7 levels (Fig. 10 (c)), and produced numerous retrogradely-labeled neurons in the BLa and BLp from the rostral pole of BLa (bregma −1.6) to the middle of the amygdala (bregma −3.3). There were also some retrogradely-labeled neurons in the lateral nucleus at these levels. As in the brains with ventral striatal injections, SMI-32-ir was intense in NPNs and very light in PNs in the BLa and BLp. No SMI-32+ NPNs were retrogradely-labeled (Fig. 10 (d)). The TMB reaction product was located in the somata and proximal dendrites of presumptive PNs that were either SMI-32-negative or had very light SMI-32-ir. The somata of the retrogradely-labeled neurons in the BLa and BLp were 15–18 μm in diameter; some of the SMI-32+ NPNs were larger than the retrogradely-labeled neurons, and some were smaller.
Two animals received bilateral injections of WGA-HRP into the temporal cortical region at two anteroposterior sites for a total of 2 injections on each side. These injections filled the perirhinal cortex, dorsolateral entorhinal cortex, and adjacent temporal cortex from bregma −6.0 to −6.7 (Fig. 11 (a)). Some injections also spread medially to involve the underlying hippocampus. A low density of retrogradely-labeled neurons were restricted to the lateral nucleus from bregma levels −2.3 to −3.3. SMI-32-ir was intense in NPNs, but none were retrogradely-labeled (Fig. 11 (b)). The TMB reaction product was located in the somata and proximal dendrites of presumptive PNs that were SMI-32-negative. The somata of most retrogradely-labeled neurons in the lateral nucleus were about 13 μm in diameter, and most SMI-32+ NPNs were about the same size.
Fig. 11.
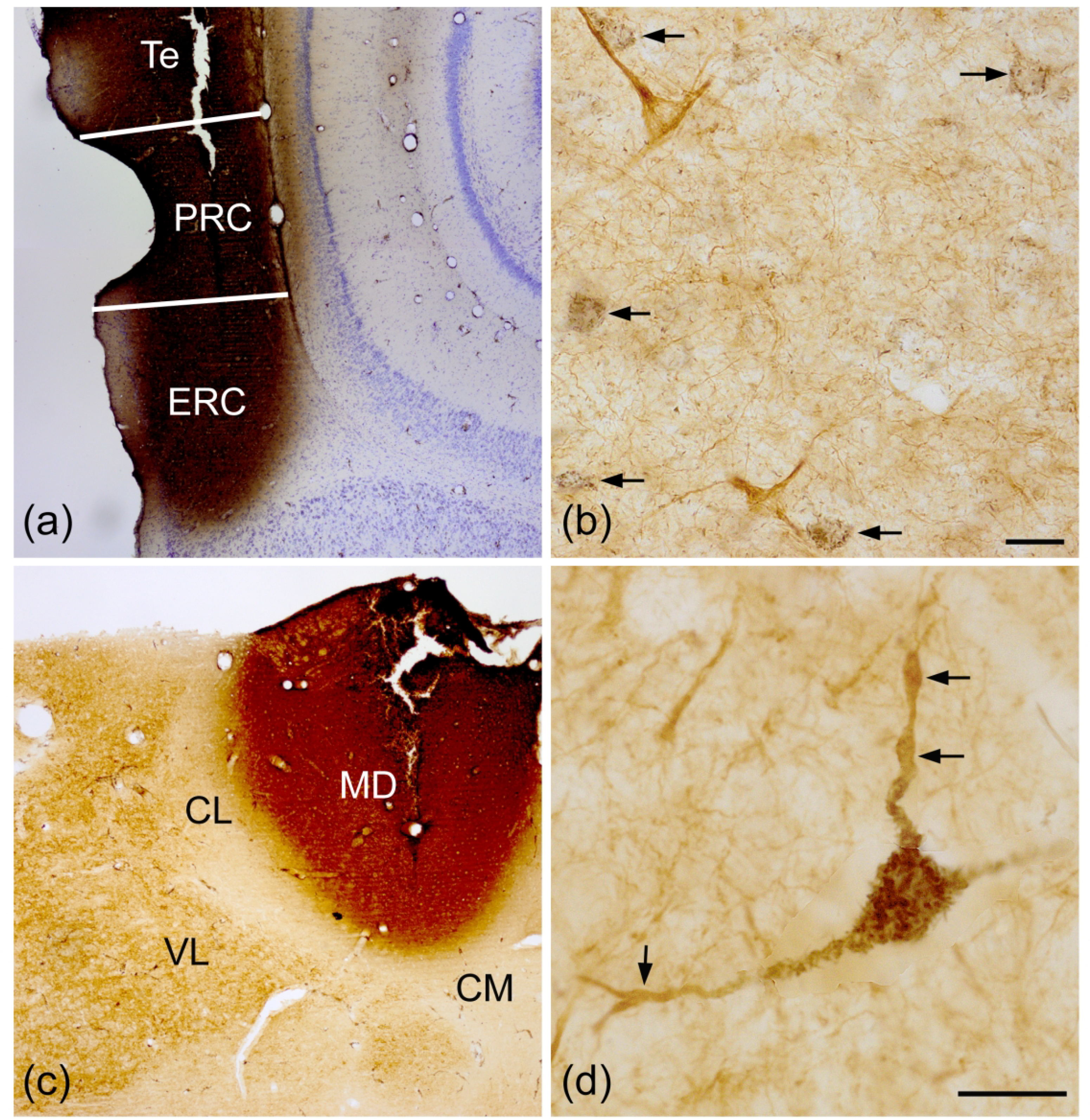
(a) Injections of WGA-HRP into the parahippocampal/temporal cortices, including the entorhinal cortex (ERC), perirhinal cortex (PRC), and temporal cortex (Te). (b) Presumptive PNs in the Lvm retrogradely-labeled by parahippocampal/temporal injections contain black granular TMB reaction product (arrows). Two SMI-32+ NPNs (brown) in this field are not retrogradely-labeled. (c) Injection of WGA-HRP into the mediodorsal thalamic nucleus (MD). (d) A large SMI-32+ NPN in the BLp retrogradely-labeled by injections into the MD contains black granular TMB reaction product in its soma and proximal dendrites. More distal dendritic segments (arrows) are SMI-32+ but do not contain TMB reaction product. Abbreviations: CL, centrolateral thalamic nucleus; CM, central medial thalamic nucleus; VL, ventrolateral thalamic nucleus. Scale bars = 20 μm in (b) and (d).
Two animals received bilateral injections of WGA-HRP into the MD thalamic nucleus at two anteroposterior sites for a total of 2 injections on each side. These injections filled the MD from rostral (bregma −2.3) to caudal (bregma −3.3) on each side (Fig. 11(c)), and produced numerous large retrogradely-labeled neurons in BLp and BM from the bregma −2.3 level to the caudal pole of the amygdala (bregma −4.5). The somata of these large retrogradely-labeled neurons were 17–25 μm long and 13–17 μm wide, and they typically had 3 primary dendrites. The TMB reaction product was restricted to the somata and proximal dendrites of these neurons. Most of these retrogradely-labeled neurons were SMI-32+, with SMI-32+ distal dendrites extending beyond the retrogradely-labeled proximal dendrites (Fig. 11 (d)). Counts performed in the BLp and BM combined revealed that approximately 70% of MD-projecting neurons in the BNC were SMI-32+ (Brain SMD1: 73.3% [55/75]; Brain SMD2: 65.3% [49/75]).
4. DISCUSSION
This is the first investigation to study the expression of SMI-32+ non-phosphorylated neurofilaments (NNFs) in the BNC. The main findings are: (1) some NPNs in all nuclei of the BNC have high levels of NNFs in their somata, dendrites, and axons; (2) most of these SMI-32+ NPNs are PV+ or SOM+, but not VIP+ or NPY+; (3) all BNC neurons projecting to the MD thalamic nucleus are large NPNs, and most are SMI-32+; (4) all BNC neurons projecting to the medial and lateral prefrontal cortices, dorsolateral entorhinal cortex, perirhinal cortex and ventral striatum are PNs; some of these PNs in the BLa and BLp express very light SMI-32-ir when Triton X-100 levels in the antibody diluent are kept low (0.2%); (5) PNs in the basolateral nucleus, but not in the lateral or basomedial nuclei, have significant levels of SMI-32-ir in their somata and proximal dendrites, but staining in these cells was greatly attenuated when the antibody diluent contained Triton X-100 detergent.
4.1. SMI-32-ir in BNC interneurons
In both the basolateral and lateral nuclei about 90% of PV+ neurons were SMI-32+. In the basolateral nucleus these PV+/SMI-32+ constituted over 80% of all SMI-32+ neurons, but in the lateral nucleus they made up only about 60% of all SMI-32+ neurons. Interestingly, SOM+ neurons constituted about 30% of SMI-32+ neurons in the lateral nucleus, but very few SOM+ neurons were SMI-32+ in the basolateral nucleus. Thus, SOM+/SMI-32+ NPNs appear to “compensate” for the lower percentage of SMI-32+ NPNs in the lateral nucleus that are PV+, so that in both nuclei 80%−90% of SMI-32+ NPNs express PV or SOM. The lower percentage of PV+/SMI-32+ NPNs in the lateral versus the basolateral nucleus (58% versus 84%) may be related to the fact that PV+ NPNs constitute about 40% of GABAergic NPNs in the basolateral nucleus, but only about 20% of GABAergic NPNs in the lateral nucleus (Mascagni McDonald, 2003). The overwhelming majority of PV+ and SOM+ NPNs in the BNC are interneurons (INs), although a small number have axonal branches that extend beyond the amygdala (Bienvenu et al., 2012; McDonald et al., 2012; McDonald and Zaric, 2015).
All VIP+ neurons and the great majority of NPY+ neurons are INs, but these IN subpopulations did not express NNFs in the BNC. Almost all NPY+ neurons in the lateral nucleus are SOM+, but 20% of SOM+ neurons in the lateral nucleus are not NPY+ (McDonald, 1989). This data, in conjunction with the results of the present study that about 20% of SOM+ neurons in the lateral nucleus are SMI-32+, suggests that all SOM+/NPY-negative neurons are SMI-32+ in this nucleus. Thus, the lateral nucleus appears to contain two separate subpopulations of SOM+ neurons, SOM+/NPY+ neurons and SOM+/SMI-32+ neurons.
About 10–15% of SMI-32+ NPNs in the basolateral and lateral nuclei are not PV+ or SOM+. Although some of these may be false negatives, it is also possible that some may be large CCK+ INs (CCKL neurons of Mascagni and McDonald, 2003), most of which do not coexpress SOM. In addition, some of the marker-negative NPNs in the basolateral nucleus may be MD-projecting neurons.
4.2. Projections of SMI-32+ neurons
Since previous retrograde tract tracing studies had shown that virtually all BNC neurons projecting to the cerebral cortex and striatum are PNs (McDonald, 1987, 1991a, 1991b, 1992, 1996), it was not surprising that all injections of WGA-HRP into the cortex or striatum in the present study retrogradely-labeled neurons that appeared to be PNs. All of these PNs in the lateral and basomedial nuclei were SMI-32-negative. However, with antibody diluents with 0.2% Triton X-100 (rather than the 0.3% Triton X-100 levels used in the single-labeling studies) many retrogradely-labeled PNs in the BLa and BLp had somata and proximal dendrites that exhibited light SMI-32-ir. None of the SMI-32+ NPNs with intense staining of somata and distal (as well as proximal) dendrites were retrogradely labeled.
In contrast, approximately 70% of MD-projecting neurons in the BNC were SMI-32+. Most of these MD-projecting neurons, including those that express SMI-32-ir, were located in the basolateral and basomedial nuclei. Previous studies have shown that amygdalar MD-projecting neurons are part of an array of similar neurons scattered throughout the ventral forebrain in both rat and monkey (Price and Slotnick, 1983; Russchen et al., 1987). These neurons in all regions, including the BNC, are large NPNs. The axon terminals of these BNC NPNs form asymmetrical synapses in the MD (Kuroda and Price, 1991), and express VGLUT2 in both rodents and primates, suggesting that they are glutamatergic (Mátyás et al., 2014; Timbie and Barbas, 2015). However, there is other evidence which suggests that they may not be glutamatergic (Ray et al., 1992; McDonald, 1996).
4.3. Comparisons with the neocortex
The cell types in the BNC are very similar to those of the neocortex (see Introduction). In all mammals PNs are the main neuronal population in the neocortex that expresses NNFs (Vickers and Costa, 1992; Hof et al., 1990, 1996; Hof and Morrison, 1990; Budinger et al., 2000; Kirkcaldie et al., 2002; Boire et al., 2005; Baldauf et al., 2005; Ouda et al., 2012). This is the exact opposite to what is observed in the lateral and basomedial amygdalar nuclei in the present study, where only NPNs express NNFs. PNs were stained in the BLa and BLp in the present study, but significant levels of SMI-32-ir were only seen in sections that were incubated in Triton X-100-free antibody diluents, or diluents with low levels of Triton X-100. However, even in those specimens, SMI-32-ir was restricted to somata and proximal dendrites; the intense dendritic staining seen in neocortical PNs was never observed in BL PNs. However, there is evidence that the axons of BL PNs are SMI-32+ regardless of diluent Triton-X-100 levels. With both diluents there were bundles of SMI-32+ axons extending from the BL to the stria terminalis, as well as many SMI-32+ axons within the stria (see Fig. 1 (b)). These axons were first described by Leonard and Scott (1972) using the Fink-Heimer silver degeneration technique with lesions of BL. The fibers coursed dorsomedially around the central nucleus to enter the stria terminalis, and terminated in the bed nucleus of the stria terminalis. We have labeled these same axons using the Phaseolus vulgaris leucoagglutinin (PHA-L) anterograde tract tracing technique with injections of PHA-L into the BLp (personal observations of AJM). With this technique neurons at the injection site that take up the PHA-L and give rise to the projections are labeled; virtually all appeared to be PNs. Moreover, re-examination of previous experiments performed in this laboratory in which WGA-HRP was injected into the bed nucleus of the stria terminalis (McDonald, 1991a) revealed that all retrogradely-labeled neurons in the BLp appeared to be PNs.
It is difficult to understand why somata of PNs in the BLa and BLp, but not in the lateral or basomedial nucleus, exhibit robust SMI-32-ir when Triton-free antibody diluents were used. We did not notice any differences in SMI-32-ir in other forebrain structures in our amygdalar sections with varying levels of Triton, although a comprehensive detailed analysis of non-amygdalar structures was not performed. These findings suggest that Triton exposure might somehow mask SMI-32+ epitopes in BL PNs. Alternatively, it is possible that the phosphorylation status of NFs in BL somata are in some way different from other regions, and that Triton may have affected either the phosphorylation or solubility of NFs in these neurons (personal communication from Dr. Aidong Yuan, Center for Dementia Research, Nathan Kline Institute, Orangeburg, New York).
The robust expression of SMI-32+ NNFs in INs in the rat BNC is in stark contrast to the results of previous studies of the rodent neocortex, which reported that very few INs are SMI-32+ (Budinger et al., 2000; Boire et al., 2005; Ouda et al., 2012). In the rat neocortex the same neurons stained by the SMI-32 antibody are also stained with antibodies that recognize neurofilament triplet proteins independent of whether they are phosphorylated, and mainly PNs are co-labeled by these antibodies (Kirkcaldie et al., 2002). Dual-labeling studies using phosphorylation-independent neurofilament antibodies demonstrated that there was little or no expression of neurofilament proteins in the somata of NPY+, VIP+, SOM+, or calbindin+ INs in the guinea pig somatosensory cortex (Vickers and Costa, 1992). Notably, antibodies to PV were not used in the latter study. We examined the somatosensory cortex found at amygdalar levels of the rat brains used in the present study and found no VIP+/SMI-32+ or SOM+/SMI-32+ double-labeled neurons, and just a few NPY+/SMI-32+ neurons. However, about half of the PV+ neurons were also SMI-32+ (data not shown). Unlike the PV+/SMI-32+ double-labeled neurons in the BNC, however, only the somata and proximal dendrites were SMI-32+, and the staining intensity of most of these INs was much less than that seen in surrounding PV-negative PNs. Thus, the relative numbers of SMI-32+ INs, as well as their dendritic content of NNFs, are major differences between the BNC and the neocortex.
4.4. Functional and Clinical Implications
NFs constitute a major component of the neuronal cytoskeleton, but are mainly found in axons where they are critical for effective action potential conduction and organelle distribution (Peters et al., 1991; Yuan et al., 2017; Bott and Winkler, 2020). In the present study SMI-32+ NFs were also seen in somata and dendrites where their functional roles are less well understood. In general, NFs in these neuronal compartments are less dense and less phosphorylated than in axons (Bott and Winkler, 2020). Recent studies indicate that NFs located in postsynaptic regions of dendrites are involved in synaptic transmission, including binding to postsynaptic NMDA and dopamine D1 receptors (D1Rs) (Yuan and Nixon, 2016). All major NF proteins are found at synapses in rodent forebrain, and higher levels are found at postsynaptic dendritic regions compared to non-synaptic dendritic regions or presynaptic axon terminals (Jordan et al., 2006; Yuan et al., 2015). Some postsynaptic NF proteins are in the form of short 10-nm filaments but others may be protofilaments. Studies of the NF-M subunit have shown that it is less phosphorylated at synapses compared to general brain phosphorylation levels (Yuan et al., 2015), which suggests that it might be recognized by the SMI-32 antibody. There is evidence that NF-M directly interacts with the third cytoplasmic loop of D1Rs (Kim et al., 2002). NF-M anchors D1R-containing endosomes in postsynaptic regions, which increases their availability for rapid recycling to the postsynaptic plasma membrane following dopamine release. Deletion of NF-M alters D1R-mediated LTP and D1R-mediated locomotor behavior (Yuan et al., 2015). These recent studies of the role of synaptic NFs, including low-phosphorylated dendritic NFs, suggest that the SMI-32+ NNFs in BNC PV+ INs, which receive an especially robust dopaminergic innervation from the ventral tegmental area and substantia nigra (Brinley-Reed and McDonald, 1999; Pinard et al., 2008), may play a role in modulating dopaminergic neurotransmission in these neurons. Dopamine release in the BNC is critical for emotional learning, and alterations in release in this region has been implicated in neuropsychiatric diseases (Reynolds, 1983; Davis, 1993; Borowski and Kokkinidis, 1996; Rosenkranz and Grace, 2002, 2003; Laviolette and Grace, 2006). It is of interest, therefore, that alterations in NF subunit proteins in the brain have been seen in schizophrenia, bipolar disorder, depression, and drug addiction (Ferrer-Alcon et al., 2000, 2003; Reines et al., 2004; Pennington et al., 2008; Yuan and Nixon, 2016).
Alzheimer’s disease (AD) is another important disorder associated with alterations of NFs, especially SMI-32+ NNFs. In the human frontal and temporal association cortices SMI-32+ PNs are the neurons most vulnerable to the formation of degenerative neurofibrillary tangles (NFTs) in AD (Morrison et al., 1987; Hof et al., 1990). The increase in NFTs, but not amyloid plaques, is correlated with the extent of dementia (Wilcock and Esiri, 1982). In contrast PV+, calbindin+, and most calretinin+ (CR+) INs are relatively resistant to degeneration in AD (Hof et al., 1991a, 1991b, 1993; Fonseca and Soriano, 1995; Mitew et al., 2013), and this resistance appears to be associated with the absence of SMI-32+ NNFs in these neuronal subpopulations (Sampson et al., 1997). Thus, less than 1% of PV+ INs in normal human cortex express SMI-32-ir, and less than 1% of PV+ INs develop NFTs in AD. Likewise, the great majority of CR+ INs, which are a distinct PV-negative IN subpopulation in the cortex, are not SMI-32+ and do not exhibit NFTs in AD. However, a distinct subpopulation of cortical CR+ INs in layer I exhibited immunoreactivity for SMI-32, and these CR+ INs did contain NFTs in AD. These data, therefore, suggest that the expression of NNFs in some cortical INs is linked to their vulnerability to the pathological processes underlying AD (Sampson et al., 1997).
There is a very significant loss of neurons and overall nuclear volumes in the BNC in AD (Herzog and Kemper, 1980; Scott et al., 1991, 1992). This atrophy and degeneration is associated with high densities of NFTs and amyloid plaques (Van Hoesen et al., 1986; Brashear et al., 1987; Brady and Mufson, 1990; Kromer Vogt et al., 1990; Price et al., 1991). In fact, NFTs are first seen in the BNC at the earliest stages of AD (Braak et al., 1996), and correlates with the extent of neuropsychiatric symptomatology in these patients (Poulin et al., 2011). Since the neurons of the BNC are very similar to those of the cortex, the studies associating the expression of NNFs with NFTs in cortical neurons suggests the possibility that SMI-32+ neurons in the BNC may also be especially vulnerable to degeneration in AD. These neurons would include PV+ INs in the LAT and basal nucleus (human homolog of the rodent BL), SOM+ INs in the LAT, and MD-projecting NPNs in the basal and basomedial nucleus. It is not clear if BL PNs might be vulnerable since these PNs differ from cortical SMI-32+ PNs in two ways. Firstly, SOM-32-ir in BL PNs is confined to somata and primary dendrites whereas cortical SMI-32+ PNs have very robust distal dendritic SOM-32-ir. Secondly, the NNFs in BL PNs also appear to be different from those in the cortex in that SOM-32-ir is not seen in BL PNs when 0.3% Triton X-100 is added to antibody diluents, but is seen in human and rodent cortical SMI-32+ PNs when 0.3% Triton X-100 is added to antibody diluents (Hof et al., 1990; Kirkcaldie et al., 2002).”
The present study of the rat BNC, in conjunction with previous studies demonstrating the association of SMI-32-ir and NFTs in the cortex, suggests that the SMI-32+ cell types most vulnerable to NFTs in the BNC may be PV+ INs, SOM+ INs, MD-projecting neurons, and perhaps some PNs in the human basal nucleus, providing that similar NPN cell types are SMI-32+ in the human BNC. The BNC is notable for extensive interconnections with frontotemporal association cortices and the hippocampal/parahippocampal region, whereas the projection to MD provides an indirect pathway to the prefrontal cortex (McDonald, 1998, 2020). Disruption in these circuits in AD undoubtedly accounts for the cognitive and affective symptoms seen in AD (Kromer Vogt et al., 1990). Although the SMI-32+ PV INs in the BNC by definition do not give rise to these projections, through their perisomatic innervation of neighboring PNs they are known to be critical for synchronous oscillations of PNs essential for the normal operation of BNC-hippocampal-prefrontal circuits involved in mnemonic functions (Woodruff and Sah, 2007; Ryan et al., 2012; Davis et al., 2017; Ozawa et al., 2020). Thus, although these neurons are INs, their degeneration could indirectly affect BNC-hippocampal-prefrontal circuit activity.
highlights text.
Dual-labeling immunohistochemistry and retrograde tracing were used to characterize neurons in the basolateral amygdala that express SMI-32+ non-phosphorylated neurofilaments (NNFs). Most of the neurons with strong somatic and dendritic expression of NNFs were parvalbumin+ (PV+) interneurons, as well as nonpyramidal neurons that project to the mediodorsal thalamic nucleus. The graphical abstract shows six SMI-32+ neurons (diffuse brown staining); five are also stained for PV (blue particulate staining), and one is PV-negative (arrow). Studies in the cortex suggest that the SMI-32+ neurons identified in this study may be especially vulnerable to degeneration in Alzheimer’s disease.
Acknowledgements:
The authors are grateful for the technical assistance of Patricia Hamilton. This work was supported by NIH grant R01NS19733 (awarded to AJM) and NIH grant R01MH104638 (awarded to AJM and Dr. David D. Mott, University of South Carolina School of Medicine).
Footnotes
CONFLICT OF INTEREST
The authors declare that there are no potential sources of conflict of interest.
DATA ACCESSIBILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Baldauf ZB (2005) SMI-32 parcellates the visual cortical areas of the marmoset. Neurosci Lett. 383:109–114. [DOI] [PubMed] [Google Scholar]
- Beyeler A, Namburi P, Glober GF, Simonnet C, Calhoon GG, Conyers GF, Luck R, Wildes CP, Tye KM (2016) Divergent Routing of Positive and Negative Information from the Amygdala during Memory Retrieval. Neuron 90:348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyeler A, Dabrowska J (2020) Neuronal diversity of the amygdala and the bed nucleus of the stria terminalis. Handb Behav Neurosci. 26:63–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu TC, Busti D, Magill PJ, Ferraguti F, Capogna M (2012) Cell-type-specific recruitment of amygdala interneurons to hippocampal theta rhythm and noxious stimuli in vivo. Neuron 74:1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boire D, Desgent S, Matteau I, Ptito M (2005) Regional analysis of neurofilament protein immunoreactivity in the hamster’s cortex. J Chem Neuroanat. 29:193–208. [DOI] [PubMed] [Google Scholar]
- Borowski TB, Kokkinidis L (1996) Contribution of ventral tegmental area dopamine neurons to expression of conditional fear: effects of electrical stimulation, excitotoxin lesions, and quinpirole infusion on potentiated startle in rats. Behav Neurosci.110:1349–1364. [DOI] [PubMed] [Google Scholar]
- Bott CJ, Winckler B (2020) Intermediate filaments in developing neurons: Beyond structure. Cytoskeleton (Hoboken) 77:110–128. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Yilmazer D, de Vos RA, Jansen EN, Bohl J (1996) Pattern of brain destruction in Parkinson’s and Alzheimer’s diseases. J Neural Transm (Vienna). 103:455–90. [DOI] [PubMed] [Google Scholar]
- Brady DR, Mufson EJ (1990) Amygdaloid pathology in Alzheimer’s disease: Qualitative and Quantitative analysis. Dementia 1:5–17. [Google Scholar]
- Brashear HR, Godec MS, Carlsen J (1988) The distribution of neuritic plaques and acetylcholinesterase staining in the amygdala in Alzheimer’s disease. Neurology 38:1694–1699. [DOI] [PubMed] [Google Scholar]
- Brinley-Reed M, McDonald AJ (1999) Evidence that dopaminergic axons provide a dense innervation of specific neuronal subpopulations in the rat basolateral amygdala. Brain Res. 850: 127–135. [DOI] [PubMed] [Google Scholar]
- Budinger E, Heil P, Scheich H (2000) Functional organization of auditory cortex in the Mongolian gerbil (Meriones unguiculatus). III. Anatomical subdivisions and corticocortical connections Eur J Neurosci 12:2425–2451. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH (1989) Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol. 282:191–205. [DOI] [PubMed] [Google Scholar]
- Capogna M (2014) GABAergic cell type diversity in the basolateral amygdala. Curr Opin Neurobiol. 26:110–116.Carden et al., 1985; [DOI] [PubMed] [Google Scholar]
- Carlsen J, Heimer L (1988) The basolateral amygdaloid complex as a cortical-like structure. Brain Res 441: 377–380. [DOI] [PubMed] [Google Scholar]
- Davis M (1993) Pharmacological analysis of fear-potentiated startle. Braz J Med Biol Res. 26:235–260. [PubMed] [Google Scholar]
- Davis P, Zaki Y, Maguire J, Reijmers LG (2017) Cellular and oscillatory substrates of fear extinction learning. Nat Neurosci. 20:1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Olmos J, Hardy H, Heimer L (1978) The afferent connections of the main and the accessory olfactory bulb formations in the rat: an experimental HRP-study. J Comp Neurol 181:213–244. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A (2009) Amygdala inhibitory circuits and the control of fear memory. Neuron. 62:757–771. [DOI] [PubMed] [Google Scholar]
- Ferrer-Alcón M, García-Sevilla JA, Jaquet PE, La Harpe R, Riederer BM, Walzer C, Guimón J (2000) Regulation of nonphosphorylated and phosphorylated forms of neurofilament proteins in the prefrontal cortex of human opioid addicts. J Neurosci Res. 61:338–349. [DOI] [PubMed] [Google Scholar]
- Ferrer-Alcón M, La Harpe R, Guimón J, García-Sevilla JA (2003) Downregulation of neuronal cdk5/p35 in opioid addicts and opiate-treated rats: relation to neurofilament phosphorylation. Neuropsychopharmacology 28:947–955. [DOI] [PubMed] [Google Scholar]
- Fonseca M, Soriano E (1995) Calretinin-immunoreactive neurons in the normal human temporal cortex and in Alzheimer’s disease. Brain Res. 691:83–91. [DOI] [PubMed] [Google Scholar]
- Gustafson EL, Card JP, Moore RY (1986) Neuropeptide Y localization in the rat amygdaloid complex. J Comp Neurol. 251:349–362. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A (2008) Switching on and off fear by distinct neuronal circuits. Nature 454:600–606. [DOI] [PubMed] [Google Scholar]
- Herzog AG, Kemper TL (1980) Amygdaloid changes in aging and dementia. Arch Neurol 37:625–629. [DOI] [PubMed] [Google Scholar]
- Hof PR, Cox K, Morrison JH (1990) Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease: I. Superior frontal and inferior temporal cortex. J Comp Neurol. 301:44–54. [DOI] [PubMed] [Google Scholar]
- Hof PR, Cox K, Young WG, Celio MR, Rogers J, Morrison JH (1991a) Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J Neuropathol Exp Neurol. 50:451–462. [DOI] [PubMed] [Google Scholar]
- Hof PR, Morrison JH (1991b) Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer’s disease. Exp Neurol. 111:293–301. [DOI] [PubMed] [Google Scholar]
- Hof PR, Nimchinsky EA, Celio MR, Bouras C, Morrison JH (1993) Calretinin-immunoreactive neocortical interneurons are unaffected in Alzheimer’s disease. Neurosci Lett. 152:145–148. [DOI] [PubMed] [Google Scholar]
- Hof PR, Rosenthal RE, Fiskum G (1996) Distribution of neurofilament protein and calcium-binding proteins parvalbumin, calbindin, and calretinin in the canine hippocampus. J Chem Neuroanat. 11:1–12. [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE (1982) Multiple phosphorylation sites in mammalian neurofilament polypeptides. J Biol Chem. 257:10467–10470. [PubMed] [Google Scholar]
- Kim OJ, Ariano MA, Lazzarini RA, Levine MS, Sibley DR (2002) Neurofilament-M interacts with the D1 dopamine receptor to regulate cell surface expression and desensitization. J Neurosci. 22:5920–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Pignatelli M, Xu S, Itohara S, Tonegawa S (2016) Antagonistic negative and positive neurons of the basolateral amygdala. Nat Neurosci. 19:1636–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Zhang X, Muralidhar S, LeBlanc SA, Tonegawa S (2017) Basolateral to Central Amygdala Neural Circuits for Appetitive Behaviors. Neuron 93:1464–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkcaldie MT, Dickson TC, King CE, Grasby D, Riederer BM, Vickers JC (2002) Neurofilament triplet proteins are restricted to a subset of neurons in the rat neocortex. J Chem Neuroanat. 24:163–171. [DOI] [PubMed] [Google Scholar]
- Klosen P, Anderton BH, Brion JP, van den Bosch de Aguilar P (1990). Perikaryal neurofilament phosphorylation in axotomized and 6-OH-dopamine-lesioned CNS neurons. Brain Res. 526:259–269. [DOI] [PubMed] [Google Scholar]
- Krabbe S, Gründemann J, Lüthi A (2018) Amygdala Inhibitory Circuits Regulate Associative Fear Conditioning. Biol Psychiatry. 83:800–809. [DOI] [PubMed] [Google Scholar]
- Kromer Vogt LJ, Hyman BT, Van Hoesen GW, Damasio AR (1990) Pathological alterations in the amygdala in Alzheimer’s disease. Neuroscience 37:377–385. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA (2006) The roles of cannabinoid and dopamine receptor systems in neural emotional learning circuits: implications for schizophrenia and addiction. Cell Mol Life Sci. 63:1597–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Otvos L Jr, Carden MJ, Hollosi M, Dietzschold B, Lazzarini RA (1988) Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc Natl Acad Sci U S A. 85:1998–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CM, Scott JW (1971) Origin and distribution of the amygdalofugal pathways in the rat: an experimental neuroanatomical study. J Comp Neurol. 141:313–329. [DOI] [PubMed] [Google Scholar]
- Lewis SE, Nixon RA (1988) Multiple phosphorylated variants of the high molecular mass subunit of neurofilaments in axons of retinal cell neurons: characterization and evidence for their differential association with stationary and moving neurofilaments. J Cell Biol. 107:2689–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Clem RL (2018) GABAergic interneurons: The orchestra or the conductor in fear learning and memory? Brain Res Bull. 141:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascagni F, McDonald AJ (2003) Immunohistochemical characterization of cholecystokinin containing neurons in the rat basolateral amygdala. Brain Res. 976:171–184. [DOI] [PubMed] [Google Scholar]
- Matus A (1988) Neurofilament protein phosphorylation--where, when and why. Trends Neurosci 11:291–292. [DOI] [PubMed] [Google Scholar]
- Mátyás F, Lee J, Shin HS, Acsády L (2014) The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur J Neurosci. 39:1810–1823. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1985) Morphology of peptide-containing neurons in the rat basolateral amygdaloid nucleus. Brain Res. 338:186–191. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1987) Organization of amygdaloid projections to the mediodorsal thalamus and prefrontal cortex: a fluorescence retrograde transport study in the rat. J. Comp. Neurol. 262:46–58. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1989) Coexistence of somatostatin with neuropeptide Y, but not with cholecystokinin or vasoactive intestinal peptide, in neurons of the rat amygdala. Brain Res. 500:37–45. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1991a) Topographical organization of amygdaloid projections to the caudatoputamen, nucleus accumbens, and related striatal-like areas of the rat brain. Neuroscience 14:15–33. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1991b) Organization of amygdaloid projections to the prefrontal cortex and associated rostral striatum in the rat. Neuroscience 44:1–14. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1992) Projection neurons of the basolateral amygdala: a correlative Golgi and retrograde tract tracing study. Brain Res. Bull. 28:179–185. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1996) Glutamate and aspartate immunoreactive neurons of the rat basolateral amygdala: colocalization of excitatory amino acids and projections to the limbic circuit. J. Comp. Neurol. 365:367–379. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1998) Cortical pathways to the mammalian amygdala. Prog Neurobiol. 55:257–332. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (2020) Functional neuroanatomy of the basolateral amygdala: neurons, neurotransmitters, and circuits. In: Handbook of amygdala structure and function, (Urban JH, Rosenkranz JA, eds), pp. 1–38. San Diego: Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ, Pearson JC (1989) Coexistence of GABA and peptide immunoreactivity in nonpyramidal neurons of the basolateral amygdala. Neurosci. Lett. 100:53–58. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Zaric V (2012) Subpopulations of somatostatin-immunoreactive non-pyramidal neurons in the amygdala and adjacent external capsule project to the basal forebrain: evidence for the existence of GABAergic projection neurons in the cortical nuclei and basolateral nuclear complex. Front Neural Circuits. July 24;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ and Zaric V (2015) GABAergic somatostatin-immunoreactive neurons in the amygdala project to the entorhinal cortex. Neuroscience. 290:227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitew S, Kirkcaldie MT, Dickson TC, Vickers JC (2013) Neurites containing the neurofilament-triplet proteins are selectively vulnerable to cytoskeletal pathology in Alzheimer’s disease and transgenic mouse models. Front Neuroanat. September 26;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Lewis DA, Campbell MJ, Huntley GW, Benson DL, Bouras C (1987) A monoclonal antibody to non-phosphorylated neurofilament protein marks the vulnerable cortical neurons in Alzheimer’s disease. Brain Res. 416:331–336. [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ (2005) Coupled networks of parvalbumin-immunoreactive interneurons in the rat basolateral amygdala. J Neurosci. 25:7366–7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ (2006) Pyramidal cells of the rat basolateral amygdala: Synaptology and innervation by parvalbumin-immunoreactive interneurons. J. Comp. Neurol. 494:635–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ (2007) Postsynaptic targets of somatostatin-containing interneurons in the rat basolateral amygdala. J. Comp.Neurol. 500:513–529. [DOI] [PubMed] [Google Scholar]
- Namburi P, Beyeler A, Yorozu S, Calhoon GG, Halbert SA, Wichmann R, Holden SS, Mertens KL, Anahtar M, Felix-Ortiz AC, Wickersham IR, Gray JM, Tye KM (2015) A circuit mechanism for differentiating positive and negative associations. Nature 520:675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouda L, Druga R, Syka J (2012) Distribution of SMI-32-immunoreactive neurons in the central auditory system of the rat. Brain Struct Funct. 217:19–36. [DOI] [PubMed] [Google Scholar]
- Ozawa M, Davis P, Ni J, Maguire J, Papouin T, Reijmers L (2020) Experience-dependent resonance in amygdalo-cortical circuits supports fear memory retrieval following extinction. Nat Commun. 11:4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (2007) The Rat Brain in Stereotaxic Coordinates. New York: Academic Press. [Google Scholar]
- Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, Wait R, Dunn MJ, Cotter DR (2008) Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol. Psychiatry 13: 1102–1117. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD (1991) The fine structure of the nervous system. New York: Oxford University Press. [Google Scholar]
- Pinard CR, Muller JF, Mascagni F, McDonald AJ (2008) Dopaminergic innervation of interneurons in the rat basolateral amygdala. Neuroscience 157:850–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin SP, Dautoff R, Morris JC, Barrett LF, Dickerson BC (2011) Amygdala atrophy is prominent in early Alzheimer’s disease and relates to symptom severity. Alzheimer’s Disease Neuroimaging Initiative. Psychiatry Res. 194:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Slotnick BM (1983) Dual olfactory representation in the rat thalamus: an anatomical and electrophysiological study. J Comp Neurol. 215:63–77. [DOI] [PubMed] [Google Scholar]
- Price JL, Davis PB, Morris JC, White DL (1991) The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging. 12:295–312. [DOI] [PubMed] [Google Scholar]
- Ray JP, Russchen FT, Fuller TA, Price JL (1992) Sources of presumptive glutamatergic/aspartatergic afferents to the mediodorsal nucleus of the thalamus in the rat. J Comp Neurol. 320:435–456. [DOI] [PubMed] [Google Scholar]
- Reines A, Cereseto M, Ferrero A, Bonavita C, Wikinski S. (2004) Neuronal cytoskeletal alterations in an experimental model of depression. Neuroscience 129:529–538. [DOI] [PubMed] [Google Scholar]
- Reynolds GP (1983) Increased concentrations and lateral asymmetry of amygdala dopamine in schizophrenia. Nature 305:527–529. [DOI] [PubMed] [Google Scholar]
- Rhomberg T, Rovira-Esteban L, Vikór A, Paradiso E, Kremser C, Nagy-Pál P, Papp OI, Tasan R, Erdélyi F, Szabó G, Ferraguti F, Hájos N (2018) Vasoactive intestinal polypeptide-immunoreactive interneurons within circuits of the mouse basolateral amygdala. J Neurosci. 2018 38:6983–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA (2002) Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci 22: 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA (2003) Affective conditioning in the basolateral amygdala of anesthetized rats is modulated by dopamine and prefrontal cortical inputs. Ann N Y Acad Sci. 985:488–491. [DOI] [PubMed] [Google Scholar]
- Rovira-Esteban L, Péterfi Z, Vikór A, Máté Z, Szabó G, Hájos N (2017) Morphological and physiological properties of CCK/CB1R-expressing interneurons in the basal amygdala. Brain Struct Funct. 222:3543–3565. [DOI] [PubMed] [Google Scholar]
- Russchen FT, Amaral DG, Price JL (1987) The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J Comp Neurol. 256:175–210. [DOI] [PubMed] [Google Scholar]
- Ryan SJ, Ehrlich DE, Jasnow AM, Daftary S, Madsen TE, Rainnie DG (2012) Spike-timing precision and neuronal synchrony are enhanced by an interaction between synaptic inhibition and membrane oscillations in the amygdala. PLoS One 7:e35320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye DB, Saper CB, Wainer BH (1984) Stabilization of the tetramethylbenzidine (TMB) reaction product: application for retrograde and anterograde tracing, and combination with immunohistochemistry. J Histochem Cytochem. 32:1145–1153. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J (2003) The amygdaloid complex: anatomy and physiology. Physiol Rev 83:803–834. [DOI] [PubMed] [Google Scholar]
- Sampson VL, Morrison JH, Vickers JC (1997) The cellular basis for the relative resistance of parvalbumin and calretinin immunoreactive neocortical neurons to the pathology of Alzheimer’s disease. Exp Neurol. 145:295–302. [DOI] [PubMed] [Google Scholar]
- Scott SA, DeKosky ST, Scheff SW (1991) Volumetric atrophy of the amygdala in Alzheimer’s disease: quantitative serial reconstruction. Neurology. 41:351–356. [DOI] [PubMed] [Google Scholar]
- Scott SA, DeKosky ST, Sparks DL, Knox CA, Scheff SW (1992) Amygdala cell loss and atrophy in Alzheimer’s disease. Ann Neurol. 32:555–63. [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, Fadok JP, Müller C, Letzkus JJ, Lüthi A (2014) Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron. 81:428–437. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Polepalli J, Sah P (2011) Interneurons in the basolateral amygdala. Neuropharmacology 60:765–773. [DOI] [PubMed] [Google Scholar]
- Sternberger LA, Sternberger NH (1983) Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci U S A. 80:6126–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timbie C, Barbas H (2015) Pathways for emotions: specializations in the amygdalar, mediodorsal thalamic, and posterior orbitofrontal network. J Neurosci. 35:11976–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JH (2020) Neuropeptide Y and amygdala circuitry: Modulation of stress-related behavior. In: Handbook of amygdala structure and function, (Urban JH, Rosenkranz JA, eds), pp. 141–160. San Diego: Academic Press. pp. 141–160. [Google Scholar]
- Van Hoesen GW, Hyman BT, Damasio AR (1986) Cell-specific pathology in neural systems of the temporal lobe in Alzheimer’s disease. Prog Brain Res. 70:321–335. [DOI] [PubMed] [Google Scholar]
- Vickers JC, Costa M (1992) The neurofilament triplet is present in distinct subpopulations of neurons in the central nervous system of the guinea-pig. Neuroscience. 49:73–100. [DOI] [PubMed] [Google Scholar]
- Voelker CC, Garin N, Taylor JS, Gähwiler BH, Hornung JP, Molnár Z (2004) Selective neurofilament (SMI-32, FNP-7 and N200) expression in subpopulations of layer V pyramidal neurons in vivo and in vitro. Cereb Cortex. 14:1276–1286. [DOI] [PubMed] [Google Scholar]
- Wilcock GK, Esiri MM (1982) Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 56:343–356. [DOI] [PubMed] [Google Scholar]
- Woodruff AR, Sah P (2007) Inhibition and synchronization of basal amygdala principal neuron spiking by parvalbumin-positive interneurons. J Neurophysiol. 98:2956–29561. [DOI] [PubMed] [Google Scholar]
- Yuan A, Sershen H, Veeranna, Basavarajappa BS, Kumar A, Hashim A, Berg M, Lee JH, Sato Y, Rao MV, Mohan PS, Dyakin V, Julien JP, Lee VM, Nixon RA (2015) Functions of neurofilaments in synapses. Mol Psychiatry. 20:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Nixon RA (2016) Specialized roles of neurofilament proteins in synapses: Relevance to neuropsychiatric disorders. Brain Res Bull. 126:334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Veeranna, Nixon RA (2017) Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb Perspect Biol. 9:a018309. [DOI] [PMC free article] [PubMed] [Google Scholar]