

Abstract
Gene regulation using RNA interference therapy has been developed as one of the frontiers in cancer treatment. The ability to tailor the expression of genes by delivering synthetic oligonucleotides to tumor cells has transformed the way scientists think about treating cancer. However, its clinical application has been limited due to the need to deliver synthetic RNAi oligonucleotides efficiently and effectively to target cells. Advances in nanotechnology and biomaterials have begun to address the limitations to RNAi therapeutic delivery, increasing the likelihood of RNAi therapeutics for cancer treatment in clinical settings. Herein, we discuss innovations in the design of nanocarriers for the delivery of oligonucleotides for successful RNAi therapy.
Keywords: cancer, RNA interference, liposomal delivery, polymeric nanocarrier, biomaterial, nanomedicine
Graphical Abstract
Gene regulation using RNA interference therapy has been developed as one of the frontiers in cancer treatment. However, its clinical application has been limited due to the need to deliver synthetic RNAi oligonucleotides efficiently and effectively to target cells. Herein, we discuss innovations in the design of nanocarriers for the delivery of oligonucleotides for successful RNAi therapy.
1. Introduction
Cancer is a global health concern, with it being the second leading cause of death worldwide. According to the World Cancer Report, cancer accounted for an estimated 9.6 million deaths in 2018.[1] Traditional treatment options for cancer, including surgery, radiotherapy, and chemotherapy, have the goal of prolonging and improving patient quality of life. However, as early as 1993, multidrug resistance of cancer cells to chemotherapeutics and anti-cancer drugs has been a clinical concern.[2] In response to this concern, new research thrusts toward investigating new chemotherapeutic carrier systems and alternative treatment strategies that can overcome this multidrug resistance have been launched.
One such strategy, touted as one of the frontiers in cancer therapeutics, is RNA interference (RNAi). RNAi is a mechanism that is present in all mammalian cells. First, a double-stranded RNA (dsRNA) or hairpin microRNA precursor is cleaved by the Dicer enzyme. The resulting oligonucleotides are incorporated into the RNA-induced silencing complex (RISC), where the oligonucleotide is activated and pairs with target mRNA strands to trigger gene silencing.
In RNAi therapy, synthetic oligonucleotides are introduced to the cell, where they are then incorporated into the RISC to promote specific gene regulation (Figure 1). The most commonly used oligonucleotides for this purpose are short interfering RNAs (siRNAs) and microRNAs (miRNAs). Formed from the cleavage of dsRNA into 21-base pair units, siRNAs are a powerful tool for mediated gene silencing through perfect pairing with target mRNA strands. The endogenously expressed miRNAs are small, non-coding RNAs, ~21-25 nucleotides in length, that base pair with specific miRNAs to either inhibit translation or promote mRNA degradation. These endogenous RNAs play a vital gene-regulatory role in animals and plants. By pairing with target mRNAs to specify post-transcriptional repression of messages, miRNAs can regulate temporal and tissue-specific gene function. Unlike siRNA, miRNA triggers gene silencing through imperfect pairing with the target mRNA strand. Due to its need to only partially pair with its mRNA target, an individual miRNA can repress hundreds of genes. As regulators of gene expression, miRNAs can control several functions and roles within the cell cycle, including fat metabolism, hematopoietic differentiation, cell proliferation, and apoptosis. Because of their role as regulators, dysregulation of miRNAs has been associated with the onset and progression of several diseases.
Figure 1.

Mechanisms of RNA interference undergone by siRNA and miRNA therapeutics for specific gene silencing. In both siRNA and miRNA-based RNA interference, the oligonucleotides are loaded into nanocarriers before being internalized by target cells. Once internalized, the oligonucleotides are incorporated into the RNA-induced silencing complex (RISC), where it is paired with a target mRNA strand for translational repression or gene silencing.
RNAi therapy has been proposed as a cancer treatment, in particular, due to the discovery that dysregulations of miRNA biogenesis enzymes and tumor suppressive miRNA are both correlated with increases in tumorigenesis.[3] Further, miRNAs have a distinct role in cancer onset and progression that is emphasized by their nonrandom distribution in the human genome and its frequent location at fragile sites and genomic regions involved in cancers.[4] Several studies have shown specific miRNA signatures that allow miRNAs to be used as prognostic and diagnostic markers for varying cancer types, including lung cancer, breast cancer, bladder cancer, oral cancer, colorectal cancer, and glioblastoma multiforme.[5-11] Similarly, siRNA, due to its gene degradation ability, has been identified as a strong candidate for RNAi cancer therapy. In these therapeutic applications, a specifically designed siRNA is used to silence oncogenes and other genes involved with cancer proliferation.[12]
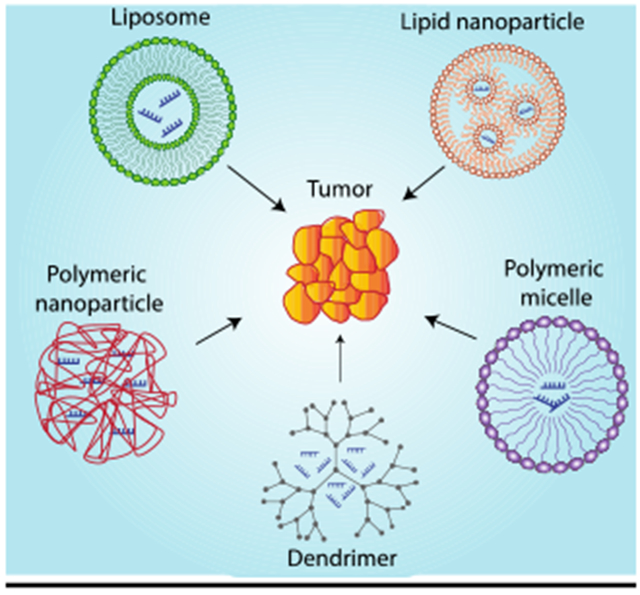
In these RNAi systems, synthetic miRNA or siRNA are uptaken by cells before being incorporated into the RISC to impact target mRNA strands to trigger gene silencing. While RNAi is an intriguing strategy for cancer therapy, there are several biological barriers that must be overcome before it can be considered a viable gene regulation therapy. Specifically, RNAi molecules must avoid degradation, fast renal clearance after systemic administration, and innate immune system activation. These challenges associated with RNAi delivery have severely inhibited its clinical application. The clear option, then, is to develop a drug delivery system that allows one to harness the full therapeutic potential of RNAi therapy. Here, we intend to discuss the various liposomal, polymeric, and peptide-based delivery systems developed for RNAi delivery as well as recent advances toward their clinical application (Figure 2). Other systems that have been developed for RNAi delivery including protein-based therapeutics, spherical nucleic acids, and DNA nanostructures are beyond the scope of this review.
Figure 2.

Various nanoparticulate delivery systems for RNA interference.
2. RNAi Therapeutic Delivery Systems
2.1. Lipid-Based Delivery Systems
2.1.1. Liposomes
Several lipid-based systems have been investigated in recent years for RNAi delivery. The most commonly studied lipid-based systems are those using liposomes. Initial formulations were mostly cationic liposomes, however, due to adverse effects associated with the use of cationic formulations in vivo, alternative approaches using anionic and neutral liposomal delivery have been developed.
The trend in the development of drug delivery systems, including liposomal delivery systems, is the “smart” carrier. These carriers are stimuli-responsive with targeting abilities that enhance specificity and cellular uptake. Figure 3 outlines the design of an intelligent liposomal delivery system. In this system, a photo-responsive liposome decorated with VEGFR2 monoclonal antibodies (mAb) is used to encapsulate DNA. The light sensitivity allows for degradation of the liposomal carrier upon illumination. The presence of VEGFR2 monoclonal antibodies allows for specific VEGFR2 receptor targeting. This specific, controlled release allows for more effective cancer therapy, while maximizing tumor targeting capabilities.[13]
Figure 3.

Construction of an intelligent liposomal delivery system. Reproduced with permission.[13] Copyright 2020, American Chemical Society.
Cationic Liposomal Systems for RNAi Delivery
Cationic liposomes are appealing for RNAi delivery for both its vascular targeting and, more importantly, their simple RNAi complexation. In our experience, cationic liposomes can be prepared by a variety of techniques. We discuss here steps that may be important. Cationic liposomes are prepared using the lipid film method, where cationic lipids, dissolved in an organic solvent mixture, are evaporated to a thin film layer using a rotary evaporator. After removing any trace solvent, the lipid film is rehydrated in a buffer to allow for liposome assembly. Once this liposome suspension is passed through an extruder, RNAi complexation can occur.[14,15] The ease in complexation of RNAi molecules with cationic liposomes makes it very appealing for RNAi delivery systems. To form RNAi-cationic liposome complexes, oligonucleotides are simply incubated with the cationic liposomes. Complexation occurs as a result of electrostatic interactions between the cationic liposomes and the anionic RNAi oligonucleic acids.
Common cationic lipids used for lipoplex formation include 1,2-diolyoxypropyl-3-trimethylammonium chloride (DOTMA), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 2-{3-[Bis-(3-amino-propyl)-amino]-propylaminopropylamine}-N- ditetradecyl carbamoyl methyl-acetamide (DMAPAP), Staramine, and Stearylamine (Figure 4). However, cationic liposomes do not allow for the most efficient transfection of oligonucleotides into cells. Because of this inefficiency, cationic lipids are often mixed with helper lipids, like 1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine (DOPE) and cholesterol (Chol), to improve cellular transfection.[16]
Figure 4.

Structures of common cationic lipids used in liposomal formulations: (a) DOTAP (b) DOTMA (c) DMAPAP (d) Staramine (e) Stearylamine
In their 2010 study, Kim et al. showcased the strengths of a cationic liposomal siRNA delivery system when they investigated the effect of cationic liposome/siRNA complexes on GFP expression (Figure 5).[14] In their study, they compared the transfection efficacy and cell viability of DOTAP-based liposome/siRNA complexes to the commercially available Lipofectamine™ standard. Results indicated that cytotoxicity was a function of cationic liposome composition. Naked siRNA delivery was the most cytotoxic, followed by non-PEGylated cationic liposome/siRNA complexes. Model cells treated with PEGylated liposome/siRNA complexes maintained the highest cell viability. Further, results showed that complexation of siRNA allows for an enhanced gene silencing effect compared to delivery of siRNA alone. It is this behavioral response that has made cationic liposomal delivery systems appealing for RNAi therapeutics.
Figure 5.

GFP expression following delivery of GFP-specific siRNA with cationic liposomes. H4II-E cells were treated with GFP-specific siRNA alone or cationic liposome/siRNA complexes. (a) Untreated H4II-E cells, (b) treated with siRNA alone, (c) treated with Lipofectamine®/siRNA complex, (d) treated with conventional liposome/siRNA complex, (e) treated with PEGylated liposome/siRNA complex, and (f) treated with PLR-PEGylated liposome/siRNA complex. Reproduced with permission.[14] Copyright 2020, Elsevier.
While cationic lipid-based delivery systems show great promise for RNAi delivery, there are several issues associated with their use when applied intravenously or by inhalation. In these scenarios, it has been shown that cationic lipid complexes can induce immune activation in vivo, resulting in an inflammatory response. This toxicity, in turn, diminishes the transfection efficacy of the lipoplex and hinders transgene expression.[17-19] However, several approaches have been taken to overcome these limitations of cationic liposomal delivery.
One approach has been the incorporation of an anionic component into the liposomal formulation, in many cases an anionic polymer, to reduce the toxicities associated with the cationic lipoplexes. In their investigation into 2-{3-[Bis-(3-amino-propyl)-amino]-propylamine}-N- ditetradecyl carbamoyl methyl-acetamide (DMAPAP) liposomes as siRNA carriers for melanoma treatment, Schlegel et al found that lipoplexes formed using the FDA-approved anionic polymer, polyglutamate, led to a more efficient lipoplex formation as well as a reduction in cellular toxicity as the dose of siRNA increased.[21-24] Other anionic polymers are definitely suitable for this application, but due to the wide usage of polyglutamate in biomedical applications, this choice made clinical sense. A similar approach was taken by Hattori et al in which they developed poly-L-glutamic acid (PGA)-coated DOTAP/Chol lipoplexes and found that PGA-coated siRNA lipoplexes could deliver siRNA and suppress ApoB expression with a fraction of the dose necessary of naked siRNA without hepatotoxicity.[25-27] Ultimately, this modification of cationic liposomes is a great leap toward intravenous delivery of cationic siRNA lipoplexes in vivo.
A route of administration that is still being investigated for RNAi delivery using cationic liposomes is the topical route of administration. Generally, the delivery of siRNAs topically has been challenging because of its low permeability through the stratum corneum. However, through the use of deformable, flexible vesicles, like cationic liposomes, transdermal delivery potential is enhanced. In their 2009 study, Geusens et al. investigated DOTAP-based liposome/siRNA complexes for transdermal melanoma treatment. Ultradeformable complexes were synthesized by incorporating the edge activator, sodium cholate, to increase lipid bilayer flexibility and permeability. Cytotoxicity experiments with these liposomes showed that blank liposomes in a 6:1 ratio of DOTAP to sodium cholate had a very minimal impact on cell viability.[28] In their 2016 study, Dorrani et al. expanded on this work when they investigated the skin permeability and internalization of edge-activated DOTAP liposome/siRNA complexes. In their studies, they investigated DOTAP to sodium cholate ratios of 6:1, 8:1, and 10:1. These liposomes were then complexed with siRNA at ratios of 4:1 to 16:1. Results indicated that penetration through the stratum corneum and into the epidermis is a function of edge-activator content, with increased penetration as sodium cholate concentration increased. Lower edge-activator content may be unable to penetrate as deeply due to reduced flexibility of the resulting liposome, preventing diffusion. Further, Dorrani et al. found that as the ratio of siRNA complexed with the liposomes increased, deposition into the basal epidermis and upper dermis increased. This deposition has positive implications for cellular internalization of the DOTAP liposome/siRNA complexes.[15]
In their 2016 study, Ghatek et al developed antihypoxamiR functionalized gramicidin lipid nanoparticles for cutaneous wound healing.[30] In this approach, cationic lipoplexes are used to deliver siRNAs. While it was observed that these lipid-based approaches were effective for wound healing, the efficiency of RNAi delivery following topical application is questionable and further research into the transport mechanisms of RNAi topical delivery is needed.
To reduce the systemic toxicity and innate immune response associated with the delivery of oligonucleotides using cationic lipoplexes, researchers have modified the lipids in a manner that would alter its behavior in vivo. One promising means by which researchers have modified lipids to enable intravenous administration is by functionalizing the lipopolyamine, Staramine, with methoxy polyethylene glycol (mPEG). In this system, administration of mPEG-modified Staramine complexes resulted in no significant change in expression of the cytokines typically induced via the innate immune response, suggesting that this functionalization of the Staramine results in improved safety of the formulation for in vivo RNAi delivery.[31] This conclusion is further supported by McClendon et al where mPEG-modified Staramine was used to deliver antimiR-145 to mice to treat pulmonary hypertension. In this study, McClendon et al observed no toxicity following antimiR-145 treatment.[32] These results are encouraging for the development of cationic lipid-based RNAi delivery systems that do not invoke an innate immune response in vivo.
Anionic Liposomal Systems for RNAi Delivery
In response to the systemic toxicity associated with positively charged lipid nanoparticles, many alternative approaches to cationic lipid-based formulations have been developed. One such method has been the development of anionic liposomal RNAi delivery systems. Anionic liposomal delivery systems are compelling because of the improved biodistribution, efficacy, and reduced toxicity compared to their cationic counterparts.
In these systems, oligonucleotides are generally encapsulated by the liposome formulation, not complexed as is done in cationic liposomal delivery systems. The degree of encapsulation is dependent on the molar charge ratio between the anionic lipid and the siRNA (Figure 6). At low anionic lipid/siRNA molar charge ratios, siRNA incorporation is dependent on complexation with cationic formulation components, particularly Ca2+. This complexation differs from that seen in the cationic liposome systems since it does not directly involve the anionic liposome. This complexation is dependent on the presence of the cationic component, calcium. However, as the molar charge ratio increases, the amount of encapsulated siRNA increases.[60] Additionally, by coating the surface of siRNA complexes with anionic lipids, it is possible to partially decomplex the siRNA to tune siRNA release and, thus, have a more efficient uptake and endosomal escape of the carrier system [33]. Compared to the dose-dependent toxicity associated with the use of cationic liposomal formulations, anionic liposomes generally do not have cellular toxicities associated with their administration. [34]
Figure 6.

Schematic representation of ternary anionic siRNA lipoplexes prepared with: (a) low anionic lipid/siRNA molar charge ratios; and (b) high anionic lipid/siRNA molar charge ratios Reproduced with permission. [60] Copyright 2012, Elsevier.
In anionic liposomal systems, there is a loss in the electrostatic binding ability, and thus, the self-assembling ability of the siRNA complexes. This limitation is often overcome by incorporating a cationic moiety to the complexes. In their 2014 study, Tagalakis et al did just that when they developed a self-assembling anionic peptide-lipid nanocomplex using the anionic lipid, 1,2-dioleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (DOPG), and a cationic, sixteen-lysine peptide for siRNA delivery (Figure 7).[35] Similarly, in their 2018 study, Yu et al developed an anionic liposome-encapsulated siRNA-polyethylenimine (PEI) complex.[36] Their lipopolyplexes were significantly less cytotoxic than the siRNA-PEI complex delivered alone. This highlights the improved therapeutic potential associated with the incorporation of anionic liposomes to cationic siRNA complexes for RNAi therapy.
Figure 7.

Common lipids used in anionic liposomal systems (a) DOPG (b) DSPE (c) CHEMS
Neutral Liposomal Systems for RNAi Delivery
Another promising method to overcome the limitations of cationic liposomal systems is the development of neutral liposomal RNAi delivery systems. One such system is carriers from hyaluronan-grafted lipid-based nanoparticles. In this structure, small, uni-lamellar liposomes are coated with hyaluronan for particle stabilization and selective targeting to tumors expressing hyaluronan receptors. Because of the inclusion of this targeting ligand, hyaluronan-grafted nanoparticles are specifically uptaken by cancer cells. Also, unlike the cationic lipid-based nanoparticles, the hyaluronan-grafted lipid nanoparticles do not induce a pro-inflammatory response in the human peripheral blood mononuclear cells they were cultured with. [37]
pH-Sensitive Liposomal Systems for RNAi Delivery
Recently, pH-responsive liposomal systems have been developed for tumor-targeted drug delivery. The most common pH-sensitive liposomal systems are based on cholesteryl hemisuccinate (CHEMS) liposomes, which facilitate drug release in the acidic tumor microenvironment (pH: 6.4-7.4). [38] The systems developed thus far have been used to deliver traditional chemotherapeutics like doxorubicin. [39] However, due to the translatable nature of these systems, there is great promise for these pH-sensitive systems toward the development of RNAi therapeutics.
2.1.2. Solid Lipid Nanoparticles
Solid Lipid Nanoparticles (SLN) have gained recent interest as a potential RNAi carrier for cancer therapeutics. These particles are attractive due to their small size, their drug protection capabilities, and their increased and efficient cellular uptake compared to the free counterpart. [67] Several studies have been conducted in recent years toward the development of SLNs for cancer therapy. Initial investigations looked into their utility in delivering chemotherapeutics such as paclitaxel and tamoxifen.[67-70] Investigations into the efficacy of co-delivery of RNAi oligonucleotides with these chemotherapeutics were launched, and it was generally found that coadministration led to enhanced cancer therapy with more potent inhibition of tumor growth and elimination of cancer cell populations compared to delivery of the chemotherapeutic, alone.[67] In their 2013 study, Bae et al. investigated siRNA and paclitaxel-loaded SLNs for synergistic chemotherapy and in situ imaging. SLNs were of particular interest for this application as it allowed for complexation of the siRNA, as well as encapsulation of both paclitaxel and quantum dots for cancer therapy and imaging (Figure 8).[68] The promising results of these initial SLN-RNAi formulations highlight the great potential for these lipid-based carriers for RNAi cancer therapy.
Figure 8.

Schematic illustration of SLNs loaded with paclitaxel and quantum dots, and the formation of polyelectrolyte complex of SLNs with siRNA for synergistic paclitaxel-siRNA combination therapy. Reproduced with permission.[68] Copyright 2013, Wiley.
2.1.3. Nanostructured Lipid Carriers
Nanostructured lipid carriers (NLCs) refer to the next generation of SLNs – a structure in which there is a solid lipid matrix with a certain liquid lipid content. These systems are beneficial for their improved drug loading and controlled drug release, similar to SLNs and unlike the other lipid-based systems. Thus far, these systems have only been explored using traditional chemotherapeutics with success. [71-73] Because of the promise that NLC-based systems have for the delivery of chemotherapeutics with higher encapsulation and drug loading efficiencies compared to traditional liposomal systems, further investigation into their potential as RNAi carriers for cancer therapy would be ideal.
2.2. Peptide-Based Delivery Systems
Cell penetrating peptides (CPPs) have been the main base for peptide-based RNAi delivery systems. CPPs, such as oligoarginines, are compounds that can enhance permeation for delivery of therapeutic macromolecules. For example, recently we showed that the co-administration of therapeutic proteins with oligoarginines, such as hexa-arginine, a peptide comprising six residues of arginine, significantly contributed to the intestinal absorption of therapeutic peptides and proteins, which are generally poorly absorbed in the gastrointestinal tract. [74,75] This process did not affect the epithelial cellular integrity.
Two general methods for CPP-RNAi complexation have been investigated – covalent conjugation to siRNA and noncovalent complexation based on electrostatic and hydrophobic interactions with the amino acids present in the peptide structures.
CPPs can be covalently complexed to RNAi through disulfide bond formation between the amino acids and the oligonucleotides. However, several issues have been associated with the covalent complexation methods. This covalent linkage between the CPP and oligonucleotide inhibits the incorporation of the oligonucleotide into the RNA-induced silencing complex (RISC), leading to ineffective gene silencing. [76,77] Further, covalent complexation methods are associated with a loss in siRNA activity due to charge neutralization during disulfide bond formation. Additionally, CPP-RNAi complexes formed via covalent conjugation have activated innate immunity in vivo, limiting their potential therapeutic efficacy. [78]
Because of the limitations associated with covalent CPP-RNAi conjugation, the development of CPP-RNAi delivery systems based on noncovalent complexation methods are the main focus. In these systems, electrostatic and hydrophobic interactions mask, not neutralize, the negative charge of the oligonucleotides, allowing for protection from degradation and increased cellular uptake. Because of the polyanionic nature of siRNA, cellular uptake of siRNAs is normally restrained, limiting their gene regulation potential. However, due to this charge masking, noncovalent CPP-siRNA complexes allow for cell transduction, internalization, and activity retention greater than that observed with naked siRNAs or covalently conjugated complexes. [79]
CPP-RNAi complexes have been created using various peptides. Some of the most commonly investigated peptides for this application include MPG, TAT, and low molecular weight protamine (LMWP).
2.2.1. MPG Peptide
Named after May, Pierre, and Gilles – the scientists who discovered the peptide, MPG is one of the initial peptides investigated for RNAi complexation. In 2003, Simeoni et al investigated the mechanism through which MPG promotes gene delivery and found that the nuclear localization sequence (NLS) of the MPG peptide is involved with the electrostatic interactions between the peptide and nucleic acids, along with being involved with nuclear targeting. When there is a mutation involving the NLS, rapid release of the siRNA into the cytoplasm of cells is enabled, promoting downregulation of target mRNAs. [80] The findings in this study prompted further investigation into CPP-RNAi complexes based on MPG and MPG-derivatives. In their 2016 study, Liu et al developed luteinizing hormone-releasing hormone (LHRH)-targeted MPG-siRNA polyplexes for hepatocellular carcinoma treatment. [81] These polyplexes were prepared by mixing the siRNA targeting gene siRNA-GAPDH with LHRH-MPGΔNLS and vortexing. The LHRH analogue was chosen due to previous reports showing that LHRH-targeted drug carriers reduce tumor size at a significantly greater rate compared to non-targeted drug carriers. This functionalization was chosen because the LHRH receptor is often overexpressed in hepatic cancer cells. Because of the incorporation of both an LHRH analogue and MPG to the carrier system, both selective targeting and efficient siRNA delivery into the cytoplasm was achieved. [81]
In their 2019 study, Schachner-Nedherer et al developed an MPG-based drug delivery system for miR-27a delivery. Similar to other studies, it was observed that incorporation of an amphipathic peptide like MPG to the drug delivery system promotes RNAi uptake across cellular membranes. Additionally, Schachner-Nedherer et al gained some insight into the RNAi release mechanism and found that after bypassing the cellular membrane, there was an intracellular miRNA release triggered by interactions between the hydrophobic regions in the MPG-miRNA complexes and cell membrane phospholipids.[82] While their system was looking at the impact of miR-27a on adipocyte development to prevent or treat obesity, the gene regulation insights gained in this study are still promising for its potential applications to the development of anti-cancer MPG-RNAi complexes based on the same mechanisms of action.
2.2.2. TAT peptide
The HIV-derived CPP, TAT, is widely studied for drug delivery due to its ability to aid in intracellular uptake of small molecules. This internalization is a result of electrostatic interactions with heparan sulfate proteoglycans found in cell membranes.[83] As aides to drug delivery, TAT peptides can be conjugated with multifunctional biopolymers to create efficient cellular delivery vehicles for antisense and siRNA and miRNA oligonucleotides. In many of the systems developed for in vivo delivery of anti-cancer siRNAs, the TAT-conjugated systems are PEGylated to enhance blood circulation times. However, PEGylation often compromises targeted cellular uptake. In their 2018 study, Majumdar et al attempted to overcome this limitation by developing a PEG-free system involving the TAT-derivative, Tissue Infiltrating Peptide Amphiphile (TIPA), for siRNA delivery. TIPA, an amphiphilic peptide containing the TAT motif, was used to decorate the surface of a nanosized liposome system. When the efficacy of this system was investigated, it was found that TIPA incorporation replaced the need for PEGylated agents, allowing for efficient VEGF siRNA delivery to tumors and its vasculature and efficient tumor reduction.[84]
2.2.3. Low molecular weight protamine
Low molecular weight protamine (LMWP) has also been investigated for RNAi delivery. This non-toxic, arginine-rich peptide promotes the condensation of RNAi complexes due to interactions between the positively charged arginine structures and the negatively charged oligonucleotides being delivered. [85] The resulting masking of the polyanionic charges carried by the oligonucleotides allows for increased cellular uptake of the RNAi complexes and a suppression of tumor growth in vivo. The discovery that polyarginine peptides possessed cell translocation activity and internalization pathways similar to TAT prompted further investigation into its potential efficacy for miRNA and siRNA delivery. [86] In their 2010 study, Choi et al found that LMWP was able to achieve transduction into cells with an efficiency equivalent to TAT peptides. Additionally, the gene regulation ability of the siRNA was preserved during LMWP-mediated delivery as they observed significant downregulation of the model protein luciferase when delivering vascular endothelial growth factor siRNA (VEGF siRNA) using LMWP.[87] This finding is further supported in a similar study where suppression of osteogenic marker levels was observed after LMWP-mediated delivery of miR-29b.[17] Ultimately, because of its non-toxic nature and its ability to retain oligonucleotide activity, LMWP shows great potential as a RNAi delivery system.
2.3. Polymer-Based Delivery System
Polymers are attractive for use in biopharmaceuticals due to their tunability, biocompatibility, and ability to serve as protective carriers for the delivery of active agents into the body.[88] Various kinds of polymers have been utilized due to specific properties. They may arise from natural or synthetic origins, be charged or neutral, or hydrophobic or hydrophilic. [89] There is a wide array of materials to select from and herein we highlight the most commonly used polymeric carriers used for the delivery of anti-cancer RNA based medicines in recent years (Table 2).
Table 2.
Summary of polymers used in RNA-based nanomedicines for applications in cancer. This table was constructed by the authors based on previous experimental data of a large number of investigators.
| Polymer | Cancer | Oligonucleotide | Refs |
|---|---|---|---|
| Polyethylenimine (PEI) | Breast | anti-miR 21 | [90] |
| Liver | miR-148b | [91] | |
| Chitosan | Breast | miR-145 | [92] |
| Liver | miR-122 | [93] | |
| Poly (amidoamine) (PAMAM) | Breast | plasmid DNA/ anti-miR-21/DOX | [95] |
| Liver | miR-122 | [96] | |
| Gastric | miR-34a | [97] | |
| Poly (β- amino ester) (PBAE) | Brain | miR-148a/ miR-296-5p | [98] |
| Poly(amino ester) | Brain | anti-miR-21 | [99] |
| PEG-B-PAEBEA | Pancreatic | anti-miR-34a | [100] |
| PEG-b-P(Gu/Hb)/Ang-PEG-b-PGu | Brain | anti-VEGFR2 siRNA/ anti-PLK1 siRNA | [101] |
Cationic polymers possess positively charged functional groups and are able to complex with negatively charged RNA molecules through electrostatic interactions. Of all the materials utilized for gene silencing applications, cationic materials have shown the most promise. This is due to their ability to “condense” multiple RNA molecules. These materials can also permeate the lipid bilayer of cells and localize within the cellular cytosol for improved transfection efficacies in RNA interference. [104]
2.3.1. Polyethylenimine
Numerous cationic polymers have been explored for their utility as non-viral gene transfection agents and one of the most successful candidates is polyethylenimine (PEI). This positively charged material comprises multiple repeating ethylene amine units (Figure 9). The amine groups within its structure are readily ionizable, imparting good aqueous solubility and buffering capacity within the physiological pH range. [105] This polymer can be synthesized in a linear or branched conformation as well as with varying molecular weights (MW). It is widely reported that higher MWs of PEI elicit better gene transfection efficacies, however, there are accompanying cellular toxicities due to its strong cationic charge density. [106,107]
Figure 9.

Chemical structures of cationic polymers a) polyethylenimine and b) poly (amidoamine)
Conversely, lower MW PEI is less toxic and less effective at gene transfection. An approach that has been explored to overcome this limitation is to merge lower MW fragments of the material via degradable linkages to produce higher MW PEI. In doing so, the improved transfection efficacies of the higher MW polymer are harnessed. Upon internalization and degradation in the cells, the lower MW degradation products can then be excreted without eliciting significant cytotoxicity. [105] A recent study that exploited this approach was exploring the efficacy of what the authors referred to as GLUT-1 targeted “nanopompons” delivering anti-miR21 in the treatment of breast cancer. [90] About half a million tandem copies of anti-miR21 were constructed into a nanoball using rolling circle transcription technology. [108]
This “nanoball” is easily cleaved by dicer to generate therapeutic RNA strands upon cellular uptake. To protect this nanoball, ethylenediamine branched polyethylenimine (OEI) was imparted with degradable disulfide linkages to generate glutathione reducible pOEI. Glutathione is present at high concentrations within the tumor microenvironment and readily reduces disulfide bonds.[109] This reaction facilitates the release of the anti-miR payload and the clearance of the polymeric degradation products. Furthermore, an active targeting agent dehydroascorbic acid (DHA) was conjugated to pOEI via a PEG surface linker to promote localization in tumor cells through interactions with the glucose transporter 1 (GLUT1). The targeted nanopompons were generated by mixing pOEI-PEG-DHA with the anti-miR21 nanospheres at room temperature (Figure 10).
Figure 10.

Schematic of nanopompon system used to deliver anti-miR21 to breast cancer cells. The delivery system utilized a condensed nanoball of anti-miR21, polyethylenimine and targeting agent dehydroascorbic acid. Reproduced with permission.[90] Copyright 2019, Elsevier
Complexation of the anti-miR21 nanospheres led to a significant improvement in their stability from only 8 hours to 3 days. In vitro experiments were conducted using the MDA-MB-231 model cell line derived from triple negative breast cancer (TNBC), an aggressive, metastatic and chemo-resistant form of the disease. The cytotoxicity of the material was appraised to be minimal and the endocytosis of DHA functionalized anti-miR21 carrying nanopompons elicited knockdown in the expression levels of the miR-21 target. This was accompanied by an inversely correlated increase in the expression of tumor suppressors PTEN and PCD4 at both the mRNA and protein levels in vitro and in vivo. [110,111] Moreover, in vitro cellular uptake was effectively hindered by the presence of glucose, a competing ligand for the GLUT1 receptor and the targeted nanopompons displayed improved sequestration within the tumors of TNBC-bearing nude mice. These findings indicate that the nanopompons preferentially associate with GLUT1 overexpressing cells and the efficacy of DHA as a targeting agent is not attenuated in vivo.
Anti-tumor efficacy of these delivery vectors was assessed based on ability to diminish tumor growth and disease progression. Orthotopic tumors from mice were treated with nanopompons These tumors exhibited the smallest volumes after treatment with the nanopompons relative to the control groups but targeting with DHA further heightened anti-tumor effects. Also, there was decreased cell proliferation and increased apoptosis as seen with the increased levels of cleaved Caspase-3. [112]
Using similar principles to overcome barriers to successful RNAi in hepatocellular carcinoma (HCC), Cheng et al synthesized bioreducible, disulfide linked PEI polymers (SSBPEI) conjugated to tumor targeting CC9 (CRGDKGPDC) peptide via PEG.[91] The CC9 peptide binds to integrins in the tumor endothelium and also the neuropilin-1 (NRP-1) receptor which promotes penetration into tumor tissue and cells.[113-115] CC9-SS-BPEI polymers were applied for the delivery of miR-148b which is downregulated in HCC cell lines such as HuH-7 and involved in the regulation of characteristics such as tumor drug resistance, metastasis and tumorigenesis.[116]
It must be noted that, there is a direct association between miR-148b and the NRP-1 receptor such that the downregulation of the former leads to an upregulation of the latter.[116] This correlating behavior was harnessed in the study such that cells under-expressing miR-148b were recognized by the CC9 targeted polymers due to their heightened expression of NRP-1 on the tumor cell surface. Cell studies evaluated the toxicity, uptake, gene silencing and antimetastatic properties of the CC9-SS-BPEI/miR-148b nanocomplexes relative to control groups. Overall, the CC9-SS-BPEI/miR-148b nanocomplexes exhibited minimal toxicity and excellent cellular uptake.
They also exhibited the ability to maintain the functionality of the payload as the tumor targeted nanocomplexes aided the rehabilitation of miR-148b expression intracellularly. Consequently, there was a decrease in the expression levels of NRP-1. The researchers also explored the effects of the CC9-SS-BPEI/miR-148b nanocomplexes on tumor cell migration using Transwell and cell scratch assays. The results showed that in both studies, groups treated with the targeted CC9-SS-BPEI/miR-148b nanocomplexes showed the least amount of cell migration and motility. This was evident with the least number of longitudinally migrating cells observed in the Transwell assay and maintenance of the largest scratch area in the cell scratch assay relative to other study groups.
These studies emphasize a number of key innovations that have improved the utility of PEI as a gene transfection agent. First, reducible analogues of PEI were implemented in the studies in order to obtain both the desirable transfection properties of the higher MW PEI but the diminished toxicities of lower MW PEI degradation products. Also, this degradability of the PEI carrier facilitates the decomplexation and release of the RNA payload for incorporation into the gene silencing machinery. Next, PEG was commonly used as a linker to conjugate active targeting agents, however, it also served the purpose of shielding the strong cationic charges of PEI to diminish its associated toxicities. Finally, active targeting moieties were imparted onto the surface of the nanocomplexes to promote internalization at the target sites and minimize off target toxicities and localization.
2.3.2. Chitosan
Another cationic material that has been studied for RNAi applications is chitosan, a naturally occurring polysaccharide of D-glucosamine and N-acetyl-D-glucosamine. [117] Its precursor, chitin, is derived from the exoskeleton of crustaceans and can be economically sourced. [118] Chitosan has gained attention for use in drug and gene delivery applications due to its numerous beneficial properties. To list a few, chitosan is non- toxic, does not elicit immunogenic responses upon introduction into the body, is easily biodegradable to form harmless waste products, and possesses charged groups that facilitate the loading and delivery of RNAi therapeutics. [117]
These qualities were harnessed for cancer therapy using a combinatorial approach for the delivery of chemotherapeutic agent and miRNA. A macromolecular prodrug/gene codelivery system of galactosylated-chitosan-5-fluorouracil (GC-FU) and liver-specific expression gene, miR-122 was developed to target HCC. [93] The approach of using a pro-drug versus encapsulating the active chemotherapeutic compound was adopted to prevent premature drug leakage prior to arrival at the desired site of delivery. The liver targeting efficacy of this delivery system was improved by incorporating galactose as a targeting a ligand due to its association with the asialoglycoprotein receptor (ASGP-R), a lectin receptor abundant in hepatocytes and hepatic cancers. [119,120] miR-122 was chosen as the therapeutic agent due to its significant expression in hepatocytes but minimal expression in other tissues. [121]
Furthermore, it mediates the regulation of hepatocyte development and could inhibit hepatocarcinogenesis through target genes such as Bcl-2, Wnt1, ADAM17, AKT3 among others.[93] Nanosized polyplexes of GC-FU/miR-122 were produced through electrostatic complexation and results indicated low loading densities.[93] The ratio of polymer to miRNA by mass that provided effective miRNA condensation and protection was 256:1. Even higher relative ratios of polymer to miRNA by mass were required to elicit gene transfection. At a ratio of 768:1, only 40% transfection was reported, and 95% transfection efficacy was achieved at 1024:1 in HepG2 cells. These findings indicate that although chitosan poses numerous benefits for intracellular drug delivery, its inability to load greater amounts of miRNA hinders its performance as a RNAi therapeutic. Nonetheless, the GC-FU/miR-122 nanoparticles were able to elicit the knockdown of oncogenes Bcl-2 and ADAM17 which regulate apoptosis and cell adhesion respectively. [122-124]
In another study utilizing carbohydrate-based polymers for miRNA delivery, thiolated dextran complexed with miR-145 (TD-miR) was mixed with chitosan to form nanoparticle polyelectrolyte complexes (PECs).[92] The PECs were conjugated to aptamer AS1411 which is known to specifically bind to the nucleolin receptors overexpressed in cancer cells such as the human breast cancer cell line, MCF-7.[125] The outcome of this study emphasized that the delivery of miR-145 was mainly dependent on the MW of chitosan used and the ratio of thiolated dextran to chitosan (D/Ch ratio) in the PECs. Higher MWs of chitosan impeded the delivery of miR-145 in vitro due to the slow rate of dissociation of the PECs. To overcome this issue, the D/Ch ratio was increased such that electrostatic repulsion between the condensed miRNA and thiolated dextran promoted decomplexation. The PECs were able to hinder the expression of MUC-1, a membrane bound protein overexpressed in breast cancer, in MCF-7 cells but did not elicit significant apoptotic or anti-proliferative effects. [126,127]
The RNA loading efficacy of chitosan may be improved by varying the degree of deacetylation (DD) to increase its charge density. [117] It is important to keep in mind however that the DD must not be exceedingly high that the complexes formed are overly stable and unable to release their payload. Also, the use of lower MW chitosan may hasten the dissociation of nanocomplexes and enhance delivery of miRNA to the cells of interest. [117]
2.3.3. Poly (amidoamine)
Polymers of varying architectures have been explored for their promise in gene delivery applications. One of such materials that has garnered much interest are poly (amidoamine) (PAMAM) dendrimers due to their controllable size and structure, abundance of reactive groups for functionalization and ionizable amine groups for RNA complexation.[128] They possess hyperbranched structures emanating from a central core and the generation of the dendrimer is determined based on the degree of couplings of the branched units (Figure 9).[128] PAMAMs of higher generations are effective at transfection agents however they pose significant toxicities. Moreover, they are extremely time, labor and cost intensive to generate. [129]
Therefore, researchers have explored alternative avenues to take advantage of the transfection capabilities of higher generation PAMAMs without the accompanying toxicities. One approach was to couple 3rd and 5th generation (G3 and G5) dendrimers using a molecular recognition system based on β-cyclodextrin (CD) and adamantane (Ad).[95,130] The core-shell tecto dendrimers (CSTDs) that were derived possessed similar properties as higher generation PAMAMs, and were capable of doxorubicin (DOX)/anti-miR21 co-delivery to cancer cells.[95] In a separate study, 2nd generation (G2) megamers were covalently linked using disulfide bonds to form a degradable megamer core and conjugated to PEG using an acid labile linker (Figure 11).[96]
Figure 11.

Biodegradable, oncosensitive, megamer-based (BOMB) nanoparticle system for miR-122 in liver cancer. BOMB system is acid-sensitive and will shed PEG surface grafts in tumor microenvironment to enhance interactions of PAMAM dendrimer core with cell membranes. Reproduced with permission. [96] Copyright 2019, The Royal Society of Chemistry.
This biodegradable, oncosensitive, megamer-based (BOMB) nanoparticle system was utilized in the delivery of miR-122, a downregulated miRNA in liver cancer. The slightly acidic tumor microenvironment will aid the removal of the PEG surface grafts and de-shield the charged groups of the PAMAM dendrimer core for heightened cellular interactions.[131] In Huh-7 xenograft tumors, the BOMB/miR-122 nanoparticles showed localization and prolonged retention for over 16 hours. BOMB/miR-122 nanoparticles also exhibited anti-tumorigenic effects with the least mean tumor volume of all the tested groups. Furthermore, intratumoral expression of miR-122 was significantly increased with a correlating decrease in the expression of Bcl-9. The reactivity of PAMAM dendrimers has also been utilized to impart targeting moieties.
Phenylboronic acid (PBA) has been reported to specifically bind to sialic acid residues on the carbohydrate antigens frequently expressed in various cancer cells.[132,133] This binding activity was harnessed in a study that successfully conjugated PAMAM to PBA through a PEG linker to obtain a PAMAM derivative named PPP.[97] The therapeutic agent of choice in the study was miR-34a, which is usually downregulated in gastric cancers.[134] Restoration of its expression levels induces cancer cell apoptosis and prevents cell metastasis among other tumor suppressing capabilities.[135] Results showed that PPP/miR-34a complexes administered in nude mice localized within BGC-823 xenograft tumors and withstood renal excretion for up to 24 hours. In comparison, free miR-34a was no longer detectable at 6 hours post administration (Figure 12). The localization of the PPP/miR-34a complexes in the tumor tissues was attributed to the targeting capabilities of PBA. Additionally, the slower renal clearance of the targeted nanoparticles highlighted the improved resistance of the PPP carrier against the excretion of the miRNA payload.
Figure 12.

Biodistribution studies of Cy3-miR-34a loaded nanoparticles at 1, 6 and 24 h after the intravenous injection. Localization of Cy3-miR-34a in the tumor tissue and various organs was measured via in vivo fluorescence. Reproduced with permission.[97] Copyright 2019, The Royal Society of Chemistry.
Finally, the PBA targeted complexes hindered tumor growth in vivo and triggered apoptotic responses in the tumor tissues. In summary, these studies emphasize the flexibility in tailoring PAMAM design to the exploit the conditions within the targeted tissue for gene delivery in cancer.
2.3.4. Poly(β-amino ester)s
Poly(β-amino ester)s (PBAEs) are a unique class of polymers utilized in gene delivery efforts because they possess multiple positively charged amine groups for complexation with genetic material, readily cleavable ester bonds within the backbone for enhanced degradation, pH buffering capacity and minimal cytotoxicity.[136] In a recent study, Green and associates explored the use of newly developed PBAEs with bioreducible and non-reducible properties to deliver miRNA mimics to glioblastoma (GBM) tumors (Figure 13).[98]
Figure 13.

Structures of the different PBAE polymers utilized for the combination therapy of miR-148a and miR-296-5p in glioblastoma. Reproduced with permission.[98] Copyright 2018, American Chemical Society.
It was previously reported that the repression of miRNAs miR-148a and miR-296-5p led to the induction of tumor promoting transcripts Oct4 and Sox2 which induce a cancer stem cell like state.[137,138] Therefore, study researchers posited that restoring the expression levels of these miRNAs through PBAE mediated delivery could hinder the progression of GBM cells to the stem-like phenotype. Findings indicated that R646, a bioreducible PBAE was the best performing candidate overall, exhibiting excellent cellular uptake, minimal toxicity and functional reconstitution of the miRNA expression levels in vitro.[136] Conversely, 646 the non-reducible analog of R646, and C32 a PBAE known for excellent DNA delivery did not reduce the expression of Dmnt1 and Hmga1, targets of the two miRNAs.
In pre-established GBM tumor xenografts, the R646 nano-miRNAs perfused up to 60% of the tumor tissue and alleviated the tumor burdens in mice treated with the combination of miR-148a/miR-296-5p. The success of the R646 polymer in delivering the miRNA mimics may be attributed to its bioreducible nature. The disulfide bonds are cleaved upon exposure to high concentrations of glutathione in the intracellular compartments. This facilitates rapid release of the payload and clearance of degradation products from the cell. In contrast, the escape of miRNA is dependent on the hydrolytic cleavage of the ester bonds in PBAEs 646 and C32, hindering the onset of RNAi effects.
In a separate study targeting GBM, antisense oligonucleotides against miR-21 (anti-miR21) were complexed with poly (amino ester) (PACE) based carriers and delivered to gliomas using convection-enhanced delivery.[99] The specificity of the PACE-anti-miR21 polyplexes was enhanced by conjugation to apolipoprotein E (APoE) which promoted sequestration into the tumor environment.[139,140] PACE-anti-miR21 nanoparticles were supplemented with chemotherapeutic drug temozolomide (TMZ) as miR-21 suppression has been reported to increase sensitivity to chemotherapy.[141] In summary, the PACE-anti-miR21 polyplexes effectively hindered the expression of miR-21 up to 90% in U87 cells. Animals bearing intracranial GBM tumors were treated with PACE-anti-miR21 and supplemented with TMZ injections. This treatment regime led to a 108% increase in survival relative to the untreated group.
Beyond the popular classes of polymers already discussed, researchers have developed other specialty materials with unique properties for gene delivery applications. Xin et al developed reactive oxygen species (ROS) responsive polymers comprising poly (ethylene glycol)–poly [aspartamidoethyl (p-boronobenzyl) diethyl ammonium bromide] (PEG-B-PAEBEA) for RNAi therapy in pancreatic ductal adenocarcinoma (PDAC).[100] The premise behind the study was that combination treatment of PDAC with miR-34a, a miRNA under-expressed in the disease, and volasertib, a polo-like kinase 1 (PLK1) inhibitor, would lead to better therapeutic outcomes. miR-34a plays a major role chemosensitization and an increase in its expression elicits apoptotic and anti-proliferative responses in tumor cells.[142-144]
On the other hand, the overexpression of PLK1 in cancer correlates with tumor aggressiveness, therefore, its suppression is attractive to improve patient prognosis.[145] PEG-B-PAEBEA polymers formed micelles with miR-34a and volasertib in aqueous solution which were stabilized by favorable charge interactions between the amines of the polymer and phosphates in the miRNA as well as the strong coordination between boronic acid and tertiary amines of the PEG-B-PAEBEA. Localization of PEG-B-PAEBEA micelles within the tumor microenvironment triggered the release of boronic acid and subsequent degradation of the polymer to facilitate miR-34a and volasertib delivery. These polymers exhibited minimal toxicity and ROS-dependent drug release as seen with dampened ability to deliver miRNA payload in the presence of ROS scavenger dithiothreitol. PEG-B-PAEBEA micelles successfully transfected cells with results indicating an increase in the expression of miR-34a as well as decreased expression of PLK1, the target of volasertib.
Finally, these polymeric formulations were shown to be well tolerated in vivo as well as elicited antitumor effects in orthotopic models of pancreatic cancer in mice. PLK1 inhibition was also utilized in the treatment of GBM tumors through the systemic delivery of PLK1 and VEGFR2 siRNA using polymeric nanocomplexes based on poly(ethylene glycol)-block-poly[(N-(3-methacrylamidopropyl) guanidinium-co-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl acrylate)] (PEG-b-P(Gu/Hb))/Ang-poly(ethylene glycol)-block-poly(N-(3-methacrylamidopropyl) guanidinium) (Ang-PEG-b-PGu).[101] Besides the electrostatic interactions typically used to stabilize RNAi based medicines, these nanocomplexes were imparted with guanidinium functional groups to enable the formation of Gu+/PO43− salt bridges with the phosphate groups in siRNA (Figure 14).
Figure 14.

Peptide-targeted, ROS-responsive nanoparticles were stabilized by electrostatic, hydrogen bond and hydrophobic interactions for enhanced glioblastoma targeting. These nanoparticles exhibited enhanced BBB permeation and successfully co-delivered PLK1 and VEGFR2 siRNA to gliomas. Reproduced with permission.[101] Copyright 2019, Wiley.
The electrostatic and hydrogen bond interactions of the salt bridge as well as the hydrophobic interactions of the phenylboronic ester work in tandem to generate 3I-NM nanocomplexes stabilized by three different interactive forces. These 3I-NM nanoparticles were also ROS responsive as a result of the oxidation of the phenylboronic esters within the polymeric structure. Additionally, the nanocomplexes were decorated with angiopep-2 peptide (Ang-3I-NM) for enhanced BBB permeation and GBM targeting.[146] Research findings indicated that mice bearing intracranial gliomas experienced retardation in tumor growth when treated with Ang-3I-NM co-loaded with PLK1 and VEGFR2 siRNA. Similarly, animals treated with Ang-3I-NM +siPLK1+siVEGFR2 had notably increased median survival times (36 days) relative to the non-targeted controls (18 days). Analysis of tumor tissue excised from the Ang-3I-NM +siPLK1+siVEGFR2 receiving group showed heightened necrosis and apoptosis. These results indicate that targeting strategies may significantly improve the transport of RNAi therapeutics across the BBB for improved RNAi results.
3. Conclusions
RNAi shows great promise as a cancer therapy strategy. Several strides have been made toward the development of clinically viable RNAi delivery systems – particularly, liposomal, peptide-based, and polymeric systems. Cationic delivery systems are particularly useful for RNA-based therapies based on the ease in complexation with the anionic oligonucleotides.
The versatility of the delivery systems developed for RNAi delivery makes them robust and widely applicable to a variety of cancers. By tuning the properties of the carriers, these systems can be delivered intravenously, transdermally, orally, and by inhalation. The high efficiency of gene silencing as a result of RNAi is broadly applicable outside of cancer. Because of this, these delivery systems can be applied to applications beyond cancer, including, but not limited to, imaging and diagnostics, cardiovascular disease, and inflammatory conditions.
4. Future Directions
Based on the analysis presented before, we conclude that there are quite a few new areas that can be pursued. They address the design of specialty carriers that will be able to provide better RNAi and siRNA therapies.
More specifically, novel cationic carriers must be molecularly designed to allow for selective transport and delivery of RNAi and siRNA. Since the carriers will have to be eventually eliminated from the cells and perfused from the body, it is important to provide advanced biodegradable carriers that are chronologically controlled. The meaning of this is that the degradation process has to be extremely well controlled and probably in several stages so that the elimination of the carrier can be done in a programmable way.
In addition, multifunctional cationic carriers need to be molecularly designed so that they can probably carry and direct two different types of siRNAs that can be delivered at two significantly different rates. That will allow for progressive treatment of specific diseases in a chronological order. Several new methods of achieving that are presently studied in our laboratories and we hope to be able to report on them in the near future.
Finally, nanosized carriers need to be better developed so that they can provide adequate stability over the therapeutic period of the systems. Associated with that is the perennial problem of the toxicity of cationic carriers which will have to be addressed in the near future.
In conclusion, there are still quite a few problems that have to be addressed despite the valiant and formidable efforts in the field.
Table 1.
Common lipids used in liposomal RNAi delivery systems for cancer therapy. This table was constructed by the authors based on previous experimental data of a large number of investigators.
| Charge | Lipid | Cancer | Oligonucleotide | Ref. |
|---|---|---|---|---|
| Cationic | DOTAP | Pancreatic | siRNA | [40] |
| Skin | siRNA | [15], [28], [29], [41], [42] | ||
| Breast | siRNA | [43] | ||
| Ovarian | siRNA | [44] | ||
| Leukemia | Plasmid DNA | [45] | ||
| Lung | miRNA | [46], [47] | ||
| siRNA | [47] | |||
| Plasmid DNA | [48] | |||
| Liver | miRNA | [49] | ||
| siRNA | [14], [25], [50] | |||
| Colorectal | miRNA | [51], [52] | ||
| Brain | miRNA | [53] | ||
| DOTMA | Lung | siRNA | [54], [55] | |
| Liver | siRNA | [56] | ||
| miRNA | [56] | |||
| Colon | siRNA | [57] | ||
| Neuroblastoma | siRNA | [58] | ||
| DMAPAP | Skin | siRNA | [21-23] | |
| Staramine | Lung | siRNA | [31] | |
| Stearylamine | Breast | miRNA | [59] | |
| Colorectal | miRNA | [52] | ||
| Anionic | DOPG | Breast | siRNA | [60, 61] |
| Neuroblastoma | siRNA | [35] | ||
| DSPE | Liver | miRNA | [62] | |
| CHEMS | Lung | siRNA | [63] | |
| Liver | siRNA | [36] | ||
| Neutral | DOPC | Ovarian | siRNA | [64-66] |
| DPPE | Ovarian | siRNA | [37] |
Acknowledgements
NAP also acknowledges support from the NIH (R01-EB022025), Cockrell Family Chair Foundation, the Office of the Dean of the Cockrell School of Engineering at the University of Texas at Austin (UT) for the Institute for Biomaterials, Drug Delivery, and Regenerative Medicine, and the UT-Portugal Collaborative Research Program. During this work, ABS was supported by a Cockrell Graduate School Engineering Fellowship.
Biography

Deidra M. Ward is a Ph.D student in the McKetta Department of Chemical Engineering at the University of Texas at Austin. Her doctoral work is focused on developing nanoparticles for targeted miRNA delivery. Prior to her arrival in Austin, she received her B. S. in Chemical and Biomolecular Engineering at Clemson University.

Aaliyah B. Shodeinde is a graduate research assistant working under the supervision of Dr. Peppas at the University of Texas at Austin. Her current research interests include the development of novel intelligent drug delivery systems for gene therapy in inflammatory conditions. Prior to her arrival in Austin, she received her B.S. in Chemical and Biomolecular Engineering at Lafayette College and performed research on the utility of targeted silver nanoparticles as anti-angiogenic agents.

Nicholas A. Peppas is the Cockrell Family Regents Chair in Engineering, Professor in the Departments of Chemical, Biomedical Engineering, Surgery and Pediatrics, and Pharmacy, and Director of the Institute of Biomaterials, Drug Delivery and Regenerative Medicine of the University of Texas at Austin. Member of the National Academy of Engineering, National Academy of Medicine, American Academy of Arts and Sciences, Academy of Athens, Chinese and Indian Academies of Engineering, Canadian Academy of Engineering, Korean Academy of Science and Technology, National Academy of France, Royal Academy of Spain, National Academy of Inventors, and International Academy of Medical and Biological Engineers. Work on multidisciplinary approach by blending modern molecular and cellular biology with engineering principles to design the next generation of medical systems and devices for patient treatment.
Contributor Information
Deidra M. Ward, McKetta Department of Chemical Engineering, 200 E. Dean Keeton St. Stop C0400, Austin, TX, USA, 78712; Institute for Biomaterials, Drug Delivery, and Regenerative Medicine, The University of Texas at Austin, 107 W Dean Keeton Street Stop C0800, Austin, TX, USA, 78712
Aaliyah B. Shodeinde, McKetta Department of Chemical Engineering, 200 E. Dean Keeton St. Stop C0400, Austin, TX, USA, 78712; Institute for Biomaterials, Drug Delivery, and Regenerative Medicine, The University of Texas at Austin, 107 W Dean Keeton Street Stop C0800, Austin, TX, USA, 78712
Nicholas A. Peppas, McKetta Department of Chemical Engineering, 200 E. Dean Keeton St. Stop C0400, Austin, TX, USA, 78712; Institute for Biomaterials, Drug Delivery, and Regenerative Medicine, The University of Texas at Austin, 107 W Dean Keeton Street Stop C0800, Austin, TX, USA, 78712; Department of Biomedical Engineering, The University of Texas at Austin, 107 W Dean Keeton Street Stop C0800, Austin, TX, USA, 78712; Division of Molecular Pharmaceutics and Drug Delivery, College of Pharmacy, The University of Texas at Austin, 2409 University Ave. Stop A1900, Austin, TX, USA, 78712; Department of Pediatrics, Surgery and Perioperative Care, Dell Medical School, 1601 Trinity St., Bldg. B, Stop Z0800, Austin, TX, USA, 78712
References
- [1].WORLD HEALTH ORGANIZATION: REGIONAL OFFICE FOR EUROPE, WORLD CANCER REPORT: Cancer Research for Cancer Development., IARC, Place of Publication Not Identified, 2020. [Google Scholar]
- [2].Gottesman MM, Pastan I, Annu. Rev. Biochem 1993, 62, 385. [DOI] [PubMed] [Google Scholar]
- [3].Rupaimoole R, Slack FJ, Nat. Rev. Drug Discov 2017, 16, 203. [DOI] [PubMed] [Google Scholar]
- [4].Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. , Proc. Natl. Acad. Sci 2004, 101, 2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, et al. , Cancer Cell 2006, 9, 189. [DOI] [PubMed] [Google Scholar]
- [6].Iorio MV, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. , Cancer Res. 2005, 65, 7065. [DOI] [PubMed] [Google Scholar]
- [7].Akers JC, Hua W, Li H, Ramakrishnan V, Yang Z, Quan K, Zhu W, Li J, Figueroa J, Hirshman BR, et al. , Oncotarget 2017, 8, 68769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mazumder S, Datta S, Ray JG, Chaudhuri K, Chatterjee R, Cancer Epidemiol. 2019, 58, 137. [DOI] [PubMed] [Google Scholar]
- [9].Zhang H, Zhu M, Shan X, Zhou X, Wang T, Zhang J, Tao J, Cheng W, Chen G, Li J, et al. , Gene 2019, 687, 246. [DOI] [PubMed] [Google Scholar]
- [10].Luo Y, Chen L, Wang G, Xiao Y, Ju L, Wang X, J. Cell. Biochem 2019, 120, 13751. [DOI] [PubMed] [Google Scholar]
- [11].Zhang Y, Han T, Feng D, Li J, Wu M, Peng X, Wang B, Zhan X, Fu P, Carcinogenesis 2020, 41, 582. [DOI] [PubMed] [Google Scholar]
- [12].Ozpolat B, Sood AK, Lopez-Berestein G, Adv. Drug Deliv. Rev 2014, 66, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fan J, Liu Y, Liu L, Huang Y, Li X, Huang W. ACS Synth. Biol 2020, 9, 343. [DOI] [PubMed] [Google Scholar]
- [14].Kim H-K, Davaa E, Myung C-S, Park J-S, Int. J. Pharm 2010, 392, 141. [DOI] [PubMed] [Google Scholar]
- [15].Dorrani M, Garbuzenko OB, Minko T, Michniak-Kohn B, J. Controlled Release 2016, 228, 150. [DOI] [PubMed] [Google Scholar]
- [16].Wasungu L, Hoekstra D, J. Controlled Release 2006, 116, 255. [DOI] [PubMed] [Google Scholar]
- [17].Suh JS, Lee JY, Choi YS, Chong PC, Park YJ, Biomaterials 2013, 34, 4347. [DOI] [PubMed] [Google Scholar]
- [18].Chien P-Y, Wang J, Carbonaro D, Lei S, Miller B, Sheikh S, Ali SM, Ahmad MU, Ahmad I, Cancer Gene Ther. 2005, 12, 321. [DOI] [PubMed] [Google Scholar]
- [19].Bauer M, Lautenschlaeger C, Kempe K, Tauhardt L, Schubert US, Fischer D, Macromol. Biosci 2012, 12, 986. [DOI] [PubMed] [Google Scholar]
- [20].Bouxsein NF, McAllister CS, Ewert KK, Samuel CE, Safinya CR, Biochemistry 2007, 46, 4785. [DOI] [PubMed] [Google Scholar]
- [21].Schlegel A, Largeau C, Bigey P, Bessodes M, Lebozec K, Scherman D, Escriou V, J. Controlled Release 2011, 152, 393. [DOI] [PubMed] [Google Scholar]
- [22].Schlegel A, Bigey P, Dhotel H, Scherman D, Escriou V, J. Controlled Release 2013, 165, 1. [DOI] [PubMed] [Google Scholar]
- [23].Arruda DC, Schlegel A, Bigey P, Escriou V, in Non-Viral Gene Deliv. Vectors (Ed: Candiani G), Springer New York, New York, NY, 2016, pp. 137–148. [Google Scholar]
- [24].Arruda DC, Gonzalez IJ, Finet S, Cordova L, Trichet V, Andrade GF, Hoffmann C, Bigey P, de Almeida Macedo WA, Da Silva Cunha A, et al. , J. Colloid Interface Sci 2019, 540, 342. [DOI] [PubMed] [Google Scholar]
- [25].Hattori Y, Nakamura A, Arai S, Nishigaki M, Ohkura H, Kawano K, Maitani Y, Yonemochi E, Results Pharma Sci. 2014, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hattori Y, Arai S, Okamoto R, Hamada M, Kawano K, Yonemochi E, Int. J. Pharm 2014, 476, 289. [DOI] [PubMed] [Google Scholar]
- [27].Hattori Y, Arai S, Kikuchi T, Ozaki K, Kawano K, Yonemochi E, J. Drug Target. 2016, 24, 309. [DOI] [PubMed] [Google Scholar]
- [28].Geusens B, Lambert J, De Smedt SC, Buyens K, Sanders NN, Van Gele M, J. Controlled Release 2009, 133, 214. [DOI] [PubMed] [Google Scholar]
- [29].Geusens B, Van Gele M, Braat S, De Smedt SC, Stuart MCA, Prow TW, Sanchez W, Roberts MS, Sanders NN, Lambert J, Adv. Funct. Mater 2010, 20, 4077. [Google Scholar]
- [30].Ghatak S, Li J, Chan YC, Gnyawali SC, Steen E, Yung BC, Khanna S, Roy S, Lee RJ, Sen CK, Nanomedicine Nanotechnol. Biol. Med 2016, 12, 1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Polach KJ, Matar M, Rice J, Slobodkin G, Sparks J, Congo R, Rea-Ramsey A, McClure D, Brunhoeber E, Krampert M, et al. , Mol. Ther 2012, 20, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].McLendon JM, Joshi SR, Sparks J, Matar M, Fewell JG, Abe K, Oka M, McMurtry IF, Gerthoffer WT, J. Controlled Release 2015, 210, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zeng Y, Zhou Z, Fan M, Gong T, Zhang Z, Sun X, Mol. Pharm 2017, 14, 81. [DOI] [PubMed] [Google Scholar]
- [34].Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y, n.d., 5. [DOI] [PubMed] [Google Scholar]
- [35].Tagalakis AD, Lee DHD, Bienemann AS, Zhou H, Munye MM, Saraiva L, McCarthy D, Du Z, Vink CA, Maeshima R, et al. , Biomaterials 2014, 35, 8406. [DOI] [PubMed] [Google Scholar]
- [36].Yu Q, Zhang B, Zhou Y, Ge Q, Chang J, Chen Y, Zhang K, Peng D, Chen W, J. Liposome Res. 2019, 29, 322. [DOI] [PubMed] [Google Scholar]
- [37].Landesman-Milo D, Goldsmith M, Leviatan Ben-Arye S, Witenberg B, Brown E, Leibovitch S, Azriel S, Tabak S, Morad V, Peer D, Cancer Lett. 2013, 334, 221. [DOI] [PubMed] [Google Scholar]
- [38].Kanamala M, Palmer BD, Jamieson SM, Wilson WR, Wu Z, Nanomed. 2019, 14, 1971. [DOI] [PubMed] [Google Scholar]
- [39].Li Y, Zhai Y, Liu W, Zhang K, Liu J, Shi J, Zhang Z, J. Nanobiotechnology 2019, 17, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pirollo KF, Zon G, Rait A, Zhou Q, Yu W, Hogrefe R, Chang DEH, n.d., 8. [Google Scholar]
- [41].Chono S, Li S-D, Conwell CC, Huang L, J. Controlled Release 2008, 131, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gonçalves C, Berchel M, Gosselin M-P, Malard V, Cheradame H, Jaffrès P-A, Guégan P, Pichon C, Midoux P, Int. J. Pharm. 2014, 460, 264. [DOI] [PubMed] [Google Scholar]
- [43].Mikhaylova M, Stasinopoulos I, Kato Y, Artemov D, Bhujwalla ZM, Cancer Gene Ther. 2009, 16, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen Y, Zhu X, Zhang X, Liu B, Huang L, Mol. Ther 2010, 18, 1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Meissner JM, Toporkiewicz M, Czogalla A, Matusewicz L, Kuliczkowski K, Sikorski AF, J. Controlled Release 2015, 220, 515. [DOI] [PubMed] [Google Scholar]
- [46].Yung BC, Li J, Zhang M, Cheng X, Li H, Yung EM, Kang C, Cosby LE, Liu Y, Teng L, et al. , Mol. Pharm 2016, 13, 653. [DOI] [PubMed] [Google Scholar]
- [47].Gentile E, Oba T, Lin J, Shao R, Meng F, Cao X, Lin HY, Mourad M, Pataer A, Baladandayuthapani V, et al. , Oncotarget 2017, 8, 48222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, Nunez MI, Wistuba II, Erasmus JJ, Hicks ME, et al. , PLoS ONE 2012, 7, e34833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tao J, Ding W-F, Che X-H, Chen Y-C, Chen F, Chen X-D, Ye X-L, Xiong S-B, Int. J. Mol. Med 2016, 37, 1345. [DOI] [PubMed] [Google Scholar]
- [50].Zhang Q-Y, Ho PY, Tu M-J, Jilek JL, Chen Q-X, Zeng S, Yu A-M, Int. J. Pharm 2018, 547, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhao Y, Xu J, Le VM, Gong Q, Li S, Gao F, Ni L, Liu J, Liang X, Mol. Pharm 2019, 16, 4696. [DOI] [PubMed] [Google Scholar]
- [52].Nagachinta S, Bouzo BL, Vazquez-Rios AJ, Lopez R, de la Fuente M, Pharmaceutics 2020, 12, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Campani V, Zappavigna S, Scotti L, Abate M, Porru M, Leonetti C, Caraglia M, De Rosa G, Int. J. Pharm 2020, 588, 119693. [DOI] [PubMed] [Google Scholar]
- [54].Fernando O, Tagalakis AD, Awwad S, Brocchini S, Khaw PT, Hart SL, Yu-Wai-Man C, Mol. Ther 2018, 26, 2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Abd Elwakil MM, Khalil IA, Elewa YHA, Kusumoto K, Sato Y, Shobaki N, Kon Y, Harashima H, Adv. Funct. Mater 2019, 29, 1807677. [Google Scholar]
- [56].Yu B, Wang X, Zhou C, Teng L, Ren W, Yang Z, Shih C-H, Wang T, Lee RJ, Tang S, et al. , Pharm. Res 2014, 31, 2685. [DOI] [PubMed] [Google Scholar]
- [57].Kodama Y, Harauchi S, Kawanabe S, Ichikawa N, Nakagawa H, Muro T, Higuchi N, Nakamura T, Kitahara T, Sasaki H, Biol. Pharm. Bull 2013, 36, 995. [DOI] [PubMed] [Google Scholar]
- [58].Tagalakis AD, He L, Saraiva L, Gustafsson KT, Hart SL, Biomaterials 2011, 32, 6302. [DOI] [PubMed] [Google Scholar]
- [59].Sharma S, Rajendran V, Kulshreshtha R, Ghosh PC, Int. J. Pharm 2017, 530, 387. [DOI] [PubMed] [Google Scholar]
- [60].Kapoor M, Burgess DJ, Biochim. Biophys. Acta BBA - Biomembr 2012, 1818, 1603. [DOI] [PubMed] [Google Scholar]
- [61].Kapoor M, Burgess DJ, Int. J. Pharm 2012, 432, 80. [DOI] [PubMed] [Google Scholar]
- [62].Zhang W, Peng F, Zhou T, Huang Y, Zhang L, Ye P, Lu M, Yang G, Gai Y, Yang T, et al. , Int. J. Nanomedicine n.d., 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zeng X, de Groot AM, Blenke EO, Colombo S, van Eden W, Franzyk H, Nielsen HM, Foged C, 2015, 12. [DOI] [PubMed] [Google Scholar]
- [64].Landen CN, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK, Cancer Res. 2005, 65, 6910. [DOI] [PubMed] [Google Scholar]
- [65].Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han H-D, Shahzad MMK, Liu X, Bhavane R, et al. , Cancer Res. 2010, 70, 3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ahmed AA, Lu Z, Jennings NB, Etemadmoghadam D, Capalbo L, Jacamo RO, Barbosa-Morais N, Le X-F, Vivas-Mejia P, Lopez-Berestein G, et al. , Cancer Cell 2010, 18, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shi S, Han L, Deng L, Zhang Y, Shen H, Gong T, Zhang Z, Sun X, J. Controlled Release 2014, 194, 228. [DOI] [PubMed] [Google Scholar]
- [68].Bae KH, Lee JY, Lee SH, Park TG, Nam YS, Adv. Healthc. Mater 2013, 2, 576. [DOI] [PubMed] [Google Scholar]
- [69].Liu HY, Yu X, Liu H, Wu D, She J-X, Sci. Rep 2016, 6, 30346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Li T, Zhang Y, Meng Y-P, Bo L-S, Ke W-B, Pharm. Res 2017, 34, 2710. [DOI] [PubMed] [Google Scholar]
- [71].Olerile LD, Liu Y, Zhang B, Wang T, Mu S, Zhang J, Selotlegeng L, Zhang N, Colloids Surf. B Biointerfaces 2017, 150, 121. [DOI] [PubMed] [Google Scholar]
- [72].Zhao X, Tang D, Yang T, Wang C, Colloids Surf. B Biointerfaces 2018, 170, 355. [DOI] [PubMed] [Google Scholar]
- [73].Oner E, Kotmakci M, Kantarci AG, Pharm. Dev. Technol 2020, 25, 936. [DOI] [PubMed] [Google Scholar]
- [74].Morishita M, Kamei N, Ehara J, Isowa K, Takayama K, J. Controlled Release 2007, 118, 177. [DOI] [PubMed] [Google Scholar]
- [75].Kamei N, Morishita M, Chiba H, Kavimandan NJ, Peppas NA, Takayama K, J. Controlled Release 2009, 134, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA, Gait MJ, Blood Cells. Mol. Dis 2007, 38, 1. [DOI] [PubMed] [Google Scholar]
- [77].Meade BR, Dowdy SF, Adv. Drug Deliv. Rev 2008, 60, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, Barnes PJ, Sproat BS, Gait MJ, Lindsay MA, Bioconjug. Chem 2007, 18, 1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].van Asbeck AH, Beyerle A, McNeill H, Bovee-Geurts PHM, Lindberg S, Verdurmen WPR, Hällbrink M, Langel Ü, Heidenreich O, Brock R, ACS Nano 2013, 7, 3797. [DOI] [PubMed] [Google Scholar]
- [80].Simeoni F, Nucleic Acids Res. 2003, 31, 2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liu X, Zhu L, Ma J, Qiao X, Zhu D, Liu L, Leng X, Drug Deliv. Transl. Res 2017, 7, 147. [DOI] [PubMed] [Google Scholar]
- [82].Schachner-Nedherer A-L, Werzer O, Kornmueller K, Prassl R, Zimmer A, Int. J. Nanomedicine 2019, Volume 14, 7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tyagi M, Rusnati M, Presta M, Giacca M. J. Biol. Chem 2001, 276, 3254. [DOI] [PubMed] [Google Scholar]
- [84].Majumder P, Bhunia S, Chaudhuri A, Chem. Commun 2018, 54, 1489. [DOI] [PubMed] [Google Scholar]
- [85].Park YJ, Liang JF, Ko KS, Kim SW, Yang VC, Gene Med J. 2003, 5, 700. [DOI] [PubMed] [Google Scholar]
- [86].Tung C-H, Mueller S, Weissleder R, Bioorg. Med. Chem 2002, 10, 3609. [DOI] [PubMed] [Google Scholar]
- [87].Choi Y-S, Lee JY, Suh JS, Kwon Y-M, Lee S-J, Chung J-K, Lee D-S, Yang VC, Chung C-P, Park Y-J, Biomaterials 2010, 31, 1429. [DOI] [PubMed] [Google Scholar]
- [88].Cavallaro G, Sardo C, Craparo EF, Porsio B, Giammona G, Int. J. Pharm 2017, 525, 313. [DOI] [PubMed] [Google Scholar]
- [89].Kapadia CH, Luo B, Dang MN, Irvin-Choy N, Valcourt DM, Day ES, J. Appl. Polym. Sci 2020, 137, 48651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Guo Q, Li C, Zhou W, Chen X, Zhang Y, Lu Y, Zhang Y, Chen Q, Liang D, Sun T, et al. , Acta Pharm. Sin. B 2019, 9, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Cheng C, Zhang Z, Wang S, Chen L, Liu Q, Colloids Surf. B Biointerfaces 2019, 183, 110412. [DOI] [PubMed] [Google Scholar]
- [92].Tekie FSM, Soleimani M, Zakerian A, Dinarvand M, Amini M, Dinarvand R, Arefian E, Atyabi F, Carbohydr. Polym 2018, 201, 131. [DOI] [PubMed] [Google Scholar]
- [93].Ning Q, Liu Y-F, Ye P-J, Gao P, Li Z-P, Tang S-Y, He D-X, Tang S-S, Wei H, Yu C-Y, ACS Appl. Mater. Interfaces 2019, 11, 10578. [DOI] [PubMed] [Google Scholar]
- [94].Phung CD, Nguyen HT, Choi JY, Pham TT, Acharya S, Timilshina M, Chang J-H, Kim J-H, Jeong J-H, Ku SK, et al. , J. Controlled Release 2019, 315, 126. [DOI] [PubMed] [Google Scholar]
- [95].Song C, Xiao Y, Ouyang Z, Shen M, Shi X, J. Mater. Chem. B 2020, 8, 2768. [DOI] [PubMed] [Google Scholar]
- [96].Wu W, Hu Q, Wang M, Shao S, Zhao X, Bai H, Huang J, Tang G, Liang T, Chem. Commun 2019, 55, 9363. [DOI] [PubMed] [Google Scholar]
- [97].Song Z, Liang X, Wang Y, Han H, Yang J, Fang X, Li Q, Biomater. Sci 2019, 7, 1632. [DOI] [PubMed] [Google Scholar]
- [98].Lopez-Bertoni H, Kozielski KL, Rui Y, Lal B, Vaughan H, Wilson DR, Mihelson N, Eberhart CG, Laterra J, Green JJ, Nano Lett. 2018, 18, 4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Seo Y-E, Suh H-W, Bahal R, Josowitz A, Zhang J, Song E, Cui J, Noorbakhsh S, Jackson C, Bu T, et al. , Biomaterials 2019, 201, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Xin X, Lin F, Wang Q, Yin L, Mahato RI, ACS Appl. Mater. Interfaces 2019, 11, 14647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zheng M, Liu Y, Wang Y, Zhang D, Zou Y, Ruan W, Yin J, Tao W, Park JB, Shi B, Adv. Mater 2019, 31, 1903277. [DOI] [PubMed] [Google Scholar]
- [102].Uz M, Kalaga M, Pothuraju R, Ju J, Junker WM, Batra SK, Mallapragada S, Rachagani S, J. Controlled Release 2019, 294, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Teo J, McCarroll JA, Boyer C, Youkhana J, Sagnella SM, Duong HTT, Liu J, Sharbeen G, Goldstein D, Davis TP, et al. , Biomacromolecules 2016, 17, 2337. [DOI] [PubMed] [Google Scholar]
- [104].van der Aa MAEM, Huth US, Häfele SY, Schubert R, Oosting RS, Mastrobattista E, Hennink WE, Peschka-Süss R, Koning GA, Crommelin DJA, Pharm. Res 2007, 24, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Neuberg P, Kichler A, in Adv. Genet, Elsevier, 2014, pp. 263–288. [DOI] [PubMed] [Google Scholar]
- [106].Zakeri A, Kouhbanani MAJ, Beheshtkhoo N, Beigi V, Mousavi SM, Hashemi SAR, Karimi Zade A, Amani AM, Savardashtaki A, Mirzaei E, et al. , Nano Rev. Exp 2018, 9, 1488497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP, Proc. Natl. Acad. Sci 1995, 92, 7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].An S, Jiang X, Shi J, He X, Li J, Guo Y, Zhang Y, Ma H, Lu Y, Jiang C, Biomaterials 2015, 53, 330. [DOI] [PubMed] [Google Scholar]
- [109].Bansal A, Simon MC, J. Cell Biol. 2018, 217, 2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH, J. Biol. Chem 2008, 283, 1026. [DOI] [PubMed] [Google Scholar]
- [111].Qi L, Bart J, Tan LP, Platteel I, van der Sluis T, Huitema S, Harms G, Fu L, Hollema H, van den Berg A, BMC Cancer 2009, 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].McComb S, Chan PK, Guinot A, Hartmannsdottir H, Jenni S, Dobay MP, Bourquin J, Bornhauser BC, Sci. Adv 2019, 5, eaau9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hu QL, Jiang QY, Jin X, Shen J, Wang K, Li YB, Xu FJ, Tang GP, Li ZH, Biomaterials 2013, 34, 2265. [DOI] [PubMed] [Google Scholar]
- [114].Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E, Cancer Cell 2009, 16, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E, Science 2010, 328, 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Liu Q, Xu Y, Wei S, Gao W, Chen L, Zhou T, Wang Z, Ying M, Zheng Q, Biosci. Rep 2015, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Raftery R, O’Brien F, Cryan S-A, Molecules 2013, 18, 5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Chuan D, Jin T, Fan R, Zhou L, Guo G, Adv. Colloid Interface Sci 2019, 268, 25. [DOI] [PubMed] [Google Scholar]
- [119].J. Controlled Release 2015, 203, 126. [DOI] [PubMed] [Google Scholar]
- [120].Shi B, Abrams M, Sepp-Lorenzino L, J. Histochem. Cytochem 2013, 61, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tsai W-C, Hsu S-D, Hsu C-S, Lai T-C, Chen S-J, Shen R, Huang Y, Chen H-C, Lee C-H, Tsai T-F, et al. , J. Clin. Invest 2012, 122, 2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Adams JM, Cory S, Science 1998, 281, 1322. [DOI] [PubMed] [Google Scholar]
- [123].Cavadas M, Oikonomidi I, Gaspar CJ, Burbridge E, Badenes M, Félix I, Bolado A, Hu T, Bileck A, Gerner C, et al. , Cell Rep. 2017, 21, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Veit M, Koyro KI, Ahrens B, Bleibaum F, Munz M, Rövekamp H, Andrä J, Schreiber R, Kunzelmann K, Sommer A, et al. , Biochim. Biophys. Acta BBA - Mol. Cell Res 2018, 1865, 1598. [DOI] [PubMed] [Google Scholar]
- [125].Shieh Y-A, Yang S-J, Wei M-F, Shieh M-J, ACS Nano 2010, 4, 1433. [DOI] [PubMed] [Google Scholar]
- [126].Regimbald LH, Pilarski LM, Longenecker BM, Reddish MA, Zimmermann G, Hugh JC, Cancer Res. 1996, 56, 4244. [PubMed] [Google Scholar]
- [127].Singh PK, Hollingsworth MA, Trends Cell Biol. 2006, 16, 467. [DOI] [PubMed] [Google Scholar]
- [128].Xu Q, Wang C-H, Wayne Pack D, Curr. Pharm. Des 2010, 16, 2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Li J, Liang H, Liu J, Wang Z, Int. J. Pharm 2018, 546, 215. [DOI] [PubMed] [Google Scholar]
- [130].Chen F, Kong L, Wang L, Fan Y, Shen M, Shi X, J. Mater. Chem. B 2017, 5, 8459. [DOI] [PubMed] [Google Scholar]
- [131].Du J-Z, Li H-J, Wang J, Acc. Chem. Res 2018, 51, 2848. [DOI] [PubMed] [Google Scholar]
- [132].Jia H, Zhu J, Wang X, Cheng H, Chen G, Zhao Y, Zeng X, Feng J, Zhang X, Zhuo R, Biomaterials 2014, 35, 5240. [DOI] [PubMed] [Google Scholar]
- [133].Zhang X, Zhang Z, Su X, Cai M, Zhuo R, Zhong Z, Biomaterials 2013, 34, 10296. [DOI] [PubMed] [Google Scholar]
- [134].Deng X, Cao M, Zhang J, Hu K, Yin Z, Zhou Z, Xiao X, Yang Y, Sheng W, Wu Y, et al. , Biomaterials 2014, 35, 4333. [DOI] [PubMed] [Google Scholar]
- [135].Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, et al. , Nat. Med 2011, 17, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Cordeiro RA, Serra A, Coelho JFJ, Faneca H, J. Controlled Release 2019, 310, 155. [DOI] [PubMed] [Google Scholar]
- [137].Lopez-Bertoni H, Lal B, Li A, Caplan M, Guerrero-Cázares H, Eberhart CG, Quiñones-Hinojosa A, Glas M, Scheffler B, Laterra J, et al. , Oncogene 2015, 34, 3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lopez-Bertoni H, Lal B, Michelson N, Guerrero-Cázares H, Quiñones-Hinojosa A, Li Y, Laterra J, Oncogene 2016, 35, 4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Neves AR, Queiroz JF, Lima SAC, Reis S, Bioconjug. Chem 2017, 28, 995. [DOI] [PubMed] [Google Scholar]
- [140].Wagner S, Zensi A, Wien SL, Tschickardt SE, Maier W, Vogel T, Worek F, Pietrzik CU, Kreuter J, von Briesen H, PLOS ONE 2012, 7, e32568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Qian X, Ren Y, Shi Z, Long L, Pu P, Sheng J, Yuan X, Kang C, Mol. Pharm 2012, 9, 2636. [DOI] [PubMed] [Google Scholar]
- [142].Ling H, Fabbri M, Calin GA, Nat. Rev. Drug Discov 2013, 12, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Li XJ, Ren ZJ, Tang JH, Cell Death Dis. 2014, 5, e1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. , Curr. Biol 2007, 17, 1298. [DOI] [PubMed] [Google Scholar]
- [145].Liu Z, Sun Q, Wang X, Transl. Oncol 2017, 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Gao S, Tian H, Xing Z, Zhang D, Guo Y, Guo Z, Zhu X, Chen X, J. Controlled Release 2016, 243, 357. [DOI] [PubMed] [Google Scholar]