

Abstract
Fragment screening is a powerful drug discovery approach particularly useful for enzymes difficult to inhibit selectively such as the thiol/selenol-dependent thioredoxin reductases (TrxRs), which are essential and druggable in several infectious diseases. Several known inhibitors are reactive electrophiles targeting the selenocysteine-containing C-terminus and thus often suffering from off-target reactivity in vivo. The lack of structural information on the interaction modalities of the C-terminus-targeting inhibitors, due to the high mobility of this domain, and the lack of alternative druggable sites prevents the development of selective inhibitors for TrxRs. In this work, fragments selected from actives identified in a large screen carried out against Thioredoxin Glutathione Reductase from Schistosoma mansoni (SmTGR), were probed by X-ray crystallography. SmTGR is one of the most promising drug targets for schistosomiasis, a devastating, neglected disease. Utilizing a multi-crystal method to analyze electron density maps, structural analysis, and functional studies, three binding sites were characterized in SmTGR: two sites are close to or partially superposable with the NADPH binding site, while the third one is found between two symmetry related SmTGR subunits of the crystal lattice. Surprisingly, one compound bound to this latter site stabilizes, through allosteric effects mediated by the so-called guiding bar residues, the crucial redox active C-terminus of SmTGR, making it finally visible at high resolution. These results further promote fragments as small molecule probes for investigating functional aspects of the target protein, exemplified by the allosteric effect on the C-terminus, and providing fundamental chemical information exploitable in drug discovery.
Keywords: secondary site, allosteric effect, enzyme inhibitor, NADPH:disulfide oxidoreductase, schistosomiasis, fragment
Graphical Abstract
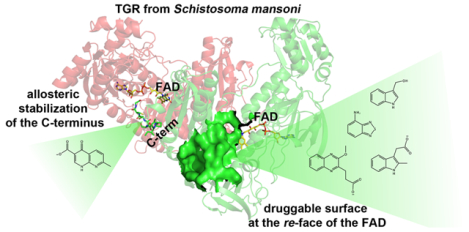
The Fragment-Based Drug Discovery (FBDD) approach is a powerful approach used in target-based drug discovery.1 Inhibitors for strategic therapeutic targets have been developed using this approach, resulting in the identification of 4 newly approved drugs with about 45 additional molecules currently in clinical trials (http://practicalfragments.blogspot.com/search/label/clinical%20candidate). Fragments are low molecular weight soluble compounds (<300 Da) characterized by low binding affinities to the target (in μM-mM range) and by simple chemical architectures. An advantage of using chemical fragments as a starting point for drug discovery over larger compounds resides in their ability to explore the chemical space of the target surface more efficiently. FBDD, when coupled to structural studies, is an attractive and promising approach for developing drugs against targets difficult to inhibit in a selective manner,2 due to its unique capacity to identify novel druggable hotspots.3–5 In this context, X-ray crystallography is the technique of choice for FBDD; it can detect compounds even with low binding affinities and, at the same time, provides strategic information required to drive the improvement of inhibitor efficiency. Moreover, such chemical probing of the target may reveal insights into structural features, protein dynamics and function not easily obtained through other experimental approaches.4
Homodimeric flavoenzymes, such as glutathione reductase (GR) and thioredoxin reductase (TrxR), are essential for maintaining cellular redox homeostasis and regulating DNA synthesis, cell growth, and apoptosis.6–7 Members of the TrxR subfamily are crucial targets for anti-cancer and anti-inflammatory therapeutics in humans8–9 and for the development of therapeutic strategies against pathogenic microorganisms.10–11 Mammalian TrxRs are selenocysteine (Sec, U)-containing enzymes exerting their function via the reduction of the small protein thioredoxin (Trx).12 The presence of Sec provides enzyme resistance to oxidative stress and an extremely high nucleophilic reactivity at physiological pH, mostly due to its low pKa = 5.6.13–14 Among enzymes of the TrxR subfamily, thioredoxin glutathione reductase (TGR) is unusual being a fusion of a glutaredoxin (Grx) domain at the N-terminus of the TrxR homodimer (Figure 1).15–16 The presence of a Grx domain confers to the modular assembly the unique ability to reduce both oxidized Trx and glutathione disulfide. This peculiarity has a significant biological relevance as in parasitic platyhelminths, such as Schistosoma spp. where authentic TrxR and GR are absent, TGR is the only enzyme able to provide electrons to both the Trx and glutathione (GSH) pathways.17 SmTGR is essential for the survival of all the schistosome species and it has been validated as a promising target for schistosomiasis chemotherapy,11, 18 a neglected disease afflicting more than 200 million people in the tropical and sub-tropical areas.19 Currently, schistosomiasis therapy relies on a single drug, praziquantel. The use of monotherapy for treatment facilitates the selection of praziquantel-resistant schistosome strains,20 making the development of alternative therapeutic strategies urgently needed. In addition, all parasitic flatworms rely on TGR and are the causative agents of important human and veterinary diseases.17
Figure 1. Three-dimensional structure and catalytic cycle of SmTGR.
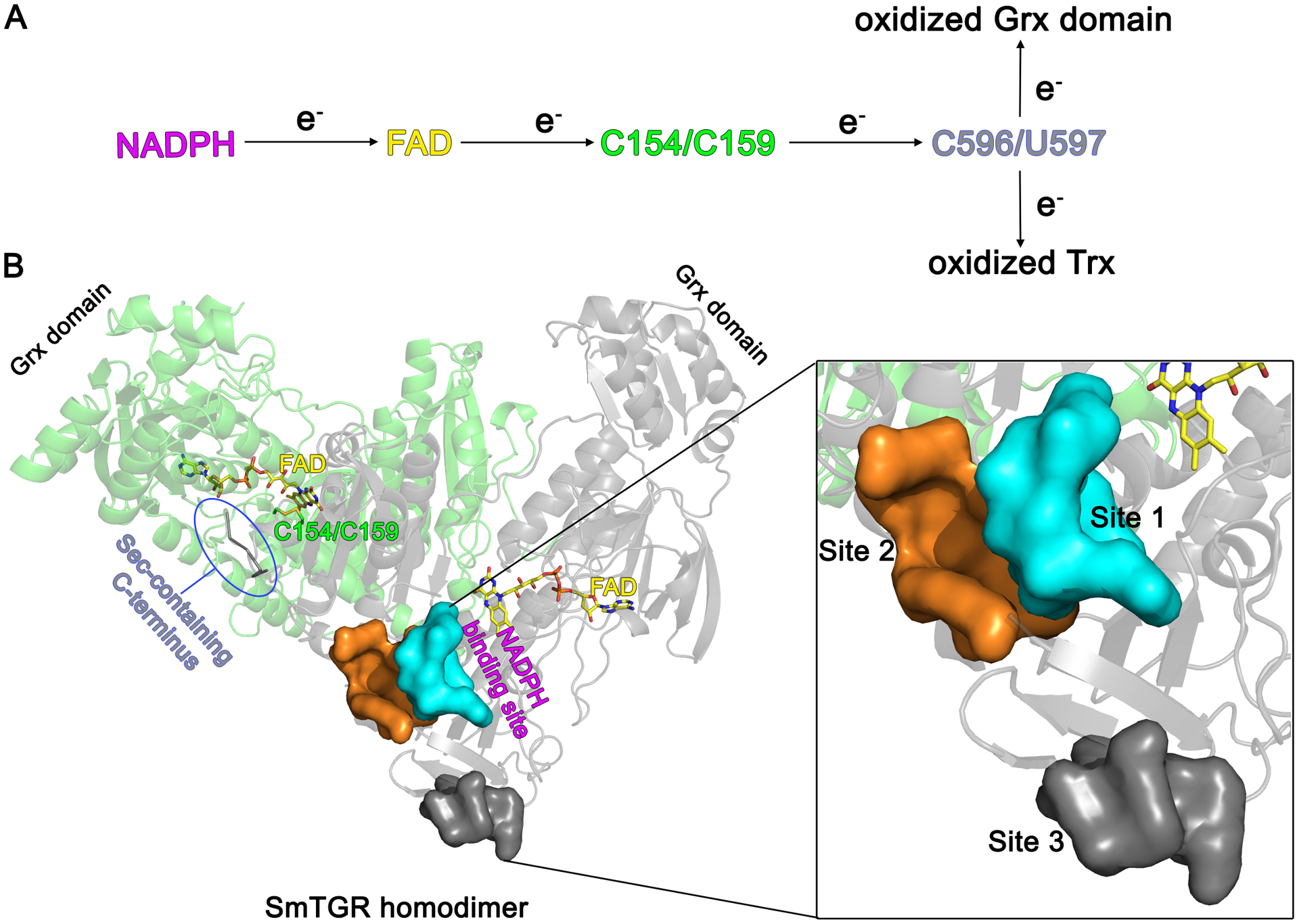
A. The electron flow within the active sites of SmTGR starting from NADPH and ending either with oxidized Trx or oxidized Grx domain is depicted. B. The subunits of the SmTGR homodimer are shown in green and grey cartoons together with the position of the main redox centers of the enzyme. The NADPH binding site extends from the re-face of the FAD, while C154/C159 and the C-terminus are found at the si-face of the co-factor. Localization of the three secondary sites described in this work with respect to the position of the FAD cofactor and of NADPH binding site is displayed in one subunit. The three secondary sites are shown by their solvent exposed surfaces in the magnification on the right.
The catalytic cycle of SmTGR starts when reduced nicotinamide adenine dinucleotide phosphate (NADPH) binds to the protein at the re-face of the flavin adenine dinucleotide (FAD) cofactor, transferring electrons to it. FAD, in turn, reduces the disulfide bond C154/C159 at its si-face allowing the passage of the reducing equivalents to the oxidized and highly mobile Sec-containing C-terminus, which belongs to the other subunit of the obligate homodimer. This latter redox center, containing residues C596 and U597 can i) transfer electrons to oxidized Trx, following the usual path found in TrxRs and ii) reduce the Grx domain of the symmetrical subunit where binding and reduction of glutathione disulfide occurs, unique to TGR proteins (Figure 1).21–22 It has been shown in human TrxR (hTrxR) that the movements of its flexible C-terminus, which adopts different conformations during catalysis, are orchestrated by the so-called “guiding bar” residues (W407, N418 and N419 in hTrxR) that contribute stabilization of the catalytically-competent conformers of this redox center.23–24
To date, most of the inhibitors identified for TrxR subfamily members are reactive electrophilic species,25–27 primarily targeting the Sec-containing C-terminus and thus often suffering from off-target activity when they are tested in vivo due to the high abundance of free thiols.9 Development of novel avenues for optimization of these inhibitors is hindered, in part, by the intrinsic mobility of the C-terminus, making visualization of the inhibitor interactions challenging, and by the lack of druggable secondary binding sites that can be targeted by reversible, nonreactive molecules.25–27 To overcome these barriers, we conducted an X-ray crystallographic study with low molecular weight fragments originally identified as hits in a quantitative high throughput screen (qHTS) against SmTGR (Pubchem BioAssay: AID 485364). To increase the hit rate, we utilized a recently developed multi-crystal method (Pan-Dataset Density Analysis: PanDDa) that makes the resulting electron density maps easier and more automatic to explore.28 The findings presented in this work can contribute to development of novel schistosomicidal agents and open new avenues for targeting TrxR subfamily in other infectious pathogens responsible for numerous important and neglected diseases.
RESULTS AND DISCUSSION
Forty-nine active and commercially available fragments selected from the qHTS have been tested against SmTGR by X-ray crystallography in both soaking and co-crystallization experiments (Table S1), considering that these two methods may generate different outcomes.29–30 In general, hit rates in a fragment X-ray screening strongly depend on the solubility of the compounds in the crystallization conditions and on the nature of the targets, due to the different accessibility of the solvent channels inside a preformed crystal lattice when soaking is carried out,31 resulting in few molecules bound to some proteins and many to others.32 Six crystal structures of SmTGR in complex with compounds reported in Table 1 have been solved at resolutions ranging from 1.45 to 2.7 Å (Table S2), yielding a fairly good hit rate (~ 10%), and identifying three secondary binding sites, namely Site 1, Site 2, and Site 3 (Figure 1). Stereo images of the compounds in their relative protein environments are reported in Figure S1, while their omit electron density maps are reported in Figure S2. Ligand placement into the X-ray derived electron density has been validated considering the ratio of the B-factor of the ligand (Bligand) versus the B-factor of the surrounding residues (atoms within 4 Å; Bsurroundings), the estimated occupancy from PanDDA analysis (see below and Table S3 legend) and the real space correlation coefficients (RSCCs), the latter being indicative of the similarity between the calculated and the X-ray-derived electron densities (Table S3). These measures fall into genuine intervals (e.g., Bligand/Bsurroundings< 2; RSCC > 0.85; see the legend of Table S3), indicating that the structures of SmTGR in complex with compounds 1–5 can provide suitable platforms for future drug-design efforts. The SmTGR-U597C mutant was employed for structural studies, while functional characterization was carried out using the Sec-containing forms of the enzyme.
Table 1.
Fragments found in the crystal structure of SmTGR (shown as dominant ionization state at pH = 7.4; pKa calculations have been carried out at http://chemicalize.com)
| Compound ID | Structure | Apparent IC50 (DTNB assay) | Binding site |
|---|---|---|---|
| CID 1520179 lb |
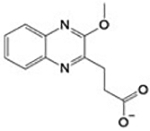 |
>3 mM | Site 1 (both insoakingand in co-crystallization experiments) |
| CID 589107 2b |
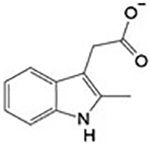 |
>1 mM | Site 1 |
| CID 69845 3 |
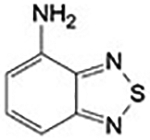 |
~ 500 μM | Site 2 |
| CID 3712 4 |
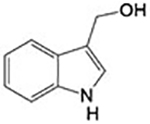 |
284±12 μM | Site 2 |
| CID 5373672 5 |
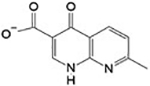 |
n.d.c | Site 3 |
Positive events in PanDDa
Not determined
Site 1 partially overlaps with the NADPH binding site
Applying PanDDa for the identification of ligand binding and structural events, binding of compound 1 [3-(3-methoxyquinoxalin-2-yl)propanoic acid] and 2 (2-methylindole-3-acetic acid) in a subpocket (Site 1) of the NADPH binding site in SmTGR was detected (Figure 2). PanDDa, recently introduced in macromolecular crystallography, makes the electron density inspection from hundreds of crystals (e.g., in a fragment screen) less time-consuming, automatic, and capable of detecting structural events characterized by weak and noisy signals, for example the presence of a ligand at low occupancy inside the crystal.28 Co-crystallization of SmTGR with 1 and soaking of SmTGR crystals with 2 for 72 h gave very weak and uninterpretable electron densities in the NADPH binding site (Panels A of Figure S3 and Figure S4). The use of PanDDa facilitated identification of ligand binding events in these sites (Panels B of Figure S3 and Figure S4). After placing the ligands as indicated and structure refinement, the RSCC values of 0.87 and 0.88 for 1 and 2, respectively, indicate that the ligands are bound as identified by PanDDa (Figure 2). To further test the validity of PanDDa prediction, we performed additional soaking experiments with 1, at twice of the concentration used in the co-crystallization experiment, and with 2, at longer incubation times, obtaining in both cases clearer electron density signals without any data averaging (Figure 2 and panels C and D of Figure S3 and S4).
Figure 2. Site 1 and steady-state competition experiments.
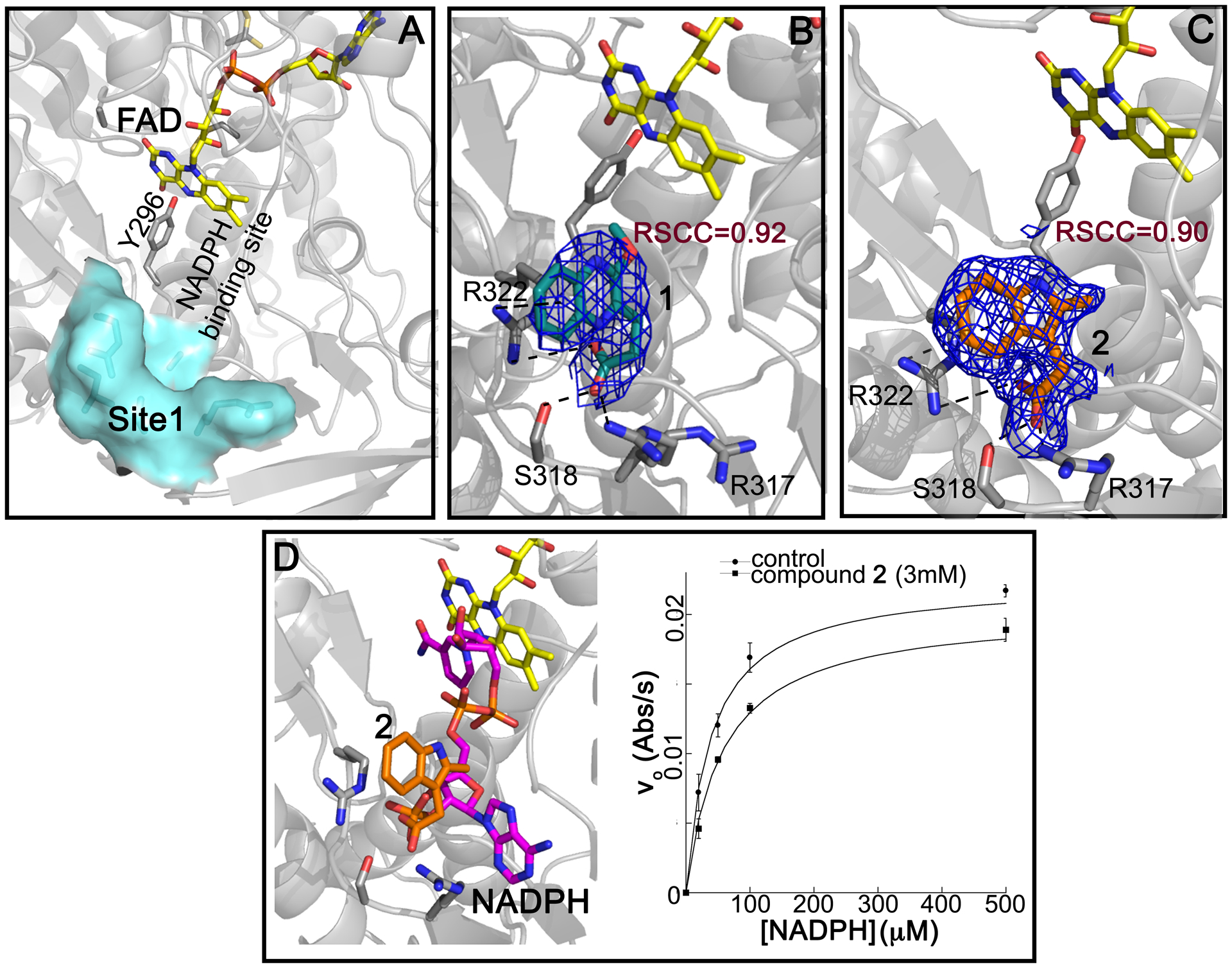
A, surface representation of Site 1 in SmTGR. In B and C, the 2Fo-Fc electron density contoured at 1 σ for 1 (in deep teal sticks in B) and for 2 (orange sticks in C). FAD and Y296, known to delimit the NADPH binding site, are shown in yellow and grey sticks, respectively. The interactions between 1 or 2 and their surrounding residues are indicated as dashed lines. D, the superposition of SmTGR structure in complex with 2 (PDB:6ZP3) and SmTGR in complex with NADPH (in magenta sticks; PDB ID: 2X99) shows that the two molecules would clash if they bind simultaneously; the resulting competitive effect of 2 versus NADPH in steady state experiments is shown on the right of the panel [KM = 35.8 ± 2.0 μM for the control; KM = 59.4 ± 5.3 μM at 3 mM of 2; the error bars correspond to the standard errors].
Compounds 1 and 2 superpose with the phosphate in position 2’ of NADPH (Figure 2, Panels A and D) making similar interactions with the surrounding residues. The carboxylate of 1 makes an H-bond with the side chain of S318, and electrostatic interactions with positive nitrogen atoms of the side chains of R317 and R322. A cation-π interaction between the positive nitrogen of R322 and the aromatic ring of 1 is also present (Figure 2, Panel B and Figure S1). The interacting amino acids are conserved in hTrxR but not in human GR, where S318 is replaced by H219 and the main chain has an amino acidic insertion in the loop contacting the 2’-phosphate of NADPH.33 Steady-state kinetic experiments performed at saturating concentrations of 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB) and varying NADPH confirmed competition between 2 and the reducing substrate (Figure 2, Panel D), supporting the information obtained by X-ray crystallography. Unfortunately, it was not possible to increase concentration of 2 above 3 mM, due to sample precipitation, to determine the inhibition constants. However, the data show both an increase of KM and a slight decrease of Vmax (~ 10%) upon increasing the concentration of 2 from 0 to 3 mM. The decrease of Vmax can be explained by a mixed type of inhibition, where NADPH and 2 simultaneously bind. This can be possible, because 2 occupies a small portion of the NADPH binding site and NADPH may undergo conformational changes upon binding exposing its 2’phosphate to solvent, allowing access to Site 1 by 2. Otherwise, the decrease in Vmax can be also explained by a concomitant precipitation of SmTGR with 2, as we detected some visible aggregation when we incubated SmTGR, at micromolar concentrations, with the compounds during co-crystallization trials. Steady-state experiments of TGR in presence of 1 were also carried out, but, again, it was not possible to further increase the concentration of 1 above 3 mM; in these conditions only a decrease of the Vmax was detected, likely due to protein aggregation induced by this compound (results not shown).
Site 2 is a reservoir of reactive species and an anchoring site exploitable in future compound development
Two different fragments have been found in a solvent-exposed pocket (Site 2; Figure 3 and Figure S1) adjacent to the NADPH binding site. Compounds 3 (4-aminopiazthiole) and 4 (indole-3-carbinol) enter the crystal in soaking experiments and make similar interactions with residues of Site 2 though displaying different apparent IC50 (Table 1). However, both compounds inhibit the enzyme in a NADPH-dependent manner, and the inhibition appears to be irreversible even upon extensive dilution of the inhibited protein, with the measured IC50 representing in these cases, a kinetic property rather than an equilibrium parameter.34
Figure 3. Site 2.
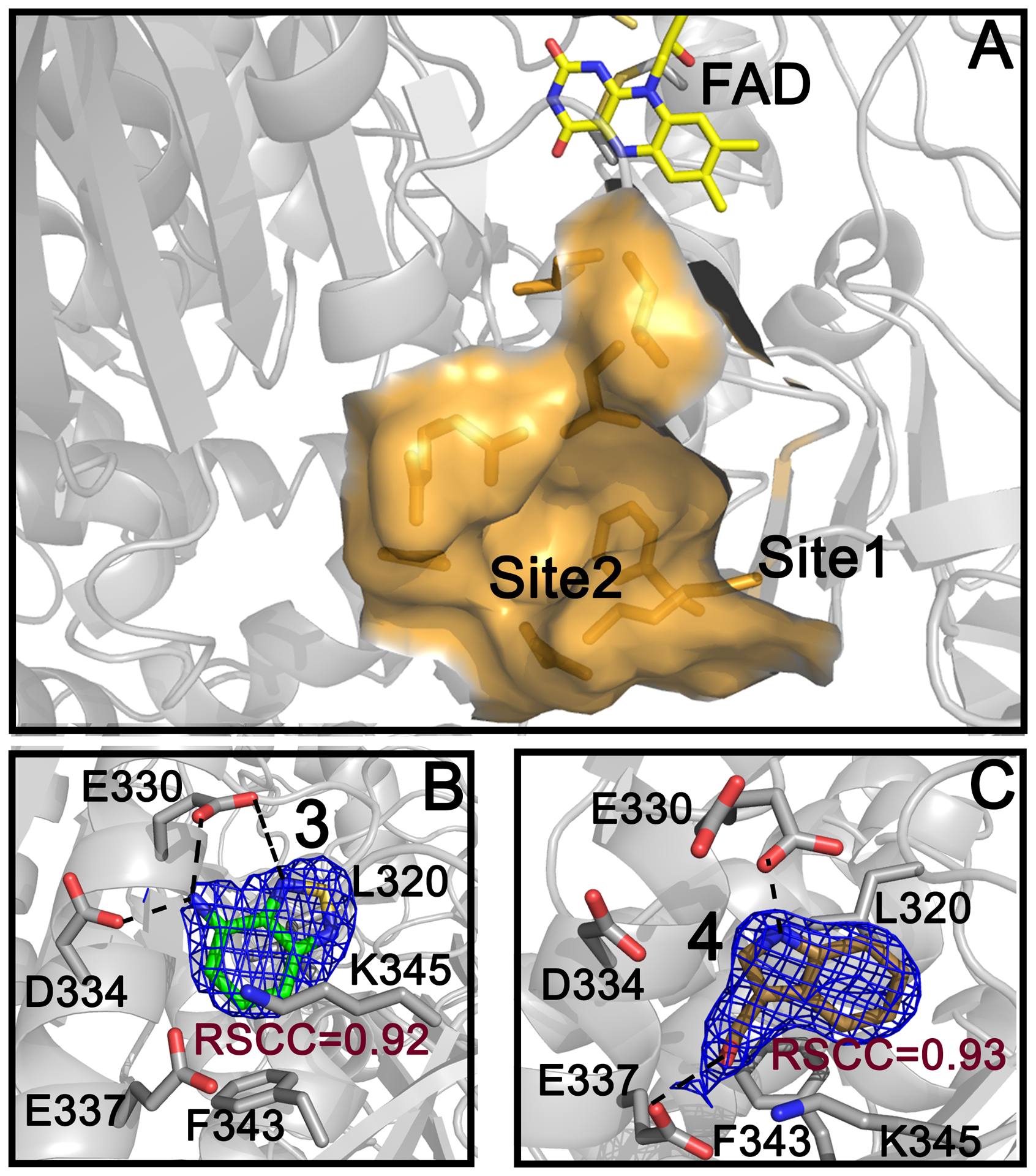
Solvent exposed surface of Site 2 in SmTGR and its proximity to Site 1 are shown (A). The 2Fo-Fc electron density contoured at 1 σ of 3 (in green sticks) and 4 (in gold sticks) are reported in B and C, respectively. FAD is shown as yellow sticks and H-bonds are indicated as dashed lines.
Compound 3 makes several hydrophobic interactions with residues in Site 2 (V316, L320, F343) and several H-bonds. The nitrogen of its thiadiazole ring contacts the side chain of E330, while its amino group interacts with D334 and E330 side chains. A cation-π interaction between its aromatic ring and the positive nitrogen of K345 is also present (Figure 3, Panel B and Figure S1). Its apparent IC50, around 500 μM (% of inhibition at 500 μM = 51.4 ± 4.1), was not possible to determine accurately given that 3 induces protein precipitation at 1 mM, as assessed by SDS page of the collected precipitate (result not shown). However, considering that the inhibition is strongly dependent on the NADPH incubation time (Figure S5) and irreversible, it is likely that the inhibition mechanism is independent of its binding to Site 2. In general, 1,2,5-chalcogenadiazoles, including 3, can behave as Lewis acids, taking part in various charge transfer processes, are redox active and can undergo polymerization.35–36 Therefore, it is possible that these species covalently modify the nucleophilic centers of SmTGR enzymes, once the enzyme has been reduced by NADPH. Moreover, the N-chalcogen bond of 3 resembles that found in ebselen (characterized by a Se-N bond), a well-known selenol/thiol reacting compound and inhibitor of the high molecular weight TrxR subfamily.37 To completely rule out any functional role of Site 2 in the mechanism of inhibition of 3, a double mutant of the enzyme (SmTGR-E330A/D334A), where two key residues interacting with 3 have been changed to alanine (see above and Figure 3), was incubated with different concentrations of the inhibitor and its activity tested, measuring an indistinguishable percentage of inhibition at 500 μM (48.1 ± 6.2) from that of the wild type enzyme, indicating that the inhibition is not related to the binding of the compound to Site 2.
Compound 4 is in clinical trials for the prevention of several classes of solid tumors (e.g., NCT00033345, NCT00100958, NCT00607932, NCT00988845). Compound 4 makes two H-bonds, one between the nitrogen of its indole ring and the side chain of E330 and the other between its OH group and the side chain of E337 (Figure 3, Panel C and Figure S1), and several hydrophobic interactions with residues at the bottom of the pocket. The apparent IC50 value of 4 for DTNB is 284 ± 12 μM. Its inhibition is, as compound 3, dependent on the NADPH incubation time (Figure S5) and irreversible; indeed, it is known that compound 4 and its analogs are not stable in buffered aqueous solutions forming several electrophilic reactive species, such as imine methide,38–39 which can covalently modify the nucleophilic centers of the reduced SmTGR enzyme. To support this, E337, an interacting residue of 4 (see Figure 3), was mutated to alanine (E337A). Compound 4 displays IC50 for the mutant enzyme [IC50 (E337A) = 283 ± 15 μM] almost identical to that of the wild type enzyme, demonstrating also in this case the absence of a role for Site 2 in its inhibition mechanism.
Compounds 3 and 4 exert their inhibition mechanism through formation of reactive species. Site 2 has been described previously by us, characterizing its involvement in the transformation of suicide inhibitors, i.e., 2-naphtholmethylamino compounds, into covalent modifiers of the Sec-containing C-terminus of SmTGR (PDB IDs: 6RTJ, 6RTM, 6RTO).39 Considering that the mobile C-terminus can interact with compounds tethered here,39 Site 2 can be seen as a druggable hotspot functioning as a reservoir for precursors of covalent modifiers of the C-terminal redox center. With the finding of two new fragments bound in this site, its surface is more finely mapped from a chemical point of view and the information gathered will be useful in the design of more selective suicide inhibitors (See Figure S6 and Conclusion). In addition, this site could be used in a fragment linking drug design strategy exploiting its proximity to Site 1 (Figure 4). The identification of Site 1 fills the gap between Site 2 and a regulatory site adjacent to the FAD, called the “doorstop” pocket. Compounds bound here interfere with the conformational changes associated with NADPH entry.40 Thus, a large druggable surface area extending from the re-face of the FAD encompassing the doorstop pocket, Site1, and Site 2 has been identified and chemically mapped (Figure 4). This surface is located far from the reactive electrophilic sites of the enzyme and represents a possible alternative site to target members of the TrxR subfamily with non-covalent inhibitors.
Figure 4. Sites 1 and 2 with the doorstop pocket create an extended druggable surface.
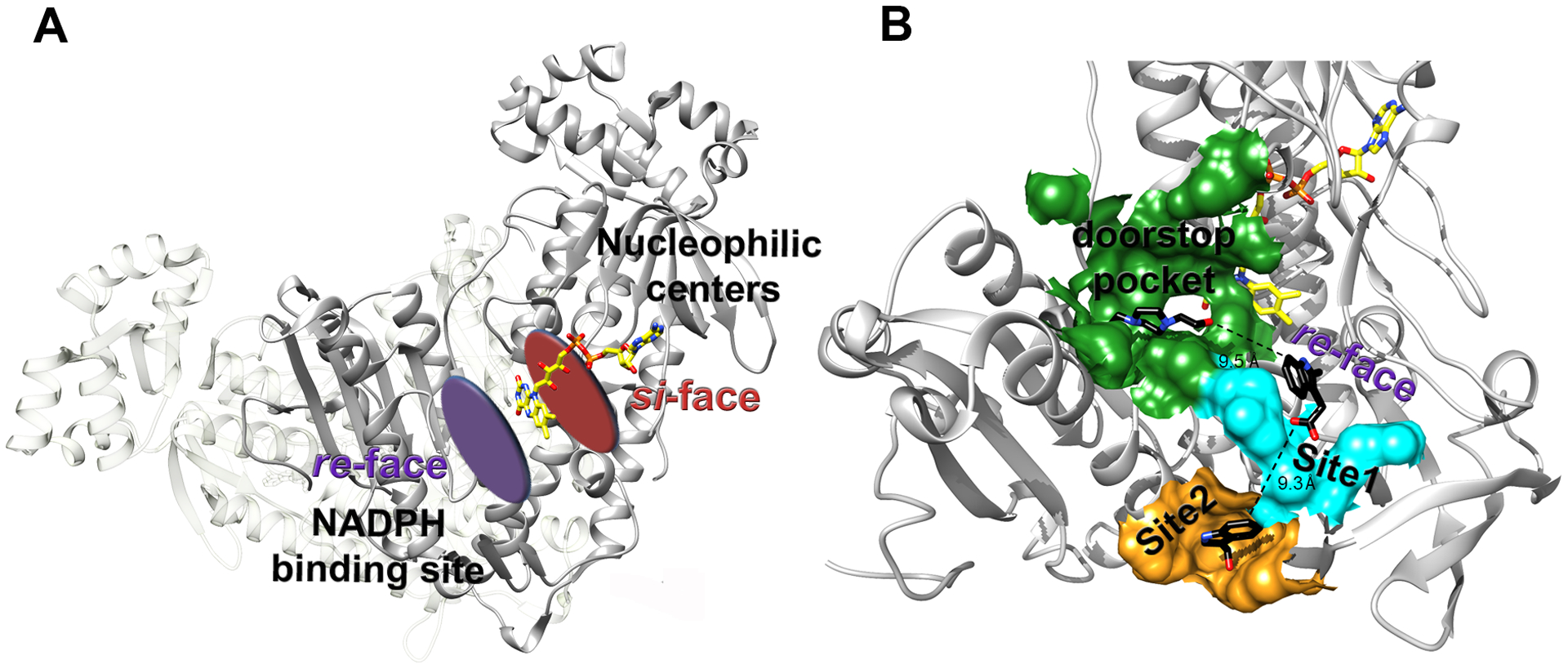
A. The re-face and si-face of the FAD are indicated. All the nucleophilic redox centers of the enzyme localize at the si-face of the cofactor, while the NADPH binding site is at the re-face. B. Druggable sites identified by X-ray fragment screening in SmTGR extend from the re-face side of the FAD and are close to each other. Distances between the closest atom of the ligands found in the three adjacent secondary sites are reported. The “doorstop pocket”, identified in a previous report,40 is highlighted by a green surface. Site 1 and Site 2 are shown by cyan and orange surfaces, respectively.
Site 3 and C-terminus stabilization due to allosteric effects of compound 5 propagated through crystal contacts
Site 3 is generated by crystallographic contacts between two SmTGR subunits related by crystallographic screw axis of the C2 space group (Figure 5). This interface creates a shallow pocket responsible for binding of 5 (4-hydroxy-7-methyl-1,8-naphthyridine-3-carboxylic acid), which can enter this site both in soaking and in co-crystallization experiments. Contacts between a ligand and atoms belonging to different, neighboring asymmetric units are occasionally observed in protein X-ray structures.29, 41 Compound 5 binds to a narrow gap between subunits of the crystal lattice, identified in Figure 5 as “c” and “d”; it makes hydrophobic interactions with L369(c) and L355(c), one H-bond with the carboxylic oxygen of the side chain of E383(c) (with the N11 atom of the naphthyridine ring), a salt bridge between the negative oxygen of the carboxylic group of the compound and the positive nitrogen of the K353(c) side chain, and a cation-π interaction between the positive nitrogen of K105(d) of the symmetric molecule and the π electrons of the aromatic ring of 5 (Figure 5, Panel E and Figure S1).
Figure 5. Site 3 and C-terminus stabilization.
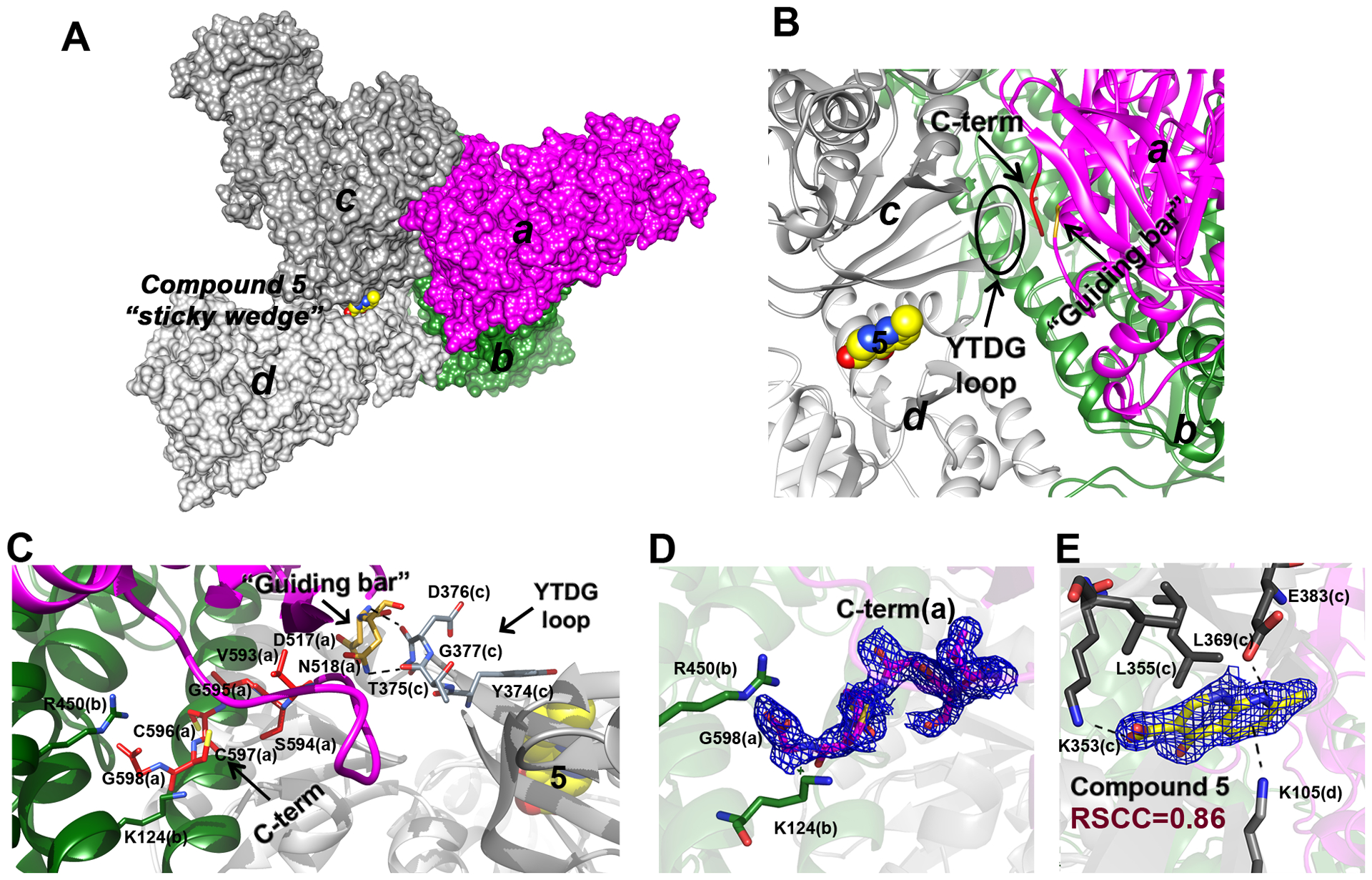
A. A subset of SmTGR subunits, “a” (in magenta), “b” (in deep green), “c” (in dark grey) and “d” (in light grey) as visualized in the crystal lattice of SmTGR in complex with 5 in Site 3, are shown as solvent-exposed surface. Subunits “a” and “b” belong to a physiological SmTGR dimer, while “c” and “d” belong to different physiological dimers. Compound 5 fits between subunits “c” and “d” as a sticky wedge. B. The relative position of 5, with respect to the YTDG loop of subunit “c”, to the guiding bar and to the C-terminus of subunit “a” is depicted. C. The YTDG loop of subunit “c”, the guiding bar residues (D517 and N518) and the last 6 residues of the C-terminus (V593-G598) of subunit “a” are shown in sticks. Dotted lines represent H-bonds between the YTDG loop and the guiding bar and between the guiding bar and the C-terminus (see main text). D. The 2Fo-Fc electron density of the C-terminus of subunit “a” is shown together with the R450 and K124 of subunit “b” (in sticks) that sandwiched the G597 carboxylate. E. The 2Fo-Fc electron density of 5 (here in yellow sticks) and its contacting residues (in grey sticks) belonging to “c” and “d” subunits are shown.
Surprisingly, in the structures of SmTGR complexed with 5, the elusive C-terminus containing a fundamental active center of the enzyme can be completely built (Figure 5, Panel D). To date, we have solved about 400 structures of different SmTGR variants (wild type, U597C mutant, and a C-terminal truncated form lacking the last two residues) in the C2 space group, but the last six residues have never been visible in the crystals. The structure of the protein in complex with 5 clearly shows the entire C-terminus, with cysteines in the reduced state, exposed to the solvent, and interacting with amino acids of the same and partner subunit belonging to the physiological dimer. The nitrogen atom of V593(a) of the main chain interacts with the oxygen atom of the side chain of N518(a), while the terminal carboxylate of G598(a) is sandwiched between the two positively charged amino acids R450(b) and K124(b) (Figure 5, panel B and C; Figure 6). The presence of two positively charged residues interacting with the C-terminus possibly reflects the necessity to stabilize the negative charges present on the carboxylate and on Sec in order to maintain the redox center in a reduced state. C597 and C596 are in trans position with respect to the main chain. This conformation is maintained in SmTGR by a H-bond between C597 and S595 that bends the C-terminus in a turn-like conformation (Figure 6).
Figure 6. Stereo images of superpositions between different TrxR structures with a visible C-terminus and SmTGR-U597C in complex with 5.
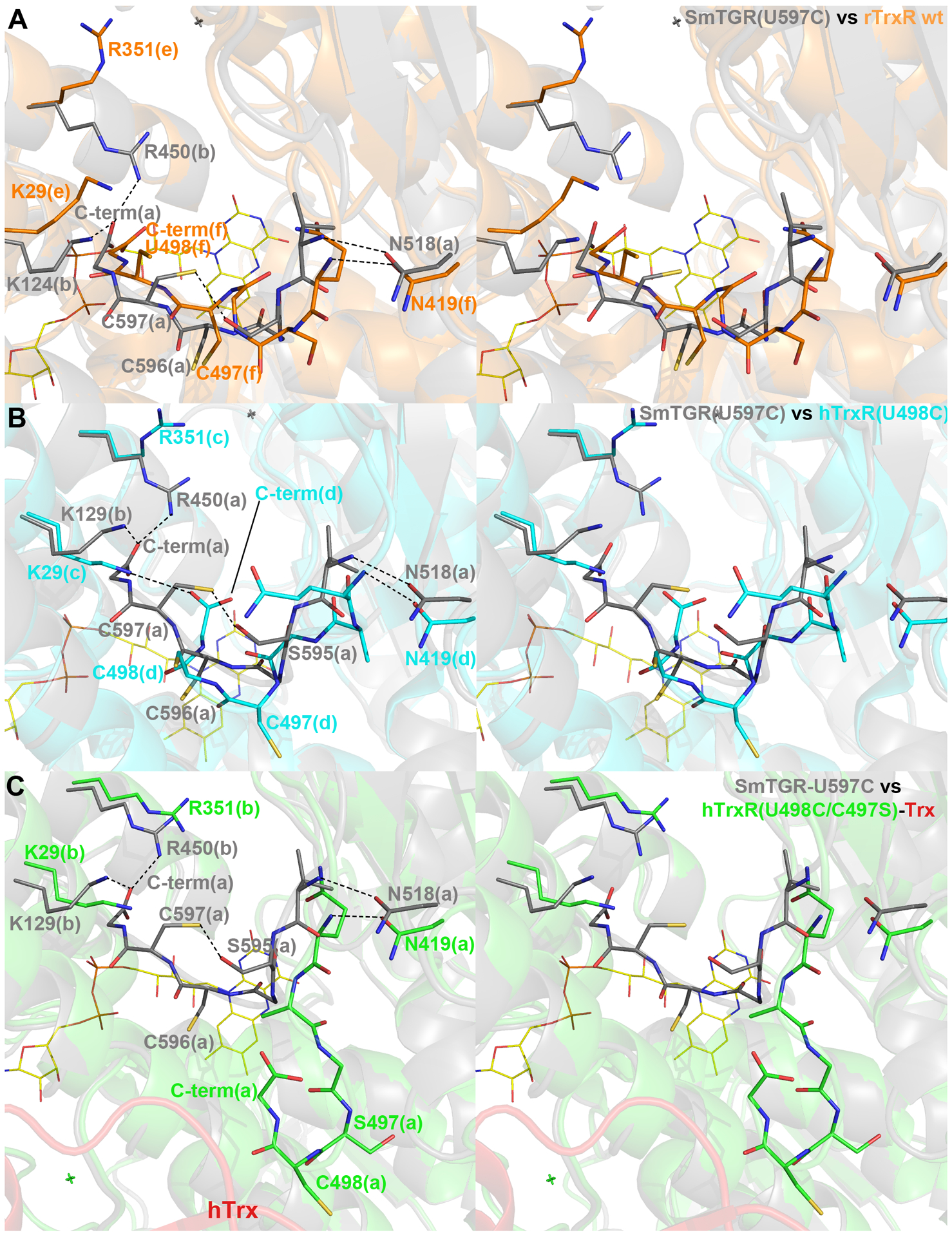
SmTGR is displayed in each panel in grey cartoon and sticks. A, the conformation of subunit “f” of the “e/f” physiological dimer in the crystal structure of the wild type rat TrxR (orange cartoon and sticks; PDB ID: 3EAN; 2.75 Å resolution), in which the electron density of the terminal tail is more continuous and less ambiguous than in other subunits of the asymmetric unit. The C-terminus of SmTGR does not present the 3 cis-peptides within the last 6 residues, as found in rTrxR; its terminal carboxylate is held between R450 and K124, while in rTrxR the carboxylate seems not to be stabilized by any interaction, being in this structure the R351 side chain turned away and K29 close to U498. B, the conformation of hTrxR-U498C as found in subunit “d” of the “c/d” physiological dimer (in cyan carton and sticks; PDB ID: 2J3N; 2.8 Å resolution), the best defined in terms of electron density.24 The C-terminal carboxylate of hTrxR is stabilized by a salt-bridge with K29. C, the conformation of the C-terminus of subunit “a” of the “a/b” physiological dimer in the crystal structure of the hTrxR(U498C/C497S)-Trx complex (hTrxR in green carton and sticks, hTrx in red cartoon; PDB ID: 3QFA; 2.2 Å resolution. The conformation of the C-terminus found in the crystal structure of in hTrxR-Trx complex is less similar to SmTGR, being more distant from the FAD redox center and more exposed to the solvent due to the disulfide bridge with Trx. However, it conserves the trans position of the two reduced cysteines with respect to the main chain. The interaction between N518 (in SmTGR) or N419 (in TrxRs) and the main chains of the C-termini of the same subunit is conserved in all the structures. Polar interactions are displayed as dotted lines.
Of the numerous structures of TrxRs present in the protein data bank, four, solved at medium-low resolution, show the entire C-terminus: the wild type TrxR from rat (rTrxR), the U498C mutant of hTrxR, the U498C/C497S double mutant of hTrxR in complex with hTrx23–24, 42 and the U597C mutant of SmTGR solved in a less-frequent space group for this enzyme.21 In the structures of TrxRs, in which the reduced C-terminus has been detected, the last six residues are characterized by high B-factors (~100 Å2) and ambiguous electron density;24, 42 this fact is also reflected by ensemble of conformations of C-terminus observed in the six subunits present in the asymmetric unit of the crystal lattice of rTrxR and hTrxR (PDB IDs: 3EAN and 2J3N, respectively). The exception is represented by the structure of the hTrxR-Trx complex in which the C-terminus has low B-factors and a clearer electron density. However, here, the C-terminus is stabilized by a mixed disulfide with Trx artificially induced by site-directed mutagenesis of hTrxR and hTrx.23 The conformation most similar to that found in SmTGR is the one seen of the C-terminus of subunit F of the rTrxR crystal structure (Figure 6, panel A; the RMSD calculated on the C-alpha of the last six residues is 2.3 Å). In both these structures, this redox active center is about 13 Å distant from C159 of the FAD redox center, in an intermediate position between the FAD redox center and the position it adopts when it interacts with Trx, as seen in the structure of the hTrxR-Trx complex (Figure 6, Panel C). Conformations like this and that found in hTrxR-U498C (PDB ID: 2j3n; Figure 6, panel B), are designated as a “waiting” position24 because the reduced C-terminus can access Trx by adopting a conformation more solvent exposed (Figure 6). The “guiding bar” is an important feature of TrxRs and three residues W407, N418 and N419 are known to orchestrate movements of the C-terminus.23 Amongst the three residues, N419 has been recognized as the most important in stabilizing the C-terminus, as demonstrated by site directed mutagenesis of hTrxR.23 The putative guiding bar residues in SmTGR, K506, D517 and N518, are partially conserved, with K506 and D517 interacting by a salt-bridge possibly replacing the pi-amide interaction between W407 and N418 in TrxRs.
The C-terminus seen in the highly defined electron density of SmTGR is likely due to compound 5 playing the role of a “sticky wedge” that can stiffen not only the intermolecular motions between dimers and single subunits but also the intramolecular motions within the subunits (Figure 6, panel A). PanDDa analysis indicates that two protein portions (L346-I352 and V370-E383), partially overlapping with the surface binding of 5, are in a slightly different conformation with respect to the same residues found in a subset of apo forms [5 structures out of the 25 structures in apo form used for PanDDa analysis, see Methods; RMSD (L346-I352) = 1.1 Å; RMSD (V370-E383) = 1.2 Å], suggesting flexibility of this region. The amino acids, interacting with 5, belong to a β-sheet made of 3-β-strands (residue ranges: 347–356, 367–374, 377–383); two of them are connected by a loop (374-YTDG-377) that form two H-bonds with the guiding bar residues of an adjacent subunit [D376(c)-D517(a) = 2.9 Å; T375(c)-N518(a) = 2.9 Å] (Figure 6, panel C). Comparison of B-factors, a measure of protein disorder inside a crystal, indicates that binding of 5 induces stabilization of (i) the residues contacting the compounds, (ii) the YTDG loop, and (iii) the guiding bar of an adjacent subunit. We used normalized B-factors, instead of simpler averaged B-factors, given that they are particularly useful to highlight small structural changes and are independent of the structural refinement protocols (see Table S4).43 Such comparison has been carried out between a published SmTGR structure in apo form (PDB ID: 2X8G),21 and SmTGR in complex with 5, where both the YTDG loop and the guiding bar maintain the same conformation and contacts. To understand the role of 5 as sticky wedge, considering that B-factors can only provide a rough estimate of the fluctuations,44 we carried out molecular dynamics simulations of a subset of TGR subunits shown in Figure 5 panel A (a dimer and two subunits) with and without 5. The complex was solvated with water and 0.1 M solution of NaCl to mimic the natural crystallization conditions and subjected to minimization, heating to 300 K, and equilibration followed by a 200 ns MD production step. For reproducibility, the simulations were repeated three times using somewhat different initial coordinates. In all the MD trajectories, the TGR subunits remained associated with only a minimal deviation from its original X-ray location, with RMSD values ranging from 4.3 Å to 4.7 Å, depending on the simulation run. On the other hand, 5 failed to maintain a stable complex with TGR. Instead, 5 traversed between the TGR subunits a-d while forming intermittent complexes with the hydrophobic portions of the subunits and maintaining its overall location between a-d subunits in proximity to its location found in the X-ray structure. These findings suggest that 5 can maintain stable binding to Site 3 only in crystal lattice.
We propose that the stabilization of the C-terminus is induced by subtle allosteric effects, here detected by a B-factor analysis, similar to those described by Nussinov and collaborators,45–46 triggered by the binding of 5 to Site 3 and propagated by the crystallographic interfaces through the interaction between the YTDG loop of one subunit and guiding bar residues of another (Figure 6). Therefore, stabilization of the guiding bar can be sufficient to stabilize the C-terminus, making it visible in the electron density maps. This view is in agreement with previous studies carried out on hTrxR in which destabilization of the guiding bar by mutagenesis makes the protein more active in reducing Trx likely due to a more mobile C-terminus.23 We further theorize that the crystallographic contact within subunits involving the guiding bar residues mimics the initial contact of the oxidized Trx to its binding surface on TGRs (or TrxRs), resulting in stabilization of the C-terminus in a catalytically-competent position.
CONCLUSIONS
Our X-ray crystallography study with 49 low molecular weight fragments identified five compounds (1–5) bound to SmTGR at three binding sites on the protein surface, Sites 1–3. Compounds bound to Site 1 are competitive with NADPH, occupying a small portion of the reductant binding site, while compounds bound to Site 2 exert their inhibition effect through an irreversible mechanism. Site 2 has been previously characterized by us for its ability to catalytically transform suicide inhibitors (2-naphtholmethylamino compounds) into reactive electrophilic species (quinone methides).39 Thus, Site 2 is a reservoir of covalent modifiers of the C-terminal redox center of the enzyme. In principle, an increased specificity of the precursors for this site coupled with a slower generation of the reactive species could saturate the protein reactive nucleophiles in a more selective manner. Moreover, Sites 1 and 2 encompass a region, that extends from the re-face of the FAD cofactor in close proximity to the previously identified allosteric site, the “doorstop pocket”. The new fragments combined with our previous work39–40 chemically and structurally probe a large druggable surface extending from the re-face of the FAD that can potentially be used in fragment linking drug design strategies (Figure 4). This large area is distant from the nucleophilic centers of SmTGR and has not previously been considered as druggable in other pharmaceutically relevant TrxRs, thus providing alternative therapeutic approaches for this enzyme family.
The third site, Site 3, is located in a shallow pocket between two symmetry related SmTGR subunits of the crystal lattice. The SmTGR structure in complex with 5 is the first to be solved at high resolution where the C-terminal peptide is not biased by covalent bond with any partner and that displays the clearest electron density of the entire C-terminal tail. Compound 5 was found to serve as a “sticky wedge”, inducing stabilization of the C-terminus via intra- and intermolecular interactions with the YTDG loop and the guiding bar residues. To the best of our knowledge, this is the first example of an allosteric effect induced by a small molecule bound within crystallographic symmetry-related subunits and mediated by crystal contacts. We believe that this information provides a strategy to identify inhibitors targeting the C-terminus and will facilitate future drug discovery approaches.
The fragment-based X-ray crystallography approach described herein characterizes both unique ligand binding sites and the detailed structure of the catalytically important C-terminus of SmTGR. These findings open new avenues for development of novel therapeutics against Schistosoma spp. and other infectious agents for which related proteins are essential.
METHODS
Materials and reagents –
NADPH was purchased from Sigma and Cayman Chemicals. Polyethylene glycol 3350 (PEG 3350), bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane (bis-Tris) were purchased from Molecular Dimension. EDTA was from Euroclone. All other reagents were purchased from Sigma-Aldrich.
Protein expression and purification
SmTGR wildtype (WT), SmTGR-U597C, SmTGR-E337A and SmTGR-E330A/D334A mutants–
SmTGR WT protein was expressed in BL21 (DE3) Escherichia coli cells co-transformed with pSUABC and purified as previously described.11 The SmTGR-U597C mutant was prepared as described as a fusion with the SUMO protein.22 The SUMO fusion technology is a successful strategy for the purification of recombinant proteins in heterologous systems that facilitates production of elevated amounts of protein.47 His-tag-SUMO-SmTGR Sec597Cys fusion protein was expressed in BL21 (DE3) E. coli induced with 1 mM isopropyl-β-D-thiogalactoside at O.D.=0.6. After 3 h of induction at 37°C, the cell pellet was recovered and frozen at −20°C. For the purification, the cell pellet was sonicated in 50 mM potassium phosphate pH 7.4, 500 mM NaCl, 30 mM imidazole, 100 μM FAD, 1 mM phenylmethylsulfonyl fluoride and 20 μg/mL lysozyme as lysis buffer. The extract was clarified (15 min. at 20k rpm), passed through 0.4 μm syringe filter and then applied to a nickel-affinity column (His-Trap Chelating, GE Healthcare) equilibrated with 20 mM tris(hydroxymethyl)aminomethane (Tris) pH 8, 350 mM NaCl, 20 mM imidazole and 1mM β-mercaptoethanol. The fusion protein was eluted with 400 mM imidazole in binding buffer. The His-tag-SUMO component of the fusion protein was removed by addition of Ulp1, the SUMO protease,48 to the protein stock at a ratio 1:1000 (Ulp1:fusion protein) in 50 mM Tris pH 7.5, 100 mM NaCl, 1 mM 1,4-dithiothreitol (DTT) and 10% glycerol as reaction buffer at 4°C for 20 h. Ulp1 was expressed in BL21 (DE3) with a His-tag and purified as reported.49 Then, the reaction mix was applied to a second nickel-affinity column to remove Ulp1 and the uncut fusion protein. The SmTGR-U597C was collected and used for the structural studies. The SmTGR-E337A mutant was generated using NEBase Changer and the Q5 Site-Directed Mutagenesis Kit and expressed and purified following the same procedure used for the WT protein.11, 40 The SmTGR-E330A/D334A mutant was prepared as reported previously.39 Protein concentrations were determined by FAD absorption at 463 nm (ε463 = 11.3 mM−1 cm−1).
Fragment selection.
We selected almost 1000 fragments from the qHTS bioassay (AID 485364)50 characterized by an inhibition activity against SmTGR and a molecular weight <350 Da. These hits were screened also against human GR and any hits that showed activity at 57 μM were eliminated. The subset of compounds used in this study were chosen based on their commercial availability. All the compounds were re-tested in the DTNB assay against SmTGR to confirm the initial activity found in the qHTS. Soluble, active hits (see Table S1) were then used in both co-crystallization and soaking experiments.
Functional studies
Steady state characterization of the SmTGR WT in complex with compounds 1 and 2.
All the functional assays were carried out using 40 nM of enzyme. Steady state experiments were carried out in 50 mM sodium phosphate (pH 7.4), 250 mM NaCl, 10 mM EDTA, 0.05% Tween and 3 mM DTNB varying NADPH concentrations from 10 μM to 2 mM. Both compounds were tested at concentrations ranging from 0 to 5 mM and were incubated with the protein for 15 min at room temperature before the assay in presence of 100 μM NADPH. The reaction was monitored by DTNB reduction at 412 nm (ε412nm =13.6 mM−1·cm−1). All assays were carried out in triplicate.
IC50 measurements –
SmTGR WT (40 nM), SmTGR-E337A (40 nM) or SmTGR-E330A/D334A (40nM) were incubated with 100 μM NADPH and different concentrations of the compounds (or DMSO as control) at room temperature for 15 min in 100 mM potassium phosphate (pH 7.4) and 10 mM EDTA. Then the DTNB assay was performed adding a second aliquot of 100 μM NADPH and 3 mM DTNB. The reaction was monitored by recording the absorbance increase at 412 nm. The assay was performed on Thermo Multiskan Spectrum plate reader. All assays were done in triplicate.
Irreversibility of inhibition –
SmTGR WT (500 nM) was incubated with 100 μM NADPH and 50 μM inhibitors for 30 minutes. After incubation samples were desalted using 7.0 kDa-cutoff spin Zeba desalting column (Thermo Scientific) and the DTNB assay was carried out.
Time-dependent inactivation –
SmTGR WT (40 nM) was incubated at room temperature with 250 and 500 μM of 3 and with 200 and 500 μM of 4 in presence of NADPH (100 μM) in 50 mM sodium phosphate pH 7.4, 10 mM EDTA plus 100 mM NaCl. The residual enzyme activities were reported as percentage of the controls, in which the compounds were replaced by DMSO. Reactions were monitored, taking aliquots at different time intervals, by the addition of fresh NADPH (100 μM) and DTNB (3 mM) in disposable plastic cuvettes. Reduction of DTNB was followed for 120 s by absorbance increase at 412 nm. The data presented in Figure S5 are the average of 3 or 6 independent measurements. Data analysis was carried out using KaleidaGraph (Synergy Software).
Structural studies
Co-crystallization and soaking experiments –
All the structural studies were performed with the SmTGR-U597C mutant. Crystals were obtained as previously reported.40 For co-crystallization experiments, the protein was equilibrated in 20 mM Tris pH 7.4 and 50 mM NaCl at a concentration of 4.5 mg/mL and the ligand in DMSO was added to 25 mM followed by incubation for 15 min at room temperature. The ligand-protein solution was mixed with equal amounts of a solution containing 20% PEG3350, 0.2 M KI, 100 mM BisTris 7.0, 2% glycerol (v/v), and 5 mM DTT for the crystallization reaction using the sitting drop method, resulting in a ligand final concentration of 12.5 mM. For soaking experiments, preformed crystals were placed in contact with a solution of 20% PEG3350, 0.2 M KI, 100 mM BisTris 7.0 containing each compound at 25 mM. Crystals were picked at different soaking times and flash frozen in liquid nitrogen, adding 35% (v/v) PEG3350 as cryoprotectant.
X-ray data collections, structure refinement and analysis –
X-ray data were collected both at Elettra (XRD1 and XRD2 beamlines - Trieste, Italy)51 and at ESRF (MASSIF-3 and ID23–1 beamlines - Grenoble, France) synchrotrons on Pilatus and Eiger 4M hybrid-pixel area detectors (Dectris, CH) at 100 K and a wavelength of 1.00 Å. As reported in Table S2, all the crystals belong to the C2 space group with very similar unit cells dimensions and with one subunit of the physiological dimer, except the structure of SmTGR with compound 1 entered in soaking, whose crystal belongs to the P212121 space group, with one physiological dimer in the asymmetric unit. The diffraction data were processed with XDS52 and reduced with CCP4.53 Structures were solved by the molecular replacement method (Phaser)54 using the structure of oxidized SmTGR (pdb code: 2V6O)16 as a search model. Model building and refinement were performed using COOT,55 PHENIX/Refine and Refmac.56–57 Waters have been automatically and manually added with COOT. All the reported structures have been refined using automatic weighting and with isotropic B-factors, except for the structure of SmTGR in complex with 5, which was refined with anisotropic B-factors. Ligands were fitted into the electron density only when the convergence of the refinement procedures has been obtained. RSCC values have been calculated by the PDB validation tool.58 RMSD have been calculated with the program Superpose of the CCP4 suite.59 Coordinates and structure factors for data sets with bound fragments have been deposited in the PDB, with the following PDB IDs: 6ZST and 7NPX (SmTGR in complex with 1), 6ZP3 (SmTGR in complex with 2), 6ZLP (SmTGR in complex with 3), 6ZLB (SmTGR in complex with 4), 7B02(SmTGR in complex with 5); refinement statistics are reported in Table S2.
Multi dataset crystallographic analysis (PanDDa) –
25 data collections of the apo form of SmTGR were collected from a pool of crystals grown under identical conditions with resolution in the range 1.7–3.0 Å. Apo dataset have been used to define protein ground state, i.e., a background to highlight ligand binding events, as implemented in PanDDa electron density analysis algorithm (Version 0.2.12).28 Default settings have been used for PanDDA.analyse with adjustment on the minimum number of datasets averaged to generate the mean map of the ground state crystal per shell. Dataset with resolution worse than 3 Å have been excluded. Event maps generated by subtracting the estimated unbound fraction of the crystal identified fragment hits that were manually analyzed using PanDDA.inspect interface embedded in COOT.55 Ligands have been fitted in PanDDa event maps and then the corresponding model was exported and refined in Refmac.57
Molecular dynamics
The X-ray coordinates were those of the structure shown in Figure 5. All the structure preparation steps were performed in Molecular Operating Environment (MOE).60 The proteins were subjected to the “structure preparation” procedure. Hydrogen atoms were added using the Protonate 3D algorithm. SmTGR protein was solvated in a periodic box with water and 0.1 M NaCl using MOE “Solvate” module. The water box was extended at least 10 Å from the protein. The “Molecular Dynamics” module in MOE was used to prepare the resulting structure for MD simulations using NAMD software, version 2.14, Linux-x86_64-multicore-CUDA (http://www.ks.uiuc.edu/Research/namd/).61 Three different conformation of the solvated protein-buffer complex were generated by either proceeding to NAMD calculations directly or minimizing the energy of the solvated protein-buffer system with periodic boundary conditions (PBC) enabled and utilizing AMBER14:EHT forcefield in MOE62–63 until the RMS gradient reached 0.1 or 0.01 kcal/mol/Å.61 Each prepared in this way complex was minimized in NAMD, heated to 300 K for 5000–10,000 ps, equilibrated for 10,000 ps in the NVT ensemble at 1 atm and 300 K. The non-bonded interactions were switched at 8 Å and zero smoothly at 10 Å (cutoff 10 Å, switchdist 8 Å, nonbondedScaling 1, pairlistdist 11.5 Å, limitdist 0.5 Å). The temperature was maintained using of Langevin dynamics with a damping coefficient of 5.0 ps−1. In PBC, the wrapAll parameter for used to calculate all the coordinates around periodic boundaries. Electrostatic interactions in PBC were treated using the Particle Mesh Ewald (PME) method and PMEGridSpacing set at 1.0 Å. Covalent bonds with hydrogen atoms were kept rigid using ShakeH with the following parameters rigidbonds - all, rigidtolerance – 10−6 Å, and water molecules were kept rigid using Settle algorithm. The time step size for integration of each step of the simulation was 2 fs. All the other parameters were unchanged from the default settings in NAMD. The final production run was obtained from the last 200 ns of the simulation. The MD trajectories were analyzed in VMD, version 1.9.4a43.64 The RMSD of the protein was analyzed using RMSD Trajectory Tool in VMD. Before analysis, the snapshots were aligned to the first snapshot using the “backbone” option (atoms C, CA, and N).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH/NIAID grant R33AI127635 to DLW, GRJT, PAP, and FA. We are grateful to Elettra-Sincrotrone Trieste (Italy) and to the European synchrotron facility (ESRF, Grenoble, France) for granting beam time. M.A. has been supported by MIUR - Ministero dell’Istruzione Ministero dell’Università e della Ricerca (Ministry of Education, University and Research) under the national project FSE/FESR - PON Ricerca e Innovazione 2014–2020 (N° AIM1887574, CUP: E18H19000350007).
ABBREVIATIONS USED
- GR
glutathione reductase
- TrxR
thioredoxin reductase
- TGR
thioredoxin glutathione reductase
- SmTGR
TGR from Schistosoma mansoni
- FBDD
Fragment-Based Drug Discovery
- Sec, U
selenocysteine
- Trx
thioredoxin
- Grx
glutaredoxin
- GSH
glutathione
- NADPH
nicotinamide adenine dinucleotide phosphate
- FAD
flavin adenine dinucleotide
- qHTS
quantitative high throughput screen
- PanDDa
Pan-Dataset Density Analysis
- RSCCs
real space correlation coefficients
- DTNB
5,5-dithio-bis-(2-nitrobenzoic acid)
- bis-Tris
bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane
- Tris
tris(hydroxymethyl)aminomethane
- DTT
1,4-dithiothreitol
- MOE
Molecular Operating Environment
- PBC
periodic boundary conditions
- PME
Particle Mesh Ewald
Footnotes
SUPPORTING INFORMATION.
This information is available free of charge on the ACS Publications website.
Table S1. The fragments tested.
Table S2. Summary of X-ray data reduction and structural refinement statistics.
Table S3. B-factor analysis, estimated occupancy from the BDC values and the RSCC of the compounds bound to the SmTGR crystal structures presented in this study.
Table S4. Normalized B-factors (B’) of SmTGR in complex with 5 bound in Site 3 and in apo-form.
Figure S1. Stereo images of the compounds described in this study and their surrounding interacting residues.
Figure S2. Omit electron density maps of the compounds found in the crystal structures of SmTGR described in this study.
Figure S3. PanDDa on compound 1 bound to Site 1
Figure S4. PanDDa on compound 2 bound to Site 1
Figure S5. Time-dependent inactivation of SmTGR WT by compounds 3 and 4
Figure S6. Superposition of the SmTGR structures in complex with all the fragments bound in Site2 identified in this study and in previous studies.39
PDB ID Codes (compound number): 6ZST (1 in co-crystallization at 12.5 mM); 7NPX (1 in soaking at 25mM for 24h); 6ZP3 (2); 6ZLP (3); 6ZLB (4); 7B02 (5). Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
REFERENCES
- 1.Erlanson DA; Fesik SW; Hubbard RE; Jahnke W; Jhoti H, Twenty years on: the impact of fragments on drug discovery. Nature reviews. Drug discovery 2016, 15 (9), 605–619. DOI: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- 2.Price AJ; Howard S; Cons BD, Fragment-based drug discovery and its application to challenging drug targets. Essays in biochemistry 2017, 61 (5), 475–484. DOI: 10.1042/EBC20170029. [DOI] [PubMed] [Google Scholar]
- 3.Di Santo R, Inhibiting the HIV integration process: past, present, and the future. J Med Chem 2014, 57 (3), 539–66. DOI: 10.1021/jm400674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludlow RF; Verdonk ML; Saini HK; Tickle IJ; Jhoti H, Detection of secondary binding sites in proteins using fragment screening. Proc Natl Acad Sci U S A 2015, 112 (52), 15910–5. DOI: 10.1073/pnas.1518946112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel D; Bauman JD; Arnold E, Advantages of crystallographic fragment screening: functional and mechanistic insights from a powerful platform for efficient drug discovery. Progress in biophysics and molecular biology 2014, 116 (2–3), 92–100. DOI: 10.1016/j.pbiomolbio.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arner ES; Holmgren A, The thioredoxin system in cancer. Seminars in cancer biology 2006, 16 (6), 420–6. DOI: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Mustacich D; Powis G, Thioredoxin reductase. The Biochemical journal 2000, 346 Pt 1, 1–8. [PMC free article] [PubMed] [Google Scholar]
- 8.Arner ESJ, Targeting the Selenoprotein Thioredoxin Reductase 1 for Anticancer Therapy. Advances in cancer research 2017, 136, 139–151. DOI: 10.1016/bs.acr.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J; Li X; Han X; Liu R; Fang J, Targeting the Thioredoxin System for Cancer Therapy. Trends in pharmacological sciences 2017, 38 (9), 794–808. DOI: 10.1016/j.tips.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Boumis G; Giardina G; Angelucci F; Bellelli A; Brunori M; Dimastrogiovanni D; Saccoccia F; Miele AE, Crystal structure of Plasmodium falciparum thioredoxin reductase, a validated drug target. Biochemical and biophysical research communications 2012, 425 (4), 806–11. DOI: 10.1016/j.bbrc.2012.07.156. [DOI] [PubMed] [Google Scholar]
- 11.Kuntz AN; Davioud-Charvet E; Sayed AA; Califf LL; Dessolin J; Arnér ESJ; Williams DL, Thioredoxin Glutathione Reductase from Schistosoma mansoni: An Essential Parasite Enzyme and a Key Drug Target. PLOS Medicine 2007, 4 (6), e206. DOI: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boumis G; Angelucci F; Bellelli A; Brunori M; Dimastrogiovanni D; Miele AE, Structural and functional characterization of Schistosoma mansoni Thioredoxin. Protein science : a publication of the Protein Society 2011, 20 (6), 1069–76. DOI: 10.1002/pro.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimastrogiovanni D; Anselmi M; Miele AE; Boumis G; Petersson L; Angelucci F; Nola AD; Brunori M; Bellelli A, Combining crystallography and molecular dynamics: the case of Schistosoma mansoni phospholipid glutathione peroxidase. Proteins 2010, 78 (2), 259–70. DOI: 10.1002/prot.22536. [DOI] [PubMed] [Google Scholar]
- 14.Reich HJ; Hondal RJ, Why Nature Chose Selenium. ACS Chem Biol 2016, 11 (4), 821–41. DOI: 10.1021/acschembio.6b00031. [DOI] [PubMed] [Google Scholar]
- 15.Alger HM; Williams DL, The disulfide redox system of Schistosomamansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Molecular and Biochemical Parasitology 2002, 121 (1), 129–139. DOI: 10.1016/S0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 16.Angelucci F; Miele AE; Boumis G; Dimastrogiovanni D; Brunori M; Bellelli A, Glutathione reductase and thioredoxin reductase at the crossroad: The structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins: Structure, Function, and Bioinformatics 2008, 72 (3), 936–945. DOI: 10.1002/prot.21986. [DOI] [PubMed] [Google Scholar]
- 17.Williams DL; Bonilla M; Gladyshev VN; Salinas G, Thioredoxin glutathione reductase-dependent redox networks in platyhelminth parasites. Antioxid Redox Signal 2013, 19 (7), 735–45. DOI: 10.1089/ars.2012.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sayed AA; Simeonov A; Thomas CJ; Inglese J; Austin CP; Williams DL, Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nat Med 2008, 14 (4), 407–12. DOI: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.WHO WHO fact sheet no.115. http://www.who.int/mediacentre/factsheets/fs115/en/. (accessed November 1, 2020).
- 20.Vale N; Gouveia MJ; Rinaldi G; Brindley PJ; Gartner F; Correia da Costa JM, Praziquantel for Schistosomiasis: Single-Drug Metabolism Revisited, Mode of Action, and Resistance. Antimicrob Agents Chemother 2017, 61 (5). DOI: 10.1128/AAC.02582-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angelucci F; Dimastrogiovanni D; Boumis G; Brunori M; Miele AE; Saccoccia F; Bellelli A, Mapping the Catalytic Cycle of Schistosoma mansoni Thioredoxin Glutathione Reductase by X-ray Crystallography. Journal of Biological Chemistry 2010, 285 (42), 32557–32567. DOI: 10.1074/jbc.M110.141960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H-H; Day L; Cass CL; Ballou DP; Williams CH; Williams DL, Investigations of the Catalytic Mechanism of Thioredoxin Glutathione Reductase from Schistosoma mansoni. Biochemistry 2011, 50 (26), 5870–5882. DOI: 10.1021/bi200107n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fritz-Wolf K; Kehr S; Stumpf M; Rahlfs S; Becker K, Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat Commun 2011, 2, 383. DOI: 10.1038/ncomms1382. [DOI] [PubMed] [Google Scholar]
- 24.Fritz-Wolf K; Urig S; Becker K, The Structure of Human Thioredoxin Reductase 1 Provides Insights into C-terminal Rearrangements During Catalysis. Journal of Molecular Biology 2007, 370 (1), 116–127. DOI: 10.1016/j.jmb.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 25.Busker S; Qian W; Haraldsson M; Espinosa B; Johansson L; Attarha S; Kolosenko I; Liu J; Dagnell M; Grander D; Arner ESJ; Tamm KP; Page BDG, Irreversible TrxR1 inhibitors block STAT3 activity and induce cancer cell death. Science advances 2020, 6 (12), eaax7945. DOI: 10.1126/sciadv.aax7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyu H; Petukhov PA; Banta PR; Jadhav A; Lea W; Cheng Q; Arner ESJ; Simeonov A; Thatcher GR; Angelucci F; Williams DL, Characterization of lead compounds targeting the selenoprotein thioredoxin glutathione reductase for treatment of schistosomiasis. ACS Infect Dis 2020, 6 (3), 393–405. DOI: 10.1021/acsinfecdis.9b00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stafford WC; Peng X; Olofsson MH; Zhang X; Luci DK; Lu L; Cheng Q; Trésaugues L; Dexheimer TS; Coussens NP; Augsten M; Ahlzén H-SM; Orwar O; Östman A; Stone-Elander S; Maloney DJ; Jadhav A; Simeonov A; Linder S; Arnér ESJ, Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Science Translational Medicine 2018, 10 (428), eaaf7444. DOI: 10.1126/scitranslmed.aaf7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearce NM; Krojer T; Bradley AR; Collins P; Nowak RP; Talon R; Marsden BD; Kelm S; Shi J; Deane CM; von Delft F, A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat Commun 2017, 8, 15123. DOI: 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cousido-Siah A; Petrova T; Hazemann I; Mitschler A; Ruiz FX; Howard E; Ginell S; Atmanene C; Van Dorsselaer A; Sanglier-Cianferani S; Joachimiak A; Podjarny A, Crystal packing modifies ligand binding affinity: the case of aldose reductase. Proteins 2012, 80 (11), 2552–61. DOI: 10.1002/prot.24136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrmann FR; Stojko J; Metz A; Debaene F; Barandun LJ; Heine A; Diederich F; Cianferani S; Reuter K; Klebe G, Soaking suggests “alternative facts”: Only co-crystallization discloses major ligand-induced interface rearrangements of a homodimeric tRNA-binding protein indicating a novel mode-of-inhibition. PLoS One 2017, 12 (4), e0175723. DOI: 10.1371/journal.pone.0175723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geremia S; Campagnolo M; Demitri N; Johnson LN, Simulation of diffusion time of small molecules in protein crystals. Structure 2006, 14 (3), 393–400. DOI: 10.1016/j.str.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Mullard A, Fragment-based screening sees the light. Nature reviews. Drug discovery 2020, 19 (11), 742–743. DOI: 10.1038/d41573-020-00181-2. [DOI] [PubMed] [Google Scholar]
- 33.Berkholz DS; Faber HR; Savvides SN; Karplus PA, Catalytic cycle of human glutathione reductase near 1 A resolution. J Mol Biol 2008, 382 (2), 371–84. DOI: 10.1016/j.jmb.2008.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellelli A; Carey J, Reversible Ligand Binding: Theory and Experiment. John Wiley & Sons Ltd.: Hoboken, NJ, 2018. DOI: 10.1002/9781119238508. [DOI] [Google Scholar]
- 35.Cozzolino AF; Vargas-Baca I; Mansour S; Mahmoudkhani AH, The nature of the supramolecular association of 1,2,5-chalcogenadiazoles. Journal of the American Chemical Society 2005, 127 (9), 3184–90. DOI: 10.1021/ja044005y. [DOI] [PubMed] [Google Scholar]
- 36.Rakitin OA; Zibarev AV, Synthesis and Applications of 5-Membered Chalcogen-Nitrogen π-Heterocycles with Three Heteroatoms. Asian Journal of Organic Chemistry 2018, 7 (12), 2397–2416. DOI: 10.1002/ajoc.201800536. [DOI] [Google Scholar]
- 37.Zhao R; Masayasu H; Holmgren A, Ebselen: a substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proc Natl Acad Sci U S A 2002, 99 (13), 8579–84. DOI: 10.1073/pnas.122061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eggler AL; Savinov SN, Chemical and biological mechanisms of phytochemical activation of Nrf2 and importance in disease prevention. Recent advances in phytochemistry 2013, 43, 121–155. DOI: 10.1007/978-3-319-00581-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silvestri I; Lyu H; Fata F; Banta PR; Mattei B; Ippoliti R; Bellelli A; Pitari G; Ardini M; Petukhova V; Thatcher GRJ; Petukhov PA; Williams DL; Angelucci F, Ectopic suicide inhibition of thioredoxin glutathione reductase. Free Radic Biol Med 2019, 147, 200–211. DOI: 10.1016/j.freeradbiomed.2019.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silvestri I; Lyu H; Fata F; Boumis G; Miele AE; Ardini M; Ippoliti R; Bellelli A; Jadhav A; Lea WA; Simeonov A; Cheng Q; Arner ESJ; Thatcher GRJ; Petukhov PA; Williams DL; Angelucci F, Fragment-Based Discovery of a Regulatory Site in Thioredoxin Glutathione Reductase Acting as “Doorstop” for NADPH Entry. ACS Chem Biol 2018, 13 (8), 2190–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sondergaard CR; Garrett AE; Carstensen T; Pollastri G; Nielsen JE, Structural artifacts in protein-ligand X-ray structures: implications for the development of docking scoring functions. J Med Chem 2009, 52 (18), 5673–84. DOI: 10.1021/jm8016464. [DOI] [PubMed] [Google Scholar]
- 42.Cheng Q; Sandalova T; Lindqvist Y; Arner ES, Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J Biol Chem 2009, 284 (6), 3998–4008. DOI: 10.1074/jbc.M807068200. [DOI] [PubMed] [Google Scholar]
- 43.Johnson TW; Gallego RA; Brooun A; Gehlhaar D; McTigue M, Reviving B-Factors: Retrospective Normalized B-Factor Analysis of c-ros Oncogene 1 Receptor Tyrosine Kinase and Anaplastic Lymphoma Kinase L1196M with Crizotinib and Lorlatinib. ACS Med Chem Lett 2018, 9 (9), 878–883. DOI: 10.1021/acsmedchemlett.8b00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuzmanic A; Pannu NS; Zagrovic B, X-ray refinement significantly underestimates the level of microscopic heterogeneity in biomolecular crystals. Nat Commun 2014, 5, 3220. DOI: 10.1038/ncomms4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nussinov R; Tsai CJ, Allostery without a conformational change? Revisiting the paradigm. Current opinion in structural biology 2015, 30, 17–24. DOI: 10.1016/j.sbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 46.Tsai CJ; del Sol A; Nussinov R, Allostery: absence of a change in shape does not imply that allostery is not at play. J Mol Biol 2008, 378 (1), 1–11. DOI: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butt TR; Edavettal SC; Hall JP; Mattern MR, SUMO fusion technology for difficult-to-express proteins. Protein expression and purification 2005, 43 (1), 1–9. DOI: 10.1016/j.pep.2005.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mossessova E; Lima CD, Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Molecular cell 2000, 5 (5), 865–76. DOI: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 49.Reverter D; Lima CD, Preparation of SUMO proteases and kinetic analysis using endogenous substrates. Methods Mol Biol 2009, 497, 225–39. DOI: 10.1007/978-1-59745-566-4_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inglese J; Auld DS; Jadhav A; Johnson RL; Simeonov A; Yasgar A; Zheng W; Austin CP, Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A 2006, 103 (31), 11473–8. DOI: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lausi A; Polentarutti M; Onesti S; Plaisier JR; Busetto E; Bais G; Barba L; Cassetta A; Campi G; Lamba D; Pifferi A; Mande SC; Sarma DD; Sharma SM; Paolucci G, Status of the crystallography beamlines at Elettra. The European Physical Journal Plus 2015, 130 (3), 43. DOI: 10.1140/epjp/i2015-15043-3. [DOI] [Google Scholar]
- 52.Kabsch W, Xds. Acta crystallographica. Section D, Biological crystallography 2010, 66 (Pt 2), 125–32. DOI: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography 2011, 67 (Pt 4), 235–42. DOI: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. Journal of applied crystallography 2007, 40 (Pt 4), 658–674. DOI: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta crystallographica. Section D, Biological crystallography 2010, 66 (Pt 4), 486–501. DOI: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Echols N; Moriarty NW; Klei HE; Afonine PV; Bunkoczi G; Headd JJ; McCoy AJ; Oeffner RD; Read RJ; Terwilliger TC; Adams PD, Automating crystallographic structure solution and refinement of protein-ligand complexes. Acta crystallographica. Section D, Biological crystallography 2014, 70 (Pt 1), 144–54. DOI: 10.1107/S139900471302748X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA, REFMAC5 for the refinement of macromolecular crystal structures. Acta crystallographica. Section D, Biological crystallography 2011, 67 (Pt 4), 355–67. DOI: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gore S; Sanz Garcia E; Hendrickx PMS; Gutmanas A; Westbrook JD; Yang H; Feng Z; Baskaran K; Berrisford JM; Hudson BP; Ikegawa Y; Kobayashi N; Lawson CL; Mading S; Mak L; Mukhopadhyay A; Oldfield TJ; Patwardhan A; Peisach E; Sahni G; Sekharan MR; Sen S; Shao C; Smart OS; Ulrich EL; Yamashita R; Quesada M; Young JY; Nakamura H; Markley JL; Berman HM; Burley SK; Velankar S; Kleywegt GJ, Validation of Structures in the Protein Data Bank. Structure 2017, 25 (12), 1916–1927. DOI: 10.1016/j.str.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krissinel E; Henrick K, Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta crystallographica. Section D, Biological crystallography 2004, 60 (Pt 12 Pt 1), 2256–68. DOI: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 60.LLC, C. C. G. Molecular Operating Environment (MOE), Chemical Computing Group LLC: 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2020. [Google Scholar]
- 61.Phillips JC; Hardy DJ; Maia JDC; Stone JE; Ribeiro JV; Bernardi RC; Buch R; Fiorin G; Henin J; Jiang W; McGreevy R; Melo MCR; Radak BK; Skeel RD; Singharoy A; Wang Y; Roux B; Aksimentiev A; Luthey-Schulten Z; Kale LV; Schulten K; Chipot C; Tajkhorshid E, Scalable molecular dynamics on CPU and GPU architectures with NAMD. The Journal of chemical physics 2020, 153 (4), 044130. DOI: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerber PR; Muller K, MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. Journal of computer-aided molecular design 1995, 9 (3), 251–68. DOI: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- 63.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of chemical theory and computation 2015, 11 (8), 3696–713. DOI: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humphrey W; Dalke A; Schulten K, VMD: visual molecular dynamics. Journal of molecular graphics 1996, 14 (1), 33–8, 27–8. DOI: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.