

Abstract
Regeneration of large bones remains a challenge in surgery. Recent developmental engineering efforts aim to recapitulate endochondral ossification (EO), a critical step in bone formation. However, this process entails the condensation of mesenchymal stem cells (MSCs) into cartilaginous templates, which requires long-term cultures and is challenging to scale up. Here, we develop a biomimetic scaffold that allows rapid and self-sustained EO without initial hypertrophic chondrogenesis. The design comprises a porous chondroitin sulfate (CS) cryogel decorated with whitlockite calcium phosphate nanoparticles, and a soft hydrogel occupying the porous space. This composite scaffold enables human endothelial colony-forming cells (ECFCs) and MSCs to rapidly assemble into osteovascular niches in immunodeficient mice. These niches contain ECFC-lined blood vessels and perivascular MSCs that differentiate into RUNX2+OSX+ pre-osteoblasts after one week in vivo. Subsequently, multiple ossification centers are formed, leading to de novo bone tissue formation by eight weeks, including mature human OCN+OPN+ osteoblasts, collagen-rich mineralized extracellular matrix, hydroxyapatite, osteoclast activity, and gradual mechanical competence. The early establishment of blood vessels are essential, and grafts that do not contain ECFCs fail to produce osteovascular niches and ossification centers. Our findings suggest a novel bioengineering approach to recapitulate EO in the context of human bone regeneration.
Keywords: bone tissue engineering, endochondral ossification, osteovascular niche, ossification center, developmental engineering
Graphical Abstract
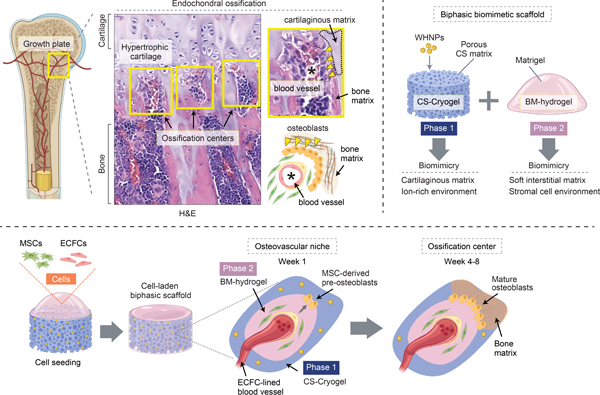
This article introduces a biomimetic biphasic scaffold that allows self-sustained endochondral ossification (EO), a critical step in bone formation. Following seeding with human vascular and mesenchymal stem cells, this biphasic material enabled rapid formation of osteovascular niches and ossification centers, ultimately leading to de novo bone tissue formation in vivo. This novel biomaterial could facilitate human bone regeneration.
1. Introduction
Regeneration of massive bone defects remains an enormous challenge in surgery. Current approaches to defect repair primarily include bone autografts and allografts.[1] However, these practices are limited by donor tissue availability and site morbidity (autografts) and the risks of immunological rejection and transfer of diseases (allografts). Alternative approaches include the use of artificial bone scaffolds, which have the advantage of unlimited supply.[2] However, synthetic materials often underperform in critical-sized bone defects due to poor integration, insufficient vascularization, and slow bone growth, impeding the regeneration of a functional bone.[3]
Replacing traditional non-degradable materials (e.g., metal and non-bioceramics) with bioengineered bone tissues generated from patients’ autologous cells could transform the treatment of critical-sized bone defects.[4] Synthetic scaffolds containing in vitro expanded osteoprogenitor cells – primarily, mesenchymal stem cells (MSCs) - and osteoinductive agents such as bone morphogenetic protein-2 (BMP2), have been extensively studied in the context of bone tissue engineering.[5] These approaches generate bone tissue by a process resembling intramembranous ossification (i.e., by direct osteoblastic differentiation), which primarily achieves mineralization of the materials.[6] Unfortunately, the resulting mineralized constructs generally lack proper vascularization and fail to integrate as bone grafts [7]. Moreover, high doses of bone morphogenetic factors can undesirably cause heterotopic bone formation and soft tissue inflammation, and their clinical use remains under debate.[8]
Postnatally, natural bone healing occurs by reactivating endochondral ossification (EO), a developmental process by which bone tissue develops from a cartilage intermediate.[9] EO is a critical step in mammalian skeleton formation, especially long bones (Figure 1A). During EO, cartilage is first formed as a framework to guide the subsequent ossification. Cellular condensation and hypertrophic chondrogenesis progressively produce a semi-solid matrix consisting of chondroitin sulfate, hyaluronic acid, and collagen fibers. As the matrix accumulates, hypertrophic chondrocytes die, leaving behind a macroporous space. Blood vessels and osteoprogenitor cells are recruited into the porous cartilaginous matrix, forming a niche that initiates the formation of a bone collar by perichondral ossification.[9] Each of these endochondral niches contains a vascular network, osteoprogenitor cells, and chondrocyte-derived matrix, forming the primary osteogenic center for rapid bone growth and regeneration (Figure 1A).
Figure 1.
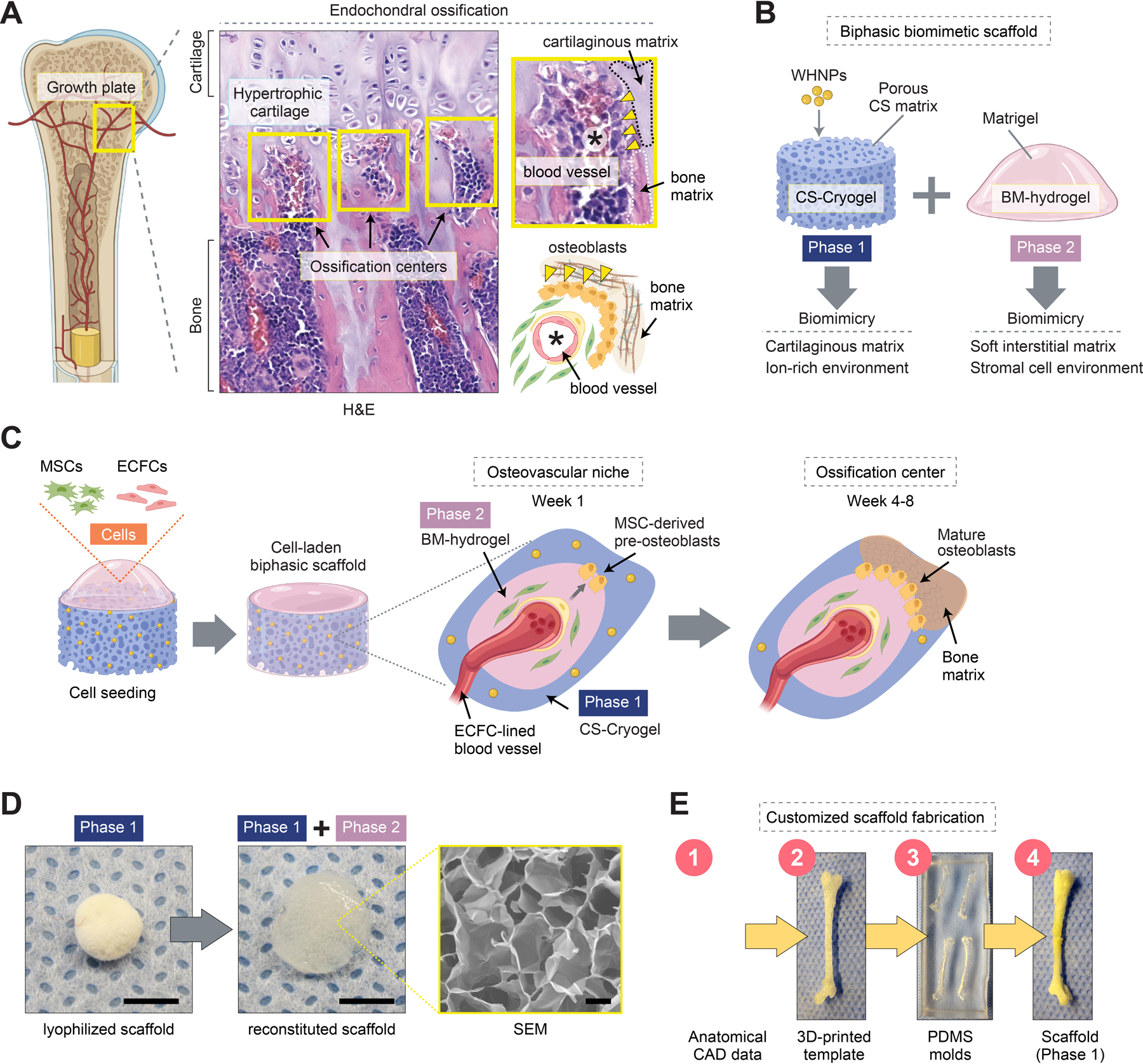
A biomimetic biphasic scaffold to recapitulate endochondral ossification. A) Schematic of endochondral ossification at the growth plate of a long bone. Histological (H&E) staining of a mouse femur reveals ossification centers (yellow boxes) adjacent to the hypertrophic cartilaginous matrix. Ossification centers contain central blood vessels, osteoblast lining, and bone matrix. B) Schematic depicting the composition of the biphasic biomimetic scaffold. Phase 1 is composed of a chondroitin sulfate (CS) matrix decorated with whitlockite calcium phosphate nanoparticles (WHNPs). Phase 2 is composed of Matrigel, a basement membrane (BM) hydrogel. C) Schematic depicting cell seeding. The cells (MSCs + ECFCs) occupy the scaffold’s porous space and mediate the progressive formation of osteovascular niches and then ossification centers in vivo. D) Scaffold fabricated using a cylindrical mold (4 mm in diameter and 1 mm in height). Macroscopic photographs of the lyophilized (Phase 1) and reconstituted (Phase 1 + Phase 2) scaffold. E) Schematic depicting the fabrication process of a scaffold with a customized anatomical geometry.
In the context of bone tissue engineering, recent efforts have been directed towards trying to recapitulate the EO process - a paradigm referred to as developmental engineering.[10] In this strategy, MSCs are condensed into aggregates that form cartilage templates that then undergo hypertrophy and progress through subsequent EO steps after implantation in vivo. Indeed, multiple studies have shown that this approach faithfully reproduces the EO process from the onset.[10a, 11] However, the process of chondrogenic priming is cumbersome and requires prolonged periods of in vitro culture (> 4 weeks) in the presence of exogenous growth factors. Furthermore, MSC condensation tends to form aggregates with a spherical shape, making it challenging to fabricate grafts with different configurations and scales.[12] From a translational standpoint, these limitations are problematic, and thus the search for alternative developmental engineering approaches to EO continues to be a priority in osteoregenerative medicine [10b, 13].
In the present study, we demonstrate that the early stages of EO (i.e., mesenchymal condensation, chondrogenesis, and cartilage hypertrophy) can be effectively bypassed by strategically combining biomaterials and cells (Figure 1B). First, instead of relying on lengthy MSC condensations to form the initial cartilage, we show that a porous cryogel scaffold that is composed of chondroitin sulfate (CS) and decorated with whitlockite calcium phosphate nanoparticles can serve as a functional cartilaginous template. Of note, this composite scaffold does not require in vitro preconditioning and could be used as an off-the-shelf material for cell seeding. Second, we show that by occupying the scaffold’s porous space with a basement membrane (BM)-based hydrogel loaded with vascular and osteoprogenitor cells, one can effectively initiate EO at the stage at which blood vessels and osteoblasts come together to form the initial osteovascular niches (Figure 1C). Indeed, in this study we show that, upon implantation in vivo, our composite grafts can recapitulate the cellular, morphogenic, and mechanical cues necessary for the formation of functional bone via the endochondral route. We envision this bioengineering approach could be harnessed to regenerate sizeable segmental bone defects.
2. Results and Discussion
2.1. Fabrication of a functional biphasic scaffold for EO
The composition of the cryogel scaffold material is critical, including both the core CS matrix and the decorating whitlockite (Ca18Mg2(HPO4)2(PO4)12) nanoparticles (referred to as WHNPs). Previously, we have shown that these WHNPs (100–150 nm size), which are synthesized by a wet precipitation method, are capable of sustaining bone formation by stimulating osteogenic differentiation, preventing osteoclastic activity, and enabling the formation of mechanically enhanced hydroxyapatite via a continuous supply of Ca2+, PO43− and Mg2+.[14] Here, we selected an concentration of WHNPs (6% w/v) to match ions compositions comparable to those during bone remodeling [15] (see Figure S1, Supporting Information, for total release of calcium, magnesium, and phosphorous ions from the WHNPs-doped scaffolds). We incorporated the WHNPs into a methacrylated CS polymer solution to fabricate the CS-cryogel. To this end, we used a thermal initiator (ammonium persulfate, APS) and an accelerator (tetramethylethylenediamine, TEMED) as crosslinking agents. For this proof-of-concept study, we polymerized the CS-cryogel into disk shape scaffolds using a simple cylindrical mold (4 mm in diameter and 1 mm in height) (Figure 1D). However, using more advanced 3D-printed mold casting, we also showed that the same fabrication approach could be applied to generate surgical grafts with the complex structures of actual bones, including murine femurs (Figure 1E).
The provision of an interconnected porous network within the scaffold is an equally essential part of the design.[16] We created this porosity by first freezing down the CS-cryogel at −20°C for 20 h and then lyophilizing them ahead of their laboratory usage. The lyophilization process created highly porous structures within the scaffold that partially resembled the zonal cartilage and woven bone of the native growth plate, although with lower porosity (Figure 1A). The lyophilized CS-cryogel was reconstituted and swelled back to its original size upon incubation with a cold Matrigel solution, which served as the BM-based hydrogel filling up the porous structures (see Figure 1D for macroscopic view of the scaffolds prior to and after hydrogel incorporation). The final reconstituted CS-cryogel/BM-hydrogel composite scaffolds had a fixed shape but were sufficiently flexible for surgical handling and manipulation.
We evaluated our reconstituted cell-laden composite scaffolds by several means. First, we examined the capacity to release ions and demonstrated that, under physiological conditions, the presence of the WHNPs enabled a sustained mineral ion-rich environment for up to 8 weeks (Figure S1, Supporting Information), providing an essential substrate for bone formation. The reconstituted scaffolds exhibited a distinct interconnected porous network, with an average Feret’s pore size of 207.69 ± 201.99 µm and a porosity of 40.59 ± 14.08% (Figure S2, Supporting Information; note that cryogel formation process produces scaffolds with smaller pores in the periphery than in the center, thus the wide range of pore size distribution), providing a morphological trabeculae-like architecture critical for facilitating cell migration, vascular morphogenesis and vascularization, and creating longitudinal septa to guide ossification. Measurements of the local stiffness by nanoindentation revealed the intended biphasic mechanical structure of our CS-cryogel/BM-hydrogel composite scaffold (Figure S1, Supporting Information). On the one hand, the soft BM-hydrogel phase had a hardness of 0.15 ± 0.13 GPa, which permitted vascular cell motility and self-assembly into functional blood vessels.[17] The softness of the hydrogel, which filled all the porous channels of the scaffold, also facilitated the infiltration of host cells that are known to mediate vascularization.[18] On the other hand, the CS-cryogel exhibited a higher stiffness (0.34 ± 0.19 GPa), similar to that of the cartilaginous matrix during EO.[19] Together, the biphasic mechanical properties (i.e., soft for blood vessel formation and stiffer for osteoinduction) and the matrix-derived biological cues (BM-hydrogel and CS-cryogel components inducing angiogenesis and osteogenesis, respectively) enabled our composite scaffold to serve as a biomimetic template for EO (Figure 1C).
2.2. Graft vascularization and formation of functional osteovascular niches
Next, we assessed the progressive formation of blood vessels and bone tissue within the scaffolds following implantation in vivo (Figure 2A). To this end, the scaffolds were seeded with human vascular progenitor and osteoprogenitor cells embedded into the BM-hydrogel phase. Specifically, we used human blood-derived endothelial colony-forming cells (ECFCs) and bone marrow-derived MSCs, both accessible progenitor cell sources in an autologous clinical setting.[20] Previously, we have shown that this combination of cells displays robust vasculogenic properties in various soft hydrogels, including Matrigel.[18a, 18b, 20–21] We compared seeding MSCs with ECFCs (ratio 3:2, total cell number 2×106) vs. MSCs alone (total cell number 1.2×106). In both groups, the cells were resuspended in 50-µL of Matrigel, and the mixture loaded into the lyophilized CS-cryogel (4–5 mg weight). The scaffolds completely absorbed the cell-laden Matrigel within 30 s, and cells were uniformly distributed throughout the interconnect porous spaces. The cell-seeded scaffolds were incubated at 37°C for 10 min to allow Matrigel’s gelation before implantation.
Figure 2.
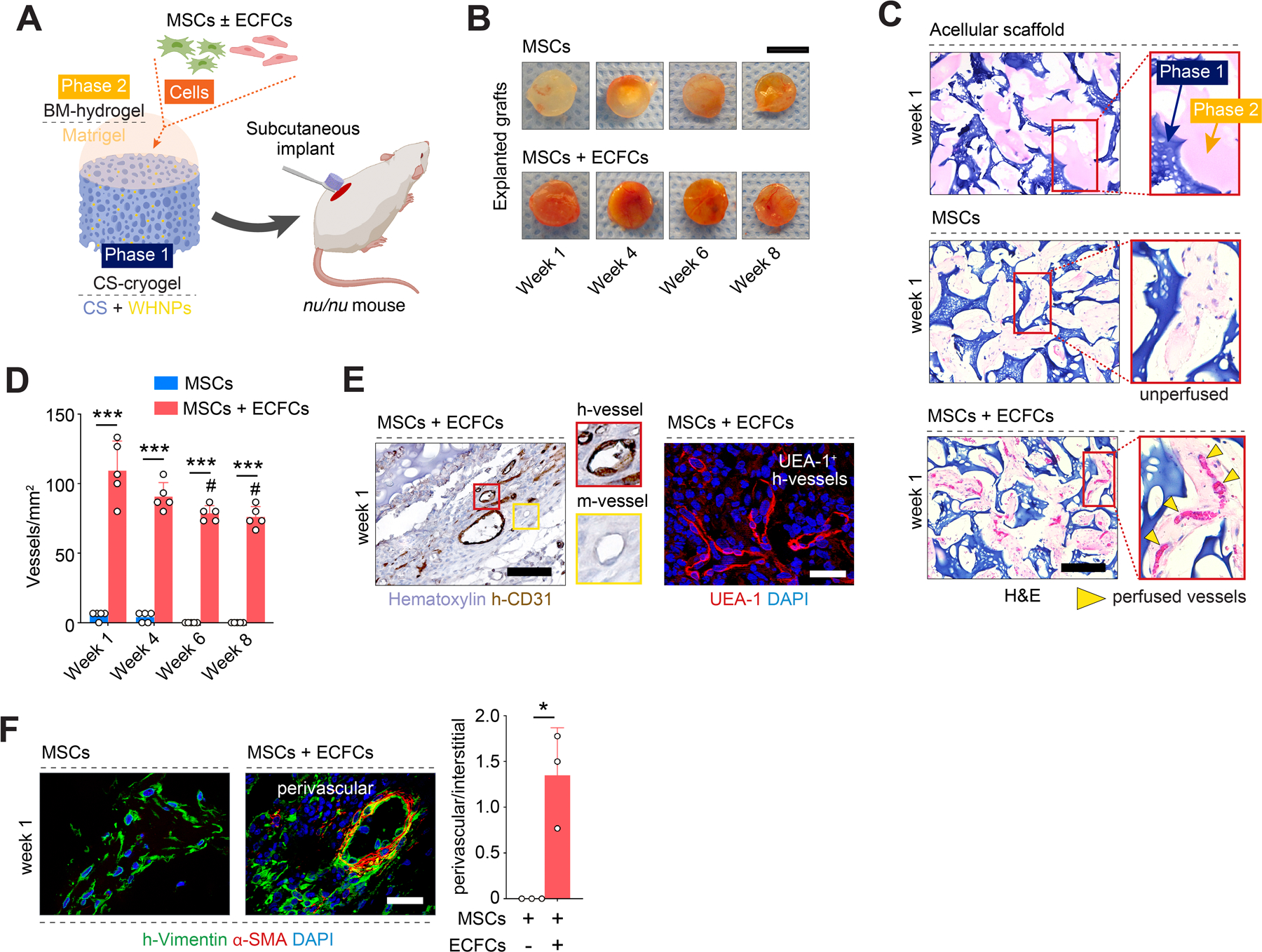
Rapid formation of osteovascular niches in vivo. A) Schematic illustration depicting composite scaffolds loaded with human MSCs with or without ECFCs and implanted into immunodeficient mice. B) Macroscopic views of the explanted grafts. C) Histological (H&E) staining of grafts at week 1. Blue and pink colors represent the CS-cryogel (Phase 1) and BM-hydrogel (Phase 2) of the scaffold. Grafts seeded with MSCs + ECFCs exhibited numerous perfused blood vessels containing erythrocytes (yellow arrowheads). MSCs grafts were largely unperfused. D) Total perfused microvessel density quantified in explanted grafts at weeks 1–8. E) Human microvessels identified in grafts seeded with MSCs + ECFCs at week 1 by immunostaining for human-specific CD31 (h-CD31) and UEA-1 lectin binding. F) Engraftment of MSCs as either interstitial (h-vimentin+α-SMA-) or perivascular (h-vimentin+α-SMA+) cells visualized and quantified by immunofluorescent staining at week 1. In all quantitative panels, bars represent mean ± s.d. (n ≥ 3). *p < 0.05, *** p < 0.001. # p < 0.01 compared to MSCs + ECFCs at week 1. Statistical methods: ANOVA followed by Bonferroni’s post-test analysis (D), and unpaired two-tailed Student’s t-tests (F). Scale bars = 4 mm (B), 100 µm (C, E, F).
We implanted the composite scaffolds into the subcutaneous space of immunodeficient nude mice (Figure 2A). We intentionally chose an ectopic transplantation site as it enabled studying EO without interference from endogenous osteogenic signals present at orthotopic bone sites. First, we evaluated initial cell engraftment and scaffold vascularization on day 7. Macroscopic examination of the explants suggested robust vascularization of grafts that were seeded with MSCs + ECFCs (red color usually indicates perfused tissues) (Figure 2B). Indeed, histological (hematoxylin and eosin, H&E) analysis revealed that scaffolds with MSCs + ECFCs had an extensive network of microvessels that contained murine erythrocytes in their lumens, indicating perfusion (Figure 2C and S3, Supporting Information. Note the presence of Matrigel in the porous space of the cryogel in H&E-stained sections, where blue and pink colors represent the CS-cryogel and BM-hydrogel phases of the scaffold, respectively. Also note that acellular scaffolds exhibited minimal infiltration of host cells and completely lack blood vessels). The differences in microvessels density between grafts with MSCs + ECFCs (109.33 ± 21.40 vessels/mm2) and those with MSCs alone (5.34 ± 2.98 vessels/mm2) were significant at week 1 and remained significant up to week 8 (Figure 2D).
The blood vessels in the MSCs + ECFCs grafts were exclusively located in the scaffolds’ soft BM-hydrogel phase (pink in H&E micrographs; Figure 2C), without any noticeable ingrowth into the CS-cryogel phase (blue). Of note, microvessels were primarily lined by human ECFCs, as confirmed by the expression of human-specific endothelial marker CD31 as well as by UEA-1, a lectin that binds with high affinity to human, but not murine, endothelial cells (Figure 2E and S4, Supporting Information). The presence of perfused human vessels indicated that ECFCs had assembled into lumenal structures and formed functional anastomoses with the host circulatory system. The human microvessels were uniformly distributed throughout the scaffold’s interconnect porous structures - each porous chamber had an average of 3.43 ± 1.17 blood vessels in the center (Figure S2, Supporting Information). There was no evidence of inflammatory cells (i.e., Ly6G+ polymorphonuclear cells) on day 7 (Figure S5, Supporting Information). However, the grafts contained F4/80+ macrophages (Figure S5, Supporting Information), consistent with previous descriptions of active post-vascularization tissue remodeling in hydrogel grafts [18c]. Also, apoptotic cell presence was minimal at week 1 (Figure S5, Supporting Information), suggesting stable cell engraftment.
The importance of having a robust vascular network in the grafts is two-fold. First, blood vessels are critical for maintaining adequate oxygenation, nutrients distribution, and waste removal in the grafts.[22] Second, the vasculature plays a central role in EO; osteogenesis is tightly coupled to vascularization as blood vessels provide a critical niche for the osteoprogenitor cells.[23] Previously, we showed that vessels lined by ECFCs could serve as functional niches for MSCs in the context of osteogenesis, a process mediated via PDGF-BB/PDGFR-β signaling.[24] We demonstrated that when MSCs engraft in perivascular positions, they retain their osteogenic differentiation potential. However, when MSCs engrafted interstitially, without blood vessels’ proximity, they rapidly become fibroblast-like cells and lose most of their regenerative capacity. Here, in grafts seeded with ECFCs, ∼57% of human MSCs were at perivascular locations at week 1, as revealed by the expression of human-specific vimentin and alpha-smooth muscle actin (α-SMA, a perivascular cell marker) (Figure 2F). In contrast, in the absence of ECFCs, grafts notably lacked blood vessels (Figure 2B-D and S4, Supporting Information), and MSCs occupied interstitial locations (Figure 2F).
The presence of ECFCs (and the vessels they form) was of central importance to recapitulate EO as they mediated the engraftment mode of MSCs (perivascular vs. interstitial), which, in turn, had a significant effect on preserving their osteogenic potential. Also, the proximity of the blood vessels and their perivascular MSCs to the cartilaginous CS-cryogel phase of the scaffold (marked by safranin O staining in Figure 3A) resembled the stage in EO at which capillaries infiltrate into the hypertrophic cartilage to form the first osteovascular niches. Indeed, grafts seeded with ECFCs, but not those with only MSCs, displayed signs of early-stage osteogenic differentiation after just one week in vivo, including significant upregulation of human-specific Runt-related transcription factor 2 (RUNX2), Osterix (OSX), and alkaline phosphatase (ALP) expression (Figure 3B,C and S6, Supporting Information). The abundance of these human RUNX2+OSX+ pre-osteoblasts was significantly higher in the presence of ECFCs compared to grafts with only MSCs (44.48 ± 6.51% vs 12.94 ± 4.26% for RUNX2, and 24.92 ± 5.57% vs. 14.89 ± 2.50% for OSX) (Figure S6, Supporting Information). It is important to note that the appearance of pre-osteoblasts occurred without the addition of any exogenous osteoinductive growth factor such as BMP2. Instead, the composition of the scaffold itself provided a permissive environment to initiate EO. The composite material enabled human ECFCs and MSCs to rapidly assemble into functional osteovascular niches after ectopic implantation in immunodeficient mice. After one week in vivo, these osteovascular niches contained ECFC-lined blood vessels and perivascular MSCs that had effectively differentiated into RUNX2+OSX+ pre-osteoblasts (Figure 3C, D).
Figure 3.
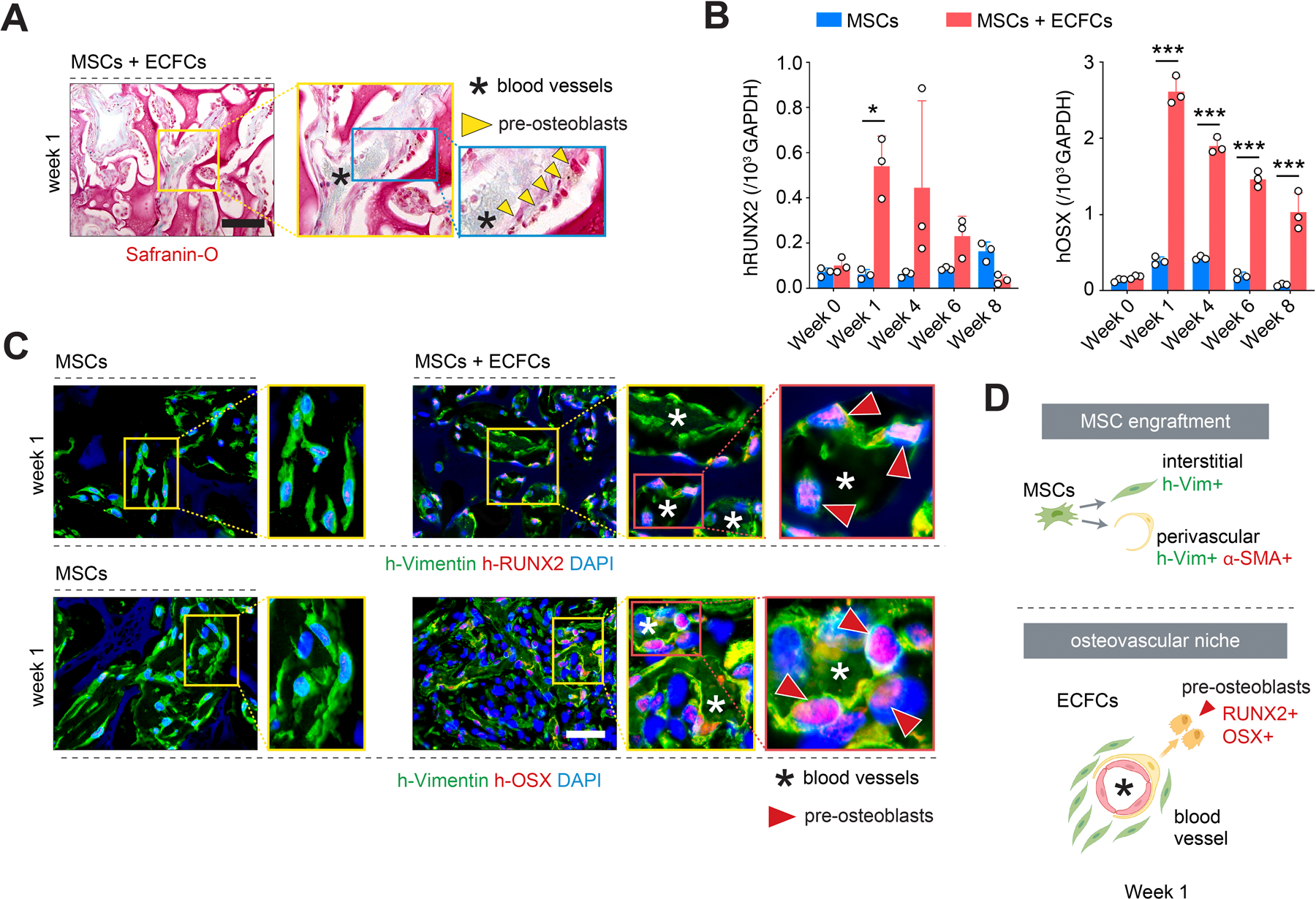
Differentiation of MSCs into pre-osteoblasts at the osteovascular niches. A) Safranin O staining of grafts seeded with MSCs + ECFCs revealed numerous central blood vessels (asterisks) surrounded by a lining of osteoblast-like cells (yellow arrowheads) in the proximity of the cartilaginous CS-cryogel phase (red color) of the scaffold. B) Quantitative real-time PCR analysis of human RUNX2 and OSX mRNA expression levels in explanted grafts at weeks 1–8. Data normalized to human GAPDH. All primers recognized human-specific transcripts. C) Immunofluorescent staining for pre-osteoblast markers RUNX2 and OSX at week 1. Human pre-osteoblasts (red arrowheads) were identified in grafts seeded with MSCs + ECFCs but not in grafts with only MSCs. D) Schematic illustration depicting the dichotomous engraftment mode of MSCs (top) and the formation of osteovascular niches (bottom) containing ECFC-lined blood vessels, perivascular MSCs, and RUNX2+OSX+ pre-osteoblast. In all quantitative panels, bars represent mean ± s.d. (n ≥ 3). *p < 0.05, *** p < 0.001. Statistical method: ANOVA followed by Bonferroni’s post-test analysis. Scale bars = 100 µm (A,C).
2.3. Formation of ossification centers and tissue mineralization
Next, we studied the ossification of our grafts over 8 weeks in vivo (Figure 4A). During this period, grafts seeded with MSCs + ECFCs remained thoroughly vascularized, although there was a gradual decrease in microvessels density (Figure 2D), consistent with adjustments in metabolic demand during tissue remodeling [20b]. In contrast, grafts with only MSCs continuously failed to recruit blood vessels and remodeled into avascular fibrotic tissues over time. Between weeks 4 to 6, histological (H&E) examination of the MSCs + ECFCs grafts revealed distinct modular ossification centers residing in the BM-hydrogel phase of the scaffolds (Figure 4B and S7, Supporting Information). Each of these osteogenic units contained at least one central ECFC-lined blood vessel that was invested by h-vimentin+α-SMA+ perivascular cells. The vessels were surrounded by concentrically aligned human h-vimentin+α-SMAdim interstitial cells, consistent with the layers found in bona fide ossification center structures.[25] Notably, these osteogenic units had a distinct lining of human oval cells at the interface between the scaffold’s CS-cryogel and the soft BM-hydrogel phase, resembling osteoblasts found at the ossification center cortex (Figure 4B, inset). These osteoblast-like cells had low expression of h-vimentin, expressed late-stage osteogenic markers osteocalcin (OCN) and osteopontin (OPN; Figure 4C), and originated from the human MSCs as confirmed by immunostaining with a human-specific mitochondria antibody (Figure S8, Supporting Information). Together, these data suggested a functional differentiation of MSCs into mature osteoblasts in grafts containing ECFCs. This differentiation from pre- to mature osteoblasts occurred progressively over the eight weeks in vivo, as revealed by a gradual downregulation of early osteogenic markers (RUNX2, OSX, and ALP; Figure 3B and S6, Supporting Information) that coincided with an increased presence of mature OCN+ OPN+ osteoblasts (Figure 4C,D and S9, Supporting Information). In contrast, grafts seeded with MSCs alone lack ossification centers and showed no induction of osteogenic markers, and no presence of osteoblasts throughout the entire period in vivo.
Figure 4.
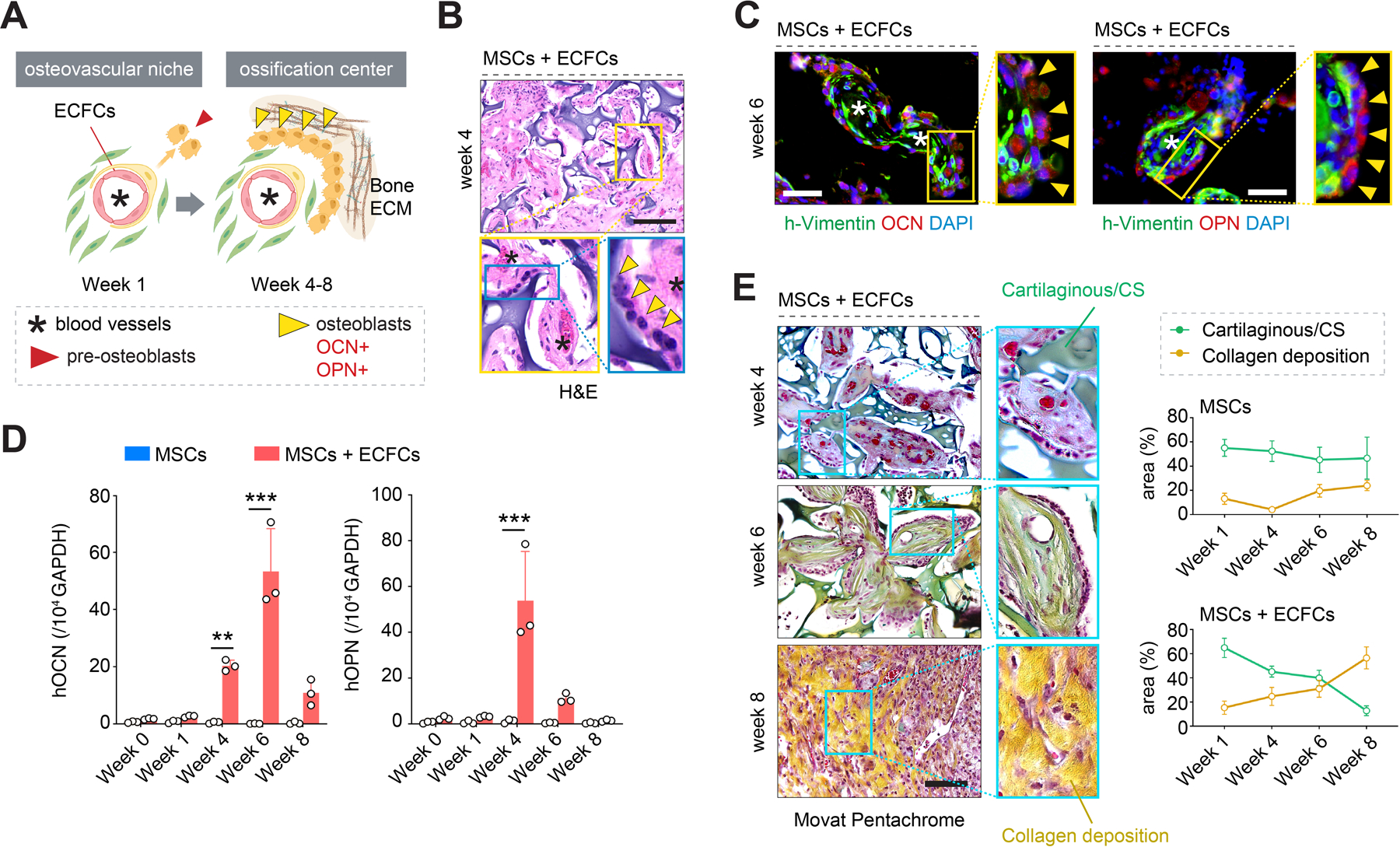
Generation of functional ossification centers. A) Schematic illustration depicting an initial osteovascular niche and its progression towards a functional ossification center. B) Histological (H&E) staining of grafts seeded with MSCs + ECFCs at week 4 revealed ossification centers containing central vessels (asterisks) and osteoblast lining (yellow arrowheads). C) Immunofluorescent staining of the ossification centers revealed mature human osteoblasts expressing osteocalcin (OCN) and osteopontin (OPN) (yellow arrowheads) as well as blood vessels (asterisks) at week 6. D) Quantitative real-time PCR analysis of human OCN and OPN mRNA expression levels in explanted grafts at weeks 0–8. Data normalized to human GAPDH. All primers recognized human-specific transcripts. E) Movat pentachrome staining of explanted grafts from weeks 4 to 8. Colorimetric analysis revealed a simultaneous increase in collagen deposition (yellow) and the gradual disappearance of the cartilaginous CS-cryogel phase (green) in grafts seeded with MSCs + ECFCs but not in those with only MSCs. In all quantitative panels, bars represent mean ± s.d. (n ≥ 3). **p < 0.01, *** p < 0.001. Statistical method: ANOVA followed by Bonferroni’s post-test analysis. Scale bars = 100 µm (B, C, E).
Remodeling of the extracellular matrix (ECM) around the ossification centers was also consistent with EO. Movat pentachrome analysis of the grafts from weeks 4 to 8 confirmed a simultaneous increase in collagen deposition (yellow staining in Figure 4E) and gradual disappearance of the cartilaginous CS-cryogel phase (green) of the scaffold, which was virtually undetectable by week 8 (Figure 4E). Newly deposited collagen fibers substituted the spaces initially occupied by the CS-cryogel; the overall size of implants did not change significantly (Figure 2B). This type of ECM remodeling is consistent with the role of a cartilaginous matrix serving as a template to guide bone formation during EO. Moreover, histological evaluation (von Kossa staining) of the explanted MSCs + ECFCs grafts revealed gradual accumulation of calcium between weeks 6 and 8, indicating bone mineral formation (Figure 5A). Sections of the MSCs + ECFCs explants obtained after eight weeks exhibited uniform dark von Kossa staining, indicating intense calcification, and consistent with the appearance of bone fragments observed during that period (Figure 5B and S10, Supporting Information). Also, images taken with a scanning electron microscope (SEM) confirmed the formation of hydroxyapatite (HA) microstructures (Figure 5C). Meanwhile, grafts that were seeded with only MSCs showed little evidence of bone tissue formation in all the histological evaluations, including H&E (Figure S10), Movat pentachrome (Figure 4E and S11) and von Kossa staining (Figure 5A and S14).
Figure 5.
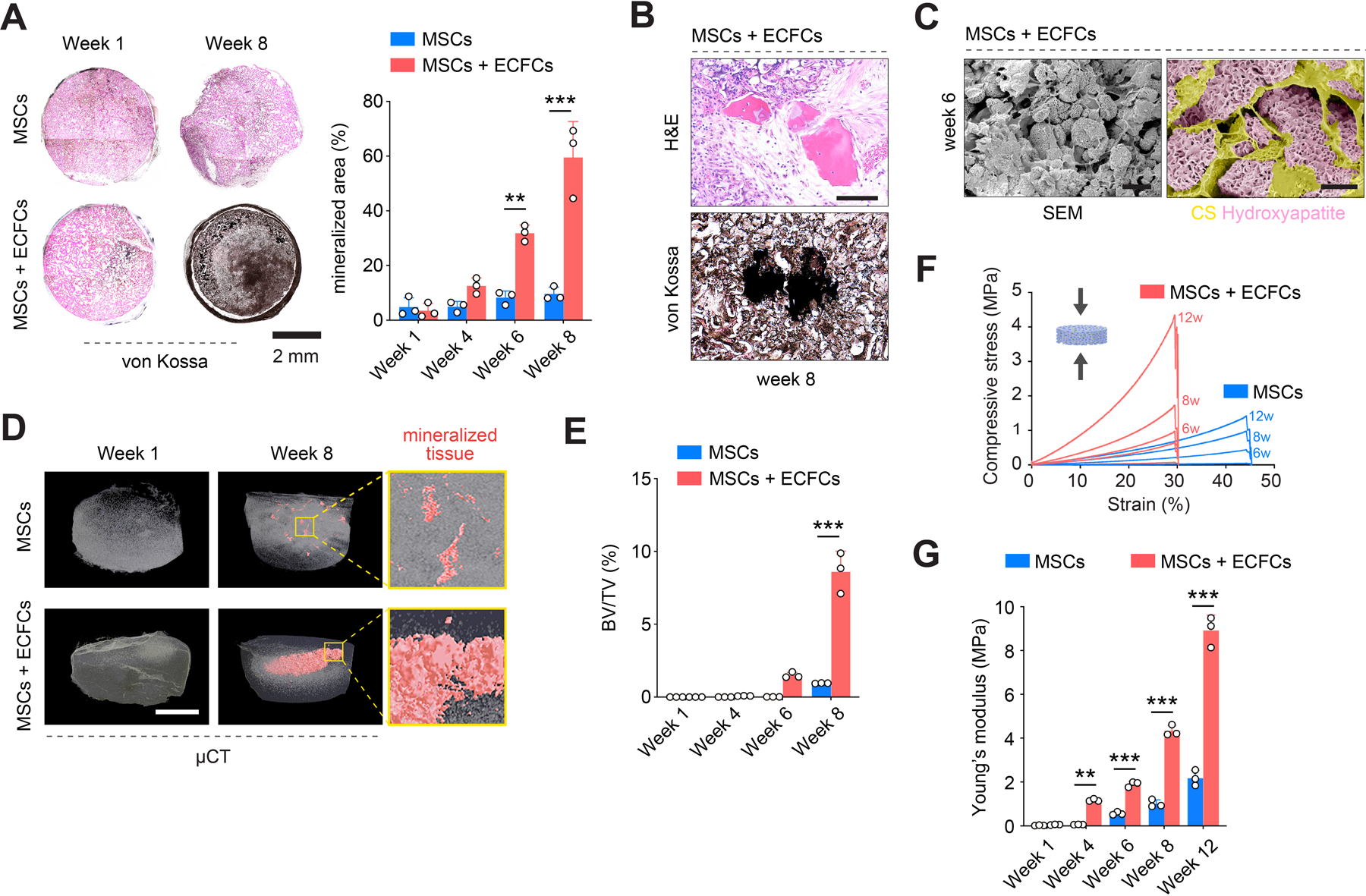
De novo formation of mineralized bone tissue. A) von Kossa staining of explanted grafts revealed gradual accumulation of calcium between weeks 6 and 8 in grafts seeded with MSCs + ECFCs, indicating bone mineral formation. B) Representative mineralized bone fragments identified at week 8 by H&E and von Kossa staining of consecutive sections from a graft seeded with MSCs + ECFCs. C) Scanning electron microscope (SEM) imaging confirmed the formation of hydroxyapatite (HA) microstructures in grafts with MSCs + ECFCs at week 6. D) Quantitative microtomography (μCT) revealed a gradual appearance of mineralized tissues within the MSCs + ECFCs grafts. Grafts with only MSCs exhibited minimal mineralization. E) Quantification of bone volume fraction (BV/TV) in grafts explanted from weeks 1 to 8. F) Compressive stress-strain curves and G) quantification of Young’s Modulus in grafts explanted from weeks 1 to 12. In all quantitative panels, bars represent mean ± s.d. (n ≥ 3). **p < 0.01, *** p < 0.001. Statistical method: ANOVA followed by Bonferroni’s post-test analysis. Scale bars = 100 µm (B), 400 µm (C, left), 60 µm (C, right), 2 mm (A, D).
It is important to note that bone tissue developed in the MSCs + ECFCs grafts without the addition of factors such as BMP2. Instead, the osteogenic differentiation of the MSCs occurred spontaneously, indicating a permissive environment for self-sustained EO. Nonetheless, the addition of WHNPs (for the release of mineral ions) and the presence of the cartilaginous CS-cryogel (which provides a strong negative charge that is important for creating an ion-rich environment [26]) were both critical - removing either of these components did not affect vascularization but completely abrogated the formation of bone tissue (Figure S11 and S12, Supporting Information). In other words, both the presence of ECFCs and the biphasic scaffold were necessary to effectively drive the osteogenic differentiation of MSCs in vivo.
Beyond the formation of a calcified ECM, morphological analysis of the MSCs + ECFCs grafts provided evidence for hematopoietic foci in certain regions neighboring the bone matrix (Figure S13, Supporting Information), which is consistent with the generation of bone marrow microenvironments during EO. Moreover, the tartrate-resistant acid phosphatase (TRAP) assay revealed osteoclastic activity along with bone matrix deposition (Figure S13, Supporting Information), indicating a metabolically active remodeling of the newly formed bone tissues.
Lastly, quantitative microtomography (μCT) confirmed the gradual formation of mineralized tissues within the MSCs + ECFCs scaffolds (Figure 5D and S14, Supporting Information). Indeed, the three-dimensionally deposition of the mineralized matrix was evident starting from week 4; by week 8, there was an extensive interconnected network of trabeculae throughout the core of scaffolds. Quantification of bone volume fraction (BV/TV), bone surface area fraction (BS/TS), and bone surface densities (BS/TV) in the mineralized matrix suggested an advanced stage of maturation (Figure 5E and S14, Supporting Information). Once again, μCT confirmed a negligible mineralized matrix in grafts seeded only with MSCs (Figure 5D). The differences in the abundance of mineralized tissue in both implants (i.e., with and without ECFCs) were also evident in the grafts’ mechanical properties. Grafts seeded with MSCs + ECFCs progressively improved their mechanical properties, including their compressive strength (Figure 5F) and Young’s Modulus, which increased from 0.06 ± 0.02 MPa at week 1 to 8.92 ± 0.70 MPa at week 12 (Figure 5G). Throughout the entire period in vivo, the compressive strength of grafts with ECFCs were significantly higher than those with only MSCs.
3. Conclusion
Collectively, we demonstrated that our cell-laden biphasic biomimetic scaffold enables a rapid and self-sustained EO process. Essentially, our approach allows initiating EO at an intermediate stage, at the point where the cartilage-like matrix and blood vessels act together to drive ossification. The scaffolds generated de novo bone tissue at ectopic subcutaneous locations within eight weeks, which is a relatively short period compared to other studies in the field (typically 16 weeks).[14b, 27] We designed a biomimetic construct that could recapitulate the critical cascade of developmental processes present during EO, an approach that is in line with the concept of developmental engineering. Our study improves previous efforts in EO bioengineering in several respects. First, we demonstrate that our CS-cryogel/BM-hydrogel biphasic scaffold could serve as a ready-made surrogate template of a hypertrophic cartilaginous matrix, which would otherwise take several weeks to form by conventional chondrogenesis. We show that the presence of CS is critical - removing CS completely abrogated the formation of bone tissue. Importantly, in addition to the cartilaginous matrix, our templates provide a porous network of soft tissue (BM-hydrogel phase) that is permissive for the cells to self-organize into first functional osteovascular niches and then into proper ossification centers. Second, we show that the combination of endothelial progenitor (ECFCs) and osteoprogenitor (MSCs) cells is sufficient to initiate and then sustain EO, leading to mechanically competent bone tissue formation. Of note, both of these progenitor cells can be derived from readily accessible autologous cell sources by minimally invasive means (i.e., blood and bone marrow, respectively), facilitating clinical translation.[20] Lastly, other efforts in the field rely on adding high doses of osteoinductive agents (such as BMPs) to prime osteogenic differentiation. However, this approach generally produces mineralization but lacks proper vascularization, adequate graft integration, and carries the risk of heterotopic bone formation [8c]. In contrast, we show that our biomimetic approach can self-initiate and sustain the osteogenic differentiation of the MSCs without the need for exogenous growth factors, thus eliminating the clinical debate about the use of BMPs.[8]
In summary, we have developed a cell-laden biphasic biomimetic scaffold to recapitulate EO in vivo. After approximately 8 weeks, our approach produces adequately vascularized and well-integrated bone grafts, providing a significant advantage over other bioengineering approaches that rely on ceramic and mineral substrates [28]. This proof-of-concept study warrants further investigations, including those related to (i) scaling-up of the grafts, (ii) fabricating scaffolds for orthotopic implantation using 3D printing technology based on patients’ desired customized anatomy, (iii) elucidating host responses at specific orthotopic injury sites and in immunocompetent animal models, and (iv) establishing the influence of biomechanical loading. We envision our research provides an opportunity to generate off-the-shelf biomimetic grafts with clinical translational potential to treat massive bone defects.
4. Experimental Section/Methods
Whitlockite nanoparticle synthesis
Whitlockite (Ca18Mg2(HPO4)2(PO4)12) nanoparticles were synthesized as previously described [14a]. Briefly, 0.37 M calcium hydroxide [Ca(OH)2] and 0.13 M magnesium hydroxide [Mg(OH)2] was originally mixed in distilled water at 80°C for 1 hr. While vigorously stirring, 0.5 M phosphoric acid (H3PO4) was incorporated into a Ca(OH)2 and Mg(OH)2 solution. The precipitant was collected after 24 hours with a 0.22-μm filter membrane (Durapore® membrane filters, Millipore) and freeze-dried. Whitlockite nanoparticles (WHNPs) between 100 to 150 nm were collected.
Ion release measurement of whitlockite nanoparticles
WHNPs were vigorously washed in distilled water five times to remove remaining ions at the surface of the particle. After completely drying, 1 weight percent of WHNPs aqueous solutions were aged on the shaker at room temperature. After 1, 2, 6, and 8 weeks, to collect filtrate ions, solutions were first centrifuged at 4,000 rpm for 30 min and also filtered through a syringe membrane (0.2 µm Acrodisc®, Sigma-Aldrich). The amount of calcium, magnesium, and phosphorous ions were measured with an inductively coupled plasma atomic emission spectrometer (ICP-AES, OPTIMA 8300, Perkin-Elmer) with argon plasma.
Methacrylation of chondroitin sulfate
Methacrylated chondroitin sulfate (CSMA) were synthesized as previously described [14b]. Briefly, 1.0 g of chondroitin sulfate A sodium salt (Sigma-Aldrich) was dissolved in phosphate-buffered saline (PBS; pH 7.4, Gibco) and reacted with 1 mL of glycidyl methacrylate (GMA, 73 mM, Sigma-Aldrich) for 10 days. CSMA was then purified by dialysis membrane cassette (2K MW, ThermoFisher) for 48 h, and then put in deionized water for 24 hours, followed by freeze-drying, and then preserved at −20°C for long term storage.
Chondroitin sulfate cryogel scaffold fabrication
Chondroitin sulfate cryogel (CS-cryogel) was fabricated by mixing CSMA (Methacrylated chondroitin sulfate) solution at 10% (w/v) in deionized water at room temperature. 6% (w/v) of WHNPs were added to the polymer solution and vortexed for particle dispersion. The mix was placed in a plastic mold (the inside of a regular Eppendorf tube cap) with the desired geometry. A thermos-initiator (100 mg/ml, ammonium persulfate; APS, Sigma-Aldrich) at 2.5% (v/v) and accelerator (N, N, N’, N’-Tetramethylethylenediamine; TEMED, Sigma-Aldrich) at 0.12% (v/v) were used as crosslinking agents for polymerization, and later cryogels were frozen at −20°C (20 h) and lyophilized for in vivo usage. For the smaller pore sizes of cryogel (10, 50, and 100 µm), longer crosslinking time is required (36, 30, and 26 h respectively). For the bigger pore sizes of cryogel (500 µm), crosslinking time can be reduced (12 h).
Fabrication of 3D printed bone types mold
For 3D printing, we used commercially available PLA filament (Hatchbox) with a diameter of 1.75 ± 0.03 mm. All PLA bones were designed with 3D modeling software (Solidworks). The 3D model file generated by the software was exported to a convenient file format (STL) and produced with a 3D printer (Monoprice). PDMS mixtures (Sylgard 184, Dow Corning) with pre-polymer: curing agent ratios of 10:1 were poured into 3D printed femur or mandible to fabricate a 3D mold for manufacturing the biphasic scaffolds. Prepared CSMA solution with WHNPs was added to the mold and crosslinked. The 3D-shaped biphasic scaffolds were frozen at −20°C (20 h) and lyophilized in the same way.
Scanning electron microscopy of explanted scaffolds
Samples were processed for scanning electron microscopy (SEM) imaging as previously described [29]. Briefly, scaffolds were rinsed with cold distilled water and then serially dehydrated with cold ethanol. Samples were then incubated with 50% ethanol and 50% hexamethyldisilazane (HMDS, Sigma-Aldrich) for five minutes and substituted with 100% HMDS for 10 transactions. Before imaging, samples were coated with platinum for 100 seconds at 20 mA. Field emission SEM images were obtained with a JEOL 7900F (JEOL, ltd) instrument.
Isolation and culture of human ECFCs and MSCs
Human endothelial colony-forming cells (ECFCs) were isolated from umbilical cord blood samples by an Institutional Review Board-approved protocol as previously describe [24]. ECFCs were isolated from the mononuclear cell fractions from the blood samples after density gradient centrifugation. Cells were plated on fibronectin (10 µg/ml in PBS, Chemicon) coated culture plates in endothelial cell medium (PromoCell). Medium was replenished every 2 days. ECFC colonies appeared in culture after 2–3 weeks and were purified using CD31-coated magnetic beads. ECFCs were then cultured on 1% gelatin-coated plates using endothelial cell medium. ECFCs were used before passage 12 in all experiments. Human mesenchymal stem cells (MSCs) were isolated from bone-marrow samples as previously described [24]. MSCs were cultured on uncoated plates using MSC-medium: MSCGM (Lonza) supplemented with 10% fetal bovine serum (FBS) and 1 x glutamine-penicillin-streptomycin (GPS). All experiments were performed with MSCs before passage 8.
Cell seeding into the biphasic scaffolds
Single-cell suspensions were centrifuged to form a pellet and then suspended in ice-cold phenol red-free Matrigel (BD Bioscience) at 4°C. For seeding with MSCs + ECFCs (ECFC:MSC ratio = 2:3, as previously described [20b]) a total of 2×106 cells was used for each construct. For seeding with only MSCs a total of 1.2×106 cells was used. In both cases, cells were suspended in Matrigel solution. The cell-Matrigel mixture was dispensed as 50 µl to swell the CS-cryogel to make the final biphasic scaffold. Final gelation was achieved by placing the scaffold into a 37°C incubator for 15 min. Thereafter, the cell-laden biphasic scaffolds were prepared for subcutaneous implant surgery. Please note that the rheological behavior of both CS hydrogel and Matrigel have been extensively documented in previous studies [30].
Subcutaneous implantation of scaffolds
Five to six-week-old athymic nude (nu/nu) mice were purchased from Envigo RMS, Inc. Grafts seeded with MSCs alone and with MSCs + ECFCs were implanted for up to 8 weeks in at least three separate mice for each time point analyzed, and all experiments were independently repeated. Mice were housed in compliance with Boston Children’s Hospital guidelines, and all animal-related protocols were approved by the Institutional Animal Care and Use Committee. The scaffolds were surgically implanted into the back subcutaneous space of the mice via a minimal incision on the skin. All surgical procedures were performed under sterile conditions and with mice under anesthesia.
Quantitative real-time PCR
Total RNA was extracted from cells using a RNeasy mini kit (Qiagen). RNA concentration was obtained with a Nanodrop and RNA purity evaluated by the ratio of absorbance at 260 nm and 280 nm. cDNA was synthesized from total RNA using a high-capacity cDNA reverse transcription kit (ThermoFisher). Quantitative real-time PCR was carried out using SYBR Green Master Mix (ThermoFisher). GAPDH served as the housekeeping gene. Sequences of primers for real-time PCR are listed in Table S1.
Micro-computed tomography (micro-CT)
Micro-CT analysis was used to quantify the volume of bone formation within the scaffolds as previously described [14b]. Briefly, the tomography was performed using SkyScan 1272 (Bruker) at 59 kV, 167 μA, and an exposure time of 40 ms through a non-filter. X-ray projections were acquired in 0.6° intervals with a scanning angular rotation of 360°. An automated threshold algorithm segmented the reconstructed dataset. The projected images were reconstructed into three-dimensional images using ReCon Micro-CT software from Bruker for 10 μm resolution.
Histological assessment
Explanted scaffolds were fixed overnight in 10% buffered formalin, embedded in paraffin, and sectioned (7 µm-thick). Each sectioned slide was deparaffinized and hydrate to buffer. Hematoxylin and eosin (H&E)-stained sections were examined for the presence of microvessels. Microvessels were detected by the evaluation of H&E-stained sections taken from the middle part of the implants. Microvessel density was reported as the average number of erythrocyte-filled vessels (vessels/mm2) and the average number of vessels per pore using ImageJ software (National Institutes of Health). Movat pentachrome-stained sections were used to identify collagen (bone: yellow), glycosaminoglycans (cartilage: green), muscle (red), mucin (blue), and fibrin (bright red) (Dana-Farber/Harvard Cancer Center Specialized Histopathology Core). Especially, Movat stained sections were used to measure the collagen deposition as well as CS degradation in the scaffolds. For von Kossa staining (Diagnostic Biosystems), slides were incubated in silver nitrate solution (5%) for 60 min with exposure to UV light. After rinsing, slides were incubated in sodium thiosulfate solution (5%) for 2 min. Von Kossa-stained sections were used to measure tissue mineralization (% of section area mineralized) using image color summarizer (v0.76, Martin Krzywinski). For Safranin O staining (ScienceCell), slides were incubated in Fast green FCF solution (0.1%) for 10 min. After rinsing, slides were incubated in Safranin O staining solution (1 mg/ml) for 30 min. Safranin O stained sections were used to examine the proximity of the blood vessels and their perivascular MSCs to the cartilaginous phase of the scaffold. For tartrate-resistant acid phosphatase staining (TRAP, Sigma-Aldrich), slides were incubated 1 h in a mixture of 45 ml of deionized water, 1.0 ml of Diazotized Fast Garnet GBC, 0.5 ml Naphitol AS-BI Phosphate, 2.0 ml of acetate, and 1.0 ml of tartrate solution at 37°C. After rinsing, counterstained 2 min in Hematoxylin solution. TRAP-stained sections were examined for the presence of osteoclasts activity in the scaffold. For TUNEL assay (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling, ThermoFisher), 50 μl of TUNEL Reaction cocktail (Click-iT™) was added to each slide, and allow the solution to spread. Incubate for 30 min at 37°C, protected from light and rinsing with 1 X PBS. For TUNEL stained sections were used to examine for in situ apoptosis detection in the scaffold.
Immunostaining analysis
For immunostaining, sections were deparaffinized and antigen retrieval was carried out with tris– EDTA buffer (10 mM Tris-Base, 2 mM EDTA, 0.05 % Tween-20, pH 9.0). Sections were then blocked for 30 min in 5% blocking serum followed by incubation with primary antibodies overnight at 4°C. Primary and secondary antibodies used are detailed in Table S2. Briefly, Horseradish peroxidase-conjugated mouse secondary antibody (1:200, Vector Laboratories) and 3,3’-diaminobenzidine (DAB) were used for the detection of hCD31 (Agilent). Hematoxylin was used for counterstaining and Permount for mounting (ThermoFisher). Fluorescent staining was carried out using either Texas Red- or FITC-conjugated secondary antibodies followed by DAPI counterstaining. On indicated experiments, additional fluorescent staining was performed using rhodamine-conjugated Ulex europaeus agglutinin I (UEA-1) lectin (1:200, Vector Laboratories).
Mechanical properties
For mechanical characterization, scaffolds were explanted from the mice at different time point (week 1, 4, 6, 8, and 12) and their stress-strain curves were obtained using a universal mechanical testing apparatus (EZ-Test, Shimadzu) with a 10 kN load cell. The compressive modulus (Young’s modulus) was quantified from the linear region of each stress-strain curves.
Nanoindentation analysis
A nanoindentation method was used to measure the elastic modulus and contact ratio of hardness of the scaffold as previously described [14b]. Briefly, the biphasic scaffolds were fixed for 24 h in 4% paraformaldehyde and embedded with acrylic resin (Ortho-Jet, Lang). Next, the resin blocks were sectioned with a low-speed diamond saw (Isomet, Buehler Lake Bluff) into 2 mm thickness slices. Sections were polished with silicon carbide abrasive paper and aluminum oxide paste. Sections were then glued onto a stainless-steel holder and mounted on a nanoindenter (Nano-XP, MTS). All indentations were conducted up to 500 nm depths with loading and unloading displacement rates of 10 nm/s. The indentation force-displacement curves were then used to obtain the contact hardness by dividing the peak indentation force by the projected area at the end of loading, and the elastic modulus using the unloading slope. The distance between indent locations was at least 30 μm to avoid any interruptions from the adjacent indents.
Microscopy
Images were acquired with an Axio Observer Z1 inverted microscope (Carl Zeiss) and AxioVision Rel. 4.8 software. Fluorescent images were acquired using a 20x objective lens. Non-fluorescent images were acquired using an AxioCam MRc5 camera with a 5x and 20x objective lens.
Statistical analysis
All statistical analyses were performed using the GraphPad Prism v.5 software (GraphPad Software Inc.). Unless otherwise stated, data were expressed as mean ± standard deviation of the mean (s.d.) and there was no pre-processing of data applied. Comparisons between multiple groups were performed by ANOVA followed by Bonferroni’s post-test analysis. Unpaired two-tailed Student’s t-tests was used for comparisons between two groups. Samples size, including number of mice per group, was chosen to ensure adequate power and were based on historical laboratory data. No exclusion criteria were applied for all analyses. A value of p<0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgements
We thank Drs. Jeremy Goss, Christopher Sudduth, and Mohammed Alomari (Department of Plastic and Oral Surgery, Boston Children’s Hospital) for facilitating acquisition of discarded human trabecular bone tissue. We thank Joseph Peine (Department of Cardiac Surgery, Boston Children’s Hospital) for assistance with the 3D printing. We thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided Movat Pentachrome staining service. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516. This work was supported by grants from the National Institutes of Health (R01AR069038 and R01HL128452 to J.M.M.-M.). This work was also partially supported by the Ministry of Science and ICT of Korea (NRF-2017M3A9C6031786 to N.S.H.)
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Data availability
The authors declare that all data supporting the findings of this study are available in the paper and its Supplementary Data.
Reference
- [1].a) Delloye C, Cornu O, Druez V, Barbier O, J Bone Joint Surg Br 2007, 89, 574; [DOI] [PubMed] [Google Scholar]; b) Wang W, Yeung KWK, Bioact Mater 2017, 2, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Agarwal R, Garcia AJ, Adv Drug Deliv Rev 2015, 94, 53; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Burg KJ, Porter S, Kellam JF, Biomaterials 2000, 21, 2347; [DOI] [PubMed] [Google Scholar]; c) Kolk A, Handschel J, Drescher W, Rothamel D, Kloss F, Blessmann M, Heiland M, Wolff KD, Smeets R, J Craniomaxillofac Surg 2012, 40, 706; [DOI] [PubMed] [Google Scholar]; d) Mistry AS, Mikos AG, Adv Biochem Eng Biotechnol 2005, 94, 1. [DOI] [PubMed] [Google Scholar]
- [3].a) Amini AR, Laurencin CT, Nukavarapu SP, Crit Rev Biomed Eng 2012, 40, 363; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bose S, Roy M, Bandyopadhyay A, Trends Biotechnol 2012, 30, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Hutmacher DW, Biomaterials 2000, 21, 2529; [DOI] [PubMed] [Google Scholar]; b) Meijer GJ, de Bruijn JD, Koole R, van Blitterswijk CA, PLoS Med 2007, 4, e9; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR, Biomaterials 2006, 27, 3413; [DOI] [PubMed] [Google Scholar]; d) Shen J, Nair A, Saxena R, Zhang CC, Borrelli J Jr., Tang L, PLoS One 2014, 9, e93514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Kempen DH, Lu L, Hefferan TE, Creemers LB, Maran A, Classic KL, Dhert WJ, Yaszemski MJ, Biomaterials 2008, 29, 3245; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meinel L, Karageorgiou V, Fajardo R, Snyder B, Shinde-Patil V, Zichner L, Kaplan D, Langer R, Vunjak-Novakovic G, Ann Biomed Eng 2004, 32, 112; [DOI] [PubMed] [Google Scholar]; c) Reichert JC, Cipitria A, Epari DR, Saifzadeh S, Krishnakanth P, Berner A, Woodruff MA, Schell H, Mehta M, Schuetz MA, Duda GN, Hutmacher DW, Sci Transl Med 2012, 4, 141ra93; [DOI] [PubMed] [Google Scholar]; d) Vo TN, Kasper FK, Mikos AG, Adv Drug Deliv Rev 2012, 64, 1292; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gonzalez-Fernandez T, Tierney EG, Cunniffe GM, O’Brien FJ, Kelly DJ, Tissue Eng Part A 2016, 22, 776. [DOI] [PubMed] [Google Scholar]
- [6].Chen G, Xu H, Yao Y, Xu T, Yuan M, Zhang X, Lv Z, Wu M, Front Cell Dev Biol 2020, 8, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Akita S, Tamai N, Myoui A, Nishikawa M, Kaito T, Takaoka K, Yoshikawa H, Tissue Eng 2004, 10, 789; [DOI] [PubMed] [Google Scholar]; b) Rouwkema J, Westerweel PE, de Boer J, Verhaar MC, van Blitterswijk CA, Tissue Eng Part A 2009, 15, 2015. [DOI] [PubMed] [Google Scholar]
- [8].a) Krishnakumar GS, Roffi A, Reale D, Kon E, Filardo G, Int Orthop 2017, 41, 1073; [DOI] [PubMed] [Google Scholar]; b) Carreira AC, Lojudice FH, Halcsik E, Navarro RD, Sogayar MC, Granjeiro JM, J Dent Res 2014, 93, 335; [DOI] [PubMed] [Google Scholar]; c) James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, Ting K, Soo C, Tissue Eng Part B Rev 2016, 22, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Long F, Ornitz DM, Cold Spring Harb Perspect Biol 2013, 5, a008334; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lefebvre V, Bhattaram P, Curr Top Dev Biol 2010, 90, 291; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N, Nat Med 1999, 5, 623. [DOI] [PubMed] [Google Scholar]
- [10].a) Scotti C, Tonnarelli B, Papadimitropoulos A, Scherberich A, Schaeren S, Schauerte A, Lopez-Rios J, Zeller R, Barbero A, Martin I, Proc Natl Acad Sci U S A 2010, 107, 7251; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lenas P, Moos M, Luyten FP, Tissue Eng Part B Rev 2009, 15, 395. [DOI] [PubMed] [Google Scholar]
- [11].a) Scotti C, Piccinini E, Takizawa H, Todorov A, Bourgine P, Papadimitropoulos A, Barbero A, Manz MG, Martin I, Proc Natl Acad Sci U S A 2013, 110, 3997; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McDermott AM, Herberg S, Mason DE, Collins JM, Pearson HB, Dawahare JH, Tang R, Patwa AN, Grinstaff MW, Kelly DJ, Alsberg E, Boerckel JD, Sci Transl Med 2019, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dang PN, Herberg S, Varghai D, Riazi H, Varghai D, McMillan A, Awadallah A, Phillips LM, Jeon O, Nguyen MK, Dwivedi N, Yu X, Murphy WL, Alsberg E, Stem Cells Transl Med 2017, 6, 1644; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Thompson EM, Matsiko A, Kelly DJ, Gleeson JP, O’Brien FJ, Tissue Eng Part A 2016, 22, 556. [DOI] [PubMed] [Google Scholar]
- [12].a) Jakab K, Neagu A, Mironov V, Markwald RR, Forgacs G, Proc Natl Acad Sci U S A 2004, 101, 2864; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin RZ, Chang HY, Biotechnol J 2008, 3, 1172. [DOI] [PubMed] [Google Scholar]
- [13].Ho-Shui-Ling A, Bolander J, Rustom LE, Johnson AW, Luyten FP, Picart C, Biomaterials 2018, 180, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Jang HL, Jin K, Lee J, Kim Y, Nahm SH, Hong KS, Nam KT, ACS Nano 2014, 8, 634; [DOI] [PubMed] [Google Scholar]; b) Kim HD, Jang HL, Ahn HY, Lee HK, Park J, Lee ES, Lee EA, Jeong YH, Kim DG, Nam KT, Hwang NS, Biomaterials 2017, 112, 31. [DOI] [PubMed] [Google Scholar]
- [15].a) Fontes-Pereira A, Rosa P, Barboza T, Matusin D, Freire AS, Braz BF, Machado CB, von Kruger MA, Souza SAL, Santelli RE, Pereira WCA, Sci Rep 2018, 8, 11963; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cross NA, Hillman LS, Allen SH, Krause GF, J Bone Miner Res 1995, 10, 1312. [DOI] [PubMed] [Google Scholar]
- [16].Huebsch N, Lippens E, Lee K, Mehta M, Koshy ST, Darnell MC, Desai RM, Madl CM, Xu M, Zhao X, Chaudhuri O, Verbeke C, Kim WS, Alim K, Mammoto A, Ingber DE, Duda GN, Mooney DJ, Nat Mater 2015, 14, 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prein C, Warmbold N, Farkas Z, Schieker M, Aszodi A, Clausen-Schaumann H, Matrix Biol 2016, 50, 1. [DOI] [PubMed] [Google Scholar]
- [18].a) Chen YC, Lin RZ, Qi H, Yang Y, Bae H, Melero-Martin JM, Khademhosseini A, Adv Funct Mater 2012, 22, 2027; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin RZ, Chen YC, Moreno-Luna R, Khademhosseini A, Melero-Martin JM, Biomaterials 2013, 34, 6785; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lin RZ, Lee CN, Moreno-Luna R, Neumeyer J, Piekarski B, Zhou P, Moses MA, Sachdev M, Pu WT, Emani S, Melero-Martin JM, Nat Biomed Eng 2017, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677; [DOI] [PubMed] [Google Scholar]; b) Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Nat Mater 2013, 12, 458; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, Chen S, Engler AJ, Nat Mater 2014, 13, 979; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ortega N, Behonick DJ, Werb Z, Trends Cell Biol 2004, 14, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S, Bischoff J, Blood 2007, 109, 4761; [DOI] [PubMed] [Google Scholar]; b) Melero-Martin JM, De Obaldia ME, Kang SY, Khan ZA, Yuan L, Oettgen P, Bischoff J, Circ Res 2008, 103, 194; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lin RZ, Moreno-Luna R, Munoz-Hernandez R, Li D, Jaminet SC, Greene AK, Melero-Martin JM, Angiogenesis 2013, 16, 735; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lin RZ, Moreno-Luna R, Zhou B, Pu WT, Melero-Martin JM, Angiogenesis 2012, 15, 443; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lin RZ, Dreyzin A, Aamodt K, Dudley AC, Melero-Martin JM, Cell Transplant 2011, 20, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Lin RZ, Melero-Martin JM, J Vis Exp 2011, DOI: 10.3791/3065e3065; [DOI] [PMC free article] [PubMed]; b) Lin RZ, Melero-Martin JM, Methods 2012, 56, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Ramasamy SK, Stem Cells Int 2017, 2017, 5046953; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stegen S, Carmeliet G, Bone 2018, 115, 50; [DOI] [PubMed] [Google Scholar]; c) Portal-Nunez S, Lozano D, Esbrit P, Histol Histopathol 2012, 27, 559. [DOI] [PubMed] [Google Scholar]
- [23].a) Kusumbe AP, Ramasamy SK, Adams RH, Nature 2014, 507, 323; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ramasamy SK, Kusumbe AP, Wang L, Adams RH, Nature 2014, 507, 376; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Watson EC, Adams RH, Cold Spring Harb Perspect Med 2018, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Towler DA, Curr Osteoporos Rep 2008, 6, 67; [DOI] [PubMed] [Google Scholar]; e) Sivaraj KK, Adams RH, Development 2016, 143, 2706; [DOI] [PubMed] [Google Scholar]; f) Greenhill C, Nat Rev Endocrinol 2014, 10, 250. [DOI] [PubMed] [Google Scholar]
- [24].Lin RZ, Moreno-Luna R, Li D, Jaminet SC, Greene AK, Melero-Martin JM, Proc Natl Acad Sci U S A 2014, 111, 10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Olsen BR, Reginato AM, Wang W, Annu Rev Cell Dev Biol 2000, 16, 191. [DOI] [PubMed] [Google Scholar]
- [26].a) Kim HD, Lee EA, An YH, Kim SL, Lee SS, Yu SJ, Jang HL, Nam KT, Im SG, Hwang NS, ACS Appl Mater Interfaces 2017, 9, 21639; [DOI] [PubMed] [Google Scholar]; b) Wang T, Yang F, Stem Cell Res Ther 2017, 8, 284; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cheng H, Chabok R, Guan X, Chawla A, Li Y, Khademhosseini A, Jang HL, Acta Biomater 2018, 69, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Egawa S, Miura S, Yokoyama H, Endo T, Tamura K, Dev Growth Differ 2014, 56, 410; [DOI] [PubMed] [Google Scholar]; b) Aghajanian P, Mohan S, Bone Res 2018, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Guda T, Walker JA, Singleton BM, Hernandez JW, Son JS, Kim SG, Oh DS, Appleford MR, Ong JL, Wenke JC, Tissue Eng Part A 2013, 19, 1879; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee JH, Ryu MY, Baek HR, Lee KM, Seo JH, Lee HK, Ryu HS, J Mater Sci Mater Med 2013, 24, 2117; [DOI] [PubMed] [Google Scholar]; c) Wang MM, Flores RL, Witek L, Torroni A, Ibrahim A, Wang Z, Liss HA, Cronstein BN, Lopez CD, Maliha SG, Coelho PG, Sci Rep 2019, 9, 18439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kang BJ, Kim H, Lee SK, Kim J, Shen Y, Jung S, Kang KS, Im SG, Lee SY, Choi M, Hwang NS, Cho JY, Acta Biomater 2014, 10, 3007. [DOI] [PubMed] [Google Scholar]
- [30].a) Allen P, Melero-Martin J, Bischoff J, J Tissue Eng Regen Med 2011, 5, e74; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ponsubha S, Jaiswal AK, Carbohydr Polym 2020, 238, 116179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available in the paper and its Supplementary Data.