

Abstract
Background
The generic term “exacerbation” does not reflect the heterogeneity of acute exacerbations of COPD (AECOPD). We utilised a novel algorithmic strategy to profile exacerbation phenotypes based on underlying aetiologies.
Methods
Patients hospitalised for AECOPD (n=146) were investigated for aetiological contributors summarised in a mnemonic acronym ABCDEFGX (A: airway virus; B: bacterial; C: co-infection; D: depression/anxiety; E: eosinophils; F: failure (cardiac); G: general environment; X: unknown). Results from clinical investigations were combined to construct AECOPD phenotypes. Relationships to clinical outcomes were examined for both composite phenotypes and their specific aetiological components. Aetiologies identified at exacerbation were reassessed at outpatient follow-up.
Results
Hospitalised AECOPDs were remarkably diverse, with 26 distinct phenotypes identified. Multiple aetiologies were common (70%) and unidentifiable aetiology rare (4.1%). If viruses were detected (29.5%), patients had longer hospitalisation (7.7±5.6 versus 6.0±3.9 days, p=0.03) despite fewer “frequent exacerbators” (9.3% versus 37%, p=0.001) and lower mortality at 1 year (p=0.03). If bacterial infection was found (40.4%), patients were commonly “frequent exacerbators” (44% versus 18.4%, p=0.001). Eosinophilic exacerbations (28%) were associated with lower pH (7.32±0.06 versus 7.36±0.09, p=0.04), higher venous carbon dioxide tension (PvCO2) (53.7±10.5 versus 48.8±12.8, p=0.04), greater noninvasive ventilation (NIV) usage (34.1% versus 18.1%) but shorter hospitalisation (4 (3–5) versus 6 (4–9) days, p<0.001) and lower infection rates (41.4% versus 80.9%, p<0.0001). Cardiac dysfunction and severe anxiety/depression were common in both infective and non-infective exacerbations. Characteristics identified at exacerbation often persisted after recovery.
Conclusions
Hospitalised AECOPDs have numerous causes, often in combination, that converge in complex, multi-faceted phenotypes. Clinically important differences in outcomes suggest that a phenotyping strategy based on aetiologies can enhance AECOPD management.
Short abstract
Hospitalised #AECOPD present as complex multidimensional clinical phenotypes, often comprising multiple distinct aetiologies. Profiling AECOPDs according to their multifactorial aetiological components has important prognostic and therapeutic implications. https://bit.ly/3nIHEnO
Introduction
COPD is a complex and heterogeneous disease. During acute exacerbations of COPD (AECOPDs), additional complexity ensues given diverse exacerbation aetiologies and comorbidities. Stable COPD is increasingly recognised as encompassing diverse disease phenotypes [1]; however the term “acute exacerbation of COPD” remains generic with a nonspecific definition [2]. The complexity and heterogeneity of AECOPDs is not yet reflected in either clinical practice or clinical research.
We have previously proposed individualised phenotyping of hospitalised AECOPDs according to aetiological contributors [3, 4]. While studies have explored phenotyping strategies based on simple microbiological classification [5, 6], a comprehensive personalised approach encompassing additional factors such as cardiac disease, mood disorders or inadequate social support has not been reported.
We hypothesised that an algorithmic approach using simple investigations would elucidate the multifactorial complexity of AECOPD aetiology, with prognostic and therapeutic implications.
Methods
Study population
Patients hospitalised for AECOPD were recruited to a prospective observational study approved by our hospital's Human Research Ethics Committee (HREC13134A). Written informed consent was obtained.
Inclusion required a post-bronchodilator forced expiratory ratio <0.7 verified by spirometry performed when clinically stable [2]. Exclusion criteria included overt left ventricular failure or acute myocardial infarction. Owing to the need for informed consent, patients mechanically ventilated at initial presentation were excluded. Sufficient cognitive capacity to complete questionnaires was required. Infiltrates on chest radiograph (CXR) were permitted.
Clinical outcomes recorded included rates of noninvasive ventilation (NIV), mechanical ventilation, inpatient mortality, length of hospital stay, readmissions and survival for 12 months following hospital discharge. The research team did not influence clinical care, which was at the discretion of the attending physicians. A follow-up assessment when clinically stable was offered to all patients. Hospitalisations in the 12-month period after hospital discharge were identified by review of electronic health records. Survival at 12 months post hospital discharge was determined by review of electronic health records and patient phone calls.
Study design
Patient recruitment and participation are shown in figure 1.
FIGURE 1.

Consolidated Standards of Reporting Trials diagram of patient recruitment and participation. AECOPD: acute exacerbations of COPD.
We assessed attributable causes of hospitalised AECOPDs using the mnemonic acronym ABCDEFGX (A: airway virus; B: bacterial; C: co-infection; D: depression/anxiety; E: eosinophils; F: failure (cardiac); G: general environment; X: unknown) [3]. Since routine computed tomography (CT) pulmonary angiography may not be justified in AECOPD, we revised our originally published acronym [3] by substituting “E: eosinophils” in place of the former “E: embolism (pulmonary)”. Patients were assigned a final composite phenotype by combining all aetiological factors that were identified (e.g. bacterial infection (B) and severe depression/anxiety symptoms (D): phenotype “BD”).
Phenotyping strategy
Demographic variables, comorbidities, exacerbation history and pharmacotherapy were obtained from patient interviews and hospital case records. Study investigators administered the COPD Assessment Tool (CAT) [7], Hospital Anxiety and Depression Scale (HADS) [8] and Medical Research Council Dyspnoea (MRCD) scale [9]. Results from nasopharyngeal virus PCR (146 out of 146), spontaneously expectorated sputum culture (136 out of 146), C-reactive protein (CRP) and fever (≥38°C) (both 146 out of 146) were used to identify aetiologies “A” (airway virus), “B” (bacteria) or “C” (co-infection) (figure 2). Inevitably, many AECOPDs have clinical features of infection without identification of a specific microorganism. We assigned putative “B” (bacterial) aetiology if fever or CRP ≥20 mg·dL−1 [10] was recorded and virus negative. Identification of “D” (depression/anxiety) was based on HADS scores at hospital admission (HADS A/D ≥15 or combined HADS Total ≥27, successfully completed for 134 out of 146). Although not specifically validated for AECOPD hospitalisations, we chose high threshold values for HADS previously shown to have 95% specificity for verified anxiety/depression in an inpatient population [11]. Eosinophils were measured on the first full blood count in 146 out of 146 with “E” assigned when eosinophils >2% total white cell count [5]. Investigation for pulmonary embolism was at the discretion of the treating team and not part of the algorithmic investigational approach. Cardiac biomarker analysis was performed on blood taken at initial hospital presentation where sufficient serum was available (119 out of 146). We identified cardiac dysfunction “F” (failure) when high-sensitivity troponin I (hs-TnI) and/or N-terminal pro-brain natriuretic peptide (NT-proBNP) [12] were above age and gender adjusted upper limits of normal (ULN) [13, 14]. Acute disruption to the patient's physical, social or therapeutic environment was assessed in 146 out of 146 and aetiology “G” (general environment) assigned if deemed causative to hospitalisation. Exacerbations with no aetiological factor identifiable were categorised “X” (unknown).
FIGURE 2.

Aetiologies identified based on ABCDEFGX acronym in patients hospitalised with acute exacerbations of COPD (AECOPD). MCS: microscopy and culture of sputum; CRP: C-reactive protein; HADS: Hospital Anxiety and Depression Scale; WCC: white cell count; NT-proBNP: N-terminal pro-brain natriuretic peptide; hs-TnI: high sensitivity troponin I; Bi: sputum culture positive; Bii: fever or CRP >20 mg·dL−1 with negative virus PCR; ULN: upper limits of normal.
Analysis
Comparisons between groups were made employing unpaired t-tests and one-way analysis of variance (ANOVA) for normally distributed data or Mann–Whitney and Kruskal–Wallis testing for non-parametric data. Chi-square analyses were used for categorical data. Blood and questionnaire results from acute versus stable disease state were analysed via Pearson correlation coefficients. Time-to-event survival analyses were conducted using Kaplan–Meier methods and log-rank tests. Data are presented as number (percentage), mean±standard deviation (sd) or median (interquartile range, IQR), where appropriate. Statistical significance was accepted at p<0.05. Analyses were conducted on Stata MP 14.1 (Statacorp, Texas, USA).
Results
Study cohort, aetiologies and phenotypes
Overall 169 AECOPD admissions were enrolled, with 146 patients included (figure 1). Twenty-three patients were excluded, chiefly because they failed to meet spirometric criteria for a diagnosis of COPD or lacked viral swab results. Demographics, comorbidities and pharmacotherapy are shown (table 1).
TABLE 1.
Baseline characteristics of 146 patients enrolled during acute exacerbations of COPD (AECOPD)
| Demographics | Comorbidities | Medications | |||
| Age years | 71.8±10.4 | Bronchiectasis | 16 (10.3) | LAMA | 133 (85.8) |
| Male | 97 (62.6) | OSA | 14 (9.0) | LABA | 129 (83.2) |
| BMI kg·m−2 | 24.8±6.5 | Hypertension | 71 (45.8) | ICS | 126 (81.3) |
| FEV1 L | 1.1±0.5 | AF/flutter | 19 (12.3) | OCSƒ | 13 (8.4) |
| FEV1 % pred | 45.2±18.6 | IHD | 43 (27.7) | Antibioticƒ | 5 (3.2) |
| TLCO % pred | 38.3±16.2 | Cardiac failure | 32 (20.6) | Antiplatelet | 55 (35.5) |
| LTOT | 19 (12) | CVD | 15 (9.7) | Anticoagulant | 18 (11.6) |
| mMRCD score median (interquartile range) | 4 (3–5) | Diabetes | 29 (18.7) | β-blocker | 19 (12.3) |
| Current smoker | 48 (31) | Malignancy¶ | 15 (9.7) | Ivabradine | 4 (2.6) |
| Former smoker | 117 (69) | Renal failure+ | 3 (1.9) | Ca2+RA | 13 (8.2) |
| Smoking history pack-years | 44±26 | Anxiety | 36 (23.2) | ACE-I/ARB | 52 (33.5) |
| AECOPDs in previous year | Depression | 35 (22.6) | Statin | 57 (36.8) | |
| Hospital | 1.5±2.3 | Alcohol misuse | 12 (7.7) | Loop diuretic | 33 (21.3) |
| Community | 1.7±2.5 | Substance misuse | 2 (1.3) | Benzodiazepine## | 20 (12.6) |
| Frequent exacerbator# | 46 (29.7) | Other psychiatric disorder§ | 3 (1.9) | Antidepressant/antipsychotic | 39 (24.5) |
Data are presented as mean±sd or n (%), unless otherwise stated. BMI: body mass index; FEV1: forced expiratory volume in 1 s; TLCO: transfer factor of the lung for carbon monoxide; LTOT: long-term oxygen therapy; mMRCD: modified Medical Research Council dyspnoea scale; OSA: obstructive sleep apnoea; AF: atrial fibrillation; IHD: ischaemic heart disease; CVD: cerebrovascular disease; LAMA: long-acting muscarinic antagonist; LABA: long-acting β2-agonist; ICS: inhaled corticosteroid; OCS: oral corticosteroid; Ca2+RA: calcium receptor antagonist; ACE-I: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker. #: two or more AECOPD hospitalisations in the previous year; ¶: receiving treatment or palliation; +: estimated glomerular filtration rate <30 mL·min−1; §: bipolar affective disorder, schizophrenia or post-traumatic stress disorder; ƒ: maintenance; ##: excluding nocte temazepam.
A flow diagram for assigning aetiological components of phenotypes is shown (figure 2). The process identified a large number of distinct phenotypes (total 26) based on various combinations of six underlying aetiologies (figure 3). Phenotypes consisting of a single aetiology were noted in a minority (38 out of 146, 26.0%). Two (74 out of 146 patients, 50.7%) or three aetiologies (27 out of 146 patients, 18.5%) were common and no identifiable aetiology was rare (6 out of 146, 4.1%).
FIGURE 3.

Distribution and combinations of identified phenotypes among 146 hospitalised acute exacerbations of COPD. A: airway virus; B: bacterial; C: co-infection; D: depression/anxiety; E: eosinophils; F: failure (cardiac); G: general environment; X: unknown.
Outcomes associated with phenotypes
We first evaluated whether composite phenotypes (rather than individual aetiologies) were associated with clinical outcomes. The large number of phenotypes and resultant small populations in each phenotype precluded meaningful statistical analyses, but descriptive summary data are shown for the 10 most common phenotypes (representing 72.6% of study cohort, supplementary table S1). A higher cumulative number of aetiologies did not show association with clinical outcomes.
Outcomes associated with individual aetiologies
Individual aetiologies (rather than complex phenotypes) are likely to be more informative in a smaller cohort. We therefore compared exacerbations with versus without individual aetiological components.
Patients with virus infection (43 out of 146, 29.5%) were less commonly frequent hospitalised exacerbators (9.3% versus 37%, p=0.001), had lower baseline MRCD scores (3 (2–4) versus 4 (3–5), p=0.0007) and less domiciliary oxygen use (7% versus 32%, p=0.001). Despite this favourable profile, they had longer hospitalisation (7.7±5.6 versus 6.0±3.9, p=0.02), even after exclusion of those with bacterial co-infection (n=8, 7.9±6.1 versus 6.0±3.9, p=0.03). Their mortality at 12 months post hospital discharge was lower (2 out of 43 (4.7%) versus 20 out of 103 (19.4%), p=0.02). Survival curves over 12 months post-discharge are shown for virus (including co-infection) (n=43) versus bacterial only (n=59) versus non-infective (n=44) AECOPDs (figure 4, p=0.03).
FIGURE 4.
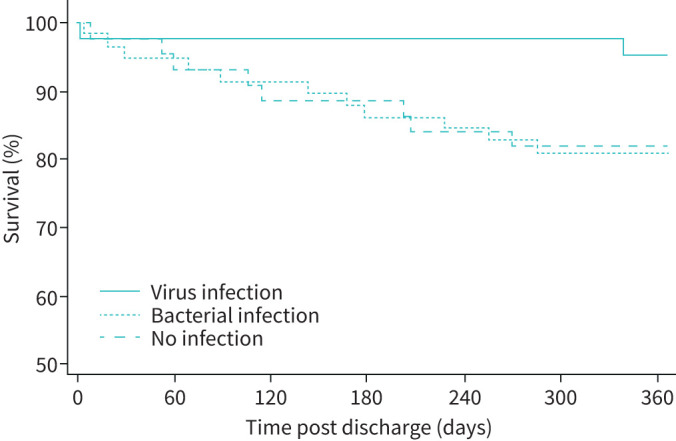
Survival 12 months post hospital discharge based on presence or absence of infection in acute exacerbations of COPD.
Bacterial aetiology was assigned in 59 out of 146 patients (40.4%), of whom 24 out of 59 patients (40.7%) had positive sputum culture (Bi, figure 2). These patients were more likely to be frequent hospitalised exacerbators (44% versus 18.4%, p=0.001), had higher baseline MRCD scores (5 (4–5) versus 4 (3–5), p=0.002) and higher prevalence of diagnosed ischaemic heart disease (53.8% versus 35.6%, p=0.045) and cardiac failure (57.1% versus 36.4%, p=0.045).
Those with combined virus and bacterial infection (co-infection) had higher white cell count (WCC) (14.7±3.7 versus 11.4±4.4, p=0.008), neutrophils (11.7±2.6 versus 8.6±3.4, p=0.01) and CRP (112 (65–167) versus 18 (4.6–69), p=0.004). Specific viruses and bacteria detected are shown supplementary table S2.
HADS scores were higher in those with diagnosed psychiatric comorbidity (20.4±8.7 versus 14.6±8.1, p=0.0001, supplementary table S3). HADS scores were above the threshold to assign aetiology “D” in 33 out of 136 patients (24.3%), of whom only 14 out of 33 (42.4%) were taking antidepressant or anxiolytic medication. Patients featuring aetiology (D) reported higher (“worse”) total CAT scores (34 (30–37) versus 29 (24–33), p=0.0001). Their responses to CAT items 1–5 (physical symptoms) did not differ, whereas CAT item responses 6 (“confidence” 4.3±1.2 versus 3.3±1.9, p=0.026), 7 (“sleep” 3.9±1.6 versus 2.9±1.6, p=0.015) and 8 (“energy” 3.9±1.6 versus 2.9±1.6, p=0.005) were higher. There were no significant differences in clinical outcomes based on aetiology “D”.
Blood eosinophils >2% (aetiology “E”) was present in 41 out of 146 (28%). Pre-hospital oral corticosteroid had been prescribed in 9.8% of those >2% and 30.5% with ≤2% eosinophils. Inhaled corticosteroids prescription was similar (75% versus 85%, p=0.34). Infection was less common in AECOPD with eosinophils >2% (41.4% versus 80.9%, p<0.0001). “Eosinophilic exacerbations” were associated with lower blood pH (7.32±0.06 versus 7.36±0.09, p=0.04), higher venous carbon dioxide tension (PvCO2) (53.7±10.5 versus 48.8±12.8, p=0.04) and NIV usage (34.1% versus 18.1%). Despite this, patients with eosinophils >2% had a shorter hospital stay (4 (3–5) versus 6 (4–9) days, p<0.001). Systemic corticosteroid prescription during hospitalisation was similar in the >/≤2% eosinophil groups (97.6% versus 97.1%).
An elevated cardiac biomarker (either/both, aetiology “F”) was noted in 85 out of 119 patients (71.2%), NT-proBNP in 83 out of 119 (69.7%) and hs-TnI in 32 out of 119 patients (26.9%). Patients with established diagnoses of cardiovascular disease tended to have higher levels (supplementary table S4), with NT-proBNP significantly higher in those with a past history of cardiac failure (618 ng·L−1 (18.5–2016) versus 321 ng·L−1 (117–693), p=0.03). Among patients with an elevated cardiac biomarker, cardiac medication use was notably low: antiplatelets (34.1%), anticoagulants (16.5%), β-blockers (17.7%), angiotensin-converting enzyme (ACE)-I/angiotensin receptor blocker (ARB) (38.8%), statins (32.9%) and loop diuretics (21.1%). Aetiology “F” was not associated with significant differences in short-term clinical outcomes, survival at 12 months or readmission rates. Using a threshold considered more definitive for cardiac failure (NT-proBNP>900 ng·L−1) was associated with longer hospital stay (7 (5–10) versus 5 (4–7) days, p=0.018).
General environmental factors contributing to hospitalisation (aetiology “G”) were rarely identified (3 out of 146 patients, 2.1%). Factors included running out of medication and failure of home air conditioning during an extreme heatwave.
No aetiology was identified in 6 out of 146 cases (aetiology “X”, 4.1%).
AECOPDs with versus without evidence of infection
Finally, we compared AECOPDs associated with infection (69.9%) versus no infection (30.1%). No differences in demographics, comorbidities or clinical outcomes were found (tables 2 and 3). Total CAT scores were similar (30 (26–33) versus 30 (25–34), p=0.98) with only CAT item 2 (“phlegm”) differing in infective exacerbations (4 (2–5) versus 3 (1–3), p=0.01). Severe anxiety/depression symptoms (23.7% versus 24.5%, p=0.92), hs-TnI (9 (5–32) versus 8 (5–20), p=0.37) and NT-proBNP (395 (164–1221) versus 263 (152–853), p=0.45) did not differ. Non-infective exacerbations featured higher blood eosinophil counts (0.25 per μL (0.08–0.46) versus 0.04 per μL (0.0–0.14), p<0.001). This observation persisted after excluding patients who had received pre-hospital oral corticosteroids (0.28 per μL (0.11–0.46) versus 0.05 per μL (0.0–0.2), p<0.001).
TABLE 2.
Characteristics of patients with and without evidence of infection
| Infection | No infection | p-value | |
| Patients | 102 (69.9) | 44 (30.1) | |
| Age years | 72.3±10.3 | 69.8±10.7 | 0.26 |
| Male | 65 (63.7) | 26 (59.1) | 0.6 |
| Frequent exacerbator, hospital | 30 (29.4) | 12 (27.3) | 0.79 |
| Current smoker | 32 (31.4) | 15 (34.1) | 0.75 |
| Smoking history pack-years | 46.7±28.5 | 38.7±21.6 | 0.1 |
| BMI kg·m−2 | 25.1±5.8 | 25.1±5.3 | 0.97 |
| FEV1 L | 1.21±0.49 | 1.10±0.67 | 0.42 |
| FEV1 % pred | 51.2±18.0 | 44.0±20.1 | 0.12 |
| TLCO % pred | 37.3±14.8 | 42.7±19.7 | 0.1 |
| mMRCD score | 4 (3–5) | 4 (3–5) | 0.81 |
| Days since symptom onset | 5 (3–7) | 4 (2–14) | 0.94 |
| Prior contact with HCP | 68 (66.7) | 24 (54.5) | 0.17 |
| Pre-hospital antibiotics | 51 (50) | 16 (36.4) | 0.13 |
| Pre-hospital OCS | 30 (29.4) | 6 (13.6) | 0.042 |
| CAT total score | 30 (25–34) | 30 (26–33) | 0.98 |
| HADS total score | 16 (10–22) | 18.5 (9–24) | 0.53 |
| BAP-65 class | 3 (2–5) | 2 (2–3) | 0.13 |
| CXR infiltrate | 30.4 | 20.9 | 0.24 |
| Fever ≥38°C | 23.5 | 0 | <0.001 |
| WCC ×109 per L | 12.2±4.8 | 10.0±3.0 | 0.006 |
| Neutrophils ×109 per L | 9.5±4.3 | 7.0±2.4 | <0.001 |
| Eosinophils ×109 per L | 0.04 (0.0–0.14) | 0.25 (0.08–0.46) | <0.001 |
| CRP mg·dL−1 | 53 (18.7–117) | 4 (1.7–7.0) | <0.001 |
| pH | 7.36±0.08 | 7.33±0.08 | 0.04 |
| PvCO2 mmHg | 48.1±11.2 | 54.5±13.8 | 0.006 |
| Bicarbonate mmol·L−1 | 27.5±4.2 | 29.3±5.7 | 0.13 |
| Base excess mmol·L−1 | 2.7±3.8 | 4.1±5.2 | 0.11 |
| NT-proBNP ng·L−1 | 395 (164–1221) | 263 (152–853) | 0.45 |
| hs-TnI ng·L−1 | 9 (5–32) | 8 (5–20) | 0.38 |
Data are presented as n (%), mean±sd or median (interquartile range), unless otherwise stated. BMI: body mass index; FEV1: forced expiratory volume in 1 s; TLCO: transfer factor of the lung for carbon monoxide; mMRCD: modified Medical Research Council dyspnoea scale; HCP: healthcare provider; OCS: oral corticosteroid; CAT: COPD Assessment Test; HADS: Hospital Anxiety and Depression Scale; BAP-65: blood urea nitrogen ≥25 mg·dL−1, altered mental status, pulse 109 beats per min, age 65 years; CXR: chest radiography; WCC: white cell count; CRP: C-reactive protein; PvCO2: venous carbon dioxide tension; NT-proBNP: N-terminal pro-brain natriuretic peptide; hs-TnI: high-sensitivity troponin I.
TABLE 3.
Management and clinical outcomes of patients with and without evidence of infection
| Infective | Non-infective | p-value | |
| Antibiotics, inpatient | 102 (100) | 40 (90.9) | 0.002 |
| Systemic CS, inpatient | 100 (98.0) | 42 (95.5) | 0.38 |
| NIV | 8 (7.8) | 10 (22.7) | 0.01 |
| HDU/ICU | 27 (26.4) | 18 (40.1) | 0.08 |
| Mechanical ventilation | 4 (3.9) | 1 (2.5) | 0.62 |
| Length of stay days | 5 (4–8) | 5 (3–8) | 0.2 |
| Mortality at 6 months | 10 (9.8) | 5 (11.4) | 0.89 |
| Mortality at 12 months | 14 (13.7) | 8 (18.2) | 0.49 |
| Readmitted within 12 months | 40 (39.2) | 22 (50.0) | 0.39 |
| Time to readmission days | 44.5 (18–195) | 78 (34–246) | 0.39 |
Data are presented as n (%) or median (interquartile range), unless otherwise stated. CS: corticosteroid; NIV: noninvasive ventilation; HDU: high-dependency unit; ICU: intensive care unit.
Repeat evaluation at stable outpatient review
Outpatient review was attended by 68 out of 146 patients (46.6%) at a median of 63 (59–98) days. Those who did versus did not attend follow-up showed no differences in demographics or spirometry with the only difference in comorbidities being less diagnosed anxiety disorder (14.9% versus 29.9%, p=0.03, supplementary table S5). At outpatient review, there was no difference in CRP (3.7 (1.3–8) versus 2.9 (1.4–5.5), p=0.69), WCC (9.2±2. versus 9.4±2.5, p=0.74) or neutrophils (6.4±2.7 versus 6.3±2.2, p=0.89) between those who had experienced infective versus non-infective exacerbations, including reanalysis based on individual infective exacerbation aetiologies (A, B or C).
In contrast, HADS scores at exacerbation and recovery were correlated (r=0.56, p<0.0001). Patients assigned aetiology “D” at exacerbation had significantly higher HADS scores at follow-up (22 (13–28) versus 9 (4–15), p=0.004).
Blood eosinophils at exacerbation correlated with eosinophil counts at recovery (r=0.54, p<0.0001). Patients with eosinophils >2% at exacerbation (aetiology “E”) had significantly higher blood eosinophils at recovery (0.3 (0.2–0.6) versus 0.11 (0.02–0.28), p=0.0003).
Correlations between exacerbation and recovery measurements were significant for NT-proBNP (r=0.39, p=0.004) but not for hs-TnI (r=0.23, p=0.14). Patients who were aetiology “F” were far more likely to have an elevated cardiac biomarker at recovery (70.6% versus 5.9%, p<0.0001) with both hs-TnI (5 (4–9) versus 3.5 (1.5–5), p=0.01) and NT-proBNP (269 (151–692) versus 67 (25–108), p<0.0001) higher at follow-up.
Discussion
The current study demonstrates that hospitalised AECOPDs comprise a remarkably heterogeneous group of events, often featuring multiple distinct aetiological contributors. This heterogeneity is a barrier to progress in the field, since interventions targeting a specific aetiology or pathology may not show benefit if applied to an unselected group. Focussing attention on exacerbation aetiology and constructing AECOPD phenotypes is a logical approach and may be the most suitable prospective strategy to identify patients eligible for targeted interventions.
AECOPD phenotyping will only gain traction if it can ultimately lead to individualisation of treatment decisions. Aetiologies that are prevalent and responsive to treatment are therefore the most important to target. We based our phenotyping strategy on examining six key putative aetiological factors (virus infection, bacterial infection, depression/anxiety, eosinophil-associated inflammation, cardiac dysfunction, environmental factors). This strategy identified many combinations (26 distinct phenotypes within a cohort of 146 patients) with only around a quarter of AECOPDs associated with a single aetiology. Clearly each AECOPD event may have multiple aetiological “ingredients”, each with distinct implications for individualised management and prognosis. The current study provides proof-of-concept evidence for a practical phenotyping strategy and demonstrates that using a relatively small number of commonly available investigations makes it possible to unravel some of the complexity of AECOPD.
Identifying viral infection appears to have prognostic implications, and multiplex virus PCR will likely become standard of care for hospitalised AECOPD due to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic. Our virus detection rate (30%) was consistent with previous research [15–17]. Despite more prolonged hospitalisation, a history of frequent severe exacerbations was less common and 12-month survival was better in the viral group. It may be that virus infections are sporadic events, whereas AECOPD linked to bacterial infection or high blood eosinophil exacerbations reflect a more “committed” phenotype [6].
Given the limitations of sputum culture and frequent pre-hospital antibiotic use (45.2% overall in our cohort) we assigned bacterial aetiology to exacerbations where infection was evident, but virus PCR testing was negative. A more precise methodology is difficult within the limitations of routine clinical investigations. Broad-range 16S rDNA PCR of sputum samples would enhance the sensitivity for detection of bacteria on sputum but is not routinely available, and its clinical interpretation remains uncertain. We chose a sensitive CRP threshold of >20 mg·dL−1 previously suggested as optimal threshold to identify bacteria in sputum at AECOPD [11]. Putative bacterial exacerbations were associated with frequent hospitalisation, comorbid cardiac disease and reduced survival at 1 year. The frequent identification of Pseudomonas aeruginosa on sputum culture (supplementary table S2) may reflect advanced structural lung disease and a propensity to recurrent bacterial infection with airway colonisation. Reduced survival has previously been associated with lung dysbiosis identified on sputum culture at the time of hospitalised exacerbation [18].
Secondary bacterial infection is known to be a frequent sequel of virus infection in AECOPD [19], and our low prevalence of confirmed co-infection is likely an underestimate reflecting prior antibiotic use, limitations of sputum culture and phenotyping based on admission samples only. In keeping with previous studies we found the highest inflammatory markers during co-infections [20].
The impact of non-infective aetiologies on AECOPD has been less extensively studied. Anxiety and depression have been associated with increased hospitalisation rates, longer hospitalisation and increased mortality in COPD [21, 22]. In our study severe symptoms of anxiety and depression were common and often untreated. Importantly, HADS scores at AECOPD and recovery were strongly correlated. Future studies could explore the role for identification of psychological morbidity and initiation of appropriate interventions prior to hospital discharge.
Blood eosinophils appear to identify an important AECOPD phenotype. Infection was less common with higher eosinophils. While patients with eosinophilic exacerbations had lower blood pH, higher PvCO2 and greater need for NIV, they had a shorter hospital length of stay, a finding that may reflect corticosteroid responsiveness [23]. Eosinophil counts were still higher after recovery suggesting association between the exacerbation and “stable” phenotype. Given the key benefit of anti-interleukin (IL)-5 therapies is reduction of exacerbations, patients hospitalised with an eosinophilic exacerbation may be the ideal candidates for future trials of anti-IL-5 therapies in COPD.
Finally, we observed biochemical evidence of acute cardiac dysfunction in a majority of AECOPDs [24–27]. The higher levels observed in patients with established cardiovascular disease suggests cardiac biomarkers reflect underlying cardiovascular health. At the same time, cardiac biomarkers were often high even amongst those without an established diagnosis of cardiovascular disease. This suggests that cardiac disease may be a crucial underdiagnosed “treatable trait” [28] which can be fortuitously detected during AECOPD. Delineation of the multitude of cardiac pathologies identifiable in a hospitalised AECOPD population was beyond the scope of this paper. While the relationship between an elevated cardiac biomarker at exacerbation and the likelihood of an identifiable treatable cardiac comorbidity requires further study, the low prescription rates of cardiac therapies observed in our cohort suggests a potential need for increased recognition and treatment of cardiovascular pathology in COPD.
Our study has a number of limitations. Our sample size was not adequate to examine associations between clusters of aetiologies (phenotypes) and pertinent outcomes. This will likely require very large-scale multicentre studies of exacerbation characteristics, which have been identified as a priority for future AECOPD research [29, 30]. The very low inpatient mortality (1 out of 146, 0.7%) may reflect exclusion of patients mechanically ventilated at the time of admission and the requirement for adequate cognition to complete questionnaires. We employed routine clinical investigations to define phenotypes, which limits the precision and reliability of diagnosing bacterial infection. However, this strategy was intentionally applicable to “real-life” practice. Virus and bacterial detection at AECOPD may potentially be “false positives” reflecting colonisation, and even virus PCR may give “false negatives” [11]. Pre-hospital antibiotic (45.2%) and/or oral corticosteroid (23.2%) use were common and may have influenced phenotypes. We phenotyped AECOPDs only once at hospital admission, but “evolution” of AECOPD phenotypes over the course of an exacerbation is an area for future study. Finally, other important AECOPD aetiologies may not have been captured in our methodology. While our strategy focussed on treatable aetiological components of the acute hospitalisation episode, phenotyping the chronic disease state (e.g. emphysema predominant) is also key to individualisation of care. Understanding relationships between the chronic disease phenotype and the acute exacerbation phenotype is an area for future research.
In conclusion, better prevention and management of AECOPD will be challenging since there are numerous causes, often in combination, that converge as complex, multi-faceted phenotypes. Identifying the individual contributory aetiologies is feasible and relates to important clinical outcomes. Large prospective studies employing phenotyping can enhance understanding of disease mechanisms and ultimately drive “personalised medicine” in AECOPD.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
TABLE S1 Characteristics of 10 most common phenotypes arranged in order of descending frequency 00198-2021.TABLES1 (186.9KB, pdf)
TABLE S2 Viruses and bacteria identified in 146 patients with AECOPD 00198-2021.TABLES2 (12.6KB, pdf)
TABLE S3 HADS scores within subgroups with or without psychological comorbidity 00198-2021.TABLES3 (87.6KB, pdf)
TABLE S4 Cardiac biomarker measurements in patients with and without a history of cardiovascular disease 00198-2021.TABLES4 (88.8KB, pdf)
TABLE S5 Characteristics of patients who did versus did not undergo follow-up outpatient (stable) assessment 00198-2021.TABLES5 (78KB, pdf)
Footnotes
Author contributions: Conception and design: M.I. MacDonald, P.T. King and P.G. Bardin; data acquisition: M.I. MacDonald, C.R. Osadnik, L. Bulfin and E. Leahy; statistical analysis and interpretation: M.I. MacDonald, C.R. Osadnik, P. Leong, K. Hamza, P.T. King and P.G. Bardin; drafting the manuscript for important intellectual content: M.I. MacDonald, C.R. Osadnik, P. Leong, E. Shafuddin, P.T. King and P.G. Bardin; final review of manuscript: all authors.
This article has supplementary material available from openres.ersjournals.com
Editorial comment in ERJ Open Res 2021; 7: 00362-2021 [https://doi.org/10.1183/23120541.00362-2021].
Conflict of interest: M.I. MacDonald reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Conflict of interest: C.R. Osadnik has nothing to disclose.
Conflict of interest: L. Bulfin reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Conflict of interest: E. Leahy reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Conflict of interest: P. Leong reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Conflict of interest: E. Shafuddin reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Conflict of interest: K. Hamza has nothing to disclose.
Conflict of interest: P.T. King reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Conflict of interest: P.G. Bardin reports financial support for this study from GlaxoSmithKline: payments made to support this project via an unrestricted educational grant. GlaxoSmithKline had no involvement in study design, patient recruitment, data analysis or manuscript preparation. No further disclosures provided.
Support statement: Funding support for this study was provided by an unrestricted educational grant from GlaxoSmithKline. The funder did not play any role in study design, data collection, analysis and interpretation of data or the decision to submit the paper for publication. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Han MK, Agusti A, Calverley PM, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010; 182: 598–604. doi: 10.1164/rccm.200912-1843CC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med 2017; 195: 557–582. doi: 10.1164/rccm.201701-0218PP [DOI] [PubMed] [Google Scholar]
- 3.MacDonald M, Beasley RW, Irving L, et al. A hypothesis to phenotype COPD exacerbations by aetiology. Respirology 2011; 16: 264–268. doi: 10.1111/j.1440-1843.2010.01908.x [DOI] [PubMed] [Google Scholar]
- 4.MacDonald M, Korman T, King P, et al. Exacerbation phenotyping in chronic obstructive pulmonary disease. Respirology 2013; 18: 1280–1281. doi: 10.1111/resp.12197 [DOI] [PubMed] [Google Scholar]
- 5.Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184: 662–671. doi: 10.1164/rccm.201104-0597OC [DOI] [PubMed] [Google Scholar]
- 6.Mayhew D, Devos N, Lambert C, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax 2018; 73: 422–430. doi: 10.1136/thoraxjnl-2017-210408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones PW. COPD assessment test: rationale, development, validation and performance. COPD 2013; 10: 269–271. doi: 10.3109/15412555.2013.776920 [DOI] [PubMed] [Google Scholar]
- 8.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983; 67: 361–370. doi: 10.1111/j.1600-0447.1983.tb09716.x [DOI] [PubMed] [Google Scholar]
- 9.Hsu KY, Lin JR, Lin MS, et al. The modified Medical Research Council dyspnoea scale is a good indicator of health-related quality of life in patients with chronic obstructive pulmonary disease. Singapore Med J 2013; 54: 321–327. doi: 10.11622/smedj.2013125 [DOI] [PubMed] [Google Scholar]
- 10.Peng C, Tian C, Zhang Y, et al. C-reactive protein levels predict bacterial exacerbation in patients with chronic obstructive pulmonary disease. Am J Med Sci 2013; 345: 190–194. doi: 10.1097/MAJ.0b013e318253c921 [DOI] [PubMed] [Google Scholar]
- 11.Singer S, Kuhnt S, Gotze H, et al. Hospital anxiety and depression scale cutoff scores for cancer patients in acute care. Br J Cancer 2009; 100: 908–912. doi: 10.1038/sj.bjc.6604952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang Robinson, SC, Mills GD, et al. Biochemical markers of cardiac dysfunction predict mortality in acute exacerbations of COPD. Thorax 2011; 66: 764–768. doi: 10.1136/thx.2010.155333 [DOI] [PubMed] [Google Scholar]
- 13.Galasko GI, Lahiri A, Barnes SC, et al. What is the normal range for N-terminal pro-brain natriuretic peptide? How well does this normal range screen for cardiovascular disease? Eur Heart J 2005; 26: 2269–2276. doi: 10.1093/eurheartj/ehi410 [DOI] [PubMed] [Google Scholar]
- 14.14 Trambas C, Pickering JW, Than M, et al. Impact of high-sensitivity troponin i testing with sex-specific cutoffs on the diagnosis of acute myocardial infarction. Clin Chem 2016; 62: 831–838. doi: 10.1373/clinchem.2015.252569 [DOI] [PubMed] [Google Scholar]
- 15.Sapey E, Stockley RA. COPD exacerbations. 2: aetiology. Thorax 2006; 61: 250–258. doi: 10.1136/thx.2005.041822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitt R, Farne H, Ritchie A, et al. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther Adv Respir Dis 2016; 10: 158–174. doi: 10.1177/1753465815618113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zwaans WA, Mallia P, van Winden ME, et al. The relevance of respiratory viral infections in the exacerbations of chronic obstructive pulmonary disease: a systematic review. J Clin Virol 2014; 61: 181–188. doi: 10.1016/j.jcv.2014.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filho FSL, Alotaibi NM, Ngan D, et al. Sputum microbiome is associated with 1-year mortality after chronic obstructive pulmonary disease hospitalizations. Am J Respir Crit Care Med 2019; 199: 1205–1213. doi: 10.1164/rccm.201806-1135OC [DOI] [PubMed] [Google Scholar]
- 19.Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186: 1117–1124. doi: 10.1164/rccm.201205-0806OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilkinson TM, Hurst JR, Perera WR, et al. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest 2006; 129: 317–324. doi: 10.1378/chest.129.2.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laurin C, Moullec G, Bacon SL, et al. Impact of anxiety and depression on chronic obstructive pulmonary disease exacerbation risk. Am J Respir Crit Care Med 2012; 185: 918–923. doi: 10.1164/rccm.201105-0939PP [DOI] [PubMed] [Google Scholar]
- 22.Eisner MD, Blanc PD, Yelin EH, et al. Influence of anxiety on health outcomes in COPD. Thorax 2010; 65: 229–234. doi: 10.1136/thx.2009.126201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacDonald MI, Osadnik CR, Bulfin L, et al. Low and high blood Eosinophil counts as biomarkers in hospitalized acute exacerbations of COPD. Chest 2019; 156: 92–100. doi: 10.1016/j.chest.2019.02.406 [DOI] [PubMed] [Google Scholar]
- 24.Aliyali M, Mehravaran H, Abedi S, et al. Impact of comorbid ischemic heart disease on short-term outcomes of patients hospitalized for acute exacerbations of COPD. Tanaffos 2015; 14: 165–171. [PMC free article] [PubMed] [Google Scholar]
- 25.Almagro P, Cabrera FJ, Diez J, et al. Comorbidities and short-term prognosis in patients hospitalized for acute exacerbation of COPD: the EPOC en Servicios de Medicina Interna (ESMI) study. Chest 2012; 142: 1126–1133. doi: 10.1378/chest.11-2413 [DOI] [PubMed] [Google Scholar]
- 26.Antonelli Incalzi R, Fuso L, De Rosa M, et al. Co-morbidity contributes to predict mortality of patients with chronic obstructive pulmonary disease. Eur Respir J 1997; 10: 2794–2800. doi: 10.1183/09031936.97.10122794 [DOI] [PubMed] [Google Scholar]
- 27.Marcun R, Sustic A, Brguljan PM, et al. Cardiac biomarkers predict outcome after hospitalisation for an acute exacerbation of chronic obstructive pulmonary disease. Int J Cardiol 2012; 161: 156–159. doi: 10.1016/j.ijcard.2012.05.044 [DOI] [PubMed] [Google Scholar]
- 28.Agusti A, Bel E, Thomas M, et al. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J 2016; 47: 410–419. doi: 10.1183/13993003.01359-2015 [DOI] [PubMed] [Google Scholar]
- 29.Mathioudakis AG, Sivapalan P, Papi A, et al. The DisEntangling Chronic Obstructive pulmonary Disease Exacerbations clinical trials NETwork (DECODE-NET): rationale and vision. Eur Respir J 2020; 56: 2000627. doi: 10.1183/13993003.00627-2020 [DOI] [PubMed] [Google Scholar]
- 30.Janssens W, Bafadhel M. The CICERO (Collaboration In COPD ExaceRbatiOns) Clinical Research Collaboration. Eur Respir J 2020; 55: 2000079. doi: 10.1183/13993003.00079-2020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
TABLE S1 Characteristics of 10 most common phenotypes arranged in order of descending frequency 00198-2021.TABLES1 (186.9KB, pdf)
TABLE S2 Viruses and bacteria identified in 146 patients with AECOPD 00198-2021.TABLES2 (12.6KB, pdf)
TABLE S3 HADS scores within subgroups with or without psychological comorbidity 00198-2021.TABLES3 (87.6KB, pdf)
TABLE S4 Cardiac biomarker measurements in patients with and without a history of cardiovascular disease 00198-2021.TABLES4 (88.8KB, pdf)
TABLE S5 Characteristics of patients who did versus did not undergo follow-up outpatient (stable) assessment 00198-2021.TABLES5 (78KB, pdf)