

Summary
Quantitative assessment of neuropathological changes is essential for the characterization of animal models of neurodegenerative disease. Here, we describe a detailed protocol for the detection and quantification of key neuropathological changes in Alzheimer's mouse models. The protocol covers detailed methods including perfusion, dissection, and paraffinization of the brain, preparation of serial brain sections, immunohistochemical analysis, stereological quantification, and sample coding methods for genotype blind analysis. This protocol may be applied to the analysis of neuropathological changes of other neurological disorders.
For complete details on the use and execution of this protocol, please refer to Lee et al. (2020), Kang and Shen (2020), Giaime et al. (2017), Xia et al. (2015), Watanabe et al. (2012, 2014), Wines-Samuelson et al. (2010), and Saura et al. (2004).
Subject areas: Microbiology, Model Organisms, Neuroscience
Graphical abstract
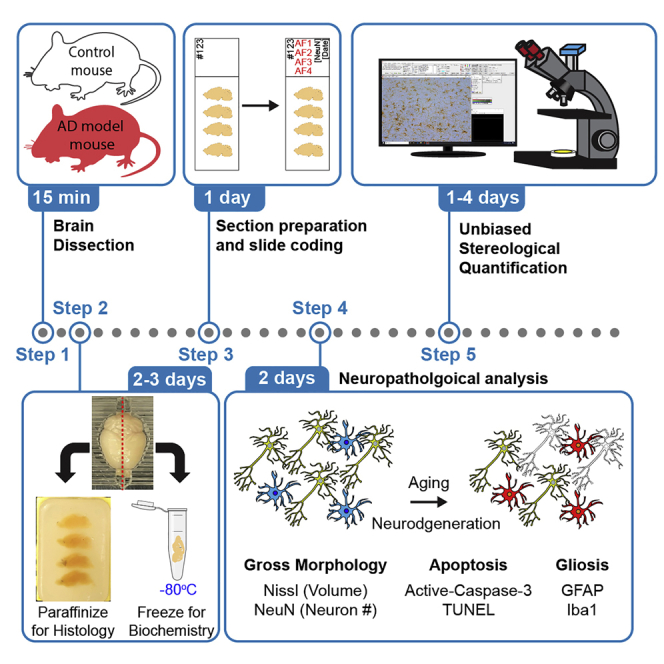
Highlights
-
•
Perfusion, dissection, paraffinization, and section preparation of the mouse brain
-
•
Sample coding methods for genotype blind analysis
-
•
Protocol for immunohistochemical analysis of neuropathology in AD mouse models
-
•
Stereological quantification method of neuropathological changes in the mouse brain
Quantitative assessment of neuropathological changes is essential for the characterization of animal models of neurodegenerative disease. Here, we describe a detailed protocol for the detection and quantification of key neuropathological changes in Alzheimer's mouse models. The protocol covers detailed methods including perfusion, dissection, and paraffinization of the brain, preparation of serial brain sections, immunohistochemical analysis, stereological quantification, and sample coding methods for genotype blind analysis. This protocol may be applied to the analysis of neuropathological changes of other neurological disorders.
Before you begin
Fixation of the mouse brain by transcardial perfusion
Timing: 1 day
Intracardiac perfusion is the best way to remove blood from brain capillaries. If you want brain materials for both histological and biochemical analysis, you should omit fixatives in perfusion solutions. After intracardiac perfusion with PBS, you dissect out both hemispheres, one undergoing post-fixation for histology and the other frozen for biochemical or molecular analysis.
-
1.
Assemble a syringe (> 20 mL capacity) with a plastic tube (> 50 cm in height) filled ice-cold PBS perfusion solution (or Ringer solution) containing procaine (5 mg/mL) and heparin (0.25 mg/mL) at least 20 mL/mouse.
-
2.
Anesthetize a mouse via intraperitoneal injection with IACUC approved anesthesia protocol. In this protocol, we used ketamine supplemented with acepromazine and xylazine (ketamine: 100 mg/kg body weight, xylazine: 10 mg/kg body weight, acepromazine: 3 mg/kg body weight) to anesthetize either male or female mice in the age range of 2–24 months old.
Note: Please follow the approved anesthesia protocol of your institute/lab. After administering an anesthesia cocktail, monitor the adequacy or depth of anesthesia by checking respiration rate and toe pinch reflection. It usually takes 5 min to complete anesthesia.
-
3.
Lay the anesthetized mouse with the abdominal side up and dissect the cavitas thoracis for heart exposure.
Note: Do not cut the middle of costae, because there are large aortas in this region.
-
4.Prick the left ventricle with a needle (e.g., Winged Infusion Set; 23Gx3/4”, TERUMO) and cut out the auricle of the right atrium to drain blood.
-
a.Left ventricle looks pale compared with other components due to the higher level of oxygen.
-
b.Insert the needle at approximately 2 mm in depth.
-
c.(Option) Using a 1 mL syringe, squeeze blood from the right ventricle (darker side) to collect venous blood, if needed. We can usually obtain 0.7–1.0 mL blood from one mouse. Mix immediately with 10 μL of 0.5 M EDTA to prevent agglutination of blood and leave on ice until all procedures have been done. If plasma specimens are needed, centrifuge blood samples at 1000 × g for 10 min at 4°C, and transfer supernatant to the new tube.
-
a.
-
5.
Perfuse with PBS perfusion solution, 20 mL per mouse.
Note: Pay attention to the flow speed (about 5–10 mL/min) during the perfusion. Alternatively, a peristatic pump can be used for perfusion. The gradual pale color change of the lung and liver is an indication of a successful perfusion.
Note: Alternatively, if you need the entire brain for histological experiments, go to step 12.
-
6.
Dissect out the whole brain carefully.
-
7.
Cut with a razor blade (AccuTec Blades. Inc., Cat #; 9404910000) in the midline using a brain matrix (e.g., Braintree Scientific Inc. BS-5000C/BS-5000S for mouse, 1-mm at interval) for sagittal brain sections (Figure 1A).
-
8.
Put one hemisphere of the brain into a 15-mL tube with 4% Paraformaldehyde (PFA)/PBS for histological analysis for ∼16 h with gentle shaking, ∼50 rpm.
-
9.
Dissect out specific brain subregions of the other hemisphere using microforceps (e.g., the neocortex, hippocampus, striatum, cerebellum, olfactory bulb, etc.) if needed. Be careful, because the brain tissue is soft and fragile.
-
10.
Put the dissected brain tissue into the Eppendorf tube for biochemical or molecular analysis. Drop the tissue quickly in liquid nitrogen or dry-ice following a measurement of tissue weight.
-
11.
Store fixed brain materials in PBS at 4°C (can be stored up to 1 week in PBS solution before paraffin processing). Store frozen brain materials at −80°C until use.
Note: Use filtered or autoclaved PBS solution with 0.1% sodium azide for long-term storage of the brain.
Figure 1.

Preparation of mouse brains for sagittal or coronal sections
(A) Red lines indicate the position of the cutting plane for sagittal or coronal brain sections.
(B) Images of paraffin blocks containing multiple brains for sagittal or coronal sections.
Scale bar: 5 mm.
Perfusion with fixative followed by post-fixation of brain sample for the brain histology.
-
12.
Change PBS perfusion solution to 4% PFA/PBS (20 mL/mouse).
-
13.
After perfusion with fixative, dissect out the whole brain carefully.
-
14.
Cut with a razor blade in the midline using a brain matrix (e.g., Braintree Scientific Inc. BS-5000C/BS-5000S for mouse, 1-mm at interval) for sagittal brain sections. In the case of coronal section preparation, put the fixed brain in the brain matrix upside down, and cut at the 5th position (5-mm) from the rostral end (so as to cut just half of the cortex) (Figure 1A).
-
15.
Immerse the hemibrain in 4% PFA/PBS for post-fixation at 4°C for 2–16 h.
Note: Gently invert every 20 min or use horizontal shaker at 50 rpm. For the frozen section, immerse the brains at 4°C for at least 2 h. For the paraffin section, immerse the brain for at least 3 h.
-
16.
Change the fixative solution to PBS, and store it at 4°C for at most 1 week.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-NeuN; 1/400 dilution | Millipore | Cat#MAB377, RRID: AB_2298772 |
| Mouse anti-GFAP (clone G-A-5); 1/500 dilution | Sigma | Cat#G6171, RRID: AB_1840893 |
| Rabbit anti-Iba1; 1/500 dilution | Wako | Cat#019–19741, RRID: AB_839504 |
| Rabbit anti-cleaved caspase3 (Asp175); 1/250 dilution | Cell Signaling Technology | Cat#9661S, RRID: AB_2341188 |
| Goat biotinylated anti-rabbit IgG (H&L); 1/400 dilution | Vector Laboratories | Cat#BA-1000, RRID: AB_2313606 |
| Goat biotinylated anti-mouse IgG (H&L); 1/400 dilution | Vector Laboratories | Cat#BA-9200, RRID: AB_2336171 |
| Alexa Fluor® 488 Goat Anti-Rabbit IgG (H+L); 1/300 dilution | Thermo Fisher Scientific | Cat#A-11034, RRDI: AB_2576217 |
| Alexa Fluor® 555 Goat Anti-Mouse IgG (H+L); 1/300 dilution | Thermo Fisher Scientific | Cat#A-21424,RRID: AB_141780 |
| Chemicals, peptides, and recombinant proteins | ||
| PBS | Thermo Fisher Scientific | AAJ75889K8 |
| Heparin sodium salt | Sigma-Aldrich | H4784 |
| Procaine hydrochloride | Fluka | 81666 |
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 |
| DAB peroxidase substrate | Vector Laboratories | Cat#SK-4100, RRID: AB_2336382 |
| Normal Goat Serum blocking solution | Vector laboratories | S-1000 |
| Tissue-Tek O.C.T. Compound | Sakura | 4583 |
| Histoclear | National Diagnostics | HS-200 |
| Absolute ethanol | Fisher Scientific | 04-355-223 |
| VECTASHIELD Mounting Medium | Vector Laboratories | H-1000 |
| DAPI | Thermo Fisher Scientific | D1306 |
| Cresyl Violet | Sigma-Aldrich | D5042 |
| Acetic acid (glacial) | Sigma-Aldrich | 1000631011 |
| Sudan Black B | Sigma-Aldrich | 199664-25G |
| Xylenes | Sigma-Aldrich | 534056-4L |
| Harris Modified Method Hematoxylin Stains | Fisher Scientific | SH30-500D |
| Paraplast Plus | Leica | 39602004 |
| 30% Hydrogen peroxide | Sigma-Aldrich | H1009 |
| Citric acid (anhydrous) | Sigma-Aldrich | 1370021000 |
| Sodium citrate tribasic dihydrate | Sigma-Aldrich | C8532-1Kg |
| Critical commercial assays | ||
| In Situ Cell Death Detection Kit | Roche | 11684795910 |
| TUNEL dilution buffer | Sigma | 11966006001 |
| VECTASTAIN Elite ABC Kit | Vector Laboratories | Cat#PK-6100, RRID: AB_2336819 |
| Experimental models: Organisms/strains | ||
| Mouse: EX-PS cDKO | Saura et al., 2004 | N/A |
| Mouse: APP cTKO | Lee et al., 2020 | N/A |
| Mouse: IN-PS cDKO | Kang and Shen, 2020 | N/A |
| Software and algorithms | ||
| BioQuant image analysis software | BIOQUANT Life Science | RRID: SCR_016423 |
| Other | ||
| Millex-GV Filter, 0.22 μm | Millipore | SLGV004SL |
| Olympus BX-51 microscope | Olympus | BX-51 |
| Olympus BX-40 microscope | Olympus | BX-40 |
| Olympus IX-51 microscope | Olympus | IX-51 |
| Nikon Ci-L microscope | Nikon | Ci-L |
| Excelsior™ AS Tissue Processor | Thermo Fisher Scientific | A82300001 |
| HistoCore Arcadia H - Heated Paraffin Embedding Station | Leica | 14039357258 |
| Tissue-Tek® Base Molds for Embedding Rings | EMS | 62528-38 |
| HistoCore Arcadia C - Cold Plate | Leica | 14039380101 |
| 20-Slide Staining Dish Complete Set | EMS | 71420-25 |
| Rotary microtome | Leica | RM2125 RT |
| Cryostat | Leica | CM1950 |
| Fisherbrand ColorFrost Plus Microscope Slides | Fisher Scientific | 12-550-17 |
| Fisherbrand Tissue Path MACROSETTE Processing/Embedding Cassettes | Fisher Scientific | 15-182-706 |
Materials and equipment
PBS perfusion solution: use 20 mL PBS perfusion solution per mouse. Store a solution at 4°C after making. It can be stored up to 1 month.
| Reagent | Final concentration | Amount |
|---|---|---|
| 1× PBS | 1× | 100 mL |
| Heparin | 0.25 mg/mL | 25 mg |
| Procaine | 5 mg/mL | 0.5 g |
| Total | n/a | 100 mL |
4% Paraformaldehyde solution for perfusion and post-fixation: use 20 mL fixative for perfusion per mouse, and 10 mL for post-fixation. It can be stored up to 1 months at 4°C, protected from light.
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% Paraformaldehyde | 4% | 10 mL |
| 20× PBS | 1× | 2 mL |
| Distilled water | n/a | 28 mL |
| Total | n/a | 40 mL |
-
•Cresyl violet solution for Nissl staining:
-
○Dissolve 1 g cresyl violet acetate in 200 mL distilled water.
-
○Add 500 μL of glacial acetic acid and filter the solution with 3mm Wattman paper.
-
○Store at 22°C–25°C. It can be stored up to six months but use a fresh batch of cresyl violet solution if possible.
-
○
-
•Antigen retrieval solution:
-
○Dissolve 1.92 g Citric acid, anhydrate in 900 mL distilled water.
-
○Adjust pH 6.0 with 10N NaOH (it requires approximately 2.6 mL).
-
○Adjust volume to 1 L at final concentration of 10 mM.
-
○Store at 22°C–25°C. It can be stored up to six months.
-
○
-
•Sudan Black (Fat Black) solution:
-
○Dissolve Sudan Black in 70% Ethanol at final concentration of 0.3% (w/v).
-
○Mix the solution with stirrer bar for more than 1 h (keep the bottle with aluminum foil).
-
○Filtrate the solution twice with 0.22 μm filter unit (Millipore,Cat #; SLGV004SL) with syringe.
-
○Keep the solution at 22°C–25°C with foil rapping to avoid the light. It can be stored up to six months.
-
○
-
•
Paraffin processing machine: This protocol uses Thermo Scientific Excelsior AS tissue processor. Other tissue processors can be used as well.
-
•
Microscope: This protocol uses Olympus BX-51, Olympus BX-40, Nikon Ci-L or Olympus IX-51 microscope for imaging. Alternative microscopes can be used as well.
-
•
Stereological quantification software: This protocol uses a BioQuant Image analysis software for stereological quantification. Alternative program can be used as well, for example, Stereo Investigator (MicroBrightField Inc.) or NewCAST/VisioMorph (Visiopharm).
Step-by-step method details
Paraffinization of the brain tissue
Paraffinization of fixed tissues depends on the tissue and species. The following protocol is optimized for the mouse brain.
-
1.
Place the fixed brain in a large size plastic paraffin cassette (FisherScientific, Cat #; 15-182-706).
-
2.Set the cassette to the tissue processor.
-
a.Processing method is as shown below in the case of the adult mouse brains.
-
a.
| Step | Solution | Temperature | Immersion | Drain |
|---|---|---|---|---|
| 1 | 75% EtOH | ambient | 1:20:00 | 0:15 |
| 2 | 75% EtOH | ambient | 1:20:00 | 0:15 |
| 3 | 80% EtOH | ambient | 1:20:00 | 0:45 |
| 4 | 80% EtOH | ambient | 1:20:00 | 0:45 |
| 5 | 95% EtOH | ambient | 1:20:00 | 0:15 |
| 6 | 95% EtOH | ambient | 1:20:00 | 0:15 |
| 7 | 100% EtOH | ambient | 1:20:00 | 0:15 |
| 8 | 100% EtOH | ambient | 1:20:00 | 0:15 |
| 9 | 100% EtOH | ambient | 1:20:00 | 0:15 |
| 10 | Xylene | ambient | 1:20:00 | 1:00 |
| 11 | Xylene | ambient | 1:20:00 | 2:00 |
| 12 | Xylene | ambient | 1:20:00 | 2:00 |
| 13 | Paraffin (Paraplast plus) | 60°C | 1:20:00 | 2:00 |
| 14 | Paraffin (Paraplast plus) | 60°C | 1:20:00 | 2:00 |
| 15 | Paraffin (Paraplast plus) | 60°C | 1:20:00 | 2:00 |
-
3.
Transfer the plastic cassette to the embedding center and immerse in paraffin pool (Lecia, Cat #; 39602004).
-
4.Take out the brain samples from the cassette and place them in the metal base molds for embedding.
-
a.Use 38 × 25 × 12 mm (Electron Microscope Science, Cat #; 62528-38). This mold can fit 4–5 brains on one block.
-
a.
-
5.
Put a small amount (∼1/3) of paraffin in the molds beforehand.
-
6.
Place multiple paraffinized brains in the mold (Figure 1B).
-
7.
Gently move the mold onto the cold plate for 5–10 s to form a thin layer at the bottom of the mold to anchor the brain in position. Pour more paraffin to the top of the mold.
-
8.
Put the lid of the plastic cassette on the mold and fill with paraffin until the cassette’s surface is completely covered.
-
9.
Cool on the cold plate or ice for at least 30 min.
-
10.
Cut extra paraffin attached on the side and gently peel off the paraffin block from the mold.
-
11.
Confirm an absence of cracks in the paraffin block. Troubleshooting 1
-
12.
Store the paraffin blocks at ambient temperature until use.
CRITICAL: When you embed multiple brains in one block, you should include both controls and experimental groups in the same block. Do not embed multiple brains of the same genotype in the same block.
Sectioning of the brain
-
13.Set the paraffin block in a microtome (Lecia, Cat #; RM2235) at an appropriate cutting angle (5 degree) and in a parallel position to the blade.
-
a.We cut sagittal sections in 10 μm thickness and coronal sections in 16 μm thickness.
-
b.Section with right handle; course tune with the left handle.
-
c.When not sectioning, secure the blade cover and lock the handle to avoid injuries.
-
a.
-
14.
Fill deionized water in the flotation workstation water bath (Fischer Scientific, Cat #; 22-047-234), and set the temperature depending on the block condition and thickness of sections. We use 42°C for 10 μm sections and 44°C for 16 μm sections.
-
15.Prepare the paraffin block for sectioning.
-
a.Section the paraffin block (usually 20–30 sagittal sections for the cerebral cortex) until you can see the entire brain structure of at least one sample on the section, which will be labeled as section #1.
-
a.
-
16.Section slowly and pick up each section at a time using a small brush in the left hand. Bring the section to the water bath by putting the section on the curved forceps and gently float the section on the water surface. Troubleshooting 2
-
a.Let the section extend for a few seconds.
-
b.Do not let it float in water for an extended period as paraffin may melt completely.
-
a.
-
17.
Pick the section on the slide and leave it vertically against the wall for a minute to drain residual water off the slide.
Pause point: Sectioning can be paused for up to 1 h if needed. Do not move or take out the paraffin block from a microtome, and cover the paraffin block with a piece of Saran wrap to prevent it from drying up. When you resume sectioning, the block should be wetted with a piece of wet Kimwipes.
-
18.
Collect all sections from the block (for sagittal section blocks, we usually cut at least 300 sections at 10 μm; for coronal section blocks we usually collect 320 sections at 16 μm).
-
19.
After drying completely > 12 h at room temperature (22°C–25°C), store the slides in plastic slide cases (e.g., BOX F/100 slides lab craft band, Fisher, Cat #; 03–446) until use.
Sample coding method for genotype blind experiments and data analysis
To avoid experimenter bias, we recommend performing experiments in a genotype blind manner. Following is a practical method to code and decode slides for histological experiments (Figure 2).
-
20.
Select the slides for histological analysis by Experimenter 1. Usually 6–9 sections from 1 block (every 30–40th sections, 300–400 μm apart from each other). Do not mark any genotype or set the group on the slides. Mark only section number.
-
21.
Provide information about the slides (e.g., mouse ID, genotype, age, type of staining) to Experimenter 2.
-
22.Code slides by Experimenter 2.
-
a.Code should omit all pertinent information of the sample.
-
b.Mark only code and type of experiment/immunostaining on the slide. Following is a coding example.
-
a.
| Original slide information | → | Coding by experimenter 2 | |
|---|---|---|---|
| Brain block #1 | Age: 2M | ||
| Mouse ID | Genotype | Code | |
| AC4313 | Control | AF1 | |
| AC4315 | Mutant | AF2 | |
| AC4322 | Mutant | AF3 | |
| AC4325 | Control | AF4 | |
| Brain block #2 | Age: 12M | ||
| Mouse ID | Genotype | Code | |
| AC4433 | Mutant | AG1 | |
| AC4434 | Control | AG2 | |
| AC4436 | Mutant | AG3 | |
| AC4437 | Control | AG4 | |
-
23.
Perform all histological analysis and quantification by Experimenter 1.
-
24.
After finishing the experiment and data analysis, Experimenter 1 provides raw data file to Experimenter 2 via email.
-
25.
Experimenter 2 then releases all coding information, such as block number, age, mouse ID, genotypes, etc. For the record, we strongly recommend keeping all unprocessed raw data file.
Figure 2.

A practical method of sample coding for genotype blind analysis
Alternatively, Experimenter 1 performs all histological experiments, and Experimenter 2 performs counting/analysis in a genotype blind manner. After finishing all experiments and data analysis, coding information can be released.
Alternatively, samples can be coded in early stages. For example, brain samples can be coded when mice are sacrificed, when brains are embedded in the paraffin or frozen block, or when brain sections are generated.
Nissl staining of brain sections
Cresyl violet is an organic compound that can stain the nucleoli and the rough endoplasmic reticulum of neurons. Nissl staining followed by stereological quantification can be used to measure the area or calculate volume of the region of interest in the brain, e.g., the cerebral cortex for AD neuropathology (Figure 3).
-
26.
Pick 6–7 slides of sagittal brain sections (every 40th, 400 μm apart) from each block, and bake the slides for 1 h at 55°C–60°C to remove extra paraffin from sections.
-
27.Deparaffinize and rehydrate the sections by sequential immersion in the following reagents. We usually use 200 mL of each solution in glass staining dishes (Electron Microscope Science, Cat #; 71420-25). Completely immerse the sections for 4 min each, and gently lift and submerge the sections every 1 min in each solution. All procedure should be performed under the fume hood.
-
a.Histoclear (or Xylene) twice
-
b.50% Histoclear/50% Ethanol
-
c.100% Ethanol twice
-
d.95% Ethanol in distilled water
-
e.70% Ethanol in distilled water
-
f.50% Ethanol in distilled water
-
g.Distilled water (2 min, only soak)
-
a.
-
28.
Immerse the rehydrated sections in 0.5% cresyl violet acetate (8 min, soak under the fume hood).
-
29.
Rinse in distilled water.
-
30.
Rinse sequentially in 50% EtOH, 75% EtOH, 90% EtOH, 100% EtOH twice, and 50% Histo-clear/50% EtOH. Troubleshooting 3
-
31.
Dehydrate the sections on Histoclear for 2 min, twice.
-
32.Mounting with Permount mounting solution (Fisher Scientific Permount Mounting Medium, Cat#; SP15-500).
-
a.Drop off Histoclear as much as possible and wipe out Histoclear solution using Kimtex wipers without touching brain specimens.
-
b.Apply 4–5 drops of Permount mounting solution on the slide.
-
c.Put cover glass (Fisher Scientific, Cat #; 12-548-5M) on the slide.
-
d.Push off bubbles and remove extra Mounting Medium.
-
a.
-
33.
Dry the mounted slide glass under the hood for > 16 h.
Note: If you are using a new batch of cresyl violet acetate solution, prepare 1–2 extra slides and optimize staining time for desired staining intensity. To avoid dye stains on glass slides, you could also use a 50-mL tube to rinse after cresyl violet staining.
Figure 3.

A Nissl-stained sagittal mouse brain section showing the area of the neocortex outlined by a blue line and the area of the hippocampus outlined by a red line
Scale bar: 1 mm.
Immunohistochemical analysis of for NeuN, GFAP, and Iba1
Immunohistological analysis followed by stereological quantification is critical for evaluating the degree of pathological neurodegenerative changes and associated gliosis in the mouse brain. For quantitative analysis, immunostaining with good quality antibodies is critical to obtain conclusive results. We use an anti-NeuN antibody to stain the nucleus of neurons in the brain. Anti-GFAP antibody, an astrocyte-specific marker, can be used to quantify astrogliosis in the brain. In the case of progressive astrogliosis, upregulated GFAP-immunoreactivity is shown in an intracellular intermediate filament of astrocytes, and its expression is upregulated in several disease conditions. Iba1, a microglia-specific marker, can be used to detect and evaluate progressive microgliosis in the brain. Since antibodies for NeuN, GFAP, and Iba1 are very specific and expression levels of NeuN, GFAP, and Iba1 are relatively high in the brain, no antigen retrieval step is necessary.
-
34.
Pick 6–7 slides of sagittal brain sections (every 40th, 400 μm apart) from each block, and bake the slides for 1 h at 55°C–60°C to remove extra paraffin from sections.
-
35.
Deparaffinize and rehydrate the sections as described above (Step 27).
-
36.
Permeabilize the tissue on TBSTx (TBS in 0.1% TritonX-100) for 15 min with gentle shaking (50 rpm).
-
37.
Rinse the slides with distilled water twice and then TBS once, for 2 min each.
-
38.
Quench endogenous peroxidase activity with 0.3% H2O2 in Methanol (use a fresh batch) at room temperature for 20 min with gentle shaking (50 rpm).
-
39.
Rinse the slides in distilled water (X2) and TBS for 5 min each.
-
40.Place the slides on Immuno Stain Moisture Chamber (Electron Microscope Science, Cat #; 62010-37).
-
a.Put pieces of paper towel and soak it with distilled water to humidify the chamber.
-
b.Isolate brain sections on the slide with PAP Pen (Electron Microscope Science, Cat #; 71310) to save antibody solutions in the following steps.
-
a.
-
41.
Apply ∼300 μL blocking solution (e.g., 5% normal goat serum in TBS) on sections at room temperature for 1 h.
-
42.Remove the blocking solution and apply primary antibody (200–250 μL to cover 4 sagittal section brains in one slide). Incubate the slides at 4°C for 16 h.
-
a.We use 1/400 dilution for NeuN antibody (Millipore, Cat #; MAB377), 1/500 dilution for GFAP (Sigma-Aldrich, Cat #; G6171), and 1/500 dilution for Iba1 (Wako, Cat #; 019–19741) in TBSTx solution.
-
a.
-
43.
Rinse the slides in TBS at room temperature for 5 min, 3 times.
-
44.
Apply 1/400 diluted biotinylated secondary antibody in 1% normal goat serum/TBS on the brain sections (250 μL per slide) and incubate at room temperature for 1.5 h.
-
45.
After 1 h (at least 30 min before finishing secondary antibody application), prepare ABC solution (Avidin/Biotin-HPR Kit, Vector Laboratories, Cat #; PK-6100): Reagent A two drops + Reagent B two drops in TBS 5 mL (300–400 μL per slide).
-
46.
Rinse the slide in TBS for 5 min, 3 times.
-
47.
Apply ABC solution (300–400 μL per slide) and incubate the slide at room temperature for 1 h.
-
48.
Rinse the slide in TBS for 5 min, 3 times.
-
49.Prepare DAB/Peroxidase solution (Vector Laboratories, Cat #; SK-4100) just before use. 400–500 μL of DAB solution is needed per slide.
-
a.Add 2 drops of Buffer Stock solution in 5 mL distilled water and mix well.
-
b.Add 4 drops of DAB and 2 drops of H2O2.
-
c.(Option) add 2 drops of Nickel Solution for black colorization for the positive signal.
-
a.
-
50.
Incubate slides in DAB solution, at room temperature for 3–10 min.
-
51.
Rinse the slide in distilled water, briefly twice.
-
52.
(Option) Counterstaining with hematoxylin (Fisher Scientific, Cat #; SH26-500D) for GFAP or Iba1 staining for 5 s, and rinse in distilled water, twice.
-
53.
Dehydrate the sections as described above (2 min each; the opposite direction of step #27).
-
54.
Mounting with Permount mounting solution as we described above (Step 32).
-
55.
Dry the mounted slide glass under the hood (usually for 16 h).
Immunostaining of active caspase-3
Examination of apoptosis in the brain is a critical step to determine whether there is ongoing programmed cell death. Active Caspase-3 immunoreactivity labels the cleaved form of Caspase-3, which is the main executioner of apoptosis (Riedl and Shi, 2004). The active Caspase-3 staining requires antigen retrieval, which is necessary to achieve good quality immunostaining for stereological quantification.
-
56.
Pick 7–9 slides (every 30th section, 300 μm apart) from each block, and bake the slides for 1 h at 55°C–60°C to remove extra paraffin from sections.
-
57.
Deparaffinize and rehydrate the sections as described above (Step 27).
-
58.
Permeabilize the tissue on PBSTx (PBS in 0.1% TritonX-100) with 0.1% Sodium citrate for 15 min with gentle shaking (50 rpm).
-
59.
Rinse the slide with distilled water twice and PBS, for 2 min each.
-
60.Antigen retrieval procedure
-
a.Pour 10 mM citric acid antigen retrieval buffer into a plastic jar.
-
b.Put the slides in the jar, and boil in the microwave for seconds.
-
c.Add fresh antigen retrieval buffer on the top (to prevent the section from drying up).
-
d.Boil in a microwave with 60% power (∼600W) for 3 min.
-
e.Boil in a microwave with 20% power (∼200W) for 2 min.
-
f.Boil in a microwave with 10% power (∼100W) for 10 min.
-
g.Cool down slide at room temperature for 20–30 min.
-
a.
-
61.
Follow the remaining steps for immunostaining as described above. We use anti-active Caspase-3 primary antibody (Cell Signaling, Cat #; 9661S) in 1/250 dilution.
TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay
TUNEL assay, a method to detect DNA fragmentation in the late stage apoptotic cells (Gavrieli et al., 1992), is performed in addition to active Caspase-3 immunostaining to confirm apoptotic cell death. Immunostaining can be used following the TUNEL assay to identify specific cell types of apoptotic cells. It is highly recommended to include positive controls (e.g., brain sections of model mouse showing robust neurodegeneration or DNaseI treated brain sections).
-
62.
Pick 7–9 slides (every 30th section, 300 μm apart) from each block, and bake the slides for 1 h at 55°C–60°C.
-
63.
Deparaffinize and rehydrate the sections as described above (Step 27).
-
64.
Permeabilize the tissue on PBSTx (PBS in 0.1% TritonX-100) with 0.1% sodium citrate for 15 min with gentle shaking (50 rpm).
-
65.
Rinse the slides with distilled water (X2) and PBS, for 2 min each.
Optional: If you are going to perform immunostaining after the TUNEL assay, you could perform an antigen retrieval step here, if needed.
-
66.Place the slides on Immuno Stain Moisture Chamber (Electron Microscope Science, Cat #; 62010-37) or a similar incubation chamber with aluminum foil to avoid light exposure during the following procedures.
-
a.Put pieces of paper towels and sock them with distilled water to humidify the chamber.
-
b.Isolate sections on the slide using a PAP Pen (Electron Microscope Science, Cat #; 71310) to save the TUNEL reaction solution in the following steps.
-
a.
-
67.
Apply ∼300 μL blocking solution (e.g., 5% normal goat serum in TBS) on sections at room temperature for 1 h.
-
68.Prepare TUNEL reaction mixture (In Situ Cell Death Detection Kit, Roche, Cat #; 11684795910):
-
a.Mix 5 μL of TdT enzyme solution with 45 μL of Label solution. 50 μL of TUNEL reaction mixture is needed for each slide. Troubleshooting 4
-
b.For negative reaction control, prepare 50 μL of Label solution only.
-
a.
-
69.
Apply 50 μL of TUNEL reaction mixture on the brain section.
-
70.
Incubate the section at 37°C for 1 h.
-
71.
Rinse the slide with PBS twice, for 2 min each.
Optional: If you are going to perform immunostaining after the TUNEL assay, start from here and follow the standard or specific immunostaining protocol for each primary antibody.
-
72.
Apply 300 μL of DAPI staining solution and incubate at room temperature for 10 min.
-
73.
Rinse the slide with PBS twice, for 2 min each.
-
74.
Mount with VECTASHIELD mounting media (Vector Laboratories, Cat #; H-1000-10) or any compatible antifade mounting medium and keep in the dark at 4°C until analysis.
Making cryo-preserved blocks for frozen brain sections
Compared to paraffin brain sections, immunoreactivity or immunosensitivity is higher in frozen brain sections, so it is more likely to succeed in immunofluorescent double-labeling, which allows visualization of two different target antigens using different fluorophore combinations. We recommend using this protocol to achieve high quality fluorescent double labeling. Images can be analyzed on confocal or fluorescent microscopes. However, compared with colorimetric IHC on paraffin sections, fluorescent signals usually bleach out quickly, so long-term storage is not feasible with this method. It is recommended to perform the experiment using a small number of sections in order to achieve high quality, consistent results.
-
75.
After perfusion, dissection, and post-fixation step as described above, replace fixative solution with 15% sucrose/PBS at 4°C, 3–6 h.
-
76.
Exchange solution to 30% sucrose/PBS at 4°C. It usually takes the brain more than one night to sink to the bottom (completely cryo-preserved).
-
77.
At least 30 min before embedding, prechill ethanol or isopentane on dry ice.
-
78.
Wash the brain tissues briefly with PBS and OCT compound.
-
79.Place brains in the disposable plastic base mold (Electron Microscope Science, Cat #; 62352-37).
-
a.Keep the mold on ice.
-
b.Usually, 4 brains in the same mold.
-
a.
-
80.
Fill up OCT compound in the mold.
-
81.
Quickly freeze it in Dry ice/EtOH or Dry ice/isopentane freezing medium, and store at −80°C until further use.
Cryosectioning of mouse brains
-
82.Set the blade in the cryostat (Lecia, Cat #; CM1860) at an appropriate angle and plane.
-
a.Temperature setting is around -18°C–20°C.
-
b.The cutting angle should be around 5 degrees.
-
c.Section thickness is usually 12–16 μm.
-
d.Blade safeguard should be placed, and the handle should be locked until sectioning for your safety.
-
e.Avoid opening the chamber shield for an extended period.
-
a.
-
83.
Prepare slide warmer and set the temperature at ∼40°C.
-
84.
Prepare dry ice and place a slide box on top of it.
-
85.
Place the cryo-preserved block int the cryostat 1 h prior to sectioning.
-
86.
Cut the section slowly and pick up the sections one at a time using a small brush in the left hand. Troubleshooting 5
-
87.
Pick the section on a slide and store it in a pre-chilled slide box.
-
88.
Collect all sections from the block.
-
89.
Store slide in a slide box at −80°C until use.
Immunofluorescent staining of frozen brain sections
-
90.
Pick 6–7 slides (every 40th, 400 μm apart) from each block, and leave on the bench (room temperature) for 5 min.
-
91.
Rinse in PBS wash at room temperature for 5–10 min to remove extra OCT compounds.
-
92.(Option) Antigen retrieval
-
a.Pour 10 mM Sodium citrate in plastic jar (∼300 mL).
-
b.Put the slides in the jar, and boil in microwave for 5 min. Occasionally add fresh citric acid on top to prevent the sections from drying up.
-
c.Cool to room temperature (usually takes 15–20 min).
-
d.Dip in PBS.
-
a.
-
93.
Place slides in Immuno Stain Moisture Chamber (Electron Microscope Science, Cat #; 62010-37).
-
94.
Apply ∼300 μL blocking solution (e.g., 5% normal goat serum/1% BSA/0.1% Triton-X 100 in PBS) and incubate at room temperature for 1 h.
-
95.
Remove the blocking solution and apply primary antibody with proper dilution depending on the antibody (200–250 μL to cover 4 sagittal section brains on one slide). Incubate the slide at 4°C for 16 h.
-
96.
Rinse the slide in PBS at room temperature for 5 min, 3 times.
-
97.
Apply 1/300 diluted Alexa Fluor-labeled secondary antibody in 3% normal goat serum/PBS on the brain sections (300 μL per slide) and incubate at room temperature for 1.5 h.
-
98.
Rinse the slide in PBS at room temperature for 5 min, 3 times.
-
99.
(optional) Apply 300 μL of DAPI staining solution (1/1000 dilution in PBS from 300 mM DAPI stock solution) and incubate at room temperature for 10 min.
-
100.
Rinse the slide in PBS at room temperature for 5 min, 3 times.
-
101.
(Optional) To remove autofluorescence, apply 500 μL Sudan Black solution for 10 min. This step is not required for embryonic brain immunostaining. After that, rinse the slides in PBS at room temperature for 5 min, 3 times.
-
102.
Mounting with VECTASHIELD mounting media (Vector Laboratories, Cat #; H-1000-10) or any compatible antifade mounting medium and keep in the dark at 4°C until analysis.
Quantitative analysis of cortical volume using stereological methods
Age-dependent volumetric changes in the cerebral cortex are a key neuropathological feature of AD. We use Nissl-stained serial sections to measure the area of the neocortex or the hippocampus of each brain section using the BioQuant Lifesciences software, and then calculate the entire neocortical or hippocampal volume (Figure 3). Alternative methods include ImageJ or other software that measures the brain subregion of interest. The measurement of other brain subregions can be done by specific immunostaining, e.g., DARPP-32 staining to label medium spiny neurons in the striatum, Calbindin staining to label Purkinje cells in the cerebellum, TH staining to label dopaminergic neurons in the substantia nigra.
-
103.
Start “BIOQUANT Lifesciences” application.
-
104.
Place the slide of Nissl stained brain sections under the 4× Objective lens with a live camera, and move to the first brain section.
-
105.
Open ‘Stereology Tools’ and make a new file on an optical fractionator project.
-
106.On the ‘Measure’ menu, select manual measurements, and set up the ‘Parameters’.
-
a.Check ‘Save TOPO’ option.
-
b.Select TOPO Array, D1.
-
c.Set Magnification as 4×.
-
a.
-
107.
Set the A1 (sampling Area) option by double clicking on ‘Select Arrays’ menu.
-
108.
On ‘Stereology Tools’ windows, click ‘1st’ icon to load Stereology defaults and ‘∗’ to set a landmark.
-
109.
Click the ‘Manually Trace Area’ icon and draw the area of neocortex (e.g., blue lines in Figure 3), hippocampus (e.g., red lines in Figure 3) or region of interest. If you need two screens to complete the drawings, first click “W” and move the stage, followed by clicking “R”. Then you can restart the drawings. Click “SPACE” key if you almost finished in order to engulf the drawing.
-
110.
Area will be displayed on the data panel window.
-
111.Volume of the neocortex is calculated as follows.
-
a.Average volume of the neocortex per section = [average area of the neocortex in the section] × [thickness of the section]. For example, if the section is 10 μm in thickness, we would multiple 10 with the average area of the neocortex per section, which is obtained by averaging the area of the neocortex quantified in all sections (typically 7 sections).
-
b.Volume of the neocortex = [average volume of the neocortex per section] × [number of sections containing the neocortex]. We typically collect ∼300 sections from wild-type mice, and we quantify the area of the neocortex in every 40th section (total 7 sections).
-
c.Then convert to mm3 as unit.
-
d.Multiply 2 to convert to the volume of the neocortex representing both hemispheres.
-
a.
Quantitative analysis of cortical neuron number using stereological methods
Neurons can be differentiated from other cell types by the presence of neuron-specific proteins such as MAP2 and NeuN, but immunostaining using an antibody against a nucleus protein (NeuN) makes it easier to count accurate cell number compared to an antibody against neurite proteins (MAP2). We use an anti-NeuN antibody to count the number of cortical neurons. This methodology can be broadened to other subtypes of neurons when you choose an appropriate antibody against a specific protein (e.g., GAD67 for inhibitory neurons, Tbr1 for deep layer cortical neurons, or TH for dopaminergic neurons). We use the Bioquant Lifesciences software for the stereological quantification.
-
112.
Start “BIOQUANT Lifesciences” application.
-
113.
Place the slide of NeuN-immunostained brain sections under the 4× Objective lens with a live camera, and move to the first brain section.
-
114.
Find and set a suitable landmark point (a reference place for the measurement).
-
115.
Delineate the region of interest (e.g., neocortical area for cortical neuron, and hippocampal CA1 region) and measure an area of the delineated region as indicated in the previous section (Figures 4A and 4C).
-
116.Select the size of the grid, and place the grid randomly in the entire specified region. (Figures 4A and 4C)
-
a.We use 500 × 500 μm for cortical neuron or 100 × 100 μm for small brain subregions, such as hippocampal CA1 region.
-
b.The grid size can be changed depending on the size of region of interest and distribution of target cell type.
-
c.Approximately 40 and 15 optical dissectors were used for the entire neocortex area and hippocampal CA1 region, respectively.
-
a.
-
117.
Change to the 40× objective lens for neuron counting.
-
118.
Select the size of the dissectors. We use 100 × 100 μm (20% of area) for cortical neurons or 50 × 50 μm (25% of area) for hippocampal CA1 region (Figures 4B and 4D).
-
119.
Count the NeuN-positive cells in the clear focus according to user’s own rules. For example, if the nucleus of the neuron is outside of the left and bottom Red line, do not count it. If the nucleus of the neuron is within the top and right Green Line, count it.
-
120.
Go to the next counting frames on the same section, and count the cells in the same way.
-
121.
Repeat these steps for the rest of dissectors. Write down the number for the current array and get the total number for this section.
-
122.The total number of neurons is calculated as follows.
-
a.Q × t/h × 1/asf × 1/ssfQ = the total number of cells actually counted in the counting dissectors of all the sections analyzedh = the height of the dissectort = the mean thickness of the sectionsasf = the area sampling fraction (the ratio of the counting frame area to the sampling grid area)ssf = the section sampling fraction (the ratio of the number of sections analyzed to the total number of sections in the entire structure).
-
b.The coefficient of error should be < 0.10.
-
a.
Figure 4.

Stereological counting of NeuN-immunoreactive neurons in the cerebral cortex
(A) Representative image of the neocortex (marked with a blue outline) overlaid with 500 × 500 μm sampling distance.
(B) Representative image of one dissector (100 × 100 μm) containing NeuN+ neurons that were counted (marked by yellow X).
(C) Representative image of hippocampal CA1 area (marked with a blue outline) overlaid with 100 × 100 μm sampling distance.
(D) Representative image of one dissector (50 × 50 μm) containing NeuN+ neurons that were counted (marked by yellow X).
Scale bar: 100 μm.
Quantification of apoptotic cells
In contrast to reductions of cortical neurons and cortical volume, which accumulate over time, identification of increases of apoptosis is a quicker way to reveal earlier signs of neuronal loss in the brain due to the low basal apoptosis in the adult brain. For example, the cortical neuron number and the cortical volume are unaltered in the cerebral cortex of excitatory neuron-specific Presenilin conditional double knockout (EX-PS cDKO) mice at 2 months of age (Saura et al., 2004). However, we detected 8-fold increases of apoptotic cells in the cerebral cortex of EX-PS cDKO mice at 2 months of age relative to controls, though the apoptotic cells represent only 0.1% of neurons in the cerebral cortex (Saura et al., 2004, Wines-Samuelson et al., 2010).
We typically find 0–2 and 3–10 active Caspase-3-immunoreactive cells (Figure 5A) or TUNEL-positive (Figure 5B) apoptotic cells per brain section in the cerebral cortex of controls and AD mouse models, respectively. Thus, it is feasible and more accurate to count all active Caspase-3-immunoreactive cells or TUNEL-positive cells in the cerebral cortex rather than sampling areas using a dissector.
-
123.
Place the slide on the stereomicroscope (e.g., Olympus BX-51) with a CCD camera for active Caspase-3-stained brain sections or in the fluorescence microscope (e.g., Olympus IX-51) with a CCD camera for TUNEL assay.
-
124.
Under the 10× objective lens of the microscope, scan the brain region from anterior to posterior and dorsal to ventral. And count all active Caspase-3 immunoreactive or TUNEL+ cells per section.
-
125.The total number of apoptotic cells is calculated as follows.
-
a.[Average of number of apoptotic cells per section] × [number of entire brain section of region of interest]
-
a.
Figure 5.

Apoptotic cells labeled by active Caspase-3 immunoreactivity and the TUNEL assay
(A) Representative image of active Caspase-3-immunoreactive cells in the neocortex.
(B) Representative image of TUNEL+ cells in the neocortex.
Scale bar: 100 μm.
Quantification of astrogliosis
Astrocytes are a major glial cell type in the mammalian brain. The number of astrocytes is estimated at approximately 25 billion in the adult human brain (Pelvig et al., 2008). Astrocytes are activated under disease conditions because of neuroinflammatory responses, and we previously found astrogliosis accompanying neurodegeneration in the brains of Alzheimer's and Parkinson's mouse models (Saura et al., 2004, Beglopoulos et al., 2004, Wines-Samuelson et al., 2010, Watanabe et al., 2012, Watanabe et al., 2014, Xia et al., 2015, Giaime et al., 2017, Lee et al., 2020, Kang and Shen, 2020). GFAP is an intracellular intermediate filament, and its expression is upregulated during neurodegeneration and astrogliosis. Thus, astrogliosis can be assessed by measuring GFAP-immunoreactive areas in the cerebral cortex. (Figure 6)
-
126.Place the slide of GFAP-immunostained brain sections on the microscope with a CCD camera.
-
a.We usually use 4× objective lens of the microscope for cerebral cortex.
-
a.
-
127.Using any quantitative image analysis software (e.g., in our laboratory, BioQuant Lifesciences software), measure the area of the neocortex or the hippocampus and GFAP+ area within the specified brain subregion.
-
a.We usually measure 6–7 sections, 400 μm apart, per brain.
-
b.The area of the region of interest can be measured as we described above in the ‘Quantitative analysis of cortical volume using stereological methods’ section.
-
c.Set the threshold value to detect all GFAP-immunoreactive areas (Figures 6A’–6D′).
-
d.Apply the threshold value to measure GFAP+ area within the area of the brain subregion of interest. GFAP+ area will be automatically quantified and displayed on the data panel window.
-
a.
-
128.Percentage of GFAP+ area is calculated as follows.
-
a.Percentage of GFAP+ in the brain subregion per section = [GFAP+ area in the brain subregion of each section] / [total area of the brain subregion of each section]
-
b.Then, calculate average percentage of GFAP+ area in all sections (typically 7 sections for neocortex and 6 sections for hippocampus) for each brain.
-
a.
Figure 6.

Quantification of GFAP-immunoreactive areas in the cerebral cortex
(A and A′) Representative images of GFAP immunoreactivity in the neocortex. The neocortex is outlined by a red line (A). The red marked areas in (A′) indicate the GFAP-immunoreactive area in the neocortex.
(B and B′) Higher power views of GFAP immunoreactivity in the neocortex. Highlighted magenta areas in (B′) indicate the GFAP-immunoreactive area.
(C and C′) Representative images of GFAP immunoreactivity in the hippocampus. The hippocampus is outlined by a red line (C). The red marked areas in (C′) indicate the GFAP-immunoreactive area in the hippocampus.
(D and D′) Higher power views of GFAP immunoreactivity in the hippocampus. Highlighted magenta areas in (D′) indicate the GFAP-immunoreactive area.
Scale bar: 100 μm.
Quantification of microgliosis
In addition to astrogliosis, microgliosis also accompanies neurodegeneration. Microgliosis is an early non-specific proliferation and migration of macrophage-like microglia cells in the central nervous system as the first response to brain damage or pathogenesis of the neurodegenerative disease. Activated microglia enhance expression of microglia-specific genes, such as Iba1, which can be detected easily by immunostaining with an anti-Iba1 antibody. Counting the number of Iba1-immunoreactive microglia in the entire cerebral cortex can assess the degree of microgliosis (Figure 7).
-
129.Place the slide of Iba1-stained sections on the microscope with a CCD camera.
-
a.We usually use a 4× objective lens of the microscope for area measurement, and a 40× and 10× objective lens for the Iba1+ cell counting in the neocortex and hippocampus, respectively.
-
a.
-
130.Using the BioQuant Lifesciences software, measure the area of the neocortex or hippocampus and count the number of Iba1+ cells within each brain subregion.
-
a.We usually count 6–7 sections, 400 μm apart, per brain.
-
b.Measure area of the region of interest as we described above in the ‘Quantitative analysis of cortical volume using stereological methods’ section.
-
c.The number of Iba1+ cells can be counted as we described above in the ‘Quantitative analysis of cortical neuron number using stereological methods’ section. The grid size is usually set at 500 × 500 μm for neocortical area and the dissector size is usually set at 100 × 100 μm. For the hippocampal area, the grid size is set at 500 × 500 μm and the dissector size is set at 250 × 250 μm.
-
a.
-
131.Iba1+ signals in the specific brain subregion is calculated as follows.
-
a.Iba1+ cells per mm2 per section = [number of Iba1+ cells in the brain subregion of each section] / [total area of the brain subregion of each section]
-
b.Then, calculate average number of Iba1+ cells in all sections (typically 7 sections for neocortex and 6 sections for hippocampus) for each brain.
-
a.
-
132.
Alternatively, total Iba1+ cell number in the cerebral cortex from one hemisphere can be calculated as described above in the section of ‘Quantitative analysis of cortical neuron number using stereological methods’. As an example, we typically find ∼3 × 105 Iba1+ cells in the neocortex of one hemisphere of control mice.
Figure 7.

Quantification of Iba1-immunopositive cells in the cerebral cortex
(A) Representative image of Iba1 immunostaining in the neocortex.
(A′) Higher power view of Iba1+ cells in the neocortex showing one dissector (100 × 100 μm) under the 40× objective lens.
(B) Representative image of Iba1 immunostaining in the hippocampus.
(B′) Higher power view of Iba1+ cells in the hippocampus showing one dissector (250 × 250 μm) under the 10× objective lens.
Scale bar: 100 μm.
Expected outcomes
During the progression of neurodegeneration in AD mouse models, various neuropathological changes take place simultaneously and continuously in the brain. Depending on the mouse model, the age of onset differs, and the progression of neuropathological changes varies. In the case of EX-PS cDKO mice, in which the Presenilin genes are inactivated in excitatory neurons of the forebrain from postnatal day 18, apoptotic cells marked by activated Caspase-3-immunoreactivity or the TUNEL assay are greatly increased (8-fold) in the cerebral cortex of EX-PS cDKO mice compared to littermate controls at 2 months of age (Saura et al., 2004, Wines-Samuelson et al., 2010). However, at this young age, there is no significant reduction of neuron number or volume of the cerebral cortex in EX-PS cDKO mice (Saura et al., 2004). At 4 months of age, EX-PS cDKO mice show a significant decrease in neuron number (∼10%) in the cerebral cortex compared to controls. By 6–9 months of age, EX-PS cDKO mice show marked loss of cortical volume (33%) and neurons (18%–24%) in the cerebral cortex, accompanied with loss of dendritic complexity and spines as well as enlargement of lateral ventricles of EX-PS cDKO mice (Saura et al., 2004, Wines-Samuelson et al., 2010). The surviving EX-PS cDKO mice at 16 months of age display dramatic reduction (∼50%) in cortical volume and neuron number (Watanabe et al., 2014).
Gliosis, astrogliosis or microgliosis, accompanies normal aging as well as age-dependent neurodegeneration. For example, in the brain of EX-PS cDKO mice at 4 months of age, GFAP+ areas in the neocortex and hippocampus are increased by ∼17 fold and 3.5-fold, respectively, compared to littermate controls (Watanabe et al., 2014). Astrogliosis also occurs during normal aging; GFAP+ areas in the neocortex of wild-type mice are increased by 3-fold and 6-fold at 12 months and 22 months, respectively, compared to 3 months old mice (Lee et al., 2020). In addition, age-dependent microgliosis accompanies neuronal loss in the neocortex and hippocampus of EX-PS cDKO and inhibitory neuron-specific PS cDKO (IN-PS cDKO) mice, as shown by significant increases of Iba1+ microglia in the cerebral cortex of aged EX-PS cDKO and IN-PS cDKO mice (Watanabe et al., 2014, Kang and Shen, 2020).
In summary, employing a comprehensive and complementary approach, summarized in this protocol, we were able to identify and quantify neuropathological changes during aging in EX-PS cDKO mice.
Limitations
Evaluation of neuropathological changes in the brain is an essential characterization of a newly generate AD mouse model, which is invaluable tool to investigate disease mechanisms or test novel therapeutic strategies. Stereological quantification of neurodegenerative phenotypes in the mouse brain provide a quantitative assessment of neuropathological changes in the 3D structure of interest (e.g., the neocortex or the hippocampus), which is far superior to the assessment of neuropathological changes in the 2D brain structure using a few sections. However, stereological quantification is highly labor-intensive. Furthermore, it requires a high-end motorized automated station on a microscope with a high-resolution camera and specialized software.
Astrocytes exhibit diverse and heterogenous morphologies during gliosis in different brain subregions (Matyash and Kettenmann, 2010, Anderson et al., 2014). In this protocol, our assessment of astrogliosis is limited to quantification of the area of GFAP-immunoreactivity in the cerebral cortex without detailed examination of morphologically distinct subtypes of reactive astrocytes. To analyze morphologically distinct reactive astrocytes in greater detail, please refer to the following publications (Bagheri et al., 2015, Taylor et al., 2020). Alternatively, other astrocyte markers, such as Serpina3n and Ctsb, may be used to evaluate astrogliosis (Habib et al., 2020).
This protocol can be used for other types of immunohistochemical analysis and stereological quantification. However, accurate assessment of cells and immunoreactive areas is limited when specific target proteins are expressed at low levels or when there is a lack of specific antibodies. Also, every effort should be made to reduce variability among independent experiments. Ideally, before you perform any histological experiment or stereological counting, you should first perform a practice experiment using a well-characterized mouse model with progressive neurodegeneration. It is highly recommended to include such positive controls in your experiments. If phenotypes are subtle, then large numbers of mice will be needed to determine if there are statistically significant changes in the mutant brain.
Troubleshooting
Problem 1
Cracks in paraffin blocks (step #11)
Potential solution
To avoid cracks in the paraffin block, do not leave the aluminum base mold on the cold plate for a long time. Once you can see the solidified paraffin on top of the cassette, move the aluminum base mold on ice or 4°C refrigerator. In addition, do not apply too much pressure when you remove the block from the aluminum base mold. If there is any crack in the paraffin block, it is better to melt the paraffin block and re-do the embedding step (see the steps #3–12 in the ‘Paraffinization of the brain tissue’ section).
Problem 2
Breakages of paraffin sections (step #16)
Potential solution
When it is difficult to section without breakage, the block should be wetted with a piece of wet Kimwipes. If the paraffin block is very old, you can humidify the block on ice for a few hours. If this does not work, you can melt the paraffin block and re-do the embedding step (see the steps #3–12 in the ‘Paraffinization of the brain tissue’ section).
Problem 3
Over- or under-staining of Nissl (step #30)
Potential solution
You need to check the intensity of Nissl staining after submerging in 95% EtOH. If brain sections are over-stained, submerge the section on a differentiation solution (1% glacial acetic acid in 95% EtOH) for 1–2 min. If brain sections are under-stained, submerge the section on cresyl violet solution for 1–2 min more. After that, proceed to the dehydration step again.
Problem 4
Non-specific TUNEL+ background signal (step #68)
Potential solution
If there are high levels of non-specific TUNEL+ signals, dilute TUNEL enzyme (TdT) in TUNEL Dilution Buffer (Sigma, Cat #; 11966006001) in 1:2 to 1:10 dilution.
Problem 5
Difficulties on cutting high quality frozen brain sections (step #86)
Potential solution
When it is difficult to section in a smooth thin sheet without breakage, it is usually because of the dullness of the blade. Replace the blade and wait 30 min until the blade temperature reaches the setting temperature of cryostat, then continue cutting sections.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jie Shen (jshen@bwh.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
Acknowledgments
We thank all members of the Shen Laboratory for their helpful discussions. We also thank the Rodent Histopathology Core at Harvard Medical School for their technical support. This study was supported by grants from the National Institutes of Health in the United States (R01NS041783, RF1AG063520, R01NS101745, and R37NS071251 to J.S.).
Author contributions
J.K. and H.W. optimized the protocols based on the methods used in the Shen Lab over the last two decades and wrote the manuscript. J.S. conceptualized and edited the manuscript.
Declaration of interests
J.S. has financial interests in Apres Therapeutics and iNeuro Therapeutics, both companies developing therapies for Alzheimer disease. J.S.’s interests were reviewed and are managed by Mass General Brigham in accordance with their conflict of interest policies.
Contributor Information
Jongkyun Kang, Email: jkang@bwh.harvard.edu.
Jie Shen, Email: jshen@bwh.harvard.edu.
References
- Anderson M.A., Ao Y., Sofroniew M.V. Heterogeneity of reactive astrocytes. Neurosci Lett. 2014;565:23–29. doi: 10.1016/j.neulet.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri M., Rezakhani A., Roghani M., Joghataei M.T., Mohseni S. Protocol for three-dimensional confocal morphometric analysis of astrocytes. J Vis Exp. 2015:e53113. doi: 10.3791/53113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglopoulos V., Sun X., Saura C.A., Lemere C.A., Kim R.D., Shen J. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem. 2004;279:46907–46914. doi: 10.1074/jbc.M409544200. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y., Sherman Y., Ben-Sasson S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaime E., Tong Y., Wagner L.K., Yuan Y., Huang G., Shen J. Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron. 2017;96:796–807.e6. doi: 10.1016/j.neuron.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N., Mccabe C., Medina S., Varshavsky M., Kitsberg D., Dvir-Szternfeld R., Green G., Dionne D., Nguyen L., Marshall J.L. Disease-associated astrocytes in Alzheimer's disease and aging. Nat Neurosci. 2020;23:701–706. doi: 10.1038/s41593-020-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J., Shen J. Cell-autonomous role of Presenilin in age-dependent survival of cortical interneurons. Mol Neurodegener. 2020;15:72. doi: 10.1186/s13024-020-00419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Kang J., Ho A., Watanabe H., Bolshakov V.Y., Shen J. APP family regulates neuronal excitability and synaptic plasticity but not neuronal survival. Neuron. 2020;108:676–690.e8. doi: 10.1016/j.neuron.2020.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V., Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Res Rev. 2010;63:2–10. doi: 10.1016/j.brainresrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Pelvig D.P., Pakkenberg H., Stark A.K., Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. 2008;29:1754–1762. doi: 10.1016/j.neurobiolaging.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Riedl S.J., Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Saura C.A., Choi S.Y., Beglopoulos V., Malkani S., Zhang D., Shankaranarayana Rao B.S., Chattarji S., Kelleher R.J., 3rd, Kandel E.R., Duff K. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Taylor X., Cisternas P., You Y., You Y., Xiang S., Marambio Y., Zhang J., Vidal R., Lasagna-Reeves C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J Neuroinflammation. 2020;17:223. doi: 10.1186/s12974-020-01900-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Xia D., Kanekiyo T., Kelleher R.J., 3rd, Shen J. Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. J Neurosci. 2012;32:5085–5096. doi: 10.1523/JNEUROSCI.0317-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Iqbal M., Zheng J., Wines-Samuelson M., Shen J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J Neurosci. 2014;34:15912–15922. doi: 10.1523/JNEUROSCI.3261-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wines-Samuelson M., Schulte E.C., Smith M.J., Aoki C., Liu X., Kelleher R.J., 3rd, Shen J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS One. 2010;5:e10195. doi: 10.1371/journal.pone.0010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D., Watanabe H., Wu B., Lee S.H., Li Y., Tsvetkov E., Bolshakov V.Y., Shen J., Kelleher R.J., 3rd Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. Neuron. 2015;85:967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.