

Abstract
Genetic degeneration is an extraordinary feature of sex chromosomes, with the loss of functions of Y-linked genes in species with XY systems, and W-linked genes in ZW systems, eventually affecting almost all genes. Although degeneration is familiar to most biologists, important aspects are not yet well understood, including how quickly a Y or W chromosome can become completely degenerated. I review the current understanding of the time-course of degeneration. Degeneration starts after crossing over between the sex chromosome pair stops, and theoretical models predict an initially fast degeneration rate and a later much slower one. It has become possible to estimate the two quantities that the models suggest are the most important in determining degeneration rates—the size of the sex-linked region, and the time when recombination became suppressed (which can be estimated using Y–X or W–Z sequence divergence). However, quantifying degeneration is still difficult. I review evidence on gene losses (based on coverage analysis) or loss of function (by classifying coding sequences into functional alleles and pseudogenes). I also review evidence about whether small genome regions degenerate, or only large ones, whether selective constraints on the genes in a sex-linked region also strongly affect degeneration rates, and about how long it takes before all (or almost all) genes are lost.
This article is part of the theme issue ‘Challenging the paradigm in sex chromosome evolution: empirical and theoretical insights with a focus on vertebrates (Part I)’.
Keywords: evolutionary strata, chromosome fusion, dosage compensation
1. Introduction
Theoretical modelling of Y chromosome degeneration suggests that rapid loss of functional genes starts immediately after recombination becomes suppressed across a region carrying many genes, initiating a Muller's ratchet process [1]. However, the rate of loss should decrease as the number of functional genes that can undergo deleterious mutations declines. After about half the genes in the ancestral chromosome region are lost, the models require a further very large number of generations to lose the remaining functional genes (as in the ancient human and Drosophila Y chromosomes). The slowing is predicted because, once few functional genes remain to cause the ratchet process, degeneration mainly involves rare advantageous mutations spreading in the population, dragging low-frequency deleterious mutations in the population to higher frequencies. This occurs very infrequently as genes that might undergo beneficial mutations are lost, or lose functions; the model therefore predicts that hitchhiking produces the highest rate of degeneration when an intermediate number of functional genes still remain present, and that complete loss of genes will take many generations [1], and may never occur in small sex-linked regions. Although this model represents an advance over earlier views of degeneration, it too is an over-simplification, like all models, and further model development will be valuable (see below).
It is important to test these theoretical predictions. If the prediction is upheld that highly degenerated sex chromosomes have long histories of evolving as sex chromosomes, without recombination, then, when an extremely degenerated Y (or W) chromosome is found in a species, this would suggest a long evolutionary history as an XY or ZW pair, which is sometimes impossible to determine in any other way. Quantification of ages using sequence divergence between members of sex chromosome pairs is described in box 1, but suitable data are often difficult to obtain. Phylogenetic analyses can suggest ages of sex chromosome systems, but can be inconclusive. Taxa in which an XY or ZW system is widespread may share it because they inherited it from a common ancestor, as has been demonstrated in mammals [9,10] and birds [11], or they may have evolved independently. If a homologous chromosome carries the sex-determining locus in several species, this may suggest that their common ancestor had a sex-determining region on this chromosome. However, the same chromosome may evolve sex-determining regions independently; for example, the homologue of the medaka autosome 10 carries the sex-determining gene in two groups of Oryzias species, and the homologue of autosome 12 is involved in another Oryzias species [12], and also in two Poecilia species [13,14]; similar cases are known in cichlid fish [6].
Box 1. Estimating the ages of strata.
Estimating relative divergence times of strata requires sequence divergence estimates, ideally KS values for synonymous sites in coding sequences (to ensure that the sequences used can be aligned reliably and that the variants are unlikely to experience strong selective constraints). The time when recombination became suppressed can be estimated from such divergence between pairs of XY genes (or W–Z pairs), or from phylogenetic analyses that rely on similar sequence data [2,3]. However, this is difficult unless the divergence is large enough that one can reliably separate (or ‘phase’) Y versus X (or W versus Z) sequences.
The times when regions evolved complete sex linkage should ideally be expressed in terms of numbers of generations, the scale used in the theoretical modelling of degeneration. Estimates in terms of years are less suitable, because they cannot be translated into numbers of generations, as generation times are often not accurately known. Numbers of substitutions per neutral site, estimated as KS values, increase roughly linearly with evolutionary divergence times [4]. These estimates therefore avoid the need for an estimated molecular clock rate to infer the relative ages of strata within a species, or in different organisms.
FST values are often reported, rather than divergence values. However, FST expresses the proportion of variability between the two populations sampled [5]: in this case, the populations of X- and Y-linked sequences (or Z and W ones), or often simply the two sexes. FST is not expected to increase in a clock-like manner with the time since the members of the sex chromosome pair became isolated (i.e. stopped recombining). Such values are excellent for detecting the presence of strata with different levels of differentiation between a sex chromosome pair [2,6,7], but not for estimating the time since recombination stopped.
A young stratum is sometimes inferred without estimating divergence, based on higher nucleotide diversity (or SNP density) in a sex chromosome region in the heterogametic sex; this suggests that the region still carries diverged Y (or W) sequences, implying that sequences have not been completely lost from the Y- or W-linked regions. However, in such cases, sequence divergence can and should be estimated. Finally, it is worth noting that sex differences in either divergence or FST levels do not necessarily imply that non-recombining strata have evolved, but can indicate that the members of a sex chromosome pair originated in different populations that have hybridized [8].
Currently, the time course of degeneration is understood largely based on theoretical models. The time until degeneration becomes very slow (see above) depends remarkably little on the fitness effects assumed for the mutations modelled, or on the effective population size, and even on the number of genes initially present. More important is the gene number, and small regions are not expected to degenerate rapidly [1]. However, empirical data are needed, both to test whether the expected general shape of the time-course is actually seen, and to relate it quantitatively to the values of measurable parameters. Actual estimates of the times when different sex chromosomes stopped recombining, and how degenerated they have become over those times, are still very scarce. In reviewing current data here, I also highlight major gaps that exist in knowledge about degeneration and suggest promising approaches for obtaining relevant information.
Estimates of the time when Y- or W-linked regions evolved, and the extent to which genes within the regions have lost their functions, will also help understand the evolution of dosage compensation that may evolve in response to hemizygosity, which often reduces expression in the heterogametic sex [15]; for some genes, a departure from the relative gene numbers needed for the proper function of multi-protein complexes may also play a role [16]. There is good evidence that dosage compensation can evolve rapidly once many genes of a Drosophila chromosome become hemizygous [17], but definitive tests for dosage compensation in many organisms are hampered by the evolution of sex-biased gene expression and enrichment of the sex chromosome for certain types of genes, and because dosage compensation appears sometimes to be partial [18,19]. Changes in gene expression after the initial evolution of dosage compensation, including the evolution of sex-biased gene expression, cause further uncertainties, even in very well-studied organisms, such as Caenorhabditid nematodes [20], Drosophila [21] and the chicken [19,22,23]. X inactivation in female mammals may also obscure an earlier dosage compensation system [24]. Conclusions about less well-studied organisms, such as snakes, are also uncertain [18,25]. I will not review dosage compensation here, as it has recently been comprehensively reviewed [24].
2. Evolutionary strata
An important advance in understanding sex chromosome evolution was the discovery that mammalian XY chromosome pairs do not have a single ‘age’, but stopped recombining in several events, at different times in their evolutionary past.
The first careful and detailed examination of the sequences of the sex chromosomes from the human genome sequencing work revealed several Y-linked sequences that are alleles of X-linked ones, allowing Y–X sequence divergence to be estimated [26]. This showed that genes in the X-linked region of Eutherian mammals corresponding to the Marsupial XY pair (part of the Xp arm) have much higher divergence than genes in regions that were added to the sex chromosomes in a fusion event after the split from the Marsupials [27]. Moreover, in the ‘younger’, so-called ‘X-added’ region of the Eutherian XY pair, sequence divergence between Y- and X-linked copies is smaller when the X-linked gene is closer to the pseudo-autosomal region (the PAR, which still undergoes XY recombination) than for genes further from the PAR [26,28]. The different divergence values indicate different times when Y-linked regions stopped recombining with their X-linked homologues and are classified into at least four or five so-called ‘evolutionary strata’ [26,28]; the gene numbers in the corresponding X-linked regions are also shown in figure 2 below. Box 1 outlines how divergence is estimated, and box 2 describes data that can be used to measure genetic degeneration.
Figure 2.

Genetic degeneration of the human Y, based on the comprehensive list of genes inferred to have been present on the ancestral mammalian X chromosome, including stratum 1, which is sex-linked in marsupials [29]. The grey bars and right-hand y axis show the estimated proportions of non-functional genes in different strata. The left-hand y axis shows Y–X synonymous site divergence estimates (Ks values) for each stratum. The figure also illustrates the loss of genes, as the numbers of XY pairs remaining for estimating Ks values are much smaller for the oldest stratum. The numbers of ancestrally X-linked genes in each stratum are also indicated (in the boxes; strata 4 and 5 are combined, because they include few genes). The horizontal white and black lines are the median and mean Ks values, respectively.
Box 2. Quantifying genetic degeneration of Y and W chromosomes (or Y- and W-linked genome regions).
To describe the time-course of genetic degeneration and to ask how long it may take until a Y or W chromosome becomes completely degenerated, one must be able to quantify degeneration. Much work to date provides evidence that degeneration has occurred, for example documenting reduced effectiveness of purifying selection after a Y- or W-linked region stops recombining [30–33], and distinguishing these effects from effects of selection favouring X-linked female-benefit mutations, rather than to less effectiveness of purifying selection due to sex chromosomes’ lower effective population sizes [15,34,35]. Many interesting studies have also documented repetitive sequence accumulation [36], expanding Y- and W-linked regions; low gene densities in such regions therefore need not reflect actual loss of genes. Transposable element (TE) insertions in non-coding regions may, however, affect gene expression, either by directly interrupting functional sequences, or through the side effects of epigenetic control of TE activity by the host cells [37]. Analyses of coding sequences alone therefore probably underestimate the extent of degeneration, although TE insertions can contribute to changes in expression, sometimes increasing it [38]: they might therefore reduce Y-linked gene expression or sometimes cause deleterious over-expression.
However, such data do not quantify degeneration, so most studies ask what proportion of genes (out of those carried by the X or Z chromosome) have clearly lost function, including sequences lacking the start codon, or with a premature stop codon of frame shift mutations. Getting even this information requires well-assembled genomes and also phased sequences, so that the alleles can be identified as X versus Y (or W versus Z), and the ability to detect genes recently transposed to the non-recombining region, such as the two added to the human Y [28]. Care is also needed to avoid bias towards low degeneration values, which could arise if only genes that can be reliably detected on both members of the sex chromosome pair are analysed. Degeneration may also be underestimated if changes in non-coding regions affecting function are not included. Such changes might explain some degeneration rate differences. For example, in the homologous X-degenerate regions of humans and chimpanzees, four of 16 genes have inactivating mutations in the chimpanzee, but appear to have remained functional in humans [39].
Pseudogenization gives only a partial picture, and it is also valuable to estimate the proportions of genes that have been lost. These are expected to increase with the age of the fully non-recombining region, as the loss of a gene is probably often unopposed by selection after it has lost functionality, and TE accumulation can contribute to loss of genes through small deletions of Y- or W-linked regions.
Genes with X-linked copies but no Y-linked copy can be identified in RNA-Seq data, without a complete genome assembly, using sequence variants in a family [40–42]. However, this loses potentially valuable information about how the losses occurred. For example, if multiple genes were lost in a single deletion event, the rate of loss in the lineage would accelerate. Moreover, the sample sizes of sex-linked genes are limited by the difficulty of ascertaining them. In an XY system, for example, hemizygous genes can be identified only when an X-linked polymorphism is detected, whereas both X polymorphisms and accumulated differences between the X and Y chromosomes can identify genes present on both the X and the Y, making XY genes often much easier to detect. Estimated percentages of hemizygous genes must therefore use only sex-linked genes that were identified from the more limited set of X polymorphisms. Genome sequencing and coverage analyses are likely to be necessary to get more accurate estimates, although assembly errors of non-single-copy sequences may cause problems.
Strata have been discovered in sex chromosome systems of several organisms, including ZW systems of birds [11,43,44] and snakes [45], and the XY systems of the stickleback [29] and two plants. In Silene latifolia, strata are inferred from the wide range of divergence values of genes between the highly heteromorphic Y and X chromosomes [40,46–48]. In the papaya, Carica papaya, two strata were probably created by inversions covering a small region carrying the male-determining factor [49].
The threespine stickleback is one of the few non-mammalian animals in which Y–X divergence has been estimated (see box 1). Two evolutionary strata were revealed, with an estimated 88% of 486 genes in the older stratum estimated to have become hemizygous [50]. Although this extensive degeneration might suggest an ancient system, the median synonymous site divergence (Ks values) for 75 genes that have not been lost from the Y is only about 1.8% [29], not much more than the estimate (1.35%, based on 582 genes) in the inferred younger stratum, where only 5% of 379 genes are estimated to have become hemizygous [50]. Expression levels of the Y-linked copies are also similar in the two strata. Apart from this case, it is unclear what proportion of fish and reptiles have sex-linked regions that have undergone genetic degeneration, and the timescale of degeneration has so far been studied in few species. Extensive Y-linked regions that are likely to contain many genes and would be predicted to degenerate, given enough time are, however, documented in a number of fish, including some cichlids and reptiles [6].
3. Detecting strata using coverage analysis and degeneration estimates
As already mentioned, studies of species and taxa with the ideal type of data (incompletely degenerated sex chromosomes, or strata whose degeneration levels and times of recombination suppression have been estimated) are scarce, apart from the mammal data in figure 2, which illustrate the large loss of genes from the older strata.
Data are available from the early-branching Paleognathous bird taxa. These ZW sex chromosomes are homologous with those of the later-evolved Neognathous birds, but have physically much larger recombining regions that are detectable cytologically, with differing sizes in different species [51], implying that new strata have evolved in some taxa. Genome sequences [11] are confirming these species' variably sized pseudo-autosomal recombining regions, or ‘PARs’, based on similar coverage in both sexes (figure 1). All birds appear to share an ancient, highly degenerated W region (electronic supplementary material, table S1) with female coverage values half those in males, and new strata have subsequently evolved that have intermediate female/male coverage ratios, indicating lesser degeneration [11]. This analysis does not require W-linked genes to estimate divergence from their Z-linked alleles, and the ancient divergence is inferred from the sharing between bird lineages. For example, in the group of Paleognath birds that includes the kiwi (Apteryx), coverage analysis suggests that its fully sex-linked region includes an Apteryx-specific stratum [52]. Although such regions should carry W–Z pairs whose sequence divergence can be estimated, this appears not yet to have been attempted for this bird. Genome sequences are, however, starting to be used to estimate ages of the strata, though it remains difficult to separate sequences into their Z and W (or X and Y) haplotypes (see box 1), and datings currently remain scarce.
Figure 1.

Schematic diagram of sex chromosome evolution: (a) shows a young XY pair with a fully sex-linked region (solid blue) that has not yet degenerated and a partially sex-linked region (or pseudo-autosomal region or PAR, white); (b) shows a later stage, in a pair that has evolved a second, younger fully non-recombining stratum that has not yet degenerated, while the older stratum has become partially degenerated, with the loss of genes symbolized by grey regions; the evolution of this new stratum involved part of the PAR stopping recombination, so that this XY pair has a smaller PAR than the younger one. The diagram shows the Y chromosome as the same size as the X, but the deletion of degenerated regions might occur, and/or accumulation of repetitive sequences in the fully sex-linked strata causing a physical expansion. (Online version in colour.)
Figure 3 summarizes degeneration data from species where age estimates are available (electronic supplementary material, table S1 gives full details). Within taxa where data suitable for comparisons exist, the data are consistent with the prediction that, other things being equal, shorter times since recombination stopped should tend to correlate with less complete degeneration. Before describing examples in the next two sections, it is important to emphasize that most species in which strata have been inferred lack full information. The number of genes in an X (or Z) region corresponding to a stratum is rarely known, and often the number remaining in the corresponding Y (or W) region has not been determined.
Figure 3.

Currently available genetic degeneration level estimates for XY and ZW systems with quantitative data. Except for the two dots for old and young strata in humans, the threespine stickleback, and Silene latifolia, each dot represents a named species. Most data, apart from those from humans, Drosophila miranda, and the plant Carica papaya (see text), are estimates of the proportions of genes that are hemizygous (see electronic supplementary material, table S1 for details, including the full species names and the sources of the data).
Strata have also been detected by coverage analysis in schistosomes [53]. Older and younger strata were inferred, because a phylogeny suggests that one stratum evolved after a species split, and is not found in all of the species studied. However, the putatively younger stratum appears highly degenerated (see electronic supplementary material, table S1), making it difficult to determine the time-course of degeneration in these species.
It is important to discover degenerated regions of sex chromosomes in species other than just the ‘classical’ mammalian and Drosophila systems and to obtain evidence about the repeated evolution of completely sex-linked regions and which species have evolved extensive regions, versus which sex-determining systems have remained physically small. However, evolutionary strata offer opportunities to relate the extent of genetic degeneration to the most important factors likely to affect degeneration rates—the sizes of non-recombining strata (or the numbers of genes they contain) and the times since recombination was suppressed between the pair of sex-linked regions. Such data will allow tests of whether younger, and smaller, strata are less degenerated, as predicted, and whether other factors also have major effects.
4. Partially degenerated sex-linked regions
To understand the likely time-course of genetic degeneration, the ideal systems are sex-linked regions with less than complete degeneration. However, if Y and W chromosomes very quickly lose all genes that were initially present (and are still present on the X or Z), species with partial degeneration are unlikely to be studied. This will make it difficult to estimate the time-course in detail, and, conversely, if extensive future studies detect no, or very few, cases of intermediate degeneration, this will suggest that complete degeneration evolves very rapidly.
Consistent with their different ages, different evolutionary strata in Eutherian mammals appear to have reached different stages of genetic degeneration. The oldest mammalian stratum has lost almost all genes from the Y chromosome; the proportions of genes still present are higher in the younger strata (figure 2), although most are non-functional [28,53]. Data from other organisms are scarce, though degeneration is documented in lizards (electronic supplementary material, table S1). Reptiles are particularly interesting, because ZW systems probably evolved independently in different lizard groups [54], and in turtles ZW and XY systems are found and both macro- and micro-chromosomes can be involved [55–57]. Micro-chromosomes might be expected to degenerate more slowly, but I am not aware of any studies in this area.
Flowering plants may include many species with sex-linked regions in intermediate stages of genetic degeneration, although degeneration may be slower than in animals due to gene expression in the haploid pollen stage. However, the plant studies illustrate the approaches that can be used to study degeneration, as well as some interesting findings.
In S. latifolia, the XY pair are both large. The X may carry around one 7th of the plant's genes, and recombination probably occurs in only a small proportion of the chromosome. The most recent study estimated that about half of Y-linked genes are non-functional [58]. Out of 909 Y-linked genes analysed, 433 (47.6%) had premature stop codons and 12 (1.3%) had lost the start codon (versus, respectively, 12 and 0 out of 945 X-linked genes). Y–X sequence divergence suggests about 7 million generations for the older stratum, and around 4 million for the younger stratum, or about 11 and 6 million years (MY), respectively [58]. This is slightly older than previous estimates of the time when separate sexes evolved in this Silene species and its close relatives, from a hermaphroditic ancestor, based on sequence divergence between species [59]. The relative sizes of the two S. latifolia strata are not yet completely clear, as the current Y–X divergence estimate does not clearly demarcate them. However, 69% of the codons used for the divergence estimates were assigned to genes in the older stratum. The estimated overall proportions of genes that have lost function differ only slightly between the two strata (figure 3), but the predicted slowing down of degeneration may be occurring, as the degeneration rates for the older and younger strata are, respectively, 7% versus 10.5% per million years, despite the latter's smaller size [58].
The proportion of genes completely lost from this plant's Y chromosome has also been estimated in this species, using RNA-Seq (see box 2). The moderately short total evolutionary time since the older stratum evolved is consistent with the estimated nearly 50% gene loss [31,32,40–42], as the theoretical prediction outlined above is that Y chromosomes will quickly lose many genes after Y–X recombination stops, but that complete loss of genes from the Y will take many generations.
The papaya sex-linked region spans only about 10% of one chromosome, and Y–X divergence is smaller than in S. latifolia. Both these differences predict less degeneration in papaya, whose degeneration indeed appears slight [33,49]. For 55 X/Y gene pairs with sequences available from outgroup species, validating that they are stable elements of the plant's genome, 46 have apparently functional copies on both the X and Y, and, overall, the Y carries only two more non-functional genes than the X [33]. Purifying selection appears less effective in papaya Y- than X-linked coding sequences, which might eventually lead to more pronounced degeneration. Finally, plant sex-linked regions that include few genes show little evidence of degeneration [49,60–64].
Degeneration has been compared in two Rumex species [65]. Both species have neo-Y chromosomes due to X-autosome fusions (see box 3). The recently evolved Rumex hastatulus neo-Y yielded only slight evidence for degeneration [67]. A new study compared the ‘ancestral’ Y of this species (with 1686 sex-linked genes) and the non-homologous Y-linked region of Rumex rothschildianus, with only 553 sex-linked genes [65]. Definitive Y–X divergence estimates are not yet available, so the times when recombination between the sex chromosomes became suppressed, and strata have not been detected. The existence of some highly diverged XY pairs suggests that the R. rothschildianus fully sex-linked region could be older than that in R. hastatulus, and the gene loss estimates are 92% versus 18%, also suggesting an older system in R. rothschildianus. However, excluding the many sequences with less than 10% Y–X divergence suggested similar times when recombination stopped in the two species. The considerably larger non-recombining region in R. hastatulus might then be expected to generate faster degeneration of its Y.
Box 3. Fusions between sex chromosomes and autosomes and new evolutionary strata.
Another situation potentially informative about degeneration arises when an autosomal region stops recombining after being translocated to or fused with a fully sex-linked region. Rapid degeneration is predicted whenever substantial non-recombining regions (strata) are added to a Y or W chromosome. In organisms where one sex is achiasmate and does not undergo crossing over in meiosis, a fusion between an autosome and either sex chromosome immediately suppresses recombination with the sex-determining locus (Box 3 Figure A).
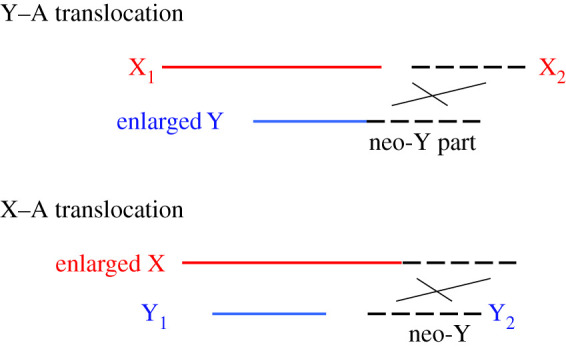
Box 3 Figure A. Diagram of fusions between an autosome (dashed lines) and the Y or X of an XY sex chromosome system in a species in which males are achiasmate. Clearly, fusion to the Y (shown the top diagram) creates a neo-Y that cannot recombine with the former autosome because it is inherited only by males, along with the ancestral Y. In the X–A case (the lower diagram), despite the former autosome having no physical attachment to the Y, a non-recombining neo-Y is again created, because it segregates from the fused chromosome attached to the X, and is therefore again inherited only by the male lineage.
However, the formation of a new fully sex-linked stratum is not inevitable. In many organisms, including mammals, crossing over occurs in meiosis of both sexes. When an autosomal region fuses to a sex chromosome, it can continue to recombine with its unfused autosomal homologue becoming partially sex-linked, not a fully sex-linked region that is expected to start degenerating. Rearrangements are known in many XY and ZW species where both sexes undergo crossovers, including in fish, lizards [66] and plants (e.g. [67]), producing systems where an autosome is physically attached to an ancestral X or Y (see Box 3 Figure B).
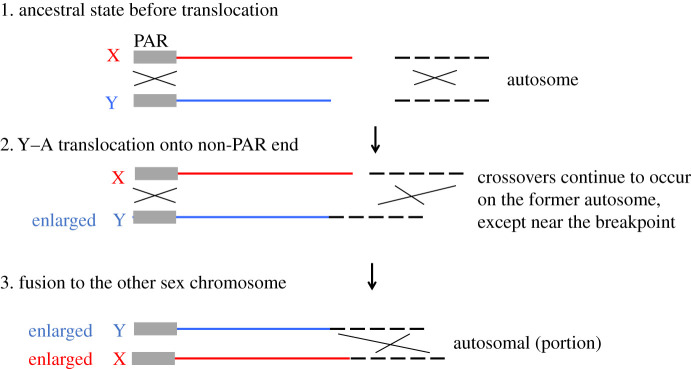
Box 3 Figure B. Diagram of fusions between an autosome and the Y or X of an XY sex chromosome system in a species in which crossing over occurs in both sexes. The figure shows the case when the autosome is added to the end of the Y that is non-recombining in males (rather than to the PAR end). The autosome is nevertheless expected to continue to pair with its homologue and undergo recombination, except possibly in a region near the breakpoint (see §5). If the free autosome subsequently fuses to the X, this restores an equal chromosome number in both sexes, making it less easy to detect that a fusion has occurred, though the number is reduced by one.
The unfused chromosome may later fuse with the other sex chromosome, or recombine onto it, if the fusion occurs onto the PAR. Whether this occurs or not, the former autosome pair can often continue to recombine, and no new stratum need arise.
A fusion in such a species may, however, directly cause failure of pairing in the region adjoining the pre-existing sex chromosome and stop crossing over, leading to this part differentiating in sequence between the sexes, like the original sex chromosome. It may be difficult to determine whether suppressed recombination in part of the former autosome is a direct effect of the rearrangement, or evolved subsequently, perhaps involving a sexually antagonistic polymorphism in the added arm near the fusion breakpoint that creates an advantage for close linkage with the sex-determining gene [68]. Such selection may explain the subsequent loss of recombination that led to the strata detected within the X-added region of Eutherian mammal sex chromosomes (see Box 1 and §3 above).
The degeneration rate appears to be slower in R. rothschildianus than in R. hastatulus [65]. This might reflect different functional categories of genes being present, which could occur by chance ‘sampling’ of genes in non-homologous regions (although sampling effects could be minor, as the sex-linked regions in both species carry many genes). Interestingly, R. rothschildianus appears to have lost many genes showing evidence of being under low selective constraint, unlike the R. hastatulus Y [65]; the remaining R. rothschildianus Y genes show several features indicating stronger purifying selection than the lost genes (including enrichment for fundamental functional categories, higher expression levels and greater frequencies of pollen expression). This is consistent with predictions that genes with important functions in males are especially likely to be retained [69]. Deleterious mutations may thus now be accumulating more slowly than in the R. hastatulus Y, suggesting a further reason for a slowing down of genetic degeneration. The R. rothschildianus results also suggest male-specific adaptation of some genes, with higher expression of Y- than X-linked alleles, which could have resulted in selective sweeps [65], as predicted in the later stages of Y chromosome evolution.
Slowing of degeneration is also inferred in humans [70]. This study compared 16 human-chimpanzee Y-linked gene sequences in the 9.5 Mb of the chimpanzee genome where X-linked genes have degenerated or become pseudogenes; all 11 pseudogenes in the region were shared by the chimpanzee and humans, and no chimpanzee-specific transcription units, without human Y counterparts, were found. Therefore, whereas it is estimated that the human Y has lost, on average, about five genes per million years, no human gene appears to have been lost or become non-transcribed during the six million years since these species separated. However, some chimpanzee genes have lost functionality since the split, due to point mutations that either disrupted splice sites or introduced stop codons, illustrating the possibility of different degeneration rates in different species.
5. Fusions between sex chromosomes and autosomes and new evolutionary strata
Fusions and reciprocal translocations can potentially stop recombining, creating new strata and so-called ‘neo-Y’ or ‘neo-W’ chromosomes (box 3). Such newly non-recombining regions may give information about the time course of genetic degeneration. In the Eutherian mammals, as described above, the ‘X-added’ region is less degenerated than the ancestral part [26,28]. Y-linked sequences are recognizable for about half the ancestral X genes in strata 2 and 3, and almost all in strata 4 and 5, allowing Y–X divergence estimates, but very few genes remain functional [53], even though these strata include few ancestral genes (figure 2). Species with rearrangements more recent than that in the Eutherian mammals can be more informative about the time course over which some functional genes remain.
In organisms where one sex does not undergo crossing over in meiosis, such as Drosophila males, fusions between sex chromosomes and autosomes immediately suppress recombination with the sex-determining locus (box 3, Figure A). Drosophila miranda underwent Y-autosome fusion about 1.5 million years, or roughly 4 million generations ago, based on estimates of divergence of neo-Y-linked sequences from their alleles on the former autosome [71], and about half of its genes are estimated to have lost their functions; consistent with the prediction that shorter times since recombination stopped should correlate with less degeneration, this neo-Y is much less degenerated than the older-established neo-Y of Drosophila pseudo-obscura, which is highly degenerated and has lost most ancestral genes (figure 3). On the other hand, Drosophila busckii is an intriguing example in which the prediction is not upheld; greater degeneration has occurred, in a shorter evolutionary time, in the D. busckii neo-Y than that of D. miranda [72]. This may perhaps be because (like the R. rothschildianus Y discussed above) a high proportion of genes on the former autosome (the D. busckii neo-Y was formed from the small ‘dot’ chromosome, with less than 100 genes) evolve under unusually low selective constraint [72]. It is well-established that genes with high expression levels (suggesting that they are likely to be under strong selective constraint), and/or dosage sensitive genes are preferentially retained on mammal Y chromosomes [9,10] and on the D. miranda neo-Y [73], and that genes on the dot chromosomes have lesser constraint levels [74].
An X-autosome fusion (or Z-autosome fusion in a ZW system) in a species that lacks crossing over in one sex also immediately causes suppressed crossing over of the neo-sex chromosome with its autosomal homologue, because the non-fused chromosome becomes confined to the heterogametic sex. Such fusions are known in several Drosophila lineages (reviewed in [75]). Many of them occurred much longer ago than in D. miranda, and, as expected, the neo-Y-chromosomes appear to be extremely degenerated, having lost most genes that were carried on the ancestral autosomes involved (figure 2).
Achiasmate meiosis is known from a few other sexually reproducing taxa, including the well-studied Lepidoptera (with WZ systems) and some related taxa [76,77]. Autosomes that have fused to a butterfly or moth sex chromosome are reported to be degenerated, but proportions of genes lost have not been estimated (electronic supplementary material, table S1). The extent of degeneration is thus mostly unknown, except when it is extremely high and the W chromosome is heterochromatic. The times when some rearrangements occurred have mostly been inferred from phylogenies of related species whose chromosome arrangements differ [78]. Although most studied cases of rearrangements appear to have occurred long ago and involve highly degenerated W chromosomes, younger systems suitable for studying the time taken for lesser degeneration may exist (see electronic supplementary material, table S1). Dosage compensation of the neo-W chromosome appears to have evolved after the rearrangement in the monarch butterfly, implying that the former autosome had degenerated; interestingly, however, it appears to have evolved de novo, as the mechanism differs from that of the ancestral W [79].
Rearrangements in other taxa in which one sex is achiasmate [80] have so far been little studied. Caddis flies (Trichoptera, with 11 000 species), the sister group to the Lepidoptera, also have female heterogamety and are apparently Z0 [81], but no further information appears to be available. In species with achiasmy in one sex, fusions involving the X of an XX/X0 system, or the Z of a ZZ/Z0 system, can also lead to a non-recombining neo-sex chromosome that would be predicted to start degenerating. I am not aware of studies of the degeneration of such chromosomes. In some taxa, achiasmatic male meiosis appears to have evolved independently several times, including in the sister group to the Diptera (Scorpion or hang flies, Mecoptera) [80], which includes one X1X2Y species [82]. It remains uncertain whether fusions exist in any achiasmatic species of this group, or in the mantid order.
In most organisms, however, both sexes undergo crossing over, and recombination will continue, even if an autosome fuses to a Y or W chromosome with a large completely non-recombining region (box 3, Figure B). Currently, there appear to be few data concerning degeneration from the many species in which such rearrangements have been reported [66]. It is rarely clear whether the former autosome has acquired, or (as in Eutherian mammals) subsequently evolved, suppressed recombination. Examples are known in which the former autosome is heterochromatic, such as some fish [83,84] and a grasshopper [85], suggesting rapid degeneration; however, gene loss has not yet been quantified, and the heterochromatin might represent repetitive sequence accumulation without major gene loss. Genome sequencing may in the future quantify both neo-sex chromosome degeneration and estimate the times since recombination between them stopped.
As explained in box 3, a rearrangement might directly suppress recombination near the breakpoint. Little information is currently available about the extent of degeneration in such regions. Threespine stickleback (Gasterosteus aculeatus) populations from the Japan Sea show high differentiation between males and females for sequences within a 7 Mb region of a 20 Mb neo-Y chromosome (autosome 9) that became attached to the older evolutionary stratum of the XY pair. This region therefore appears to have formed a new stratum at the opposite end from the previously established younger stratum adjacent to the PAR. None of the 24 loci examined in this region were hemizygous in a male sample, so the region has probably not undergone major degeneration, consistent with the fairly short time involved—estimated to be about 2 million years [2]. As far as I am aware, all genes studied within these regions of other species with such rearrangements, such as lark species [86,87], appear to be diploid, with no evidence of degeneration, despite the estimated fusing being dated to at least 37 million years ago.
6. Discussion and future prospects
The examples reviewed here show how existing molecular evolutionary analyses can detect degenerated regions of sex chromosomes, and how some theoretical predictions about degeneration might be tested. However, estimating Y–X and W–Z divergence remains difficult, because this requires assembling separate Y and X (or W and Z) haplotypes. If the sex chromosomes are ancient, and sequence divergence is high, as in humans (figure 1), it is possible to infer the sequences carried by the two haplotypes, especially if parent–offspring sets are available for studying segregation of sequence variants. Long-read sequencing promises to make it possible to construct phased haplotypes in more species without the need for genetic studies (e.g. [69]). Genome sequence data will probably reveal non-recombining regions in many more organisms with genetic sex-determination, along with their gene contents and densities, and will probably reveal evolutionary strata in some of them, clarifying the time-course of degeneration.
Further theoretical modelling will also be valuable, as the present conclusions about the time-course of degeneration involve several simplifying assumptions. Although the models discussed in §1 can explain degeneration during the history of the Therian mammal sex chromosome evolutionary strata [1], including the stratum shared by Eutherians and Marsupials [1], the time involved is estimated to be between 180 and 220 million years [9,10]. Changes in some of the assumptions can increase or decrease the predicted rates [1]. In the well-studied mammals, the Muller's ratchet could be responsible for between 40% and 75% of the observed degeneration. Clearly, more empirical data are needed from other organisms, including ones with fully sex-linked regions carrying only a few genes. If these are found to degenerate, this may suggest mechanisms other than those discussed here. Evidence from plants suggests that such physically small sex-linked regions show little degeneration [49,60–64], though they may have evolved too recently for this to have evolved yet.
As discussed above, differences in the genes carried in a fully sex-linked region could sometimes have important effects on degeneration rates. Genes important for male functions may be retained on Y chromosomes, for example, as may genes evolving under stronger constraint. However, degeneration rate differences due to such effects are most likely to arise when the fully sex-linked region or sex chromosome includes just a small number of genes, for instance in the case of a bird or lizard micro-chromosome, or a dot chromosome of Drosophila. A similar situation may arise in a sex chromosome region that has already lost most genes; given enough time, the only remaining genes will be ones that degenerate slowly, even if the region (and its X counterpart) initially contained many genes. This may have contributed to reducing the degeneration rate in the plant genus Rumex. However, since Y chromosomes in diverse taxa have lost almost all the ancestral genes (figure 3), such effects probably cannot prevent eventual complete genetic degeneration. However, it remains unclear how much evolutionary time is required for such complete degeneration.
Another particularly interesting question is whether dosage compensation evolves independently for individual genes, or is acquired across Y-linked regions carrying multiple genes. In the latter situation, an X- (or W-) linked gene or genes must experience selection for increased expression that outweighs any deleterious effects of changing the other genes' expression, allowing dosage compensation to evolve. The selection might then favour reduced expression from functional Y-linked genes in the region. If this can occur, degeneration might not slow down in the manner outlined above [1] and could perhaps become complete very rapidly, possibly overthrowing the paradigm outlined above. All-in-all, much work remains to be done to better understand degeneration rates.
Data accessibility
This article does not contain any additional data.
Competing interests
I declare I have no competing interests.
Funding
I received no funding for this study.
References
- 1.Bachtrog D. 2008. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics 179, 1513-1525. ( 10.1534/genetics.107.084012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Natri HM, Shikano T, Merilä J. 2013. Progressive recombination suppression and differentiation in recently evolved neo-sex chromosomes. Mol. Biol. Evol. 30, 1131-1144. ( 10.1093/molbev/mst035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawson-Handley LJ, Ceplitis H, Ellegren H. 2004. Evolutionary strata on the chicken Z chromosome: implications for sex chromosome evolution. Genetics 167, 367-376. ( 10.1534/genetics.167.1.367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimura M. 1983. The neutral theory of molecular evolution. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 5.Hudson RR, Boos DD, Kaplan NL. 1992. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9, 138-151. ( 10.1093/oxfordjournals.molbev.a040703) [DOI] [PubMed] [Google Scholar]
- 6.Gammerdinger W, Kocher T. 2018. Unusual diversity of sex chromosomes in African Cichlid fishes. Genes 9, 480. ( 10.3390/genes9100480) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon G, Kitano J, Kirkpatrick M. 2018. The origin of a new sex chromosome by introgression between two stickleback fishes. Mol. Biol. Evol. 36, 28-38. ( 10.1093/molbev/msy181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Natri H, Merilä J, Shikano T. 2019. The evolution of sex determination associated with a chromosomal inversion. Nat. Commun. 10, 145. ( 10.1038/s41467-018-08014-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellott DW et al. 2014. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508, 494-499. ( 10.1038/nature13206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, Grützner F, Kaessmann H. 2014. Origins and functional evolution of Y chromosomes across mammals. Nature 508, 488-493. ( 10.1038/nature13151) [DOI] [PubMed] [Google Scholar]
- 11.Zhou Q, Zhang J, Bachtrog D, An N, Huang Q, Jarvis ED, Gilbert MTP, Zhang G. 2014. Complex evolutionary trajectories of sex chromosomes across bird taxa. Science 346, 1332. ( 10.1126/science.1246338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myosho T, Takehana Y, Hamaguchi S, Sakaizumi M. 2015. Turnover of sex chromosomes in celebensis group medaka fishes. G3-Genes Genomes Genetics 5, 2685-2691. ( 10.1534/g3.115.021543) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nanda I, Schories S, Tripathi N, Dreyer C, Haaf T, Schmid M, Schartl M. 2014. Sex chromosome polymorphism in guppies. Chromosoma 123, 373-383. ( 10.1007/s00412-014-0455-z) [DOI] [PubMed] [Google Scholar]
- 14.Darolti I et al. 2019. Extreme heterogeneity in sex chromosome differentiation and dosage compensation in livebearers. Proc. Natl Acad. Sci. USA 116, 19 031-19 036. ( 10.1073/pnas.1905298116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicoso B, Charlesworth B. 2006. Evolution on the X chromosome: unusual patterns and processes. Nat. Rev. Genet. 7, 645-653. ( 10.1038/nrg1914) [DOI] [PubMed] [Google Scholar]
- 16.Pessia E, Makino T, Bailly-Bechet M, Mclysaght A, Marais GAB. 2012. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc. Natl Acad. Sci. USA 109, 5346-5351. ( 10.1073/pnas.1116763109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Q, Ellison C, Kaiser V, Alekseyenko A, Gorchakov A, Bachtrog D. 2013. The epigenome of evolving Drosophila neo-sex chromosomes: dosage compensation and heterochromatin formation. PLoS Biol. 11, e1001711. ( 10.1371/journal.pbio.1001711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schield DR et al. 2019. The origins and evolution of chromosomes, dosage compensation, and mechanisms underlying venom regulation in snakes. Genome Res. 29, 590-601. ( 10.1101/gr.240952.118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmer F, Harrison PW, Dessimoz C, Mank JE. 2016. Compensation of dosage-sensitive genes on the chicken Z chromosome. Genome Biol. Evol. 8, 1233-1242. ( 10.1093/gbe/evw075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kramer M, Rao P, Ercan S. 2016. Untangling the contributions of sex-specific gene regulation and X chromosome dosage to sex-biased gene expression in C. elegans. Genetics 204, 355. ( 10.1534/genetics.116.190298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nozawa M, Fukuda N, Ikeo K, Gojobori T. 2014. Tissue- and stage-dependent dosage compensation on the neo-X chromosome in Drosophila pseudoobscura. Mol. Biol. Evol. 31, 614-624. ( 10.1093/molbev/mst239) [DOI] [PubMed] [Google Scholar]
- 22.Itoh Y, Replogle K, Kim Y, Wade J, Clayton D, Arnold A. 2010. Sex bias and dosage compensation in the zebra finch versus chicken genomes: general and specialized patterns among birds. Genome Res. 20, 512-518. ( 10.1101/gr.102343.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellott DW et al. 2017. Avian W and mammalian Y chromosomes convergently retained dosage sensitive regulators. Nat. Genet. 49, 387-394. ( 10.1038/ng.3778) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng X et al. 2011. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat. Genet. 43, 1179-1185. ( 10.1038/ng.948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vicoso B, Emerson J, Zektser Y, Mahajan S, Bachtrog D. 2013. Comparative sex chromosome genomics in snakes: differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol. 11, e1001643. ( 10.1371/journal.pbio.1001643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lahn BT, Page DC. 1999. Four evolutionary strata on the human X chromosome. Science 286, 964-967. ( 10.1126/science.286.5441.964) [DOI] [PubMed] [Google Scholar]
- 27.Waters PD, Duffy B, Frost CJ, Delbridge ML, Graves JAM. 2001. The human Y chromosome derives largely from a single autosomal region added to the sex chromosomes 80–130 million years ago. Cytogenet. Cell Genet. 92, 74-79. ( 10.1159/000056872) [DOI] [PubMed] [Google Scholar]
- 28.Skaletsky H et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825-837. ( 10.1038/nature01722) [DOI] [PubMed] [Google Scholar]
- 29.Sayres M, Makova K. 2013. Gene survival and death on the human Y chromosome. Mol. Biol. Evol. 30, 781-787. ( 10.1093/molbev/mss267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White M, Kitano J, Peichel C. 2015. Purifying selection maintains dosage-sensitive genes during degeneration of the threespine stickleback Y chromosome. Mol. Biol. Evol. 32, 1981-1995. ( 10.1093/molbev/msv078) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergero R, Charlesworth D. 2011. Preservation of the Y transcriptome in a 10MY old plant sex chromosome system. Curr. Biol. 21, 1470-1474. ( 10.1016/j.cub.2011.07.032) [DOI] [PubMed] [Google Scholar]
- 32.Chibalina M, Filatov D. 2011. Plant Y chromosome degeneration is retarded by haploid purifying selection. Curr. Biol. 21, 1475-1479. ( 10.1016/j.cub.2011.07.045) [DOI] [PubMed] [Google Scholar]
- 33.Wu M, Moore PH. 2015. The evolutionary tempo of sex chromosome degradation in Carica papaya. J. Mol. Evol. 80, 265-277. ( 10.1007/s00239-015-9680-1) [DOI] [PubMed] [Google Scholar]
- 34.Mank JE, Vicoso B, Berlin S, Charlesworth B. 2010. Effective population size and the faster-X effect: empirical results and their interpretation. Evolution 64, 663-674. ( 10.1111/j.1558-5646.2009.00853.x) [DOI] [PubMed] [Google Scholar]
- 35.Meisel RP, Connallon T. 2013. The faster-X effect: integrating theory and data. Trends Genet. 29, 537-544. ( 10.1016/j.tig.2013.05.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachtrog D. 2003. Accumulation of Spock and Worf, two novel non-LTR retrotransposons, on the neo-Y chromosome of Drosophila miranda. Mol. Biol. Evol. 20, 173-181. ( 10.1093/molbev/msg035) [DOI] [PubMed] [Google Scholar]
- 37.Hollister JD, Gaut BS. 2009. Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 19, 1419-1428. ( 10.1101/gr.091678.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uzunovic J, Josephs EB, Stinchcombe JR, Wright SI. 2017. Transposable elements are important contributors to standing variation in gene expression in Capsella grandiflora. Mol. Biol. Evol. 36, 1734-1745. ( 10.1093/molbev/msz098) [DOI] [PubMed] [Google Scholar]
- 39.Hughes JF, Skaletsky H, Pyntikova T, Minx PJ, Graves T, Rozen S, Wilson RK, Page DC. 2005. Conservation of Y-linked genes during human evolution revealed by comparative sequencing in chimpanzee. Nature 437, 100. ( 10.1038/nature04101) [DOI] [PubMed] [Google Scholar]
- 40.Papadopulos AST, Chester M, Ridout K, Filatov DA. 2015. Rapid Y degeneration and dosage compensation in plant sex chromosomes. Proc. Natl Acad. Sci. USA 112, 13 021-13 026. ( 10.1073/pnas.1508454112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergero R, Qiu S, Charlesworth D. 2015. Gene loss from a plant sex chromosome system. Curr. Biol. 25, 1234-1240. ( 10.1016/j.cub.2015.03.015) [DOI] [PubMed] [Google Scholar]
- 42.Muyle A, Zemp N, Deschamps C, Mousset S, Widmer A, Marais G. 2012. Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLoS Biol. 10, e1001308. ( 10.1371/journal.pbio.1001308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L et al. 2019. Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nat. Ecol. Evol. 3, 834-844. ( 10.1038/s41559-019-0850-1) [DOI] [PubMed] [Google Scholar]
- 44.Nam K, Ellegren H. 2008. Scrambled eggs: the chicken (Gallus gallus) Z chromsome contains at least three non-linear evolutionary strata. Genetics 180, 1131-1136. ( 10.1534/genetics.108.090324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schield DR et al. 2020. Snake recombination landscapes are concentrated in functional regions despite PRDM9. Mol. Biol. Evol. 37, 1272-1294. ( 10.1093/molbev/msaa003) [DOI] [PubMed] [Google Scholar]
- 46.Bergero R, Forrest A, Kamau E, Charlesworth D. 2007. Evolutionary strata on the X chromosomes of the dioecious plant Silene latifolia: evidence from new sex-linked genes. Genetics 175, 1945-1954. ( 10.1534/genetics.106.070110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atanassov I, Delichère C, Filatov DA, Charlesworth D, Negrutiu I, Monéger F. 2001. A putative monofunctional fructose-2,6-bisphosphatase gene has functional copies located on the X and Y sex chromosomes in white campion (Silene latifolia). Mol. Biol. Evol. 18, 2162-2168. ( 10.1093/oxfordjournals.molbev.a003762) [DOI] [PubMed] [Google Scholar]
- 48.Nicolas M et al. 2005. A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biol. 3, 47-56. ( 10.1371/journal.pbio.0030047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J et al. 2012. Sequencing papaya X and Yh chromosomes reveals molecular basis of incipient sex chromosome evolution. Proc. Natl Acad. Sci. USA 109, 13 710-13 715. ( 10.1073/pnas.1207833109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schultheiß R, Viitaniemi H, Leder E. 2015. Spatial dynamics of evolving dosage compensation in a young sex chromosome system. Genome Biol. Evol. 7, 581-590. ( 10.1093/gbe/evv013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pigozzi M. 2011. Diverse stages of sex-chromosome differentiation in tinamid birds: evidence from crossover analysis in Eudromia elegans and Crypturellus tataupa. Genetica 139, 771-777. ( 10.1007/s10709-011-9581-1) [DOI] [PubMed] [Google Scholar]
- 52.Ramstad K, Miller H, Kolle G. 2016. Sixteen kiwi (Apteryx spp) transcriptomes provide a wealth of genetic markers and insight into sex chromosome evolution in birds. BMC Genomics 17, 410. ( 10.1186/s12864-016-2714-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Picard MAL, Cosseau C, Ferré S, Quack T, Grevelding CG, Couté Y, Vicoso B. 2018. Evolution of gene dosage on the Z-chromosome of schistosome parasites. eLife 7, e35684. ( 10.7554/eLife.35684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pokorna M, Kratochvil L, Kejnovsky E. 2011. Microsatellite distribution on sex chromosomes at different stages of heteromorphism and heterochromatinization in two lizard species (Squamata: Eublepharidae: Coleonyx elegans and Lacertidae: Eremias velox). BMC Genet. 12, 90. ( 10.1186/1471-2156-12-90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Badenhorst D, Stanyon R, Engstrom N, Valenzuela N. 2013. A ZZ/ZW microchromosome system in the spiny softshell turtle, Apalone spinifera, reveals an intriguing sex chromosome conservation in Trionychidae. Chromosome Res. 21, 137-147. ( 10.1007/s10577-013-9343-2) [DOI] [PubMed] [Google Scholar]
- 56.Montiel E, Badenhorst D, Tamplin J, Burke R, Valenzuela N. 2017. Discovery of the youngest sex chromosomes reveals first case of convergent co-option of ancestral autosomes in turtles. Chromosoma 126, 105-113. ( 10.1007/s00412-016-0576-7) [DOI] [PubMed] [Google Scholar]
- 57.Lee L, Montiel E, Valenzuela N. 2019. Discovery of putative XX/XY male heterogamety in Emydura subglobosa turtles exposes a novel trajectory of sex chromosome evolution in Emydura. Cytogenet. Genome Res. 158, 160-169. ( 10.1159/000501891) [DOI] [PubMed] [Google Scholar]
- 58.Krasovec M, Chester M, Ridout K, Filatov D. 2018. The mutation rate and the age of the sex chromosomes in Silene latifolia. Curr. Biol. 28, 1832-1838.E4. ( 10.1016/j.cub.2018.04.069) [DOI] [PubMed] [Google Scholar]
- 59.Rautenberg A, Hathaway L, Oxelman B, Prentice HC. 2010. Geographic and phylogenetic patterns in Silene section Melandrium (Caryophyllaceae) as inferred from chloroplast and nuclear DNA sequences. Mol. Phylogenet. Evol. 57, 978-991. ( 10.1016/j.ympev.2010.08.003) [DOI] [PubMed] [Google Scholar]
- 60.Harkess A, Huang K, Hulst R, Tissen B, Caplan JL, Koppula A, Batish M, Meyers BC, Leebens-Mack J. 2020. Sex determination by two Y-linked genes in garden asparagus. Plant Cell 32, 1790-1796. ( 10.1105/tpc.19.00859) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harkess A et al. 2017. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 8, 1279. ( 10.1038/s41467-017-01064-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akagi T et al. 2019. Two Y-chromosome-encoded genes determine sex in kiwifruit. Nat. Plants 5, 801-809. ( 10.1038/s41477-019-0489-6) [DOI] [PubMed] [Google Scholar]
- 63.Okazaki Y, Takahata S, Hirakawa H, Suzuki Y, Onodera Y. 2020. Molecular evidence for recent divergence of X- and Y-linked gene pairs in Spinacia oleracea L. PLoS ONE 14, e0214949. ( 10.1371/journal.pone.0214949) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Picq S et al. 2014. A small XY chromosomal region explains sex determination in wild dioecious V. vinifera and the reversal to hermaphroditism in domesticated grapevines. BMC Plant Biol. 14, 229. ( 10.1186/s12870-014-0229-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crowson D, Barrett SCH, Wright SI. 2017. Purifying and positive selection influence patterns of gene loss and gene expression in the evolution of a plant sex chromosome system. Mol. Biol. Evol. 34, 1140-1154. ( 10.1093/molbev/msx064) [DOI] [PubMed] [Google Scholar]
- 66.Pennell MW, Kirkpatrick M, Otto SP, Vamosi JC, Peichel CL, Valenzuela N, Kitano J. 2015. Y fuse? Sex chromosome fusions in fishes and reptiles. PLoS Genet. 11, e1005237. ( 10.1371/journal.pgen.1005237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hough J, Hollister JD, Wang W, Barrett SCH, Otto SP. 2014. Genetic degeneration of old and young Y chromosomes in the flowering plant Rumex hastatulus. Proc. Natl Acad. Sci. USA 111, 7713-7718. ( 10.1073/pnas.1319227111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Doorn G, Kirkpatrick M. 2007. Turnover of sex chromosomes induced by sexual conflict. Nature 449, 909-912. ( 10.1038/nature06178) [DOI] [PubMed] [Google Scholar]
- 69.Wei KH-C, Bachtrog D. 2019. Ancestral male recombination in Drosophila albomicans produced geographically restricted neo-Y chromosome haplotypes varying in age and onset of decay. PLoS Genet. 15, e1008502. ( 10.1371/journal.pgen.1008502) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hughes JF et al. 2010. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463, 536-539. ( 10.1038/nature08700) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bachtrog D, Hom E, Wong K, Maside X, Pd J. 2008. Genomic degradation of a young Y chromosome in Drosophila miranda. Genome Biol. 9, R30. ( 10.1186/gb-2008-9-2-r30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou Q, Bachtrog D. 2015. Ancestral chromatin configuration constrains chromatin evolution on differentiating sex chromosomes in Drosophila. PLoS Genet. 11, e1005331. ( 10.1371/journal.pgen.1005331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaiser V, Zhou Q, Bachtrog D. 2011. Nonrandom gene loss from the Drosophila miranda neo-Y chromosome. Genome Biol. Evol. 3, 1329-1337. ( 10.1093/gbe/evr103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaiser VB, Charlesworth B. 2010. Muller's ratchet and the degeneration of the Drosophila miranda neo-Y chromosome. Genetics 185, 339-348. ( 10.1534/genetics.109.112789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dupim EG et al. 2018. An investigation of Y chromosome incorporations in 400 species of Drosophila and related genera. PLoS Genet. 14, e1007770. ( 10.1371/journal.pgen.1007770) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fraisse C, Picard M, Vicoso B. 2017. The deep conservation of the Lepidoptera Z chromosome suggests a non-canonical origin of the W. Nat. Commun. 8, 1486. ( 10.1038/s41467-017-01663-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Traut W, Marec F. 1996. Sex chromatin in Lepidoptera. Q. Rev. Biol. 71, 239-256. ( 10.1086/419371) [DOI] [PubMed] [Google Scholar]
- 78.Mongue AJ, Nguyen P, Volenikova A, Walters JR. 2017. A neo-sex chromosome in the monarch butterfly, Danaus plexippus. G3 (Bethesda) 7, 3281-3294. ( 10.1534/g3.117.300187) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gu L, Reilly PF, Lewis JJ, Reed RD, Andolfatto P, Walters JR. 2019. Dichotomy of dosage compensation along the neo Z chromosome of the monarch butterfly. Curr. Biol. 29, 4071-4077. ( 10.1016/j.cub.2019.09.056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blackmon H, Ross L, Bachtrog D. 2017. Sex determination, sex chromosomes, and karyotype evolution in insects. J. Hered. 106, 78-93. ( 10.1093/jhered/esw047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marec F, Novak K. 1998. Absence of sex chromatin corresponds with a sex-chromosome univalent in females of Trichoptera. Eur. J. Entomol. 95, 197-209. [Google Scholar]
- 82.Xu B, Li Y, Hua B. 2013. A chromosomal investigation of four species of Chinese Panorpidae (Insecta, Mecoptera). Comp. Cytogenet. 7, 229-239. ( 10.3897/compcytogen.v7i3.5500) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sember A, Bertollo LAC, RáB P, Yano CF, Hatanaka T, De Oliveira EA, Cioffi MB. 2018. Sex chromosome evolution and genomic divergence in the fish Hoplias malabaricus (Characiformes, Erythrinidae). Front. Genet. 9, 71. ( 10.3389/fgene.2018.00071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yano CF, Bertollo LAC, Ezaz T, Trifonov V, Sember A, Liehr T, Cioffi MB. 2017. Highly conserved Z and molecularly diverged W chromosomes in the fish genus Triportheus (Characiformes, Triportheidae. Heredity 118, 276-283. ( 10.1038/hdy.2016.83) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Palacios-Gimenez OM, Milani D, Lemos B, Castillo ER, Martã DA, Ramos E, Martins C, Cabral-De-Mello DC. 2018. Uncovering the evolutionary history of neo-XY sex chromosomes in the grasshopper Ronderosia bergii (Orthoptera, Melanoplinae) through satellite DNA analysis. BMC Evol. Biol. 18, 2. ( 10.1186/s12862-017-1113-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pala I, Naurin S, Stervander M, Hasselquist D, Bensch S, Hansson B. 2012. Evidence of a neo-sex chromosome in birds. Heredity 108, 264-272. ( 10.1038/hdy.2011.70) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pala I, Hasselquist D, Bensch S, Hansson B. 2012. Patterns of molecular evolution of an Avian neo-sex chromosome. Mol. Biol. Evol. 29, 3741-3754. ( 10.1093/molbev/mss177) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article does not contain any additional data.