

Abstract
An understanding of the genetic variation underlying transcript splicing is essential to dissect the molecular mechanisms of common disease. The available evidence from splicing quantitative trait locus (sQTL) studies has been limited to small samples. We performed genome-wide screening to identify SNPs that might control mRNA splicing in whole blood collected from 5,257 Framingham Heart Study participants. We identified 572,333 cis sQTLs involving 2,650 unique genes. Many sQTL-associated genes (40%) undergo alternative splicing. Using the National Human Genome Research Institute (NHGRI) genome-wide association study (GWAS) catalog, we determined that 528 unique sQTLs were significantly enriched for 8,845 SNPs associated with traits in previous GWAS. In particular, we found 395 (4.5%) GWAS SNPs with evidence of cis sQTLs but not gene-level cis expression quantitative trait loci (eQTLs), suggesting that sQTL analysis could provide additional insights into the functional mechanism underlying GWAS results. Our findings provide an informative sQTL resource for further characterizing the potential functional roles of SNPs that control transcript isoforms relevant to common diseases.
GWAS have found many SNPs associated with various traits and diseases1. eQTL studies have been employed to identify SNPs that may influence the expression levels of a particular gene, thereby lending credibility to the SNP-disease association2. However, only a moderate proportion of GWAS-identified loci have been shown to be strong eQTLs3, which might in part be owing to the tissues studied, small sample sizes and a focus on whole-gene level expression measurements without consideration of transcript isoforms.
Alternative splicing is a process by which identical pre-mRNA molecules can give rise to multiple distinct mRNA or transcript isoforms. It affects ~80% of human genes and often occurs during tissue or cell development4–7. Alternative splicing contributes to phenotypic differences within and between individuals by increasing the multiplicity and diversity of the translated proteins arising from the same gene8,9. Available evidence suggests that at least 20–30% of disease-causing mutations may affect pre-mRNA splicing10,11. Thus, identification of genetic variants that affect the generation of transcript isoforms of the same genes (sQTLs) might represent an important step toward fully understanding the contribution of genetic variants in disease development.
Thus far, sQTL studies have been conducted largely by use of Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines (LCLs)8,12,13. LCLs may not represent the in vivo state owing to their transformation by EBV, and they also may not exhibit the spectrum of alternative splicing incidents that define tissue specificity. Recently, an sQTL study performed in 922 whole-blood samples by combining RNA sequencing expression and genetic data increased the number of known sQTLs by nearly an order of magnitude14. However, because these samples were derived from a major depressive disorder case-control study, it is possible that their expression profile might not be representative of the general community of men and women. Additional sQTL studies using large numbers of human primary tissue samples from community-based cohorts are needed to comprehensively catalog sQTLs and their correlations with GWAS signals in unbiased samples. Here we hypothesize that strong sQTL signals, colocalized with GWAS signals for disease, can provide insights into how genetic regulatory effects on the pre-mRNA splicing of genes close to GWAS SNPs might be the causal molecular mechanisms underlying respective traits or diseases.
Our current study aimed to create and characterize an sQTL catalog for whole blood collected from a single large cohort and to further examine the relationships between specific sQTLs and disease-associated loci from previous GWAS. To accomplish this, we first performed a genome-wide screen to identify common SNPs controlling transcript isoform variations (cis sQTLs) using 5,257 samples with both expression data and SNP data imputed using the 1000 Genomes Project15. To the best of our knowledge, this is the largest sQTL study thus far. We then categorized the identified cis sQTLs on the basis of their position relative to the differentially spliced transcript (exonic, intronic, etc.) and annotated the alternative splicing category for each individual cis sQTL. Lastly, we assessed the overlap between known GWAS signals and cis sQTLs. Of note, we were able to identify many GWAS hits for disease traits with cis-sQTL associations, which were not significantly associated with gene expression at the whole-gene level (eQTLs). Our findings may provide insights into the mechanisms through which genetic variation influences transcript isoforms and, ultimately, disease development.
RESULTS
Cis-QTL association with gene- and exon-level expression
A total of 5,257 Framingham Heart Study (FHS) participants with whole-blood expression data and 1000 Genomes Project15 imputed genotype data were included in this analysis. The clinical characteristics of these study participants are presented in Supplementary Table 1. A schematic showing coverage of Affymetrix Exon array probe sets across the entire length of the transcript is shown in Supplementary Figure 1. A total of 17,873 genes were included in this analysis; on average, there were ~553 SNPs located within 50 kb of each gene and 16 ‘core’ exon-level probe sets for each gene (Supplementary Fig. 2).
Applying a conservative Bonferroni-corrected P < 5.2 × 10−9 (0.05/9,623,954) to the whole-gene level eQTL analysis, we observed 6,137 genes that were significantly associated with at least one SNP. Of the genes without statistically significant associations of any SNPs with whole-gene expression, 2,864 (24.4%) unique genes were found to contain at least one exon (corresponding to 7,410 unique exons) that was significantly associated with a neighboring SNP at P < 2.8 × 10−10 (0.05/178,799,891). A total of 672,845 exon-SNP pairs (cis sQTLs) were identified at this significance threshold. After excluding 214 (7.5%) genes with possible sQTL signals due to SNPs residing in probes (corresponding to 760 unique exons), a final set of 572,333 cis sQTLs in 2,650 genes was identified. A flowchart of the analysis to identify cis sQTLs is shown in Figure 1, including the total number of tests performed and the Bonferroni-corrected P-value thresholds for both gene-level and exon-level analysis. The most significant cis-sQTL results for each of the 2,650 genes are shown in Supplementary Table 2. For the 2,650 genes, the distribution of fold differences in gene and exon expression levels between the two homozygous genotypes for each SNP is shown in Supplementary Figure 3. The mean of the gene-level fold difference in expression was near 0. The distribution for fold differences in exon-level expression was bimodal, representing positive and negative fold changes.
Figure 1.
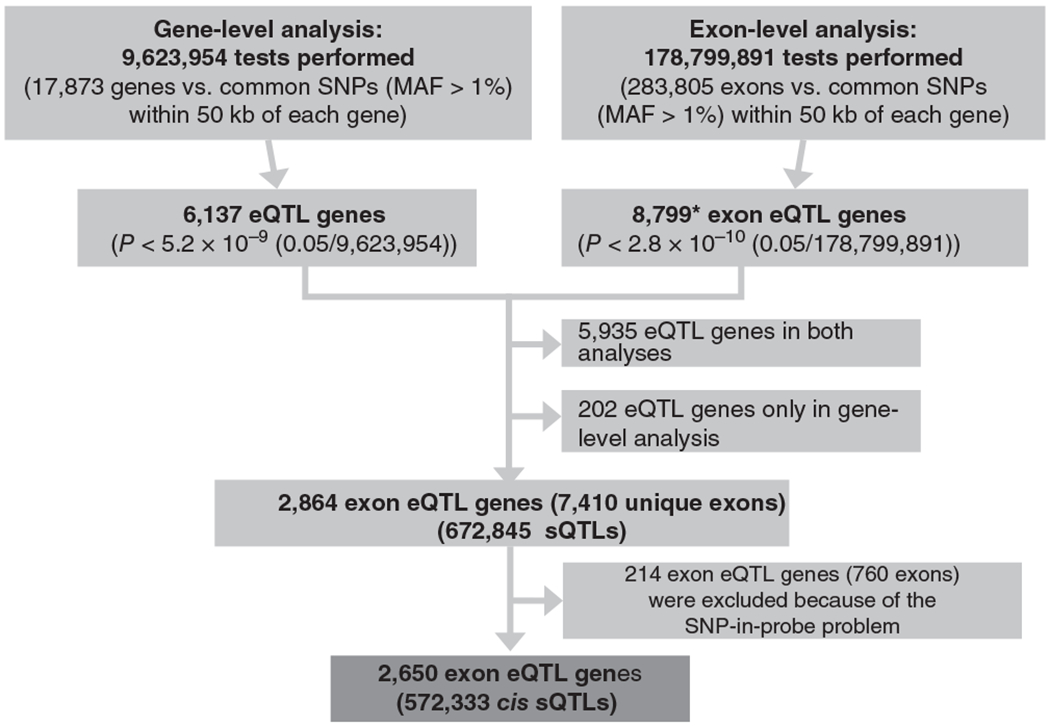
Overview of sQTL analysis. Flowchart of the gene-level and exon-level cis-QTL analysis used to identify cis splicing QTL associations (cis sQTLs) using 5,257 whole-blood samples and 9,623,954 common SNPs (minor allele frequency (MAF) > 1%) within 50 kb of each gene. *Among the 8,799 genes identified by exon-level QTL analysis, 5,935 overlapped with 6,137 genes from the gene-level analysis. Therefore, 2,864 genes with only QTL associations at the exon level were defined as genes with sQTL associations, including 672,845 significant probe set–SNP pairs. After excluding 214 (7.5%) genes with suspect sQTL signals due to the SNP-in-probe problem (corresponding to 760 (10%) unique exons), 572,333 cis sQTLs (2,650 genes) were finally identified.
Functional categories of cis sQTLs
Among the 572,333 cis sQTLs associated with 2,650 genes only at the exon level of expression, 258,113 SNPs were unique. Functional annotation for these unique SNPs is reported in Table 1. As noted there, a large percentage (~90%) of the cis sQTLs were located within intergenic or intronic regions (32% and 58%, respectively).
Table 1.
Functional annotation of 258,113 unique cis sQTLs obtained from a total of 572,333 cis sQTLs
| SNP functional class | Count | Proportion |
|---|---|---|
| Intronic | 150,440 | 0.58 |
| Intergenic | 81,370 | 0.32 |
| 5′ near gene | 10,795 | 0.04 |
| 3′ near gene | 2,251 | 0.01 |
| 5′ UTR | 1,233 | 0.005 |
| 3′ UTR | 5,908 | 0.02 |
| Synonymous | 2,926 | 0.01 |
| Missense | 2,235 | 0.01 |
| Noncoding RNA | 909 | 0.004 |
| 5′ splice site | 20 | 7.75 × 10−5 |
| 3′ splice site | 12 | 4.65 × 10−5 |
| Stop loss | 1 | 3.87 × 10−6 |
| Stop gain | 12 | 4.70 × 10−5 |
| Frameshift | 1 | 3.87 × 10−6 |
Four percent of cis sQTLs were located 5′ to a gene (‘5′ near gene’) or close to a 5′ promoter region of a gene, whereas only 1% were located 3′ to a gene (‘3′ near gene’). This fourfold difference is not surprising, as the UCSC database asymmetrically defines ‘5′ near gene’ as 2 kb upstream of a gene, whereas ‘3′ near gene’ is defined as 0.5 kb downstream of a gene. Contrasting results were observed for UTRs. In comparison to cis sQTLs found in the 5′ UTR of a gene (0.5% overall), four times more SNPs (2% overall) were found in the 3′ UTR, Although the 3′ UTR is known to serve as the functional regulatory locus of small RNA post-transcriptional regulatory pathways and is involved in nonsense-mediated mRNA decay surveillance pathways16, after normalizing sQTLs by region size, we found that there was no relative enrichment of sQTLs in the 3′ UTRs of genes.
Interestingly, 0.4% of our cis sQTLs were in noncoding RNA gene regions. The majority of noncoding RNA genes have not been well characterized. Recent studies have shown that many noncoding RNA genes exhibit a structure with multiple exons similar to that of protein-coding genes and that their noncoding transcripts bear many similarities to mRNAs, including 5′ capping, splicing and polyadenylation17. A small percentage of our cis sQTLs (~1%) were missense variants. Approximately 3% of our cis sQTLs were coding synonymous mutations that could affect mRNA splicing or stability and could have substantial effects on protein function18,19.
Using the canonical definition of splice-site regions from the NCBI dbSNP database, we identified a small number (n = 32) of unique cis sQTLs located in a 5′ splice donor site (2-base region at the 5′ end of an intron) or 3′ splice acceptor site (2-base region at the 3′ end of an intron) (Table 2). As an example, the rs3842788 splice-3 variant of the PTGS1 gene (encoding prostaglandin-endoperoxide synthase 1) was significantly associated with exon 3 of PTGS1 (probe set 3188118) at P < 2.2 × 10−25 with a coefficient of 0.55 (fold change = 1.1). The box plot of exon-level expression versus the most significant cis sQTL is shown in Figure 2b, and a visualization of the probe set expression level for this gene against the most significant sQTL is shown in Figure 2a. The gene structure of PTGS1 and seven of its RefSeq transcript isoforms are shown in Figure 2c. This sQTL is located in a 3′ splice acceptor site, and its associated exon-level probe set is absent in one of the seven transcript isoforms (NM_001271367.1; Fig. 2c).
Table 2.
Thirty-two unique cis-sQTL associations found to be in a canonical splice site (NCBI dbSNP v137 database)
| Gene | Affymetrix probe set | rs ID | Chr. | Position (bp) | Effect allele | Non-effect allele | Observed EAF | Estimate | SE | PsQTL | Function |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IRF5 | 3023264 | rs2004640 | 7 | 128,578,301 | G | T | 0.47 | 0.549 | 0.015 | 1.46 × 10−262 | Splice-5 |
| KIF20B | 3257353 | rs12264026 | 10 | 91,534,702 | T | C | 0.78 | −0.424 | 0.0128 | 9.07 × 10−220 | Splice-5 |
| MS4A14 | 3332343 | rs4938941 | 11 | 60,173,360 | G | A | 0.67 | −0.338 | 0.012 | 7.48 × 10−163 | Splice-3 |
| WDR82 | 2676049 | rs2276834 | 3 | 52,325,759 | G | A | 0.52 | −0.374 | 0.0175 | 1.16 × 10−97 | Splice-3 |
| NME2 | 3726971 | rs115848216 | 17 | 49,243,859 | G | A | 0.96 | 0.441 | 0.0209 | 2.45 × 10−95 | Splice-5 |
| PNPLA2 | 3316308 | rs61876746 | 11 | 827,713 | A | T | 0.69 | 0.49 | 0.0235 | 7.74 × 10−93 | Splice-5 |
| MTMR9 | 3085893 | rs2736277 | 8 | 11,218,893 | A | G | 0.52 | −0.214 | 0.0117 | 4.73 × 10−72 | Splice-3 |
| ADNP2 | 3795520 | rs7233511 | 18 | 77,915,113 | C | T | 0.79 | −0.202 | 0.0133 | 1.93 × 10−51 | Splice-5 |
| PPIP5K1 | 3621297 | rs7169097 | 15 | 43,925,134 | T | A | 0.71 | 0.291 | 0.0202 | 3.89 × 10−46 | Splice-3 |
| UBTF | 3758978 | rs7220138 | 17 | 42,254,011 | G | C | 0.31 | −0.0939 | 0.00812 | 1.56 × 10−30 | Splice-3 |
| PTDSS2 | 3315872 | rs12419209 | 11 | 494,510 | T | C | 0.89 | 0.179 | 0.0155 | 2.44 × 10−30 | Splice-5 |
| C19orf53 | 3822350 | rs8108058 | 19 | 13,889,589 | C | T | 0.74 | −0.1 | 0.00895 | 1.04 × 10−28 | Splice-5 |
| HNRNPF | 3286287 | rs10899795 | 10 | 43,869,097 | C | A | 0.76 | −0.101 | 0.0094 | 9.36 × 10−27 | splice-3 |
| PTGS1a | 3188118 | rs3842788 | 9 | 125,140,206 | G | A | 0.96 | 0.547 | 0.0523 | 2.24 × 10−25 | Splice-3 |
| RNF216P1 | 2988420 | rs9689983 | 7 | 5,024,540 | G | A | 0.95 | 0.361 | 0.0359 | 1.31 × 10−23 | Splice-3 |
| RHOT2 | 3643270 | rs35915772 | 16 | 679,274 | G | A | 0.78 | −0.171 | 0.0176 | 6.45 × 10−22 | Splice-5 |
| APOL6 | 3944259 | rs67090823 | 22 | 36,064,455 | R | D | 0.93 | 0.163 | 0.0178 | 7.21 × 10−20 | Splice-5 |
| GOLGA2P5 | 3467744 | rs11110268 | 12 | 100,560,416 | T | C | 0.94 | 0.197 | 0.0219 | 3.03 × 10−19 | Splice-3 |
| RNF213 | 3737392 | rs41298712 | 17 | 78,396,004 | A | G | 0.95 | −0.261 | 0.0297 | 1.67 × 10−18 | Splice-3 |
| MOK | 3580293 | rs56377169 | 14 | 102,729,881 | A | G | 0.93 | 0.202 | 0.0233 | 7.69 × 10−18 | Splice-5 |
| ASB16 | 3722813 | rs7220138 | 17 | 42,254,011 | G | C | 0.31 | −0.0648 | 0.00751 | 8.11 × 10−18 | Splice-3 |
| TMUB2 | 3722846 | rs7220138 | 17 | 42,254,011 | G | C | 0.31 | −0.0559 | 0.00673 | 1.20 × 10−16 | Splice-3 |
| CYP2D7P1 | 3962303 | rs3892097 | 22 | 42,524,947 | C | T | 0.81 | −0.121 | 0.0154 | 5.39 × 10−15 | Splice-3 |
| STRC | 3621380 | rs7169097 | 15 | 43,925,134 | T | A | 0.71 | −0.0713 | 0.0091 | 5.59 × 10−15 | Splice-3 |
| TMEM8B | 3168220 | rs2381409 | 9 | 35,829,390 | T | C | 0.62 | 0.069 | 0.00938 | 2.16 × 10−13 | Splice-5 |
| NCL | 2603490 | rs28685161 | 2 | 232,378,792 | C | T | 0.76 | 0.0534 | 0.00729 | 2.85 × 10−13 | Splice-5 |
| BZW1 | 2522472 | rs62623595 | 2 | 201,637,798 | G | A | 0.89 | −0.0889 | 0.0126 | 2.34 × 10−12 | Splice-5 |
| IFIH1 | 2584224 | rs35337543 | 2 | 163,136,505 | C | G | 0.98 | 0.69 | 0.0995 | 4.49 × 10−12 | Splice-5 |
| LLGL2 | 3734961 | rs73998360 | 17 | 73,586,325 | G | A | 0.93 | 0.136 | 0.0207 | 6.66 × 10−11 | Splice-5 |
| WDR46 | 2950587 | rs3130099 | 6 | 33,282,181 | G | A | 0.51 | −0.0655 | 0.0101 | 1.18 × 10−10 | Splice-5 |
| ISG15 | 4053546 | rs3813194 | 1 | 998,582 | C | G | 0.53 | −0.0941 | 0.0146 | 1.27 × 10−10 | Splice-5 |
| BMP8A | 2331542 | rs79473113 | 1 | 40,020,020 | C | A | 0.96 | 0.261 | 0.0413 | 2.67 × 10−10 | Splice-5 |
Chr., chromosome; EAF, effect allele frequency; SE, standard error.
An example of the PTGS1 gene with its associated cis sQTL is shown in Figure 2.
Figure 2.
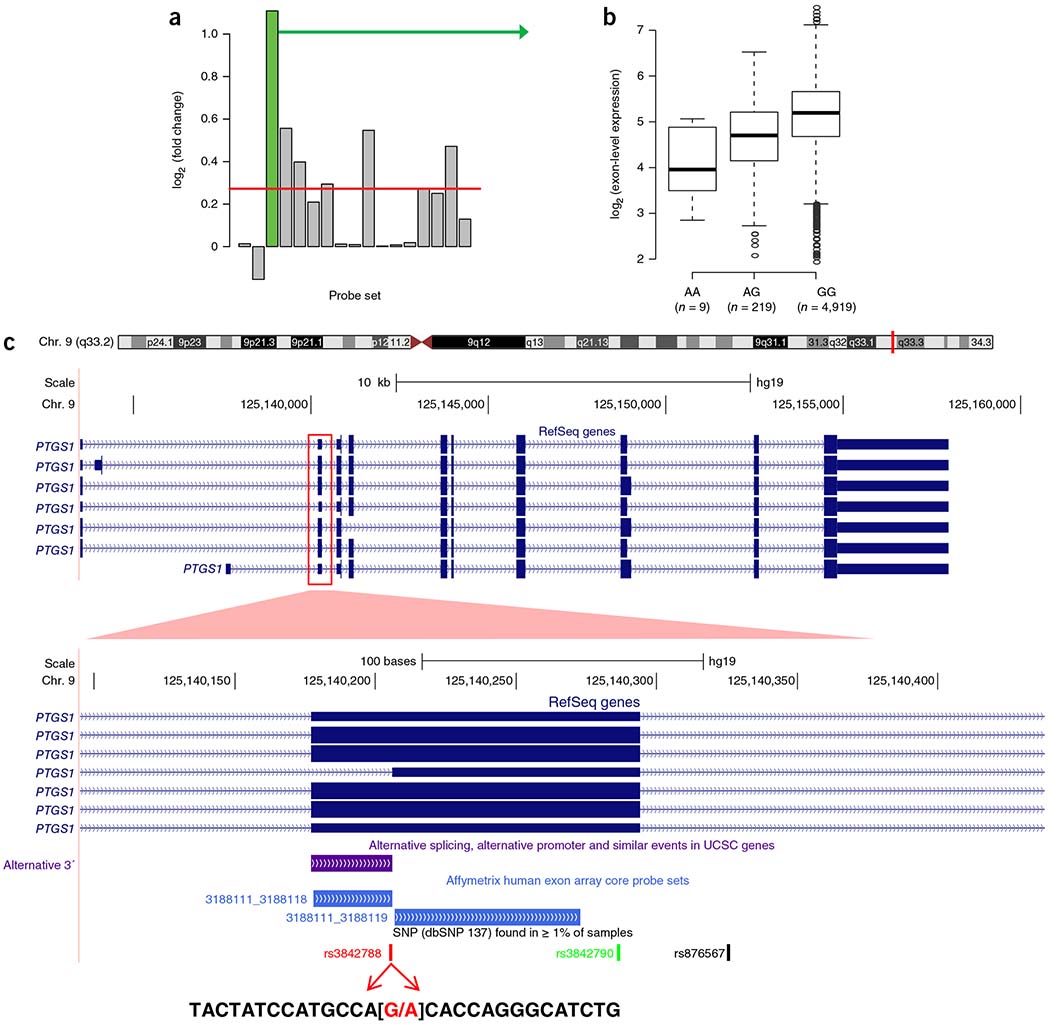
An example of the PTGS1 gene with its associated cis sQTL located in the 3′ acceptor splice site, (a) Visualization of probe set 3188118 in the context of all other probe sets belonging to the PTGS1 gene (Affymetrix transcript ID 3188111). For each probe set, the fold change between the mean expression scores of the two homozygous genotypes (mean(AA)/mean(GG)) at rs3842788 is shown by vertical gray bars (probe set 3188118 is highlighted in green). The horizontal red line represents the fold change in expression at the whole-gene-level against SNP rs3842788. (b) Box plot of the expression signals for probe set 3188118 with the indicated genotypes at SNP rs3842788, giving a P value for association of 2.2 × 10−25 (P value = 0.001 for the association between rs3842788 genotype and the whole-gene expression level of PTGS1). The sample size analyzed is shown under each genotype. The solid horizontal line within the box represents the median. The interquartile range (IQR) is defined as Q3-Q1 with whiskers that extend 1.5 times the IQR from the box edges, (c) Schematic of the seven transcript isoforms of PTGS1 in the NCBI RefSeq database. A zoomed-in view of exon 3 of PTGS1 with Affymetrix Exon array core probe sets is shown below the exon. The significant probe set 3188118 is highlighted in blue and corresponds to alternative 3′ acceptor splice site usage that results in a shorter exon for transcript NM_001271367.1. The SNP rs3842788 is located in the last position of the intron and is a G>A substitution that disrupts the consensus splice-site sequence.
The regulation of alternative splicing might involve additional features, such as splice-site disruptions, exonic and intronic splicing enhancers and silencers, or RNA secondary structures9. To address these potential complexities, we extended our splice-site definition to sequence variants that were either within 1–3 bases of an exon or 3–8 bases of an intron and within a branch point—a branch-point sequence of 20–50 nt upstream of acceptor sites that affects splice-site selection20,21. As expected, using this expanded definition, many more cis sQTLs identified in this study overlapped with the extended functional splicing-relevant components (Supplementary Table 3), suggesting that expanding consensus splice sites to other regulatory splicing sites could provide additional insights into the functional regulation of alternative splicing by common genetic variants and their associations with disease.
In addition to identifying functional categories, we analyzed a total of 258,113 unique cis sQTLs for functional enrichment. It is known that mRNA-binding proteins (RBPs) have an important role in post-transcriptional processes in which RBPs recognize and bind to multiple mRNA targets to facilitate gene expression patterns22. Because genetic variants might change the binding affinity of RBPs, we examined whether sQTLs were significantly enriched for mapping to RBP-binding sites using Encyclopedia of DNA Elements (ENCODE) RIP-seq data23. sQTLs mapped to regions for two RBPs (ELAVL1 and PABPC1) in the GM12878 (lymphoid) and K562 (myeloid) cell lines in comparison to the signals observed in the permuted data (Fig. 3). Enrichment of RBP-binding sites in myeloid and lymphoid cells provides support for the validity of our blood-derived sQTLs.
Figure 3.
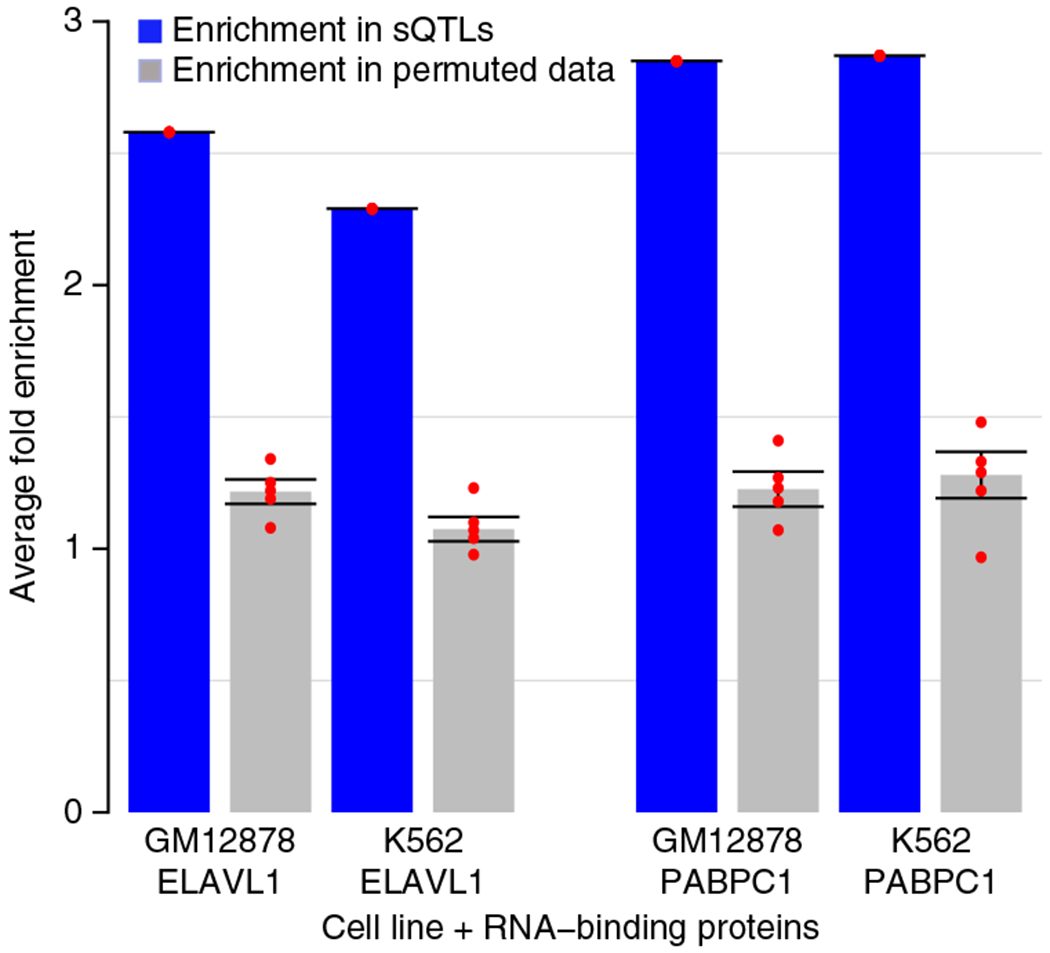
Cis sQTLs are highly enriched in binding sites for RNA-binding proteins. Examples are shown for the RNA-binding proteins ELAVL1 and PABPC1 that are present in the ENCODE GM12878 (lymphoid) and K562 (myeloid) cell lines, respectively. Error bars, s.d. Red data points represent minimum values, first quarter, median, third quarter and maximum value, which provide an overall distribution for the enrichment score obtained through 200 permutation tests.
Alternative splicing categories of cis-sQTL associations
We aimed to understand the biological functions of the genes for which transcript isoform-level expression was associated with nearby SNPs. Thus, we conducted a gene ontology (GO) enrichment analysis for the 2,650 sQTL-associated genes. We found that 121 GO categories surpassed the Bonferroni-corrected P-value threshold of <0.05 (Supplementary Table 4). The GO categories ‘phosphoprotein’, ‘acetylation’ and ‘nucleus’ were the top three most significant terms. Other highly enriched GO categories included ‘alternative splicing’, ‘splice variant’, ‘transcription’, ‘transcription regulation’, ‘RNA binding’ and ‘mRNA processing’. Of note, the GO category ‘alternative splicing’ identified genes whose protein products themselves had multiple isoforms. Of the 2,650 sQTL-associated genes, 47.8% (n 1,267) had more than one transcript isoform, resulting in a highly significant enrichment of GO genes involved in alternative splicing in comparison to all human genes (P < 7.6 × 10−23, Bonferroni-corrected P < 5.8 × 10−20).
Furthermore, for each of the 2,650 unique sQTL-associated genes (6,650 unique probe sets or exons), we checked for known alternative splicing events in the UCSC database. We found 43.7% of the genes with specific cis-sQTL associations (n = 1,159) in the alternative splicing database by mapping the probe set positions of the sQTL-associated exons to 8 types of alternative splicing events. Of these 1,159 genes, about half (52.2%) were classified as an ‘exon-skipping/cassette exon’ event (n = 605) (Fig. 4a). The large number of cassette exon events is expected, as they are the most common, well-characterized and most easily validated alternative splicing type4. An additional 30% (n = 349) of sQTL-associated exons were classified as ‘bleeding exon’ events. Examples of the different types of transcript isoform events are shown in Figure 4b and Supplementary Figure 4a–c.
Figure 4.
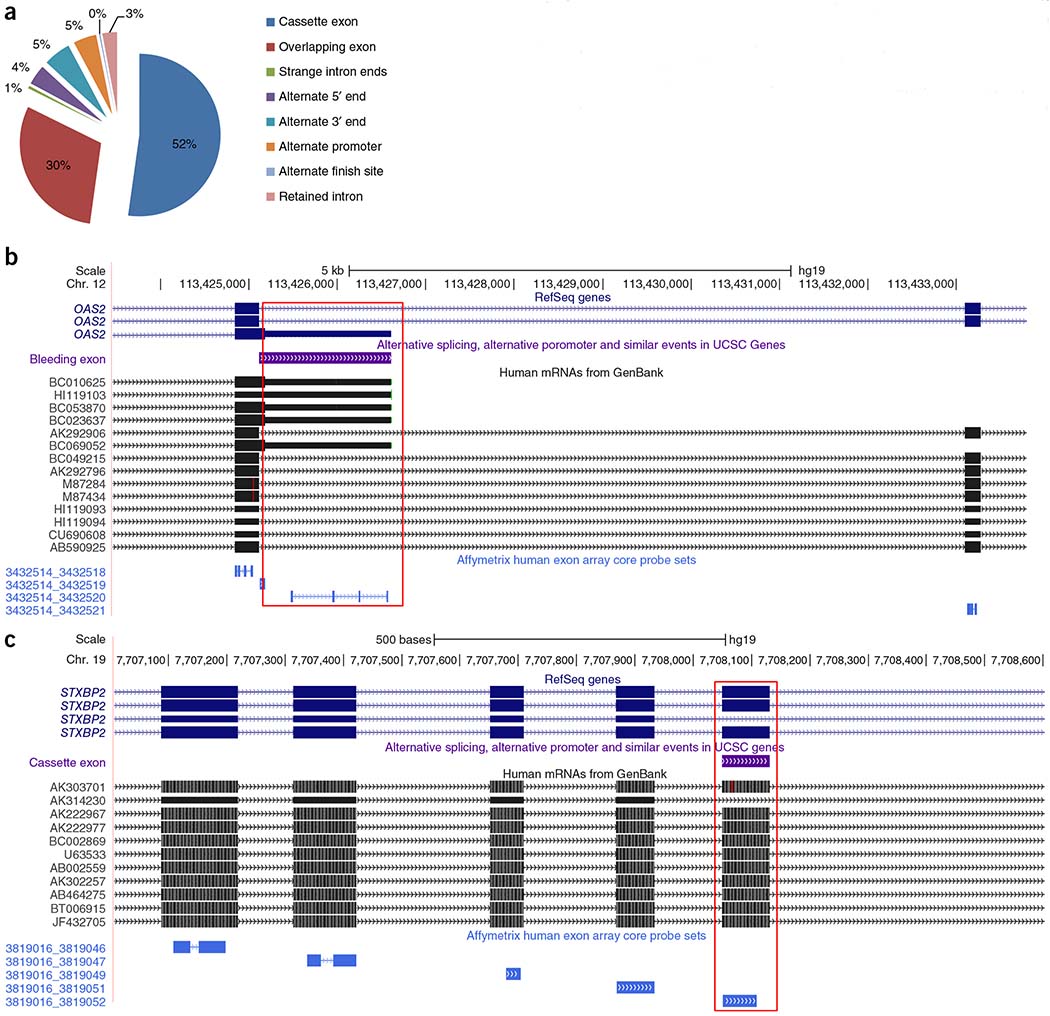
Functional annotation of genes and exons with cis-sQTL associations, (a) Concordance of sQTL-associated exons and known alternative splicing events. (b) Example of the bleeding exon type of transcript isoform event: OAS2, probe set 3432520 versus rs117666908. (c) Example of the cassette exon type of transcript isoform event: STXBP2, probe set 3819052 versus rs72994460.
Co-occurrence of cis sQTLs with GWAS trait SNPs
Recent studies have shown that eQTLs are highly enriched for SNPs that are strongly associated with a trait or disease phenotype24. sQTLs may represent additional functional alleles implicated in disease risk. To test this hypothesis, we investigated whether our cis sQTLs were previously reported to be associated with clinical diseases or with traits in previous GWAS in the NHGRI GWAS catalog25.
By cross-referencing the rs IDs of our cis sQTLs to the 8,845 unique trait-associated GWAS SNPs, we found 528 unique cis sQTLs (corresponding to 367 unique genes) that were associated with 238 phenotypes and disease traits in the GWAS catalog. After restricting to SNPs that were genome-wide significant (P < 5.0 × 10−8), we obtained 202 genes (corresponding to 304 unique sQTLs) with both cis-sQTL and strong GWAS-identified trait associations (Supplementary Table 5). The majority of the intergenic GWAS SNPs were mapped to genes that differed from those we identified as being associated with our cis sQTLs. This difference may be due in part to the fact that genes reported in GWAS are identified largely on the basis of their proximity to an associated SNP, whereas sQTLs benefit from associations with mRNA expression levels. Thus, sQTLs use additional sources of information to identify genes through which GWAS SNPs might confer their effects.
Because alternate splicing is known to be tissue-type specific, we further examined whether the cis sQTLs detected in our whole-blood samples were enriched for GWAS signals for blood-relevant diseases or traits, including hemostatic factors, hematology traits, lipid traits and coronary heart disease (CHD). Several cis-sQTL genes were associated with lipid traits, for example, LDLR and CELSR2. Other traits with cis-sQTL evidence included metabolic traits (for example, bloodpressure, diabetes and body mass index), CHD, C-reactive protein levels and hematology traits such as red blood cell traits, mean platelet volume and platelet counts (Supplementary Table 5).
In exploring biological functions and pathways in which the genes with both cis-sQTL and strong GWAS-identified trait associations might act, we found that annotations of these 202 genes (304 unique sQTLs) were highly enriched for several GO categories, including ‘alternative splicing’, ‘splice variants’, ‘ATPase activity’ and ‘cellular response to stress’, and with one Online Mendelian Inheritance in Man (OMIM) disease category called ‘loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts’ (Table 3). Of the 202 genes, 7 (DNAH11, LDLR, HMGCR, TOMM40, CELSR2, ABCA1 and TRIB1) contributed to the enrichment of this GO disease category with Benjamini-corrected P < 5.2 × 10−4.
Table 3.
GO enrichment analysis for 202 genes with significant cis-sQTL associations and genome-wide significant GWAS trait associations in the NHGRI GWAS catalog
| Category | Term | Count | Percent | P | Fold enrichment | Benjamini-corrected P |
|---|---|---|---|---|---|---|
| SP_PIR_KEYWORDS | Phosphoprotein | 110 | 56.70 | 6.21 × 10−8 | 1.51 | 1.95 × 10−5 |
| OMIM_DISEASE | Loci influencing lipid levels and CHD risk in 16 European population cohorts | 7 | 3.61 | 3.86 × 10−6 | 15.28 | 5.25 × 10−4 |
| SP_PIR_KEYWORDS | Acetylation | 51 | 26.29 | 3.79 × 10−6 | 1.93 | 5.94 × 10−4 |
| SP_PIR_KEYWORDS | Magnesium | 16 | 8.25 | 3.95 × 10−5 | 3.61 | 4.12 × 10−3 |
| SP_PIR_KEYWORDS | ATP binding | 30 | 15.46 | 5.42 × 10−5 | 2.25 | 4.25 × 10−3 |
| SP_PIR_KEYWORDS | Nucleotide binding | 33 | 17.01 | 3.02 × 10−4 | 1.95 | 0.0189 |
| GOTERM_MF_FAT | GO:0016887, ATPase activity | 13 | 6.70 | 3.65 × 10−4 | 3.46 | 0.02 |
| SP_PIR_KEYWORDS | Cell cycle | 14 | 7.22 | 7.61 × 10−4 | 3.03 | 0.039 |
| GOTERM_MF_FAT | GO:0004518, nuclease activity | 8 | 4.12 | 0.001967 | 4.50 | 0.047 |
| SP_PIR_KEYWORDS | Alternative splicing | 97 | 50.00 | 0.001129 | 1.29 | 0.049 |
Among the 528 unique cis sQTLs with a co-occurring GWAS signal, 133 (25.2%) also had cis-eQTL associations detected in our samples. About one-third (n = 39) of the 133 SNPs were the top SNP or were in close linkage disequilibrium (LD; r2 > 0.8) with the top cis sQTL associated with a target gene and the top cis eQTL associated with a separate, neighboring gene, suggesting that these SNPs and their proxy SNPs might truly serve as both a cis sQTL for the target gene and a cis eQTL for the neighboring gene. Examples of these 39 SNPs are shown in Supplementary Figure 6a,b. For the remaining 395 GWAS hits, there was evidence of splicing regulatory variants (cis sQTLs) but no evidence of cis eQTLs. After sorting the sQTLs by the frequency of association with different disease traits in the GWAS catalog, we found that nine sQTLs were associated with at least four GWAS disease traits. About 42 sQTLs were associated with at least 2 disease traits (Supplementary Table 6a), and the remaining 353 sQTLs were associated with only 1 GWAS trait (Supplementary Table 6b). Notably, several sQTLs were associated with CHD, lipids, blood pressure, red blood cell traits and platelet counts (Supplementary Table 6a). For example, a cis sQTL, rs4420638, which is 16 kb 3′ to TOMM40 and 350 bp 3′ to APOC1, was associated with 12 GWAS traits, including lipoprotein-associated phospholipase A2 activity and mass, C-reactive protein levels, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol levels, triglyceride levels and Alzheimer’s disease (Supplementary Table 6a). TOMM40 is located in the APOC cluster region on chromosome 19 that harbors well-known lipid risk loci, including APOE, APOC1, APOC1P1, APOC2 and APOC4. TOMM40 has three well-annotated, well-supported transcript isoforms in the RefSeq database, is 1 of 116 genes contributing to enrichment of the GO category ‘alternative splicing’ and is also 1 of 7 genes enriched in disease-associated loci influencing lipids and CHD risk that we identified by GO enrichment analysis (Table 3). The observed unique sQTL evidence for GWAS signals provides insights into how genetic effects on the regulation of alternative splicing may affect genes close to GWAS signals. New knowledge regarding these regulatory changes may improve the ability to functionally characterize susceptibility variants associated with diseases and related risk factors.
Replication of cis sQTLs
For replication, we downloaded a recent sQTL database with the largest available sample size14 in which RNA sequencing was applied to measure expression levels in whole-blood samples. This paper reported a total of 1,763 unique cis sQTLs. Of these, 1,464 were in line with our findings at P < 2.8 × 10−5 (0.05/1,763), providing replication evidence for 83% of the cis sQTLs reported in the RNA sequencing study (Supplementary Table 7).
DISCUSSION
Although GWAS have identified many candidate eQTLs, emerging research suggests that eQTL signals may not explain most GWAS associations26. Among the factors that might account for this discrepancy, transcript isoform variation is poorly understood but may underlie a substantial proportion of the phenotypic variation explained by GWAS variants in humans27. However, thus far, available data from large-scale sQTL studies using primary human tissues have been sparse, thus limiting understanding of the role of transcript isoforms associated with GWAS variants.
We generated sQTL results from exon array expression data and 1000 Genomes Project imputed genotype data in a single large, well-described population (n = 5,257). Through sQTL analysis of 17,873 genes and 283,805 exons, we found a large number of cis sQTLs (n = 572,333) associated with alternative splicing, exceeding the numbers reported previously. We found that 2,650 genes not detected in eQTL analysis had alternatively spliced transcript isoforms that were genetically controlled. We identified 14.8% (n = 2,650) of the examined genes with specific exon expression levels highly associated with nearby cis-acting SNPs, indicating that these genes experience alternative splicing due to genetic variation. This finding is consistent with a recent estimate that ~21% of annotated alternatively spliced genes may be associated with SNPs regulating the relative abundance of alternative transcript isoforms28.
Our examination of the functional categories of cis sQTLs identified 32 cis sQTLs located within 5′ or 3′ essential splice sites, providing new evidence for functional sQTLs that affect the pattern of splicing in whole-blood samples. For example, PTGS1 (prostaglandin-endoperoxide synthase 1), a prostaglandin G/H synthase and cyclooxygenase, encodes a group of enzymes that regulate angiogenesis in endothelial cells and are inhibited by non-steroidal anti-inflammatory drugs such as aspirin. There were seven alternatively spliced transcript isoforms of PTGS1 in the NCBI RefSeq database. The PTGS1 sQTL we identified was within a known splice site, which may be attributed to the polymorphism in this gene region associated with aspirin resistance29 and decreased incidence of myocardial infarction in individuals with coronary artery disease30. This finding provides a possible molecular mechanism for disease-associated variants in individuals with PTGS1-related diseases.
Of the unique sQTLs we identified, 528 were associated with 238 diseases or traits listed in the NHGRI GWAS catalog. The majority (75%; n = 395) of these disease-associated sQTLs were detected only in sQTL analysis and not in gene-level eQTL analysis. This observation suggests that there are many disease-associated variants that might contribute to disease etiology by affecting pre-mRNA splicing. These findings may also explain the lack of overlap of many eQTL signals with GWAS associations, as the former do not take into consideration splicing variants. For example, rs4420638, a cis sQTL near the TOMM40 gene, was associated with 12 diseases and traits in the GWAS catalog, including lipid traits, red blood cell traits and CHD (Supplementary Table 6a). The impact of these genetic associations on the development of cardiovascular disease has yet to be investigated. Results from our whole-blood sQTL study could help identify and functionally characterize susceptibility genes for cardiovascular diseases and related risk factors.
Additionally, our sQTL analysis can help exclude false positive eQTL signals identified in previous eQTL studies that used 3′-targeting expression platforms to measure expression traits at the whole-gene level. For example, the IRF5 gene has five transcript isoforms and five corresponding protein isoforms according to RefSeq. After broadly checking 53 eQTL data sets in ref. 31, we found over 35 data sets with cis-eQTL associations with IRF5, using data sets where expression traits were measured on platforms targeting 3′ UTRs, such as the Affymetrix U133A or U133 plus2.0 expression arrays, in which probes were designed to target only the short isoform12. By contrast, in two studies using the Affymetrix Exon expression array, in which probes were designed to target the whole transcript region of a gene, negative eQTL associations of IRF5 were reported32. This observation was based on a comprehensive comparison of eQTL data across various tissues and cell types and is in line with our expectation. In our study, we identified no SNPs that were significantly associated with the gene-level expression of IRF5 measured on the Affymetrix Exon expression array; however, there was an sQTL signal associated with specific exons of IRF5 that had been validated previously by RT-PCR in LCL cell lines12. The sQTL rs10954213 located in the 3′ UTR is associated with a different 3′ UTR change in IRF5 (ref. 12), and rs10488630 near the 3′ end of the gene is also associated with the choice of a proximal or distal polyadenylation site in IRF5 (ref. 33). However, despite our application of a very stringent significance threshold for sQTLs, it may still be difficult to distinguish between a splicing mechanism and a gene-level effect for some sQTLs that manifest significant statistical evidence for association at the gene level (Supplementary Table 2).
Potential limitations of our sQTL resource include its conduct using RNA derived from whole blood rather than specific blood cell types. To understand the potential confounding effects from the use of specific blood cell types on our sQTL results, for all 572,333 cis sQTLs identified, we reran our QTL analysis pipeline using gene and exon expression data after adjustment for seven cell types comprising whole blood (total white blood cell counts, total platelet counts and subfractions (percentages) of neutrophils, lymphocytes, monocytes, eosinophils and basophils). Then, we compared the β values and −log10 (P values) for these sQTLs generated using cell type–unadjusted expression data to those obtained using cell type–adjusted expression data. sQTL associations with and without adjustment for blood cell counts were highly correlated (r = 0.99; Supplementary Fig. 5), indicating the robustness of our cis-sQTL results to differences in blood cell counts.
Thus far, the majority of sQTL studies have been conducted with blood-derived cells or cell lines. It is reasonable to assume that the amount of splicing may vary in different tissues and cell types. Future studies examining sQTLs in multiple tissue types beyond blood will provide a more complete picture of the role of genetic variants in disease mechanisms by pairwise comparison of splicing in many tissues between individuals. The Genotype-Tissue Expression (GTEx) project34 may serve as a first step to examining this issue.
In summary, we identified 572,333 sQTLs in whole blood that were missed using conventional eQTL analysis, providing a catalog of sQTL variants that will be a publicly available resource for future studies and for the scientific community. Our findings provide insights into the genomic locations of sQTL SNPs, and many of the sQTL-associated genes were previously identified to be involved in alternative splicing. As the largest sQTL study thus far, our study had sufficient statistical power to identify and functionally characterize many cis sQTLs, including SNPs that were significantly associated with complex diseases and traits in GWAS. In particular, we found 4.5% of published GWAS SNPs with cis-sQTL but not gene-level cis-eQTL evidence, suggesting that sQTL analysis provides additional insights into the functional mechanisms underlying GWAS results. Our findings lay the groundwork for studies to define the role of mRNA splicing in the prevention and treatment of common diseases.
URLs.
The gene annotations used for each probe set were from the annotation file obtained from Affymetrix at http://www.affymetrix.com/. The database of known human alternative splicing events was downloaded from the UCSC Genome Browser for hg19 at http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=383408861_aoFr9g8KQoWF1FmaxEpYXni4qKtB&c=chr9&g=knownAlt. Results in the NHGRI GWAS catalog (22 March 2013) were downloaded from http://www.genome.gov/gwastudies/. The deposition of expression-level data into the database of Genotypes and Phenotypes (dbGaP) for the FHS is described on the dbGaP website at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000363.v10.p8 under SABRe CVD Project 3, expression dataset phe000002.v2. The deposition of data for genotyped and imputed SNPs into dbGaP for the FHS has been described on the dbGaP website at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000342.v4.p6.
ONLINE METHODS
Study population and sample collection.
The FHS started in 1948 with 5,209 randomly ascertained participants from Framingham, Massachusetts, who underwent biennial examinations to investigate cardiovascular disease and its risk factors35. In 1971, the Offspring cohort36,37 (comprising 5,124 children of the original cohort and the children’s spouses) and, in 2002, the Third Generation (consisting of 4,095 children of the Offspring cohort) were recruited38. Participants of the FHS Offspring cohort who attended examination 8 (n = 2,446) and the Third Generation cohort who attended examination 2 (n = 3,180) were included, constituting a total of 5,626 individuals. The clinical characteristics of the FHS Offspring and Third Generation cohorts are presented in Supplementary Table 1. FHS participants in this study are mainly of European ancestry. The study protocol was reviewed by the Boston University Medical Center Institutional Review Board, and all participants gave written informed consent.
Microarray data acquisition and preprocessing.
Fasting peripheral whole-blood samples (2.5 ml) were collected in PAXgene tubes (PreAnalytiX), incubated at room temperature for 4 h for RNA stabilization and then stored at −80 °C. Total RNA was isolated from frozen PAXgene blood tubes by Asuragen, according to the company’s standard operating procedures for the automated isolation of RNA from 96 samples in a single batch on a KingFisher 96 robot. RNA samples (50 ng) were then amplified using the WT-Ovation Pico RNA Amplification System (NuGEN) as recommended by the manufacturer in an automated manner using the GeneChip Array Station (GCAS). This treated RNA was then converted into cDNA and subsequently processed, labeled and hybridized onto Affymetrix Human Exon Array ST 1.0 arrays. After hybridization, each array was washed and stained according to the standard Affymetrix protocol. Stained arrays were scanned using the Affymetrix 7G GCS3000 scanner, resulting in a raw data CEL file for each array (details of RNA isolation, preparation of cDNA from RNA and microarray processing are available in the Supplementary Note).
Microarray preprocessing.
Over 1 million core probes were used to obtain gene-level and exon-level expression values, which were derived from CEL files by quantile sketch normalization using the model-based Robust Multichip Average (RMA) method39 as implemented in Affymetrix APT software version 1.12.0 (the full pipeline is described in the Supplementary Note). Gene-level analysis was conducted on 17,873 empirically supported transcripts (RefSeq and full-length GenBank mRNAs). Exon-level analysis was conducted on 283,805 ‘core’ exon-level probe sets from the Affymetrix Human Exon Array ST 1.0 that mapped to these core transcripts with a high degree of confidence. The gene annotations used for each probe set were from the annotation file obtained from Affymetrix.
Technical data adjustment.
Ten technical covariates fulfilled our selection criteria (Supplementary Table 8). These technical covariates and the first principal component (PC1) were used to adjust the data in all analyses.
Blood cell count adjustment.
Because blood cell count information was only available for the Third Generation cohort, values for samples in the Offspring cohort were imputed on the basis of the actual measurements for samples within the former. Five white blood cell subfractions, including neutrophils, lymphocytes, monocytes, eosinophils and basophils, in addition to the total white blood cell count and the total platelet count were adjusted for the gene-level and exon-level expression data.
Detailed methodology is provided in a companion paper submitted by joehanes et al.
Genotyping.
Genotyping was carried out as a part of the SNP Health Association Resource (SHARe) project using the Affymetrix 500K mapping array (250K Nsp and 250K Sty arrays) and the Affymetrix 50K supplemental gene-focused array on 9,274 individuals. Genotyping resulted in 503,551 SNPs with successful call rate > 95% and Hardy-Weinberg equilibrium P > 1 × 10−6. Finally, 8,481 individuals (91.4%) remained with call rate > 97%. Imputation of ~36.3 million autosomal and X-chromosome SNPs in 1000 Genomes Project Phase 1 SNP data15 was conducted using MACH40. There were 5,257 Offspring and Third Generation participants with genotype imputation and expression measurements available for the analysis.
Statistical analysis.
For our cis-eQTL analysis, we first filtered for common SNPs (MAF ≥ 1%) with good imputation quality (ratio > 0.1) in the 1000 Genomes Project imputed SNP data set. Furthermore, for each gene, SNPs were included in analysis if they were within 50 kb of the gene boundary according to NCBI human reference genome Build 37/hg19 and calculated as the maximum union of overlapping transcript isoforms based on RefSeq genes. The 50-kb distance was based on previous studies12,14 that indicated that sQTLs tend to be more gene-centric than gene-level eQTLs.
In both gene-level and exon-level eQTL analyses, we applied a linear mixed-effects model in which the residual from expression data adjusted for technical covariates (as described above) was the outcome (dependent variable), the SNP was the predictor (a fixed-effects term), and age and sex were included as covariates. Family structure was modeled as a random-effects term by including the matrix of the kinship coefficients in the genetic covariance matrix as previously described41. We applied the lmekin function in the R 2.15.1 package kinship (version 1.1.3) to estimate the SNP effects using an additive genetic model.
Gene-level eQTL analysis.
A total of 9,623,954 tests were performed examining the associations between each of the 17,873 genes and their neighboring SNPs (±50 kb). Following a conservative Bonferroni correction for multiple testing, adjusted P < 5.2 × 10−9 (0.05/9,623,954) was used to define significant associations between the expression level of genes and their neighboring SNPs (cis eQTLs).
Exon-level eQTL analysis.
All SNPs within 50 kb of a gene boundary were tested for association with all core probe sets/exons of that gene. For the 17,873 genes included in this analysis, on average, there were 16 core exon-level probe sets neighboring each gene. A total of 178,799,891 tests were performed for each probe set and its neighboring SNP pair. A Bonferroni-corrected P < 2.8 × 10−10 (0.05/178,799,891) was used to define significant associations between exon expression levels and neighboring SNPs (cis eQTLs).
Definition and classification of cis sQTLs.
Of the genes without detectable gene-level expression associations with any nearby SNPs, 2,864 genes were found to have at least one exon that was significantly associated with neighboring SNPs at P < 2.8 × 10−10. Associations between the expression level of exons and their neighboring SNPs are referred to as cis sQTLs.
Removal of suspect sQTLs due to the SNP-in-probe problem.
We removed cis-sQTL probe sets/exons that were susceptible to the effect of the SNP located within the probe. In the most recent study42, using the 1000 Genomes Project (March 2012) and NationL heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project data (9,025,738 common SNPs from the European panel), Ramasamy et al. identified 16,426 (5.5%) Affymetrix core probe sets/exons with the SNP in the 25-mer probe sequence for at least 2 probes, which the authors suggested should be discarded. Of 7,410 cis-sQTL probe sets/exons, 760 (10%) were found in the above 16,426 core probe sets and were discarded.
Comparison of the effects of cell count on cis sQTLs.
For all 572,333 cis sQTLs, we compared the P values and the estimated β values of the sQTL results generated by using gene-level and exon-level expression data before and after adjustment of the whole-blood complete blood cell counts (CBCs).
Comparison of statistical methods for detecting alternative splicing.
There are several methods of detecting alternative splicing events available using Affymetrix Exon Array data. Before deciding on the final model used in this study, we tested four different methods in our pilot data sets (see the Supplementary Note for more details). After conducting this comparison, we opted to examine variation at the exon level and identified exon-level signals without concurrent whole-transcript change, following the method of Kwan and Majewski12.
Functional classification of cis sQTLs.
SNP annotation data were downloaded from NCBI for dbSNP 137 (37.3), which contains >50 million SNPs. Each sQTL was classified as intergenic, intron, splice site, near gene, missense, UTR, etc. directly by matching SNP rs IDs.
To determine whether our significant cis sQTLs were enriched for the binding sites for RBPs, we downloaded the RBP-associated mRNA sequencing track (ENCODE RIP-seq) from the UCSC Genome Browser (March 2012 freeze), which includes transcriptional fragments associated with RBPs in the K562 and GM12878 cell lines identified using ribonomic profiling23,43. For the total of 258,113 unique cis sQTLs identified in this study, we first tested whether they were enriched in each RIP-seq data set (peak calls) applying a binomial test. We then selected equally sized sets of SNPs within 50 kb of each gene from each chromosome in the 1000 Genomes Project Phase 1 imputed SNP data as permuted cis sQTLs. The same functional enrichment analysis was conducted for this permuted cis-sQTL data to obtain the enrichment score; the realistic null distribution of RBP enrichments was then obtained by 200 permutations.
Annotation of alternative splicing events.
The database of known human alternative splicing was downloaded from the UCSC Genome Browser for hg19. We annotated the cis-sQTL probe sets by alternative splicing types if they overlapped with regions of known alternative splicing events.
Comparison of expression findings to the GWAS catalog.
Results in the NHGRI GWAS catalog (22 March 2013) included data from 1,595 publications and 11,124 SNPs, of which 8,845 SNPs were unique. Specific exon-level associated cis sQTLs were mapped to the GWAS catalog directly by matching SNP rs IDs.
Gene ontology enrichment analysis.
For the 2,650 genes with at least 1 exon significantly associated with at least 1 neighboring SNP, we submitted their unique gene symbol to the DAVID website44,45 to perform functional GO enrichment analysis using all human genes as the background. We accounted for multiple testing using Bonferroni-corrected significance levels: 0.05/1,472 = 0.00003 (cellular component) or 0.05/8,972 = 0.000006 (biological process). The same analysis was conducted for the 202 genes with the strongest sQTLs and reported as genome-wide significant (P < 5.0 × 10−8) in the NHGRI GWAS catalog.
Supplementary Material
ACKNOWLEDGMENTS
This research was conducted in part using data and resources from the FHS of the National Heart, Lung, and Blood Institute (NHLBI) of the US National Institutes of Health (NIH) and the Boston University School of Medicine. The analyses reflect intellectual input and resource development from the FHS investigators participating in the SNP Health Association Resource (SHARe) project and in the Systems Approach to Biomarker Research in Cardiovascular Disease (SABRe) project.
We thank J. Zhu and Y. Yang at the DNA Sequencing and Genomics Core of the NHLBI for detailed review of our manuscript and helpful suggestions. We also thank J. Dupuis at the Boston University School of Public Health for her statistical suggestions.
This study used the high-performance computational capabilities of the Biowulf Linux cluster at the US NIH (http://biowulf.nih.gov/).
The FHS is funded by US NIH contract N01-HC-25195; this work was also supported by the NHLBI, Division of Intramural Research.
Footnotes
Accession codes. The expression levels for the 17,873 genes and 283,805 probe sets (exons) were deposited in NCBI dbGaP under the data set name expression dataset phe000002.v2 and the data set accession phs000363.v10.p8. SNP genotype data were previously deposited in dbGaP under the Framingham SNP Health Association Resource (SHARe) project.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Hindorff LA et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 106, 9362–9367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cookson W, Liang L, Abecasis G, Moffatt M & Lathrop M Mapping complex disease traits with global gene expression. Nat. Rev. Genet 10, 184–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Lee JA & Black DL Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci 8, 819–831 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Yeo G, Holste D, Kreiman G & Burge CB Variation in alternative splicing across human tissues. Genome Biol. 5, R74 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ET et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merkin J, Russell C, Chen P & Burge CB Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338, 1593–1599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coulombe-Huntington J, Lam KC, Dias C & Majewski J Fine-scale variation and genetic determinants of alternative splicing across individuals. PLoS Genet. 5, e1000766 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwan T et al. Heritability of alternative splicing in the human genome. Genome Res. 17, 1210–1218 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faustino NA & Cooper TA Pre-mRNA splicing and human disease. Genes Dev. 17, 419–437 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Nissim-Rafinia M & Kerem B The splicing machinery is a genetic modifier of disease severity. Trends Genet. 21, 480–483 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Kwan T et al. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet 40, 225–231 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Montgomery SB et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464, 773–777 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Battle A et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 24, 14–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F & Dietz HC Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet 36, 1073–1078 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Carninci P et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–1563 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Hunt R, Sauna ZE, Ambudkar SV, Gottesman MM & Kimchi-Sarfaty C Silent (synonymous) SNPs: should we care about them? Methods Mol. Biol 578, 23–39 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Carlini DB & Genut JE Synonymous SNPs provide evidence for selective constraint on human exonic splicing enhancers. J. Mol. Evol 62, 89–98 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Taggart AJ, DeSimone AM, Shih JS, Filloux ME & Fairbrother WG Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol 19, 719–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corvelo A, Hallegger M, Smith CW & Eyras E Genome-wide association between branch point properties and alternative splicing. PLoS Comput. Biol 6, e1001016 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keene JD & Tenenbaum SA Eukaryotic mRNPs may represent posttranscriptional operons. Mol. Cell 9, 1161–1167 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Jayaseelan S, Doyle F, Currenti S & Tenenbaum SA RIP: an mRNA localization technique. Methods Mol. Biol 714, 407–422 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Nicolae DL et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6, e1000888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welter D et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X et al. Genetic associations with expression for genes implicated in GWAS studies for atherosclerotic cardiovascular disease and blood phenotypes. Hum. Mol. Genet 23, 782–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graveley BR The haplo-spliceo-transcriptome: common variations in alternative splicing in the human population. Trends Genet. 24, 5–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nembaware V, Wolfe KH, Bettoni F, Kelso J & Seoighe C Allele-specific transcript isoforms in human. FEBS Lett. 577, 233–238 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Bondar TN & Kravchenko NA Cyclooxigenase-1 gene polymorphism and aspirin resistance. Tsitol. Genet 46, 66–72 (2012). [PubMed] [Google Scholar]
- 30.Licis N, Krivmane B, Latkovskis G & Erglis A A common promoter variant of the gene encoding cyclooxygenase-1 (PTGS1) is related to decreased incidence of myocardial infarction in patients with coronary artery disease. Thromb. Res 127, 600–602 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Zhang X et al. Synthesis of 53 tissue and cell line expression QTL datasets reveals master eQTLs. BMC Genomics 15, 532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinzen EL et al. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 6, e1 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhernakova DV et al. DeepSAGE reveals genetic variants associated with alternative polyadenylation and expression of coding and non-coding transcripts. PLoS Genet. 9, e1003594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium GTEx. The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawber TR, Kannel WB & Lyell LP An approach to longitudinal studies in a community: the Framingham Study. Ann. NY Acad. Sci 107, 539–556 (1963). [DOI] [PubMed] [Google Scholar]
- 36.Feinleib M, Kannel WB, Garrison RJ, McNamara PM & Castelli WP The Framingham Offspring Study. Design and preliminary data. Prev. Med 4, 518–525 (1975). [DOI] [PubMed] [Google Scholar]
- 37.Kannel WB, Feinleib M, McNamara PM, Garrison RJ & Castelli WP An investigation of coronary heart disease in families. The Framingham offspring study. Am. J. Epidemiol 110, 281–290 (1979). [DOI] [PubMed] [Google Scholar]
- 38.Splansky GL et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am. J. Epidemiol 165, 1328–1335 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Irizarry RA et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Willer CJ, Ding J, Scheet P & Abecasis GR MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol 34, 816–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lange K Mathematical and Statistical Methods for Genetic Analysis (Springer, 2002). [Google Scholar]
- 42.Ramasamy A et al. Resolving the polymorphism-in-probe problem is critical for correct interpretation of expression QTL studies. Nucleic Acids Res. 41, e88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tenenbaum SA, Lager PJ., Carson CC & Keene JD Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 26, 191–198 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Huang W, Sherman BT & Lempicki RA Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.