

Abstract
Diabetic kidney disease (DKD) has been the leading cause of chronic kidney disease for over 20 years. Yet, over these two decades, the clinical approach to this condition has not much improved beyond the administration of glucose-lowering agents, renin-angiotensin-aldosterone system blockers for blood pressure control, and lipid-lowering agents. The proportion of diabetic patients who develop DKD and progress to end-stage renal disease has remained nearly the same. This unmet need for DKD treatment is caused by the complex pathophysiology of DKD, and the difficulty of translating treatment from bench to bed, which further adds to the growing argument that DKD is not a homogeneous disease. To better capture the full spectrum of DKD in our design of treatment regimens, we need improved diagnostic tools that can better distinguish the subgroups within the condition. For instance, DKD is typically placed in the broad category of a non-inflammatory kidney disease. However, genome-wide transcriptome analysis studies consistently indicate the inflammatory signaling pathway activation in DKD. This review will utilize human data in discussing the potential for redefining the role of inflammation in DKD. We also comment on the therapeutic potential of targeted anti-inflammatory therapy for DKD.
Keywords: Diabetic nephropathies, Pathogenesis, Inflammation
INTRODUCTION
The number of immuno-oncology (IO) medications in the development pipeline in 2020 grew to 4,720, a 22% increase compared to 3,876 in 2019, and a 233% increase when compared to 2017 numbers [1]. Surprisingly the number of disease targets in the pipeline also jumped from 265 in 2017 to 504 in 2020. This quantum leap in IO medications is driven by progress in histological analysis. Over the last decade, the researchers have been able to secure the needed volume of data from tumor specimens and normal adjacent tissue during cancer operations to now proceed with multi-omics analysis in a substantial way. The disease-specific expression of IO targets even within the same cancer type has now become visible thanks to this technique, which in turn, can pave the way for a truly personalized oncological therapy.
Clinical findings such as the estimated glomerular filtration rate (eGFR) and the urinary albumin excretion (UAE) can be used to diagnose diabetic kidney disease (DKD) in diabetic patients without kidney biopsy [2-5]. These clinical indicators for DKD are helpful to recognize the DKD; however, it has limitations. A kidney biopsy can refine the diagnosis, but the existing diabetic nephropathy classification provides only an organ injury severity scale of the kidney [6], not indications of the various mechanisms involved in DKD pathogenesis. It is the reason that our understanding and treatment options for DKD are limited and DKD is main reason of renal replacement therapy [7-10].
DKD’s diverse pathophysiological pathways activate a wide range of intracellular signaling and transcriptional factors such as nuclear factor kappa-B (NF-κB), Janus kinase-signal transducers and activators of transcription (JAK/STAT), and mitogen- activated kinase (MAPK). Which and how these pathophysiological factors involve the progression of DKD at with which point in the sequence of events remain unclear. Though DKD arises from metabolic derangements, pathophysiological feature of DKD in diabetes patients is accompanied by chronic and sterile inflammation such as the observation of significant inflammatory cell infiltration in kidney biopsy results. Inflammation continues to present itself as an important pathophysiological factor that needs to be examined for its potential as a causative factor, not simply an insignificant byproduct. In this review, we will discuss the role of inflammation in DKD initiation and progression in human DKD. We also comment on the therapeutic potentials which targeting for anti-inflammatory therapy in DKD.
THE ROLE OF INFLAMMATION IN THE PATHOPHYSIOLOGY OF DKD
The pathogenesis of DKD is complex, and multiple pathways involve several years before clinical diagnosis of DKD. The pro-inflammatory and pro-fibrotic processes during DKD development and progression result from metabolic alterations, hyperfiltration, reactive oxidative stress (ROS), immune and inflammation activation, and subsequent fibrosis [11-15]. DKD usually is classified as a non-inflammatory glomerular disease; however, genome-wide transcriptome analysis studies consistently indicate the strong presence of inflammatory signaling pathways [16]. This evidence is supported by recent single nucleus RNA sequencing results with type 2 diabetes (T2D) patient’s kidney biopsy specimens. Diabetic kidney had an approximate 7- to 8-fold increase in leukocytes compared to controls, and among total 347 immune cells consisted of 49% T cells, 21% B cells, 23% monocytes, and 7% plasma cells. The expression of TNFRSF21 was upregulated in the infiltrating diabetic CD14+ monocyte subset, interleukin 1 receptor type 1 (IL1R1) was increased in CD16+ monocytes and antigen-presenting cells, and interleukin 18 receptor 1 (IL18R1) was increased in CD4+ and CD8+ T cells [17]. Sodium-glucose cotransporter 2 (SGLT2) inhibitors and incretin therapies, including glucagon-like peptide 1 receptor (GLP-1R) agonists and dipeptidyl peptidase 4 (DPP4)-inhibitors, also act not only by lowering glucose level, but also by blocking the mechanism of kidney injury related with innate immune cell activation and inflammatory response [18-21]. To better understand the role of inflammation in the pathophysiology of DKD, we approach immune activation in DKD patients, including innate and adaptive immune responses. We also discuss the inflammatory response in DKD from human studies (Fig. 1).
Figure 1.

Activation of Inflammation in diabetic kidney disease. Activation of Inflammation in diabetic kidney disease. Various inflammatory factors such as pattern recognition receptor (PRR), inflammatory cytokines, chemokines, innate immune cells, complement pathways, and adaptive immune cells are linked to activate the immune and inflammatory responses in diabetic kidney disease. ATP, adenosine triphosphate; AGE, advanced glycation end product; CSF-1, colony-stimulating factor-1; TLR, Toll-like receptor; NLRP, nucleotide-binding oligomerization domain-, leucine rich repeat-, and pyrin domain-containing; IL, interleukin; TNF-α, tumor necrosis factor α; IFN-γ, interferon-γ; TGF-β, transforming growth factor-β; EC, endothelial cell; TEC, tubular epithelial cell; CCL, C-C motif chemokine ligand; CX3CL1, C-X3-C motif chemokine 1; eGFR, estimated glomerular filtration rate.
Pattern recognition receptors
Toll-like receptors
Membrane Toll-like receptors (TLRs) and cytoplasmic Nod-like receptors (NLRs) are the two major sensors of immune cells that have a pivotal role in initiating the innate immune response by sensing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). TLRs are expressed in various immune cells such as macrophages, dendritic cells, T cells, B cells, and natural killer cells. TLRs also are expressed on non-immune cells, including kidney tubular epithelial cells, endothelial cells, podocytes, and mesangial cells [22]. TLR1, 2, 4, 5, and 6 are located on the cell surface and detect PAMPs and DAMPs such as bacterial lipopolysaccharide, lipopeptides, flagellin, bacterial DNA, double-stranded RNA, and high-mobility group box 1 proteins [23]. Once triggered, TLRs signal via MyD88/interleukin (IL) receptor-associated kinases 1 and 4, activating MAPK, NF-κB, and interferon regulatory factor. In DKD patients, TLR2 and TLR4 have been reported to contribute to the inflammatory pathogenesis. Systemic monocytes have higher expression levels of TLR2 and TLR4 in type 1 diabetes (T1D) and T2D patients, and these TLR expressions are correlated positively with hemoglobin A1C and insulin resistance levels [24-26]. Renal tubular TLR4 is overexpressed and associated with interstitial macrophage infiltration in T2D patients [27]. This tubular TLR4 expression is correlated negatively with renal function. Glomerular TLR4 is increased in T2D patients with UAE or overt DKD and has prognostic value in chronic kidney disease (CKD) progression [28].
Nod-like receptors
NLRs are expressed in the cytoplasm and sense intracellular PAMPs and DAMPs. NLRs, including nucleotide-binding oligomerization domain-, leucine rich repeat-, and pyrin domain-containing 1 (NLRP1), NLRP3, NLRP6, NLRP12, and NLRC4 (C for CARD, caspase activation and recruitment domain), oligomerize to form inflammasome complexes with an adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase-activating recruitment domain) and an effector protein, pro-caspase 1. These initiate inflammatory cascades that lead to activation of caspase 1 and secretion of IL-1β and IL-18 [29]. NLRP3 is the best studied example of NLRs in diabetes and DKD. Hyperglycemia and its related metabolic rearrangement could act as DAMPs detected by NLRP3 [30,31]. NLRP3 is activated by several ligands, including fatty acid, uric acid, uromodulin, extracellular adenosine triphosphate, hyperglycemia, serum amyloid A, and mitochondrial reactive oxygen species [32-35]. Renal resident mononuclear cells (MNCs), such as macrophages and dendritic cells, contain all parts of the NLRP3 inflammasome and are able to secrete mature pro-inflammatory cytokines; therefore, renal MNCs undergo caspase-1-dependent pyroptosis [36,37]. In addition to MNCs, non-immune cells in the kidney including tubular epithelial cells, podocytes, glomerular endothelial cells, and mesangial cells contain a substantial amount of NLRP3 [30,38-40]. A beautiful study evaluating the role of NLRP3 in non-hematopoietic cells undergoing chimeric bone marrow transplantation revealed that NLRP3 is important in exacerbating diabetic nephropathy [38]. The transcripts of NLRP3 inflammasome components and pro-inflammatory cytokines increased in systemic monocytes of patients with T2D, and these transcript levels are attenuated by metformin treatment [41,42]. In the human diabetic kidney, glomerular mRNA for NLRP3, caspase-1, IL-1β, and IL-18 was increased [43]. Podocytes and endothelial cells were identified as the primary source of IL-1β in glomerulosclerosis in human DKD. Interestingly, inhibition of IL-1β by anakinra, an IL-1 receptor antagonist, to prevent gout attacks improved serum creatinine in a small number of enrolled diabetic patients with moderate to severe CKD [44].
Innate immune cells
Briefly, macrophages and dendritic cells infiltrate into the diabetic kidney, and the number of macrophages and dendritic cells correlated with histologic damage and clinical measures such as UAE and kidney function decline [45,46]. We face critical questions in assessment of this process. First, how are these innate immune cells activated by metabolic alteration and glomerular hyperfiltration under diabetic conditions? The inflammatory response could be induced by multiple aberrant metabolic products in diabetes, including hyperglycemia-induced cell death, mitochondrial ROS, hyperuricemia, and lipid metabolites that serve as DAMPs sensed by TLRs and NLRs to trigger innate immune cells [47,48]. Dysregulated metabolic products could evoke immune cells directly and affect various non-immune cells in the kidney to secrete chemokines and cytokines or recruit immune cells indirectly. The second question involves the source of expanded innate immune cells in the diabetic kidney. The proliferation and maintenance of MNCs require colony-stimulating factor-1 (CSF-1), which acts exclusively through the c-fms receptor. The serum level of macrophage CSF is elevated in hemodialysis patients [49], and CSF-1 is secreted from proximal tubular epithelial cells in the CKD model, including DKD [50-52]. Given these findings, increased systemic and renal CSF-1 in diabetes contributes to the proliferation and activation of renal MNCs. Homing of circulating macrophages to kidneys also is an issue in DKD. The response to increased expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and monocyte chemoattractant protein 1 (MCP-1)/C-C motif chemokine ligand 2 (CCL2) is reported to promote immune cell infiltration in the diabetic kidney [53-55]. The last consideration is the diversity of innate immune cells and the difficulty in defining the status of immune cells. The renal MNCs population is not divided simply as macrophages and dendritic cells since classic, specific surface markers such as CD11b, F4/80, and CD68 for macrophages or CD11c, myosin heavy chain II, and CD80/86 for dendritic cells are co-expressed on renal MNCs. This suggests difficulty in differentiating function between macrophages and dendritic cells. This is because the in vivo environment is regulated by complex and plastic chemokines, cytokines, and intercellular cross-talking. Sub-identification and functional characterization of human renal MNCs are limited by lack of information on specific receptors for the differentiating subset. This creates an obstacle for developing the treatment target for renal and systemic MNCs in DKD [56].
Inflammatory cytokines
Inflammatory cytokines are polypeptide molecules produced by immune cells, endothelial cells, epithelial cells, and fibroblasts in autocrine, paracrine, and juxtacrine manners. IL-1, IL-18, IL-6, tumor necrosis factor (TNF), and IL-17 are major pro-inflammatory cytokines that have been studied in DKD development and progression [57,58]. The sterile inflammatory response is dependent on IL-1α signaling via the IL-1 receptor. IL-1β and IL-18 amplify the immune response by acting as costimulatory molecules for T cells and B cells. Urinary and plasma IL-1 levels in T2D patients are associated with podocyte and proximal tubular epithelial cell injury markers [59]. IL-18 is a member of the IL-1 superfamily and stimulates interferon-γ (IFN-γ) release and modulates innate and adaptive immune cells. Both serum and urinary levels of IL-18 were significantly increased in type 2 DKD patients [60,61]. Renal tubular IL-18 expression was increased in type 2 DKD patients, and this overexpression was induced by transforming growth factor-β-mediated MAPK pathway activation [62]. Serum IL-18 level in Japanese diabetic patients with normoalbuminuria showed a predictive role for developing albuminuria and rapid loss of eGFR [63].
IL-6 acts as a costimulatory molecule and acute phase reactant in targeting T cells and B cells for activation. Serum level of IL-6 is increased in patients with DKD [64,65] and correlates with glomerular basement membrane width in diabetic glomerulopathy [66]. In a cohort of patients with T2D, circulating IL-6 level correlated with atherosclerotic changes, and urinary IL-6 was associated with DKD progression [67]. Urinary inflammatory cytokines, especially IL-6 and IL-10, can assist in identifying DKD in T2D patients, even in the absence of UAE [68]. IL-6 mRNA is expressed on glomerular cells and interstitial cells in type 2 DKD patients, and its expression might be associated with mesangial proliferation and involvement in renal injury [69].
TNF-α has multiple pro-inflammatory actions in DKD, including induction and differentiation of inflammatory cells, cytotoxicity to kidney cells, activation of apoptosis, altered glomerular hemodynamics, increased vascular endothelial permeability, and increased oxidative stress. TNF-α activates biological signaling through TNFR1 and TNFR2. In patients with DKD, the whole-genome DNA methylation maps for human kidney highlighted coordinated changes in immune signaling through methylation and gene expression, including that of TNF-α, and changes in TNF methylation correlated with kidney function decline [70]. In patients with DKD, serum and urinary concentrations of TNF are significantly elevated and closely correlate with the UAE [71,72]. Glomerular TNF expression, but neither tumor necrosis factor receptor 1 (TNFR1) nor TNFR2 expression, inversely correlated with eGFR in DKD patients enrolled in the NEPTUNE study [73]. Intriguingly, serum TNF levels did not correlate with glomerular TNF expression suggests intrarenal TNF production. In a cohort of normo- and micro-albuminuric T1D patients (the 2nd Joslin Kidney Study), serum soluble TNFR1 and TNFR2 were associated strongly with decreased eGFR, independent of UAE [74]. Cohorts of incident type 2 DKD (ACCORD) and progressive type 2 DKD (VA-Nephron-D) revealed associations of doubling in TNFR1 and TNFR2 with kidney injury molecule-1 (KIM-1) levels; risk of adverse renal outcomes was significant for both cohorts [75].
IL-17 is the key cytokine produced by CD4+ IL-17+ cells known as T helper 17 cells and binds to the IL-17 receptor. IL-17 plays a critical role in clearance of bacterial and fungal infection. Dysregulated IL-17 in autoimmune disease activates several signaling cascades that lead to induction of IL-6, TNF-α, CCL2, and CCL5. Acting as chemoattractants, these cytokines and chemokines recruit immune cells, such as monocytes and neutrophils, to the site of inflammation. The role of IL-17 has been studied well in T1D development. Patients with T1D present with elevated plasma level of IL-17, increased circulating IL-17-producing T cells, and islet antigen-specific Th17 cells [76]. Plasma IL-17 level decreased with progression from normal glucose tolerance to T2D with DKD [77]. However, CD4+ IL-17+ T cells were found in T2D patient kidneys, and the number of CD4+ IL-17+ T cells was correlated positively with deterioration in eGFR [78]. In this aspect, systemic and local IL-17 production can differ in DKD.
Chemokines and their receptors
The name chemokine derives from the ability of these small cytokines or signaling proteins to recruit cells by chemotaxis. Chemokines exert biological effects by binding receptors on the surface of target cells. Chemokines are activated in non-immune kidney cells in response to hemodynamic and metabolic alteration and play a critical role in inflammatory cell recruitment, migration, and cellular adhesion in DKD [79]. CCL2 (MCP-1), CCL5 (RANTES), and C-X3-C motif chemokine 1 (CX3CL1, Fractalkine) have been studied as major pro-inflammatory chemokines in DKD. CCL2, which is produced by renal tubular epithelial cells and podocytes in diabetic kidneys, recruits mononuclear cells and memory T cells to the sites of inflammation [80]. We investigated the impact of CCL2 polymorphism (-2518 A/G genotype, A carriage) on DKD progression in T2D patients [81]. In logistic regression analysis, the carriage of the A allele retained a significant association with progression to end-stage renal disease (ESRD). Urine CCL2 level in T2D patients was significantly higher than that in healthy adults and gradually increased with CKD stage [82,83]. Tubulointerstitial CCL2 expression significantly increased in T2D kidneys. Moreover, urinary level of CCL2 was well correlated with the number of CD68-positive infiltrating cells in the interstitium. In contrast, serum CCL2 level remained similar to those of healthy volunteers. The CCL2 receptor, C-C chemokine receptor 2 (CCR2), is expressed on MNCs and differentiated podocytes. CCL2 exerts influence on the actin cytoskeleton through CCR2 on podocytes, affecting foot process effacement and subsequent albuminuria development [84,85]. CCL5 recruits monocytes and T cells and plays an active role in homing leukocytes into inflammatory sites. In the Finnish Diabetes Prevention Study, progression to T2D in overweight individuals and in those with impaired glucose tolerance was significantly higher in subjects with the highest RANTES concentration and lower in subjects with the highest macrophage migration inhibitory factor levels [86]. Biopsy specimens from patients with T2D and overt nephropathy showed strong upregulation of CCL2 and CCL5, mainly in tubular cells. There was a strong correlation between the expression of these chemokines and NF-κB activation in the same cells [87]. CX3CL1-CX3CR1 axis activation in T2D patients has been studied in relation to atherogenic chemokines since they are associated with angiogenesis, cardiovascular mortality, and monocyte adhesion to adipocytes [88,89]. Plasma CX3CL1 level was significantly higher in T2D patients compared with non-diabetics [90,91]. In addition, plasma CX3CL1 level correlated positively with inflammatory chemokines and cytokines in T2D patients.
Complement system
The complement system is a potent effector in the innate immune system and is involved in various infection and inflammatory diseases such as autoimmune disease, atypical hemolytic uremic syndrome, and paroxysmal nocturnal hematuria. The complement system is constituted of many soluble and membrane-bound proteins, mostly proteases that respond to danger signals and generate excess immune effectors. In the kidney, proximal tubular epithelial cells express complement C3 and membrane-bound C3 convertase, which could activate intrarenal complement pathway in various kidney diseases [92-94].
Two critical roles of the complement pathway in DKD pathogenesis are activation of the lectin pathway in response to glycated proteins on the cell surface and dysfunctional complement regulatory proteins by glycation under long-lasting hyperglycemia [95]. A cohort study of 95,202 individuals from the general population followed for 10 years revealed that high baseline concentration of complement C3 was associated with increased risk of diabetic retinopathy, nephropathy, and neuropathy [96]. Differential transcriptome analysis of early diabetic kidneys and matched non-diabetic controls revealed five distinct canonical pathways. Among these pathways, the complement pathway was most significantly changed in early DKD [97]. In a cohort of 326 T2D patients followed for 15 years, higher mannose-binding lectin (MBL) value was associated with a 2.6 hazard ratio for development of proteinuria and kidney function decline [98]. The same group published data from 1,564 T1D patients with a median follow-up of 5.8 years; MBL values significantly correlated with UAE and predicted the onset of end-stage kidney disease [99]. Urine samples from T2D patients with proteinuria were analyzed using targeted mass spectrometry and showed a low risk of ESRD with high urine CD59, an inhibitor of terminal complement complex formation (hazard ratio [HR], 0.50; 95% confidence interval [CI], 0.29 to 0.87) [100]. In biopsy specimens from T2D patients, C1q deposits were associated with interstitial fibrosis and tubular atrophy (IFTA), interstitial inflammation, and vascular lesions. In comparison, patients with C3c deposits scored high in IFTA and global sclerosis [101]. Patients with C1q deposition had a significantly higher urinary protein level and significantly lower eGFR. Renal tubular C5a expression increased in DKD patients, and the expression intensity of C5a correlated with DKD progression [102].
Adaptive immune cells
T cell and B cell infiltration in the diabetic kidney has not been observed as widely as innate immune cell infiltration. Recent study results have shed light on the role of adaptive immune cells in metabolic disease, including DKD; particularly, increase of Th1 and Th17 cells and decrease of Treg cells in response to hyperglycemia are distinct features of adaptive immune cells in DKD. Enhanced activation of circulatory Th1 and suppressed Th2 profiles were reported in patients with T2D with proteinuria [103]. In people with T2D, circulating Th1 and Th17 increased proportional to albuminuria. Serum cytokines characteristic of Th1 (IFN-γ, TNF-α, IL-2) and Th17 (IL-17) subsets were increased in people with DKD in correlation with albuminuria [104]. We demonstrated that marked increase of CD4+, CD8+, and CD20+ cells in the glomerular and tubulointerstitial area and the number of CD4+ and CD20+ cells correlated with the amount of proteinuria in T2D patients (Fig. 2) [105]. Juxtaglomerular T cell infiltration appears to play a role in hemodynamic alteration in T1D related with local angiotensin system activity [106,107].
Figure 2.
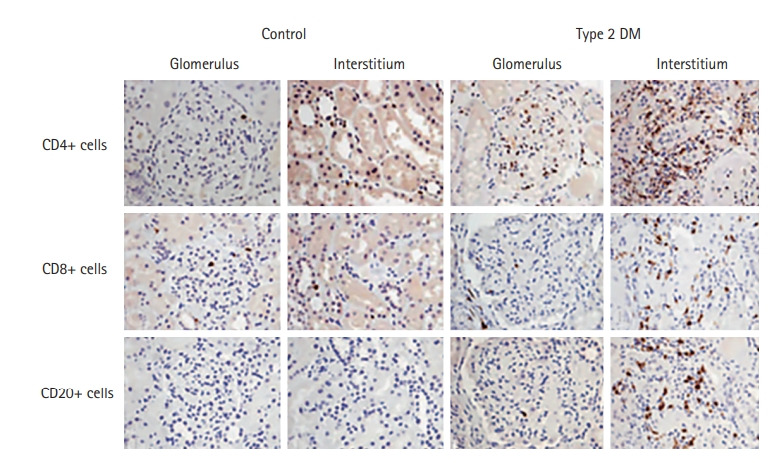
Kidney lymphocytes infiltration in type 2 diabetic human kidneys. Immunostaining for CD4+, CD8+ T cells, and CD20+ cells in control patients and type 2 diabetes mellitus (DM) patients. In comparison, there is significant infiltration of CD4+, CD8+T cells, and CD20+ cells to interstitium in diabetic kidney. Adapted from Moon et al., with permission from Karger Publishers [105].
NEW TRIALS FOR REGULATING INFLAMMATION IN DKD
Pentoxifylline (PTX) is a methylxanthine derivative with diverse effects, including preventing platelet aggregation, improving blood flow, and immune modulation [108,109]. The mechanism of action of PTX is inhibiting phosphodiesterases 3 and 4 that primarily regulate intracellular secondary messenger cyclic adenosine monophosphate (cAMP). Increased cAMP by PTX activates protein kinase A, leading to attenuated production of inflammatory cytokines including IL-1, IL-6, and TNF-α [110,111]. The PREDIAN trial [112] evaluated the renoprotective effects of PTX and renin-angiotensin system (RAS) blockade therapy in 169 white patients with T2D, stage 3 or 4 CKD, and UAE greater than 30 mg/day for 2 years. The PTX (1,200 mg/day) treatment group had decreased proteinuria and urinary concentration of TNF-α and slowed eGFR decline (2.1 mL/min/1.73 m2 in the PTX group vs. 6.5 mL/min/1.73 m2 in the placebo group). The unresolved questions of PTX clinical trials stem from the small number of patients and short study duration. The hard renal outcome, ESRD or renal death, also was not evaluated. An ongoing clinical trial with PTX (NCT03625648) targets high-risk patients according to the “heat map” of the Kidney Disease Improving Global Outcomes in patients with T2D and CKD stage 3 or 4 [113]. The primary outcome of this study is time to ESRD or all-cause mortality in 2,510 enrolled patients. The results could answer questions regarding the current issues of PTX in DKD (Table 1).
Table 1.
Targeting inflammation as therapeutics in human diabetic kidney disease
| Agent, Clinical trial stage | Mechanism of action | Inclusion criteria/study duration | Primary outcome | Results |
|---|---|---|---|---|
| Pentoxifylline, Phase 4 (NCT03625648) | Non-specific phosphodiesterase 3 and 4 inhibitor | Type 2 DM | Time to ESRD or death | On going |
| eGFR 15–60 mL/min/1.73 m2 | ||||
| Barticinib, Phase 2 (NCT01683409) | JAK-1,-2 inhibitor | Type 2 DM | Change from baseline in UACR at week 24 | Barcitinib decreased albuminuria by 20%–30% compared with placebo. |
| eGFR 20–70 mL/min/1.73 m2 and UACR 300–5,000 mg/g/24 weeks | ||||
| Serinsertib (GS-4997), Phase 2 (NCT02177786) | Selective, small molecule, ASK1 inhibitor | Type 2 DM | Change in eGFR from baseline at week 48 | Mean eGFR for selonsertib and placebo groups did not differ significantly at 48 weeks. After a post hoc analysis, rate of eGFR decline was reduced 71% for the 18 mg group compared with placebo between 4 and 48 weeks. |
| 15 ≤ eGFR < 60 mL/min/1.73 m2 and UACR > 100 mg/g/48 weeks | ||||
| Ematicap pegol (NOX-E36), Phase 2 (NCT01547897) | Anti-human CCL2 blocking aptamer | Type 2 DM | Effect of NOX-E36 on albuminuria as measured by UACR | After 12 weeks, albuminuria reduction by 29% relative to baseline, but no significant difference with placebo. A post hoc analysis, increased the UACR difference between the two treatment arms to 32% at week 12. |
| eGFR > 25 mL/min/1.73 m2 and UACR > 100 mg/g/12 weeks | ||||
| CCX 140-B, Phase 2 (NCT01447147) | Selective CCR2 inhibitor | Type 2 DM | Evaluate the safety and tolerability of CCX140-B | Adverse events rate are not higher in CCX 140-B compared to placebo. CCX 140-B decrease albuminuria of 18% as compared to the placebo group. |
| eGFR ≥ 25 mL/min/1.73 m2 and UACR 100–3,000 mg/g/52 weeks | ||||
| ASP8232, Phase 2 (NCT02358096) | VAP-1 inhibitor | Type 2 DM/ | Mean change of log transformed UACR from baseline to end of treatment | UACR decreased by 17.7% in the ASP8232 group and increased by 2.3% in the placebo group. |
| 25 ≤ eGFR < 75 mL/min/1.73 m2 and UACR 200–3,000 mg/g/12 weeks |
DM, diabetes mellitus; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; JAK, Janus kinase; UACR, urinary albumin to creatinine ratio; ASK1, apoptosis signal-regulating kinase 1; CCL2, C-C motif chemokine ligand 2; CCR2, C-C chemokine receptor 2; VAP-1, vascular adhesion protein-1.
The effects of barcitinib, a selective JAK-1 and -2 inhibitor, on proteinuria in T2D patients were evaluated. Barcitinib reduced the proteinuria in combination with RAS inhibitor treatment in a phase 2 study over 24 weeks. In a dose-dependent manner, barcitinib decreased albuminuria by 20% to 30% compared with placebo with no eGFR change [114]. Inflammatory biomarkers such as urine CCL2, serum TNFR1, TNFR2, vascular cell adhesion molecule (VCAM-1), VCAM-2, and serum amyloid A level were decreased in the barcitinib treatment group.
Apoptosis signal-regulating kinase 1 (ASK1) is a stress-responsive mitogen-activated protein kinase kinase kinase that signals through a cascade of downstream kinases including p38 and c-Jun N-terminal kinase. ASK1 regulates target gene expression including genes for inflammatory cytokines [115,116]. Selonsertib is a selective, small molecule ASK1 inhibitor that has been evaluated for efficacy in preventing eGFR decline in T2D patients over 48 weeks. Mean eGFR for the two groups did not differ significantly at 48 weeks; however, clinical researchers found that selonsertib inhibited creatinine secretion. After a post hoc analysis, the rate of eGFR decline was reduced 71% for the 18 mg group compared with the placebo group between 4 and 48 weeks (difference 3.11 ± 1.53 mL/min/1.73 m2 annualized over 1 year; 95% CI, 0.10 to 6.13; nominal p = 0.043). The UAE did not differ between selonsertib treatment and the placebo group [117].
CCL2 has been investigated as the anti-inflammatory target in DKD. Emapticap pegol (NOX-E36), the anti-human CCL2 blocking aptamer, specifically binds and inhibits the pro-inflammatory cytokine CCL2.
Emapticap was evaluated in a phase 2a study to confirm the safety and tolerability, as well as, the renoprotective and anti-diabetic effect in T2D patients with albuminuria taking RAS inhibitors treatment. After 12 weeks, albuminuria was reduced by 29% relative to baseline (p < 0.05) but with no significant difference from placebo [118]. A post hoc analysis excluding patients with major protocol violations showed increased UAE difference between the two treatment arms to 32% at 12 weeks (p = 0.014) and 39% at 20 weeks (p = 0.010).
CCX140-B is a selective inhibitor of CCR2 that was evaluated for an effect on proteinuria in T2D patients with albuminuria. This inhibitor was taken in combination with RAS inhibitors in a phase 2 study for 52 weeks. UAE changes from baseline during 52 weeks were –2% for placebo, –18% for 5 mg CCX140-B, and –11% for 10 mg CCX140-B. The albuminuria-lowering treatment effect persisted throughout the 52 weeks of study [119].
Vascular adhesion protein-1 (VAP-1) is an endothelial surface sialoglycoprotein that has two functions. VAP-1 is induced under inflammatory conditions and acts as an adhesion molecule for granulocyte and lymphocyte trafficking [120,121]. VAP-1, also known as amine oxidase copper-containing 3, possesses mono-amine oxidase activity and interacts with leukocyte adhesion molecules including Siglec-9 and -10. These are active in endothelial cells of vascularized tissues in the kidney. The soluble end products of its enzymatic cleavage are hydrogen peroxide and reactive aldehydes that lead to protein cross-linking and eventual oxidative stress [122]. ASP8232 is a small molecule inhibitor of VAP-1 evaluated for its effect on reducing albuminuria in T2D patients over a 12-week phase 2 study period [123]. UAE decreased by 17.7% in the ASP8232 group and increased by 2.3% in the placebo group; the placebo-adjusted difference between groups was –19.5% (95% CI, –34.0 to –1.8; p = 0.033).
CONCLUSIONS
Agents targeting inflammation in combination with RAS blockade therapy are a trend in recent clinical trials for DKD [3,124]. However, the difficulty in selecting appropriate patients who can benefit from anti-inflammatory treatment undermines most attempts to properly evaluate this treatment strategy. Additionally, not all DKD patients benefit from JAK-1, -2 inhibitor or anti-human CCL2 blocking aptamer. Some specific populations at certain stages of DKD respond efficiently to those treatments; however, the current clinical diagnostic approaches do not select for such patients. We need new biomarkers that determine the inflammation threshold for predicting progression to DKD and serve as indicators of DKD onset even in early diabetic patients. We summarized the DKD human data related to inflammation in this review. The evidence level is low due to the small number of patients studied. Integrating genetic data with kidney tissue level transcriptomic and proteomic data will be a powerful tool to search for biomarkers and treatment candidates. Based on this viewpoint, we hope future studies will expand and refine our understanding of the development and progression of DKD, so as to ultimately reduce the need for renal replacement therapy in DKD.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (Grant 2018R1D1A1B05049016).
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Upadhaya S, Hubbard-Lucey VM, Yu JX. Immuno-oncology drug development forges on despite COVID-19. Nat Rev Drug Discov. 2020;19:751–752. doi: 10.1038/d41573-020-00166-1. [DOI] [PubMed] [Google Scholar]
- 2.National Kidney Foundation KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis. 2012;60:850–886. doi: 10.1053/j.ajkd.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 3.American Diabetes Association Standards of medical care in diabetes: 2020. Abridged for primary care providers. Clin Diabetes. 2020;38:10–38. doi: 10.2337/cd20-as01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim K, Lee SH, Lee SW, Lee JP, Chin HJ; Korean GlomeruloNEphritis sTudy (KoGNET) Group. Current findings of kidney biopsy including nephropathy associated with hypertension and diabetes mellitus in Korea. Korean J Intern Med. 2020;35:1173–1187. doi: 10.3904/kjim.2020.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh KH, Kang M, Kang E, et al. The KNOW-CKD study: what we have learned about chronic kidney diseases. Kidney Res Clin Pract. 2020;39:121–135. doi: 10.23876/j.krcp.20.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tervaert TW, Mooyaart AL, Amann K, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 7.Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12:2032–2045. doi: 10.2215/CJN.11491116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park S, Kim M, Kim JE, et al. Characteristics of kidney transplantation recipients over time in South Korea. Korean J Intern Med. 2020;35:1457–1467. doi: 10.3904/kjim.2019.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoo JS, Choe EY, Kim YM, Kim SH, Won YJ. Predictive costs in medical care for Koreans with metabolic syndrome from 2009 to 2013 based on the National Health Insurance claims dataset. Korean J Intern Med. 2020;35:936–945. doi: 10.3904/kjim.2016.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yim T, Kim SU, Park S, et al. Patterns in renal diseases diagnosed by kidney biopsy: a single-center experience. Kidney Res Clin Pract. 2020;39:60–69. doi: 10.23876/j.krcp.19.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JY, Yang JW, Han BG, Choi SO, Kim JS. Adiponectin for the treatment of diabetic nephropathy. Korean J Intern Med. 2019;34:480–491. doi: 10.3904/kjim.2019.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugahara M, Pak WL, Tanaka T, Tang SC, Nangaku M. Update on diagnosis, pathophysiology, and management of diabetic kidney disease. Nephrology (Carlton) 2021;26:491–500. doi: 10.1111/nep.13860. [DOI] [PubMed] [Google Scholar]
- 13.Honda T, Hirakawa Y, Nangaku M. The role of oxidative stress and hypoxia in renal disease. Kidney Res Clin Pract. 2019;38:414–426. doi: 10.23876/j.krcp.19.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhatia D, Capili A, Choi ME. Mitochondrial dysfunction in kidney injury, inflammation, and disease: potential therapeutic approaches. Kidney Res Clin Pract. 2020;39:244–258. doi: 10.23876/j.krcp.20.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasegawa S, Inoue T, Inagi R. Neuroimmune interactions and kidney disease. Kidney Res Clin Pract. 2019;38:282–294. doi: 10.23876/j.krcp.19.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson PC, Wu H, Kirita Y, et al. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci USA. 2019;116:19619–19625. doi: 10.1073/pnas.1908706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kodera R, Shikata K, Kataoka HU, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54:965–978. doi: 10.1007/s00125-010-2028-x. [DOI] [PubMed] [Google Scholar]
- 19.Katagiri D, Hamasaki Y, Doi K, et al. Protection of glucagon-like peptide-1 in cisplatin-induced renal injury elucidates gut-kidney connection. J Am Soc Nephrol. 2013;24:2034–2043. doi: 10.1681/ASN.2013020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita H, Morii T, Fujishima H, et al. The protective roles of GLP-1R signaling in diabetic nephropathy: possible mechanism and therapeutic potential. Kidney Int. 2014;85:579–589. doi: 10.1038/ki.2013.427. [DOI] [PubMed] [Google Scholar]
- 21.Heerspink HJ, Perco P, Mulder S, et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia. 2019;62:1154–1166. doi: 10.1007/s00125-019-4859-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin M, Tang SC. Toll-like receptors: sensing and reacting to diabetic injury in the kidney. Nephrol Dial Transplant. 2014;29:746–754. doi: 10.1093/ndt/gft446. [DOI] [PubMed] [Google Scholar]
- 23.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased Toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. 2008;93:578–583. doi: 10.1210/jc.2007-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Devaraj S, Dasu MR, Park SH, Jialal I. Increased levels of ligands of Toll-like receptors 2 and 4 in type 1 diabetes. Diabetologia. 2009;52:1665–1668. doi: 10.1007/s00125-009-1394-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dasu MR, Devaraj S, Park S, Jialal I. Increased Toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33:861–868. doi: 10.2337/dc09-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin M, Yiu WH, Wu HJ, et al. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol. 2012;23:86–102. doi: 10.1681/ASN.2010111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verzola D, Cappuccino L, D’Amato E, et al. Enhanced glomerular Toll-like receptor 4 expression and signaling in patients with type 2 diabetic nephropathy and microalbuminuria. Kidney Int. 2014;86:1229–1243. doi: 10.1038/ki.2014.116. [DOI] [PubMed] [Google Scholar]
- 29.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Niemir ZI, Stein H, Dworacki G, et al. Podocytes are the major source of IL-1 alpha and IL-1 beta in human glomerulonephritides. Kidney Int. 1997;52:393–403. doi: 10.1038/ki.1997.346. [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Li Y, Fan J, et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017;8:e2937. doi: 10.1038/cddis.2017.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 2013;191:5230–5238. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kersse K, Bertrand MJ, Lamkanfi M, Vandenabeele P. NOD-like receptors and the innate immune system: coping with danger, damage and death. Cytokine Growth Factor Rev. 2011;22:257–276. doi: 10.1016/j.cytogfr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 35.Shah A, Xia L, Goldberg H, Lee KW, Quaggin SE, Fantus IG. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J Biol Chem. 2013;288:6835–6848. doi: 10.1074/jbc.M112.419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ludwig-Portugall I, Bartok E, Dhana E, et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2019;90:525–539. doi: 10.1016/j.kint.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Lau A, Chung H, Komada T, et al. Renal immune surveillance and dipeptidase-1 contribute to contrast-induced acute kidney injury. J Clin Invest. 2018;128:2894–2913. doi: 10.1172/JCI96640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shahzad K, Bock F, Dong W, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87:74–84. doi: 10.1038/ki.2014.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SM, Kim YG, Kim DJ, et al. Inflammasome-independent role of NLRP3 mediates mitochondrial regulation in renal injury. Front Immunol. 2018;9:2563. doi: 10.3389/fimmu.2018.02563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Boini KM, Xia M, et al. Activation of NOD-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension. 2012;60:154–162. doi: 10.1161/HYPERTENSIONAHA.111.189688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;11:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martini S, Eichinger F, Nair V, Kretzler M. Defining human diabetic nephropathy on the molecular level: integration of transcriptomic profiles with biological knowledge. Rev Endocr Metab Disord. 2008;9:267–274. doi: 10.1007/s11154-008-9103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balasubramaniam G, Almond M, Dasgupta B. Improved renal function in diabetic patients with acute gout treated with anakinra. Kidney Int. 2015;88:195–196. doi: 10.1038/ki.2015.125. [DOI] [PubMed] [Google Scholar]
- 45.Furuta T, Saito T, Ootaka T, et al. The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis. 1993;21:480–485. doi: 10.1016/s0272-6386(12)80393-3. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006;11:226–231. doi: 10.1111/j.1440-1797.2006.00576.x. [DOI] [PubMed] [Google Scholar]
- 47.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 48.Har R, Scholey JW, Daneman D, et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/ chemokines in patients with uncomplicated type 1 diabetes mellitus. Diabetologia. 2013;56:1166–1173. doi: 10.1007/s00125-013-2857-5. [DOI] [PubMed] [Google Scholar]
- 49.Nitta K, Akiba T, Kawashima A, et al. Serum levels of macrophage colony-stimulating factor and aortic calcification in hemodialysis patients. Am J Nephrol. 2001;21:465–470. doi: 10.1159/000046650. [DOI] [PubMed] [Google Scholar]
- 50.Moore KJ, Yeh K, Naito T, Kelley VR. TNF-alpha enhances colony-stimulating factor-1-induced macrophage accumulation in autoimmune renal disease. J Immunol. 1996;157:427–432. [PubMed] [Google Scholar]
- 51.Jang MH, Herber DM, Jiang X, et al. Distinct in vivo roles of colony-stimulating factor-1 isoforms in renal inflammation. J Immunol. 2006;177:4055–4063. doi: 10.4049/jimmunol.177.6.4055. [DOI] [PubMed] [Google Scholar]
- 52.Lim AK, Ma FY, Nikolic-Paterson DJ, Thomas MC, Hurst LA, Tesch GH. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia. 2009;52:1669–1679. doi: 10.1007/s00125-009-1399-3. [DOI] [PubMed] [Google Scholar]
- 53.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol. 2005;16:1711–1722. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 54.Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–480. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- 55.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 56.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol. 2012;23:194–203. doi: 10.1681/ASN.2011070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 58.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 59.Milas O, Gadalean F, Vlad A, et al. Pro-inflammatory cytokines are associated with podocyte damage and proximal tubular dysfunction in the early stage of diabetic kidney disease in type 2 diabetes mellitus patients. J Diabetes Complications. 2020;34:107479. doi: 10.1016/j.jdiacomp.2019.107479. [DOI] [PubMed] [Google Scholar]
- 60.Moriwaki Y, Yamamoto T, Shibutani Y, et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. 2003;52:605–608. doi: 10.1053/meta.2003.50096. [DOI] [PubMed] [Google Scholar]
- 61.Wong CK, Ho AW, Tong PC, et al. Aberrant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetic patients with nephropathy. Clin Exp Immunol. 2007;149:123–131. doi: 10.1111/j.1365-2249.2007.03389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyauchi K, Takiyama Y, Honjyo J, Tateno M, Haneda M. Upregulated IL-18 expression in type 2 diabetic subjects with nephropathy: TGF-beta1 enhanced IL-18 expression in human renal proximal tubular epithelial cells. Diabetes Res Clin Pract. 2009;83:190–199. doi: 10.1016/j.diabres.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 63.Araki S, Haneda M, Koya D, et al. Predictive impact of elevated serum level of IL-18 for early renal dysfunction in type 2 diabetes: an observational follow-up study. Diabetologia. 2007;50:867–873. doi: 10.1007/s00125-006-0586-8. [DOI] [PubMed] [Google Scholar]
- 64.Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000;67:291–300. doi: 10.1016/s0024-3205(00)00622-6. [DOI] [PubMed] [Google Scholar]
- 65.Sekizuka K, Tomino Y, Sei C, et al. Detection of serum IL-6 in patients with diabetic nephropathy. Nephron. 1994;68:284–285. doi: 10.1159/000188281. [DOI] [PubMed] [Google Scholar]
- 66.Dalla Vestra M, Mussap M, Gallina P, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. 2005;16 Suppl 1:S78–S82. doi: 10.1681/asn.2004110961. [DOI] [PubMed] [Google Scholar]
- 67.Shikano M, Sobajima H, Yoshikawa H, et al. Usefulness of a highly sensitive urinary and serum IL-6 assay in patients with diabetic nephropathy. Nephron. 2000;85:81–85. doi: 10.1159/000045634. [DOI] [PubMed] [Google Scholar]
- 68.Sangoi MB, de Carvalho JA, Tatsch E, et al. Urinary inflammatory cytokines as indicators of kidney damage in type 2 diabetic patients. Clin Chim Acta. 2016;460:178–183. doi: 10.1016/j.cca.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 69.Suzuki D, Miyazaki M, Naka R, et al. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes. 1995;44:1233–1238. doi: 10.2337/diab.44.10.1233. [DOI] [PubMed] [Google Scholar]
- 70.Park J, Guan Y, Sheng X, et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight. 2019;4:e128886. doi: 10.1172/jci.insight.128886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Navarro JF, Milena FJ, Mora C, Leon C, Garcia J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol. 2006;26:562–570. doi: 10.1159/000098004. [DOI] [PubMed] [Google Scholar]
- 72.Navarro JF, Mora C, Maca M, Garca J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am J Kidney Dis. 2003;42:53–61. doi: 10.1016/s0272-6386(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 73.Pedigo CE, Ducasa GM, Leclercq F, et al. Local TNF causes NFATc1-dependent cholesterol- mediated podocyte injury. J Clin Invest. 2016;126:3336–3350. doi: 10.1172/JCI85939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niewczas MA, Ficociello LH, Johnson AC, et al. Serum concentrations of markers of TNFalpha and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes. Clin J Am Soc Nephrol. 2009;4:62–70. doi: 10.2215/CJN.03010608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coca SG, Nadkarni GN, Huang Y, et al. Plasma biomarkers and kidney function decline in early and established diabetic kidney disease. J Am Soc Nephrol. 2017;28:2786–2793. doi: 10.1681/ASN.2016101101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baharlou R, Ahmadi-Vasmehjani A, Davami MH, et al. Elevated levels of T-helper 17-associated cytokines in diabetes type 1 patients. Indicators for following the course of disease. Immunol Invest. 2016;45:641–651. doi: 10.1080/08820139.2016.1197243. [DOI] [PubMed] [Google Scholar]
- 77.Vasanthakumar R, Mohan V, Anand G, Deepa M, Babu S, Aravindhan V. Serum IL-9, IL-17, and TGF-β levels in subjects with diabetic kidney disease (CURES-134) Cytokine. 2015;72:109–112. doi: 10.1016/j.cyto.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kuo HL, Huang CC, Lin TY, Lin CY. IL-17 and CD40 ligand synergistically stimulate the chronicity of diabetic nephropathy. Nephrol Dial Transplant. 2018;33:248–256. doi: 10.1093/ndt/gfw397. [DOI] [PubMed] [Google Scholar]
- 79.Ruster C, Wolf G. The role of chemokines and chemokine receptors in diabetic nephropathy. Front Biosci. 2008;13:944–955. doi: 10.2741/2734. [DOI] [PubMed] [Google Scholar]
- 80.Tesch GH. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2008;294:F697–F701. doi: 10.1152/ajprenal.00016.2008. [DOI] [PubMed] [Google Scholar]
- 81.Moon JY, Jeong L, Lee S, et al. Association of polymorphisms in monocyte chemoattractant protein-1 promoter with diabetic kidney failure in Korean patients with type 2 diabetes mellitus. J Korean Med Sci. 2007;22:810–814. doi: 10.3346/jkms.2007.22.5.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tashiro K, Koyanagi I, Saitoh A, et al. Urinary levels of monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8), and renal injuries in patients with type 2 diabetic nephropathy. J Clin Lab Anal. 2002;16:1–4. doi: 10.1002/jcla.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wada T, Furuichi K, Sakai N, et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 2000;58:1492–1499. doi: 10.1046/j.1523-1755.2000.00311.x. [DOI] [PubMed] [Google Scholar]
- 84.Tarabra E, Giunti S, Barutta F, et al. Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes. 2009;58:2109–2118. doi: 10.2337/db08-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee EY, Chung CH, Khoury CC, et al. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-beta, increases podocyte motility and albumin permeability. Am J Physiol Renal Physiol. 2009;297:F85–F94. doi: 10.1152/ajprenal.90642.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Herder C, Peltonen M, Koenig W, et al. Systemic immunemediators and lifestyle changes in the prevention of type2 diabetes: results from the Finnish Diabetes Prevention Study. Diabetes. 2006;55:2340–2346. doi: 10.2337/db05-1320. [DOI] [PubMed] [Google Scholar]
- 87.Mezzano S, Aros C, Droguett A, et al. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant. 2004;19:2505–2512. doi: 10.1093/ndt/gfh207. [DOI] [PubMed] [Google Scholar]
- 88.Schinzari F, Tesauro M, Campia U, Cardillo C. Increased fractalkine and vascular dysfunction in obesity and in type 2 diabetes. Effects of oral antidiabetic treatment. Vascul Pharmacol. 2020;128-129:106676. doi: 10.1016/j.vph.2020.106676. [DOI] [PubMed] [Google Scholar]
- 89.Shah R, Matthews GJ, Shah RY, et al. Serum fractalkine (CX3CL1) and cardiovascular outcomes and diabetes: findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. Am J Kidney Dis. 2015;66:266–273. doi: 10.1053/j.ajkd.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sindhu S, Akhter N, Arefanian H, et al. Increased circulatory levels of fractalkine (CX3CL1) are associated with inflammatory chemokines and cytokines in individuals with type-2 diabetes. J Diabetes Metab Disord. 2017;16:15. doi: 10.1186/s40200-017-0297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams A, Greene N, Kimbro K. Increased circulating cytokine levels in African American women with obesity and elevated HbA1c. Cytokine. 2020;128:154989. doi: 10.1016/j.cyto.2020.154989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. 2017;18:1288–1298. doi: 10.1038/ni.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang S, Zhou W, Sheerin NS, Vaughan RW, Sacks SH. Contribution of renal secreted complement C3 to the circulating pool in humans. J Immunol. 1999;162:4336–4341. [PubMed] [Google Scholar]
- 94.Biancone L, David S, Della Pietra V, Montrucchio G, Cambi V, Camussi G. Alternative pathway activation of complement by cultured human proximal tubular epithelial cells. Kidney Int. 1994;45:451–460. doi: 10.1038/ki.1994.59. [DOI] [PubMed] [Google Scholar]
- 95.Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol. 2017;13:311–318. doi: 10.1038/nrneph.2017.31. [DOI] [PubMed] [Google Scholar]
- 96.Rasmussen KL, Nordestgaard BG, Nielsen SF. Complement C3 and risk of diabetic microvascular disease: a cohort study of 95202 individuals from the general population. Clin Chem. 2018;64:1113–1124. doi: 10.1373/clinchem.2018.287581. [DOI] [PubMed] [Google Scholar]
- 97.Sircar M, Rosales IA, Selig MK, et al. Complement 7 is up-regulated in human early diabetic kidney disease. Am J Pathol. 2018;188:2147–2154. doi: 10.1016/j.ajpath.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hansen TK, Gall MA, Tarnow L, et al. Mannose-binding lectin and mortality in type 2 diabetes. Arch Intern Med. 2006;166:2007–2013. doi: 10.1001/archinte.166.18.2007. [DOI] [PubMed] [Google Scholar]
- 99.Hansen TK, Forsblom C, Saraheimo M, et al. Association between mannose-binding lectin, high-sensitivity C-reactive protein and the progression of diabetic nephropathy in type 1 diabetes. Diabetologia. 2010;53:1517–1524. doi: 10.1007/s00125-010-1742-8. [DOI] [PubMed] [Google Scholar]
- 100.Vaisar T, Durbin-Johnson B, Whitlock K, et al. Urine complement proteins and the risk of kidney disease progression and mortality in type 2 diabetes. Diabetes Care. 2018;41:2361–2369. doi: 10.2337/dc18-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun ZJ, Li XQ, Chang DY, et al. Complement deposition on renal histopathology of patients with diabetic nephropathy. Diabetes Metab. 2019;45:363–368. doi: 10.1016/j.diabet.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 102.Yiu WH, Li RX, Wong DW, et al. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol Dial Transplant. 2018;33:1323–1332. doi: 10.1093/ndt/gfx336. [DOI] [PubMed] [Google Scholar]
- 103.Anand G, Vasanthakumar R, Mohan V, Babu S, Aravindhan V. Increased IL-12 and decreased IL-33 serum levels are associated with increased Th1 and suppressed Th2 cytokine profile in patients with diabetic nephropathy (CURES-134) Int J Clin Exp Pathol. 2014;7:8008–8015. [PMC free article] [PubMed] [Google Scholar]
- 104.Zeng C, Shi X, Zhang B, et al. The imbalance of Th17/Th1/ Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med (Berl) 2012;90:175–186. doi: 10.1007/s00109-011-0816-5. [DOI] [PubMed] [Google Scholar]
- 105.Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol. 2012;35:164–174. doi: 10.1159/000334928. [DOI] [PubMed] [Google Scholar]
- 106.Moriya R, Manivel JC, Mauer M. Juxtaglomerular apparatus T-cell infiltration affects glomerular structure in type 1 diabetic patients. Diabetologia. 2004;47:82–88. doi: 10.1007/s00125-003-1253-y. [DOI] [PubMed] [Google Scholar]
- 107.Paulsen EP, Burke BA, Vernier RL, Mallare MJ, Innes DJ, Jr, Sturgill BC. Juxtaglomerular body abnormalities in youth-onset diabetic subjects. Kidney Int. 1994;45:1132–1139. doi: 10.1038/ki.1994.150. [DOI] [PubMed] [Google Scholar]
- 108.Aviado DM, Porter JM. Pentoxifylline: a new drug for the treatment of intermittent claudication. Mechanism of action, pharmacokinetics, clinical efficacy and adverse effects. Pharmacotherapy. 1984;4:297–307. doi: 10.1002/j.1875-9114.1984.tb03380.x. [DOI] [PubMed] [Google Scholar]
- 109.Rodriguez-Moran M, Guerrero-Romero F. Efficacy of pentoxifylline in the management of microalbuminuria in patients with diabetes. Curr Diabetes Rev. 2008;4:55–62. doi: 10.2174/157339908783502343. [DOI] [PubMed] [Google Scholar]
- 110.Bhanot S, Leehey DJ. Pentoxifylline for diabetic nephropathy: an important opportunity to re-purpose an old drug? Curr Hypertens Rep. 2016;18:8. doi: 10.1007/s11906-015-0612-7. [DOI] [PubMed] [Google Scholar]
- 111.Donate-Correa J, Tagua VG, Ferri C, et al. Pentoxifylline for renal protection in diabetic kidney disease. A model of old drugs for new horizons. J Clin Med. 2019;8:287. doi: 10.3390/jcm8030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, et al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J Am Soc Nephrol. 2015;26:220–229. doi: 10.1681/ASN.2014010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int. 2011;80:17–28. doi: 10.1038/ki.2010.483. [DOI] [PubMed] [Google Scholar]
- 114.Tuttle KR, Brosius FC 3rd, Adler SG, et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a phase 2 randomized controlled clinical trial. Nephrol Dial Transplant. 2018;33:1950–1959. doi: 10.1093/ndt/gfx377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang M, Yu M, Guan D, et al. ASK1-JNK signaling cascade mediates Ad-ST13-induced apoptosis in colorectal HCT116 cells. J Cell Biochem. 2010;110:581–588. doi: 10.1002/jcb.22551. [DOI] [PubMed] [Google Scholar]
- 116.Nishitoh H, Saitoh M, Mochida Y, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 117.Chertow GM, Pergola PE, Chen F, et al. Effects of selonsertib in patients with diabetic kidney disease. J Am Soc Nephrol. 2019;30:1980–1990. doi: 10.1681/ASN.2018121231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Menne J, Eulberg D, Beyer D, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. 2017;32:307–315. doi: 10.1093/ndt/gfv459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.de Zeeuw D, Bekker P, Henkel E, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. 2015;3:687–696. doi: 10.1016/S2213-8587(15)00261-2. [DOI] [PubMed] [Google Scholar]
- 120.Salmi M, Kalimo K, Jalkanen S. Induction and function of vascular adhesion protein-1 at sites of inflammation. J Exp Med. 1993;178:2255–2260. doi: 10.1084/jem.178.6.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smith DJ, Salmi M, Bono P, Hellman J, Leu T, Jalkanen S. Cloning of vascular adhesion protein 1 reveals a novel multifunctional adhesion molecule. J Exp Med. 1998;188:17–27. doi: 10.1084/jem.188.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang SH, Yu TY, Hung CS, et al. Inhibition of semicarbazide-sensitive amine oxidase reduces atherosclerosis in cholesterol-fed New Zealand white rabbits. Sci Rep. 2018;8:9249. doi: 10.1038/s41598-018-27551-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.de Zeeuw D, Renfurm RW, Bakris G, et al. Efficacy of a novel inhibitor of vascular adhesion protein-1 in reducing albuminuria in patients with diabetic kidney disease (ALBUM): a randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2018;6:925–933. doi: 10.1016/S2213-8587(18)30289-4. [DOI] [PubMed] [Google Scholar]
- 124.Mazidi M, Karimi E, Rezaie P, Ferns GA. Treatment with GLP1 receptor agonists reduce serum CRP concentrations in patients with type 2 diabetes mellitus: a systematic review and meta-analysis of randomized controlled trials. J Diabetes Complications. 2017;31:1237–1242. doi: 10.1016/j.jdiacomp.2016.05.022. [DOI] [PubMed] [Google Scholar]