

Abstract

The inhibition of the nuclear receptor retinoic-acid-receptor-related orphan receptor γt (RORγt) is a promising strategy in the treatment of autoimmune diseases. RORγt features an allosteric binding site within its ligand-binding domain that provides an opportunity to overcome drawbacks associated with orthosteric modulators. Recently, trisubstituted isoxazoles were identified as a novel class of allosteric RORγt inverse agonists. This chemotype offers new opportunities for optimization into selective and efficacious allosteric drug-like molecules. Here, we explore the structure–activity relationship profile of the isoxazole series utilizing a combination of structure-based design, X-ray crystallography, and biochemical assays. The initial lead isoxazole (FM26) was optimized, resulting in compounds with a ∼10-fold increase in potency (low nM), significant cellular activity, promising pharmacokinetic properties, and a good selectivity profile over the peroxisome-proliferated-activated receptor γ and the farnesoid X receptor. We envisage that this work will serve as a platform for the accelerated development of isoxazoles and other novel chemotypes for the effective allosteric targeting of RORγt.
1. Introduction
Nuclear receptors (NRs) are a family of ligand-dependent transcription factors and attractive drug targets because of their central role in several regulatory processes in the human body.1−4 Within the NR family, the retinoic-acid-receptor-related orphan receptor γt (RORγt) has received increased attention because of its essential role in the immune system.5 RORγt is a key regulator in the differentiation of naïve CD4+ T cells into T helper 17 (Th17) cells, and the production of the pro-inflammatory cytokine IL-17a.5−7 Elevated IL-17a levels are associated with the development of autoimmune diseases, including psoriasis, multiple sclerosis, and rheumatoid arthritis.8−11 Disrupting the Th17/IL-17a pathway shows high potential for the treatment of autoimmune disorders, which has already been validated by the clinical success of anti-IL-17a monoclonal antibodies.8,12,13 The inhibition of RORγt could pose an attractive alternative strategy to decrease IL-17a production in the treatment of autoimmune diseases.12 Many research groups have shown significant interest in the identification of small-molecule RORγt inhibitors (or, more specifically, inverse agonists), with several synthetic compounds progressing into clinical trials.14−18
With the exception of RTA-1701 (Figure 1C),18,19 all of the RORγt inverse agonists that have entered clinical trials thus far likely target the highly conserved ligand-binding pocket, termed the orthosteric-binding site, within the ligand-binding domain (LBD) of RORγt (Figure 1A).5,14 While orthosteric targeting has been highly successful, novel molecular modalities with alternative modes of action, such as allosteric modulators, offer an interesting alternative for targeting NRs and could circumvent issues related to promiscuity between NRs.20−23 Recently, an allosteric binding site was identified in the LBD of RORγt (formed by helices 3, 4, 11, and 12), at a location that is topographically distinct to the orthosteric site (Figure 1A).24 The indazole MRL-871 (1) (Figure 1C) was identified as a prototypical allosteric ligand for RORγt, acting as a potent inverse agonist and decreasing coactivator binding with a similar efficacy as orthosteric inverse agonists.24,25 The allosteric pocket of RORγt is unique within the NR family and thought not to be the target of endogenous ligands.24,26 Therapeutic compounds targeting the allosteric-binding pocket potentially have a higher selectivity profile and do not act in competition, but in synergy, with endogenous agonists.20,21,27 Therefore, these compounds would be greatly beneficial for drug discovery and chemical biology applications.
Figure 1.

(A) Co-crystal structure of the RORγt LBD in complex with allosteric ligand FM26 (shown as orange sticks) (PDB: 6SAL),28 where the allosteric site (orange circle) and the orthosteric site (blue circle) are indicated; (B) enlarged view of FM26 (shown as orange sticks) in the allosteric pocket, where H-bond interactions are indicated with orange dashes; (C) chemical structures of allosteric RORγt ligands FM26 (2), MRL-871 (1), and RTA-1701; and (D) exploration of the hit-to-lead SAR profile of FM26.
Despite the high potential of allosteric inverse agonists for RORγt, the number of examples have remained limited to compounds based on the indazole core of 1 or similar chemotypes, and the previously mentioned allosteric ligand RTA-1701 (which binds to RORγt via covalent attachment to Cys476 of helix 11 and partly overlaps with the allosteric site).19,24,25,29−34 Follow-up studies improved the pharmacokinetic (PK) and selectivity profile of 1 and its derivatives. However, off-target effects toward other NRs, most notably toward the peroxisome proliferated-activated receptor γ (PPARγ), and challenges regarding cytotoxicity and metabolic stability have been observed.24,29−32 The scarcity and lack of scaffold diversity of allosteric RORγt ligands, and the limitations regarding specificity and drug-like properties, necessitates further exploration of the allosteric pocket. Specifically, establishing structure–activity relationship (SAR) profiles for allosteric chemotypes distinct from the indazole-analogous ligand classes is of great importance.
Recently, we used an in silico pharmacophore search approach to identify a novel class of RORγt allosteric inverse agonists, featuring a trisubstituted isoxazole core.28,35 SAR studies around this novel chemotype resulted in the discovery of FM26 (2) (Figure 1C), which shows sub-micromolar potency as a RORγt inverse agonist and significant inhibition of cellular IL-17a expression levels. Initial SAR studies of 2 identified two key pharmacophore features, where the 2,6-disubstituted phenyl ring at the C-3 position and a C-4 benzoic acid moiety were shown to be optimal (Figure 1D).24,25,28,30−33 Less knowledge has been garnered on the linker at the C-4 position and the C-5 substituent. Regarding the C-4 linker, an amine linker (as is used for 2) was found to be preferred for potency over some more rigid linkers.28 Additionally, the co-crystal structure (Figure 1B) showed that the presence of a hydrogen bond donating N-heterocycle at the C-5 isoxazole position (as is the case for the pyrrole in 2) significantly increased the potency toward RORγt by the formation of an additional polar interaction with the backbone carbonyls of residues Leu353 and Lys354.28 To identify compounds suitable for focused lead optimization, crucial explorations around the C-4 and C-5 positions are needed to further explore the SAR, improve the potency of the isoxazole series, increase the RORγt specificity, and direct PK optimization.
Utilizing X-ray crystallography to guide focused library development, herein, we report on the synthesis of allosteric RORγt inverse agonists diversified at the C-4 and C-5 isoxazole positions (Figure 1D). The structure-based SAR study was specifically focused on improved RORγt activity, mitigating off-target PPARγ activity, and a first exploration of PK profiles.
2. Results and Discussion
2.1. In Silico Docking Studies Guide a SAR Study at the Isoxazole C-4 Position
The initial investigation started with expanding the SAR data around the C-4 position of the isoxazole series guided by in silico docking studies. A virtual library of derivatives around 2, containing different C-4 linkers and benzoic acid substituents, was designed with a focus on diversity and synthetic feasibility (see Table S1). This library was docked into the allosteric site of RORγt (PDB: 4YPQ(24)), using Glide, the molecular docking tool in Schrödinger (12.3, 2020-1).36,37 For each ligand, the docking pose was evaluated and given a “glide score”, which is an empirical scoring function that approximates the ligand binding free energy (the more negative the values, the higher the expected potency) but will not always translate into experimental IC50 values.36,37Table 1 shows the compounds that resulted in the most promising glide scores, indicating that linkers responsible for an increased clog P of the compounds [i.e., ether (3), thioether (4), methylated amine (5), and alkene (6 and 7)] would be beneficial for affinity. Additionally, the data suggest that a fluoro substitution at the ortho position of the benzoic acid moiety would be well tolerated (8 and 9).
Table 1. SAR Studies around the C-4 Isoxazole Positiona.


cLogP values, glide scores, TR-FRET IC50 values (μM) from coactivator recruitment and AlexaFluor-MRL-871 displacement assays, ΔTm values (°C) from thermal shift assays, and fold decrease in IL-17a mRNA expression levels relative to DMSO reverse transcriptase PCR (RT-PCR). Abbreviations: n.d., not determined. TR-FRET and TSA data are recorded in triplicate; values are representative of ≥3 repeated experiments. RT-PCR data are recorded in triplicate; values are representative of ≥2 repeated experiments. cLogP values were predicted using MarvinSketch (20.10).
2.2. Optimized Synthesis Route Allowed the Efficient Synthesis of C-4 Isoxazole Derivatives
Isoxazoles 2–9 (Table 1) were synthesized (Scheme 1) in order to biochemically evaluate the predictions from the in silico docking experiments. Common intermediate 14 was synthesized via a 1,3-dipolar cycloaddition using a nitrile oxide and alkynyl bromide, as described previously (Scheme S1).28 In order to prepare derivatives 2–9, it was important to obtain core intermediate 16 with a N-Boc-protected pyrrole moiety, instead of the free pyrrole, as was the case in the original synthesis route for 2 (Scheme S2).28 This was achieved via a Suzuki cross-coupling reaction, employing a dppf instead of a tetrakis palladium catalyst (used in previous research), which proved to be essential for maintaining the Boc-protected pyrrole 15 in an acceptable yield (Scheme 1). DIBAL was then used to selectively reduce the ethyl ester, yielding 16 without a concomitant loss of the Boc group (Scheme 1), which was previously observed with LiAlH4 (Scheme S1).28
Scheme 1. Synthesis Route for Different Trisubstituted Isoxazoles (C-4 Library).

Reagents and conditions: (a) N-Boc-pyrrole-B(pin), Pd(dppf)Cl2, Cs2CO3, and DME, 85 °C, 8 h, 50%; (b) DIBAL and CH2Cl2, −78 °C, 3 h, 80%; (c) (i) (MeSO2)2O, Et3N, and CH2Cl2, 0 °C → rt, 3 h, (ii) aniline or N-methylaniline, rt, 3 h, 36% (17) and 52% (18); (d) (i) DMP and CH2Cl2, rt, 3 h, 44%, (ii) methyl 4-amino benzoate, MeOH, and AcOH, reflux, 24 h, 42%, (iii) NaBH4 and EtOH, reflux, 3 h, 17% (19); (e) LiOH, EtOH, and H2O, 95 °C, 3 h, 29–81%; (f) methyl 4-hydroxybenzoate or methyl 2-fluoro-4-hydroxybenzoate, DIAD, PPh3, Et3N, and THF, reflux, 3 h, 55% (20), 25% (21); (g) methyl 4-mercaptobenzoate, DIAD, PPh3, Et3N, and THF, reflux, 3 h, 27%; (h) DMP and CH2Cl2, rt, 3 h, quant.; (i) (4-(methoxycarbonyl)benzyl)triphenylphosphonium (see Experimental Section for the synthetic procedure of the triphenylphosphonium), LiHMDS, and THF, −78 °C → rt, 24 h, 7% (24), 27% (25).
The primary alcohol of 16 was used as a functional handle for the derivatization of the isoxazole C-4 position. Critical to the synthesis of this series of isoxazoles was the optimization of the reductive amination step, which provided sub-optimal yields in the synthesis of 2 (Scheme S2).28 In order to improve the total yield of the synthesis, the reductive amination was substituted for a nucleophilic substitution reaction. Final compounds 2 and 5 were accessed in an efficient manner by the mesylation of alcohol 16 with methane sulfonic anhydride (monitored by NMR), the in situ addition of the substituted aniline, followed by the hydrolysis of the benzoate ester. In contrast, 8 (fluoro substituent) was synthesized via the original reductive amination route (Scheme 1).
Compounds with an ether and thioether linkage (3, 9, and 4) were synthesized from core intermediate 16via a Mitsunobu reaction with hydroxy- or mercaptobenzoate, respectively, furnishing the products in acceptable yields (Scheme 1). For the cis and trans alkene linker (6 and 7), the alcohol of compound 16 was oxidized to the aldehyde, which was subsequently used in a Wittig reaction, obtaining a 3:1 mixture of the cis/trans isomers (Scheme 1). These were separated via preparative high-performance liquid chromatography/ultraviolet (HPLC-UV) to afford 24 and 25, and followed by ester hydrolysis, which afforded final compounds 6 (trans) and 7 (cis) (for which the stereochemistry was determined based on the relative 1H NMR J-coupling values between the alkene protons).
2.3. Biochemical Assays and X-ray Crystallography Reveal a Positive Correlation between the C-4 Linker Lipophilicity and Potency
The potency of the C-4 modified compounds for RORγt was investigated in a time-resolved FRET (TR-FRET) coactivator recruitment assay (Table 1, Figure 2A, see Figure S3A for a schematic representation of the assay setup).38 RORγt is constitutively active, which means that it shows a basic level of transcriptional activity, thus allowing the partial recruitment of coactivators in the absence of an agonist.39 Reference compounds 1 and 2 showed potent inhibition of coactivator binding, with IC50 values comparable to previous studies.24,28 Interestingly, compound 3 (FM156) containing an ether linkage demonstrated a 9-fold increase in potency compared to 2 with an amine linker (IC50 of 31 ± 3 nM vs 270 ± 20 nM, respectively), resulting in an IC50 value in the same range as indazole 1. The trans-alkene linker (6, FM260) resulted in a low nM IC50 value as well (IC50 of 20 ± 2 nM), in contrast to the cis-alkene linker (7) which was 25 times less potent (IC50 of 490 ± 30 nM), as predicted by the docking scores. In contrast, a thioether linkage (4) resulted in a significantly decreased potency compared to 2 (IC50 of 6.6 ± 0.8 μM), whereas a methylated amine linker (5) resulted in a loss of activity (IC50 > 100 μM). The introduction of a fluorine substituent at the ortho position of the benzoic acid moiety proved detrimental to coactivator inhibition with a 7–13 fold decrease in potency compared to the compound without a substituent (8 vs 2 and 9 vs 3), which was also the case for an isoxazole analogue in a previous SAR study.28
Figure 2.

Biochemical analysis and X-ray crystallography data for isoxazole compounds with C-4 modifications. (A) Dose–response curves from the TR-FRET coactivator recruitment assay for 1, 2, 3, and 6; (B) dose–response curves from the ligand displacement TR-FRET assay using the AlexaFluor MRL-871 probe for 1, 2, 3, and 6; (C) melting temperatures (ΔTm in °C), as measured in TSA for 1, 2, 3, and 6. Data were recorded in ≥3 independent experiments, each recorded in triplicate (one representative dataset shown). Error bars represent the SD of the mean; (D,G) tertiary co-crystal structure of RORγt in complex with 3 (PDB: 7NPC) (D) and 9 (PDB: 7NP5) (G) (stick representation). The final 2Fo–Fc electron density map of compounds is shown as an isomesh contoured at 1σ; (E,H) overlay of the co-crystal structure of RORγt in complex with FM26 (2) (PDB: 6SAL) and RORγt bound to 3 (E), and an overlay of RORγt with 3 and 9 (H); and (F,I) enlarged view of the allosteric pocket of RORγt showing the interactions between 3 (F)/9 (I) and the protein.
To prove an allosteric mode of action, the compounds were also tested in two other TR-FRET assay formats. First, a previously described AlexaFluor 647-labeled MRL-871 probe24 (Figure S1) was used, which upon binding to RORγt shows FRET pairing with an anti-His terbium cryptate donor on the protein (see Figure S3B).24 All compounds showed displacement of the AlexaFluor-MRL allosteric probe, proving an allosteric binding mode (Table 1, Figure 2B), with the IC50 values correlating with the IC50 values from the TR-FRET coactivator recruitment assay (Table 1). In particular, ether 3 and trans-alkene 6 demonstrated efficient displacement of the probe, with IC50 values of 12 ± 1 and 7.4 ± 0.9 nM, respectively, significantly increased as compared to 2 (IC50 value of 100 ± 10 nM).
The most potent ligands 3 and 6 were also measured in a competitive TR-FRET coactivator recruitment assay, where the compounds were titrated in the presence of cholesterol, which is an orthosteric agonist (activator) for RORγt40 (Figure S2, see Figure S3C for a schematic representation of the assay setup). The binding curves for compounds 3 and 6 show that the IC50 values did not decrease upon increasing concentrations of cholesterol, demonstrating that their binding mode is independent to that of cholesterol, supporting their allosteric-binding mode. In fact, the presence of cholesterol slightly enhanced the potency of both 3 and 6, suggesting a cooperative behavior between orthosteric and allosteric ligand binding, as was observed previously.27,28
A thermal shift assay (TSA) was performed as an orthogonal assay to investigate the effect of the compounds on the thermal denaturation of the RORγt protein (Table 1, Figure 2C). Ligand binding typically improves the thermal stability of a protein by stabilizing the protein fold, as indicated by the change in the melting temperature ΔTm.41−43 The C-4 isoxazole derivatives 3–9 showed a thermal stabilization effect according to their potency, as observed in the TR-FRET assays. The most potent compounds 3 and 6 showed particularly high ΔTm values (4.9 and 6.4 °C resp.). These values are improved compared to that for 2, which provides further indication that 3 and 6 have a high-binding affinity for RORγt.
Crystallization studies provided the co-crystal structures of the RORγt LBD in complex with compounds 3 (ether linker) and 9 (fluoro substituent), with resolutions of 1.46 and 1.55 Å, respectively (Tables S4 and S5, Figure S6). The co-crystal structures of RORγt with 3 and 9 showed clear electron density for the compounds in the allosteric binding site between helices 3, 4, 11, and 12 (Figure 2D,G). In Figure 2E, an overlay of RORγt in complex with 2 and with 3 is shown, demonstrating a common binding pose, with the C-3, C-4, and C-5 isoxazole substituents anchored at the same position. The pyrrole moiety forms a hydrogen bond interaction with the main chain carbonyls of residues Leu353 and Lys354 (Figure 2F). Additionally, the carboxylic acid moiety forms hydrogen bond interactions with the side chain of Gln329 and backbone nitrogens of Phe498 and Ala497, as is also the case for 1 and 2. Interestingly, the electron density for 3 and 2 would allow two different conformations of the C-4 linker,28 but the preferred conformation for the ether linker in 3 is opposite to the preferred conformation for the amine linker in 2 (Figure 2E). For compound 9, the electron density and binding mode are highly similar to 3, with the same conformation for the ether linker present (Figure 2G–I). The ortho-fluoro substituent on the benzoic acid ring does not significantly influence the conformation of the compound, except for the disubstituted phenyl ring at the isoxazole C-3 position, which is normally fixed at the same position but is now slightly shifted (Figure 2H).
The increased potency of ether 3 and alkene 6 could be the result of increased hydrophobic effects of these more lipophilic compounds toward the hydrophobic allosteric-binding pocket (see the protein–ligand interaction plot in Figure S5). The different preferred conformations of the ether linker for 3, as seen in the co-crystal structure, might also be contributing to the potency. In contrast, the compound with a thioether linkage (4) shows a significantly lower potency, which might be caused by a slight change in the bond angle and length of the linker. The docking pose shows that the isoxazole core and pyrrole moiety are clearly shifted compared to the compound with ether linker (3) (Figure S4A). The drop in potency for 5, featuring a methylated amine linker, likely results from the restricted rotation of the linker. The electron density for the isoxazoles shows that two conformations of the linker are present, but when the linker is more rotationally restricted, as is the case for 5, only one of these linker conformations will most likely be present, potentially resulting in a higher entropic penalty and in turn a decrease in the binding affinity. The introduction of an ortho-fluoro substituent at the benzoic acid moiety leads to a slight decrease in potency (8 vs 2 and 9 vs 3). The co-crystal structure in complex with 9 shows that the fluoro substituent is placed in the same plane as the carboxylic acid moiety (Figure 2I). This unfavorable conformation is believed to be caused in order to both fit the fluoro substituent in the pocket and also allow hydrogen bond interactions between the carboxylic acid and the protein, which could lead to charge repulsion between the two moieties and a decreased potency.
2.4. SAR Studies Show the Necessity of a H-Bond Donor Moiety at the Isoxazole C-5 Position
The SAR was further explored at the isoxazole C-5 position to investigate the hydrogen bonding character of the pyrrole moiety of 2. Compound 3 with the ether linkage was chosen as a core scaffold, based on the combination of its high potency, availability of the co-crystal structure, and synthetic feasibility. A focused library of C-5 derivatives (compounds 10–13, Table 2) was designed and synthesized (the full library used for docking is shown in Table S2). Specifically, the effect of the nitrogen position (10), pKa (11) (pKa = 10.17 for 11 vs 14.99 for 3) and substitution (12) was investigated, as well as the hydrophobic space around the pyrrole (13).
Table 2. SAR Studies around the C-5 Isoxazole Positiona.


Glide scores, TR-FRET IC50 values (μM) from the coactivator recruitment and AlexaFluor-MRL-871 displacement assays, ΔTm values (°C) from TSA, and fold decrease in IL-17a expression levels relative to DMSO (RT-PCR) are shown. Abbreviations: n.d., not determined. TR-FRET and TSA data are recorded in triplicate; values are representative of ≥3 repeated experiments. RT-PCR data are recorded in triplicate; values are representative of ≥2 repeated experiments.
Compounds 10–13 were synthesized using an analogous synthesis route as shown previously (Scheme 2). Because the C-5 moiety is incorporated early in the synthesis, it was not possible to diversify from a late-stage intermediate, as for the C-4 modifications. Bromide 14 was subjected to a Suzuki reaction with the associated pinacol ester, containing a Boc-, THP-, or methyl-protected heterocycle, followed by ester hydrolysis with DIBAL to yield 30–33 (Scheme 2). For the N-methyl pyrrole, the original conditions were used (Scheme S2) (the tetrakis palladium catalyst in the Suzuki reaction and LiAlH4 for reduction) because no labile-protecting group was used in this synthesis route. Next, a Mitsunobu reaction with methyl 4-hydroxybenzoate and ester hydrolysis combined with heterocycle deprotection were conducted to afford the final compounds 10–13.
Scheme 2. Synthesis Route for Different Trisubstituted Isoxazoles (C-5 Library).
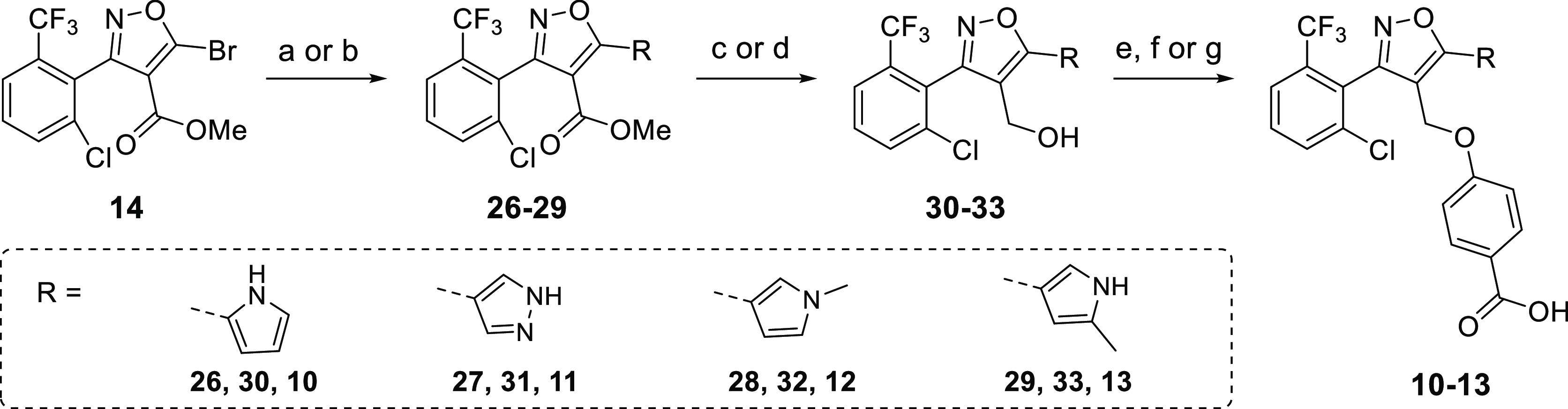
Reagents and conditions: (a) N-Boc-pyrrole-B(pin), N-THP-pyrazole-B(pin) or 5-methyl N-Boc-pyrrole-B(pin) (see Experimental Section for the synthetic procedure of this pinacol ester), Pd(dppf)Cl2, Cs2CO3, and DME, 85 °C, 8 h, 49% (26), 34% (27), and 24% (29); (b) N-methyl-pyrrole-B(pin), Pd(PPh3)4, Na2CO3, DME, and H2O, 85 °C, 8 h, 55% (28); (c) DIBAL and CH2Cl2, −78 °C, 3 h, 69% (30), 71% (31), and 58% (33); (d) LiAlH4 and THF, 0 °C → rt, 2 h, 76% (32); (e) (i) methyl 4-hydroxybenzoate, DIAD, PPh3, and THF, reflux, 3 h, 21%, (ii) LiOH, EtOH, and H2O, 95 °C, 3 h, 41% (10); (f) (i) methyl 4-hydroxybenzoate, DIAD, PPh3, Et3N, and THF, reflux, 3 h, 44–45%, (ii) LiOH, EtOH, and H2O, 95 °C, 3 h, 86% (12), 83% (13); (g) (i) methyl 4-hydroxybenzoate, DIAD, PPh3, Et3N, and THF, reflux, 3 h, 49%, (ii) TFA and CH2Cl2, 40 °C, 2 h, 87%, (iii) LiOH, EtOH, and H2O, 95 °C, 3 h, 89% (11).
To assess the SAR around the C-5 position, analogues 10–13 were evaluated in the TR-FRET coactivator recruitment assay (Figure 3A, Table 2). A nitrogen shift (2- instead of 3-position) in the pyrrole ring (10) resulted in a 4.5-fold decrease in potency (IC50 = 140 ± 10 nM) compared to 3. Pyrazole 11 (FM257) showed a slight drop in potency, with an IC50 value of 110 ± 10 nM. For compound 12 (containing a methylated pyrrole) and compound 13 (methylation of the pyrrole at the 5-position), a lower activity was observed with an IC50 value of 3.3 ± 0.3 and 2.9 ± 0.2 μM, respectively. Additionally, the TR-FRET AlexaFluor-MRL assay was performed, validating an allosteric binding mode for all compounds (Figure 3B, Table 2). Analysis of the IC50 values for this series showed that these were in line with the IC50 values from the TR-FRET coactivator assay (Table 2). In the TSA, compounds 10 and 11 induced thermal stabilization with ΔTm values of 2.8 and 2.7 °C respectively, which is lower than for 3 (ΔTm = 4.9 °C) but slightly higher than for 2 (ΔTm = 2.4 °C). Compounds 12 and 13 did not show any response (Figure 3C, Table 2).
Figure 3.

Biochemical analysis and X-ray crystallography data for isoxazole compounds with C-5 modifications. (A) Dose–response curves from the TR-FRET coactivator recruitment assay for 2, 3, 10, and 11; (B) dose–response curves from the ligand displacement TR-FRET assay using the AlexaFluor MRL-871 probe for 2, 3, 10, and 11; (C) melting temperatures (ΔTm in °C), as measured in TSA for 2, 3, 10, and 11. Data were recorded in ≥3 independent experiments, each recorded in triplicate (one representative dataset shown). Error bars represent the SD of the mean. (D,G) Tertiary co-crystal structure of RORγt in complex with 10 (D) (PDB: 7NEC) and 11 (G) (PDB: 7NP6) (stick representation). The final 2Fo–Fc electron density map of the compounds is shown as an isomesh contoured at 1σ; (E,H) overlay of the crystal structure of RORγt in complex with 3 and RORγt bound to 10 (E) or 11 (H); and (F,I) enlarged view of the allosteric pocket of RORγt showing the interactions between 10 (F)/11 (I) and the protein.
Crystallization studies provided the co-crystal structures of the RORγt LBD in complex with C-5-modified compounds 10 (2-pyrrole) and 11 (pyrazole), with resolutions of 1.95 and 1.84 Å, respectively (Tables S6 and S7, Figure S6). The co-crystal structure of RORγt with 10 reveals a clear ligand electron density in the allosteric site with the ether linker in the same preferred conformation as for 3 (Figure 3D,E). The 2-pyrrole at the C-5 position, with the nitrogen at a different position, does not establish a direct hydrogen bond interaction with the protein, as expected, potentially explaining the lower potency of the compound for RORγt. Interestingly, the NH of the pyrrole substituent of 10 forms an alternative hydrogen bond network via a water molecule in the binding pocket at a distance of 3.2 Å from the NH of the pyrrole (Figure 3F) (this water molecule appears to be present in all crystal structures with an allosteric ligand, and thus has a structural role). For pyrazole 11, the binding mode is highly similar to 3, again establishing a hydrogen bond interaction between the NH of the pyrazole and the backbone carbonyls of RORγt (Figure 3G–I).
These results show that small changes at the pyrrole C-5 moiety lead to relevant changes in the potency of the compounds. The 4.5-fold decrease in potency for the compound with a changed position of the nitrogen in the pyrrole ring (10) (compared to 3) is most probably due to the loss of the characteristic H-bond interaction between the pyrrole ring and the backbone of the protein. The additional H-bond interaction with a water molecule explains why 10 is still active on RORγt with an IC50 value < 150 nM, despite the lack of the characteristic C-5 H-bond interaction. For the pyrazole (11, FM257), the 3.5-fold lower potency indicates that the lower pKa of the pyrazole is not beneficial for H-bond formation, although other factors might also be involved, for example, the existence of the pyrazole in two tautomeric forms for which only one can establish the hydrogen bond interaction with the protein. The much lower potency for compound 12 (methylated pyrrole) suggests that the space around the pyrrole is limited, which is also supported by the low potency of the methyl substitution of the pyrrole at the 5-positon (13). The docking score predicts compound 12 to be highly active, based on the docking pose where the pyrrole ring is rotated and the methyl substituent could point toward a small cavity (Figure S4B). Together, based on the biochemical data, the methylated pyrrole does not appear to have enough space for this rotation in the allosteric pocket. Combined, these data show the need for an H-bond donor at the right position of the heterocycle at the C-5 position of the isoxazole scaffold, while additional substituents at the ring appear to be too bulky to be tolerated.
2.5. The New Isoxazole Series Show the Inhibition of IL-17a Expression in EL4 Cells
A selection of the novel isoxazole compounds was tested in a quantitative RT-PCR assay to investigate their functional effect. EL4 cells were treated with 10 μM of the compound or dimethyl sulfoxide (DMSO), after which the IL-17a mRNA levels were measured (Figure 4). Hit compound 2 showed a 9.3-fold reduction, in accordance with our previous reports.28 The optimized compounds 3 and 6 (IC50 values of 31 and 20 nM, respectively) showed a 15- and 16-fold reduction of IL-17a levels compared to DMSO, which is in line with their high biochemical potency and indicates good cellular uptake and activity. Compounds 10 and 11 induced a 12- and 6.6-fold decrease in IL-17a mRNA levels, correlating with their slightly lower biochemical potency compared to 3 and 6. Compound 12 only resulted in a 2.4-fold decrease compared to DMSO, as expected, given its significantly lower potency in the TR-FRET coactivator recruitment assay.
Figure 4.

IL-17a mRNA expression levels in EL4 cells treated with ligands 1, 2, 3, 6, 10, 11, and 12 (10 μM, 24 h) or DMSO and fold decrease of the IL-17a expression relative to DMSO. The level of IL-17a expression was normalized to that of GAPDH expression. All data are expressed as mean ± s.d. (n = 3). The relative gene expression was calculated by the 2–ΔΔCt (Livak) method using DMSO control as a calibrator. Statistical analysis was performed using a one-way analysis of variance (ANOVA) compared against the DMSO control following Dunnett’s post hoc test; ***P < 0.001 and ****P < 0.0001.
2.6. The Isoxazole Series Shows an Improved Selectivity Profile for RORγt
Previous studies have shown that 1 is selective for RORγt over other NRs, except for PPARγ on which it shows a significant cross-reactivity to the orthosteric binding site, as was also supported by the co-crystal structure.24,44 Isoxazole 2 already showed a higher selectivity for RORγt over PPARγ than 1,28 so we were interested to establish if the novel isoxazole compounds further improved this promising selectivity profile.
In a TR-FRET coactivator recruitment assay (Table 3), 1 showed PPARγ agonism with an EC50 value of 0.34 ± 0.02 μM, while the EC50 value for 2 was >20-fold higher (EC50 = 8.2 ± 0.3 μM).24,28 All novel isoxazole ligands developed in this study were at least five times less active on PPARγ than 1 (EC50 values > 1.7 μM), except for compounds 12 and 13 (both bearing a methyl substituent at the pyrrole moiety), which showed a higher cross-reactivity on PPARγ (EC50 values of 1.2 ± 0.1 and 0.79 ± 0.10 μM resp.). This might be due to the more bulky substitution pattern around the pyrrole C-5 substituent.
Table 3. EC50 Values Observed in the TR-FRET Coactivator Recruitment Assay, IC50 Values from the TR-FRET Competition Assay with PPARγ, and ΔTm Values (°C) from TSA Assaysa.
| fold-selective for
RORγt over PPARγ |
|||||
|---|---|---|---|---|---|
| compound | EC50 (μM) PPARγ | IC50 (μM) PPARγ (competition rosiglitazone) | ΔTm (°C) PPARγ | TR-FRET | TSA |
| 1 | 0.34 ± 0.02 | 6.3 ± 0.7 | 2.5 ± 0.2 | 26 | 3.1 |
| 2 | 8.2 ± 0.3 | 81 ± 7 | 0.3 ± 0.0 | 30 | 8.0 |
| 3 | 3.1 ± 0.1 | 63 ± 8 | 0.6 ± 0.0 | 100 | 8.1 |
| 4 | 18 ± 1 | >100 | 0.7 ± 0.0 | 2.7 | 0.5 |
| 5 | >100 | >100 | 0.0 ± 0.0 | ||
| 6 | 2.5 ± 0.1 | 64 ± 3 | 1.1 ± 0.2 | 125 | 5.8 |
| 7 | 7.5 ± 0.3 | >100 | 0.1 ± 0.0 | 15 | 7.0 |
| 8 | 15 ± 1 | >100 | 0.1 ± 0.0 | 1.6 | 7.0 |
| 9 | 5.8 ± 0.2 | 67 ± 6 | 0.4 ± 0.0 | 67 | 7.5 |
| 10 | 1.7 ± 0.1 | 29 ± 4 | 1.2 ± 0.0 | 7.1 | 2.3 |
| 11 | 3.6 ± 0.1 | 79 ± 11 | 0.7 ± 0.0 | 33 | 3.9 |
| 12 | 1.2 ± 0.1 | 20 ± 2 | 1.5 ± 0.0 | 0.36 | <0.1 |
| 13 | 0.79 ± 0.10 | 19 ± 3 | 1.5 ± 0.0 | 0.27 | <0.1 |
TR-FRET and TSA data are recorded in triplicate; values are representative of ≥3 repeated experiments.
In order to further explore the binding site of the isoxazole compounds on PPARγ, a TR-FRET assay was performed whereby the competition between the isoxazole ligands and a known orthosteric PPARγ agonist, rosiglitazone, was measured (Table 3).28,45 All compounds showed competition with rosiglitazone at high concentrations, indicating binding to the orthosteric site of PPARγ. Interestingly, at least a 3-fold lower competition with rosiglitazone was observed for the isoxazole compounds compared to 1 (i.e., at least a 3-fold increase of the IC50 values compared to 1), with most analogues demonstrating IC50 values > 50 μM. Overall, a similar trend was observed for the IC50 values in this competition assay as for the EC50 values in the previously mentioned TR-FRET coactivator recruitment assay (Table 2). Again, compounds 12 and 13 appeared to be less selective for RORγt than the more potent RORγt-binding isoxazoles (IC50 values of 20 ± 2 and 19 ± 3 μM resp.). Compound 10 showed some cross-reactivity with PPARγ as well, although with an IC50 value > 20 μM.
Next, the compounds were evaluated in a PPARγ TSA (Table 3). Compounds 10, 12, and 13 showed the highest thermal stabilization effect (ΔTm of 1.2 and 1.5 °C), in line with the TR-FRET data, and also 6 appeared to show a significant stabilization effect (ΔTm of 1.1 °C). Compound 1 again showed the most significant activity on PPARγ, with a ΔTm of 2.5 °C. The selectivity of the compounds for RORγt over PPARγ was calculated (Table 3) by comparing the data for PPARγ and RORγt for both the TR-FRET coactivator recruitment assay and TSA (see Tables 1–3). Interestingly, compounds 3 and 6, which are highly potent RORγt inverse agonists (IC50 values < 35 nM), show a 100- and 125-fold selectivity for RORγt over PPARγ, respectively, based on the TR-FRET results, which is a significant improvement compared to 2 (30-fold). Contrastingly, 12 and 13 containing a methyl substituent at the pyrrole moiety show higher cross-reactivity toward PPARγ, in the same range as for 1. Together, the cross-reactivity of the isoxazoles on PPARγ can be modulated by only small changes in the pyrrole substitution pattern. Gratifyingly, the most potent compounds for RORγt (3 and 6) also feature the highest selectivity for RORγt over PPARγ.
Lastly, the isoxazole compounds were tested on the farnesoid X receptor (FXR) to exclude cross-reactivity because trisubstituted isoxazole ligands, such as GW4064, have been reported as potent FXR agonists.46,47 The most potent RORγt isoxazole ligands and GW4064 were evaluated in a fluorescence polarization (FP) assay. GW4064 showed a typical agonistic dose–response curve for FXR, whereas the novel isoxazole ligands did not elicit an agonistic response on FXR (Figure S7A). Additionally, the EC50 value of GW4064 was not significantly affected by the presence of isoxazole 3 (Figure S7B), thus also excluding an antagonistic behavior of 3 on FXR.
2.7. Absorption, Distribution, Metabolism, and Excretion Profile
Previous studies on the absorption, distribution, metabolism, and excretion (ADME) parameters of the isoxazoles showed that these were promising but not optimal yet for compound 2, most probably governed by the pyrrole moiety and the amine linker, which are both likely to be prone to oxidation.28,48 The ADME profiling of the novel isoxazoles (Table 4) demonstrates that compounds 3, 6, 9, and 10 in general show comparable properties to hit compound 2, with promising values for the kinetic solubility and (passive) membrane permeability, especially because the compounds contain a carboxylic acid moiety which generally leads to lower permeability values. The metabolic stability in human liver microsomes showed that the clearance values in phase I metabolism are rather high, similar to 2. Glucuronidation assays (as the most important phase II pathway) showed promising improved values (% remain), especially for compound 10. The plasma stability (except for 6) was slightly lower than for 1, and relatively high levels of blood plasma protein binding were measured. Contrastingly, compound 11, containing a pyrazole instead of a pyrrole substituent at the C-5 position, showed a rather different ADME profile. It demonstrated a promising stability in phase I metabolism and was highly stable in plasma, although the phase II stability (glucuronidation) and permeability were lower than for the other isoxazoles.
Table 4. ADME Properties for Compounds 1, 2, 3, 6, 9, 10, and 11.
| microsomal
stability |
||||||
|---|---|---|---|---|---|---|
| compound | kinetic solubility (μM) | PAMPA (% flux) | phase 1 (Clint, μL/min/mg) | glucuronidation in vitro (% remaining) | plasma stability (% remaining) | plasma protein binding (% bound) |
| 1 | 423 | 29.7 | 0.4 | 37.7 | 95.5 | 99.7 |
| 2 | 460 | 36.1 | 31.2 | 73.0 | 79.1 | 99.9 |
| 3 | 436 | 40.9 | 57.8 | 30.7 | 74.2 | 99.7 |
| 6 | 392 | 40.3 | 69.0 | 58.3 | 59.5 | 99.9 |
| 9 | 445 | 37.3 | 77.0 | 54.6 | 97.1 | 99.8 |
| 10 | 421 | 50.5 | 68.0 | 68.9 | 89.5 | 99.3 |
| 11 | 476 | 23.0 | 8.8 | 39.1 | 100 | 98.8 |
The open-source program OSIRIS Property Explorer49 was used to predict the drug likeness and toxicity of the compounds (Table S3) to have a first indication on these aspects. All isoxazole compounds (except 5) were predicted to have no side effects (i.e., mutagenic, tumorigenic, irritant, and reproductive effects), whereas 1 was predicted to have a potentially high risk of reproductive toxicity. Additionally, all isoxazole ligands showed higher drug scores than 1, with pyrazole 11 showing the best properties, again highlighting its potential for further development.
Combined, the change of an amine to an ether/alkene linker (2 vs 3 and 6) does not significantly affect the ADME(T) properties (except for the glucuronidation of 3), while the change of a pyrrole to a pyrazole moiety (11) results in a differentiated, promising profile. The C-4/C-5 isoxazole modifications thus provide valuable entry points for optimization of the ADME properties.
3. Conclusions
The NR RORγt has an important regulatory role in the immune system, and inhibition of RORγt via allosteric inverse agonists could be a promising strategy in the treatment of autoimmune disorders. Apart from indazoles, with 1 (MRL-871) as a prominent example, there has been little chemical diversity of allosteric RORγt modulators. Trisubstituted isoxazoles constitute a distinct novel chemotype in this matter, with 2 (FM26) as an exemplary potent hit compound.
Here, we further optimized this isoxazole series, specifically focusing on the linker at the C-4 position and the heterocycle at the C-5 position to deliver lead compounds for further optimization. The optimization of the linker at the C-4 position revealed a clear correlation between potency and the lipophilicity and flexibility of the linker, with an ether or alkene linker resulting in the highest potency. The co-crystal structure of RORγt in complex with ether compound 3 revealed the role of hydrophobic interactions and conformation of the linker at the C-4 position. The most potent compounds 3 (FM156) and 6 (FM260) were highly selective for RORγt over PPARγ, which is a valuable improvement compared to the exemplary indazole 1.
The focused library of C-5 isoxazoles revealed several routes for chemical exploration. Changing the position of the hydrogen bond donor via the repositioning of the pyrrole ring (10) led only to a 3-fold lower potency, with the formation of another hydrogen bond interaction network via a water molecule in the RORγt pocket. Changing the pyrrole to a pyrazole (with a lower pKa) in compound 11 (FM257) was also tolerated and resulted in a promising PK profile, opening up routes for affinity and ADME optimization.
In conclusion, by probing the effect of the linker at the C-4 position and the importance of the polar character of the C-5 moiety, this study has led to valuable leads as allosteric inverse agonists for RORγt with improved potency, increased selectivity against PPARγ and FXR, and promising initial ADME properties. The results not only provide new insights into the SAR for this specific isoxazole class of allosteric RORγt ligands but can also most probably similarly be translated to other allosteric RORγt chemotypes. Overall, the trisubstituted isoxazole class of allosteric RORγt ligands shows high potential for chemical biology approaches as well as for future development in drug discovery programs against autoimmune diseases.
4. Experimental Section
4.1. In Silico Studies
4.1.1. Molecular Docking Studies
The receptor–ligand complex structure (PDB code: 4YPQ) was prepared using the Protein Preparation Wizard within Maestro (version 12.3 (2020-1), Schrödinger LLC, New York, NY, USA) (default parameters). A receptor grid centered on the bound ligand was generated using the Protein Preparation Wizard. All parameters were kept as the default. The examined ligands were drawn in ChemDraw. Energy-minimized 3D ligand conformations for each molecule to be investigated were generated using the Ligand Preparation Wizard within Maestro (default parameters). Ligands were docked using Glide in standard precision mode with flexible ligand sampling. The predicted binding modes of all ligands were ranked according to their glide score.
4.1.2. In Silico Drug Likeness and Toxicity Predictions
OSIRIS Property Explorer utilizes the database of traded drugs and commercially available compounds (Fluka), assumable as nondrug-like data set, to assess the occurrence frequency of each fragment in the individual structure.49 The program was used to estimate the risks of side effects, such as mutagenic, tumorigenic, irritant, and reproductive effects, as well as drug likeness and overall drug score (by combining the outcome of clog P, log S (solubility), MW, toxicity risks, and drug likeness). The drug score is a measure of the compound’s potential to meet the criteria of a possible drug candidate.
4.2. General Chemistry
All nonaqueous reactions were performed under an argon atmosphere unless otherwise stated. Water-sensitive reactions were performed in oven-dried glassware, cooled under argon before use. All solvents were supplied by Biosolve or Sigma-Aldrich and used without further purification. Dry solvents were obtained from a MBRAUN solvent purification system (MB-SPS-800). Water was purified by a Millipore purification train. Deuterated solvents were obtained from Cambridge Isotope Laboratories. Solvents were removed in vacuo using a Büchi rotary evaporator and a diaphragm pump. Commercially available starting materials were obtained from Sigma-Aldrich, TCI Chemicals and Fluorochem. Proton (1H) NMR (400 MHz), carbon (13C) NMR (100 MHz), and 2D NMR (400 MHz) were recorded on a Bruker Avance 400 MHz NMR spectrometer. Proton spectra are referenced to tetramethyl silane (TMS). Carbon spectra are referenced to TMS or the solvent peak of the deuterated spectrum. NMR spectra are reported as follows: chemical shift (δ) in parts per million (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, td = triplet of doublets, app. = apparent), coupling constant (J) in Hertz (Hz) (if applicable), and integration (proton spectra only). Peak assignments are based on additional 2D NMR techniques (COSY, HMBC, and HSQC). Analytical liquid chromatography coupled with mass spectrometry (LC–MS) was performed on a C4 Jupiter SuC4300A 150 × 2.0 mm column using ultrapure water with 0.1% formic acid (FA) and acetonitrile with 0.1% FA, in general with a gradient of 5–100% acetonitrile for over 10 min, connected to a Thermo Fisher LCQ fleet ion-trap mass spectrometer. The purity of the samples was assessed using a UV detector at 254 nm. Unless otherwise stated all final compounds were >95% pure as judged by HPLC. High-resolution mass spectra (HRMS) were recorded using a Waters ACQUITY UPLC I-Class LC system coupled to a Xevo G2 quadrupole time-of-flight (Q-TOF) mass spectrometer. Preparative HP-LC was performed on a Gemini S4 110A 150 × 21.20 mm column using ultrapure water with 0.1% FA and acetonitrile with 0.1% FA with various gradients (mentioned for each compound specifically). Column chromatography was either performed manually using silica gel (60–200 μm particle size, 60 AÅ) or using an automated Grace Reveleris X2 chromatograph with pre-packed silica columns supplied by Buchi/Grace (40 μm particle size). The reaction progress was monitored by thin-layer liquid chromatography (TLC) using Merck TLC silica gel 60 F254 plates. The visualization of the plates was achieved using an ultraviolet lamp (λmax = 254 nm).
4.2.1. General Procedure for Suzuki Coupling
Under an inert atmosphere, pinacol boronate (2.0 equiv), Cs2CO3 (2.0 equiv), and Pd(dppf)Cl2 (0.1 equiv) were added to a solution of bromide 14 (1.0 equiv) in degassed 1,2-dimethoxyethane (DME) (0.1 M). The reaction mixture was heated at 85 °C for 8 h, cooled to room temperature, diluted with H2O, and extracted with EtOAc (3×). The combined organic phase was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography using the specified eluent to afford the desired product.
4.2.2. General Procedure for the Reduction of Esters to Alcohols
Under an inert atmosphere, DIBAL (1.23 g/mL in cyclohexane, 15.0 equiv) was added dropwise to a solution of ester (1.0 equiv) in anhydrous CH2Cl2 (0.1 M) at −78 °C. The reaction mixture was followed by TLC analysis. Upon complete consumption of the starting material, the reaction mixture was warmed to room temperature, quenched by the addition of a saturated Rochelle salt (KNaC4H4O6·4H2O) solution and stirred vigorously for 60 min. Subsequently, the mixture was extracted with CH2Cl2 (3×) and separated. The combined organic phase was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo to afford the title compound which was purified as described.
4.2.3. General Procedure for Mesylation and Substitution
Under an inert atmosphere, triethylamine (3.0 equiv) was added to a solution of alcohol compound 16 (1.0 equiv) in anhydrous CH2Cl2 (0.1 M). Methanesulfonic anhydride (1.5 equiv) was added to the flask, and the reaction mixture was heated at 40 °C. The reaction was monitored using NMR analysis. Subsequently, aniline (5.0 equiv) was added, and the reaction mixture was stirred for 1 h, cooled to room temperature, and CH2Cl2 was removed in vacuo. The crude product was purified by flash column chromatography using the specified eluent.
4.2.4. General Procedure for Ester Hydrolysis and N-Boc Deprotection
LiOH·H2O (10.0 equiv) was added to a suspension of ester (1.0 equiv) in a 4:1 mixture of EtOH/H2O (0.05 M). The reaction mixture was heated to 95 °C followed by TLC analysis. Upon complete consumption of the starting material, EtOH was removed in vacuo, and the resulting aqueous mixture was acidified to pH 3 using 10% v/v aqueous HCl and extracted with a 9:1 mixture of CH2Cl2/MeOH (5×). The combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo to furnish the final compound which was purified as described.
4.2.5. General Procedure for Mitsunobu Coupling
Under an inert atmosphere, triphenylphosphine (2.0 equiv) and DIAD (2.0 equiv) were dissolved in anhydrous tetrahydrofuran (THF) (0.05 M) and stirred for 15 min at 0 °C. Subsequently, the alcohol compound (1.0 equiv), the benzoate (1.1 equiv), and triethylamine (1.0 equiv) were added, and the reaction mixture was heated at 80 °C for 3 h, cooled to room temperature, and THF was removed in vacuo. The crude product was purified by flash column chromatography using the specified eluent.
4.2.6. 4-(((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl)methyl) amino) Benzoic Acid (2)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 17 (0.035 g, 0.067 mmol) was hydrolyzed with a concomitant loss of the Boc protecting group. The crude product was purified by column chromatography, eluted with 1.5% MeOH in CH2Cl2, to furnish carboxylic acid 2 (10.3 mg, 33%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.98 (1H, br s, CO2H), 11.52 (1H, s, pyrrole-NH), 7.90 (1H, d, J = 8.0 Hz, ArH-3), 7.84 (1H, d, J = 7.8 Hz, ArH-5), 7.72 (1H, app. t, J = 8.0 Hz, ArH-4), 7.53 (2H, d, J = 8.7 Hz, benzoate C-2), 7.39 (1H, m, pyrrole H-2), 6.98 (1H, m, pyrrole H-5), 6.55 (1H, m, pyrrole H-4), 6.41 (2H, d, J = 8.7 Hz, benzoate H-3), 6.34 (1H, app. t, J = 4.4 Hz, CH2NH), 4.07 (1H, dd, J = 14.4, 5.0 Hz, CHaNH), 4.02 (1H, dd, J = 14.5, 4.5 Hz, CHbNH); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 167.9 (CO2H), 165.7 (C-5), 159.2 (C-3), 152.3 (benzoate C-4), 136.0 (ArC-2), 134.1 (ArC-3), 132.4 (ArC-4), 131.3 (benzoate C-2), 130.9 (q, J = 30.4 Hz, ArC-6), 126.8 (ArC-1), 125.8 (q, J = 5.0 Hz, ArC-5), 122.0 (q, J = 274.3 Hz, CF3), 120.5 (pyrrole C-5), 119.2 (pyrrole C-2), 117.8 (benzoate C-1), 111.2 (benzoate C-3), 110.5 (pyrrole C-3), 109.2 (C-4), 106.7 (pyrrole C-4), 36.2 (CH2NH). LC–MS (ESI): calcd for C22H15ClF3N3O3 [M + H]+, 462.08; observed, 462.17 (Rt = 6.13 min). HRMS (ESI): calcd for C22H15ClF3N3O3 [M + H]+, 462.0832; observed, 462.0835.
4.2.7. 4-((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl)methoxy) Benzoic Acid (3)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 20 (0.020 g, 0.035 mmol) was hydrolyzed with a concomitant loss of the Boc-protecting group. The crude product was purified by column chromatography, eluting with 1.5% MeOH in CH2Cl2, to furnish carboxylic acid 3 (12.0 mg, 75%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.78 (1H, br s, CO2H), 7.92 (4H, m, ArH-3, ArH-5 and benzoate C-2), 7.77 (1H, app. t, J = 8.0 Hz, ArH-4), 7.47 (1H, m, pyrrole H-2), 7.03 (1H, m, pyrrole H-5), 6.94 (2H, d, J = 8.8 Hz, benzoate H-3), 6.64 (1H, m, pyrrole H-4), 5.04 (2H, s, CH2O); 13C NMR (100 MHz, acetone-d6): δ (ppm) 167.6 (C-5), 167.3 (benzoate C-4), 163.1 (CO2H), 159.8 (C-3), 137.2 (ArC-2), 134.4 (ArC-3), 132.8 (q, J = 30.4 Hz, ArC-6), 132.6 (benzoate C-2), 132.5 (ArC-4), 127.9 (ArC-1), 126.1 (q, J = 5.0 Hz, ArC-5), 124.2 (benzoate C-1), 122.7 (q, J = 274.3 Hz, CF3), 121.0 (pyrrole C-5), 120.0 (pyrrole C-2), 115.1 (benzoate C-3), 111.6 (pyrrole C-3), 108.3 (C-4), 107.4 (pyrrole C-4), 60.8 (CH2O). LC–MS (ESI): calcd for C22H14ClF3N2O4 [M + H]+, 463.06; observed, 463.25 (Rt = 4.62 min). HRMS (ESI): calcd for C22H14ClF3N2O4 [M + H]+, 463.0672; observed, 463.0650.
4.2.8. 4-(((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl)methyl)thio) Benzoic Acid (4)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 22 (0.020 g, 0.034 mmol) was hydrolyzed with a concomitant loss of the Boc protecting group. The crude product was purified by column chromatography, eluting with 2% MeOH in CH2Cl2, to furnish carboxylic acid 4 (8.00 mg, 49%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.77 (1H, br s, CO2H), 7.89 (4H, m, ArH-3, ArH-5 and benzoate C-2), 7.79 (1H, app. t, J = 8.0 Hz, ArH-4), 7.52 (1H, m, pyrrole H-2), 7.31 (2H, d, J = 8.5 Hz, benzoate H-3), 7.03 (1H, m, pyrrole H-5), 6.68 (1H, m, pyrrole H-4), 4.22 (2H, s, CH2S); 13C NMR (100 MHz, acetone-d6): δ (ppm) 167.2 (CO2H), 166.8 (C-5), 159.8 (C-3), 144.6 (benzoate C-4), 137.2 (ArC-2), 134.5 (ArC-3), 132.7 (ArC-4), 132.5 (q, J = 30.4 Hz, ArC-6), 131.4 (benzoate C-1), 130.9 (benzoate C-2), 128.3 (ArC-1), 127.3 (benzoate C-3), 126.3 (q, J = 5.0 Hz, ArC-5), 122.7 (q, J = 274.3 Hz, CF3), 120.9 (pyrrole C-5), 119.7 (pyrrole C-2), 111.6 (pyrrole C-3), 107.3 (C-4), 107.3 (pyrrole C-4), 26.2 (CH2S). LC–MS (ESI): calcd for C22H14ClF3N2O3S [M + H]+, 479.04; observed, 479.25 (Rt = 4.86 min). HRMS (ESI): calcd for C22H14ClF3N2O3S [M + H]+, 479.0444; observed, 479.0438.
4.2.9. 4-(((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl)methyl) (methyl)amino) Benzoic Acid (5)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 18 (0.040 g, 0.068 mmol) was hydrolyzed with a concomitant loss of the Boc protecting group. The crude product was purified by column chromatography, eluting with 2% MeOH in CH2Cl2, to furnish carboxylic acid 5 (19.3 mg, 60%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 12.08 (1H, br s, CO2H), 11.60 (1H, s, pyrrole-NH), 7.80 (1H, d, J = 8.1 Hz, ArH-3), 7.76 (1H, d, J = 7.8 Hz, ArH-5), 7.64 (1H, app. t, J = 8.0 Hz, ArH-4), 7.53 (1H, m, pyrrole H-2), 7.49 (2H, d, J = 8.9 Hz, benzoate C-2), 7.02 (1H, m, pyrrole H-5), 6.63 (1H, m, pyrrole H-4), 6.33 (2H, d, J = 8.9 Hz, benzoate H-3), 4.48 (2H, s, CH2NCH3), 2.54 (3H, s, CH2NCH3); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 167.3 (CO2H), 165.0 (C-5), 158.5 (C-3), 151.7 (benzoate C-4), 135.5 (ArC-2), 133.6 (ArC-3), 131.8 (ArC-4), 130.5 (benzoate C-2), 130.1 (q, J = 30.4 Hz, ArC-6), 126.5 (ArC-1), 125.4 (q, J = 5.0 Hz, ArC-5), 124.2 (q, J = 274.3 Hz, CF3), 120.2 (pyrrole C-5), 118.8 (pyrrole C-2), 117.3 (benzoate C-1), 110.5 (benzoate C-3), 109.7 (pyrrole C-3), 109.0 (C-4), 106.3 (pyrrole C-4), 43.9 (CH2NCH3), 37.0 (CH2NCH3). LC–MS (ESI): calcd for C23H17ClF3N3O3 [M + H]+, 476.09; observed, 476.17 (Rt = 6.33 min). HRMS (ESI): calcd for C23H17ClF3N3O3 [M + H]+, 476.0989; observed, 476.0971.
4.2.10. (E)-4-(2-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl) vinyl) Benzoic Acid (6)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 24 (0.012 g, 0.021 mmol) was hydrolyzed with a concomitant loss of the Boc protecting group. The crude product was purified via preparative HPLC (gradient of 70–73% acetonitrile in H2O) to furnish carboxylic acid 6 (6.1 mg, 63%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.85 (1H, br s, CO2H), 8.00 (2H, ArH-3 and ArH-5), 7.92 (3H, m, benzoate C-2 and ArH-4), 7.62 (1H, m, pyrrole H-2), 7.39 (3H, m, benzoate H-3 and C4-HC=CH), 7.08 (1H, m, pyrrole H-5), 6.74 (1H, m, pyrrole H-4), 6.15 (1H, s, benzoate C4-HC=CH); 13C NMR (100 MHz, acetone-d6): δ (ppm) 166.3 (CO2H), 165.2 (C-5), 156.8 (C-3), 141.5 (benzoate C-4), 136.1 (ArC-2), 133.8 (ArC-3), 131.9 (ArC-4), 131.4 (q, J = 30.4 Hz, ArC-6), 130.0 (benzoate C-2), 129.4 (benzoate C-1), 128.1 (benzoate C4-HC=CH), 128.0 (ArC-1), 126.0 (benzoate C-3), 125.6 (q, J = 5.0 Hz, ArC-5), 121.9 (q, J = 274.3 Hz, CF3), 120.2 (pyrrole C-5), 119.3 (pyrrole C-2), 118.8 (C4-HC=CH), 110.9 (pyrrole C-3), 110.3 (C-4), 106.7 (pyrrole C-4). LC–MS (ESI): calcd for C23H14ClF3N2O3 [M + H]+, 459.06; observed, 459.25 (Rt = 4.90 min). HRMS (ESI): calcd for C23H14ClF3N2O3 [M + H]+, 459.0723; observed, 459.0726.
4.2.11. (Z)-4-(2-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl) vinyl) Benzoic Acid (7)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 25 (0.040 g, 0.070 mmol) was hydrolyzed with a concomitant loss of the Boc-protecting group. The crude product was purified via preparative HPLC (gradient of 70–73% acetonitrile in H2O) to furnish carboxylic acid 7 (26.0 mg, 81%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.50 (1H, br s, CO2H), 7.80 (5H, m, ArH-3, ArH-5, ArH-4 and benzoate C-2), 7.36 (2H, d, J = 8.2 Hz, benzoate H-3), 7.21 (1H, m, pyrrole H-2), 6.79 (1H, m, pyrrole H-5), 6.74 (1H, d, J = 12.2 Hz, benzoate C4-HC=CH), 6.49 (1H, m, pyrrole H-4), 6.29 (1H, d, J = 12.2 Hz, C4-HC=CH); 13C NMR (100 MHz, acetone-d6): δ (ppm) 166.3 (CO2H), 163.7 (C-5), 158.4 (C-3), 141.1 (benzoate C-4), 136.1 (ArC-2), 133.5 (benzoate C4-HC=CH), 133.5 (ArC-3), 131.6 (ArC-4), 131.3 (q, J = 30.4 Hz, ArC-6), 129.4 (benzoate C-2), 128.4 (benzoate C-3), 127.2 (benzoate C-1), 125.3 (q, J = 5.0 Hz, ArC-5), 124.5 (ArC-1), 121.8 (q, J = 274.3 Hz, CF3), 119.3 (pyrrole C-5), 118.8 (pyrrole C-2), 118.7 (C4-HC=CH), 111.2 (pyrrole C-3), 108.6 (C-4), 106.3 (pyrrole C-4). LC–MS (ESI): calcd for C23H14ClF3N2O3 [M + H]+, 459.06; observed, 459.25 (Rt = 4.87 min). HRMS (ESI): calcd for C23H14ClF3N2O3 [M + H]+, 459.0723; observed, 459.0715.
4.2.12. 4-(((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl)methyl) amino)-2-fluorobenzoic Acid (8)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 19 (0.008 g, 0.016 mmol) was hydrolyzed. The crude product was purified by column chromatography, eluting with 1.5% MeOH in CH2Cl2, to furnish carboxylic acid 8 (2.2 mg, 29%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 12.09 (1H, br s, CO2H), 11.5 (1H, s, pyrrole-NH), 7.87 (1H, d, J = 8.0 Hz, ArH-3), 7.82 (1H, d, J = 7.9 Hz, ArH-5), 7.72 (1H, app. t, J = 7.9 Hz, ArH-4), 7.47 (1H, app. t, J = 8.8 Hz, benzoate H-2), 7.38 (1H, m, pyrrole H-2), 6.97 (1H, m, pyrrole H-5), 6.64 (1H, app. t, J = 4.8 Hz, CH2NH), 6.55 (1H, m, pyrrole H-4), 6.24 (1H, dd, J = 8.8, 2.1 Hz, benzoate H-3a), 6.10 (1H, dd, J = 14.4, 1.8 Hz, benzoate H-3b), 4.06 (2H, m, CH2NH). LC–MS (ESI): calcd for C22H14ClF4N3O3 [M + H]+, 480.07; observed, 480.25 (Rt = 6.21 min). HRMS (ESI): calcd for C22H14ClF4N3O3 [M + H]+, 480.0738; observed, 480.0759.
4.2.13. 4-((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl)methoxy)-2-fluorobenzoic Acid (9)
According to the general procedure for ester hydrolysis and N-Boc deprotection, ester 21 (0.017 mg, 0.029 mmol) was hydrolyzed with a concomitant loss of the Boc protecting group. The crude product was purified by column chromatography, eluting with 1.5% MeOH in CH2Cl2, to furnish carboxylic acid 9 (8.0 mg, 57%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.81 (1H, br s, CO2H), 7.98 (3H, m, ArH-3, ArH-5 and benzoate C-2), 7.78 (1H, app. t, J = 8.0 Hz, ArH-4), 7.48 (1H, m, pyrrole H-2), 7.02 (1H, m, pyrrole H-5), 6.73 (2H, m, benzoate H-3), 6.64 (1H, m, pyrrole H-4), 5.06 (2H, s, CH2O); 13C NMR (100 MHz, acetone-d6): δ (ppm) 167.4 (C-5), 165.5 (CO2H), 164.0 (benzoate C-4), 162.9 (benzoate C–F), 159.7 (C-3), 137.2 (ArC-2), 134.4 (ArC-3), 132.7 (q, J = 30.4 Hz, ArC-6), 132.6 (Ar C-4), 132.4 (benzoate C-2), 127.8 (ArC-1), 126.1 (q, J = 5.0 Hz, ArC-5), 125.4 (benzoate C-1), 122.7 (q, J = 274.3 Hz, CF3), 121.0 (pyrrole C-5), 120.0 (pyrrole C-2), 111.7 (benzoate C-3a), 111.5 (pyrrole C-3), 108.0 (C-4), 107.4 (pyrrole C-4), 103.7 (benzoate C-3b), 61.3 (CH2O). LC–MS (ESI): calcd for C22H13ClF4N2O3 [M + H]+, 481.05; observed, 481.25 (Rt = 4.92 min). HRMS (ESI): calcd for C22H13ClF4N2O3 [M + H]+, 481.0578; observed, 481.0572.
4.2.14. 4-((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-2-yl)isoxazol-4-yl)methoxy) Benzoic Acid (10)
According to the general procedure for Mitsunobu coupling (without the addition of triethylamine), alcohol 30 (0.080 g, 0.180 mmol) was reacted with methyl 4-hydroxybenzoate (0.030 g, 0.200 mmol). The crude product was purified by column chromatography, eluting with a gradient of 5–25% EtOAc in n-heptane, to furnish the ether compound (22.0 mg, 21%). The resulting product (0.012 g, 0.021 mmol) was subject to ester hydrolysis with a concomitant loss of the Boc protecting group according to the general procedure for ester hydrolysis and N-Boc deprotection. The crude product was purified by column chromatography, eluting with 1.5% MeOH in CH2Cl2, to furnish carboxylic acid 10 (4.0 mg, 41%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 11.13 (1H, br s, CO2H), 7.91 (4H, m, ArH-3, ArH-5 and benzoate C-2), 7.80 (1H, app. t, J = 8.0 Hz, ArH-4), 7.19 (1H, m, pyrrole H-5), 6.93 (2H, d, J = 8.4 Hz, benzoate H-3), 6.79 (1H, m, pyrrole H-3), 6.34 (1H, m, pyrrole H-4), 5.09 (2H, s, CH2O); 13C NMR (100 MHz, acetone-d6): δ (ppm) 162.4 (C-5), 162.0 (CO2H), 161.2 (benzoate C-4), 159.1 (C-3), 136.2 (ArC-2), 133.5 (ArC-3), 131.9 (benzoate C-2), 131.6 (ArC-4), 131.5 (q, J = 30.4 Hz, ArC-6), 126.5 (ArC-1), 125.3 (q, J = 5.0 Hz, ArC-5), 123.2 (benzoate C-1), 122.7 (pyrrole C-5), 121.8 (q, J = 274.3 Hz, CF3), 118.7 (pyrrole C-2), 114.2 (benzoate C-3), 111.6 (pyrrole C-3), 111.3 (pyrrole C-4), 107.6 (C-4), 59.8 (CH2O). LC–MS (ESI): calcd for C22H14ClF3N2O4 [M + H]+, 463.06; observed, 463.17 (Rt = 4.87 min). HRMS (ESI): calcd for C22H14ClF3N2O4 [M + H]+, 463.0672; observed, 463.0665.
4.2.15. 4-((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrazol-4-yl)isoxazol-4-yl) methoxy) Benzoic Acid (11)
According to the general procedure for Mitsunobu coupling, alcohol 31 (0.100 g, 0.234 mmol) was reacted with methyl 4-hydroxybenzoate (0.039 g, 0.257 mmol). The crude product was purified by column chromatography, eluting with a gradient of 10–25% EtOAc in n-heptane, to furnish the ether compound (65.0 mg, 49%). The ether compound (0.045 g, 0.080 mmol), containing a THP-protected pyrazole, was diluted in a mixture of TFA/CH2Cl2 (50/50) (0.05 M), and the reaction mixture was stirred at 40 °C for 2 h. Afterward, it was cooled to room temperature, and the solvent was removed in vacuo. The crude product was purified by column chromatography, eluting with 25–40% EtOAc in n-heptane, to furnish the pyrazole product (28.0 mg, 88%). The resulting product was subjected to ester hydrolysis according to the general procedure for ester hydrolysis and N-Boc deprotection. The crude product was purified via preparative HPLC (gradient of 62–67% acetonitrile in H2O) to furnish carboxylic acid 11 (26.0 mg, 89%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 8.24 (2H, s, pyrazole H-3 and H-5), 7.92 (4H, m, ArH-3, ArH-5 and benzoate C-2), 7.80 (1H, app. t, J = 7.9 Hz, ArH-4), 6.94 (2H, d, J = 8.1 Hz, benzoate H-3), 5.12 (2H, s, CH2O); 13C NMR (100 MHz, acetone-d6): δ (ppm) 167.2 (CO2H), 164.6 (C-5), 162.9 (benzoate C-4), 159.9 (C-3), 137.1 (ArC-2), 134.4 (ArC-3), 133.6 (pyrazole C-3 and C-5), 133.0 (q, J = 30.4 Hz, ArC-6), 132.8 (ArC-4), 132.4 (benzoate C-2), 127.4 (ArC-1), 126.2 (q, J = 5.0 Hz, ArC-5), 124.3 (benzoate C-1), 122.7 (q, J = 274.3 Hz, CF3), 115.1 (benzoate C-3), 110.2 (C-4), 109.6 (pyrrole C-4). LC–MS (ESI): calcd for C21H13ClF3N3O4 [M + H]+, 464.05; observed, 464.08 (Rt = 4.45 min). HRMS (ESI): calcd for C21H13ClF3N3O4 [M + H]+, 464.0625; observed, 464.0610.
4.2.16. 4-((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1-methyl-1H-pyrrol-3-yl)isoxazol-4-yl)methoxy) Benzoic Acid (12)
According to the general procedure for Mitsunobu coupling, alcohol 32 (0.175 g, 0.490 mmol) was reacted with methyl 4-hydroxybenzoate (0.082 g, 0.540 mmol). The crude product was purified by column chromatography, eluting with a gradient of 5–20% EtOAc in n-heptane, to furnish the ether compound (109 mg, 45%). The resulting product (0.060 g, 0.122 mmol) was subjected to ester hydrolysis according to the general procedure for ester hydrolysis and N-Boc deprotection. The crude product was purified by column chromatography, eluting with 1.5% MeOH in CH2Cl2, to furnish carboxylic acid 12 (50.0 mg, 86%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 7.92 (4H, m, ArH-3, ArH-5 and benzoate C-2), 7.76 (1H, app. t, J = 8.0 Hz, ArH-4), 7.38 (1H, m, pyrrole H-2), 6.92 (2H, d, J = 8.8 Hz, benzoate H-3), 6.88 (1H, m, pyrrole H-5), 6.56 (1H, m, pyrrole H-4), 5.04 (2H, s, CH2O), 3.78 (3H, s, NCH3); 13C NMR (100 MHz, acetone-d6): δ (ppm) 167.3 (C-5), 167.1 (CO2H), 163.1 (benzoate C-4), 159.7 (C-3), 137.1 (ArC-2), 134.3 (ArC-3), 132.7 (q, J = 30.4 Hz, ArC-6), 132.5 (ArC-4), 132.4 (benzoate C-2), 127.9 (ArC-1), 126.1 (q, J = 5.1 Hz, ArC-5), 124.8 (pyrrole C-5), 124.1 (benzoate C-1), 123.3 (pyrrole C-2), 122.7 (q, J = 274.0 Hz, CF3), 115.1 (benzoate C-3), 111.6 (pyrrole C-3), 108.2 (C-4), 107.8 (pyrrole C-4), 60.8 (CH2O), 36.7 (NCH3). LC–MS (ESI): calcd for C23H16ClF3N2O4 [M + H]+, 477.08; observed, 477.33 (Rt = 5.06 min). HRMS (ESI): calcd for C23H16ClF3N2O4 [M + H]+, 477.0829; observed, 477.0813.
4.2.17. 4-((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(5-methyl-1H-pyrrol-3-yl)isoxazol-4-yl)methoxy) Benzoic Acid (13)
According to the general procedure for Mitsunobu coupling, alcohol 33 (0.175 g, 0.383 mmol) was reacted with methyl 4-hydroxybenzoate (0.064 g, 0.421 mmol). The crude product was purified by column chromatography, eluting with a gradient of 10–40% EtOAc in n-heptane, to furnish the ether compound (100 mg, 44%). The resulting product (0.030 g, 0.051 mmol) was subjected to ester hydrolysis with a concomitant loss of the Boc protecting group according to the general procedure for ester hydrolysis and N-Boc deprotection. The crude product was purified via preparative HPLC (gradient of 65–70% acetonitrile in H2O) to furnish carboxylic acid 13 (20.0 mg, 83%) as a white solid. 1H NMR (400 MHz, acetone-d6): δ (ppm) 10.47 (1H, br s, CO2H), 7.90 (4H, m, ArH-3, ArH-5 and benzoate C-2), 7.77 (1H, app. t, J = 8.0 Hz, ArH-4), 7.29 (1H, m, pyrrole H-2), 6.93 (2H, d, J = 8.5 Hz, benzoate H-3), 6.44 (1H, m, pyrrole H-4), 5.02 (2H, s, CH2O), 2.29 (3H, s, NCCH3); 13C NMR (100 MHz, acetone-d6): δ (ppm) 167.7 (C-5), 167.3 (benzoate C-4), 163.1 (CO2H), 159.7 (C-3), 137.2 (ArC-2), 134.4 (ArC-3), 132.7 (q, J = 30.4 Hz, ArC-6), 132.5 (benzoate C-2), 132.4 (ArC-4), 130.7 (pyrrole C-5), 128.0 (ArC-1), 126.1 (q, J = 5.0 Hz, ArC-5), 124.2 (benzoate C-1), 122.7 (q, J = 274.3 Hz, CF3), 119.7 (pyrrole C-2), 115.1 (benzoate C-3), 111.7 (pyrrole C-3), 108.0 (C-4), 105.2 (pyrrole C-4), 60.8 (CH2O), 12.6 (NCCH3). LC–MS (ESI): calcd for C23H16ClF3N2O4 [M + H]+, 477.08; observed, 477.25 (Rt = 4.88 min). HRMS (ESI): calcd for C23H16ClF3N2O4 [M + H]+, 477.0829; observed, 477.0830.
4.2.18. Methyl 5-Bromo-3-(2-chloro-6-(trifluoromethyl)phenyl)isoxazole-4-carboxylate (15)
According to the general procedure for Suzuki coupling, bromide 14 (0.900 g, 2.34 mmol) was reacted with tert-butyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole-1-carboxylate (1.37 g, 4.68 mmol). The crude product was purified by flash column chromatography, eluting with a gradient of 5–10% EtOAc in n-heptane, to furnish 15 (0.553 g, 50%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.44 (1H, app. t, J = 2.0 Hz, pyrrole H-2), 7.76–7.65 (2H, m, ArH-3 and ArH-5), 7.55 (1H, app. t, J = 8.0 Hz, ArH-4), 7.36 (1H, dd, J = 3.4, 2.2 Hz, pyrrole H-5), 6.99 (1H, dd, J = 3.4, 1.6 Hz, pyrrole H-4), 3.61 (3H, s, CO2CH3), 1.64 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 168.9 (C-5), 161.5 (CO2CH3), 158.9 (C-3), 148.1 (NCO2), 136.2 (ArC-2), 132.7 (ArC-3), 131.4 (q, J = 31.4 Hz, ArC-6), 130.5 (ArC-4), 127.7 (ArC-1), 124.6 (q, J = 5.0 Hz, ArC-5), 124.1 (pyrrole C-2), 121.6 (q, J = 274.4 Hz, CF3), 120.9 (pyrrole C-5), 113.7 (pyrrole C-3), 111.5 (pyrrole C-4), 107.1 (C-4), 85.1 (C(CH3)3), 51.6 (CO2CH3), 27.9 (C(CH3)3). LC–MS (ESI): calcd for C21H18ClF3N2O5 [M + H]+, 471.09; observed, 470.92 (Rt = 6.04 min).
4.2.19. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-(hydroxymethyl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (16)
Ester 15 (0.100 g, 0.212 mmol) was treated according to the general procedure for the reduction of esters to alcohols. The crude product was purified by flash column chromatography, eluting with a gradient of 10–20% EtOAc in n-heptane, to furnish alcohol 16 (0.075 g, 80%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.88 (1H, s, pyrrole H-2), 7.78–7.68 (2H, m, ArH-3 and ArH-5), 7.57 (1H, app. t, J = 8.0 Hz, ArH-4), 7.37–7.33 (1H, m, pyrrole H-5), 6.79–6.73 (1H, m, pyrrole H-4), 4.42 (2H, br s, CH2OH), 1.63 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 164.6 (C-5), 158.7 (C-3), 148.2 (NCO2), 136.5 (ArC-2), 133.1 (ArC-3), 132.2 (q, J = 31.2 Hz, ArC-6), 131.0 (ArC-4), 126.7 (ArC-1), 124.9 (q, J = 5.1 Hz, ArC-5), 121.5 (q, J = 274.5 Hz, CF3), 121.4 (pyrrole C-5), 120.0 (pyrrole C-2), 114.5 (pyrrole C-3), 113.1 (C-4), 110.3 (pyrrole C-4), 84.9 (C(CH3)3), 53.8 (CH2OH), 27.9 (C(CH3)3). LC–MS (ESI): calcd for C20H18ClF3N2O4 [M + H]+, 443.09; observed, 443.08 (Rt = 7.23 min).
4.2.20. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-(((4-(methoxycarbonyl) phenyl)amino)methyl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (17)
According to the general procedure for mesylation and substitution, alcohol 16 (0.072 g, 0.163 mmol) was mesylated and reacted with methyl 4-aminobenzoate (0.122 g, 0.805 mmol) in situ. The crude product was purified by column chromatography, eluting with a gradient of 5–20% EtOAc in n-heptane, to furnish 17 (32.2 mg, 36%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.83 (2H, d, J = 8.2 Hz, benzoate H-2), 7.69 (3H, m, ArH-3, ArH-5 and pyrrole H-2), 7.53 (1 H, app. t, J = 8.0 Hz, ArH-4), 7.34 (1H, m, pyrrole H-5), 6.65 (1H, m, pyrrole H-4), 6.51 (2H, d, J = 8.6 Hz, benzoate H-3), 4.09 (2H, m, CH2NH), 4.06 (1H, m, CH2NH), 3.84 (3H, s, OCH3), 1.52 (9H, s, (C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 167.1 (COOCH3), 164.3 (C-5), 159.1 (C-3), 150.9 (benzoate C-4), 148.0 (NCO2), 136.5 (ArC-2), 133.2 (ArC-3), 132.1 (q, J = 31.2 Hz, ArC-6), 131.5 (benzoate C-2), 131.2 (ArC-4), 126.4 (ArC-1), 125.0 (q, J = 5.0 Hz, ArC-5), 121.6 (pyrrole C-5), 121.5 (q, J = 274.5 Hz, CF3), 119.7 (pyrrole C-2), 119.2 (benzoate C-1), 114.2 (pyrrole C-3), 110.3 (C-4), 111.5 (benzoate C-3), 109.9 (pyrrole C-4), 85.0 (C(CH3)3), 51.6 (OCH3), 36.5 (CH2NH), 27.8 (C(CH3)3). LC–MS (ESI): calcd for C28H25ClF3N3O5 [M + H]+, 576.14; observed, 576.08 (Rt = 8.23 min).
4.2.21. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-(((4-(methoxycarbonyl) phenyl) (methyl)amino)methyl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (18)
According to the general procedure for mesylation and substitution, alcohol 16 (0.120 g, 0.271 mmol) was mesylated and reacted with methyl 4-(methylamino)benzoate (0.190 g, 1.15 mmol) in situ. The crude product was purified by column chromatography, eluting with a gradient of 5–20% EtOAc in n-heptane, to furnish 18 (70.3 mg, 52%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.70 (3H, m, benzoate H-2 and pyrrole H-2), 7.60 (1H, d, J = 7.8 Hz, ArH-5), 7.50 (1 H, d, J = 8.0 Hz, ArH-3), 7.41 (1H, app. t, J = 8.0 Hz, ArH-4), 7.38 (1H, m, pyrrole H-4), 6.68 (1H, m, pyrrole H-5), 6.39 (2H, d, J = 8.8 Hz, benzoate H-3), 4.42 (2H, m, CH2NCH3), 3.84 (3H, s, OCH3), 2.66 (3H, s, CH2NCH3), 1.52 (9H, s, (C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 167.2 (COOCH3), 163.3 (C-5), 159.0 (C-3), 152.2 (benzoate C-4), 148.1 (NCO2), 136.7 (ArC-2), 133.0 (ArC-3), 131.7 (q, J = 31.2 Hz, ArC-6), 131.0 (benzoate C-2), 130.8 (ArC-4), 126.9 (ArC-1), 124.9 (q, J = 5.0 Hz, ArC-5), 121.6 (q, J = 274.5 Hz, CF3), 121.5 (pyrrole C-5), 119.5 (pyrrole C-2), 117.8 (benzoate C-1), 114.3 (pyrrole C-3), 111.5 (C-4), 110.9 (benzoate C-3), 110.2 (pyrrole C-4), 85.1 (C(CH3)3), 51.5 (OCH3), 44.2 (CH2NCH3), 36.8 (CH2NCH3), 27.9 (C(CH3)3). LC–MS (ESI): calcd for C29H27ClF3N3O5 [M + H]+, 590.16; observed, 590.00 (Rt = 8.64 min).
4.2.22. Methyl 4-(((3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-3-yl)isoxazol-4-yl) methyl)amino)-2-fluorobenzoate (19)
Alcohol compound 16 (0.256 g, 0.580 mmol) was dissolved in anhydrous CH2Cl2 (8 mL). To this was added Dess–Martin periodinane (0.369 mg, 0.870 mmol), and the reaction mixture was stirred at room temperature for 3 h. The reaction mixture was quenched by the addition of 10% aqueous Na2S2O3 solution and extracted with CH2Cl2 (3×). The combined organic phase was washed with saturated aqueous NaHCO3 and H2O, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography, eluting with a gradient of 20–30% EtOAc in n-heptane, to furnish the aldehyde (112.0 mg, 44%). Methyl 4-amino-2-fluorobenzoate (0.052 g, 0.310 mmol) was added to a solution of the aldehyde (0.112 g, 0.250 mmol) and AcOH (1.43 μL, 0.025 mmol) in MeOH (2 mL). The reaction mixture was heated at the reflux for 24 h and then concentrated in vacuo. The intermediate imine was isolated by flash column chromatography, eluting with a gradient of 10–30% EtOAc in n-heptane (62.3 mg, 42%), and then dissolved in EtOH (1 mL), cooled to 0 °C (ice), and treated with NaBH4 (0.020 mg, 0.526 mmol). The reaction mixture was stirred at 86 °C for 4 h. The solvent was removed in vacuo, and the mixture was dissolved in CH2Cl2 and washed with water. The aqueous phase was further extracted with CH2Cl2 (3×), dried (MgSO4), filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography, eluting with a gradient of 30–40% EtOAc in n-heptane, to furnish compound 19 (10.5 mg, 17%). 1H NMR (400 MHz, DMSO-d6): δ 11.4 (1H, s, pyrrole-NH), 7.89 (1H, d, J = 8.0 Hz, ArH-5), 7.84 (1H, d, J = 7.5 Hz, ArH-3), 7.72 (1H, app. t, J = 8.0 Hz, ArH-4), 7.47 (1H, app. t, J = 8.7 Hz, benzoate H-2), 7.40 (1H, m, pyrrole H-2), 6.99 (1H, m, pyrrole H-5), 6.76 (1H, app. t, J = 4.6 Hz, CH2NH), 6.55 (1H, m, pyrrole H-4), 6.25 (1H, dd, J = 8.9, 1.9 Hz, benzoate H-3a), 6.10 (1H, d, J = 14.6 Hz, benzoate H-3b), 4.17 (2H, m, CH2NH). LC–MS (ESI): calcd for C23H16ClF4N3O3 [M + H]+, 494.08; observed, 494.25 (Rt = 7.14 min).
4.2.23. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-((4-(methoxycarbonyl) phenoxy)methyl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (20)
According to the general procedure for Mitsunobu coupling, alcohol 16 (0.023 g, 0.053 mmol) was reacted with methyl 4-hydroxybenzoate (0.010 g, 0.058 mmol). The crude product was purified by column chromatography, eluting with a gradient of 5–25% EtOAc in n-heptane, to furnish 20 (16.6 mg, 55%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.92 (2H, d, J = 8.9 Hz, benzoate H-2), 7.71 (3H, m, ArH-3, ArH-5 and pyrrole H-2), 7.56 (1 H, app. t, J = 8.0 Hz, ArH-4), 7.35 (1H, m, pyrrole H-5), 6.82 (2H, d, J = 8.9 Hz, benzoate H-3), 6.66 (1H, m, pyrrole H-4), 4.83 (2H, s, CH2O), 3.87 (3H, s, OCH3), 1.55 (9H, s, (C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 166.7 (COOCH3), 165.1 (C-5), 161.8 (benzoate C-4), 158.9 (C-3), 148.1 (NCO2), 136.7 (ArC-2), 133.1 (ArC-3), 132.3 (q, J = 31.2 Hz, ArC-6), 131.6 (benzoate C-2), 131.1 (ArC-4), 126.4 (ArC-1), 124.9 (q, J = 5.0 Hz, ArC-5), 123.2 (benzoate C-1), 121.6 (pyrrole C-5), 121.4 (q, J = 274.5 Hz, CF3), 120.0 (pyrrole C-2), 114.2 (pyrrole C-3), 114.1 (benzoate C-3), 110.2 (pyrrole C-4), 109.3 (C-4), 85.0 (C(CH3)3), 59.4 (CH2O), 51.9 (OCH3), 27.8 (C(CH3)3). LC–MS (ESI): calcd for C28H24ClF3N2O6 [M + H]+, 577.13; observed, 577.17 (Rt = 8.57 min).
4.2.24. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-((3-fluoro-4-(methoxy carbonyl)phenoxy)methyl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (21)
According to the general procedure for Mitsunobu coupling, alcohol 16 (0.050 g, 0.113 mmol) was reacted with methyl 2-fluoro-4-hydroxybenzoate (0.021 g, 0.124 mmol). The crude product was purified by column chromatography, eluting with a gradient of 5–25% EtOAc in n-heptane, to furnish 21 (17.0 mg, 25%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.85 (1H, app. t, J = 8.6 Hz, benzoate H-2), 7.73 (3H, m, ArH-3, ArH-5 and pyrrole H-2), 7.57 (1 H, app. t, J = 8.0 Hz, ArH-4), 7.36 (1H, m, pyrrole H-5), 6.65 (2H, m, pyrrole H-4 and benzoate H-3a), 6.52 (1H, dd, J = 12.4, 2.3 Hz, benzoate H-3b), 4.82 (2H, s, CH2O), 3.89 (3H, s, OCH3), 1.57 (9H, s, (C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 165.2 (C-5), 164.5 (COOCH3), 162.9 (benzoate C–F), 161.9 (benzoate C-4), 158.9 (C-3), 148.0 (NCO2), 136.7 (ArC-2), 133.6 (benzoate C-2), 133.2 (ArC-3), 132.3 (q, J = 31.2 Hz, ArC-6), 131.2 (ArC-4), 126.2 (ArC-1), 125.0 (q, J = 5.0 Hz, ArC-5), 121.7 (pyrrole C-5), 121.4 (q, J = 274.5 Hz, CF3), 120.0 (pyrrole C-2), 114.1 (pyrrole C-3), 111.6 (benzoate C-3a), 110.4 (benzoate C-1), 110.2 (pyrrole C-4), 108.8 (C-4), 103.1 (benzoate C-3b), 85.1 (C(CH3)3), 59.9 (CH2O), 52.0 (OCH3), 27.8 (C(CH3)3). LC–MS (ESI): calcd for C28H23ClF4N2O6 [M + H]+, 595.12; observed, 595.00 (Rt = 6.14 min).
4.2.25. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-(((4-(methoxycarbonyl) phenyl)thio)methyl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (22)
According to the general procedure for Mitsunobu coupling, alcohol 16 (0.055 g, 0.124 mmol) was reacted with methyl 4-mercaptobenzoate (0.024 g, 0.137 mmol). The crude product was purified by column chromatography, eluting with a gradient of 0–20% EtOAc in n-heptane, to furnish 21 (20.0 mg, 27%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.84 (2H, d, J = 8.5 Hz, benzoate H-2), 7.72 (3H, m, ArH-3, ArH-5 and pyrrole H-2), 7.57 (1 H, app. t, J = 8.5 Hz, ArH-4), 7.34 (1H, m, pyrrole H-5), 7.18 (2H, d, J = 8.9 Hz, benzoate H-3), 6.68 (1H, m, pyrrole H-4), 4.01 (2H, s, CH2O), 3.89 (3H, s, OCH3), 1.58 (9H, s, (C(CH3)3). LC–MS (ESI): calcd for C28H24ClF3N2O5S [M + H]+, 593.10; observed, 592.92 (Rt = 6.28 min).
4.2.26. tert-Butyl 3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-formylisoxazol-5-yl)-1H-pyrrole-1-carboxylate (23)
Alcohol compound 16 (0.150 g, 0.407 mmol) was dissolved in anhydrous CH2Cl2 (5 mL). To this was added Dess–Martin periodinane (0.216 mg, 0.610 mmol), and the reaction mixture was stirred at room temperature for 3 h. The reaction mixture was quenched by the addition of 10% aqueous Na2S2O3 solution and extracted with CH2Cl2 (3×). The combined organic phase was washed with saturated aqueous NaHCO3 and H2O, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography, eluting with a gradient of 15–30% EtOAc in n-heptane, to furnish 21 (150 mg, 100%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 9.67 (1H, s, CHO), 8.54 (1H, s, pyrrole H-2), 7.81–7.73 (2H, m, ArH-3 and ArH-5), 7.63 (1H, app. t, J = 8.0 Hz, ArH-4), 7.43–7.38 (1H, m, pyrrole H-5), 6.97–6.92 (1H, m, pyrrole H-4), 1.66 (9H, s, C(CH3)3). LC–MS (ESI): calcd for C20H16ClF3N2O4 [M + H]+, 441.08; observed, 440.92 (Rt = 5.87 min).
4.2.27. tert-Butyl (E) or (Z)-3-(3-(2-Chloro-6-(trifluoromethyl)phenyl)-4-(4-(methoxy carbonyl)styryl)isoxazol-5-yl)-1H-pyrrole-1-carboxylate (24 and 25)
Under an inert atmosphere, triphenylphosphine (0.890 g, 3.39 mmol) and methyl 4-(bromomethyl)benzoate (0.519 g, 2.27 mmol) were dissolved in acetonitrile (20 mL). The reaction mixture was stirred at 82 °C for 3 h, when TLC indicated the full conversion of the starting material. The reaction mixture was cooled to room temperature and precipitated in toluene (150 mL). The solid was filtered, collected, and dried under reduced pressure to yield (4-(methoxycarbonyl)benzyl) triphenylphosphonium as a crystalline white solid (930 mg, 100%). The resulting product (0.152 g, 0.369 mmol) was dissolved in anhydrous THF (2 mL) under an inert atmosphere. The solution was cooled to −78 °C, LiHMDS (0.4 mL, 1 M in hexane) was added and the solution was stirred at −78 °C for 2 h. Aldehyde 23 (0.125 g, 0.284 mmol), dissolved in anhydrous THF (2 mL), was added dropwise. The reaction mixture was slowly warmed to room temperature and stirred for 24 h. The reaction was quenched with H2O (10 mL) and extracted with EtOAc (3×). The combined organic phase was washed with H2O and brine, dried over MgSO4, filtrated, and concentrated in vacuo. The crude product was purified by column chromatography, eluting with a gradient of 5–20% EtOAc in n-heptane, to furnish a mixture of cis/trans isomers (3:1) (70 mg). The mixture of cis/trans isomers was separated via preparative HPLC (gradient of 80–100% acetonitrile in H2O) to furnish 24 (trans isomer, 12.0 mg, 7%) and 25 (cis isomer, 44.0 mg, 27%), both as white solids. Trans isomer (24): 1H NMR (400 MHz, CDCl3): δ 7.92 (2H, d, J = 7.9 Hz, benzoate H-2), 7.80 (3H, m, ArH-3, ArH-5 and pyrrole H-2), 7.63 (1 H, app. t, J = 7.8 Hz, ArH-4), 7.39 (1H, m, pyrrole H-5), 7.24 (2H, d, J = 7.9 Hz, benzoate H-3), 7.03 (1H, d, J = 16.5 Hz, C4-HC=CH), 6.73 (1H, m, pyrrole H-4), 6.17 (1H, d, J = 16.4 Hz, benzoate C4-HC=CH), 3.90 (3H, s, OCH3), 1.64 (9H, s, (C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 166.7 (COOCH3), 162.7 (C-5), 157.1 (C-3), 148.1 (NCO2), 141.3 (benzoate C-4), 136.7 (ArC-2), 133.2 (ArC-3), 132.2 (q, J = 31.5 Hz, ArC-6), 131.1 (ArC-4), 129.9 (benzoate C-2), 129.8 (benzoate C4-HC=CH), 129.2 (benzoate C-1), 127.6 (ArC-1), 126.0 (benzoate C-3), 125.1 (q, J = 5.0 Hz, ArC-5), 121.5 (pyrrole C-5), 121.4 (q, J = 274.5 Hz, CF3), 119.7 (pyrrole C-2), 118.0 (C4-HC=CH), 114.6 (pyrrole C-3), 112.6 (C-4), 110.4 (pyrrole C-4), 85.1 (C(CH3)3), 52.1 (OCH3), 27.9 (C(CH3)3). LC–MS (ESI): calcd for C29H24ClF3IN2O5 [M + H]+, 573.13; observed, 573.00 (Rt = 6.44 min).
Cis isomer (25): 1H NMR (400 MHz, CDCl3): δ 7.77 (2H, d, J = 8.0 Hz, benzoate H-2), 7.68 (2H, m, ArH-3 and ArH-5), 7.53 (1 H, app. t, J = 8.0 Hz, ArH-4), 7.38 (1H, br s, pyrrole H-2), 7.28 (2H, d, J = 8.0 Hz, benzoate H-3), 7.08 (1H, br s, pyrrole H-5), 6.62 (1H, d, J = 12.1 Hz, C4-HC=CH), 6.50 (1H, br s, pyrrole H-4), 6.18 (1H, d, J = 12.1 Hz, benzoate C4-HC=CH), 3.87 (3H, s, OCH3), 1.57 (9H, s, (C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 166.7 (COOCH3), 161.3 (C-5), 159.0 (C-3), 148.1 (NCO2), 140.8 (benzoate C-4), 136.6 (ArC-2), 133.9 (C4-HC=CH), 133.1 (ArC-3), 132.0 (q, J = 31.5 Hz, ArC-6), 130.8 (ArC-4), 129.5 (benzoate C-2), 129.1 (benzoate C-1), 128.2 (benzoate C-3), 126.8 (ArC-1), 124.9 (q, J = 5.0 Hz, ArC-5), 121.5 (q, J = 274.5 Hz, CF3), 120.6 (pyrrole C-5), 119.7 (pyrrole C-2), 118.2 (benzoate C4-HC=CH), 114.6 (pyrrole C-3), 110.8 (C-4), 110.0 (pyrrole C-4), 84.7 (C(CH3)3), 52.1 (OCH3), 27.9 (C(CH3)3). LC–MS (ESI): calcd for C29H24ClF3IN2O5 [M + H]+, 573.13; observed, 573.00 (Rt = 6.26 min).
4.2.28. Methyl 3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-2-yl)isoxazole-4-carboxylate (26)
According to the general procedure for Suzuki coupling, bromide 14 (0.500 g, 1.30 mmol) was coupled to tert-butyl 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole-1-carboxylate (0.762 g, 2.60 mmol). The crude product was purified by flash column chromatography, eluting with a gradient of 5–10% EtOAc in n-heptane, to furnish 26 (0.298 g, 49%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.77–7.67 (2H, m, ArH-3 and ArH-5), 7.60–7.55 (2H, m, ArH-4 and pyrrole H-5), 6.88 (1H, dd, J = 3.6, 1.7 Hz, pyrrole H-3), 6.36 (1H, app. t, J = 3.4 Hz, pyrrole H-4), 3.57 (3H, s, CO2CH3), 1.52 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 166.7 (C-5), 161.1 (CO2CH3), 158.4 (C-3), 148.3 (NCO2), 136.3 (ArC-2), 132.8 (ArC-3), 131.5 (q, J = 31.4 Hz, ArC-6), 130.7 (ArC-4), 127.1 (ArC-1), 126.2 (pyrrole C-5), 124.6 (q, J = 5.0 Hz, ArC-5), 121.6 (q, J = 274.4 Hz, CF3), 121.2 (pyrrole C-3), 118.5 (pyrrole C-2), 111.1 (pyrrole C-4), 110.2 (C-4), 85.3 (C(CH3)3), 51.7 (CO2CH3), 27.7 (C(CH3)3). LC–MS (ESI): calcd for C21H18ClF3N2O5 [M + H]+, 471.09; observed, 470.92 (Rt = 5.73 min).
4.2.29. Methyl 3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrazol-4-yl)isoxazole-4-carboxylate (27)
According to the general procedure for Suzuki coupling, bromide 14 (0.500 g, 1.30 mmol) was reacted with tert-butyl 1-(3,4-dihydro-2H-pyran-6-yl)-4-(4,4,5-trimethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.723 g, 2.60 mmol). The crude product was purified by flash column chromatography, eluting with a gradient of 5–10% EtOAc in n-heptane, to furnish 27 (0.203 g, 34%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.78 (1H, s, pyrazole H-5), 8.34 (1H, s, pyrazole H-3), 7.75–7.67 (2H, app. t, J = 9.2 Hz, ArH-3 and ArH-5), 7.56 (1H, app. t, J = 8.0 Hz, ArH-4), 5.51–5.46 (1H, m, NCHCO), 4.14–4.04 (1H, m, THP-H), 3.78–3.72 (1H, m, THP-H), 3.60 (3H, s, CO2CH3), 2.20–2.12 (2H, m, THP-H), 1.75–1.62 (4H, m, THP-H); 13C NMR (100 MHz, CDCl3): δ (ppm) 167.8 (C-5), 161.6 (CO2CH3), 158.6 (C-3), 139.7 (pyrazole C-3), 136.2 (ArC-2), 132.7 (ArC-3), 131.4 (q, J = 31.3 Hz, ArC-6), 130.6 (ArC-4), 130.6 (pyrazole C-5), 127.5 (ArC-1), 124.6 (q, J = 4.9 Hz, ArC-5), 121.6 (q, J = 274.4 Hz, CF3), 109.6 (pyrazole C-4), 106.8 (C-4), 88.0 (NCHCO), 67.8 (THP-C), 51.7 (CO2CH3), 30.6 (THP-C), 24.8 (THP-C), 20.1 (THP-C). LC–MS (ESI): calcd for C20H17ClF3N3O4 [M + H]+, 456.09; observed, 456.08 (Rt = 5.37 min).
4.2.30. Methyl 3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1-methyl-1H-pyrrol-3-yl)isoxazole-4-carboxylate (28)
Under an inert atmosphere 1-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole (0.404 g, 1.95 mmol), Na2CO3 (0.276 g, 2.6 mmol)), and Pd(PPh3)4 (0.150 g, 0.13 mmol) were added to a solution of bromide 14 (0.500 g, 1.3 mmol) in de-gassed DME/H2O (4:1) (15 mL). The reaction mixture was heated at 85 °C for 8 h, cooled to room temperature, diluted with H2O, and extracted with EtOAc (3×). The combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The crude product was purified by column chromatography, eluting with 5–15% EtOH in n-heptane, to furnish 28 (0.275 g, 55%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.97 (1H, s, pyrrole H-2), 7.75–7.64 (2H, m, ArH-3 and ArH-5), 7.52 (1H, app. t, J = 8.0 Hz, ArH-4), 6.92 (1H, br s, pyrrole H-5), 6.69 (1H, br s, pyrrole H-4), 3.74 (3H, s, NCH3), 3.56 (3H, s, CO2CH3); 13C NMR (100 MHz, CDCl3): δ (ppm) 170.4 (C-5), 162.1 (CO2CH3), 158.6 (C-3), 136.2 (ArC-2), 132.6 (ArC-3), 131.3 (q, J = 31.3 Hz, ArC-6), 130.3 (ArC-4), 128.2 (ArC-1), 126.8 (pyrrole C-2), 124.5 (q, J = 5.0 Hz, ArC-5), 123.1 (pyrrole C-5), 121.6 (q, J = 274.4 Hz, CF3), 110.6 (pyrrole C-3), 109.4 (pyrrole C-4), 104.9 (C-4), 51.4 (CO2CH3), 36.7 (NCH3). LC–MS (ESI): calcd for C17H12ClF3N2O3 [M + H]+, 385.05; observed, 385.17 (Rt = 5.27 min).
4.2.31. Methyl 3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(5-methyl-1H-pyrrol-3-yl)isoxazole-4-carboxylate (29)
Under an inert atmosphere, a Schlenk tube was charged with tert-butyl 2,4-dimethyl-1H-pyrrole-1-carboxylate (1.10 g, 6.05 mmol). Two separate flasks under argon were charged with [Ir(OMe)(COD)]2 (0.060 g, 0.091 mmol) and dtbpy (0.049 g, 0.182 mmol). HBPin (1.16 g, 9.08 mmol) was added to the [Ir(OMe)(COD)]2 containing flask. Anhydrous THF (18 mL) was added to the dtbpy-containing flask, and the solution was mixed with the [Ir(OMe)(COD)]2 and HBPin mixture. The resulting solution was transferred to the Schlenk flask, and the reaction mixture was stirred at 60 °C for 6 h. Afterward, the reaction mixture was cooled to room temperature and THF was removed in vacuo. The crude product was purified by flash column chromatography, eluting with a gradient of 0–5% EtOAc in n-heptane, to furnish tert-butyl 2-methyl-4-(4,4,5-trimethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole-1-carboxylate (1.30 g, 70%).50
Subsequently, according to the general procedure for Suzuki coupling, bromide 14 (1.09 g, 2.82 mmol) was reacted with the synthesized pinacol boronate (1.30 g, 4.23 mmol). The crude product was purified by flash column chromatography, eluting with a gradient of 30–60% CH2Cl2 in n-heptane, to furnish 29 (0.325 g, 24%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.39 (1H, s, pyrrole H-2), 7.74–7.65 (2H, m, ArH-3 and ArH-5), 7.54 (1H, app. t, J = 8.0 Hz, ArH-4), 6.70 (1H, s, pyrrole H-4), 3.61 (3H, s, CO2CH3), 2.51 (3H, s, NCCH3), 1.64 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 169.0 (C-5), 161.6 (CO2CH3), 158.9 (C-3), 148.9 (NCO2), 136.2 (ArC-2), 132.8 (pyrrole C-5), 132.6 (ArC-3), 131.3 (q, J = 31.4 Hz, ArC-6), 130.5 (ArC-4), 127.7 (ArC-1), 124.9 (pyrrole C-2), 124.5 (q, J = 4.9 Hz, ArC-5), 121.6 (q, J = 274.4 Hz, CF3), 111.6 (pyrrole C-3), 110.9 (pyrrole C-4), 106.9 (C-4), 84.7 (C(CH3)3), 51.6 (CO2CH3), 28.0 (C(CH3)3), 15.3 (NCCH3). LC–MS (ESI): calcd for C22H20ClF3N2O5 [M + H]+, 485.10; observed, 485.00 (Rt = 6.22 min).
4.2.32. (3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrrol-2-yl)isoxazol-4-yl)methanol (30)
Ester 26 (0.260 g, 0.552 mmol) was treated according to the general procedure for the reduction of esters to alcohols. The crude product was purified by flash column chromatography, eluting with a gradient of 10–20% EtOAc in n-heptane, to furnish alcohol 30 (169 mg, 69%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.77–7.69 (2H, m, ArH-3 and ArH-5), 7.57 (1H, app. t, J = 8.0 Hz, ArH-4), 7.51 (1H, dd, J = 3.2, 1.7 Hz, pyrrole H-5), 6.70 (1H, dd, J = 3.4, 1.7 Hz, pyrrole H-3), 6.34 (1H, app. t, J = 3.4 Hz, pyrrole H-4), 4.34 (2H, br s, CH2OH), 1.53 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 161.9 (C-5), 158.6 (C-3), 148.7 (NCO2), 136.5 (ArC-2), 133.2 (ArC-3), 132.1 (q, J = 31.2 Hz, ArC-6), 131.0 (ArC-4), 126.8 (ArC-1), 125.5 (pyrrole C-5), 125.0 (q, J = 5.1 Hz, ArC-5), 121.5 (q, J = 274.5 Hz, CF3), 119.9 (pyrrole C-3), 118.7 (pyrrole C-2), 116.9 (C-4), 111.3 (pyrrole C-4), 85.5 (C(CH3)3), 54.1 (CH2OH), 27.8 (C(CH3)3). LC–MS (ESI): calcd for C20H18ClF3N2O4 [M + H]+, 443.09; observed, 443.00 (Rt = 5.27 min).
4.2.33. (3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1H-pyrazol-4-yl)isoxazol-4-yl)methanol (31)
Ester 27 (0.200 g, 0.434 mmol) was treated according to the general procedure for the reduction of esters to alcohols. The crude product was purified by flash column chromatography, eluting with a gradient of 30–50% EtOAc in n-heptane, to furnish alcohol 31 (0.100 g, 71%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.25 (1H, s, pyrazole H-5), 8.09 (1H, s, pyrazole H-3), 7.80–7.69 (2H, m, ArH-3 and ArH-5), 7.59 (1H, app. t, J = 8.0 Hz, ArH-4), 5.47 (1H, t, J = 6.1 Hz, NCHCO), 4.41 (1H, br s, CH2OH), 4.14–3.98 (1H, m, THP-H), 3.76–3.71 (1H, m, THP-H), 2.17–1.98 (3H, m, THP-H), 1.78–1.58 (3H, m, THP-H); 13C NMR (100 MHz, CDCl3): δ (ppm) 163.3 (C-5), 158.6 (C-3), 137.9 (pyrazole C-3), 136.5 (ArC-2), 133.2 (ArC-3), 132.2 (q, J = 31.2 Hz, ArC-6), 131.1 (ArC-4), 127.5 (pyrazole C-5), 126.6 (ArC-1), 125.0 (q, J = 5.1 Hz, ArC-5), 121.5 (q, J = 274.4 Hz, CF3), 112.8 (C-4), 110.2 (pyrazole C-4), 87.9 (NCHCO), 67.8 (THP-C), 53.9 (CH2OH), 30.7 (THP-C), 24.9 (THP-C), 22.1 (THP-C). LC–MS (ESI): calcd for C19H17ClF3N3O3 [M + H]+, 428.09; observed, 428.08 (Rt = 4.64 min).
4.2.34. (3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(1-methyl-1H-pyrrol-3-yl)isoxazol-4-yl) Methanol (32)
Under an inert atmosphere, LiAlH4 (1 M in THF, 1.07 mL, 1.01 mmol) was added dropwise to a solution of ester 28 (0.275 g, 0.710 mmol) in anhydrous THF (6 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h. The reaction mixture was quenched by the addition of saturated aqueous NH4Cl solution and stirred vigorously for 60 min. Subsequently, the mixture was extracted with EtOAc (3×). The combined organic phase was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography, eluting with a gradient of 10–20% EtOAc in n-heptane, to furnish alcohol 32 (0.192 g, 76%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.76–7.68 (2H, m, ArH-3 and ArH-5), 7.56 (1H, app. t, J = 8.3 Hz, ArH-4), 7.30 (1H, app. t, J = 1.9 Hz, pyrrole H-2), 6.70 (1H, app. t, J = 2.5 Hz, pyrrole H-5), 6.68 (1H, app. t, J = 2.5 Hz, pyrrole H-4), 4.38 (2H, br s, CH2OH), 3.73 (3H, s, NCH3); 13C NMR (100 MHz, CDCl3): δ (ppm) 166.3 (C-5), 158.6 (C-3), 136.6 (ArC-2), 133.1 (ArC-3), 132.2 (q, J = 31.3 Hz, ArC-6), 130.9 (ArC-4), 127.1 (ArC-1), 124.9 (q, J = 5.0 Hz, ArC-5), 123.4 (pyrrole C-5), 122.5 (pyrrole C-2), 121.5 (q, J = 274.4 Hz, CF3), 111.3 (pyrrole C-3), 110.9 (C-4), 107.5 (pyrrole C-4), 54.2 (CH2OH), 36.6 (NCH3). LC–MS (ESI): calcd for C16H12ClF3N2O2 [M + H]+, 357.05; observed, 357.17 (Rt = 4.63 min).
4.2.35. (3-(2-Chloro-6-(trifluoromethyl)phenyl)-5-(5-methyl-1H-pyrrol-3-yl)isoxazol-4-yl) Methanol (33)
Ester 29 (0.400 g, 0.825 mmol) was treated according to the general procedure for the reduction of esters to alcohols. The crude product was purified by flash column chromatography, eluting with a gradient of 10–25% EtOAc in n-heptane, to furnish alcohol 33 (0.220 g, 58%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.83 (1H, s, pyrrole H-2), 7.77–7.58 (2H, m, ArH-3 and ArH-5), 7.58 (1H, app. t, J = 8.0 Hz, ArH-4), 6.48 (1H, s, pyrrole H-4), 4.42 (2H, br s, CH2OH), 2.50 (3H, s, NCCH3), 1.63 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3): δ (ppm) 164.8 (C-5), 158.7 (C-3), 149.1 (NCO2), 136.5 (ArC-2), 133.4 (pyrrole C-5), 133.1 (ArC-3), 132.2 (q, J = 31.4 Hz, ArC-6), 131.0 (ArC-4), 126.8 (ArC-1), 124.9 (q, J = 4.9 Hz, ArC-5), 121.5 (q, J = 274.4 Hz, CF3), 120.7 (pyrrole C-2), 112.9 (C-4), 112.3 (pyrrole C-3), 109.9 (pyrrole C-4), 84.6 (C(CH3)3), 53.9 (CH2OH), 28.0 (C(CH3)3), 15.4 (NCCH3). LC–MS (ESI): calcd for C21H20ClF3N2O4 [M + H]+, 457.11; observed, 456.92 (Rt = 5.53 min).
4.3. Biophysical Assays
4.3.1. RORγt-LBD Expression and Purification
His-tagged RORγt LBD was expressed and purified as described in previous studies.28
4.3.2. TR-FRET Coactivator Recruitment Assay
Assays were conducted using 100 nM N-terminal biotinylated SRC-1 box2 coactivator peptide (Biotin-N-PSSHSSLTARHKILHRLLQEGSPSD-CONH2) and 20 nM His6-RORγt-LBD in buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM dithiothreitol (DTT), 0.1% bovine serum albumin (BSA) (w/v), and 0.1 mM CHAPS. A terbium-labeled anti-His antibody (CisBio Bioassays, 61HISTLA) and D2-labeled streptavidin (CisBio Bioassays, 610SADLA) were used at the concentrations recommended by the supplier. Compounds (dissolved in DMSO) were titrated using a 2× dilution series in Corning white low volume, low binding, 384-well plates at a final volume of 10 μL. The final DMSO concentration was 1% v/v throughout. The plate was directly measured and centrifuged before reading (excitation = 340 nm; emission = 665 and 620 nm) on a Tecan infinite F500 plate reader using the parameters recommended by CisBio Bioassays. The data were analyzed with GraphPad Prism 7.0 software. The dose–response curve was fitted represented by
where y is the FRET ratio ((acceptor/donor) × 1000), A1 is the bottom asymptote, A2 is the top asymptote, p is the Hill slope, x is the ligand concentration in μM, and x0 is the IC50 value in μM. Where dose–response curves did not reach a bottom asymptote, this was fixed at the value of the negative control. The data were recorded in triplicate in three independent experiments (one representative data set shown). Error bars represent the SD of the mean.
4.3.3. Competition TR-FRET Coactivator Recruitment Assay
Competition assays were performed in an analogous fashion to that described above, only in the presence of fixed concentrations of cholesterol: 0 μM (DMSO), 0.25 μM, and 1.0 μM such that the final concentration of DMSO remained at 1.2% v/v.
4.3.4. TR-FRET AlexaFluor-MRL-871 Recruitment Assay
Assays were conducted using 100 nM AlexaFluor647-labeled MRL-871 and 20 nM His6-RORγt-LBD in buffer as described above. A terbium-labeled anti-His antibody (CisBio Bioassays, 61HISTLA) was used at the concentrations recommended by the supplier. The assay was carried out in the same manner as described above.
4.3.5. Thermal Shift Assay
TSA were performed using 40 μL samples containing 5 μM RORγt-LBD, 10 μM compound, and 2.5x SYPRO Orange (Sigma) in buffer containing 150 mM NaCl, 10 mM HEPES (pH 7.5), and 1% DMSO. Hard-Shell 96-well PCR plates (low profile, thin wall, skirted, green/white #hsp9645) were used. The samples were heated from 25 to 80 °C at a rate of 0.5 °C per 5 s in a CFX96 touch real-time PCR detection system (Bio-Rad). After each increment, Sypro Orange intensity was measured using the plate read option in Bio-Rad CFX Manager 3.1. Melting temperatures were determined by Bio-Rad CFX Manager 3.1 software in negative mode. The data were recorded in triplicate in three independent experiments (one representative data set shown). Error bars represent the SD of the mean.
4.4. Quantitative IL-17a mRNA RT-PCR Assay
Cell culture, RT-PCR experiments, and data analysis were performed as described in previous studies.28 Statistical analysis was performed using a one-way ANOVA, comparing against the DMSO control following the Dunnett’s post hoc test (GraphPad Prism 7.0 software). A P value < 0.05 was considered statistically significant. (Data recorded in triplicate from two independent experiments. Data shown are representative of two independent experiments. Error bars represent the SD of the mean.)
4.5. Protein X-ray Crystallography
4.5.1. RORγt-LBD Expression and Purification (Used for Crystallography)
His-tagged RORγt LBD containing a C455H mutation was expressed and purified as described in previous studies.28
4.5.2. X-ray Crystallography
X-ray crystallography experiments and data analysis were performed as described in previous studies.28 Diffraction data were collected at the P11 beamline of the PETRA III facility at DESY (Hamburg, Germany), the ID30B beamline of ESRF (Grenola, France), and the i04 beamline of Diamond Light Source (Didcot, UK). The structures of RORγtC455H in complex with 3, 9, 10, and 11 were deposited in the Protein Data Bank (PDB) under codes 7NPC, 7NP5, 7NEC, and 7NP6.
4.6. Selectivity TR-FRET Coactivator Recruitment Assays
4.6.1. TR-FRET Coactivator Recruitment Assays on PPARγ
TR-FRET competition assays were performed in an analogous fashion to that described above, only using 10 nM His6-PPARγ LBD instead of 20 nM His6-RORγt LBD and 200 nM N-terminal biotinylated PGC1a coactivator peptide (Biotin-N-GTPPPQEAEEPSLLKLLLAPANTQ-CONH2) instead of 100 nM SRC-1 box 2 coactivator peptide.
4.6.2. Competition TR-FRET Coactivator Recruitment Assays on PPARγ
TR-FRET competition assays were performed in an analogous fashion to that described above, only using 100 nM His6-PPARγ LBD instead of 20 nM His6-RORγt LBD. The assay was performed in the presence of 1 μM rosiglitazone, in order to initially activate PPARγ.
4.6.3. TSA on PPARγ
TSA were performed in an analogous fashion to that described above, only using PPARγ LBD instead of RORγt LBD.
4.6.4. FP Assays on FXR
Assays were conducted using 100 nM N-terminal FITC-labeled SRC-1 box2 coactivator peptide (FITC-N-PSSHSSLTARHKILHRLLQEGSPSD-CONH2) and 1 μM GST-tagged FXR-LBD in buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM DTT, 0.1% BSA (w/v), and 0.1 mM CHAPS. For the competition assay, the assay was performed in the presence of 0, 1, 10, or 50 μM of ligand 3. Compounds (dissolved in DMSO) were titrated using a 2× dilution series in Corning black round-bottom, low-volume, low-binding, 384-well plates at a final volume of 10 μL. The final DMSO concentration was 1% v/v throughout. The plate was directly measured (excitation = 485 nm; emission = 535 nm) on a Tecan infinite F500 plate reader. The data were analyzed with GraphPad Prism 7.0 software. The dose–response curve was fitted in the same way as described for the TR-FRET data on RORγt. The data were recorded in triplicate in three independent experiments (one representative data set shown). Error bars represent the SD of the mean.
4.7. ADME Experiments
4.7.1. Kinetic Solubility
Aqueous solubility of compounds was determined by the spectrophotometrical measurement of the kinetic solubility of a 500 μM compound solution in HEPES buffer pH 7.4 compared to a solution containing 50% of the organic solvent acetonitrile. To this end, saturated samples of the compounds in buffer were prepared starting from DMSO stocks, and samples were shaken for 90 min at room temperature. The precipitated material was removed by filtration and samples were further diluted with the same volume of acetonitrile. Absorbance spectra (250–500 nm) were recorded and relative solubility was calculated in comparison to the acetonitrile–buffer solution of the compounds using absorbance ratios.
4.7.2. Parallel Artificial Membrane Permeability Assay
Permeability through artificial membranes [parallel artificial membrane permeability assay (PAMPA)] was performed at an initial concentration of 500 μM of the compound in the donor compartment. After an incubation period of 20 h, the absorption of the receiver wells was measured by spectrophotometry and permeation was calculated by the normalization of the compound flux across a blank filter.
4.7.3. Microsomal Stability—Phase I
Metabolic stability under oxidative conditions was measured
using
NADPH-supplemented human liver microsomes. Compound depletion was
analyzed by liquid chromatography with tandem mass spectrometry (LC–MS/MS)
at a concentration of 1 μM over a time up to 50 min at 37 °C.
Based on compound half-life t1/2, in vitro
intrinsic clearance Clint was calculated.  , with V = assay volume
and mg = amount of microsomal protein.
, with V = assay volume
and mg = amount of microsomal protein.
4.7.4. Microsomal Stability—In Vitro Glucuronidation
Metabolic stability under conjugative conditions was measured in the glucuronidation assay by the LC–MS-based determination of % remaining of selected compounds. Prior to the assay, human liver microsomes were activated using alamethicin and further supplemented with 5 mM of the cofactor UDPGA. 5 μM compound was added to the reaction mixture, and it was incubated for 1 h at 37 °C.
4.7.5. Plasma Stability
Plasma stability was measured by the LC–MS-based determination of % remaining of selected compounds at a concentration of 5 μM after incubation in 50% plasma in phosphate-buffered saline (obtained from humans) for 1 h at 37 °C.
4.7.6. Plasma Protein Binding
Plasma protein binding was determined by equilibrium dialysis. Plasma containing 5 μM of test compound was allowed to equilibrate with the buffer compartment for 6 h at 37 °C. Compound concentrations on both sides of the semipermeable membrane were analyzed by LC–MS/MS and % bound was calculated.
Acknowledgments
We thank Iris A. Leijten-van de Gevel for expression of the RORγt protein used for biophysical assays, Auke A. Koops for the expression of the PPARγ protein, and Joost L.J. van Dongen for performing HRMS measurements. Additionally, we thank Saskia W.C. van Mil for providing the plasmid for FXR expression and for providing GW4064 (FXR agonist). We also thank the DLS-CCP4 Data Collection and Structure Solution Workshop 2021 at Diamond Light Source (Oxfordshire, UK).
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- FP
fluorescence polarization
- FXR
farnesoid X receptor
- H12
helix 12
- HPLC–UV
high-performance liquid chromatography/ultraviolet
- IPTG
isopropyl-β-d-thiogalactoside
- LBD
ligand-binding domain
- LC–MS
liquid chromatography–mass spectrometry
- NR
nuclear receptor
- PAMPA
parallel artificial membrane permeability assay
- PK
pharmacokinetic
- PPARγ
peroxisome proliferated-activated receptor γ
- Q-TOF
quadrupole time-of-flight
- RORγt
retinoic-acid-receptor-related orphan receptor γt
- RT-PCR
reverse transcriptase PCR
- SAR
structure–activity relationship
- Th17
T helper 17
- Tm
melting temperature
- TR-FRET
time-resolved FRET
- TSA
thermal shift assay
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00475.
Chemical structure of AlexaFluor-MRL probe; dose–response curves; schematic representation of TR-FRET assays; docking poses of 4 and 12 in overlay with 3; protein–ligand interaction plot for 3; electron density around compounds 3, 9, 10, and 11; fluorescence polarization assay; synthesis route for isoxazole scaffold 14; original synthesis route for isoxazole compound FM26; library of compounds used in docking studies; toxicity risks, drug likeness, and drug score for all compounds; data collection and refinement statistics (molecular replacement) of 3, 9, 10, and 11; synthesis, biochemical assays, and docking experiments; NMR and LC–UV spectra for assayed compounds; and details on crystallography experiments (PDF)
Molecular formula strings (CSV)
Accession Codes
Coordinates and structure factors for the RORγt bound to compounds 3, 9, 10, and 11 have been deposited in the Protein Data Bank under accession codes 7NPC, 7NP5, 7NEC, and 7NP6. The authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
The manuscript was written through contributions of all the authors. F.A.M. and A.O.W.M.S. performed synthesis; F.A.M. performed biochemical studies; G.J.M.O., R.M.J.M.d.V., and B.A.S. performed crystallography studies; G.J.M.O. performed the expression of the FXR protein; AU performed ADME measurements; F.A.M., R.G.D., B.K., C.O., P.J.C., and L.B. designed the studies. All the authors have given approval to the final version of the manuscript.
This work was supported by the Netherlands Organization for Scientific Research through Gravity program 024.001.035, Vici grant 016.150.366, ECHO grants 711.018.003 and 711.017.014, the European Union through a Marie Skłodowska-Curie Actions (MSCA) Individual Fellowship (R.G.D., H2020-MSCA-IEF-2016, grant no. 705188) and Eurotech Postdoctoral Fellow program (grant no. 754462).
The authors declare the following competing financial interest(s): F.A.M., R.G.D., C.O. and L.B. are coinventors of patent WO2020149740: Substituted heterocyclic compounds and their use as retinoid-related orphan receptor (ROR) gamma-t inhibitors.
Supplementary Material
References
- Robinson-Rechavi M.; Garcia H. E.; Laudet V. The nuclear receptor superfamily. J. Cell Sci. 2003, 116, 585–586. 10.1242/jcs.00247. [DOI] [PubMed] [Google Scholar]
- Huang P.; Chandra V.; Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronemeyer H.; Gustafsson J.-A.; Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discovery 2004, 3, 950–964. 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- Santos R.; Ursu O.; Gaulton A.; Bento A. P.; Donadi R. S.; Bologa C. G.; Karlsson A.; Al-Lazikani B.; Hersey A.; Oprea T. I.; Overington J. P. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 2017, 16, 19–34. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt L. A.; Burris T. P. Action of RORs and their ligands in (patho)physiology. Trends Endocrinol. Metab. 2012, 23, 619–627. 10.1016/j.tem.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I. I.; McKenzie B. S.; Zhou L.; Tadokoro C. E.; Lepelley A.; Lafaille J. J.; Cua D. J.; Littman D. R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Manel N.; Unutmaz D.; Littman D. R. The differentiation of human TH-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat. Immunol. 2008, 9, 641–649. 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P.; Kolls J. K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discovery 2012, 11, 763–776. 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- Yang J.; Sundrud M. S.; Skepner J.; Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. 10.1016/j.tips.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Burkett P. R.; Kuchroo V. K. IL-17 Blockade in Psoriasis. Cell 2016, 167, 1669. 10.1016/j.cell.2016.11.044. [DOI] [PubMed] [Google Scholar]
- Lock C.; Hermans G.; Pedotti R.; Brendolan A.; Schadt E.; Garren H.; Langer-Gould A.; Strober S.; Cannella B.; Allard J.; Klonowski P.; Austin A.; Lad N.; Kaminski N.; Galli S. J.; Oksenberg J. R.; Raine C. S.; Heller R.; Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Isono F.; Fujita-Sato S.; Ito S. Inhibiting RORγt/Th17 axis for autoimmune disorders. Drug Discov. Today 2014, 19, 1205–1211. 10.1016/j.drudis.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Hueber W.; Patel D. D.; Dryja T.; Wright A. M.; Koroleva I.; Bruin G.; Antoni C.; Draelos Z.; Gold M. H.; Durez P.; Tak P. P.; Gomez-Reino J. J.; Foster C. S.; Kim R. Y.; Samson C. M.; Falk N. S.; Chu D. S.; Callanan D.; Nguyen Q. D.; Rose K.; Haider A.; Di Padova F. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2010, 2, 52ra72. 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- Fauber B. P.; Magnuson S. Modulators of the nuclear receptor retinoic acid receptor-related orphan receptor-γ (RORγ or RORc). J. Med. Chem. 2014, 57, 5871–5892. 10.1021/jm401901d. [DOI] [PubMed] [Google Scholar]
- Cyr P.; Bronner S. M.; Crawford J. J. Recent progress on nuclear receptor RORγ modulators. Bioorg. Med. Chem. Lett 2016, 26, 4387–4393. 10.1016/j.bmcl.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Pandya V. B.; Kumar S.; Sachchidanand S.; Sharma R.; Desai R. C. Combating Autoimmune Diseases With Retinoic Acid Receptor-Related Orphan Receptor-γ (RORγ or RORc) Inhibitors: Hits and Misses. J. Med. Chem. 2018, 61, 10976–10995. 10.1021/acs.jmedchem.8b00588. [DOI] [PubMed] [Google Scholar]
- Bronner S. M.; Zbieg J. R.; Crawford J. J. RORgamma antagonists and inverse agonists: a patent review. Expert Opin. Ther. Pat. 2017, 27, 101–112. 10.1080/13543776.2017.1236918. [DOI] [PubMed] [Google Scholar]
- Sun N.; Guo H.; Wang Y. Retinoic acid receptor-related orphan receptor gamma-t (RORγt) inhibitors in clinical development for the treatment of autoimmune diseases: a patent review (2016-present). Expert Opin. Ther. Pat. 2019, 29, 663–674. 10.1080/13543776.2019.1655541. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Dulubova I.; Reisman S. A.; Hotema M.; Lee C. I.; Liu L.; McCauley L.; Trevino I.; Ferguson D. A.; Eken Y.; Wilson A. K.; Wigley W. C.; Visnick M. A novel series of cysteine-dependent, allosteric inverse agonists of the nuclear receptor RORγt. Bioorg. Med. Chem. Lett. 2020, 30, 126967. 10.1016/j.bmcl.2020.126967. [DOI] [PubMed] [Google Scholar]
- Meijer F. A.; Leijten-van de Gevel I. A.; de Vries R. M. J. M.; Brunsveld L. Allosteric small molecule modulators of nuclear receptors. Mol. Cell. Endocrinol. 2019, 485, 20–34. 10.1016/j.mce.2019.01.022. [DOI] [PubMed] [Google Scholar]
- Moore T. W.; Mayne C. G.; Katzenellenbogen J. A. Minireview: Not picking pockets: nuclear receptor alternate-site modulators (NRAMs). Mol. Endocrinol. 2010, 24, 683–695. 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboni L.; Lloyd D. G. Beyond the ligand-binding pocket: targeting alternate sites in nuclear receptors. Med. Res. Rev. 2013, 33, 1081–1118. 10.1002/med.21275. [DOI] [PubMed] [Google Scholar]
- Tice C. M.; Zheng Y.-J. Non-canonical modulators of nuclear receptors. Bioorg. Med. Chem. Lett. 2016, 26, 4157–4164. 10.1016/j.bmcl.2016.07.067. [DOI] [PubMed] [Google Scholar]
- Scheepstra M.; Leysen S.; van Almen G. C.; Miller J. R.; Piesvaux J.; Kutilek V.; van Eenennaam H.; Zhang H.; Barr K.; Nagpal S.; Soisson S. M.; Kornienko M.; Wiley K.; Elsen N.; Sharma S.; Correll C. C.; Trotter B. W.; van der Stelt M.; Oubrie A.; Ottmann C.; Parthasarathy G.; Brunsveld L. Identification of an allosteric binding site for RORγt inhibition. Nat. Commun. 2015, 6, e8833 10.1038/ncomms9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karstens W. F. J.; van der Stelt M.; Cals J.; Azevedo R. C. R. G.; Barr K. J.; Zhang H.; Beresis R. T.; Zhang D.; Duan X.. RORgammaT Inhibitors. PCT Int. Appl. WO 2012106995 A1, 2012.
- Leijten-van de Gevel I. A.; Brunsveld L. Delineation of the molecular determinants of the unique allosteric binding site of the orphan nuclear receptor RORγt. J. Biol. Chem. 2020, 295, 9183–9191. 10.1074/jbc.ra120.013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries R. M. J. M.; Meijer F. A.; Doveston R. G.; Leijten-van de Gevel I. A.; Brunsveld L. Cooperativity between the Orthosteric and Allosteric Ligand Binding Sites of RORγt. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2021287118 10.1073/pnas.2021287118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer F. A.; Doveston R. G.; De Vries R. M. J. M.; Vos G. M.; Vos A. A. A.; Leysen S.; Scheepstra M.; Ottmann C.; Milroy L.-G.; Brunsveld L. Ligand-Based Design of Allosteric Retinoic Acid Receptor-Related Orphan Receptor γt (RORγt) Inverse Agonists. J. Med. Chem. 2020, 63, 241–259. 10.1021/acs.jmedchem.9b01372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; Gobbi A.; Robarge K.; Zhou A.; Barnard A.; Cao J.; Deng Y.; Eidenschenk C.; Everett C.; Ganguli A.; Hawkins J.; Johnson A. R.; La H.; Norman M.; Salmon G.; Summerhill S.; Ouyang W.; Tang W.; Wong H. Discovery of imidazo[1,5-a]pyridines and -pyrimidines as potent and selective RORc inverse agonists. Bioorg. Med. Chem. Lett 2015, 25, 2907–2912. 10.1016/j.bmcl.2015.05.055. [DOI] [PubMed] [Google Scholar]
- Ouvry G.; Bouix-Peter C.; Ciesielski F.; Chantalat L.; Christin O.; Comino C.; Duvert D.; Feret C.; Harris C. S.; Lamy L.; Luzy A.-P.; Musicki B.; Orfila D.; Pascau J.; Parnet V.; Perrin A.; Pierre R.; Polge G.; Raffin C.; Rival Y.; Taquet N.; Thoreau E.; Hennequin L. F. Discovery of phenoxyindazoles and phenylthioindazoles as RORγ inverse agonists. Bioorg. Med. Chem. Lett 2016, 26, 5802–5808. 10.1016/j.bmcl.2016.10.023. [DOI] [PubMed] [Google Scholar]
- Shaikh N. S.; Iyer J. P.; Munot Y. S.; Mukhopadhyay P. P.; Raje A. A.; Nagaraj R.; Jamdar V.; Gavhane R.; Lohote M.; Sherkar P.; Bala M.; Petla R.; Meru A.; Umrani D.; Rouduri S.; Joshi S.; Reddy S.; Kandikere V.; Bhuniya D.; Kulkarni B.; Mookhtiar K. A. Discovery and pharmacological evaluation of indole derivatives as potent and selective RORγt inverse agonist for multiple autoimmune conditions. Bioorg. Med. Chem. Lett. 2019, 29, 2208–2217. 10.1016/j.bmcl.2019.06.044. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Lapointe B. T.; Anthony N.; Azevedo R.; Cals J.; Correll C. C.; Daniels M.; Deshmukh S.; van Eenenaam H.; Ferguson H.; Hegde L. G.; Karstens W. J.; Maclean J.; Miller J. R.; Moy L. Y.; Simov V.; Nagpal S.; Oubrie A.; Palte R. L.; Parthasarathy G.; Sciammetta N.; van der Stelt M.; Woodhouse J. D.; Trotter B. W.; Barr K. Discovery of N-(Indazol-3-yl)piperidine-4-carboxylic Acids as RORγt Allosteric Inhibitors for Autoimmune Diseases. ACS Med. Chem. Lett. 2020, 11, 114–119. 10.1021/acsmedchemlett.9b00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudari S. S.; Thomas A.; Dhone S. V.; Khairatkar-Joshi N.; Bajpai M.. Bicyclic Heterocyclic Compounds as ROR gamma Modulators. PCT Int. Appl. WO 2015008234 A1, 2015.
- Vries R. M. J. M.; Doveston R. G.; Meijer F. A.; Brunsveld L. Elucidation of an Allosteric Mode of Action for a Thienopyrazole RORγt Inverse Agonist. ChemMedChem 2020, 15, 561–565. 10.1002/cmdc.202000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunsveld L.; Doveston R. G.; Leysen S. F. R.; Milroy L.-G.; Meijer F. A.; Scheepstra M.; Ottmann C.. Substituted heterocyclic compounds and their use as retinoic acid receptor-related orphan receptor (ROR) gamma-t inhibitors. PCT Int. Appl. WO 2020149740 A1, 2020.
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Degorce F. HTRF: A Technology Tailored for Drug Discovery - A Review of Theoretical Aspects and Recent Applications. Curr. Chem. Genom. 2009, 3, 22–32. 10.2174/1875397300903010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Anderson M.; Collin D.; Muegge I.; Wan J.; Brennan D.; Kugler S.; Terenzio D.; Kennedy C.; Lin S.; Labadia M. E.; Cook B.; Hughes R.; Farrow N. A. Structural studies unravel the active conformation of apo RORγt nuclear receptor and a common inverse agonism of two diverse classes of RORγt inhibitors. J. Biol. Chem. 2017, 292, 11618–11630. 10.1074/jbc.m117.789024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroosh P.; Wu J.; Xue X.; Song J.; Sutton S. W.; Sablad M.; Yu J.; Nelen M. I.; Liu X.; Castro G.; Luna R.; Crawford S.; Banie H.; Dandridge R. A.; Deng X.; Bittner A.; Kuei C.; Tootoonchi M.; Rozenkrants N.; Herman K.; Gao J.; Yang X. V.; Sachen K.; Ngo K.; Fung-Leung W.-P.; Nguyen S.; de Leon-Tabaldo A.; Blevitt J.; Zhang Y.; Cummings M. D.; Rao T.; Mani N. S.; Liu C.; McKinnon M.; Milla M. E.; Fourie A. M.; Sun S. Oxysterols are agonist ligands of RORγt and drive Th17 cell differentiation. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 12163–12168. 10.1073/pnas.1322807111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis K. A.; Reinking J. L. Use of Differential Scanning Fluorimetry to Identify Nuclear Receptor Ligands. Methods Mol. Biol. 2016, 1443, 21–30. 10.1007/978-1-4939-3724-0_3. [DOI] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Gao K.; Oerlemans R.; Groves M. R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. 10.1007/s12551-020-00619-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijten-van de Gevel I. A.; van Herk K. H. N.; de Vries R. M. J. M.; Brunsveld L.. PDB ID: 6TDC. PPAR gamma ligand binding domain in complex with MRL-871; National Center for Biotechnology Information, 2019.
- Cuzzocrea S.; Pisano B.; Dugo L.; Ianaro A.; Maffia P.; Patel N. S. A.; Di Paola R.; Ialenti A.; Genovese T.; Chatterjee P. K.; Di Rosa M.; Caputi A. P.; Thiemermann C. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces acute inflammation. Eur. J. Pharmacol. 2004, 483, 79–93. 10.1016/j.ejphar.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Bass J. Y.; Caldwell R. D.; Caravella J. A.; Chen L.; Creech K. L.; Deaton D. N.; Madauss K. P.; Marr H. B.; McFadyen R. B.; Miller A. B.; Parks D. J.; Todd D.; Williams S. P.; Wisely G. B. Substituted isoxazole analogs of farnesoid X receptor (FXR) agonist GW4064. Bioorg. Med. Chem. Lett. 2009, 19, 2969–2973. 10.1016/j.bmcl.2009.04.047. [DOI] [PubMed] [Google Scholar]
- Sepe V.; Marchianò S.; Finamore C.; Baronissi G.; Di Leva F. S.; Carino A.; Biagioli M.; Fiorucci C.; Cassiano C.; Monti M. C.; del Gaudio F.; Novellino E.; Limongelli V.; Fiorucci S.; Zampella A. Novel Isoxazole Derivatives with Potent FXR Agonistic Activity Prevent Acetaminophen-Induced Liver Injury. ACS Med. Chem. Lett. 2019, 10, 407–412. 10.1021/acsmedchemlett.8b00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvie D. K.; Kalgutkar A. S.; Khojasteh-Bakht S. C.; Obach R. S.; O’Donnell J. P. Biotransformation Reactions of Five-Membered Aromatic Heterocyclic Rings. Chem. Res. Toxicol. 2002, 15, 269–299. 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- Molecular property explorer OSIRIS. 2021, Available from: http://www.organic-chemistry.org/prog/peo/drugScore (accessed Feb 26, 2021).
- Kallepalli V. A.; Shi F.; Paul S.; Onyeozili E. N.; Maleczka R. E.; Smith M. R. Boc Groups as Protectors and Directors for Ir-Catalyzed C–H Borylation of Heterocycles. J. Org. Chem. 2009, 74, 9199–9201. 10.1021/jo901822b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.