

Abstract
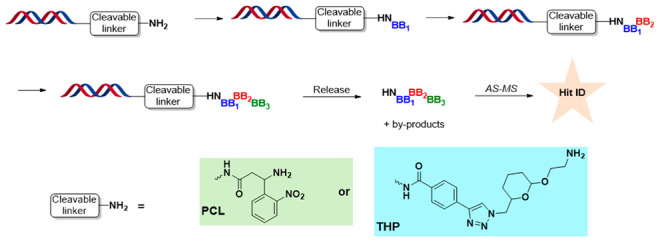
DNA-encoded library (DEL) technology is a powerful platform for hit identification in academia and the pharmaceutical industry. When conducting off-DNA resynthesis hit confirmation after affinity selection, PCR/sequencing, and data analysis, one typically assumes a “one-to-one” relationship between the DNA tag and the chemical structure of the attached small-molecule it encodes. Because library synthesis often yields a mixture, this approximation increases the risk of overlooking positive discoveries and valuable information. To address this issue, we apply a library synthesis “recipe” strategy for on-DNA resynthesis using a cleavable linker, followed by direct affinity selection mass spectrometry (AS-MS) evaluation and identification of binder(s) from the released small-molecule mixture. We validate and showcase this approach employing the receptor-interacting-protein kinase 2 (RIP2) DEL campaign. We also designed and developed two cleavable linkers to enable this method, a photocleavable linker (nitrophenyl-based) and acid-labile linker (tetrahydropyranyl ether). The strategy provides an effective means of hit identification and rapid determination of key active component(s) of the mixture.
Keywords: DNA-encoded libraries, Hit confirmation, Photocleavable linker, Tetrahyrdopyranyl, Affinity-selection mass spectrometry, Drug discovery
Drug discovery is a highly resource intensive effort,1,2 beginning with the identification of molecules that can modulate biological targets of pharmaceutical interest. DNA-encoded library (DEL) technology is a powerful hit identification platform that is synergistic with high-throughput screening (HTS) and other hit ID methods (e.g., affinity selection mass spectrometry(AS-MS), fragment-based lead discovery (FBLD)).3−7 DELs are generally constructed through two to four rounds of chemistry employing the “split-and-pool” synthesis strategy, beginning with functionalization of the free amine of a DNA headpiece (HP, a short sequence of duplex DNA stabilized by a synthetic hairpin).8 For each cycle of chemistry, suitable DNA tags are enzymatically ligated to code for the identity of the corresponding building blocks (BB) used. The affinity selection is typically performed by incubating DELs with an immobilized protein target followed by serial washes to detach weak binders. The remaining high affinity ligands are heat-eluted, collected, amplified by polymerase chain reaction (PCR) and identified by high-throughput DNA sequencing. Interpretation of the screening output is then carried out, and “putative” strong binders are synthesized off DNA for testing in biophysical and biochemical assays.9−11
When executing traditional off-DNA hit follow-up, a static view of the “encoding” is often accepted, with a “one-to-one relationship” between the DNA tag and the fully enumerated ligand it encodes. This approximation works well most of the time, but information can be missed by ignoring the “dynamic” nature of DNA-encoded chemistry. It is important to note that the DNA tag of a library member encodes the history of the on-DNA library production, including the production of intermediates and byproducts. Since library synthesis often produces a mixture, the above “one-to-one” assumption increases the risk of overlooking positive discoveries. DEL selections are ultrasensitive, arising from the combination of PCR and high throughput DNA sequencing. As such, even minor impurities or unreacted starting material can be detected as binders. DEL practitioners have indeed noticed the limitation of the “static” analysis and thus sometimes employ a “semidynamic” approach by introducing the synthesis of intermediates and expected byproducts when pursuing off-DNA follow-up chemistry. However, conducting reactions in organic solvents (in a classic medicinal chemistry fashion) does not mimic library production conditions where reagents and BBs are generally used in large excess and in aqueous media. Furthermore, in order to explore structure–activity relationships (SAR) more rapidly, the “off-DNA resynthesis” route might not follow the exact sequence or even chemistry of the original on-DNA library production.12 To bridge the gap between on-DNA and off-DNA chemistry and increase DEL campaign hit confirmation rate, we introduced the concept of a DEL “recipe” and its relation to the DNA barcode and the selected ligand.13,14 By using a recipe approach, we take advantage of established on-DNA protocols using the DNA headpiece as a handle for synthesis and purification. By employing on-DNA resynthesis, one can expect to obtain a product mixture including precursors, intermediates, byproducts, and “cycle-missing” truncates, similar to that observed during the original library production. AS-MS15,16 can then be employed to directly test the on-DNA compounds or the released mixture of all products through a cleavable linker. By applying this “nonstatic” approach, we can extract more information from DEL selections while lowering the risk of overlooking positive discoveries. Here we describe and elaborate on this unconventional DEL hit confirmation strategy and two generations of cleavable linkers designed and developed specifically to facilitate this process.
Direct inhibition of receptor-interacting-protein kinase 2 (RIP2) has great potential in the treatment of a variety of inflammatory diseases.17 We identified potent and selective inhibitors of RIP2 kinase through complementary HTS and DEL screening campaigns and progressed a hybrid lead series into a clinical candidate.18−20 Interestingly, during the off-DNA follow-up of the first “putative binder”, the synthetic route used for library production did not yield the structure recorded by the DNA barcode but generated a major byproduct not observed during prelibrary validation (Figure 1). The library which yielded the hit was constructed with three cycles of chemistry on an invariant 3-formyl-5-iodobenzoic acid core to provide 112 million compounds.21 Interpretation of the affinity selection output is shown in a cubic scatter plot, where each axis corresponds to a cycle of chemistry. An apparent plane is observed which is determined by a specific BB2 ((4-chloroquinolin-7-yl)boronic acid). On the plane, a highly enriched line focused on 5-amino-2-chlorophenol as BB3 is noticeably recognized, with no preference for BB1, suggesting that BB1 contributes very little to the binding. Guided by this analysis, we postulated a potential chemotype and selected 1, a compound lacking cycle 1 BB, as an exemplar for follow-up synthesis. Surprisingly, the exemplar 1 was inactive in a fluorescence polarization (FP)19 based binding assay (IC50 > 50 μM). One explanation could be that, under library synthesis conditions, where the cycle 3 BB is used in large excess (80 equiv), in addition to the anticipated reductive alkylation product, the amine BB could react further and displace the relative labile Cl on the quinoline which was introduced by C2 Suzuki coupling. We prepared the bis-BB3 compound 2 and found it to be quite potent in the biochemical assay (IC50 = 6 nM). A SAR exploration yielded analogue 3 which maintained potency (IC50 = 6 nM) with significantly improved physiochemical properties (increased LE from 0.28 to 0.47). Further optimization of 3 concentrated on the BB3 moiety (the unexpected SNAr product). Inspection of the DEL selection data (a neighboring line on the same plane) led to the synthesis of the equipotent lead 4 exhibiting the best selectivity profile in class.
Figure 1.

RIP2 affinity selection data analysis, hit identification, and progression of the hit to lead. Ligand efficiency (LE) = 1.4 × pIC50/N, where N is non-hydrogen/heavy atoms.
This RIP2 example, in which the off-DNA synthesis led to a compound with no desired activity, unfortunately is not uncommon to DEL technology. On-DNA library synthesis most often yields a mixture of intermediates, byproducts, and rarely (if ever) a pure desired final product for any given DNA barcode. In a thought experiment for the RIP2 case, if all three cycles of chemistry proceed with 70% yield during library synthesis, the maximum amount of desired product generated is 34% (presuming the scaffold installation is quantitative). This means that 66% of what results from the recipe is not the intended product but is available and competent to be selected. The DEL selection process is extremely sensitive, and we have observed molecules in the single digit percent of final library mixture to be the selected binder (unpublished internal results). This can cause DNA sequence translation to small molecule structure to be complex and even misleading. In order to develop a method to recreate the library recipe and deliver all library products including intermediates and byproducts, we decided to incorporate a cleavable linker for our on-DNA synthesis. In this manner, we can evaluate our DEL hits either on- or off-DNA in both biophysical and biochemical assays. We selected the RIP2 series as an example for our initial proof of concept study to test our hypothesis that the bis-BB3 compound 2 exists in the library mixture and to determine whether it is the driving force behind the affinity selection.
The initial linker we chose to test was a 3-(9-Fmoc)amino-3-(2-nitrophenyl)propionic acid (6, Scheme 1). We selected this photocleavable linker (PCL) because it has been widely studied and employed in solid-phase organic synthesis including split-and-pool combinatorial library productions.22,23 Additionally, the cleavage conditions for this linker are mild and do not require a strong acid or metal catalyst which can cause damage to the DNA. These gentle cleavage conditions are also attractive for assessing the released material in biophysical and biochemical assays where contamination from cleavage material can be detrimental. One final advantage of this cleavage technique is the cleaved product has the minimum possible “scar”, i.e., H, which mimics the DNA attachment for the investigated molecules.
Scheme 1. Synthesis of HP-PCL-NH2 (HP = Headpiece; PCL = Photocleavable Linker).
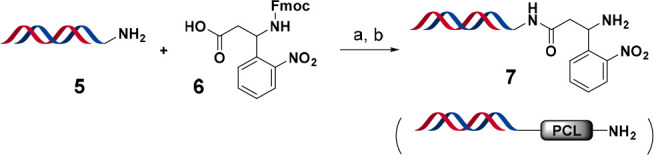
Reagents and conditions: (a) HATU, DIPEA, DMF, pH 9.4 sodium borate buffer, rt, 2 h; (b) 10% v/v piperidine, rt, 1 h.
To demonstrate the value and applicability of the method for DEL hit confirmation we followed the exact library recipe for compound 1 (Scheme 2). For the installation of the 3-formyl-5-iodobenzoic acid, we used our previously reported DMT-MM acylation conditions24 The cycle 2 Suzuki cross-coupling of (4-chloroquinolin-7-yl)boronic acid was performed using our standard Pd(PPh3)4 protocol,24 while the introduction of the cycle 3 benzo[d]thiazol-5-amine was completed using on-DNA sodium cyanoborohydride reductive alkylation. The on-DNA quality control (QC) confirmed our hypothesis that the desired final product 10 was not formed and the bis-BB3 side product (10a, on-DNA version of 12) was indeed the major component in the mixture (see Supporting Information Figure S9). Finally, the mixture was cleaved from the DNA headpiece using UV irradiation at 365 nm for 1 h at 4 °C in aqueous methanol to yield compounds 11 and 12 (see Figure S10).
Scheme 2. On-DNA “Recipe” Synthesis toward 10 Using the Photocleavable Linker.

Reagents and conditions: (a) DMT-MM, DMA, pH 9.4 sodium borate buffer, rt, 2 h; (b) Pd(PPh3)4 in ACN, Na2CO3, DMA, water, 80 °C, 5 h; (c) NaCNBH3, DMA, pH 5.5 sodium phosphate buffer, 60 °C, 16 h; (d) hν 365 nm, 4:1 (H2O:MeOH), 0 °C, 1 h. (%RBA = relative binding affinity by AS-MS.)
The cleaved products 11 and 12 were then assessed for binding using AS-MS. Both compounds exhibited high binding to the RIP2 protein (58–70% RBA), further verifying that they are the true binders in the affinity selection (Scheme 2 and Figure S18). The detection of 11 indicates that the SNAr side reaction occurred prior to the intended cycle 3 reductive alkylation chemistry. Moreover, the verification of 11 as a true binder also revealed critical direction of travel for the SAR. This proof-of-concept study demonstrates the value of applying the library “recipe” strategy for the on-DNA resynthesis using a cleavable linker. Had this method been employed when the hit confirmation was originally performed, we would have focused synthesis efforts on the active byproducts on our first attempt with clear path of SAR in mind and not extended further valuable chemistry resources.
Having demonstrated the newly developed platform to be an effective way to identify true binders resulting from DEL mixture, we recognized that the photocleavable linker (6) has some limitations. The amine attachment point of the current PCL is more sterically hindered than the amine of the headpiece 5 used in library production and is not as reactive. More importantly, while less hindered PCLs could be pursued, we recognized that some library chemistry was not compatible with the photocleavage strategy. For example, benzimidazole is a privileged scaffold in medicinal chemistry and benzimidazole DELs are well represented in our library collection. The on-DNA formation of benzimidazole scaffolds is realized by condensation of aldehydes and substituted on-DNA anilines which are derived from on-DNA nitrobenzenes.25 However, the PCL is not compatible with chemistry requiring nitro reduction, because once reduced the PCL is no longer photocleavable. The PCL also cannot be utilized for BBs containing photocleavable protecting groups such as Nvoc. The use of on-DNA photoredox chemistry26,27 may also result in premature cleavage depending on the light source. An analysis of 116 DELs identified 27 libraries that contain a step where the entire library undergoes chemistry that is not compatible with the PCL linker and another 18 libraries where a subset of the library contains an incompatible reaction, revealing that only ∼60% of our DEL libraries can be covered by the existing PCL. We thus turned our attention to the development of an alternate linker to broaden the scope of this hit confirmation approach.
The ideal linker would be stable enough under all reaction conditions used for on-DNA library synthesis and would not interfere with the desired chemistry without premature cleavage, while labile enough to be cleaved without degrading DNA and leaving a minimal “scar”. Additional benefits can be achieved if the “scar” can improve the physiochemical properties, such as solubility, of the cleaved small molecule. After thorough analysis of our on-DNA chemistry and reviewing literature on cleavable linkers, we decided a linker based on the broadly applied tetrahydropyranyl (THP) ethers (14, Scheme 3) protecting group28 could be the solution.
Scheme 3. Synthesis of HP-THP-NH2 (THP = Tetrahydropyranyl Ether).
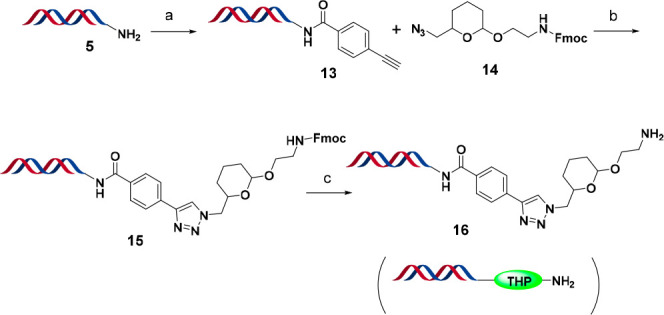
Reagents and conditions: (a) 4-ethynylbenzoic acid, HATU, DIPEA, DMF, pH 9.4 sodium borate buffer, rt, 2 h; (b) 14, copper(II) sulfate, sodium ascorbate, DMA, pH 9.4 sodium borate buffer, rt, o/n; (c) 10% v/v piperidine, rt, 1 h.
The copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) “click” reaction was postulated to be a suitable choice for installation of the THP linker. The click reaction is robust, mild, and water-friendly.29 The resulting amine is unhindered and has similar reactivity to that of headpiece 5 used in library synthesis. Furthermore, the formed triazole moiety should endure reaction conditions both for DEL library production and linker cleavage.
We introduced an alkyne on our DNA HP through amide bond formation using 4-ethynylbenzoic acid with HATU as the coupling reagent. A THP linker containing an azide functionality (14) was custom-synthesized (see the Supporting Information) and then reacted with the alkyne-HP (13) under CuAAC reaction condition to quickly assemble our HP-THP-NH-Fmoc (15) in high yield (Scheme 3). The obtained HP-THP-NH2 (16) was subjected to various DEL chemistry conditions to examine its stability and cleavability. The conditions span from basic (pH > 10) to weakly acidic (pH 5.5), elevated temperature (∼80 °C), to the presence of metal (e.g., FeSO4), as those settings widely cover conditions for a number of important on-DNA transformations such as SNAr, NO2 reduction, and reductive amination, to name a few (e.g., Scheme 4). To our delight, we observed no degradation of the DNA or small molecule warhead under the above-mentioned conditions. Encouraged by these results, we proceeded to screen a wide range of reactions and the desired chemistries worked well, covering 100% of the reactions typically used in our libraries.3 Notably, nitro reductions (Scheme 4) performed satisfactorily (>85%), allowing access to benzimidazoles previously not accessible by the PCL.
Scheme 4. On-DNA “Recipe” Synthesis of 20 Using the THP Linker Followed by Release of Compound 21.

Reagents and conditions: (a) 4-fluoro-3-nitrobenzoic acid, DMTMM, DMA, pH 9.4 sodium borate buffer, rt, 2 h; (b) 2-methoxyehtan-1-amine, acetonitrile/water = 50:50, pH 9.4 sodium borate buffer, 60 °C, 1 h; (c) FeSO4, pH 9.4 sodium borate buffer, 10% v/v 1 M NaOH, 80 °C, 2 h; (d) DMA, pH 5.5 sodium phosphate buffer, rt, o/n; (e) formic acid (0.1%), 60 °C, 1 h. (%RBA = Relative Binding Affinity by AS-MS.)
After surveying a selection of conditions, we identified multiple mild reagents to affect cleavage including dilute formic acid or LiCl in water (data not shown). Formic acid (0.1%) was chosen as the primary method because it is volatile and can be removed under gentle inert gas stream or by lyophilization, which is particularly valuable for high-throughput chemistry applications. Formic acid is also one of most common HPLC purification buffers, and hence, its impact to assays should be minimal. After cleavage, a small PEG “scar” remains, which is beneficial and in fact is designed purposely in this fashion because the PEG residue has been used in the past30 to mimic the DNA tag while conducting off-DNA follow-up and improves compound solubility.
With these promising results in hand, we conducted a case study targeting a known potent RIP2 DEL hit. Compound 20 was synthesized on-DNA using the THP linker (Scheme 4). (Notably, the PCL method was not compatible here because the benzimidazole synthetic route contains a nitro reduction, which will reduce the nitro functionality of PCL as well, rendering it no longer cleavable.) Compound 21 was released under the optimized condition and confirmed by AS-MS to be a true binder31 (30% RBA) (see Supporting Information Figures S16 and S17).
In this paper, we demonstrate a DEL “recipe” approach to hit confirmation using on-DNA cleavable linkers (PCL and THP) to enable full interrogation of selections results. The RIP2 case study showcases how application of this technique would have avoided the synthesis of inactive compounds off DNA and directly identified the true binder(s) in the library mixture. In addition to more rapidly finding the key active component(s) of the mixture, this approach is amenable to coupling with automation and creating a higher throughput hit confirmation platform12 which enables the triage of hundreds of potential chemotypes on small scale. Given that DEL selections typically produce hundreds or even thousands of chemotypes, this tactic could transform hit-to-lead (H2L) programs by providing diverse confirmed hit series rather than being limited to only a small subset of compounds generated by traditional organic synthesis. We have also employed an on-DNA version of this technique where the compounds are not cleaved and binding affinity is tested directly on the HP. Pfizer and WuXi recently communicated the validation of this approach,14 and our results on those fronts will be reported in due course. The main advantage of the cleavable linker strategy is that this method is not prone to false positives which may arise from interactions between the DNA and the target of interest, and testing the compounds as small molecules is more relevant.
To conclude, we have implemented a strategy effectively closing the gap between on-DNA and off-DNA synthesis for DEL technology and successfully designed and developed two generations of cleavable linkers, making the proposed method widely applicable for hit follow-up. This strategy has great potential beyond the described scope to better enable DEL hit identification efforts.
Acknowledgments
We thank Christopher Kollmann, Lijun Fan, and Steven Skinner for their consultation and support of this work.
Glossary
Abbreviations
- DEL
DNA-encoded library
- HP
headpiece
- BB
building blocks
- PCR
polymerase chain reaction
- SAR
structure–activity relationship
- AS-MS
affinity selection mass spectrometry
- RIP2
receptor-interacting-protein kinase 2
- FP
fluorescence polarization
- VEGFR
vascular endothelial growth factor receptor
- EGFR
epidermal growth factor receptor
- ALK
anaplastic lymphoma kinase
- LE
ligand efficiency
- PCL
photocleavable linker
- THP
tetrahydropyranyl
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- DMTMM
(4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride)
- CuAAC
copper(I)-catalyzed alkyne–azide cycloaddition
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00156.
Detailed synthetic procedures, spectroscopic data of synthesized compounds, and experimental procedure of AS-MS (PDF)
Author Present Address
† G.J.F.: Anagenex, 10 Roessler Rd. Unit N, Woburn, MA 01801, United States.
Author Present Address
‡ X.L.: Shanghai Institute of Materia Medica, Chinese Academy of Science, 501 Haike Road, Zhangjiang Hi-tech Park, Shanghai, China.
Author Present Address
§ L.C.G.: Anagenex, 10 Roessler Rd. Unit N, Woburn, MA 01801, United States.
Author Present Address
∥ E.W.: Department of Chemistry, University of Texas at Austin, 105 E 24th St., Austin, TX 78712, United States.
Author Present Address
⊥ V.B.: Intellectual Ventures, 14360 SE Eastgate Way, Bellevue, WA 98007, United States.
Author Present Address
# X.B.: UCB, 3 Preston Ct., Bedford, MA 01730, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- The Pharmaceutical Industry and Global Health Facts and Figures 2017; IFPMA. https://www.ifpma.org/wp-content/uploads/2017/02/IFPMA-Facts-And-Figures-2017.pdf.
- DiMasi J. A.; Grabowski H. G.; Hansen R. W. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J. Health Economics 2016, 47, 20. 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Arico-Muendel C. C. From haystack to needle: finding value with DNA encoded library technology at GSK. MedChemComm 2016, 7, 1898. 10.1039/C6MD00341A. [DOI] [Google Scholar]
- Goodnow R. A.; Dumelin C. E.; Keefe A. D. DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discovery 2017, 16, 131. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Leveridge M.; Chung C. W.; Gross J. W.; Phelps C. B.; Green D. Integration of lead discovery tactics and the evolution of the lead discovery toolbox. SLAS DISCOVERY: Advancing Life Sciences R&D 2018, 23, 881. 10.1177/2472555218778503. [DOI] [PubMed] [Google Scholar]
- Neri D. Twenty-five Years of DNA-Encoded Chemical Libraries. ChemBioChem 2017, 18, 827. 10.1002/cbic.201700130. [DOI] [PubMed] [Google Scholar]
- Machutta C. A.; Kollmann C. S.; Lind K. E.; Bai X.; Chan P. F.; Huang J.; Ballell L.; Belyanskaya S.; Besra G. S.; Barros-Aguirre D.; Bates R. H.; Evindar G.; et al. Prioritizing multiple therapeutic targets in parallel using automated DNA-encoded library screening. Nat. Commun. 2017, 8, 16081. 10.1038/ncomms16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; Davie C. P.; Morgan B. A.; et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 2009, 5, 647. 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Song M.; Hwang G. T. DNA-encoded library screening as core platform technology in drug discovery: its synthetic method development and applications in DEL synthesis. J. Med. Chem. 2020, 63, 6578. 10.1021/acs.jmedchem.9b01782. [DOI] [PubMed] [Google Scholar]
- Kunig V.; Potowski M.; Gohla A.; Brunschweiger A. DNA-encoded Libraries-An Efficient Small Molecule Discovery Technology for the Biomedical Sciences. Biol. Chem. 2018, 399, 691. 10.1515/hsz-2018-0119. [DOI] [PubMed] [Google Scholar]
- Reiher C. A.; Schuman D. P.; Simmons N.; Wolkenberg S. E. Trends in Hit-to-Lead Optimization Following DNA-Encoded Library Screens. ACS Med. Chem. Lett. 2021, 12, 343. 10.1021/acsmedchemlett.0c00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierski W. M.; Xia B.; Miller J.; De la Rosa M.; Favre D.; Dunham R. M.; Washio Y.; Zhu Z.; Wang F.; Mebrahtu M.; Deng H.; Samano V.; et al. DNA-Encoded Library Technology-Based Discovery, Lead Optimization, and Prodrug Strategy toward Structurally Unique Indoleamine 2, 3-Dioxygenase-1 (IDO1) Inhibitors. J. Med. Chem. 2020, 63, 3552. 10.1021/acs.jmedchem.9b01799. [DOI] [PubMed] [Google Scholar]
- Franklin J.; Bai X.; Fan L.. High-Throughput Binder Confirmation (HTBC): A New Non-Combinatorial Synthesis Platform Created to Enhance and Accelerate Hit ID. SLAS 2018 International Conference and Exhibition, San Diego, CA, February 3–7, 2018.
- a Foley T. L.; Burchett W.; Chen Q.; Flanagan M. E.; Kapinos B.; Li X.; Montgomery J. I.; Ratnayake A. S.; Zhu H.; Peakman M. C. Selecting Approaches for Hit Identification and Increasing Options by Building the Efficient Discovery of Actionable Chemical Matter from DNA-Encoded Libraries. SLAS DISCOVERY: Advancing the Science of Drug Discovery 2021, 26, 263. 10.1177/2472555220979589. [DOI] [PubMed] [Google Scholar]; b Ratnayake S. A.; Flanagan M. E.; Foley T. L.; Hutlgren S. L.; Bellenger J.; Montgomery J. I.; Lall M. S.; Liu B.; Ryder T.; Kölmel D. K.; Shavnya A.; Feng X.; Lefker B.; Byrnes L. J.; Sahasrabudhe P. V.; Farley K. A.; Chen S.; Wan J. Toward the assembly and characterization of an encoded library hit confirmation platform: Bead-Assisted Ligand Isolation Mass Spectrometry (BALI-MS). Bioorg. Med. Chem. 2021, 116205. 10.1016/j.bmc.2021.116205. [DOI] [PubMed] [Google Scholar]; c Su W.; Ge R.; Ding D.; Chen W.; Wang W.; Yan H.; Wang W.; Yuan Y.; Liu H.; Zhang M.; Zhang J.; Shu Q.; Satz A. L.; Kuai L. Triaging of DNA-Encoded Library Selection Results by High-Throughput Resynthesis of DNA–Conjugate and Affinity Selection Mass Spectrometry. Bioconjugate Chem. 2021, 32, 1001–1007. 10.1021/acs.bioconjchem.1c00170. [DOI] [PubMed] [Google Scholar]
- Annis D. A.; Nazef N.; Chuang C. C.; Scott M. P.; Nash H. M. A general technique to rank protein– ligand binding affinities and determine allosteric versus direct binding site competition in compound mixtures. J. Am. Chem. Soc. 2004, 126, 15495. 10.1021/ja048365x. [DOI] [PubMed] [Google Scholar]
- Prudent R.; Annis D. A.; Dandliker P. J.; Ortholand J. Y.; Roche D. Exploring new targets and chemical space with affinity selection-mass spectrometry. Nat. Rev. Chem. 2021, 5, 62. 10.1038/s41570-020-00229-2. [DOI] [PubMed] [Google Scholar]
- Haile P. A.; Casillas L. N.; Votta B. J.; Wang G. Z.; Charnley A. K.; Dong X.; Bury M. J.; Romano J. J.; Mehlmann J. F.; King B. W.; Erhard K. F.; Marquis R. W.; et al. Discovery of a First-in-Class Receptor Interacting Protein 2 (RIP2) Kinase Specific Clinical Candidate, 2-((4-(Benzo [d] thiazol-5-ylamino)-6-(tert-butylsulfonyl) quinazolin-7-yl) oxy) ethyl Dihydrogen Phosphate, for the Treatment of Inflammatory Diseases. J. Med. Chem. 2019, 62, 6482. 10.1021/acs.jmedchem.9b00575. [DOI] [PubMed] [Google Scholar]
- Drewry D. H.; King B. W.; Wells C. I.; Zuercher W. J.. New Screening Approaches for Kinases. In Kinase Drug Discovery: Modern Approaches; Ward R. A., Goldberg F. W., Eds.; Royal Society of Chemistry: Croydon, U.K., 2018; pp 9–33. [Google Scholar]
- Haile P. A.; Votta B. J.; Marquis R. W.; Bury M. J.; Mehlmann J. F.; Singhaus R.; Charnley A. K.; Lakdawala A. S.; Convery M. A.; Lipshutz D. B.; Desai B. M.; Casillas L. N.; et al. The Identification and Pharmacological Characterization of 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl) quinolin-4-amine (GSK583), a Highly Potent and Selective Inhibitor of RIP2 Kinase. J. Med. Chem. 2016, 59, 4867. 10.1021/acs.jmedchem.6b00211. [DOI] [PubMed] [Google Scholar]
- Haile P. A.; Casillas L. N.; Bury M. J.; Mehlmann J. F.; Singhaus R.; Charnley A. K.; Hughes T. V.; DeMartino M. P.; Wang G. Z.; Romano J. J.; Dong X.; Marquis R. W.; et al. Identification of Quinoline-Based RIP2 Kinase Inhibitors with an Improved Therapeutic Index to the hERG Ion Channel. ACS Med. Chem. Lett. 2018, 9, 1039. 10.1021/acsmedchemlett.8b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Medeiros P. F.; Raha K.; Elkins P.; Lind K. E.; Lehr R.; Adams N. D.; Burgess J. L.; Schmidt S. J.; Knight S. D.; Auger K. R.; Evindar G.; et al. Discovery of a potent class of PI3Kα inhibitors with unique binding mode via encoded library technology (ELT). ACS Med. Chem. Lett. 2015, 6, 531. 10.1021/acsmedchemlett.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown B. B.; Wagner D. S.; Geysen H. M. A single-bead decode strategy using electrospray ionization mass spectrometry and a new photolabile linker: 3-amino-3-(2-nitrophenyl) propionic acid. Mol. Diversity 1995, 1, 4. 10.1007/BF01715804. [DOI] [PubMed] [Google Scholar]
- Holmes C. P.; Jones D. G. Reagents for combinatorial organic synthesis: development of a new o-nitrobenzyl photolabile linker for solid phase synthesis. J. Org. Chem. 1995, 60, 2318. 10.1021/jo00113a004. [DOI] [Google Scholar]
- Ding Y.; Chai J.; Centrella P. A.; Gondo C.; DeLorey J. L.; Clark M. A. Development and synthesis of DNA-encoded benzimidazole library. ACS Comb. Sci. 2018, 20, 251. 10.1021/acscombsci.8b00009. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Clark M. A. Robust Suzuki–Miyaura cross-coupling on DNA-linked substrates. ACS Comb. Sci. 2015, 17, 1. 10.1021/co5001037. [DOI] [PubMed] [Google Scholar]
- Phelan J. P.; Lang S. B.; Sim J.; Berritt S.; Peat A. J.; Billings K.; Fan L.; Molander G. A. Open-air alkylation reactions in photoredox-catalyzed DNA-encoded library synthesis. J. Am. Chem. Soc. 2019, 141, 3723. 10.1021/jacs.9b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Badir S. O.; Molander G. A. Developments in Photoredox-Mediated Alkylation for DNA-Encoded Libraries. Trends in Chemistry 2021, 3, 161. 10.1016/j.trechm.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L. A.; Ellman J. A. Straightforward and general method for coupling alcohols to solid supports. Tetrahedron Lett. 1994, 35, 9333. 10.1016/S0040-4039(00)78535-2. [DOI] [Google Scholar]
- Zhu Z.; Shaginian A.; Grady L. C.; O’Keeffe T.; Shi X. E.; Davie C. P.; Simpson G. L.; Messer J. A.; Evindar G.; Bream R. N.; Thansandote P. P.; et al. Design and application of a DNA-encoded macrocyclic peptide library. ACS Chem. Biol. 2018, 13, 53. 10.1021/acschembio.7b00852. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Graybill T. L.; Zeng X.; Platchek M.; Zhang J.; Bodmer V. Q.; Wisnoski D. D.; Deng J.; Coppo F. T.; Yao G.; Tamburino A.; Israel D. I.; et al. Cell-based selection expands the utility of DNA-encoded small-molecule library technology to cell surface drug targets: Identification of novel antagonists of the NK3 tachykinin receptor. ACS Comb. Sci. 2015, 17, 722. 10.1021/acscombsci.5b00124. [DOI] [PubMed] [Google Scholar]
- Charnley A. K.; Convery M. A.; Shah A. L.; Jones E.; Hardwicke P.; Bridges A.; Ouellette M.; Totoritis R.; Schwartz B.; King B. W.; Wisnoski D. D.; Casillas L.; et al. Crystal structures of human RIP2 kinase catalytic domain complexed with ATP-competitive inhibitors: foundations for understanding inhibitor selectivity. Bioorg. Med. Chem. 2015, 23, 7000. 10.1016/j.bmc.2015.09.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.