

Abstract
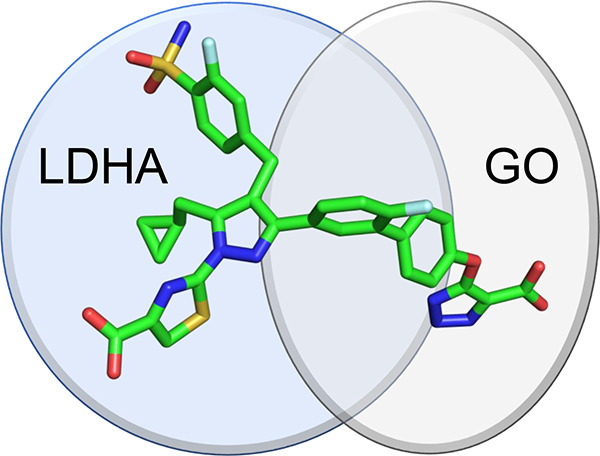
Both glycolate oxidase (GO) and lactate dehydrogenase A (LDHA) influence the endogenous synthesis of oxalate and are clinically validated targets for treatment of primary hyperoxaluria (PH). We investigated whether dual inhibition of GO and LDHA may provide advantage over single agents in treating PH. Utilizing a structure-based drug design (SBDD) approach, we developed a series of novel, potent, dual GO/LDHA inhibitors. X-ray crystal structures of compound 15 bound to individual GO and LDHA proteins validated our SBDD strategy. Dual inhibitor 7 demonstrated an IC50 of 88 nM for oxalate reduction in an Agxt-knockdown mouse hepatocyte assay. Limited by poor liver exposure, this series of dual inhibitors failed to demonstrate significant PD modulation in an in vivo mouse model. This work highlights the challenges in optimizing in vivo liver exposures for diacid containing compounds and limited benefit seen with dual GO/LDHA inhibitors over single agents alone in an in vitro setting.
Keywords: Glycolate oxidase, Lactate dehydrogenase A, Dual inhibitors, Structure-based drug design, Primary hyperoxaluria
Primary hyperoxaluria (PH) is a rare genetic disorder resulting in oxalate overproduction, formation of calcium oxalate kidney stones, and in some patients, end stage kidney failure.1 There are three types of PH, 1, 2, and 3.2 PH1 is caused by mutations in the gene AGXT, which encodes the alanine-glyoxylate aminotransferase (AGT) protein (Figure 1). PH2 and PH3 are caused by mutations in the genes which produce glyoxylate reductase (GR) and 4-hydroxy-2-oxoglutarate aldolase (HOGA) proteins, respectively. These mutations result in an autosomal recessive disorder of glyoxylate metabolism, impairing hepatic detoxification of glyoxylate and corresponding excessive oxalate production in the liver via the action of lactate dehydrogenase A (LDHA). Oxalate produced in the liver is excreted via the kidneys, but when present at high concentrations, it complexes with calcium to form calcium oxalate salt. Deposits of calcium oxalate within the kidney can lead to inflammation, interstitial fibrosis, tissue damage, urinary tract infections and, in severe cases, kidney failure.3a As kidney function deteriorates, concentrations of plasma oxalate increase, and calcium oxalate can accumulate and crystallize in other tissues such as eyes, skin, heart, bones, and brain. Two inhibition mechanisms have been the focus of therapeutic development for PH: glycolate oxidase (GO)3 and LDHA.4
Figure 1.

Metabolic pathways of oxalate synthesis.
GO is encoded by the HAO1 gene and expressed exclusively in the liver peroxisome in humans. As a flavin mononucleotide (FMN)-dependent α-hydroxyacid oxidase, GO converts glycolate into glyoxylate (Figure 1), resulting in increased production of oxalate via LDHA. PH1 patients, with deficiency in AGT, have higher levels of glycolate.5 The Salido group reported that Hao1–/– mice developed high urinary glycolate levels but were overall healthy.6a Compared to Agxt–/– mice with a hyperoxaluria phenotype, Agxt–/–-Hao1–/– double-knockout mice demonstrated reduced urinary oxalate levels. Additionally, CCPST (4-carboxy-5-[(4-chlorophenyl)sulfanyl]-1,2,3-thiadiazole), a modestly potent GO inhibitor, caused oxalate reductions in a cellular Agxt–/– mouse hepatocyte assay. When administered orally to Agxt–/– mice, CCPST reduced urinary oxalate excretion by 30–50%.6a Clinically, lumasiran, a GalNAc-conjugated GO-siRNA therapeutic developed by Alnylam, achieved robust urinary oxalate reduction in PH1 patients.3
In contrast to the indirect influence of GO on oxalate production, the LDH enzymes, encoded by LDH A–C genes, not only catalyze the reversible conversion of pyruvate and NADH to lactate and NAD+ but also convert glyoxylate to oxalate (Figure 1). Thus, inhibition of liver LDHA represents a viable approach for blocking oxalate production and, being the terminal and committed step in oxalate synthesis, may be effective in treating all three types of PH. Individuals with LDHB deficiency often do not have adverse symptoms,7 although LDHA-deficient individuals can experience muscle-related adverse events which can lead to myoglobinuria and kidney damage.8 Importantly, liver LDHA has been selectively targeted using the GalNAc–siRNA conjugate nedosiran from Dicerna. This approach is currently in phase 3 trials and has shown efficacy in reducing urinary oxalate levels in PH1 and PH2 patients.4,9 This suggests that LDHA-inhibition in the liver is sufficient for reducing urinary oxalate in PH patients.
Because GO and LDHA inhibition have been validated clinically via GalNAc-conjugated siRNA therapeutics, we sought to investigate whether additional benefit may be obtained from a dual GO/LDHA small molecule inhibitor.10 This would represent a novel and clinically derisked therapeutic approach for all three forms of PH. With multispecific drugs emerging as a powerful drug targeting approach in modern biopharmaceutical industry,11 we also envisioned that a class of bispecific small molecules would further illustrate the effectiveness of target-based therapeutic molecules. To evaluate the possibility of dual GO/LDHA inhibition by a single small molecule agent, we utilized published structural data of small molecule inhibitors bound to GO and LDHA to inform our approach.
Several potent, small molecule GO inhibitors have been reported, which are defined structurally by a nonpolar tail and polar acidic warhead. The latter resembles the GO substrate, glycolate, and forms multiple key interactions with the enzyme. Examples of well-studied acidic warheads include 3-hydroxy-1H-pyrrole-2,5-dione,12 1,2,3-thiadiazole-4-carboxylic acid,6 and 1,2,3-triazole-4-carboxylic acid13 (Figure 2). The crystal structure of GO in complex with CDST (4-carboxy-5-dodecylsulfanyl-1,2,3-triazole; Ki 15 nM) containing a triazole carboxylic acid warhead was studied by Murray et al.13a A hybrid of CDST and CCPST, compound A, was synthesized and determined as a potent GO inhibitor (Figure 2). Several patented examples of potent GO inhibitors featuring a triazole acid warhead have also been described.14 The numerous interactions between CDST and the GO active site are detailed in Figure 3A. Residues His260 and Tyr132 interact with the triazole ring, while Arg167 and Arg263 coordinate the acid moiety. The phenol moiety of Tyr26 has a long-range interaction with the sulfur atom of the thioether. We considered that a triazole warhead of a dual inhibitor could be utilized to engage the active site and the solvent exposed, hydrophobic tail could potentially be substituted with LDHA-inhibitor.
Figure 2.

Representative examples of GO and LDHA inhibitors.
Figure 3.
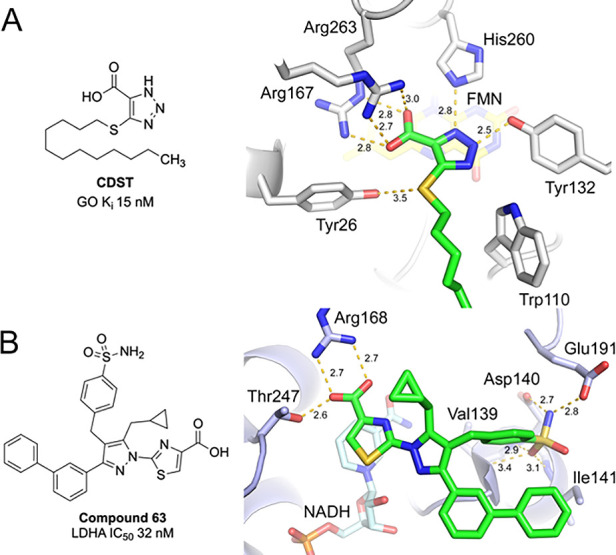
(A) Key interactions of GO with CDST (PDB 2RDT)13a and (B) LDHA with compound 63 (PDB 5W8L).19a
Given the central role of LDHA in glycolysis, LDHA inhibitors have been studied as potential therapeutics in oncology.15 Several scaffolds with potent LDHA inhibition have been developed by pharmaceutical companies, including AstraZeneca (compound 33),16 GlaxoSmithKline (GSK 2837808A),17 and Genentech (GNE-140)18 (Figure 2). Upon examining the SAR and the crystal structures of these compounds bound to LDHA, we were intrigued by the class of pyrazole-based LDHA inhibitors and their unique binding conformation.19 The nanomolar inhibitor compound 63 (Figure 3B) highlights three key interactions: (1) thiazole acid warhead with residue Arg168 and Thr247, (2) cyclopropyl π–π interaction with Tyr238 (not shown for clarity), and (3) sulfonamide with Asp140, Glu191, and Ile141. More recently, the same group reported the beneficial impact of a 2-fluorine substituent on the benzenesulfonamide ring.19b Of note, the terminal biphenyl tail points toward solvent-exposed area and does not form any key interactions with the LDHA enzyme.
On the basis of structural analysis of GO-CDST and LDHA-compound 63 complexes described above, both scaffolds contain a rather large solvent-exposed terminal moiety. Therefore, we hypothesized that upon merging the two scaffolds at the solvent-exposed fronts, a series of diacid-containing compounds may engage the required protein residues to potently inhibit both GO and LDHA (Figure 4). We sought to identify shorter linkers to minimize the overall molecular weight of the dual inhibitors and ideally maintain drug-like properties.
Figure 4.

Strategy for the generation of dual GO/LDHA inhibitors.
On the basis of the above hypothesis, a series of compounds containing both the GO warhead (triazole acid) and LDHA warhead (thiazole carboxylic acid) were synthesized (Scheme 1). The GO warhead precursor, N-PMB protected triazole carboxylate 1, was assembled following a literature method.20 The corresponding tert-butyl ester 2 was obtained upon hydrolysis of ethyl ester 1 followed by treatment with di-tert-butyl dicarbonate. After an SNAr reaction with 1-(3-sulfanylphenyl)ethanone, the sulfur-linked triazole carboxylate 3, was synthesized. The acetyl group of 3 allowed further construction of the compound 63-like scaffold, following literature procedures.19 The acetophenone 3 underwent a magnesium bromide-catalyzed soft enolization in the presence of Hünig’s base,21 yielding 1,3-diketone 4 in 51% yield, which was then alkylated with 4-(bromomethyl)-2-fluorobenzenesulfonamide. This was followed by cyclization with hydrazine, yielding the desired pyrazole intermediate 5 in a quantitative yield. When treated with sodium tert-pentoxide at 0 °C, pyrazole 5 underwent a regioselective arylation reaction with ethyl 2-fluorothiazole-4-carboxylate, favoring the desired regioisomer 6. Use of other bases and the chloro or bromo thiazole ester resulted in reduced selectivity for the desired arylated pyrazole isomer. Both tert-butyl and PMB protecting groups in compound 6 were removed in one step by reaction with trifluoroacetic acid and triethylsilane. Further hydrolysis of the resulting ethyl ester resulted in diacid 7.
Scheme 1. Synthesis of Analogue 7.

Reagents and conditions: (a) LiOH, THF, H2O, 23 °C, 4 h, 66%; (b) (Boc)2O, DMAP, iPr2NEt, CH2Cl2, 23 °C, 3 h, 83%; (c) K2CO3, DMF, 130 °C, 2 h, 87%; (d) 1-(1H-benzo[d][1,2,3]triazol-1-yl)-2-cyclopropylethan-1-one, MgBr2·Et2O, iPr2NEt, CH2Cl2, 23 °C, 2 h, 51%; (e) 4-(bromomethyl)-2-fluorobenzenesulfonamide, Cs2CO3, KI, DMSO, 23 °C, 2 h, 78%; (f) NH2NH2·H2O, EtOH, 70 °C, 2 h, 95%; (g) sodium tert-pentoxide, DMF, 0 °C, 1 h, 21%; (h) TFA, Et3SiH, 40 °C, 18 h, 18%; (i) 1 M aqueous LiOH, THF, MeOH, 23 °C, 2 h, 45%.
The corresponding oxygen linker analogue 11 (Scheme 2) was synthesized from the bromo intermediate 8, which can be accessed in the same manner as described in Scheme 1 (steps d–g), starting from 1-(3-bromo-4-fluorophenyl)ethan-1-one. The bromo intermediate 8 underwent a Miyaura borylation reaction, yielding the corresponding pinacol boronate 9, which was further oxidized to the desired phenol intermediate 10. After SNAr reaction with intermediate 2, deprotection of PMB and tert-butyl ester, and saponification of ethyl carboxylate, the oxygen linker analogue 11 was accessed.
Scheme 2. Synthesis of Analogue 11.

Reagents and conditions: (a) Pd(dppf)Cl2, B2pin2, KOAc, dioxane, 100 °C, 18 h, 46%; (b) urea hydrogen peroxide, MeOH, 23 °C, 18 h, 82%; (c) 2, K2CO3, DMF, 130 °C, 2 h, 19%; (d) TFA, Et3SiH, 40 °C, 18 h, 37%; (e) 1 M aqueous LiOH, THF, MeOH, 23 °C, 2 h, 53%.
Both sulfur and oxygen-linker analogues were further evaluated in a series of in vitro assays. Gratifyingly, both compounds exhibited good potency against both GO and LDHA enzymes, with IC50 values ranging from 0.3–11 nM (Table 1). Both compounds were further examined in a mouse primary hepatocyte assay, where hepatocytes were freshly isolated from wild-type mice (CD1 from Charles River Laboratories) and incubated with tested compound in the presence of pyruvate. Compound potency was determined by measuring inhibition of lactate production in the freshly isolated hepatocytes. Measurement of oxalate production requires ion chromatography mass spectrometry (ICMS)22 for enhanced sensitivity. Interestingly, compound 7 was about 6-fold more potent in the mouse primary hepatocyte assay than compound 11, while both compounds showed similar potency against mouse LDHA (mLDHA) and the human enzyme. Because this primary hepatocyte assay measures intracellular LDHA inhibition, their minor potency difference against GO would have little impact in this discrepancy. Compound 7 exhibits a 0.8 unit higher clogD7.4 value than that of compound 11 (Table 1) and may possess higher cellular permeability, which could explain the cellular potency differences. Additionally, structural changes from compounds 7 to 11 might have an impact on LDHA binding kinetics.19b
Table 1. In Vitro Profile of First-Generation Dual GO/LDHA Inhibitors 7 and 11a.
| enzyme
IC50 (nM) |
|||||
|---|---|---|---|---|---|
| compd | GO | LDHA | mLDHA | primary mouse hepatocyte IC50 (nM) | clogD7.4 |
| 7 | 4.7 | 0.5 | 0.7 | 136 | –0.3 |
| 11 | 11 | 0.4 | 0.3 | 732 | –1.1 |
See Supporting Information for assay conditions; clogD7.4 values were calculated using ACD laboratories (ACD/Percepta 2014 Release). All IC50 values are mean values determined from more than two replicates.
Given the improved cellular LDHA inhibition of the less hydrophilic analogue 7, we next explored the synthesis of analogues with more lipophilic moieties in the linker region (Scheme 3) with the goal of further improving the mouse hepatocyte cellular potency via improved permeability. The more lipophilic oxygen-linker analogue 14 was accessed in three steps, starting from a SNAr reaction between 3-bromophenol and triazole intermediate 1 (Scheme 3). The resulting bromide 12 underwent a Pd-catalyzed Suzuki–Miyaura coupling reaction with pinacol boronate intermediate 9, yielding the diethyl ester 13. After PMB deprotection and ester hydrolysis, 14 was obtained in 15% yield over two steps. Three more analogues, 15, 16, and 17, were synthesized in a similar manner, starting from 4-bromophenol, 4-bromothiophenol, and tert-butyl 5-(4-iodophenyl)-1H-1,2,3-triazole-4-carboxylate, respectively.
Scheme 3. Synthesis of Second-Generation Dual GO/LDHA Inhibitors.

Reagents and conditions: (a) 1, K2CO3, DMF, 130 °C, 9 h, 93%; (b) 9, Pd(dppf)Cl2, 2 M aqueous K3PO4, dioxane, 130 °C, 1 h, 60%; (c) TFA, Et3SiH, 40 °C, 18 h, 59%; (d) 1 M aqueous LiOH, THF, MeOH, 23 °C, 18 h, 25%.
These less hydrophilic compounds, with clogD7.4 values in the 0.6–1.2 range, exhibited subnanomolar IC50 values against both GO and LDHA enzymes. The direct-linked analogue 17 had an IC50 of 3.6 nM against GO (Table 2). Compared to the oxygen or sulfur-linker analogues, the moderately reduced potency observed in the direct-linker analogue 17 could be due to the polar hydroxyl group of Tyr26 in the GO active site (Figure 3A). Nonetheless, all four analogues demonstrated good potency (EC50 143–230 nM) in a mouse hepatocyte assay, further strengthening our hypothesis between lipophilicity and cell activity for the current structural series.
Table 2. In Vitro Profile of Second-Generation Dual GO/LDHA Inhibitorsa.
| enzyme
IC50 (nM) |
|||||
|---|---|---|---|---|---|
| compd | GO | LDHA | mLDHA | primary mouse hepatocyte IC50 (nM) | clogD7.4 |
| 14 | 0.9 | 0.1 | 0.2 | 143 | 0.6 |
| 15 | 1.2 | 0.07 | 0.4 | 198 | 0.6 |
| 16 | 1.1 | 0.3 | 0.2 | 230 | 1.2 |
| 17 | 3.6 | 0.3 | 0.4 | 170 | 0.7 |
See Supporting Information for enzyme and hepatocyte assay conditions; clogD7.4 values were calculated using ACD laboratories (ACD/Percepta 2014 Release). All IC50 values are mean values determined from more than two replicates.
Our strategy of utilizing a structure-based drug design (SBDD)23 led to a series of highly potent dual GO/LDHA inhibitors. To validate the mode of binding for these novel dicarboxylic acid series, we determined the crystal structure of compound 15 in complex with GO and LDHA at 2.07 and 2.50 Å resolution (Supporting Information, Table S1), respectively. In the GO-compound 15 complex, the triazole acid warhead preserves all key interactions with residues Arg167/263, Tyr26, Trp110, Tyr132, and His260 (Figure 5A), previously observed in the GO-CDST complex (Figure 3A). The remainder of the compound, similar to the lipid-like tail of CDST, was accommodated into the solvent exposed area by loop 4 movements.13a
Figure 5.
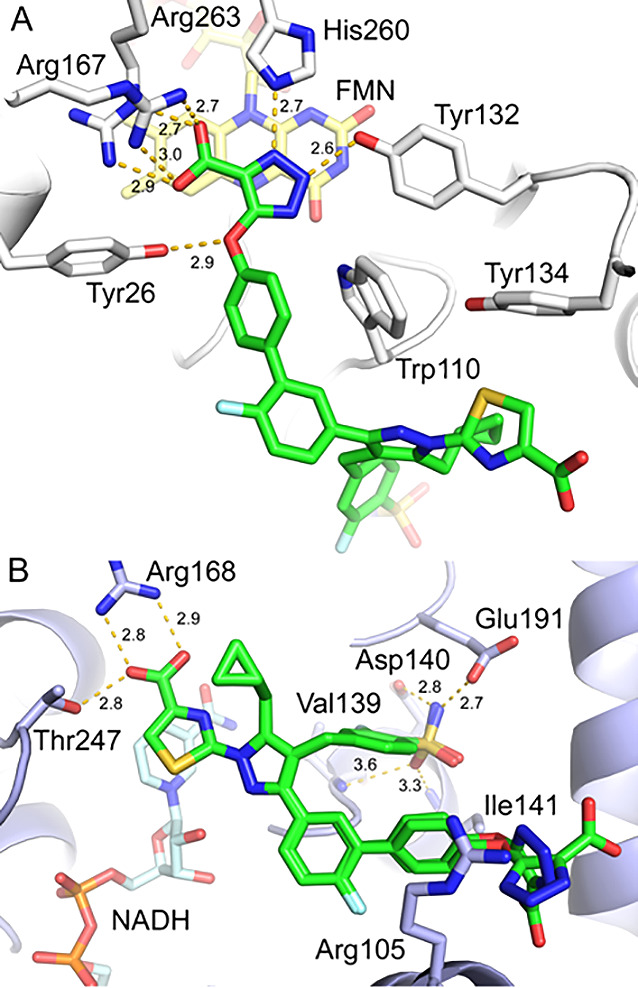
X-ray crystal structures of compound 15 in complex with (A) GO (PDB 7M2O) and (B) LDHA (PDB 7M2N).
In contrast to GO, compound 15 binds to LDHA in two slightly different but well-defined conformations (Figure 5B). In both conformations, the LDHA binding regions of compound 15 were well aligned, highlighting all three binding features observed in the LDHA-compound 63 complex (Figure 3B), including thiazole acid warhead with Arg168/Thr247, cyclopropyl moiety with Tyr238 (not shown for clarity), and the sulfonamide with Asp140/Glu191/Ile141, all of which are critical for a potent LDHA inhibition. Interestingly, the terminal triazole acid GO warhead group extended toward solvent, with a ∼90° rotation between conformers, demonstrating the considerable conformational freedom afforded by this region. The helix associated with Arg105 also moved outward to facilitate binding.
While the structural and chemical hypotheses proposed herein were validated with the new dual inhibitors, the question of whether the dual compound scaffold exhibits efficacy in cellular assays and animals remained. Therefore, we examined the profile of the current series of dual GO/LDHA inhibitors in a cellular model of PH1. In this assay, compound-mediated inhibition of glycolate to oxalate conversion was assessed in freshly isolated hepatocytes from mice deficient in AGT through the hepatic knockdown of the Agxt gene via an Agxt-siRNA encapsulated in a lipid nanoparticle (LNP) as reported in the literature.24 Conversion of glycolate to oxalate was measured using ICMS.22 The advantage of this lower throughput assay is the direct measurement of reduction of oxalate production from glycolate, which encompasses both GO and LDHA inhibitory pathways simultaneously (Figure 1). Seven days following a single intravenous administration of Agxt-siRNA LNP to CD1 mice, hepatocytes were isolated and freshly incubated with glycolate and inhibitor at various concentrations (Table 3). In this assay, compound 7 demonstrated robust inhibition of oxalate production with an IC50 of 88 nM, further validating the role of these dual GO/LDHA inhibitors in inhibiting oxalate synthesis, the etiological driver in PH. In the same oxalate reduction assay, compound 7 was only moderately more potent compared to individual GO inhibitor A or LDHA inhibitor 63, while their corresponding intrinsic mouse GO or LDHA potency are comparable, suggesting dual GO/LDHA inhibition might not be additive or synergistic compared to individual inhibition.
Table 3. Wild-Type and Agxt-KD Hepatocyte Assaysa.
| compd | mGO IC50 (nM) | mLDHA IC50 (nM) | mouse primary hepatocyte (pyruvate to lactate) IC50 (nM) | AgxtKD mouse primary hepatocyte (glycolate to oxalate) IC50 (nM) |
|---|---|---|---|---|
| 7 | 7.7 | 0.8 | 136 | 88 |
| A | 14.6 | 149 | ||
| 63 | 0.1 | 181b |
See Supporting Information for assay conditions. All IC50 values are mean values determined from more than two replicates unless otherwise specified.
IC50 value from n = 1 experiment.
Encouraged by the robust inhibition in the PH1 mouse hepatocyte assay demonstrated by compound 7, we sought to further assess the in vivo pharmacodynamic (PD) effect of this series of dual inhibitors. It is challenging to measure changes in urinary oxalate in vivo in wild-type mice, where their basal urinary oxalate is low. Measurements of urinary oxalate in Agxt-KD mice can be achieved but require 5+ days of compound treatment.6a Thus, we sought to measure plasma glycolate in wild-type mice, a convenient and noninvasive PD marker for GO inhibition, as a more acute measurement of compound PD.25 Compound 7 and the second-generation analogue compound 14, which showed superior potency in a mouse primary hepatocyte assay, were further evaluated in the mouse plasma glycolate-PD study. Compounds were orally administered at 100 mg/kg to C57BL/6N mice (n = 3 per group), and plasma glycolate levels were measured at 6 h after compound administration.26 Neither of the dual inhibitors demonstrated a significant PD effect for plasma glycolate elevation (Figure 6). In comparison, compound A produced a 4–5-fold elevation of plasma glycolate 2 h post oral dosing.
Figure 6.

Mouse PK/PD of compounds 7, 14 (6 h post oral dosing), and A (2 h post oral dosing).
We investigated the hepatic exposure of both compounds to understand the relationship between pharmacokinetic (PK) and the modest GO-driven PD effect. As a result, mouse liver samples were collected at the end of the PD experiment. Both dual inhibitors showed low μM liver exposures, which were significantly below liver exposures seen with compound A at the same dose. Poor liver exposure may be attributed to low oral absorption of the highly polar dicarboxylic acid series. Recognizing the limitations of this dicarboxylic acid series is important for further optimization and acid isosteres27 or prodrug approaches28 are possible strategies to improve the oral bioavailability and liver exposures.
In summary, we developed a series of potent dual GO/LDHA inhibitors utilizing a structure-based drug design strategy. The binding mode of these novel inhibitors was confirmed by the X-ray crystal structures of compound 15 in complex with GO and LDHA. Careful tuning of clogD7.4 led to a series of second-generation dual GO/LDHA inhibitors, which showed improved potency in a mouse primary hepatocyte assay, presumably via improved passive cellular permeability. Compound 7 demonstrated potent cellular inhibition of oxalate production in an PH1 mouse cellular model, the Agxt-knockdown primary hepatocyte assay. Further mouse PD studies with hepatic PK analysis revealed a limitation of the current series of dual inhibitors in reaching sufficient liver exposure to engage the hepatic GO enzyme. Finally, we were disappointed that serial inhibition of oxalate synthesis via dual GO/LDHA inhibition did not appear to be additive.29 Individual novel GO or LDHA small-molecule inhibitors demonstrated similar potencies as dual inhibition in the Agxt-knockdown primary hepatocyte assay (50–200 nM range). Our efforts to further develop GO and LDHA small-molecule inhibitors for primary hyperoxaluria will be presented in future publications.
Acknowledgments
We thank Chinook leadership team in supporting this project, Wake Forest School of Medicine (under the supervision of W.T.L.) for providing crystallization and X-ray diffraction facilities (Wake Forest Baptist Comprehensive Cancer Center P30 CA012197), and University of Alabama at Birmingham (under the supervision of J.K.) for ICMS facilities. We also thank Terrence L. Smalley for many helpful discussions.
Glossary
Abbreviations
- siRNA
small interfering RNA
- GalNAc
N-acetylgalactosamine
- NAD
nicotinamide adenine dinucleotide
- PMB
p-methoxybenzyl
- SNAr
nucleophilic aromatic substitution
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- TFA
trifluoroacetic acid
- LNP
lipid nanoparticle.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00196.
Detailed description of chemical synthesis, 1H NMR data, methods of protein expression and purification, X-ray crystallography, enzyme and hepatocyte (both wild-type and Agxt-knock down) assays, in vivo pharmacokinetic, pharmacodynamic studies, and analysis (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Some authors are current or former employees of Chinook Therapeutics and potentially own and/or hold stocks in the company.
Supplementary Material
References
- Lawrence J. E.; Wattenberg D. J. Primary Hyperoxaluria: The Patient and Caregiver Perspective. Clin. J. Am. Soc. Nephrol. 2020, 15, 909–911. 10.2215/CJN.13831119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Raju D. L.; Cantarovich M.; Brisson M.; Tchervenkov J.; Lipman M. L. Primary hyperoxaluria: clinical course, diagnosis, and treatment after kidney failure. Am. J. Kidney Dis. 2008, 51, e1–e5. 10.1053/j.ajkd.2007.08.027. [DOI] [PubMed] [Google Scholar]; b Hoppe B. An update on primary hyperoxaluria. Nat. Rev. Nephrol. 2012, 8, 467–475. 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]; c Bhasin B.; Ürekli H. M.; Atta M. G. Primary and secondary hyperoxaluria: Understanding the enigma. World J. Nephrol. 2015, 4, 235–244. 10.5527/wjn.v4.i2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Milliner D.; McGregor T.; Thompson A.; Dehmel B.; Knight J.; Rosskamp R.; Blank M.; Yang S.; Fargue S.; Rumsby G.; Groothoff J.; Allain M.; West M.; Hollander K.; Lowther W.; Lieske J. End Points for Clinical Trials In Primary Hyperoxaluria. Clin. J. Am. Soc. Nephrol. 2020, 15, 1056–1065. 10.2215/CJN.13821119. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liebow A.; Li X.; Racie T.; Hettinger J.; Bettencourt B. R.; Najafian N.; Haslett P.; Fitzgerald K.; Holmes R. P.; Erbe D.; Querbes W.; Knight J. An Investigational RNAi Therapeutic Targeting Glycolate Oxidase Reduces Oxalate Production in Models of Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2017, 28, 494–503. 10.1681/ASN.2016030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe B., Cochat P., Lemoine S., Lipkin G., Gentile A. M., Brown B. D., Rosskamp R., Hulton S., Groothoff J. W., Baum M. A.. PHYOX: A Safety and Tolerability Study of DCR-PHXC In Primary Hyperoxaluria Types 1 and 2; Dicerna, 2019; https://investors.dicerna.com/static-files/51c47431-1ff6-463f-8fec-67ad571da49a.
- a Frishberg Y.; Zeharia A.; Lyakhovetsky R.; Bargal R.; Belostotsky R. Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J. Med. Genet. 2014, 51, 526–529. 10.1136/jmedgenet-2014-102529. [DOI] [PubMed] [Google Scholar]; b Craigen W. J. Persistent glycolic aciduria in a healthy child with normal alanine-glyoxylate aminotransferase activity. J. Inherited Metab. Dis. 1996, 19, 793–794. 10.1007/BF01799176. [DOI] [PubMed] [Google Scholar]
- a Martin-Higueras C.; Luis-Lima S.; Salido E. Glycolate Oxidase Is a Safe and Efficient Target for Substrate Reduction Therapy in a Mouse Model of Primary Hyperoxaluria Type I. Mol. Ther. 2016, 24, 719–725. 10.1038/mt.2015.224. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen Z. W.; Vignaud C.; Jaafar A.; Lévy B.; Guéritte F.; Guénard D.; Lederer F.; Mathews F. S. High resolution crystal structure of rat long chain hydroxy acid oxidase in complex with the inhibitor 4-carboxy-5-[(4-chlorophenyl)sulfanyl]-1, 2, 3-thiadiazole. Implications for inhibitor specificity and drug design. Biochimie 2012, 94, 1172–1179. 10.1016/j.biochi.2012.02.003. [DOI] [PubMed] [Google Scholar]; c Bourhis J. M.; Vignaud C.; Pietrancosta N.; Guéritte F.; Guénard D.; Lederer F.; Lindqvist Y. Structure of human glycolate oxidase in complex with the inhibitor 4-carboxy-5-[(4-chlorophenyl)sulfanyl]-1,2,3-thiadiazole. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2009, 65, 1246–1253. 10.1107/S1744309109041670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanis R. J.; Neel J. V.; Torres de Arauz R. Two more ″private″ polymorphisms of Amerindian tribes: LDHb GUA-1 and ACP1 B GUA-1 in the Guaymi in Panama. Am. J. Human Genetics 1977, 29, 419–430. [PMC free article] [PubMed] [Google Scholar]
- a Kanno T.; Maekawa M. Lactate dehydrogenase M-subunit deficiencies: clinical features, metabolic background, and genetic heterogeneities. Muscle Nerve 1995, 18, S54–S60. 10.1002/mus.880181413. [DOI] [PubMed] [Google Scholar]; b Lactate dehydrogenase deficiency. Genetic Home Reference; U.S. National Library of Medicine, 2020; https://ghr.nlm.nih.gov/condition/lactate-dehydrogenase-deficiency.
- Hoppe B., Cochat P., Lemoine S., Lipkin G., Gentile A. M., Brown B. D., Rosskamp R., Hulton S., Groothoff J. W., Baum M. A.. PHYOX: A Safety and Tolerability Study of DCR-PHXC In Primary Hyperoxaluria Types 1 and 2; Dicerna, 2019; https://investors.dicerna.com/static-files/51c47431-1ff6-463f-8fec-67ad571da49a. [Google Scholar]
- Moya-Garzon M. D.; Gomez-Vidal J. A.; Alejo-Armijo A.; Altarejos J.; Rodriguez-Madoz J. R.; Fernandes M. X.; Salido E.; Salido S.; Diaz-Gavilan M. Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias. J. Pers. Med. 2021, 11, 74. 10.3390/jpm11020074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Deshaies R. J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. 10.1038/s41586-020-2168-1. [DOI] [PubMed] [Google Scholar]; b Wang Y.; Yang S. Multispecific drugs: the fourth wave of biopharmaceutical innovation. Sig. Transduct. Target. Ther. 2020, 5, 86. 10.1038/s41392-020-0201-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney C. S.; Randall W. C.; Streeter K. B.; Ziegler C.; Cragoe E. J. Jr.; Schwam H.; Michelson S. R.; Williams H. W. R.; Eichler E.; Duggan D. E.; Ulm E. H.; Noll R. M. Inhibitors of glycolic acid oxidase. 4-Substituted 3-hydroxy-1H-pyrrole-2,5-dione derivatives. J. Med. Chem. 1983, 26, 700–714. 10.1021/jm00359a015. [DOI] [PubMed] [Google Scholar]
- a Murray M. S.; Holmes R. P.; Lowther W. T. Active Site and Loop 4 Movements within Human Glycolate Oxidase: Implications for Substrate Specificity and Drug Design. Biochemistry 2008, 47, 2439–2449. 10.1021/bi701710r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stenberg K.; Lindqvist Y. Three-dimensional structures of glycolate oxidase with bound active-site inhibitors. Protein Sci. 1997, 6, 1009–1015. 10.1002/pro.5560060506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lowther W. T., Holmes R. P.. Glycolate oxidase inhibitors and methods of use for the treatment of kidney stones. WO2017100266A1, June 15, 2017.; b Wang B., Chao Q.. Glycolate Oxidase Inhibitors for the treatment of disease. WO2019133770, July 4, 2019.; c Keller B. T., Talley J. J.. Compounds and methods for treating oxalate related diseases, WO2019165159, August 29, 2019.; d Maag H., Fernandes M. X., Zamboni R., Akbariromani E., Beaulieu M., Leblanc Y., Thakur P.. Triazole glycolate oxidase inhibitors. WO2020010309, January 9, 2020.
- a Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Le A.; Cooper C. R.; Gouw A. M.; Dinavahi R.; Maitra A.; Deck L. M.; Royer R. E.; Vander Jagt D. L.; Semenza G. L.; Dang C. V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2037–2042. 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yu Y.; Liao M.; Liu R.; Chen J.; Feng H.; Fu Z. Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: its implication for treatment. World J. Surg. Oncol. 2014, 12, 78. 10.1186/1477-7819-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R. A.; Brassington C.; Breeze A. L.; Caputo A.; Critchlow S.; Davies G.; Goodwin L.; Hassall G.; Greenwood R.; Holdgate G. A.; Mrosek M.; Norman R. A.; Pearson S.; Tart J.; Tucker J. A.; Vogtherr M.; Whittaker D.; Wingfield J.; Winter J.; Hudson K. Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J. Med. Chem. 2012, 55, 3285–3306. 10.1021/jm201734r. [DOI] [PubMed] [Google Scholar]
- Billiard J.; Dennison J. B.; Briand J.; Annan R. S.; Chai D.; Colón M.; Dodson C. S.; Gilbert S. A.; Greshock J.; Jing J.; Lu H.; McSurdy-Freed J. E.; Orband-Miller L. A.; Mills G. B.; Quinn C. J.; Schneck J. L.; Scott G. F.; Shaw A. N.; Waitt G. M.; Wooster R. F.; Duffy K. J. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013, 1, 19. 10.1186/2049-3002-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dragovich P. S.; Fauber B. P.; Corson L. B.; Ding C. Z.; Eigenbrot C.; Ge H.; Giannetti A. M.; Hunsaker T.; Labadie S.; Liu Y.; Malek S.; Pan B.; Peterson D.; Pitts K.; Purkey H. E.; Sideris S.; Ultsch M.; VanderPorten E.; Wei B.; Xu Q.; Yen I.; Yue Q.; Zhang H.; Zhang Z. Identification of substituted 2-thio-6-oxo-1,6-dihydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23, 3186–3194. 10.1016/j.bmcl.2013.04.001. [DOI] [PubMed] [Google Scholar]; b Boudreau A.; Purkey H. E.; Hitz A.; Robarge K.; Peterson D.; Labadie S.; Kwong M.; Hong R.; Gao M.; Del Nagro C.; Pusapati R.; Ma S.; Salphati L.; Pang P.; Zhou A.; Lai T.; Li Y.; Chen Z.; Wei B.; Yen I.; Sideris S.; McCleland M.; Firestein R.; Corson L.; Vanderbilt A.; Williams S.; Daemen A.; Belvin M.; Eigenbrot C.; Jackson P. K.; Malek S.; Hatzivassiliou G.; Sampath D.; Evangelista M.; O’Brien T. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
- a Rai G.; Brimacombe K. R.; Mott B. T.; Urban D. J.; Hu X.; Yang S.; Lee T.; Cheff D. M.; Kouznetsova J.; Benavides G. A.; Pohida K.; Kuenstner E. J.; Luci D. K.; Lukacs C. M.; Davies D. R.; Dranow D. M.; Zhu H.; Sulikowski G.; Moore W. J.; Stott G. M.; Flint A. J.; Hall M. D.; Darley-Usmar V. M.; Neckers L. M.; Dang C. V.; Waterson A. G.; Simeonov A.; Jadhav A.; Maloney D. J. Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. 10.1021/acs.jmedchem.7b00941. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rai G.; Urban D. J.; Mott B. T.; Hu X.; Yang S.; Benavides G. A.; Johnson M. S.; Squadrito G. L.; Brimacombe K. R.; Lee T. D.; Cheff D. M.; Zhu H.; Henderson M. J.; Pohida K.; Sulikowski G. A.; Dranow D. M.; Kabir M.; Shah P.; Padilha E.; Tao D.; Fang Y.; Christov P. P.; Kim K.; Jana S.; Muttil P.; Anderson T.; Kunda N. K.; Hathaway H. J.; Kusewitt D. F.; Oshima N.; Cherukuri M.; Davies D. R.; Norenberg J. P.; Sklar L. A.; Moore W. J.; Dang C. V.; Stott G. M.; Neckers L.; Flint A. J.; Darley-Usmar V. M.; Simeonov A.; Waterson A. G.; Jadhav A.; Hall M. D.; Maloney D. J. Pyrazole-Based Lactate Dehydrogenase Inhibitors with Optimized Cell Activity and Pharmacokinetic Properties. J. Med. Chem. 2020, 63, 10984–11011. 10.1021/acs.jmedchem.0c00916. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Christov P. P.; Kim K.; Jana S.; Romaine I. M.; Rai G.; Mott B. T.; Allweil A. A.; Lamers A.; Brimacombe K. R.; Urban D. J.; Lee T. D.; Hu X.; Lukacs C. M.; Davies D. R.; Jadhav A.; Hall M. D.; Green N.; Moore W. J.; Stott G. M.; Flint A. J.; Maloney D. J.; Sulikowski G. A.; Waterson A. G. Optimization of ether and aniline based inhibitors of lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2021, 41, 127974. 10.1016/j.bmcl.2021.127974. [DOI] [PMC free article] [PubMed] [Google Scholar]; d LDHB potency was not profiled for the dual GO/LDHA compounds reported in this paper, however, based on reported potency profile of compound 63, we expected all compounds to be of similar potency against LDHB.
- Buckle D. R.; Rockell C. J. M. Studies on v-triazoles. Part 4. The 4-methoxybenzyl group, a versatile N-protecting group for the synthesis of N-unsubstituted v-triazoles. J. Chem. Soc., Perkin Trans. 1 1982, 0, 627–630. 10.1039/p19820000627. [DOI] [Google Scholar]
- Lim D.; Fang F.; Zhou G.; Coltart D. M. Direct Carbon–Carbon Bond Formation via Soft Enolization: A Facile and Efficient Synthesis of 1,3-Diketones. Org. Lett. 2007, 9, 4139–4142. 10.1021/ol701599v. [DOI] [PubMed] [Google Scholar]
- Fargue S.; Knight J.; Holmes R. P.; Rumsby G.; Danpure C. J. Effects of alanine:glyoxylate aminotransferase variants and pyridoxine sensitivity on oxalate metabolism in a cell-based cytotoxicity assay. Biochim. Biophys. Acta, Mol. Basis Dis. 2016, 1862, 1055–1062. 10.1016/j.bbadis.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reich S. H.; Webber S. E. Structure-based drug design (SBDD): Every structure tells a story. Perspect. Drug Discovery Des. 1993, 1, 371–390. 10.1007/BF02174536. [DOI] [Google Scholar]; b Klebe G. Recent developments in structure-based drug design. J. Mol. Med. 2000, 78, 269–281. 10.1007/s001090000084. [DOI] [PubMed] [Google Scholar]
- Harwiger P., Vornlocher H., Brown J. M., Neuman K. K. H.. Defined Multi-Conjugate Oligonucleotides. WO2016205410, December 22, 2016.
- Dutta C.; Avitahl-Curtis N.; Pursell N.; Larsson Cohen M.; Holmes B.; Diwanji R.; Zhou W.; Apponi L.; Koser M.; Ying B.; Chen D.; Shui X.; Saxena U.; Cyr W. A.; Shah A.; Nazef N.; Wang W.; Abrams M.; Dudek H.; Salido E.; Brown B. D.; Lai C. Inhibition of Glycolate Oxidase With Dicer-substrate siRNA Reduces Calcium Oxalate Deposition in a Mouse Model of Primary Hyperoxaluria Type 1. Mol. Ther. 2016, 24, 770–778. 10.1038/mt.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 6 h time point was chosen based on previous time course studies with GO inhibitors, where maximal plasma glycolate elevations (up to 10-fold) were determined 6 h post PO dosing.
- Lassalas P.; Gay B.; Lasfargeas C.; James M. J.; Tran V.; Vijayendran K. G.; Brunden K. R.; Kozlowski M. C.; Thomas C. J.; Smith A. B. III; Huryn D. M.; Ballatore C. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem. 2016, 59 (7), 3183–3203. 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Järvinen T.; Savolainen J. Prodrugs: design and clinical applications. Nat. Rev. Drug Discovery 2008, 7, 255–270. 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]; b Jana S.; Mandlekar S.; Marathe P. Prodrug design to improve pharmacokinetic and drug delivery properties: challenges to the discovery scientists. Curr. Med. Chem. 2010, 17, 3874–3908. 10.2174/092986710793205426. [DOI] [PubMed] [Google Scholar]; c Najjar A.; Najjar A.; Karaman R. Newly Developed Prodrugs and Prodrugs in Development; an Insight of the Recent Years. Molecules 2020, 25, 884. 10.3390/molecules25040884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin N.; Ma W.; Pei J.; Ouyang Q.; Tang C.; Lai L. Synergistic and Antagonistic Drug Combinations Depend on Network Topology. PLoS One 2014, 9 (4), e93960 10.1371/journal.pone.0093960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.