

Abstract
The impairment of the cystic fibrosis transmembrane conductance regulator (CFTR) activity induces intracellular chloride (Cl–) accumulation. The anion Cl–, acting as a second messenger, stimulates the secretion of interleukin‐1β (IL‐1β), which starts an autocrine positive feedback loop. Here, we show that NLR family pyrin domain containing 3 (NLRP3) and caspase 1 (CASP1) are indirectly modulated by the intracellular Cl– concentration, showing maximal expression and activity at 75 mM Cl–, in the presence of the ionophores nigericin and tributyltin. The expression of PYD and CARD domain containing (PYCARD/ASC) remained constant from 0 to 125 mM Cl–. The CASP1 inhibitor VX‐765 and the NLRP3 inflammasome inhibitor MCC950 completely blocked the Cl–‐stimulated IL‐1β mRNA expression and partially the IL‐1β secretion. DCF fluorescence (cellular reactive oxygen species, cROS) and MitoSOX fluorescence (mitochondrial ROS, mtROS) also showed maximal ROS levels at 75 mM Cl–, a response strongly inhibited by the ROS scavenger N‐acetyl‐L‐cysteine (NAC) or the NADPH oxidase (NOX) inhibitor GKT137831. These inhibitors also affected CASP1 and NLRP3 mRNA and protein expression. More importantly, the serum/glucocorticoid regulated kinase 1 (SGK1) inhibitor GSK650394, or its shRNAs, completely abrogated the IL‐1β mRNA response to Cl– and the IL‐1β secretion, interrupting the autocrine IL‐1β loop. The results suggest that Cl– effects are mediated by SGK1, in which under Cl– modulation stimulates the secretion of mature IL‐1β, in turn, responsible for the upregulation of ROS, CASP1, NLRP3 and IL‐1β itself, through autocrine signalling.
Keywords: autocrine signalling, CFTR, chloride anion, chloride channel, inflammasome, interleukin, NLRP3, ROS, second messenger, SGK1
The Cl– anion induces the SGK1 activation which in turn stimulates the IL‐1β secretion, starting an autocrine IL‐1β‐positive feedback loop that enhances the CASP1 and NLRP3 expression through IL1R1 canonical signalling and parallel IL1R1/NOX/ROS signalling that further activates the IL‐1β expression, maturation and secretion.
Abbreviations
- CASP1
caspase 1
- cROS
cellular reactive oxygen species
- CFTR
cystic fibrosis transmembrane conductance regulator
- CF
cystic fibrosis
- IL‐1β
interleukin 1 beta
- mtROS
mitochondrial ROS
- NAC
N‐acetyl‐L‐cysteine
- NOX
NADPH oxidase
- NLRP3
NLR family pyrin domain containing 3
- PYCARD/ASC
PYD and CARD domain containing
- SGK1
serum/glucocorticoid regulated kinase 1
INTRODUCTION
CFTR is a chloride channel activated by cAMP, which is responsible for the inherited disease cystic fibrosis (CF). In the initial years after the CFTR was cloned, most works focused on the non‐genomic effects of the CFTR failure. Since CF had a complex phenotype, we tested the hypothesis that this complex phenotype was due to the modulation of a net of genes under CFTR control (genomic effects). By using differential display [1], we were able to find the first molecule in the chain of the CFTR‐signalling pathway, which was the protein tyrosine kinase c‐Src [2]. Its activity was increased in CF cells and determined the overexpression of MUC1 [2]. At the same time, we explored possible regulators of CFTR expression, which was considered at that time a housekeeping gene, and find that IL‐1β (interleukin 1 beta, official HUGO symbol IL‐1B) was able to modulate its expression [3] through NF‐κB activation [4]. On the other hand, other laboratories also found differentially expressed genes by using microarrays [5, 6. Therefore, the hypothesis of the existence of a net of CFTR‐dependent genes was correct.
Later, two additional differentially expressed genes were selected for further characterization since, contrary to c‐Src, their expression was reduced in CF cells or cells treated with CFTR inhibitors. Noteworthy both genes, CISD1 (yet with an unclear function) [7] and MTND4 [8] codified mitochondrial proteins. Since MTND4 was known to be essential for the activity and assembly of the mitochondrial complex I (mCx‐I), we then decided to test the activity of this complex in CF cells. In agreement with the reduced MTND4 found in CF cells, reduced activity of mCx‐I was also found [9]. We soon realized that these results were in consonance with an earlier mitochondrial theory for CF postulated by Burton Shapiro in the late 80 s, which was completely disregarded (erroneously) when the CFTR was cloned and found to be a chloride channel, located in the plasma membrane and not in the mitochondria [10, 11, 12. The work of Shapiro's laboratory was discussed in detail elsewhere [13]. Therefore, the CFTR channel was signalling inside the cell, increasing c‐Src activity and reducing the mCx‐I activity. Further exploring the possible CFTR‐signalling molecules, we found that IL‐1β was responsible for the reduced mCx‐I activity and increased ROS production in CF cells, through an autocrine positive feedback loop [14]. Later, also using differential display, we found that Cl– could modulate the expression of specific genes (Cl–‐dependent genes) [15], acting as a second messenger to indirectly regulate gene expression [16]. Recently, we also found that IL‐1β was upstream of c‐Src in the CFTR‐signalling pathway [17] and that the increased intracellular Cl– concentration found in cells with impaired CFTR activity was responsible for the increased IL‐1β maturation and secretion [18].
The NLRP3 inflammasome is involved in IL‐1β maturation and secretion [19]. The role and relevance of the NLRP3 inflammasome in cystic fibrosis have been extensively reported [20, 21, 22. The NLRP3 inflammasome is a complex composed of three proteins, NLR family pyrin domain containing 3 (NLRP3), caspase 1 (CASP1) and apoptosis‐associated speck‐like protein containing a CARD (ASC/PYCARD). The NLRP3 inflammasome activation usually needs two signals. The first signal (signal 1 or priming) induces the upregulation and accumulation of IL‐1β and inflammasome components. The second signal activates the inflammasome, which involves the recruitment of NLRP3, ASC and pro‐CASP1 to mitochondria to form the NLRP3 inflammasome complex, with the help of cardiolipin in the external mitochondrial membrane and then follows a self‐cleavage and activation of pro‐CASP1 and the cleavage of pro‐IL‐1β. Usually, pro‐IL‐1β accumulates until the NLRP3 inflammasome is activated by different danger‐ and pathogen‐associated molecular patterns (DAMPs, PAMPs)(signal 2 or activation) [23, 24 or by noncanonical activation [25, 26; all DAMPs and PAMPs signals appear to be integrated through mtROS signalling. [23]
The precise mechanism of activation for the NLRP3 inflammasome is still unknown, and different signals can activate the NLRP3 inflammasome, including K+ efflux, Ca2+ influx and high levels of reactive oxygen species (ROS), which appear to integrate most signals [19, 27. Once IL‐1β is processed by CASP1, it is secreted by mechanisms that are not fully understood [28]. The secreted cytokine can bind to its IL1R1 receptor starting a positive autocrine/paracrine feedback loop (concepts originally applied to mammalian cells by Michael Sporn [29]) that further enhances its signalling [14]. Once the IL1R1 receptor is activated by IL‐1β, it recruits MyD88 and Rac1, initiating a cascade of events including receptor internalization, NADPH oxidase (NOX) activation [30, 31, 32, and further recruitment of proteins and events that constitutes the canonical IL‐1β pathway, eventually leading to NF‐κB activation [33].
Here, we further explored the mechanism by which Cl– modulates IL‐1β expression and secretion, considering the Cl– effects on the expression and activity of the inflammasome components. IB3‐1 bronchial epithelial CF cells were used, which constitutively overexpress and release IL‐1β without the need for external priming or activating steps [14]. Thus, LPS (priming) and ATP (activation) are not needed to prime/activate these cells, which simplifies the model system since LPS and ATP activate by themselves multiple signalling pathways that might render very complex results, hard to interpret. We found that IL‐1β, caspase 1 (CASP1) and NLRP3 expression, mRNA and protein levels, were upregulated when the intracellular Cl– was increased from 5 to 75 mM, in the presence of the tributyltin and nigericin ionophores. This regulation by Cl– occurs through a complex pathway involving the activation of the serum/glucocorticoid regulated kinase 1 (SGK1). Although SGK1 was reported to activate NF‐κB [34] and NLRP3 [35], we postulate that SGK1 activation by Cl– may also increase the secretion of IL‐1β that in turn starts an IL‐1β autocrine/paracrine positive feedback loop through the IL1R1 canonical signalling (IL1R1→MYD88 → IRAK →TRAF6 → TAK1 → IKK →IkBα → NF‐κB axis) and the parallel IL1R1 → NOX →cROS signalling. This results in further upregulation of NLRP3, CASP1 and IL‐1β mRNAs and proteins, completing the autocrine loop with more IL‐1β secretion. Thus, the intracellular Cl– and the SGK1 kinase are important regulators of the NLRP3 inflammasome and IL‐1β expression and secretion.
MATERIAL AND METHODS
Reagents
IL‐1 receptor antagonist (Cat. No. SRP3327), dimethyl sulphoxide (DMSO, culture grade), p‐coumaric acid, luminol, tributyltin, nigericin, Fura‐2 AM, N‐acetyl‐L‐cysteine, BAPTA‐AM, mesoxalonitrile 4‐trifluoromethoxyphenyl‐hydrazone (FCCP, mitochondrial uncoupler) and protease inhibitor cocktail (Cat. No. P2714) were purchased from Sigma‐Aldrich (St. Louis, MO). Trypsin was purchased from Life Technologies (GIBCO BRL, Rockville, MD). MitoSOX (Cat. No. M36008), 2´,7´‐dichlorofluorescein diacetate (DCFH‐DA, Cat. No. D399) and tetramethylrhodamine‐ethyl ester perchlorate (TMRE, Cat. No. T669) were from Molecular Probes, Life Technologies Corporation. NLRP3 inflammasome inhibitor MCC950, CASP1 inhibitor VX‐765, NOX inhibitor GKT137831 and SGK1 inhibitor GSK650394 were from MedChem Express (MCE). DiBAC4(3) (Bis‐(1,3‐dibutylbarbituric acid)trimethine oxonol, Cat. No. 61011) and BCECF‐AM (2′,7′‐Bis(2‐carboxyethyl)‐5(6)‐carboxyfluorescein acetoxymethyl ester, Cat. No. 51012) were from Biotium. All other reagents were analytical grade. Antibodies: rabbit anti‐caspase 1 (polyclonal, sc‐515) was from Santa Cruz Biotechnology Inc.; anti‐mouse antibody coupled to horseradish peroxidase (polyclonal, W402B) and anti‐rabbit antibody coupled to horseradish peroxidase (polyclonal, W401B) were from Promega; mouse anti‐IL‐1β (mAb, IgG1, I3642) and rabbit anti‐actin antibody (polyclonal, A2066) were from Sigma‐Aldrich; mouse monoclonal anti‐NLRP3 (Cryo‐2) and rabbit polyclonal anti‐ASC (AL177) were from Adipogen; and rabbit anti SGK1 (D27C11) was from Cell Signaling Technology.
Cultured cells
IB3‐1 cells (ATCC CRL‐2777, a bronchial cell line derived from a cystic fibrosis patient with a ΔF508/W1282X CFTR genotype) [36] were purchased from ATCC (www.atcc.org) (this cell line is no longer provided by ATCC; it is now kept at the John Hopkins University Cell Center). Caco‐2 cells (ATCC; human colon carcinoma epithelial cells) expressing wt‐CFTR were previously selected and cloned after transfections with four short hairpin RNA interference (shRNA) directed against different regions of CFTR [9, 14, 17, 37. The plasmids were constructed by OriGene Technologies, Inc. (Rockville, USA) and the two sense sequences were pRS25: AAGAAATATGGAAAGTTGCAGATGAGGTT; pRS26: AAATATCATCTTTGGTGTT TCCTATGATG. The sequence corresponding to pRS‐shGFP was used as a control (pRSctrl), corresponding to a non‐effective shRNA plasmid against GFP, provided by OriGene. These cells were cultured adding 1 mg/ml puromycin to the culture medium for cell expansion and without puromycin during the experiments. All cells were cultured in DMEM/F12 (Life Technologies, GIBCO BRL) supplemented with 5% FBS (Internegocios S.A., Mercedes), 100 U/ml penicillin and 100 µg/ml streptomycin (Life Technologies, GIBCO BRL,). Cultures were grown at 37 ºC in a humidified air atmosphere containing 5% CO2 and plated at a density of 20,000 cells/cm2. Before treatments, cells were cultured 24 h in serum‐free medium.
shRNA preparation for stable transfections
shRNA preparation was performed as previously described [9]. Briefly, to specifically and transiently knockdown SGK1 expression, we prepared shRNAs (short hairpin RNAs) by inserting sequences complementary to the SGK1 mRNA (the antisense sequence and its complementary strand plus a small connecting loop) into a pRNATin‐H1·2/Hygro vector (GenScript). To construct the insert, two complementary oligonucleotides containing BamH1 and HindIII restriction sites and also a connecting loop of sequence TTCAAGAGA (Oligo A: 5’‐GATCCAAGGAG AACATTGAACACAACTTCAAGA GAGTTGTGTTCAATGTTCTC CTTA‐3’; Oligo B: 5’‐GATCCAAGG TCTCCTGCAG ATCTGTCTTCAAGAGAGACAGATCTG CAGGAGA CCTTA‐3’; the target sequence is underlined). These oligonucleotides were then annealed with their complementary oligonucleotide and cloned into the BamH1 and HindIII restriction sites of the pRNATin‐H1·2/Hygro vector. The empty pRNATin‐H1·2/Hygro vector was used as a control.
shRNA stable transfections
IB3‐1 cells were cultured in DMEM/F12 medium containing 5% FBS and maintained below 70% confluence. Then, cells were collected by trypsin (0·25% trypsin, 0·02% EDTA in PBS) treatment and electroporated using a BTX ECM 830 square‐wave electroporator (Genetronix Inc). The electroporation was performed by using a cuvette plus (Genetronix Inc.) for mammalian cells. Settings for electroporation were 140 volts and 1 pulse of 17 msec, using 40‐100 µg of shRNA plasmid and 4 × 106 cells, in a final volume of 400 ml. Transfected cells were selected in DMEM/F12 plus 5% FBS, containing 50 µg/ml of hygromycin, for 30 days. To perform the experiments, cells were plated in DMEM/F12 containing 5% FBS and hygromycin 50 µg/ml, at a confluence of 70–80%. Then, the cells were cultured 24 h in serum‐free medium.
Modulation of intracellular chloride concentration ([Cl–]i)
Intracellular chloride concentration ([Cl–]i) was modulated as previously reported [15, 16, 18. Briefly, IB3‐1 cells were incubated for 1 h in the presence of different Cl– concentrations (0, 5, 25, 50, 75, 100 and 125 mM). To establish a rapid equilibrium between the [Cl–]i and the extracellular chloride concentration ([Cl–]e), independently of the chloride channel activities, a double‐ionophore strategy was used [15]. IB3‐1 cells were washed with Hank´s gluconate, to remove the remaining extracellular Cl–, and then incubated for 1 h with different [Cl–]e. The different Cl– concentrations were obtained by combining two high K+ buffers (high KCl and high KNO3) [15]. Additional treatments (inhibitors, or IL‐1RN) were performed in this 1‐h period, incubating the cells in these high K+ buffers.
Reverse transcription and quantitative real‐time PCR (qRT‐PCR) for IL‐1β, CASP1, NLRP3 and ASC
The IB3‐1 cells were cultured as indicated above. After incubation, total RNA isolation, reverse transcription and qRT‐PCR were done as previously described [14, 18. Briefly, total RNA was isolated by using a guanidinium thiocyanate–phenol–chloroform extraction solution [38]. The quality of RNA was checked by electrophoresis in denaturing formaldehyde agarose gels [39] and measuring the ratios A260/A230 (greater than 2) and A260/A280 nm (over 1·7). Reverse transcription (RT) was performed by using 4 µg of total RNA, M‐MLV reverse transcriptase (100 U, Promega) and oligo‐dT12 in a 25 µl final reaction volume, according to manufacturer´s instructions. qRT‐PCRs were performed using an ABI 7500 real‐time PCR system (Applied Biosystems Inc); the ΔΔCt method was used for comparative quantification. TBP (Tata Box Binding Protein) was used as an internal control. Primer sequences for PCR were as follows: TBP, 5´‐TGCACAGGAGCCAAGAGTGAA‐3´ (forward) and 5´‐CACATCACAGCTCCCCACCA‐3´ (reverse); IL‐1β, 5´‐ACAGATGAAGTGCTCCTTCCA‐3´ (forward) and 5´‐GTCGGAGATTCGTAGCTGGAT‐3´ (reverse); CASP1, 5’‐GAATGTCAAGCTTTGCTCCCTAGA‐3’ (forward) and 5´‐AAGACGTGTGCGGCTTGACT‐3´ (reverse); NLRP3, 5´‐TCTGTGTGTGGGACTGAAGCA‐3´ (forward) and 5´‐TACTGATGCAAGATCCTGACAACA‐3´ (reverse); and ASC 5´‐ATCCAGGCCCCTCCTCAGT‐3´ (forward) and 5´‐GTTTGTGACCCTCGCGATAAG‐3´ (reverse). The cDNA samples (10 µl of a 1:10 of cDNA from reverse‐transcribed RNA) were added to 25 µl of PCR mixture containing a final concentration of 2·5 mM MgCl2, 0·4 mM deoxynucleotide triphosphates, 1 U of Go Taq DNA polymerase (Promega), 0·1 × EvaGreen (Biotium), 50 nM ROX as a reference dye and 0·2 nM of each primer. The qRT‐PCR conditions were as follows: initial denaturation at 95ºC for 10 min, followed by 40 cycles at 95ºC for 30 s, 62ºC for 30 s and 72ºC for 30 s. The fluorescence signal was acquired at the elongation step, at the end of each cycle. qRT‐PCRs were carried out in triplicates (intra‐ and inter‐assays by triplicate). The final quantification values were obtained as the mean of the relative quantification (RQ) for each biological triplicate (n = 3).
Secretion of IL‐1β
Secreted IL‐1β was measured in conditioned media from IB3‐1 cells as previously described. [18] Briefly, IB3‐1 cells were cultured 48 h, as above indicated, in p100 dishes. The last 1 h of incubation was done in 5 ml of high K+ buffer, at different Cl– concentrations. After incubation, the buffer solutions were collected and concentrated by centrifugation at 3500 x g for 30 min at 4ºC by using Amicon Ultra‐15 centrifugal filter units (10,000 kDa cut‐off, EMD Millipore). IL‐1β was measured from frozen concentrate supernatants using the Human IL‐1β ELISA set (BD OptEIA™ ‐ Human IL‐1B ELISA Set, BD Biosciences). The detection limit for this kit is 4 pg/mL; 20X concentration was performed in the supernatants by using the Amicon filters, allowing the IL‐1β measurement within the detection range (4–256 pg/ml). Measurements were performed using a microplate reader (model Benchmark, Bio‐Rad).
Protein extraction
Cells were incubated as above indicated, washed twice with cold PBS, scraped with cold extraction buffer (10 mM Tris pH 7·4, 100 mM NaCl, 0·1% SDS, 0·5% sodium deoxycholate, 1% Triton X‐100, 10% glycerol) containing the protease inhibitor cocktail (5 ml of cocktail/20 g of cell extract) plus phosphatase inhibitors (2 mM Na3VO4, 1 mM NaF and 10 mM Na2PO7), and centrifuged at 14000 × g for 20 min at 4°C. The supernatant was stored at −80°C until use. The protein concentration was measured by using the method of Lowry et al [40, 41.
Western blot analysis
Western blots were performed as previously described [18]. Briefly, total protein extracts (30‐50 µg of proteins) were separated on a denaturing SDS/PAGE (15%) and transferred to nitrocellulose membranes. Membranes were blocked with BSA 5% in TBS 1 h and then incubated with primary monoclonal antibodies against IL‐1β, CASP1, NLRP3 or ASC (dilutions 1:1000 in TBS plus Tween‐20, 0·05% v/v) for 3 h. The membranes were washed three times with TBS plus Tween‐20 (0·05% v/v) for 5 min and incubated for 1 h with goat IgG anti‐mouse or anti‐rabbit antibody coupled to horseradish peroxidase (dilution 1:2000 in TBS plus Tween‐20, 0·05% v/v), washed three times with TBS plus Tween‐20. Finally, membranes were incubated with 2·5 ml of solution A (25 µl of luminol 250 mM, 11 µl of p‐coumaric acid 90 mM, 250 µl Tris 1 M pH 8·8 and 2·22 ml H2O) and 2·5 ml of solution B (4·55 µl H2O2 30%, 250 µl Tris 1 M pH 8·8 and 2·25 ml H2O). Results were visualized by using an ImageQuant LAS 4000 system (GE Healthcare Life Sciences).
Measurement of mitochondrial and cellular ROS levels by microplate reader
Mitochondrial and cellular ROS levels were measured by using fluorescent probes in 96‐well black plates (Greiner Bio‐One, Germany; 655090) as previously described [14, 17. The cells were cultured as above indicated and treated the last 1 h with IL1RN/anakinra, or inhibitors, at different Cl– concentrations in the presence of the T‐N ionophores. To measure mitochondrial ROS levels, at the end of incubation, the DMEM/F12 medium was changed to Hank's solution (136·9 mM NaCl, 5·4 mM KCl, 1·3 mM CaCl2, 3·7 mM NaH2PO4, 0·4 mM KH2PO4, 4·2 mM NaHCO3, 0·7 mM MgSO4, 5·5 mM D‐glucose and 10 mM HEPES) containing 5 µM of MitoSOX (stock prepared as 5 mM solution in DMSO) and incubated at 37 ºC in the 5% CO2/air incubator for 10 min. Cellular ROS levels were measured by using the fluorescent probe DCFH‐DA in Hank's solution containing 10 µM of the fluorescent probe (stock prepared as 20 mM solution in DMSO) and incubated at 37 ºC in the 5% CO2/air incubator for 40 min. Then, cells were washed with 0·2 ml of Hank's solution three times and the fluorescence was measured in a fluorescence plate reader (NOVOstar, BMG LABTECH GmbH) with incubation at 37ºC. Filters were Ex = 510 ± 10 nm, Em = 580 ± 10 nm for MitoSOX and Ex = 510 ± 10 nm, Em = 540 ± 10 nm for DCFH‐DA, and readings were performed from the bottom of the plate).
Measurement of mitochondrial and cellular ROS levels by flow cytometry
Mitochondrial and cellular ROS levels were measured by flow cytometry as previously described by Duyndam et al [42] and Kauffman et al [43] with some modifications. Briefly, the cells were cultured 48 h, as above indicated, in p100 dishes. To measure mitochondrial ROS levels, the DMEM/F12 medium was changed to Hank's solution containing 5 µM of MitoSOX (stock prepared as 5 mM solution in DMSO) and incubated at 37 ºC in the 5% CO2/air incubator for 10 min. Cellular ROS levels were measured by using the fluorescent probe DCFH‐DA in Hank's solution containing 10 µM of the fluorescent probe (stock prepared as 20 mM solution in DMSO) and incubated at 37 ºC in the 5% CO2/air incubator for 40 min. Then, cells were harvested by trypsin treatment and pelleted by centrifugation at 400 x g for 5 min. Finally, the cells were resuspended in 500 μl of the corresponding treatment (different Cl– concentrations or inhibitors) and analysed on the flow cytometer (Accuri, BD Biosciences).
Measurement of cellular membrane potential
Cellular membrane potential changes were measured as previously described by using the fluorescent probe DiBAC4(3) [44, 45. Briefly, the cells were cultured in 96‐well black plates (Greiner Bio‐One, Germany; 655090) as above indicated and the last 1 h of incubation was done in high K+ buffer, at different Cl– concentrations in the presence of the T‐N ionophores with 5 µM of DiBAC4(3) in Hank's solution (stock prepared as 5 mM solution in DMSO) at 37 ºC in the 5% CO2/air incubator. Then, cells were washed with 0·2 ml of Hank's solution three times and the fluorescence was measured in a fluorescence plate reader (NOVOstar, BMG LABTECH GmbH, Ortenberg, Germany) with incubation at 37 ºC. Filters for DiBAC4(3) were Ex = 485 ± 12 nm, Em = 520 ± 30 nm. Excitations and readings were performed from the bottom of the plate.
Measurement of mitochondrial membrane potential
The mitochondrial membrane potential (Ψm) was measured by using the fluorescent probe TMRE, as previously described [7, 46, with some modifications. Briefly, IB3‐1 cells were cultured in 96‐well black plates (Greiner Bio‐One, Germany; 655090) as above indicated and incubated with 20 nM of TMRE in DMEM/F12 medium (stock 1000 × prepared as 20 µM solution in DMSO) for 30 min at 37 ºC in the 5% CO2/air incubator. Then, cells were washed with 0·2 ml of Hank's solution three times and incubated for 1 h of in high K+ buffer, at different Cl– concentrations in the presence of the T‐N ionophores After 1 h, fluorescence was measured as above indicated, and the values corresponding to FCCP 20 µM treatment at each Cl– concentration were subtracted and plotted (to discount the background fluorescence) [47]. TMRE staining is reversible and does not affect cell proliferation and viability; it also shows the absence of dead, damaged or apoptotic cells [48]; besides, the mitochondrial membrane potential, TMRE signal reflects cell viability. The filters used for TMRE were Ex = 540 ± 10 nm, Em = 590 ± 10 nm.
Measurements of intracellular pH levels
Intracellular pH levels were measured by using the fluorescent probe BCECF‐AM (2′,7′‑bis‑(2‑carboxyethyl)‑5(6)‑carboxyfluorescein acetoxymethyl ester) as reported by Rink et al. [49, 50 Briefly, the cells were cultured in 96‐well black plates (Greiner Bio‐One, Germany; 655090) as above indicated and the last 1 h of incubation was done in high K+ buffer, at different Cl– concentrations or different pH points (6·5; 7·0; 7·35; 8·0; 8·5) in the presence of the T‐N ionophores with 0·1 µM of BCECF‐AM in Hank's solution (stock prepared as 1 mM solution in DMSO) at 37 ºC in the 5% CO2/air incubator. Then, cells were washed with 0·2 ml of Hank's solution three times and the fluorescence was measured in a fluorescence plate reader (NOVOstar, BMG LABTECH GmbH, Ortenberg, Germany) with incubation at 37 ºC. Filters were Ex = 440 ± 10 and 485 ± 12 nm, Em = 540 ± 10 nm for BCECF‐AM, and readings were performed from the bottom of the plate.
Intracellular calcium levels
Relative changes in intracellular calcium levels were estimated using the fluorescent probe Fura −2‐AM in 96‐well black plates as previously reported by Hunt and Lambert [51]. Cells were cultured as above indicated and incubated with 10 µM of Fura‐2‐AM at 37 ºC in the 5% CO2/air incubator for 1 h. Then, the cells were washed with 0·2 ml of Hank's solution three times. The fluorescence was measured in a fluorescence spectrofluorometer (NOVOstar) with incubation at 37ºC. As controls, cell in Hank's solution was treated with 10 mM of EGTA (extracellular Ca2+ chelator), 10 µM of BAPTA‐AM (intracellular Ca2+ chelator) or 10 µM of A23187 (Ca2+ ionophore). Filters were Ex = 340 ± 10 nm and Ex = 380 ± 10, Em = 510 ± 10 nm, and readings were performed from the bottom of the plate. The values were plotted as the fluorescence ratios [fluorescence at 510 exited at 340/fluorescence at 510 exited at 380], normalizing the values to have control values equal to one.
Statistics
The assays were performed at least by duplicates, and the experiments were repeated at least three times (indicated in each figure). The results were expressed as means obtained from the different independent experiments (inter‐assay comparisons). One‐way ANOVA and Tukey's test were applied to calculate significant differences among samples (α = 0·05). All values are shown as mean ±SD (n). * indicates significant differences (p < 0·05). Open circles in bars show the values of the means of each independent experiment. The bars represent the average of the open circle values.
RESULTS
Changes in the [Cl–]i modulate the mRNA and protein expression of the NLRP3 inflammasome complex
We have previously demonstrated that the [Cl–]i modulates the levels of IL‐1β mRNA and protein, in a biphasic way, with a peak of expression at 75 mM Cl– [18]. Taking into account that the inflammasome complex is a key regulator of the IL‐1β production, here we studied the response to [Cl–]i changes of the NLRP3 inflammasome subunits CASP1, NLRP3 and ASC. To change the [Cl–]i, we used a double‐ionophore strategy (nigericin and tributyltin; T‐N) that allows the intracellular and extracellular chloride and H+ concentrations to equilibrate [15, 18, 52.
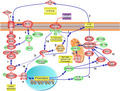
As shown in Figure 1a, the steady‐state levels of CASP1 mRNA were modulated by increasing [Cl–]i. The modulation was biphasic, with maximal mRNA levels at 75 mM Cl–, following a pattern very similar to the one obtained previously for IL‐1β [18]. Western blots (WBs) of total proteins obtained from IB3‐1 cells were then performed to determine the effects of [Cl–]i changes on the intracellular levels of CASP1. As shown in Figure 1b (WB) and 1C (quantification), both the pro‐CASP1 and the mature CASP1 levels were modulated by [Cl–]i, reaching maximal levels of pro‐ and mature CASP1 at high Cl–, in agreement with its mRNA levels (Figure 1a).
FIGURE 1.
Effects of [Cl–]i changes in the expression of inflammasome subunits. IB3‐1 cells were preincubated for 24 h in serum‐free DMEM/F12 and then incubated in the same serum‐free medium at different chloride concentrations (0, 5, 25, 50, 75, 100 and 125 mM) in the presence of the ionophores nigericin (5 μM) and tributyltin (10 μM) for 1 h. (a) Quantitative real‐time RT‐PCR of CASP1 mRNA levels in IB3‐1 cells. (b) Representative WB of CASP1, pro‐CASP1 and actin of cell lysates from IB3‐1 cells. (c) Densitometric quantification and statistical analysis of the results shown in panel B. (d) Quantitative real‐time RT‐PCR of NLRP3 mRNA levels in IB3‐1 cells. (e) Representative WB of NLRP3 and actin of cell lysates from IB3‐1 cells. (f) Densitometric quantification and statistical analysis of the results shown in panel E. (g) Quantitative real‐time RT‐PCR of ASC mRNA levels in IB3‐1 cells. (h) Representative WB of ASC and actin of cellular lysates from IB3‐1 cells. I, Densitometric quantification and statistical analysis of the results shown in panel H. The results were expressed as fold‐change over control values (0 mM chloride). Measurements were performed in triplicate (n = 3, intra‐assay), and data are expressed as mean ±SD of three independent experiments (n = 3, inter‐assay). In all figures, the open circles represent the intra‐assay mean for each independent experiment and the bars show the mean of the inter‐assay means. * indicates p < 0·05 compared with 0 mM Cl–
Equivalent results were obtained for the NLRP3 mRNA (Figure 1d,e,f). NLRP3 mRNA was modulated by [Cl–]i, again with a maximal response at 75 mM. As occurred with the CASP1 response, the NLRP3 protein expression was modulated by [Cl–]i with maximal levels at 75 mM Cl–, as shown in Figure 1e (WB) and 1e (quantification). The NLRP3 stimulation, compared to controls at 0–5 mM Cl–, was less than 1·5‐fold (nevertheless, it was significant). Noteworthy, the ASC mRNA did not show any response to [Cl–]i changes (Figure 1g). Also, in agreement with its mRNA levels, the ASC protein levels did not show changes in response to Cl– (WBs in Figure 1h; quantification in Figure 1i).
These results suggest that the [Cl–]i can regulate IL‐1β maturation, at least partially, through modulation of CASP1 expression and to a lesser extent of NLRP3 expression. More interestingly, the results show that the ASC mRNA and protein expression, contrary to CASP1 and NLRP3, do not respond to Cl–, constituting an excellent demonstration that the Cl– effects are specific for CASP1 and NLRP3.
As controls, we measured the intracellular pH (using BCECF‐AM 0·1 µM, Figure S1a and b), the membrane potential (using DiBAC4(3) 5 µM, Figure S1c) and the mitochondrial membrane potential (fluorescent probe TMRE 20 nM, Figure S1d), which were not affected from 0 to 100 mM Cl–. The viability was not compromised by the double‐ionophore strategy during 1 h incubation since the mitochondrial potential remaining unchanged from 0 to 100 mM Cl– (Figure S1d). In addition, the Ca2+ levels were measured with Fura‐2 AM and remained constant from 0 to 125 mM Cl– (Figure S2a). To confirm that Ca2+ was not involved in the IL‐1β response to Cl– in the presence of the T‐N ionophores, the cells were incubated in the presence of BAPTA‐AM and the T‐N ionophores. As shown in Figure S2a, BAPTA‐AM reduced the amount of Ca2+ but did not affect the response to Cl– of IL‐1β (Figure S2b and c protein), CASP1 (Figure S2d) or NLRP3 (Figure S2e) mRNAs. Again, the mRNA levels of ASC were not affected by BAPTA (Figure S2f).
Zhang et al. measured the intracellular Cl– concentration in cells derived from CF patients around 70 mM for the higher rank. A similar value was obtained in BEAS‐2B cells stimulated with LPS [34]. Rat RINm5F insulinoma cells, non‐CF cells, showed basal values of around 100 mM Cl– that increase up to 125 mM by using the CFTR inhibitor CFTRinh‐172·[53] Thus, concentrations between 50 and 100 mM Cl– are within the expected values under a CFTR failure.
Inhibition of the NLRP3 inflammasome blocked the IL‐1β mRNA upregulation induced by Cl–
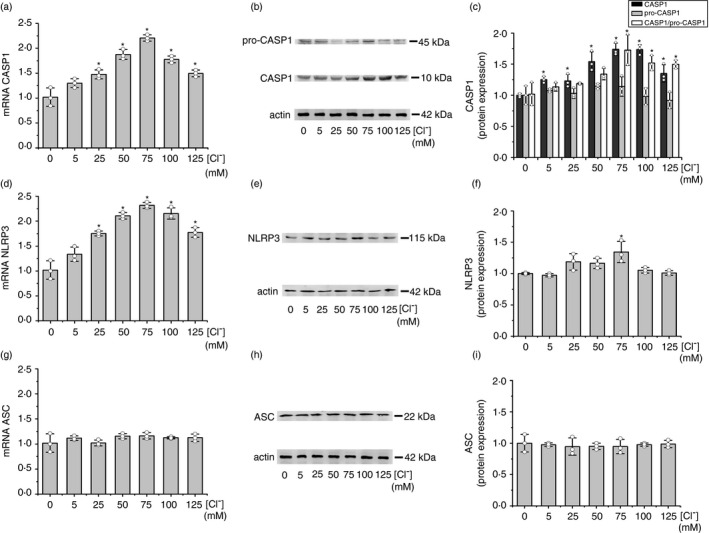
IB3‐1 cells were then incubated with the CASP1 inhibitor VX‐765 (10 μM) [54] or the NLRP3 inflammasome inhibitor MCC950 (10 μM) [55] to determine whether the NLRP3 inflammasome was involved in the IL‐1β expression induced by Cl– [18]. Noteworthy, the upregulation induced by 75 mM [Cl–]i on IL‐1β mRNA expression was completely blocked by these inhibitors (Figure 2a), reaching the basal levels of 5 mM Cl– (the basal levels were not affected). This complete inhibition of the Cl– effects with MCC950 also suggests that other inflammasomes [56] are not involved in the Cl– effects. As shown in Figure 2b, similar results were obtained on the secreted IL‐1β, except that here some response to Cl– 75 mM remains for the secreted protein (basal levels were slightly reduced without reaching significance). On the other hand, as shown in Figure 2c,d the blocking effects seen on the IL‐1β mRNA expression and the secreted IL‐1β protein were also reflected in the intracellular levels of pro‐IL‐1β and mature IL‐1β.
FIGURE 2.
Effects of CASP1 and NLRP3 inflammasome inhibitors on IL‐1β mRNA and secreted protein levels in response to [Cl–]i changes. IB3‐1 cells were incubated in serum‐free medium for 24 h and then for 1 h at 5 or 75 mM Cl– in the presence of tributyltin (10 μM) and nigericin (5 μM), treated as indicated in the presence of the CASP1 inhibitor VX‐765 (10 µM) or the NLRP3 inflammasome inhibitor: MCC950 (10 µM). (a) Quantitative real‐time RT‐PCR of IL‐1β mRNA levels in IB3‐1 cells. (b) ELISA quantification of the IL‐1β present in culture media. (c) Representative WB of pro‐IL‐1β, mature IL‐1β and actin of cell lysates from IB3‐1 cells. (d) Densitometric quantification and statistical analysis of the results shown in panel c. The results were expressed as fold‐change over control values (corresponding to 5 mM Cl–). Measurements were performed in triplicate for each experiment (n = 3, intra‐assay), and data are expressed as mean ± SD of three independent experiments (n = 3, inter‐assay). * indicates p < 0·05 compared with untreated 5 mM Cl– IB3‐1 cells
Changes in [Cl–]i modulate cellular and mitochondrial ROS Levels
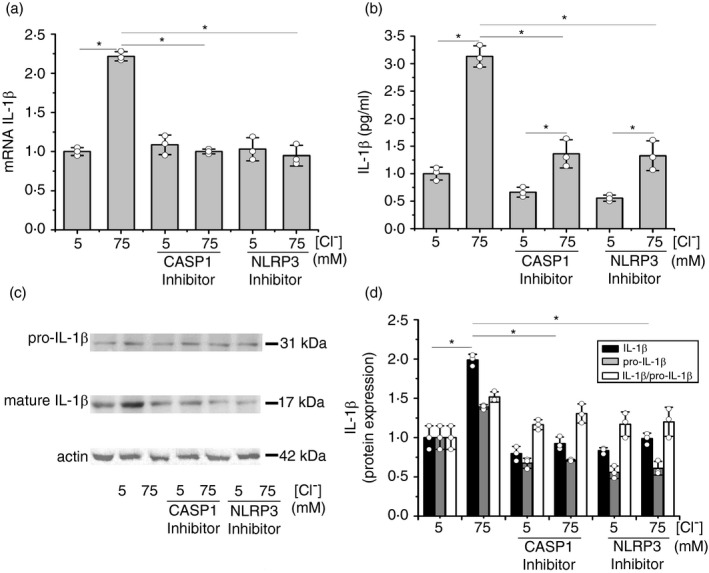
It was previously demonstrated that increased ROS levels in cells with impaired CFTR activity are mainly due to IL‐1β autocrine effects [14]. It was also demonstrated that the inhibition/mutation of the CFTR activity induces Cl– accumulation, upregulating the IL‐1β expression [18]. Thus, we hypothesized that changes in [Cl–]i might modulate ROS levels. In agreement with our hypothesis, as shown in Figure 3a, the steady‐state levels of intracellular ROS were modulated by [Cl–]i; this modulation was biphasic, with a maximal response at 75 mM of [Cl–]i, in agreement with previous results showing a biphasic expression of IL‐1β, with maximal secretion at 75 mM Cl– [18]. Also, as shown in Figure 3b, the biphasic effect can be observed in the mitochondrial ROS levels, again with a maximal response at 75 mM, which was the concentration for maximal IL‐1β, NLRP3 and CASP1 response to Cl–. The relatively small rise in cROS (~30%) and mROS (~50%) appears to be physiological significant since ROS inhibition with NAC or GKT137831, as described below, produced a significant (p < 0·05) and strong inhibition of the IL‐1β response to Cl–.
FIGURE 3.
Effects of [Cl–]i on ROS levels. IB3‐1 cells were preincubated for 24 h in serum‐free DMEM/F12. After, the cells were incubated at different chloride concentrations (0, 5, 25, 50, 75, 100 and 125 mM) in the presence of the ionophores nigericin (5 μM) and tributyltin (10 μM) for 1 h. Then, cells at 5 mM and 75 mM Cl– were treated with NAC (5 mM) or NOX Inhibitor: GKT137831 (10 µM). (a) DCF fluorescence (10 µM DCFH‐DA) of IB3‐1 cells. (b) MitoSOX fluorescence (5 µM). (c) Quantitative real‐time RT‐PCR of IL‐1β mRNA levels in IB3‐1 cells. (d) ELISA quantification of the IL‐1β present in culture media. The results were expressed in arbitrary units relative to the value corresponding to 0 mM chloride (a and b) or 5 mM chloride (c and d). Measurements were performed in triplicate for each experiment (n = 3, intra‐assay) and data are expressed as mean ±SD of three independent experiments (n = 3, inter‐assay). * indicates p < 0·05 compared to control values
Effects of NAC and the NOX inhibitor GKT137831 on IL‐1β expression
Since changes in the [Cl‐]i modulated mitochondrial and cellular ROS levels, and NOX has been detected in IB3‐1 cells [57], we then used the ROS scavenger N‐acetyl‐L‐cysteine (NAC) and the NOX inhibitor GKT137831/GKT831, to determine whether ROS were involved in the regulation of IL‐1β expression by Cl–. The GKT137831 has specificity for NOX1 (IC50 in vitro 0·14 µM), NOX4 (0·11 µM) and NOX5 (0·41 µM); NOX2 1750 µM. Data for NOX3 and DUOX1/2 are uncertain yet and it inhibits xanthine oxidase‐derived ROS formation with IC50 values over 100 µM [58].
As shown in Figure 3c, NAC (5 mM) [59] and GKT137831 (10 µM) [17] produced a significant reduction in the response of IL‐1β mRNA to [Cl–]i. These compounds also reduced basal IL‐1β mRNA levels at 5 mM of Cl–. As shown in Figure 3d, comparable effects were found on IL‐1β secretion. Thus, these results suggest that the NOX/ROS axis is involved in the IL‐1β response to Cl–, and in the maintenance of the basal IL‐1β levels, since they are not affected by IL1RN.
NAC and the NOX inhibitor GKT137831 blocked the upregulation of inflammasome complex subunits (mRNA and protein) in response to 75 mM Cl–
We then studied if cROS were able to modulate the expression of the inflammasome subunits and to affect the response to Cl–. Cells were incubated for 1 h at 5 mM or 75 mM Cl– (in the presence of tributyltin and nigericin), with NAC (5 mM) or the NOX inhibitor GKT137831 (10 µM), and mRNA and protein levels of CASP1, NLRP3 and ASC were measured. As shown in Figure 4a, NAC and GKT137831 blocked the Cl– effects on steady‐state levels of CASP1 mRNA. A similar effect was seen in CASP1 protein expression (Figure 4b,c). Equivalent results were found when the NLRP3 subunit was studied. As shown in Figure 4d,e,f both inhibitors blocked Cl– modulation of NLRP3 mRNA and protein levels. On the other hand, these inhibitors did not affect the ASC mRNA and protein expression (Figure 4g,h,i), implying that the response to Cl– is specific and not a general effect on gene/protein expression (basal levels were not affected). These results suggest that ROS induced by Cl– are involved in the response of CASP1 and NLRP3 to Cl–.
FIGURE 4.
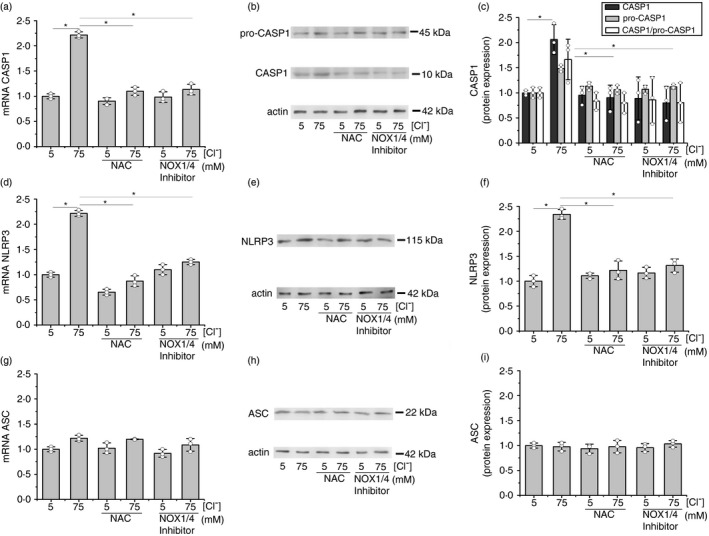
Effects of NAC and NOX inhibitors on mRNA levels of inflammasome subunits in response to different [Cl–]i. IB3‐1 cells were incubated for 1 h at 5 or 75 mM Cl–, in the presence of tributyltin (10 μM) and nigericin (5 μM) and treated with NAC (5 mM) or NOX inhibitor: GKT137831 (10 µM). (a) Quantitative real‐time RT‐PCR of CASP1 mRNA levels in IB3‐1 cells. (b) Representative WB of CASP1, pro‐CASP1 and actin of cell lysates from IB3‐1 cells. (c) Densitometric quantification and statistical analysis of the results shown in panel B. (d) Quantitative real‐time RT‐PCR of NLRP3 mRNA levels in IB3‐1 cells. (e) Representative WB of NLRP3 and actin of cell lysates from IB3‐1 cells. (f) Densitometric quantification and statistical analysis of the results shown in panel E. (g) Quantitative real‐time RT‐PCR of ASC mRNA levels in IB3‐1 cells. (h) Representative WB of ASC and actin of cell lysates from IB3‐1 cells. (i) Densitometric quantification and statistical analysis of the results shown in panel h. Results were expressed as fold‐changes compared to the values corresponding to controls (5 mM Cl–). Measurements were performed in triplicate and data are expressed as mean ±SD of three independent experiments (n = 3). * indicates p < 0·05 compared with untreated 5 mM Cl– IB3‐1 cells
IL1R1, CASP1, NLRP3, NOX and ROS inhibitors reduced the cellular and mitochondrial ROS levels that increased in response to Cl– 75 mM
Since [Cl–]i changes modulated mtROS and cROS levels (Figure 3), we then studied how ROS responded to Cl– in the presence of NLRP3 and NOX/ROS inhibitors. As shown in Figure 5a, the IL1R1 antagonist IL1RN, the CASP1 inhibitor VX‐765 and the NLRP3 inflammasome inhibitor MCC950, similarly produced a small reduction in cROS levels (DCF fluorescence) without reducing the differences between 5 and 75 mM Cl– (the reduction in cROS levels at 75 mM did not reach significance, although it was observed with IL1RN, VX‐765 and MCC950). On the other hand, NAC and the NOX inhibitor GKT137831 significantly reduced both, the basal ROS levels at 5 mM and the response to 75 mM Cl–, suggesting that both the IL‐1β loop and the ROS response to Cl– were blocked by NAC and GKT137831. As shown in Figure 5b, comparable results were found for mitochondrial ROS (mtROS) production. The inhibition in the mtROS production seen in the presence of IL1RN suggests that an active IL‐1β autocrine loop is needed for the mtROS response to Cl–.
FIGURE 5.
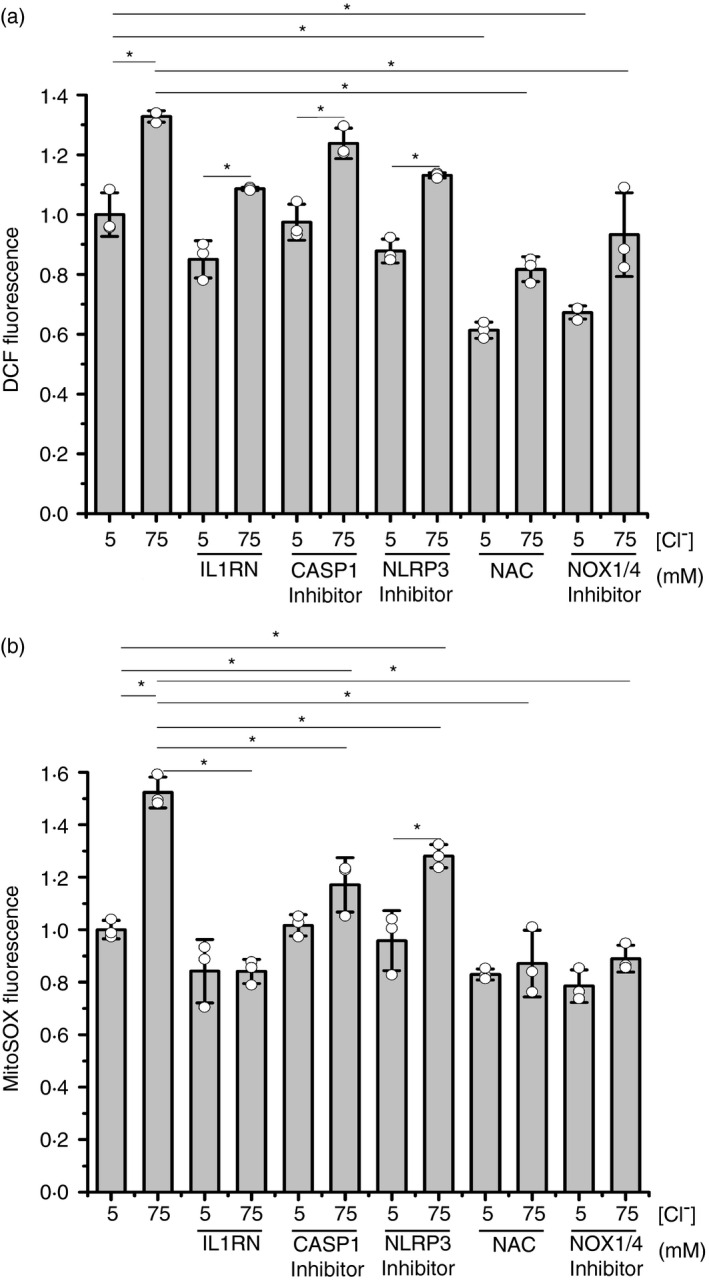
Effects of IL1RN, NAC, NOX, CASP1 and NLRP3 inflammasome inhibitors on ROS levels in response to different [Cl–]i. IB3‐1 cells were incubated for 1 h at 5 or 75 mM Cl–, in the presence of tributyltin (10 μM) and nigericin (5 μM) and treated as indicated; IL1RN (10 ng/ml), NAC (5 mM), NOX inhibitor: GKT137831 (10 µM), CASP1 inhibitor: VX‐765 (10 µM) and NLRP3 inflammasome inhibitor: MCC950 (10 µM). (a) DCF fluorescence (10 µM DCFH‐DA) in IB3‐1 cells. (b) MitoSOX fluorescence (5 µM). Results were expressed as fold‐changes relative to the values corresponding to controls (5 mM Cl–). Measurements were performed in triplicate and data are expressed as mean ±SD of three independent experiments (n = 3). * indicates p < 0·05 compared with untreated 5 mM Cl– IB3‐1 cells
SGK1 inhibition blocked the IL‐1β mRNA and secretion in response to intracellular Cl– changes
While this work was in progress, Zhang et al [34] confirmed our previous results regarding the role of Cl– in the IL‐1β secretion, producing a high [Cl–]i an sterile proinflammatory state [18]. They additionally found that recombinant SGK1 directly responds to Cl– changes in a way resembling the results seen previously [14, 18, and also here, for IL‐1β. Thus, to explore if SGK1 was also involved in the IL‐1β response to [Cl–]i in IB3‐1 cells, we used the SGK1 inhibitor GSK650394 at 0, 0·1, 1 and 10 µM in the presence of 5 or 75 mM [Cl–]i (and nigericin plus tributyltin). The results (Figure 6a) showed that increased SGK1 inhibitor concentrations progressively diminished the response of IL‐1β mRNA to changes in the [Cl–]i from 5 to 75 mM; GSK650394, at 10 µM, completely abrogated the IL‐1β mRNA response to Cl– 75 mM. As shown in Figure 6b, similar results were obtained on the secreted IL‐1β. On the other hand, as shown in Figure 6c–h, the SGK1 inhibitor significantly reduced both the cellular and mitochondrial ROS levels at 75 mM Cl–, suggesting that both, the IL‐1β loop and the ROS response to Cl–, were blocked by GSK650394. Similar results were found using a different cell line, Caco‐2 cells, stably expressing shRNA against CFTR (Figure S3). The effects of SGK1 on IL‐1β secretion were also confirmed using stable SGK1 knockdown cells (shRNAs against SGK1, Figure 7a–c). Thus, these results suggest that SGK1 has a key role in the response of IL‐1β to Cl–. Noteworthy, and resembling IL1RN, SGK1 pharmacological inhibition (or shRNA) did not affect IL‐1β basal levels. This is consistent with a location for SGK1 action over the mechanism of IL‐1β secretion rather than influencing IL‐1β expression or maturation; thus, the effects of the SGK1 inhibitors are equivalent to block the extracellular IL‐1β or the IL1R1 with IL1RN/anakinra or blocking antibodies [18].
FIGURE 6.
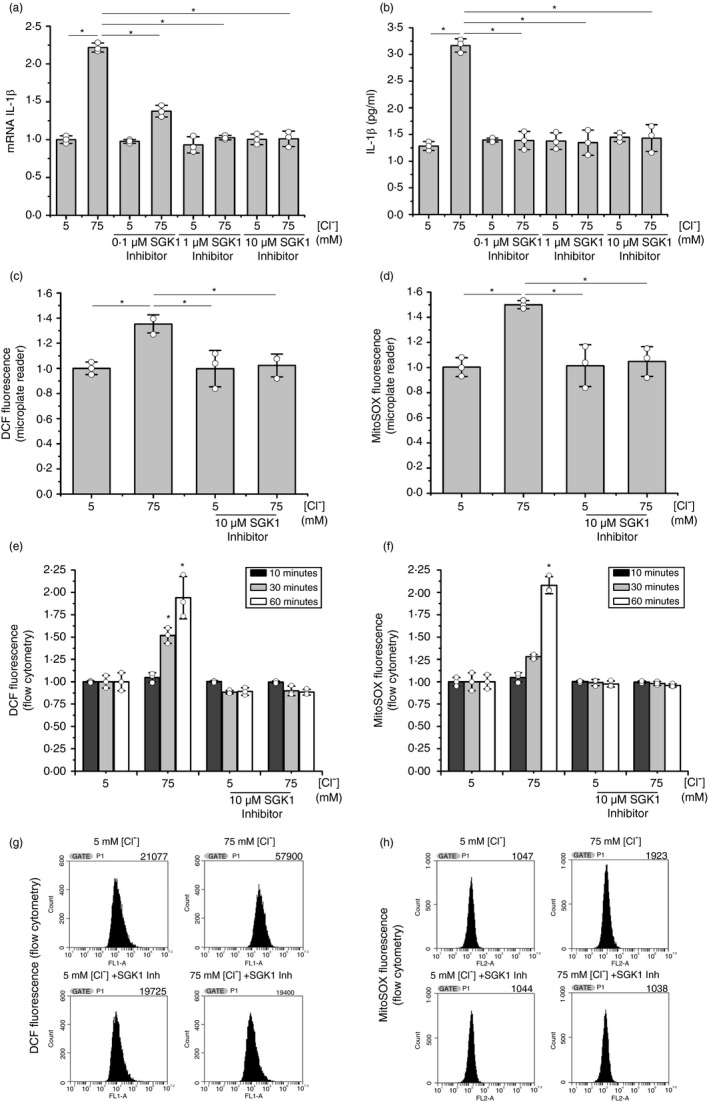
SGK1 inhibition blocked the IL‐1β mRNA response to Cl– changes. IB3‐1 cells were incubated for 1 h at 5 or 75 mM Cl–, in the presence of tributyltin (10 μM) and nigericin (5 μM), and treated with SGK1 inhibitor GSK650394 (0·1, 1 and 10 µM). (a) Quantitative real‐time RT‐PCR of IL‐1β mRNA levels in IB3‐1 cells. (b) ELISA quantification of the IL‐1β present in culture media. (c) DCF fluorescence (10 µM DCFH‐DA) by using a microplate reader. (d) MitoSOX fluorescence (5 µM) by using a microplate reader. (e) DCF fluorescence (10 µM DCFH‐DA) by flow cytometry. (f) MitoSOX fluorescence (5 µM) by flow cytometry. (g) Representative cytometry plots panel e (60 min); counts vs FL1‐A (fluorescence intensity). (h) Representative cytometry plot of panel f (60 min); counts vs FL2‐A (fluorescence intensity). Results were expressed as fold‐changes relative to the values corresponding to 5 mM Cl–. Measurements were performed in triplicate and data are expressed as mean ±SD of three independent experiments (n = 3). * indicates p < 0·05 compared with untreated 5 mM Cl– IB3‐1 cells
FIGURE 7.
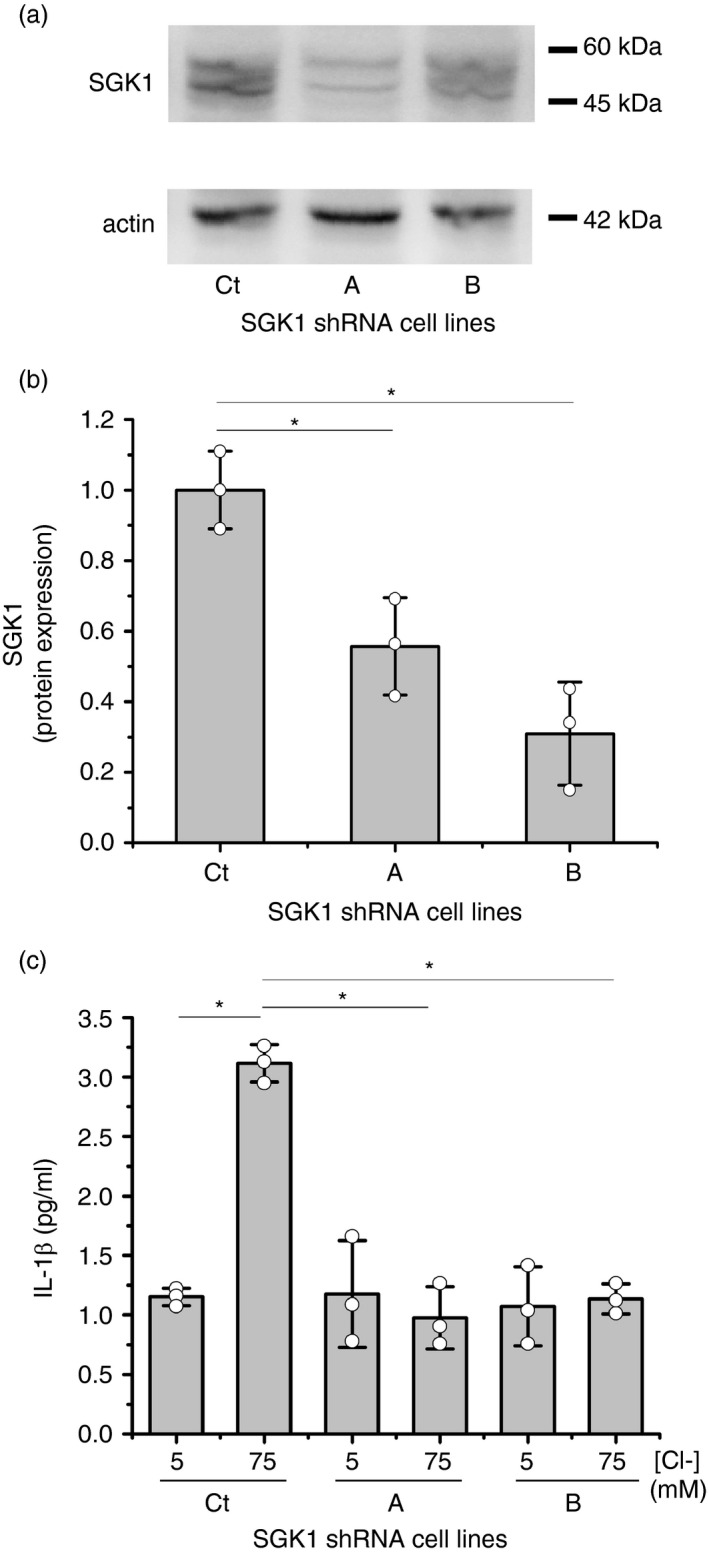
SGK1 shRNA blocked the IL‐1β response to Cl– changes. IB3‐1 control cells (Ct) (transfected with pRNATin‐H1·2 control), IB3‐1/A cells (transfected with pRNATin‐H1·2 shRNA A) and IB3‐1/B cells (transfected with pRNATin‐H1·2 shRNA B) were incubated for 1 h at 5 or 75 mM Cl– in the presence of tributyltin (10 μM) and nigericin (5 μM). (a) Representative WB of SGK1 and actin of cell lysates from the three cell lines. (b) Densitometric quantification and statistical analysis of the results shown in panel a. (c) ELISA quantification of the IL‐1β present in culture media. Results were expressed as fold‐changes relative to control values (5 mM Cl–). Measurements were performed in triplicate and data are expressed as mean ±SD of three independent experiments (n = 3). * indicates p < 0·05 compared with untreated 5 mM Cl– IB3‐1 cells
DISCUSSION
Since the NLRP3 inflammasome is involved in IL‐1β maturation, here we first studied the effects of changes in the intracellular Cl– concentration, [Cl–]i, on the expression of the NLRP3 inflammasome complex components NLRP3, CASP1 and ASC (Figure 1). Interestingly, the NLRP3 and CASP1 mRNA and protein levels were modulated by [Cl–]i changes, from 0 to 125 mM, reaching both genes a maximal and similar expression at 75 mM Cl–; this is the same concentration that we had previously reported for maximal expression of IL‐1β [18]. The response of CASP1 (protein) to Cl– was slightly higher than the response of NLRP3 (~ 1·75‐ vs 1·25‐fold increase). Noteworthy, the ASC expression remained constant in both mRNA and protein levels when [Cl–]i was changed within the same range (0–125 mM). This is an important result. ASC constitutes in this way an example of a gene product that is not under Cl– regulation. This lack of response to Cl– also indicates that the NLRP3 and CASP1 responses are Cl–‐specific and not the consequence of a general Cl– effect on gene or protein expression. We can conclude that NLRP3 and CASP1, in addition to IL‐1β [18], are Cl–‐dependent genes. This is an indirect effect, through the IL‐1β loop.
We then studied the effects of the NLRP3 inflammasome inhibitor MCC950 [55] and the CASP1 inhibitor VX‐765 [54] on IL‐1β expression at 5 mM and 75 mM [Cl–]i (Figure 2). These inhibitors completely blocked the IL‐1β mRNA response to Cl– (Figure 2a). The secreted IL‐1β was also strongly inhibited, although a residual and significant response to Cl– remained (Figure 2b); it was not enough to maintain the IL‐1β loop active (the IL‐1β mRNA levels no longer changed with Cl– changes). The results are quite similar to those obtained previously by blocking the IL‐1β autocrine/paracrine loop with the IL‐1β receptor antagonist IL1RN (anakinra) [18], without modifying the basal mRNA values. These results suggest that most of the IL‐1β upregulation by Cl– depends on the signal amplification through the IL‐1β positive feedback loop, which needs an active NLRP3 inflammasome for continuous feedback stimulation and maximal loop signalling. Noteworthy, these results also suggest that the residual IL‐1β protein response to Cl– (Figure 2b) is independent of the IL‐1β loop and, therefore, should correspond to effects of Cl– on IL‐1β secretion, as we previously suggested [18]. Most importantly, when the IL‐1β inside the cells was measured (WBs Figure 2c,d), the response to Cl– changes almost disappeared with the CASP1 inhibitor VX‐765. This effect was completely abolished with the NLRP3 inhibitor MCC950, implying that the residual response to Cl– is downstream of the NLRP3 inflammasome, within the IL‐1β secretion mechanism. This is shown in Figure 2b, where the IL‐1β secretion, in response to Cl– changes, was not completely inhibited by these two inhibitors.
We have previously seen that the increased cellular ROS (cROS) levels in IB3‐1 cells were normalized by blocking the autocrine IL‐1β signalling with either IL‐1β blocking antibodies or an NF‐kB inhibitor (using BMS‐345541, an IKK kinase inhibitor that blocks the IκB phosphorylation) [14], or by using the IL1R1 antagonist anakinra/IL1RN [17]. Thus, we then studied if [Cl–]i changes had effects on ROS levels. We observed (Figure 3a,b) that DCF fluorescence (cROS, cellular ROS) and the MitoSOX fluorescence (mtROS, mitochondrial ROS) had a biphasic response to Cl–, with a maximal response at 75 mM Cl–, as occurred with IL‐1β [18], NLRP3 and CASP1 mRNAs and proteins. Also, N‐acetyl‐L‐cysteine (NAC) and the NOX1/4 inhibitor GKT137831 had strong effects on IL‐1β expression and secretion (Figure 3c,d). Noteworthy, a significant residual response to Cl– 75 mM remains for IL‐1β expression and secretion in the presence of NAC and GKT137831, suggesting that the response to Cl– is downstream of the NOX/ROS axis.
Besides partially reducing the ROS response to Cl–, NAC and GKT137831 also reduced the basal values of IL‐1β mRNA, as it was previously reported for the IKK inhibitor III and the JNK inhibitor SP600125, but not with the IL1R1 inhibitor IL1RN/anakinra or the c‐Src inhibitor PP2·[18] Thus, it appears that the inhibition of the intracellular pathway of the IL‐β loop with NAC and GKT137831 affects the ROS levels, although the inhibition of the extracellular part of the loop by blocking of IL1R1 with IL1RN or inhibition of IL‐1β secretion with SGK1 inhibition does not affect the basal ROS levels, nor the basal IL‐1β mRNA levels.
We then studied the effects of NAC and the NOX inhibitor GKT137831 over mRNA and protein levels of the NLRP3 inflammasome subunits CASP1, ASC and NLRP3, in response to [Cl–]i changes (Figure 4a–i). Both inhibitors blocked the response to 75 mM Cl– of CASP1 and NLRP3 (mRNA and protein). ASC, again, did not show any response to these inhibitors or Cl–. These results imply that the IL‐1β loop→IL1R1→NOX/ROS axis modulates the CASP1 and NLRP3 expression (mRNA and protein) in response to Cl–. It should be taken into account that the possible influence of the Cl–→IL‐1β → IL‐1β loop axis on the activation of other inflammasomes [56] was not evaluated here. However, MCC950 completely blocked the IL‐1β mRNA upregulation induced by Cl–, implying that only the NLRP3 inflammasome is involved in the observed effects.
We then measured the effect of IL1RN, CASP1, NLRP3, NAC and NOX inhibitors over cROS and mtROS levels (Figure 5a,b, respectively). The cROS production induced by 75 mM Cl– was only partially inhibited with IL1RN (anakinra), CASP1 and NLRP3 inflammasome inhibitors; of note, some IL‐1β responses to Cl– remained in the presence of these inhibitors. A much stronger inhibition of cROS was obtained with NAC and the NOX inhibitor GKT137831, which also reduced the basal 5 mM Cl– levels. Still, some responses of cROS to Cl– remained in the presence of NAC or the NOX inhibitor. The responses to NAC or GKT137831 were strong and almost identical, suggesting that most of the cROS are formed in response to Cl– through NOX.
On the other hand, the mtROS (Figure 5b) showed an almost identical response as cROS to all the inhibitors used, excluding the response to IL1RN (anakinra), which abrogated the mtROS response to 75 mM Cl–. This implies that the increased mtROS in response to Cl– is due to the IL‐1β loop, which appears in response to the secreted IL‐1β. The mtROS levels were also strongly reduced by NAC and by the NOX inhibitor GKT137831, below basal values; the Cl– response was almost lost (but not with CASP1 and NLRP3 inhibitors). This suggests that the NOX/cROS axis is also needed to stimulate the mtROS increase with Cl– changes. In this sense, it is known that the NOX/ROS axis is needed to recruit TRAF6 and IKK, which in turn signals to activate NF‐κB through IKK [33]. However, in macrophages, TRAF6 can also migrate to mitochondria to interact with evolutionary conserved signalling intermediate in Toll pathways (ECSIT); the TRAF6‐mediated ubiquitination of ECSIT affects the mCx‐I and leads to increased mtROS [60, 61. Also, NOX4 (which is inhibited by GKT137831) localizes to mitochondria in cultured mesangial cells and kidney cortex, and siRNA‐mediated KD of NOX4 reduced mtCx‐I activity and blocks glucose‐induced mitochondrial superoxide generation [62]. Similar results were found in endothelial cells, where NOX4 reduced mCx‐I activity and induce mtROS production [63]. This might explain why GKT137831 inhibited the mtROS production in IB3‐1 cells (Figure 5b) and it might explain our earlier findings that mCx‐I is reduced in CF cells through IL‐1β signalling [9, 14. Further studies are needed to determine whether this is also the case in IB3‐1 cells.
Of note, while this work was in progress, Zhang et al [34] confirmed our earlier observations regarding the role of intracellular Cl– as a second messenger and as a proinflammatory signal through IL‐1β. [14, 15, 18 Also, they found that clamping [Cl–]i at high levels by using the same strategy (double‐ionophore tributyltin and nigericin) [15, 18 increased SGK1 phosphorylation and subsequently triggered NF‐κB activation in airway epithelial cells, whereas inhibition of SGK1 abrogated airway inflammation. Noteworthy, they also found that the activity of the recombinant SGK1 was modulated by Cl– in vitro, reaching the sigmoidal response a plateau at concentrations over 70 mM, in agreement with the concentration of 75 mM that we had found for maximal Cl– effects on IL‐1β expression and secretion [18]. Therefore, we decided to study the effects of the SGK1 inhibitor GSK650394 on the IL‐1β mRNA expression. IB3‐1 cells were incubated at 5 mM or 75 mM [Cl–] (plus tributyltin and nigericin) in the presence of GSK650394, and the IL‐1β mRNA response was measured (Figure 6). Noteworthy, the IL‐1β mRNA response to 75 mM Cl– was strongly inhibited by the SGK1 inhibitor at 0·1 and 1 µM and completely abrogated at 10 µM. However, basal IL‐1β mRNA levels were not affected by GSK650394 and, in this sense, the response to SGK1 inhibition was identical to that obtained with IL1R1, NLRP3 or CASP1 inhibitors. By contrast, NAC and the NOX1/4 inhibitor GKT137831 reduced the IL‐1β mRNA basal values, suggesting that IL1RN, MCC950, VX‐765 and GSK650394 act downstream of the NLRP3 priming step. However, it should be noted that SGK1 inhibition completely inhibited the effects of Cl– on IL‐1β secretion, without affecting IL‐1β basal levels (an effect identical to IL1RN), suggesting that SGK1 is involved in the Cl–‐stimulated IL‐1β secretion mechanism, downstream of NLRP3 inflammasome. We then transfected cells with shRNA to knockdown SGK1 expression and reinforce the results obtained with its pharmacological inhibitor GSK650394. As it is shown in Figure 7, the shRNA reduced SGK1 expression and inhibited the response to Cl– in terms of mRNA expression and IL‐1β protein secretion, confirming the results obtained with GSK650394.
Taken together, these results are consistent with the working model illustrated in Figure 8, which is essentially the canonical IL‐1β signalling pathway plus its autocrine positive feedback loop, as previously reported [18], now with the addition of the present results. We postulate that an increased intracellular Cl– concentration stimulates IL‐1β secretion through SGK1 activation, starting the autocrine positive feedback IL‐1β loop that further amplifies the signal increasing the expression of more IL‐1β (through IKK/NF‐k [14], NLRP3 and CASP1). Since SGK1 inhibition does not reduce IL‐1β basal values (mRNA or protein), it does not behave as the ROS inhibitors (NAC, NOX inhibitor), the IKK inhibitor [18], the CASP1 inhibitor or the NLRP3 inhibitor. Thus, SGK1 inhibition resembles IL1RN inhibition (Figure 3a in [18]), and for this reason, we postulate that SGK1 is affecting the IL‐1β secretion mechanism (yet unknown), downstream of the NLRP3 inflammasome.
FIGURE 8.
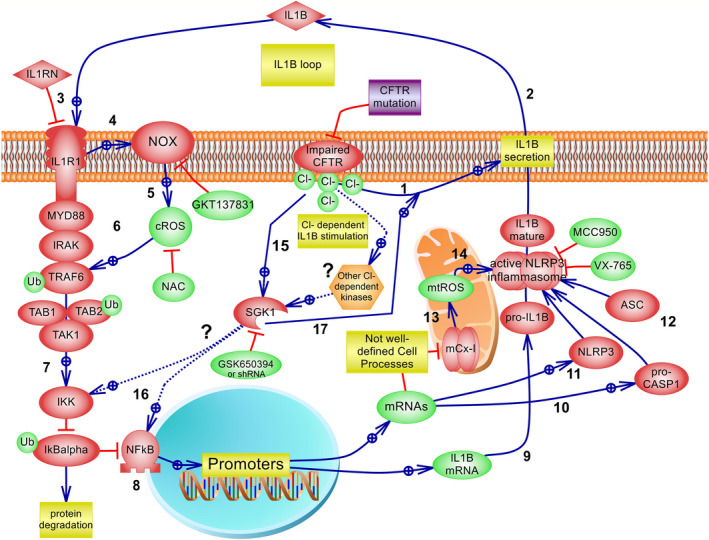
The IL‐1β autocrine/paracrine positive feedback loop initiated under Cl– signalling. Working model compatible with the results obtained here. Most interactions correspond to previous results that constitute the IL‐1β canonical pathway and the IL‐1β autocrine positive feedback loop (adapted from [18] and the references indicated below). 1: The CFTR failure in IB3‐1 CF cells induces Cl– accumulation [16]; Cl–, acting as a second messenger [15], in turn, upregulates IL‐1β secretion (Figures 2, 3, 6 and [18]). 2: IL‐1β is then secreted by a yet not well‐defined mechanism, reinforcing the autocrine positive feedback loop previously described [14, 18. 3: The IL1R1 antagonist IL1RN/anakinra inhibits the IL‐1β loop [14]. 4: Cl–→IL‐1β→IL1R1 activates the NOX/cROS axis [33]. 5: NOX produces cROS at the cell membrane or in redoxosomes [33]; NAC and the NOX1/4 inhibitor GKT137831 reduced cROS levels. 6: cROS are involved in MYD88, IRAK and TRAF6 recruitment and activation of its E3 ubiquitin ligase, self‐ubiquitination and other effectors involved in the activation of the TAK1→IKK→IkBα→NF‐κB axis [14, 70, 71 (the IL1R1→NF‐κB pathway has been simplified in the figure). 7: IKK phosphorylates the NF‐κB inhibitor IκBα leading to ubiquitin‐dependent IκBα degradation and NF‐κB translocation to the nucleus. [71] 8: Cl–→IL‐1β→IL1R1→→NF‐κB upregulates IL‐1β gene and protein expression [14, 18; this was inhibited here by NAC, the ROS inhibitor GKT137831 (Figure 4), which also reduced IL‐1β basal levels, similarly to IKK inhibition [18]. 9: Cl–→IL‐1β→IL1R1→→NF‐κB upregulates IL‐1β mRNA. 10: Cl–→IL‐1β→IL1R1→→NF‐κB upregulates pro‐CASP1 mRNA and protein levels (Figure 1). 11: Cl–→IL‐1β→IL1R1→→NF‐κB upregulates NLRP3 (Figures 1 and 2); together with IL‐1β and CASP1 upregulation constitute the priming step (signal 1) [23, 72. 12: ASC is not modulated by [Cl–]i. 13: IL‐1β signalling inhibits mCx‐I and increases mtROS by mechanisms not fully understood, probably involving TRAF6/ECSIT [73], c‐Src, NF‐κB and p38 activities [9, 14, 17. 13: IL1RN can block the production of mtROS (Figure 5B). 14: Increased mtROS levels induce NLRP3 activation to form, together with pro‐CASP1 and ASC, the NLRP3 inflammasome complex [74]; however, this was not tested yet in our model system; pro‐CASP1 is cleaved and CASP1, in turn, cleaves pro‐IL‐1β, which is then secreted by unclear mechanism(s) (Figures 3, 4, 5). 15: Intracellular Cl–, acting as a second messenger [15], stimulates SGK1 kinase activity [34]; other indirect actions of Cl– over SGK1 are still possible, such effects on WNK1 [75] or additional kinases; this needs further studies. 16: It was suggested that SGK1 mediates the Cl– ‐induced upregulation of IL‐1β mRNA (Figure 6) through IKK or NF‐κB activation [34, 64; an indirect SGK1 effect through the IL‐1β secretion and loop might also explain the effects of NF‐κB and NLRP3 activation [35]. 17: A direct effect of SGK1 over the mechanism of IL‐1β secretion seems to better fit the results. Dotted lines (…) indicate that further evidence is needed. In summary, we postulate that Cl– indirectly or directly activates SGK1, which in turn induces IL‐1β secretion, starting the IL‐1β loop. This positive feedback loop is then responsible for an amplified ROS production, NF‐kB activation, IL‐1β expression and inflammasome activation with more IL‐1β production and secretion
In parallel, the IL‐1β receptor complex induces further ROS production through NOX, as suggested by the effects of the NAC and the NOX inhibitor. It has been postulated that Cl– directly binds and activates SGK1 [34] and that SGK1 phosphorylates IKK [64] and activates NF‐κB [34]. However, the site of SGK1 action is controversial. Some authors reported that SGK1 stimulates NF‐κB activation, [34, 65 while others found an inhibitory role for SGK1·[66, 67 Also, the direct action of Cl– over SGK1 needs further confirmation. On the other hand, our results are consistent with the idea that SGK1 is acting on the secretion mechanism of IL‐1β since SGK1 inhibition completely abrogated the IL‐1β loop without affecting IL‐1β mRNA basal values. It also completely abrogated the IL‐1β secretion in response to Cl– changes, an effect that was not observed with the NLRP3 inhibitor MCC950 which blocked the Cl– induced intracellular mature IL‐1β, but was not able to completely block the secreted IL‐1β, as the SGK1 inhibitor did. The other IL‐1β loop inhibitors were not able to fully block the IL‐1β secretion mediated by Cl–. The alternative action of SGK1 over NF‐κB [34] or NLRP3 [35] activities suggested by other authors seem less likely since the inhibition of SGK1 did not affect basal IL‐1β mRNA levels, as the IKK inhibitor [18]or the MCC950 inhibitor showed. Rather, we postulate that the Cl–‐dependent SGK1 effects are downstream of the NLRP3 inflammasome, just in the mechanism of IL‐1β secretion, as illustrated in the working model of Figure 8, which is in agreement with present results. However, since the mechanism of IL‐1β secretion is a matter of strong debate, at this point we cannot be sure of the exact site of action for SGK1. Also, it should be pointed out that the results here were obtained in the presence of ionophores to isolate Cl– effects from other effects that can also potentially activate the inflammasome or the IL‐1β secretion, such changes in the intracellular Ca2+, pH, membrane potential, etc., which acting together might produce very different results.
The notion that SGK1 acts on the IL‐1β loop, independently of the exact site of action, agrees with Gan et al. which reported that the SGK1 inhibitor EMD638683 prevents angiotensin II‐induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation [68]. The model proposed in Figure 8, in which Cl– stimulates IL‐1β secretion, in turn activating the IL1R1/NOX/ROS axis, is also in agreement with the work of Wan et al [69] showing that primary bronchial epithelial cells express NOX4 and DUOX1/2; NOX4 overexpression is associated with ciliary dysfunction in these primary cells derived from patients with neutrophilic asthma [69].
In conclusion, Cl– and SGK1 are new players in the NLRP3 inflammasome activation, which connect the CFTR failure in cystic fibrosis and related diseases, in which the Cl– homeostasis is altered, with a basal proinflammatory state that occurs through modulation of the IL‐1β secretion and IL1R1 signalling.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We thank Professor Diego Battiato and Romina D’Agostino for administrative help. We also acknowledge the help of Dr. Lutz Birnbaumer and Dr. Adriana Mantegazza for the interesting suggestions and support for our work.
Funding information
This work was financed by the National Agency for the Promotion of Science and Technology (ANPCYT) [grant numbers PICT 2012‐1278 and PICT 2018‐04429 to TASC], the National Scientific and Technical Research Council of Argentina (CONICET) [grants numbers: PIP 2015‐2017 11220150100227CO and PUE 2016 22920160100129CO to TASC], the Pontifical Catholic University of Argentina (UCA) to TASC and Pablo Cassará Foundation to JMF and TASC; also by postdoctoral research fellowships from CONICET to MMMC and MC and a doctoral research fellowship to CM.
REFERENCES
- 1. Cafferata EG, Gonzalez‐Guerrico AM, Pivetta OH, Santa‐Coloma TA. Identification by differential display of a mRNA specifically induced by 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) in T84 human colon carcinoma cells. Cell Mol Biol (Noisy‐le‐grand). 1996;42:797–804. [PubMed] [Google Scholar]
- 2. Gonzalez‐Guerrico AM, Cafferata EG, Radrizzani M, Marcucci F, Gruenert D, Pivetta OH, et al. Tyrosine kinase c‐Src constitutes a bridge between cystic fibrosis transmembrane regulator channel failure and MUC1 overexpression in cystic fibrosis. J Biol Chem. 2002;277:17239–47. [DOI] [PubMed] [Google Scholar]
- 3. Cafferata EG, Gonzalez‐Guerrico AM, Giordano L, Pivetta OH, Santa‐Coloma TA. Interleukin‐1beta regulates CFTR expression in human intestinal T84 cells. Biochim Biophys Acta. 2000;1500:241–8. [DOI] [PubMed] [Google Scholar]
- 4. Cafferata EG, Guerrico AM, Pivetta OH, Santa‐Coloma TA. NF‐kappaB activation is involved in regulation of cystic fibrosis transmembrane conductance regulator (CFTR) by interleukin‐1beta. J Biol Chem. 2001;276:15441–4. [DOI] [PubMed] [Google Scholar]
- 5. Srivastava M, Eidelman O, Pollard HB. Pharmacogenomics of the cystic fibrosis transmembrane conductance regulator (CFTR) and the cystic fibrosis drug CPX using genome microarray analysis. Mol Med. 1999;5:753–67. [PMC free article] [PubMed] [Google Scholar]
- 6. Xu Y, Clark JC, Aronow BJ, Dey CR, Liu C, Wooldridge JL, et al. Transcriptional adaptation to cystic fibrosis transmembrane conductance regulator deficiency. J Biol Chem. 2003;278:7674–82. [DOI] [PubMed] [Google Scholar]
- 7. Taminelli GL, Sotomayor V, Valdivieso AG, Teiber ML, Marin MC, Santa‐Coloma TA. CISD1 codifies a mitochondrial protein upregulated by the CFTR channel. Biochem Biophys Res Commun. 2008;365:856–62. [DOI] [PubMed] [Google Scholar]
- 8. Valdivieso AG, Marcucci F, Taminelli G, Guerrico AG, Alvarez S, Teiber ML, et al. The expression of the mitochondrial gene MT‐ND4 is downregulated in cystic fibrosis. Biochem Biophys Res Commun. 2007;356:805–9. [DOI] [PubMed] [Google Scholar]
- 9. Valdivieso AG, Clauzure M, Marin MC, Taminelli GL, Massip Copiz MM, Sanchez F, et al. The mitochondrial complex I activity is reduced in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. PLoS One. 2012;7:e48059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bear CE, Duguay F, Naismith AL, Kartner N, Hanrahan JW, Riordan JR. Cl‐ channel activity in Xenopus oocytes expressing the cystic fibrosis gene. J Biol Chem. 1991;266:19142–5. [PubMed] [Google Scholar]
- 11. Collins FS, Riordan JR, Tsui LC. The cystic fibrosis gene: isolation and significance. Hosp Pract (Off Ed). 1990;25:47–57. [DOI] [PubMed] [Google Scholar]
- 12. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–73. [DOI] [PubMed] [Google Scholar]
- 13. Valdivieso AG, Santa‐Coloma TA. CFTR activity and mitochondrial function. Redox Biol. 2013;1:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clauzure M, Valdivieso AG, Massip Copiz MM, Schulman G, Teiber ML, Santa‐Coloma TA. Disruption of interleukin‐1beta autocrine signaling rescues complex I activity and improves ROS levels in immortalized epithelial cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. PLoS One. 2014;9:e99257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Valdivieso AG, Clauzure M, Massip‐Copiz M, Santa‐Coloma TA. The chloride anion acts as a second messenger in mammalian cells ‐ modifying the expression of specific genes. Cell Physiol Biochem. 2016;38:49–64. [DOI] [PubMed] [Google Scholar]
- 16. Valdivieso AG, Mori C, Clauzure M, Massip‐Copiz M, Santa‐Coloma TA. CFTR modulates RPS27 gene expression using chloride anion as signaling effector. Arch Biochem Biophys. 2017;633:103–9. [DOI] [PubMed] [Google Scholar]
- 17. Massip‐Copiz MM, Clauzure M, Valdivieso AG, Santa‐Coloma TA. CFTR impairment upregulates c‐Src activity through IL‐1beta autocrine signaling. Arch Biochem Biophys. 2017;616:1–12. [DOI] [PubMed] [Google Scholar]
- 18. Clauzure M, Valdivieso AG, Massip‐Copiz MM, Mori C, Dugour AV, Figueroa JM, et al. Intracellular chloride concentration changes modulate IL‐1beta expression and secretion in human bronchial epithelial cultured cells. J Cell Biochem. 2017;118:2131–40. [DOI] [PubMed] [Google Scholar]
- 19. Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–51. [DOI] [PubMed] [Google Scholar]
- 20. Scambler T, Jarosz‐Griffiths HH, Lara‐Reyna S, Pathak S, Wong C, Holbrook J, et al. ENaC‐mediated sodium influx exacerbates NLRP3‐dependent inflammation in cystic fibrosis. Elife. 2019;8:e49248. 10.7554/eLife.49248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McElvaney OJ, Zaslona Z, Becker‐Flegler K, Palsson‐McDermott EM, Boland F, Gunaratnam C, et al. Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am J Respir Crit Care Med. 2019;200:1381–91. [DOI] [PubMed] [Google Scholar]
- 22. Cantin AM. Cystic fibrosis lung disease and immunometabolism. Targeting the NLRP3 inflammasome. Am J Respir Crit Care Med. 2019;200:1335–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hennig P, Garstkiewicz M, Grossi S, Di Filippo M, French LE, Beer HD. The Crosstalk between Nrf2 and Inflammasomes. Int J Mol Sci. 2018;19:E562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dela Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. 2018;41:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature. 2015;526:666–71. [DOI] [PubMed] [Google Scholar]
- 26. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin‐1beta secretion. Cell Res. 2015;25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ding L, Zhang L, Kim M, Byzova T, Podrez E. Akt3 kinase suppresses pinocytosis of low‐density lipoprotein by macrophages via a novel WNK/SGK1/Cdc42 protein pathway. J Biol Chem. 2017;292:9283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sitia R, Rubartelli A. The unconventional secretion of IL‐1beta: Handling a dangerous weapon to optimize inflammatory responses. Semin Cell Dev Biol. 2018;83:12–21. [DOI] [PubMed] [Google Scholar]
- 29. Sporn MB, Roberts AB. Autocrine, paracrine and endocrine mechanisms of growth control. Cancer Surv. 1985;4:627–32. [PubMed] [Google Scholar]
- 30. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. [DOI] [PubMed] [Google Scholar]
- 31. Sorce S, Stocker R, Seredenina T, Holmdahl R, Aguzzi A, Chio A, et al. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Radic Biol Med. 2017;112:387–96. [DOI] [PubMed] [Google Scholar]
- 32. Ovrevik J, Refsnes M, Lag M, Holme JA, Schwarze PE. Activation of Proinflammatory Responses in Cells of the Airway Mucosa by Particulate Matter: Oxidant‐ and Non‐Oxidant‐Mediated Triggering Mechanisms. Biomolecules. 2015;5:1399–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spencer NY, Engelhardt JF. The basic biology of redoxosomes in cytokine‐mediated signal transduction and implications for disease‐specific therapies. Biochemistry. 2014;53:1551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang YL, Chen PX, Guan WJ, Guo HM, Qiu ZE, Xu JW, et al. Increased intracellular Cl(‐) concentration promotes ongoing inflammation in airway epithelium. Mucosal Immunol. 2018;11:1149–57. [DOI] [PubMed] [Google Scholar]
- 35. Green JP, Yu S, Martín‐Sánchez F, Pelegrin P, Lopez‐Castejon G, Lawrence CB, et al. Chloride regulates dynamic NLRP3‐dependent ASC oligomerization and inflammasome priming. Proc Nat Acad Sci USA. 2018;115:E9371–E9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zeitlin PL, Lu L, Rhim J, Cutting G, Stetten G, Kieffer KA, et al. A cystic fibrosis bronchial epithelial cell line: immortalization by adeno‐12‐SV40 infection. Am J Respir Cell Mol Biol. 1991;4:313–9. [DOI] [PubMed] [Google Scholar]
- 37. Massip‐Copiz M, Clauzure M, Valdivieso AG, Santa‐Coloma TA. Epiregulin (EREG) is upregulated through an IL‐1beta autocrine loop in Caco‐2 epithelial cells with reduced CFTR function. J Cell Biochem. 2018;119:2911–22. [DOI] [PubMed] [Google Scholar]
- 38. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Anal Biochem. 1987;162:156–9. [DOI] [PubMed] [Google Scholar]
- 39. Sambrook J, Fritsch E, Maniatis T. Molecular cloning: a laboratory manual. 2ndº: edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 40. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 41. Fryer HJ, Davis GE, Manthorpe M, Varon S. Lowry protein assay using an automatic microtiter plate spectrophotometer. Anal Biochem. 1986;153:262–6. [DOI] [PubMed] [Google Scholar]
- 42. Duyndam MC, Hulscher TM, Fontijn D, Pinedo HM, Boven E. Induction of vascular endothelial growth factor expression and hypoxia‐inducible factor 1alpha protein by the oxidative stressor arsenite. J Biol Chem. 2001;276:48066–76. [DOI] [PubMed] [Google Scholar]
- 43. Kauffman ME, Kauffman MK, Traore K, Zhu H, Trush MA, Jia Z, et al. MitoSOX‐based flow cytometry for detecting mitochondrial ROS. React Oxygen Species (Apex. N.C.). 2016;2:361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Magalhaes D, Soares‐da‐Silva P, Magro F. The effect of PRR ligands on the membrane potential of intestinal epithelial cells. Pharmacol Rep. 2017;69:978–84. [DOI] [PubMed] [Google Scholar]
- 45. Epps DE, Wolfe ML, Groppi V. Characterization of the steady‐state and dynamic fluorescence properties of the potential‐sensitive dye bis‐(1,3‐dibutylbarbituric acid)trimethine oxonol (Dibac4(3)) in model systems and cells. Chem Phys Lipids. 1994;69:137–50. [DOI] [PubMed] [Google Scholar]
- 46. Scaduto RC Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76:469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Upadhyay D, Panduri V, Ghio A, Kamp DW. Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria. Am J Respir Cell Mol Biol. 2003;29:180–7. [DOI] [PubMed] [Google Scholar]
- 48. Barteneva NS, Ponomarev ED, Tsytsykova A, Armant M, Vorobjev IA. Mitochondrial staining allows robust elimination of apoptotic and damaged cells during cell sorting. J Histochem Cytochem. 2014;62:265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rink TJ, Tsien RY, Pozzan T. Cytoplasmic pH and free Mg2+ in lymphocytes. J Cell Biol. 1982;95:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mardones P, Medina JF, Elferink RP. Activation of cyclic AMP Signaling in Ae2‐deficient mouse fibroblasts. J Biol Chem. 2008;283:12146–53. [DOI] [PubMed] [Google Scholar]
- 51. Hunt BD, Lambert DG. Ratiometric [Ca(2)(+)]i measurements in adherent cell‐lines using the NOVOstar microplate reader. Methods Mol Biol. 2013;937:111–20. [DOI] [PubMed] [Google Scholar]
- 52. Valdivieso AG, Marin MC, Clauzure M, Santa‐Coloma TA. Measurement of cystic fibrosis transmembrane conductance regulator activity using fluorescence spectrophotometry. Anal Biochem. 2011;418:231–7. [DOI] [PubMed] [Google Scholar]
- 53. Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose‐induced electrical activities and insulin secretion in pancreatic islet β‐cells are modulated by CFTR. Nat Commun. 2014;5:4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JC, et al. IL‐converting enzyme/caspase‐1 inhibitor VX‐765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol. 2005;175:2630–4. [DOI] [PubMed] [Google Scholar]
- 55. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz‐Planillo R, Inserra MC, et al. A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lopez‐Castejon G, Brough D. Understanding the mechanism of IL‐1β secretion. Cytokine Growth Factor Rev. 2011;22:189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pongnimitprasert N, Hurtado M, Lamari F, El Benna J, Dupuy C, Fay M, et al. Implication of NADPH oxidases in the early inflammation process generated by cystic fibrosis cells. ISRN Inflamm. 2012;2012:481432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal. 2015;23:406–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Valdivieso AG, Dugour AV, Sotomayor V, Clauzure M, Figueroa JM, Santa‐Coloma TA. N‐acetyl cysteine reverts the proinflammatory state induced by cigarette smoke extract in lung Calu‐3 cells. Redox Biol. 2018;16:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015;6:472–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koziel R, Pircher H, Kratochwil M, Lener B, Hermann M, Dencher NA, et al. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem J. 2013;452:231–9. [DOI] [PubMed] [Google Scholar]
- 64. Tai DJ, Su CC, Ma YL, Lee EH. SGK1 phosphorylation of IkappaB Kinase alpha and p300 Up‐regulates NF‐kappaB activity and increases N‐Methyl‐D‐aspartate receptor NR2A and NR2B expression. J Biol Chem. 2009;284:4073–89. [DOI] [PubMed] [Google Scholar]
- 65. Borst O, Schaub M, Walker B, Schmid E, Munzer P, Voelkl J, et al. Pivotal role of serum‐ and glucocorticoid‐inducible kinase 1 in vascular inflammation and atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35:547–57. [DOI] [PubMed] [Google Scholar]
- 66. Zhou H, Gao S, Duan X, Liang S, Scott DA, Lamont RJ, et al. Inhibition of serum‐ and glucocorticoid‐inducible kinase 1 enhances TLR‐mediated inflammation and promotes endotoxin‐driven organ failure. FASEB J. 2015;29:3737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lou Y, Hu M, Wang Q, Yuan M, Wang N, Le F, et al. Estradiol suppresses TLR4‐triggered apoptosis of decidual stromal cells and drives an anti‐inflammatory TH2 shift by activating SGK1. Int J Biol Sci. 2017;13:434–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gan W, Ren J, Li T, Lv S, Li C, Liu Z, et al. The SGK1 inhibitor EMD638683, prevents Angiotensin II‐induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta. 2018;1864:1–10. [DOI] [PubMed] [Google Scholar]
- 69. Wan WY, Hollins F, Haste L, Woodman L, Hirst RA, Bolton S, et al. NADPH oxidase‐4 overexpression is associated with epithelial ciliary dysfunction in neutrophilic asthma. Chest. 2016;149:1445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cheng CJ, Huang CL. Activation of PI3‐kinase stimulates endocytosis of ROMK via Akt1/SGK1‐dependent phosphorylation of WNK1. J Am Soc Nephrol. 2011;22:460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu T, Zhang L, Joo D, Sun S‐C. NF‐κB signaling in inflammation. Sig Transduction Targeted Therapy. 2017;2:e17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol. 2018;50:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument‐Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, et al. Cutting edge: Mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J Immunol. 2018;200:3047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu BE, Stippec S, Lazrak A, Huang CL, Cobb MH. WNK1 activates SGK1 by a phosphatidylinositol 3‐kinase‐dependent and non‐catalytic mechanism. J Biol Chem. 2005;280:34218–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material