

Abstract
Nucleotide‐binding domain and leucine‐rich repeat receptor (NLR)‐mediated inflammasome activation is important in host response to microbes, danger‐associated molecular patterns (DAMPs) and metabolic disease. Some NLRs have been shown to interact with distinct cell metabolic pathways and cause negative regulation, tumorigenesis and autoimmune disorders, interacting with multiple innate immune receptors to modulate disease. NLR activation is therefore crucial in host response and in the regulation of metabolic pathways that can trigger a wide range of immunometabolic diseases or syndromes. However, the exact mode by which some of the less well‐studied NLR inflammasomes are activated, interact with other metabolites and immune receptors, and the role they play in the progression of metabolic diseases is still not fully elucidated. In this study, we review up‐to‐date evidence regarding NLR function in metabolic pathways and the interplay with other immune receptors involved in GPCR signalling, gut microbiota and the complement system, in order to gain a better understanding of its link to disease processes.
Keywords: complement system, GPCR signalling, gut microbiota, immunometabolic diseases, inflammasome, metabolic reprogramming
In this study we review the current evidence with respect to inflammasome function in metabolic pathways and the interplay with other immune receptors involved in GPCR signalling, gut microbiota and the complement system, in order to gain a better understanding of its link to disease processes.
Abbreviations
- AMP
Adenosine monophosphate
- ATP
Adenosine triphosphate
- CNS
Central nervous system
- GSDMD
Gasdermin D
- GPCR
G protein‐coupled receptor
- GLUT1
Glucose transporter 1
- LAT1
L‐type amino acid transporter 1
- LPS
Lipopolysaccharide
- mTOR
Mechanistic target of rapamycin
- mtDNA
Mitochondrial DNA
- NLR
Nucleotide‐binding domain and leucine‐rich repeat receptor
- NPC
intracellular cholesterol transporter 1
- ω‐3 FA
Omega‐3 fatty acid
- OXPHOS
Oxidative phosphorylation
- PI3K
Phosphoinositide 3‐kinase
- PKA
Protein kinase A
- PRRs
Pattern recognition receptors
- ROS
Reactive oxygen species
- TCA cycle
Tricarboxylic acid cycle
- T1D
Type 1 diabetes
CROSSTALK OF CELL METABOLISM AND THE NLR INFLAMMASOME
Organisms have developed host defence mechanisms that protect from microbial pathogens. The host's immune response can also regulate metabolic responses including changes in glycolysis, oxidative phosphorylation and cholesterol metabolism [1, 2, 3]. These critical biochemical reactions play a major role in immune cell function and are vital for cell maintenance, energy, proliferation and signalling [4]. Thus, it is not surprising that these metabolic processes are regulated and controlled by immune receptors, and dysregulation at any of these pathways can have an impact on organismal health and therefore cause a number of different autoimmune diseases.
Inflammasomes are an essential part of host defence and have been also implicated in metabolic regulation [5, 6]. Inflammasomes are multiprotein complexes that become activated in response to a variety of physiological and pathogenic stimuli. Canonical inflammasome assembly through caspase 1 activation is highly regulated and two different danger signals are required for its activation, priming and activation. Priming is initiated mainly by activation of PRRs, including TLR4 and cytokine receptors, leading to nuclear translocation of nuclear factor κB (NF‐κB) and subsequent upregulation and translation of pro‐interleukin‐1β (pro‐IL‐1β) (pro‐IL‐18 is constitutively expressed). Inflammasome assembly and activation require cytosolic sensing of pathogen‐associated molecular patterns (PAMPS) or danger‐associated molecular patterns (DAMPS) by a nucleotide‐binding domain and leucine‐rich repeat receptor (NLR) or absent in melanoma 2 (AIM2)‐like receptors (ALR). NLRs and ALRs engage caspase 1, in most cases requiring the recruitment and binding of the ASC (apoptotic‐speck‐containing protein with a CARD), to catalyse proteolytic cleavage of pro‐IL‐1β and pro‐IL‐18, as well as gasdermin D (GSDMD) cleavage, forming pores on the cell membrane leading to pyroptosis (Figure 1) [7, 8]. The main NLRs that have been linked with metabolic regulation include pyrin domain‐containing protein 1 (NLRP1), NLRP3, NLRP6, NLRP12, NLR family CARD (caspase activation and recruitment) domain‐containing 4 (NLRC4), NAIP (neuronal apoptosis inhibitor protein), NLRC3 and NLRX1, and members of the PYHIN family include absence in melanoma 2 (AIM2) [6, 9].
FIGURE 1.
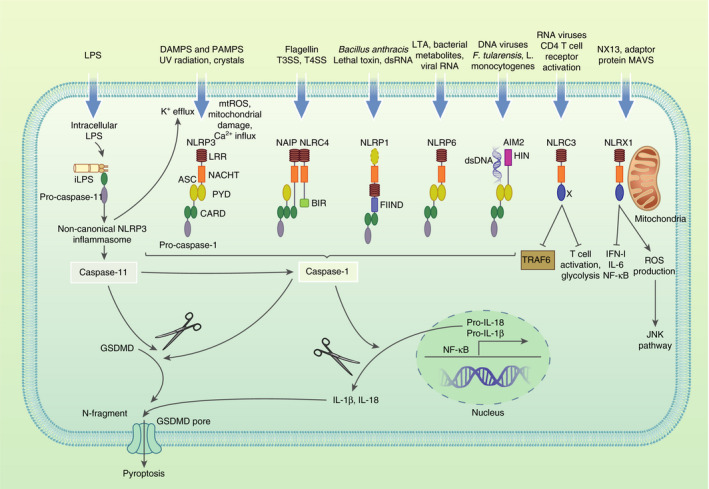
Activation of NLRs, non‐canonical and canonical inflammasomes. Structure and activating ligands of canonical NLRP3, NLRC4, NLRP1 (human NLRP1 inflammasome contains PYD, mouse NLRP1 does not), NLRP6 and AIM2 inflammasomes, as well as non‐canonical NLRP3 inflammasome, and NLRC3 and NLRX1, which are non‐inflammasome forming NLRs. Human and mouse NLRs and AIM2 get activated in response to distinct ligands. Double‐stranded RNA (dsRNA) is an activator of human NLRP1 while anthrax lethal toxin activates murine NLRP1B. NLRC4 inflammasome gets activated by T3SS/T4SS indirectly by detecting flagellin, or directly by detecting the T3SS rod protein. Mouse NAIP5 and 6 detect flagellin, mouse NAIP 1 and 2 as well as human NAIP respond to proteins of type III secretion systems (T3SS). Upon ligand binding, NAIP and NLRC4 assemble into an oligomerized inflammasome. Among several bacterial ligands of AIM2, Francisella tularensis and Listeria monocytogenes act as activators of mouse AIM2 inflammasome. DNA and RNA viruses are ligands to mostly human AIM2 inflammasome. Canonical activation of inflammasomes results in oligomerization of ASC into speck complexes, recruiting pro‐caspase 1 leading to self‐cleavage and activation of caspase 1. Caspase 1 cleaves pro‐IL‐1β (produced by NFκB‐induced upregulation) and pro‐IL‐18, as well as GSDMD cleavage. The N‐terminal fragment of GSDMD forms pores on the cell membrane and enables secretion of IL‐1β and IL‐18, with subsequent pyroptosis. Non‐canonical NLRP3 inflammasome activation occurs via human caspase 4 and 5 or murine caspase 11. Sensing of intracellular LPS by pro‐caspase 11 leads to caspase 11 activation and subsequent GSDMD cleavage, as well as K+ efflux promoting the activation of NLRP3‐ASC‐caspase 1 pathway
METABOLIC REGULATION AND INFLAMMASOME INTERACTIONS
NLRP3
Activation of the NLRP3 inflammasome can be modulated by the metabolic state of a cell and acts as a general sensor of cellular homeostasis. For instance, increased AMP leads to inhibition of inflammasome activation by activating the nutrient sensor AMP‐dependent protein kinase (AMPK), which causes a metabolic switch from glycolysis (and energy‐consuming pathways linked to high cellular activity) to OXPHOS, which is linked to anti‐inflammatory, quiescent and contracting cell responses [10]. In addition, glycolytic and Krebs cycle enzymes have been reported to induce NLRP3 inflammasome activation. The glycolytic enzyme pyruvate kinase M2 (PKM2) activates the NLRP3 inflammasome in macrophages stimulated with LPS via hypoxia‐inducible factor 1‐alpha (HIF1α) regulation, causing maintained IL‐1β production [11]. Other signals linked to cellular stress that activate NLRP3 include ATP influx, reactive oxygen species (ROS) and increased concentration of intracellular calcium ([Ca2+]i) (Figure 1) as a result of ion fluxes or changes in glucose and lipid metabolism [12]. Recent studies showing NLRP3 activation by changes in lipid metabolism, such as increased endoplasmic reticulum cholesterol levels via NPC1, or saturated fatty acids crystallization via lysosomal destabilization, are crucial to provide further key roles for inflammasomes in lipid metabolism‐related diseases [13]. Metabolic regulation of the NLRP3 inflammasome has been extensively investigated and is deeply described in the following reviews [9, 14], the focus here is on the less well‐studied inflammasomes.
NLRC4
Regulation of the NLRC4 inflammasome has been mainly linked to lipid metabolism and acts as a contributor to pathology in several immunometabolic diseases. Murine NAIP5 (NLR family, apoptosis inhibitory protein 5)/NLRC4 inflammasome activation by cytosolic delivery of bacterial flagellin has been shown to cause an eicosanoid storm, causing an uncontrolled release of signalling lipids such as prostaglandins, leading to inflammation, vascular fluid loss and death in mice. This process was dependent on the initial caspase 1 activation by NAIP5/NLRC4, triggering Ca2+ influx and cyclooxygenase 1 stimulation, a crucial enzyme in lipid metabolism responsible for prostaglandin biosynthesis, leading to the eicosanoid storm [15].
Importantly, NLRC4 inflammasome activation has been involved in several metabolic diseases such as colitis‐associated tumorigenesis [16], obesity‐associated breast cancer progression, non‐alcoholic steatohepatitis (NASH), diabetic nephropathy (DN) and type 1 diabetes (T1D). Activated NLRC4 leading to IL‐1β signalling has been reported to contribute to pathology and angiogenesis in obese mice with breast cancer via adipocyte‐mediated vascular endothelial growth factor A (VEGFA) expression [17]. In addition, the omega‐3 fatty acid (ω‐3 FA) docosahexaenoic acid (DHA), which has a protective effect against NASH, has recently been shown to alleviate the palmitate (PA)‐induced inflammatory agents and lipid accumulation via suppression of human NLRC4 expression and inhibition of caspase 1 and IL‐1β cleavage. NLRC4 knockdown inhibited PA‐induced inflammation and lipid accumulation, therefore presenting DHA as a potential antagonist for NLRC4 to provide beneficial hepatoprotective effects in NASH [18].
NLRC4‐driven IL‐1β production has also been shown to contribute to pathology in DN, where NLRC4‐deficient mice showed decreased blood glucose and albumin excretion, together with preserved renal histology and lower DN progression [19]. In addition, two recent studies identified certain human NLRC4 polymorphisms associated with markers of glucose and lipid metabolism: insulin levels and higher triglyceride levels associated with NLRC4 polymorphisms from a healthy southern Brazil population [20], and positive rate of glutamic acid decarboxylase antibody (GADA) and the onset age associated with rs385076 NLRC4 polymorphism in patients with T1D in a Chinese Han population [21], indicating a clear association between NLRC4 and clinical characteristics of T1D. Further investigation should focus in the metabolic modulators involved in NLRC4 inflammasome contribution to ND and possibly T1D. In addition, it would be interesting to determine whether similar molecular mechanisms of NLRC4 leading to eicosanoid storm and inflammation that were observed in mice, such as cyclooxygenase 1 stimulation, are also involved in NASH (Table 1).
TABLE 1.
Summary of upstream and downstream metabolic regulators interacting with NLRC4, NLRP1, AIM2, caspase 11 inflammasomes, NRLC3 and NLRX1, linked with autoimmune pathologies
| Inflammasome/NLR | Effector and function | Mechanism | Pathology | References |
|---|---|---|---|---|
| NLRC4 | DHA. Most present ω‐3 FA in the brain and retina, component of membrane glycerophospholipids, reduces circulating triglyceride levels. | DHA ⤍ NLRC4 suppression ⤍ decreased palmitate‐induced inflammation and lipid accumulation | Protective effect of DHA against non‐alcoholic steatohepatitis (NASH) | [18] |
| Cyclooxygenase 1. Enzyme responsible for prostaglandin biosynthesis. | NAIP5/NLRC4 activation ⤍ Ca2+ influx ⤍ cyclooxygenase 1 stimulation ⤍ leading to an eicosanoid storm | Eicosanoid storm leading to inflammation, vascular fluid loss and death in mice | [15] | |
| Obesity/VEGFA. VEGFA contributes to angiogenesis and tumour progression and induces proliferation and migration of vascular endothelial cells. | NLRC4 activation IL‐1β signalling ⤍ VEGFA expression ⤍ contributing to pathology | Obesity‐associated breast cancer progression | [17] | |
| NLRP1 | MCFA and SCFA. Substrates of energy metabolism mostly inhibit glycolysis and stimulate lipogenesis/gluconeogenesis. | Changes in MCFA and SCFA metabolism ⤍ dysbiosis in NLRP1‐deficient mice | IBD progression | [28] |
| High glucose and glycated albumin. Diabetes metabolites. | High glucose and glycated albumin ⤍ increased NLRP1 expression | Protective effect against diabetic kidney disease (DKD) in T1D patients. | [26] | |
| High‐fat diet (HFD) | HFD ⤍ active NLRP1, IL‐18 production ⤍ lipolysis (in mice with an activating mutation in NLRP1) | Prevention of obesity and metabolic syndrome | [34] | |
| ER stress. Engages the unfolded protein response (UPR). UPR signal activators include PERK/IRE1α. | ER stress ⤍ PERK/IRE1α ⤍ ATF4 ⤍ NLRP1 upregulation | Contribution to PERK/ATF4‐associated ISR and Chondrodysplasia. | [32, 113] | |
| Streptozotocin (STZ)‐induced diabetes. Toxic compound to pancreatic β‐cells, causing hyperglycaemia and hypoinsulinaemia. | Active NLRP1 ⤍ increased blood glucose levels ⤍ VEGF expression ⤍ pro‐inflammatory cytokine release | Contribution to pathology of diabetic retinopathy | [27] | |
| AIM2 | PKM2. Activated tetrameric PKM2 catalyses dephosphorylation of phosphoenolpyruvate (PEP) to pyruvate. | PKM2 ⤍ mediated glycolysis ⤍ PDK1‐EIF2AK2 ⤍ AIM2 activation, IL‐1β/IL‐18 ⤍ septic death in mice | Contribution to sepsis progression | [36] |
| Cholesterol‐25‐hydroxylase (Ch25h)‐induced 25‐HC. Ch25h produces 25‐HC from cholesterol. 25‐HC has antiviral activity. | Type I IFN ⤍ Ch25h, 25‐HC ⤍ suppression of SREBP2 and cholesterol biosynthesis ⤍ AIM2 inhibition | 25‐HC prevents mitochondrial dysfunction and AIM2 activation | [37] | |
| SIRT1. Deacetylation of proteins, NAD+‐dependent deacetylase and metabolic reprogramming. | SIRT1 ⤍ increased glycolysis ⤍ suppression of AIM2 expression | Contribution to pathology of cervical cancer | [38] | |
|
Ifi202b/p202. The IFN‐inducible gene Ifi202b encodes the protein 202 (p202), which binds dsDNA in the cytosol and inhibits caspase activation and AIM2 inflammasome. |
Active AIM2 ⤍ suppressed Ifi202b/p202‐prevention of high fasting glucose levels ⤍ adipogenesis ⤍ monocyte infiltration and inflammation | Protective effect against obesity and insulin resistance | [39] | |
| High glucose, ROS production | High glucose, ROS production ⤍ AIM2 activation, GSDMD signalling ⤍ cardiac dysfunction | Contribution to pathology of diabetic cardiomyopathy | [40] | |
| Caspase 11 inflammasome | L‐adrenaline‐induced cAMP. Produced from ATP by adenylate cyclase, key second messenger, anti‐inflammatory effect in several immune cells. | L‐adrenaline ⤍ ADCY4‐cAMP‐PKA activation ⤍ inhibition of caspase 11 inflammasome and pyroptosis | Protective effect of L‐adrenaline against sepsis | [41] |
| NLRC3 | TRAF6, NF‐κB signalling. TRAF6 is important for TNFR signalling and is a critical factor for the IL‐1R/TLR family. | NLRC3 activation⤍ ubiquitination of TRAF6 ⤍ NF‐κB inhibition ⤍ diminished glycolysis and oxidative phosphorylation of CD4+ T cells. | Protective effect against MS | [48] |
| Lactate dehydrogenase. Glycolytic enzyme that catalyses the reversible conversion of pyruvate and NADH to lactate and NAD+. | NLRX1 deletion⤍ increased lactate dehydrogenase⤍ increased proliferation and differentiation into inflammatory phenotype in T cells. | Protective effect against colitis | [57, 59] | |
| NLRX1 | NX‐13. Small molecule acting as an NLRX1 agonist, induces immunometabolic changes leading to decreased inflammation and has a protective role in IBD. | NX‐13‐NLRX1 activation ⤍ increased OXPHOS ⤍ decreased Th1 and Th17 differentiation ⤍ decreased ROS | Protective effect against IBD | [59] |
| Glutamate. In astrocytes, glutamate either is converted to glutamine or metabolized in the TCA cycle. | NLRX1 activation ⤍ increased astrocytic glutamate uptake ⤍ inhibition of Ca2+‐mediated glutamate exocytosis from astrocytes | Protective effect against CNS inflammation | [65] |
NLRP1
A wide range of studies has identified the NLRP1 inflammasome as a sensor of metabolic stress, leading to IL‐18 production. Human NLRP1 gets activated by double‐stranded RNA (dsRNA) and enteroviral 3C protease [22, 23], whereas NLRP1B, the murine paralog of NLRP1 that forms an inflammasome, is mainly activated in response to anthrax lethal toxin [24, 25]. NLRP1 has a protective or worsening effect in different immunometabolic diseases [26, 27] and can act as a sensor of cellular stress, glucose and lipid metabolism. In mice deficient in NLRP1B, changes in medium‐chain fatty acid (MCFA) and short‐chain fatty acid (SCFA) metabolism have been associated with microbiome dysbiosis and inflammatory bowel disease (IBD) [28]. In addition, the ω‐3 FA DHA has been shown to inhibit anthrax lethal toxin‐induced NLRP1B via β‐arrestin [29], clearly indicating the modulatory role of lipid metabolism in NLRP1B and its protective role in maintaining gut homeostasis.
NLRP1B can also act as a sensor of cellular energy stress, being activated by the glycolysis inhibitor 2‐deoxyglucose (2‐DG) and the electron transport chain inhibitor sodium azide via depletion of cytosolic ATP. Glucose‐free media and hypoxia, which lower ATP, both activated NLRP1B inflammasome transfected in human fibroblasts, and NLRP1B was suggested as a direct sensor of ATP levels [30, 31]. In addition, NLRP1 is upregulated during endoplasmic reticulum (ER) stress through PERK and IRE1α, as well as ATF4 transcription factor, which is involved in the general integrated stress response (ISR) [32].
Interestingly, NLRP1 has been shown to have a protective role in diabetes. Diabetic kidney disease (DKD) T1D patients with rs2670660 and rs11651270 NLRP1 polymorphisms (gain‐of‐function variants) were associated with having a decreased risk of developing DKD. Supporting these findings, diabetes‐associated metabolites (high glucose and glycated albumin) were used to mimic the diabetic milieu in monocytes from healthy individuals and were found to increase NLRP1 expression [26]. In addition, the same polymorphisms have recently been associated with lower susceptibility of T1D [33]. The molecular mechanisms by which NLRP1 regulates T1D, however, are still unclear and could potentially uncover new therapeutic targets for this disease.
Several studies have involved NLRP1 having either a protective or worsening role in a variety of metabolic diseases. However, the molecular and metabolic mechanisms of NLRP1 that regulate these diseases are still unclear. IL‐18 production by NLRP1B has shown a protective effect in obesity and metabolic syndrome in mice [34]. Conversely, NLRP1 is known to contribute to pathology in metastatic melanoma [35] and diabetic retinopathy, where excessive inflammation and high glucose levels seem to promote disease [27]. Overall, it is clear that NLRP1 inflammasome has a crucial role as a sensor of cellular stress, as well as glucose and lipid metabolic perturbation, maintaining cellular homeostasis to prevent a wide range of metabolic diseases. However, NLRP1 can also lead to uncontrolled inflammation contributing to disease; therefore, further investigation into the mechanisms that control the level of NLRP1 activation is needed.
AIM2
The AIM2 inflammasome can be upstream or downstream to metabolic regulation, mainly by metabolites and enzymes from glucose metabolism. Pyruvate kinase isoform M2 (PKM2)‐mediated glycolysis, measured by phosphoenolpyruvate (PEP) and lactate levels, was shown to activate pyruvate dehydrogenase kinase 1 (PDK1), which inhibits the entry of pyruvate to the TCA cycle, leading to phosphorylation of eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2) and subsequent enhanced activation of mouse and human AIM2 inflammasome by poly(dA:dT). AIM2 inflammasome contributed to the release of IL‐1β, IL‐18 and septic death in mice, which was protected by conditional knockout of PKM2, suggesting a novel mechanism of AIM2 regulation by glycolytic metabolism and a potential therapeutic strategy to treat sepsis [36].
Furthermore, elevated cholesterol in macrophages is known to impair mitochondrial metabolism and activate murine AIM2 inflammasome via mtDNA as a possible ligand. Production of the soluble cholesterol‐25‐hydroxylase (Ch25 h)‐induced 25‐HC (Ch25h produces 25‐HC from cholesterol) by macrophages was reported to inhibit AIM2 inflammasome via suppression of sterol regulatory element‐binding protein (SREBP2) activation and cholesterol biosynthesis, resulting in mitochondrial integrity and homeostasis [37].
AIM2 regulation has directly been involved with certain autoimmune diseases, mostly mediated by signalling events linked to glucose metabolism. A study using sirtuin 1 (SIRT1) knockdown showed that SIRT1 has been shown to induce metabolic reprogramming by increasing mitochondrial respiration and repress the NF‐κB‐driven transcription of AIM2, contributing to cervical cancer pathology in patients [38]. In addition, deletion of AIM2 in mice results in elevated levels of fasting glucose and insulin, leading to white adipose tissue inflammation and obesity via the IFN activated gene Ifi202b [39]. In contrast, AIM2 activation by high glucose in rats has recently been shown to contribute to pathology in diabetic cardiomyopathy via ROS production and the GSDMD pathway [40]. Overall, increased glycolysis and disruption in TCA cycle and mitochondrial dysfunction have generally been implicated in the activation of AIM2 inflammasome across different studies. It would be interesting to determine whether 25‐HC modulation of this inflammasome is involved in diseases where lipid metabolism is disrupted, as well as the involvement of other lipid metabolites. In addition, further research considering the SIRT1‐AIM2 axis as a target for cervical cancer will be crucial for the development of new therapies.
Non‐canonical caspase 11 inflammasome
A recent mouse study identified a mechanism of metabolic regulation of the caspase 11 inflammasome by regulation of cAMP, which promotes PKA leading to caspase 11 inflammasome inhibition. L‐adrenaline‐induced cAMP via the enzyme ADCY4 showed inhibition of cytosolic LPS‐induced caspase 11 inflammasome pathway and consequent pyroptosis in macrophages, and presented this novel interaction with caspase 11 inflammasome as a potential therapeutic target in sepsis [41]. Interestingly, the cAMP‐PKA axis leading to inhibition of inflammasome and pyroptosis has also been described in several studies involving the canonical NLRP3 inflammasome [42, 43], given its contributing role in chronic inflammatory diseases, it would be interesting to explore further the cAMP regulation of this inflammasome in sepsis.
NLRC3
NLRC3 whose functions are mostly undiscovered as compared to other NLRs, has been identified as a negative regulator of innate immunity and inflammatory responses [44, 45]. NLRC3 does not form an inflammasome complex and was shown to inhibit LPS‐induced Toll‐like receptor (TLR) signalling by inhibiting the adaptor protein TNF receptor‐associated factor 6 (TRAF6) [46]. Furthermore, NLRC3 blocks activation of the PI3K‐dependent Akt kinase and inhibits mTOR pathways in colon epithelial cells [45].
Interestingly, NLRC3 plays a major role in T‐cell metabolism. Aerobic glycolysis is a metabolic hallmark of activated T cells and Th1 cell differentiation [47]. NLRC3 acts as a brake, diminishing metabolic pathways to attenuate T‐cell activation in a NF‐κB‐dependent manner. NLRC3 deficiency in mice increases oxidative phosphorylation and glycolytic capacity, driving Th1 and Th17 cell differentiation, proliferation and IFN‐γ, IL‐2, TNF and IL‐17 production, thus leading to unrestrained generation of activated CD4+ T cells with maximal glycolytic and mitochondrial metabolism [48], which could lead to autoimmune disease progression. Profiling of T cells from patients with multiple sclerosis (MS) has reported increased glycolysis and mitochondrial activity [49]. Therefore, it could be possible that a deficiency in NLRC3 expression within the human population could predispose individuals with low CD4+ T‐cell NLRC3 expression to develop multiple sclerosis (MS) or MS‐related disorders.
NLRX1
NLRX1, an enigmatic modulator of immune function, is part of an NLR family subgroup which are non‐inflammasome` forming and regulate the inflammation associated with other PRRs activation. The location and trafficking of NLRX1 is debated but it is known to negatively regulate type I interferon, IL‐6, NF‐κB signalling and to induce ROS production, activating the JNK pathway leading to apoptosis [50, 51, 52, 53, 54].
Loss of function of NLRX1 has been linked with a variety of autoimmune diseases, such as cancer, colitis, inflammatory bowel disease and CNS inflammation [55]. In human breast cancer cells, NLRX1 expression is increased and, in the presence of TNF, helps maintenance of mitochondrial metabolism‐lysosomal crosstalk to regulate invasion and metastasis capability. NLRX1 deletion results in lysosomal dysfunction and decreased turnover of damaged mitochondria via mitophagy, leading to a reduction of OXPHOS‐mediated cell proliferation and migration ability [56]. In mouse T cells, loss of NLRX1 mediated increased lactate dehydrogenase activity, suggesting a preference for aerobic glycolysis, and a greater ability to proliferate and differentiate into a pro‐inflammatory phenotype, combined with a reduced sensitivity to immune checkpoint pathways. Interestingly, in the same study NLRX1 showed a protective effect against colitis, where rag2 −/− mice (an adoptive‐transfer model of colitis) receiving nlrx 1 −/− naive or effector T cells, showed increased disease activity [57]. Another study showed that NLRX1 deletion in colitis mice drives changes in gut–microbiome interactions exacerbating disease; however, restoration of WT glutamine metabolic profiles via glutamine supplementation abrogated effects in microbiome, inflammation and disease severity [58]. In addition, a novel orally active, gut‐restricted drug named NX‐13 has been shown to activate mouse NLRX1 and alleviate disease severity in IBD mouse models, as well as increased OXPHOS, decreased differentiation into Th1 and Th17 subsets and decreased ROS and NF‐κB activation in naïve CD4+ T cells. In addition, NX‐13 activation of NLRX1 also showed resistance to oxidative stress and inflammation in human primary cells from ulcerative colitis patients, therefore becoming a promising NLRX1 agonist for treating IBD [59, 60, 61].
Finally, NLRX1 has been identified as a regulator of glutamate homeostasis in the CNS. Astrocyte dysfunction leading to accumulation of extracellular glutamate and lack of NLRX1 is known to be associated with CNS trauma and excessive inflammation [62, 63, 64]. Using NLRX1‐depleted mice, NLRX1 was reported to potentiate mitochondrial function causing increased astrocytic glutamate uptake, as well as inhibition of Ca2+‐mediated glutamate exocytosis, leading to repression of glutamate release from astrocytes, suggesting a protective role of NLRX1 in CNS inflammation [65]. Overall, NLRX1 has been described to act as a brake in inflammation and negative regulator of pathology, playing a key role in the regulation of immunometabolism with links to disease. The emergence of NLRX1 agonists such as NX‐13 show the potential in targeting NLRX1 to modulate immunometabolic function in diease, and future research focusing on its involvement in colitis and gut microbiota is needed.
CROSSTALK BETWEEN METABOLISM–INFLAMMASOME INTERACTIONS AND OTHER IMMUNE PATHWAYS
GPCR signalling
G protein‐coupled receptors (GPCRs) are an extensive family of membrane receptors known to share seven‐transmembrane α‐helix domains, with an intracellular C terminus and a large extracellular N terminus. Generally, the extracellular region senses extracellular ligands such as ions, metabolites, chemokines and temperature, causing conformational changes in the receptor and subsequent transduction of the signal into intracellular responses, activating further downstream signalling pathways. Canonical GPCRs are classically modulated by the intracellular region ligands: G proteins, which activate downstream signalling, or β‐arrestins, which can control excessive activation [66, 67]. GPCRs are the most common class of targets for therapeutic drugs (~35% of approved drugs) due to their relevance in several human diseases, restricted expression and structure. However, only about 16% of the ~800 different GPCRs have been targeted for therapeutic drugs, leaving a substantial number of distinct GPCRs to be explored as potential therapeutic targets [68, 69].
Activation of inflammasomes, mainly NLRP3, can be modulated via GPCRs interacting with different metabolites, ions, neurotransmitters and hormones, indicating crosstalk between metabolism, GPCR signalling and inflammasomes. Metabolites and metabolic proteins involved in such process include ω‐3 FAs, cyclic adenosine monophosphate (cAMP), β‐hydroxybutyrate (BHB), bile acids, prostaglandin E2 (PGE2) and SCFAs such as acetate (Figure 2).
FIGURE 2.
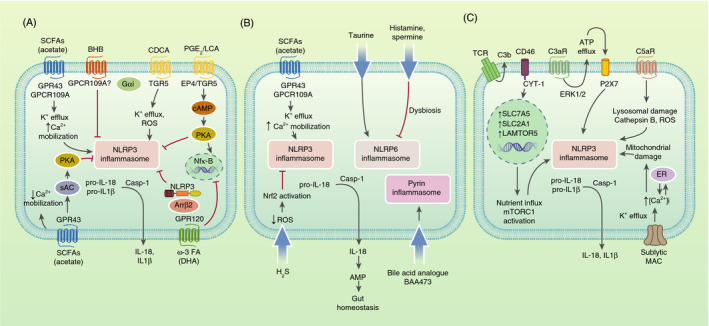
Metabolic regulators of the inflammasome in the crossroads with: GPCR signalling, host–microbiota communication and the complement system. (A) GPCRs that activate or inhibit NLRP3 inflammasome by metabolites. GPR43 and/or GPR109A act as SCFA (mainly acetate) receptors to induce NLRP3 inflammasome activation via K+ efflux, increased Ca2+ mobilization and membrane hyperpolarization in gut epithelial cells, or to repress NLRP3 inflammasome activation via decreased Ca2+ mobilization and activation of sAC and PKA in BMDMs. GPR120, receptor of the ω‐3 FA DHA, inhibits NLRP3 activation through suppression of NF‐κB and binding of β‐arrestin 2 (Arrβ2) to NLRP3, impeding its assembly. BHB inhibits NLRP3 activation via an undefined Gαi‐coupled GPCR. TGR5 and EP4, receptors of bile acids (LCA) and PGE2, respectively, both inhibit NLRP3 via cAMP‐PKA signalling, causing NLRP3 phosphorylation. PGE2 can also promote NLRP3 priming via EP4‐cAMP‐PKA signalling leading to NF‐κB activation in BMDMs. The bile acid CDCA can bind TRG5 leading to NLRP3 activation through K+ efflux and ROS production. (B) Inflammasome activation in the gut by metabolites leads to IL‐18 release which promotes AMP production and secretion by nearby cells, controlling the gut microbial composition. Upon dysbiosis induction, histamine and spermine inhibit NLRP6‐dependent IL‐18 and AMP, supporting the dysbiotic microbiota. Taurine reduces spermine and histamine levels and induces IL‐18 secretion and improved microbiota composition. H2S inhibits NLRP3 inflammasome and IL‐1β secretion in DSS‐induced colitis colons via reduced ROS and Nrf2 activation.The bile acid analogue BAA473 activates pyrin inflammasome in intestinal epithelial cells leading to IL‐18 secretion. (C) The complement system activates NLRP3 inflammasome via metabolic changes. Activation of C3aR drives ATP efflux from the cytosol and ERK1/2 phosphorylation. ATP efflux triggered by C3a activates P2X7, leading to NLRP3 activation. C5a binding to C5aR1 triggers NLRP3 inflammasome priming and activation via ROS production, lysosomal damage and cathepsin B activity. CD46‐CYT‐1 upregulated by TCR stimulation through autocrine C3b production drives upregulation of LAT1 and GLUT1, which mediate nutrient influx into the cell, as well as LAMTOR5. LAMTOR5 drives mTORC1 activation (a known NLRP3 inflammasome activator) leading to increased glycolysis and Th1 cell induction. CD46‐CYT‐1 signalling also primes NLRP3 inflammasome by upregulating IL‐1β and NLRP3. Membrane attack complex deposition drives NLRP3 activation in human lung epithelial cells via Ca2+ influx, increased [Ca2+]i in the cytosol and endoplasmic reticulum (ER) stores, subsequent mitochondrial [Ca2+]i uptake and alteration of mitochondrial membrane potential
SCFAs are derived from commensal anaerobic bacterial fermentation of dietary fibre and are known to play a crucial role in gut homeostasis and inflammation. SCFA acetate binding to metabolite‐sensing receptors GPR43 and/or GPCR109A has been shown to mediate a protective effect against colitis and drive activation of NLRP3 inflammasome in epithelial cells, showing caspase 1 activation and secretion of IL‐18. Acetate led to GPR43‐dependent inflammasome activation through K+ efflux, Ca2 + mobilization and downstream hyperpolarization [70]. Conversely, acetate interacting with GPR43 in bone marrow‐derived macrophages (BMDMs) has recently been described as a suppressor of NLRP3 inflammasome via a Ca2 +‐dependent mechanism, leading to decreased Ca2 + mobilization. The mechanism also involved adenylyl cyclase (sAC)‐PKA signalling leading to NLRP3 ubiquitination and autophagy. In addition, the study showed that acetate had a protective effect on NLRP3‐dependent peritonitis in vivo [71].
Certain GPCRs can have a dual effect on regulating the inflammasome in different situations and inflammatory states; therefore, further research is crucial to better understand their mechanisms and establish more GPCRs as new therapeutic targets. Another example of that is the PGE2 receptor 4 (EP4) and the bile acids active membrane receptor TGR5, which are known to inhibit or activate NLRP3. EP4 binding to PGE2 can inhibit or activate priming of NLRP3 via cAMP increase and subsequent PKA activation: in HEK cells, cAMP leads to phosphorylation of human NLRP3 on Ser295 and PKA decreases its ATPase function causing NLRP3 inhibition, whereas in BMDMs, PGE2 promotes NLRP3 priming through the EP4‐cAMP‐PKA mechanism leading to NF‐κB activation [43, 72]. Interestingly, TGR5 binding to lithocholic acid (LCA) can suppress NLRP3 in a similar mechanism which is also cAMP‐PKA dependent, where PKA leads to NLRP3 phosphorylation on mouse Ser291 (corresponding to human S295), causing ubiquitination of NLRP3. The same study showed prevention of high‐fat diet‐induced insulin resistance downstream of TGR5‐NLRP3 inhibition axis [42]. Conversely, TGR5 binding to the major bile acid involved in cholestatic liver injury, chenodeoxycholic acid (CDCA), leads to activation of NLRP3 by an independent cAMP‐PKA mechanism, where ROS production, K+ efflux and c‐Jun N‐terminal kinase pathways have been involved [73]. It is important to note that CDCA binding to TGR5 has been reported to both activate and inhibit NLRP3‐mediated caspase 1 activation and IL‐1β secretion in BMDMs through different mechanisms, the biological basis of these discrepancies has not been elucidated and therefore more research is needed to clarify the CDCA‐TGR5‐inflammasome axis [42, 73].
Metabolites that have been reported to only inhibit the inflammasome via GPCRs are ω‐3 FAs and β‐hydroxybutyrate. The ω‐3 FA docosahexaenoic acid (DHA) interacting with its receptor GPR120 was shown to inhibit inflammasome priming and activation, first through suppression of NF‐κB and then inhibiting IL‐1β release via increased autophagy. Interestingly, not only NLRP3 but also NAIP5/NLRC4 and AIM2 inflammasomes were involved in this process. In addition, DHA mediated β‐arrestin 2 recruitment to GPR120, leading to a β‐arrestin 2‐dependent reduction of inflammasome activation [74]. Finally, while the ketone body BHB was found to inhibit NLRP3 independently of GPCRs in macrophages and neutrophils, it has recently been reported that in C6 glioma cells, BHB can inhibit NLRP3 through a Gαi‐coupled GPCR, whether that is the BHB Gαi‐coupled receptor GPR109A or not remains to be explored [75].
It is clear that certain metabolism–inflammasome interactions with GPCR signalling are still controversial, this is especially apparent in the case of SCFAs having opposite roles as modulators of NLRP3 inflammasome in colitis and peritonitis, which could be attributed to the fact that studies were performed in different cell types and experimental conditions. Thus, further research should focus on the upstream NLRP3 activation mechanisms in disease, as well as understanding how GPCR‐driven cAMP production inhibits NLRP3, or whether other inflammasomes other than NLRP3 are involved in GPCR signalling.
Host–Microbiota communication
The gut microbiota is known to communicate with the host having a commensal interaction, where microbes and their derived metabolites resident in the gut are able to sense changes in the microbiota and act as signals to regulate a variety of signalling pathways including host PRRs, such as inflammasomes. The inflammasomes regulated by microbiota‐associated metabolites have mainly been NLRP6, NLRP3 and pyrin (Figure 2). Inflammasome activation at a basal level and production of IL‐18 in the gut are crucial to maintain homeostasis of the intestinal microbiome; however, dysregulation of inflammasome activation caused by certain host and gut‐derived metabolites can lead to dysbiosis [76, 77]. Dysregulation of the microbiota homeostasis can alter the intestinal barrier's function with unwanted leakage of pro‐inflammatory signals, as well as alteration of many metabolic products contributing to dysbiosis‐associated intestinal carcinogenesis and non‐alcoholic fatty acid disease (NAFLD) [78, 79].
Given the high expression of NLRP6 in the intestine, it is not unexpected that its ligands are mostly derived from bacterial components in the gut. Expression of NLRP6 in gut epithelial cells has been shown to regulate the intestinal microbiota and mucus secretion [80]. Furthermore, a metabolomics–metagenomics analysis of caecal content identified microbiota‐modulated metabolites as co‐modulators of NLRP6 inflammasome activation, with downstream epithelial IL‐18‐induced antimicrobial peptide (AMP) production, driving stability of the intestinal microbial community. In vitro and in vivo mice studies showed that upon dysbiosis induction, the metabolites histamine and spermine inhibited NLRP6 inflammasome‐dependent IL‐18 production, supporting the dysbiotic microbiota. Administration of taurine showed reduced spermine and histamine levels and induced IL‐18 production, improved microbiota composition and had a protective effect against colitis via NLRP6 inflammasome [76]. Interestingly, a recent study showed improvement of gut permeability and reduction of intestinal inflammation after reactivating NLRP6 through Roux‐en‐Y gastric bypass (RYGB) surgery in obese rats, housed in a specific pathogen‐free facility. After RYGB, increased taurine positively affected NLRP6 expression [81].
In contrast, studies conducted in germ‐free facilities contradicted the aforementioned studies and observed no impact of NLRP6 in gut microbiota composition, attributing this result to the fact that they used littermate‐controlled WT and NLRP6‐deficient mice to minimize non‐genetic confounders, allowing them to shape their gut microbiota naturally after birth. Such studies alleged that the results obtained by previous studies showing the opposite effect were not obtained from littermate animals; therefore, it was possible that the dysbiosis observed in the NLRP6‐deficient mice was genotype independent and acquired stochastically [82, 83].
A recent study identified a secondary metabolite of flavonoids and a common constituent of the human diet, apigenin, as a regulator of gut microbiota via NLPR6. Apigenin showed a protective effect against colitis in mice via NLRP6 signalling, which still happened in the absence of caspase 1/11 or Asc, suggesting that an alternative mechanism to the classical inflammasome pathways was involved. Mice showed protection against colitis when cohoused with apigenin‐treated mice, whereas NLRP6‐deficient mice lost the protective effect [84]. Thus, the role of NLRP6 inflammasome as a regulator of intestinal microbiota is still controversial and further research, possibly in human studies, is needed to confirm this role.
Activation of NLRP3 inflammasome in the colon has been shown to have both protective and detrimental effects in different colitis studies. NLRP3 deficiencies showed either defects in intestinal mucosa repairing, exacerbating disease, or decreased levels of secreted IL‐1β and autoinflammation, reducing disease severity [70, 85, 86, 87]. In an aforementioned study, bacterial SCFAs metabolites showed steady‐state NLRP3 inflammasome activation and IL‐18 production in epithelial cells, leading to regulation of the microbiome composition, reduction of inflammatory responses and a protective effect against colitis [70]. Conversely, the bacterial metabolite hydrogen sulphide (H2S) has been shown to inhibit NLRP3 inflammasome and IL‐1β secretion in BMDMs and in dextran sulphate sodium (DSS)‐induced colitis colons via reduced ROS and Nrf2 activation. In addition, H2S attenuated the severity of mouse colitis by NLRP3 inflammasome inhibition [88]. Different levels of tissue injury and inflammation caused by intestinal NLRP3 can determine its protective or detrimental function in the gut, which can also depend on the different experimental conditions between studies [89].
Interestingly, a screen for microbiome metabolites that may function as inflammasome activators recently identified BAA473, a bile acid analogue, as a pyrin inflammasome activator leading to IL‐18 secretion in human intestinal epithelial and myeloid cells. Given the crucial role of IL‐18 in the gut, the study suggests that microbiota‐mediated bile acid conversion might modulate intestinal homeostasis via the pyrin inflammasome pathway [90].
Finally, NLRP12 has been reported to have a protective role in IBD patients and act as a regulator of the colonic microbiota and suppressor of inflammation to microbial ligands. NLRP12 deficiencies in mice lead to IL‐6 and TNF‐driven colonic inflammation and dysbiosis via an inflammasome‐independent pathway [91]. However, whether there are any bacterial metabolites that regulate NLRP12 and its functions in the gut remain to be explored. Overall, the controversial evidence regarding the involvement of NLRP6 and its microbiota‐derived metabolites as regulators of gut homeostasis, as well as the different roles of NLRP3 against colitis, which possibly lays in different cell types and grades of inflammasome activation, indicates the need for further research in human disease settings and into the downstream mechanisms that control these effects.
The complement system
In recent decades, it has become apparent that the complement system and inflammasomes are not only pathogen sensors, but also systems that can recognize cell metabolic changes and induce reactive responses, for instance, to support cell activation or to maintain cell homeostasis. Several complement receptors and regulators have been presented as critical signals for NLRP3 inflammasome activation, either linked with signals coming from RLR or TLR activation, or independently [92]. This process can occur through metabolic changes being upstream or downstream to complement, leading to inflammasome activation (Figure 2).
Activation of the complement anaphylatoxin receptor C3aR in myeloid cells results in NLRP3 inflammasome activation. This process has been reported to occur in the presence of LPS and TLR4 activation through increased ATP efflux from the cytosol and ERK1/2 phosphorylation. ATP efflux triggered by C3a resulted in P2X purinoceptor 7 (P2X7) activation, leading to inflammasome activation [89]. Interestingly, an ATP‐inflammasome‐complement axis has recently been proposed, where complement is activated by DAMPs released by inflammasome activation, promoting sterile brain inflammation [93].
C5a generation driven by cholesterol and acid uric crystals has also been shown to act as a trigger for the NLRP3 inflammasome. Abnormally high concentrations of the metabolite cholesterol, which can be driven by defects in cholesterol metabolism and homeostasis, precipitate to form cholesterol crystals (CC) driving inflammation and contributing to pathogenesis in atherosclerosis [94]. A study showed that CC activate complement and that the resulting C5a generated, combined with TNF‐α, triggered ROS and priming of the inflammasome [95], suggesting complement inhibition as a potential therapeutic target for atherosclerosis treatment. Furthermore, studies in which inflammation was induced by the crystalized metabolite uric acid in monocytes as a model of gout disease have shown that C5aR1 triggers priming and activation of the inflammasome via lysosomal damage and cathepsin B activity [96]. This role for C5a was confirmed by other studies using neutrophils in a mouse peritonitis model [97].
Complement can not only regulate metabolic changes at a systemic level [98], but also at an intracellular level. Autocrine complement activity has been shown to induce metabolic reprogramming leading to T‐cell activation and inflammasome activation [99, 100]. Human CD4+ T cells express CD46 in two different isoforms based on their cytoplasmic tails, known as CYT‐1 and CYT‐2. CD46‐CYT‐1 is upregulated by T‐cell receptor (TCR) stimulation via autocrine C3b production, leading to increased expression of the amino acid and glucose transporters LAT1 and GLUT1, as well as MAPK and MTOR activator 5 (LAMTOR5), leading to Th1 cell induction. LAT1 and GLUT1 mediate nutrient influx, while LAMTOR5 drives mTORC1 activation and glycolysis, a metabolic hallmark in activated T cells and known NLRP3 inflammasome activator [92]. Alternatively, CD46‐CYT‐2 is expressed in resting and contracting T‐cells, where CD46‐CYT‐2 mediates a switch from glycolysis to OXPHOS metabolism [99]. Interestingly, CD46 (which binds C3b) stimulation in activated CD4+T cells was found to simultaneously upregulate IL‐1β and NLRP3 gene expression to prime the NLRP3 inflammasome, as well as increase intracellular generation of C5a and activation of C5aR1, which led to increased ROS production and subsequent NLRP3 inflammasome activation and Th1 cell induction [100]. Dysregulation of C3 and CD46 in CD4+ T cells has been reported to contribute to Th1‐mediated autoimmune diseases [101, 102, 103]. Furthermore, cytotoxic CD8+ T cells have recently been involved in the CD46 regulator costimulation signalling, leading to increased fatty acid synthesis and nutrient influx which promote optimal cell activity. However, this process was not linked with the canonical NLRP3 signalling [104].
Finally, studies in dendritic cells, C6‐deficient mice [105] or in human lung epithelial cells have elucidated that sublytic membrane attack complex (MAC) activates NLRP3 inflammasome via Ca2+ influx, subsequent mitochondrial [Ca2+]i uptake and alteration of mitochondrial membrane potential [106]. Interestingly, sublytic MAC and increased glycolysis have been implicated in rheumatoid arthritis, contributing to inflammation [107, 108]. Given that MAC is an inflammatory trigger and such stimuli have been implicated in the modulation of immunometabolic responses, perturbations in cellular metabolism downstream of sublytic MAC might be involved in this process. In conclusion, a functional complement–metabolism–inflammasome axis has been demonstrated particularly in T cells (CD46‐C3b and C5aR1‐ROS interactions). More complement components such as MAC, as well as other cell types are likely to be involved in this axis in the future, providing potential novel therapeutic targets in the treatment of autoimmune diseases.
CONCLUSIONS AND FUTURE PERSPECTIVES
Growing evidence on modulation of inflammasome activation by metabolic changes has become apparent in the recent years not only for NLRP3, but also for NLRC4, NLRP1, AIM2, NLRP6, NLRP12, pyrin, NLRC3, NLRX1 and non‐canonical caspase 11 inflammasome. Metabolic control of these inflammasomes and non‐inflammasome forming NLRs implicates a variety of perturbations in the glycolytic pathway, mitochondrial and lipid metabolism, which has been linked to several autoimmune and metabolic diseases, such as obesity, diabetes and cancer. The downstream mechanisms of the metabolic perturbations that directly regulate the inflammasome, however, remain unclear, where some independent mechanisms have been proposed but not always linked to each other. Whether such mechanisms interact with each other in a common signalling cascade or distinct pathways needs to be understood. Furthermore, novel links between inflammasomes and metabolic diseases are recently emerging, such as NLRP1 and NLRC4’s involvement in T1D and diabetic complications, which have been linked to certain metabolic markers [21, 26]. Elucidation of the mechanism of action between the metabolic alterations and inflammasomes linked to disease will be crucial to allow development of new therapeutic targets.
There is contradictory evidence regarding certain metabolism–inflammasome interactions and whether they have a protective or worsening effect in disease. SCFAs interacting with GPCR signalling have been reported to both activate and inhibit NLRP3 through different mechanisms, leading to a protective or detrimental role in colitis/peritonitis [70, 71]. The fact that metabolites often interact with Gs‐coupled GPCR signalling to regulate the inflammasome indicates potential development of new GPCR‐targeted treatments for inflammasome‐driven diseases. However, more research is needed to understand GPCR regulation of the upstream NLRP3 activation mechanisms, as well as how GPCR‐driven cAMP production inhibits NLRP3, whether other inflammasomes other than NLRP3 are involved or how to specifically modulate inflammasome activation by targeting GPCRs.
NLRP3 and NLRP6 are the main gut inflammasomes, NLRP3 and arguably NLRP6, seem to be regulated by the metabolic outputs of gut microbiota and have a crucial involvement in DSS‐colitis. The recent finding about intestinal pyrin inflammasome being activated by bile acids presents pyrin as an emerging metabolic sensor in the gut [90]. Its upstream mechanisms, role in gut microbiota and gut‐related diseases, however, remain unclear and need to be understood. In addition, gut microbiota activating intestinal NLRP3 inflammasome is known to aggravate progression of CNS diseases such as AD and has been described as the microbiota‐gut‐brain axis [109, 110, 111, 112]. Whether certain gut metabolites (SCFA catabolism [109]) have a role in NLRP3 activation in CNS diseases should be investigated. Finally, the complement–metabolism–inflammasome axis has been well described for the NLRP3 inflammasome mainly in T cells. More research is needed to unveil whether other inflammasomes are involved in this process, as well as other primary cells and tissues. Equally important will be to better understand if other complement components are involved, i.e. further elucidation of the role of MAC complex in this axis, and their downstream metabolic mechanisms leading to inflammasome activation.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
ACKNOWLEDGEMENTS
The authors’ research is supported by the Institute of Infection and Immunity from Cardiff University and the Immunology Network at GlaxoSmithKline. Lee Booty (scientist in the Immunology Network at GlaxoSmithKline) supervised the NLRX1 section of this review. A funding statement is not applicable other than what is stated in acknowledgements.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Enigmatic inflammasomes‐Sequel (Part 2). Immunology 2021, 163: 345‐347.
NLRP7: From inflammasome regulation to human disease. Immunology 2021, 163: 363‐376.
Unknown/enigmatic functions of extracellular ASC. Immunology 2021, 163: 377‐88.
REFERENCES
- 1. Lachmandas E, Beigier‐Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, et al. Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur J Immunol. 2016;46:2574–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA, et al. Microbial stimulation of different Toll‐like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. 2016;2:16246. [DOI] [PubMed] [Google Scholar]
- 3. Gleeson LE, Sheedy FJ, editors. Metabolic reprogramming & inflammation: Fuelling the host response to pathogens. In: Seminars in immunology, Vol. 28. Amsterdam, The Netherlands: Elsevier; 2016;450–468. [DOI] [PubMed] [Google Scholar]
- 4. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis‐associated sepsis. Cell Metab. 2017;25:856–67.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Próchnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26:71–93. [DOI] [PubMed] [Google Scholar]
- 7. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- 9. Yang Q, Liu R, Yu Q, Bi Y, Liu G. Metabolic regulation of inflammasomes in inflammation. Immunology. 2019;157:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palsson‐McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif‐1α activity and IL‐1β induction and is a critical determinant of the Warburg effect in LPS‐activated macrophages. Cell Metab. 2015;21:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anand PK. Lipids, inflammasomes, metabolism, and disease. Immunol Rev. 2020;297:108–22. [DOI] [PubMed] [Google Scholar]
- 14. Haneklaus M, O'Neill LA. NLRP3 at the interface of metabolism and inflammation. Immunol Rev. 2015;265:53–62. [DOI] [PubMed] [Google Scholar]
- 15. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation‐induced tumorigenesis in the colon is regulated by caspase‐1 and NLRC4. Proc Natl Acad Sci. 2010;107:21635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity‐associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun. 2016;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo X, Yang Y, Shen T, Tang X, Xiao Y, Zou T, et al. Docosahexaenoic acid ameliorates palmitate‐induced lipid accumulation and inflammation through repressing NLRC4 inflammasome activation in HepG2 cells. Nutr Metab. 2012;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan F, Kolb R, Pandey G, Li W, Sun L, Liu F, et al. Involvement of the NLRC4‐inflammasome in diabetic nephropathy. PLoS One. 2016;11:e0164135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Torres ACMG, Leite N, Tureck LV, de Souza RLR, Titski ACK, Milano‐Gai GE, et al. Association between Toll‐like receptors (TLR) and NOD‐like receptor (NLR) polymorphisms and lipid and glucose metabolism. Gene. 2019;685:211–21. [DOI] [PubMed] [Google Scholar]
- 21. Xu L, Sun X, Xia Y, Luo S, Lin J, Xiao Y, et al. Polymorphisms of the NLRC4 gene are associated with the onset age, positive rate of GADA and 2‐h postprandial C‐peptide in patients with type 1 diabetes. Diabetes Metab Syndr Obes. 2020;13:811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double‐stranded RNA. Science. 2020;371(6528):eabd0811. [DOI] [PubMed] [Google Scholar]
- 23. Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, Lim CK, et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science. 2020;370(6521):eaay2002. [DOI] [PubMed] [Google Scholar]
- 24. Neiman‐Zenevich J, Liao K‐C, Mogridge J. Distinct regions of NLRP1B are required to respond to anthrax lethal toxin and metabolic inhibition. Infect Immun. 2014;82:3697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012;8:e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Soares JL, Fernandes FP, Patente TA, Monteiro MB, Parisi MC, Giannella‐Neto D, et al. Gain‐of‐function variants in NLRP1 protect against the development of diabetic kidney disease: NLRP1 inflammasome role in metabolic stress sensing? Clin Immunol. 2018;187:46–9. [DOI] [PubMed] [Google Scholar]
- 27. Li Y, Liu C, Wan X‐S, Li S‐W. NLRP1 deficiency attenuates diabetic retinopathy (DR) in mice through suppressing inflammation response. Biochem Biophys Res Comm. 2018;501:351–7. [DOI] [PubMed] [Google Scholar]
- 28. Ringel‐Scaia VM, Qin Y, Thomas CA, Huie KE, McDaniel DK, Eden K, et al. Maternal influence and murine housing confound impact of NLRP1 inflammasome on microbiome composition. J Innate Immun. 2019;11:416–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, et al. Omega‐3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154–63. [DOI] [PubMed] [Google Scholar]
- 30. Liao K‐C, Mogridge J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun. 2013;81:570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taabazuing CY, Griswold AR, Bachovchin DA. The NLRP1 and CARD8 inflammasomes. Immunol Rev. 2020;297:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. D’Osualdo A, Anania VG, Yu K, Lill JR, Kaufman RJ, Matsuzawa S‐I, et al. Transcription factor ATF4 induces NLRP1 inflammasome expression during endoplasmic reticulum stress. PLoS One. 2015;10:e0130635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun X, Xia Y, Liu Y, Wang Y, Luo S, Lin J, et al. Polymorphisms in NLRP1 gene are associated with type 1 diabetes. J Diabetes Res. 2019;2019:9 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MK, Lawlor KE, et al. IL‐18 production from the NLRP1 inflammasome prevents obesity and metabolic syndrome. Cell Metab. 2016;23:155–64. [DOI] [PubMed] [Google Scholar]
- 35. Zhai Z, Liu W, Kaur M, Luo Y, Domenico J, Samson JM, et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene. 2017;36:3820–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2‐dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 2017;171:1057–71.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. So D, Shin H‐W, Kim J, Lee M, Myeong J, Chun Y‐S, et al. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene. 2018;37:5191–204. [DOI] [PubMed] [Google Scholar]
- 39. Gong Z, Zhang X, Su K, Jiang R, Sun Z, Chen W, et al. Deficiency in AIM2 induces inflammation and adipogenesis in white adipose tissue leading to obesity and insulin resistance. Diabetologia. 2019;62:2325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X, Pan J, Liu H, Zhang M, Liu D, Lu L, et al. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019;221:249–58. [DOI] [PubMed] [Google Scholar]
- 41. Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, Kroemer G, et al. cAMP metabolism controls caspase‐11 inflammasome activation and pyroptosis in sepsis. Sci Adv. 2019;5:eaav5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–16. [DOI] [PubMed] [Google Scholar]
- 43. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto‐inflammatory disease CAPS mutations. Nat Immunol. 2016;17:1176–86. [DOI] [PubMed] [Google Scholar]
- 44. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. 2014;40:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karki R, Man SM, Malireddi RKS, Kesavardhana S, Zhu Q, Burton AR, et al. NLRC3 is an inhibitory sensor of PI3K‐mTOR pathways in cancer. Nature. 2016;540:583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll‐like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF‐kappaB. Nat Immunol. 2012;13:823–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. 2015;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uchimura T, Oyama Y, Deng M, Guo H, Wilson JE, Rampanelli E, et al. The innate immune sensor NLRC3 acts as a rheostat that fine‐tunes T cell responses in infection and autoimmunity. Immunity. 2018;49:1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Riccardis L, Rizzello A, Ferramosca A, Urso E, De Robertis F, Danieli A, et al. Bioenergetics profile of CD4(+) T cells in relapsing remitting multiple sclerosis subjects. J Biotechnol. 2015;202:31–9. [DOI] [PubMed] [Google Scholar]
- 50. Nagai‐Singer MA, Morrison HA, Allen IC. NLRX1 Is a multifaceted and enigmatic regulator of immune system function. Front Immunol. 2019;10:2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR‐induced NF‐κB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yin H, Sun G, Yang Q, Chen C, Qi Q, Wang H, et al. NLRX1 accelerates cisplatin‐induced ototoxity in HEI‐OC1 cells via promoting generation of ROS and activation of JNK signaling pathway. Sci Rep. 2017;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy. 2013;9:432–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–7. [DOI] [PubMed] [Google Scholar]
- 55. Pickering RJ, Booty LM. NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology. 2021;162(3):268–280. 10.1111/imm.13291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Singh K, Roy M, Prajapati P, Lipatova A, Sripada L, Gohel D, et al. NLRX1 regulates TNF‐α‐induced mitochondria‐lysosomal crosstalk to maintain the invasive and metastatic potential of breast cancer cells. Biochim Biophys Acta (BBA)‐Mol Basis Dis. 2019;1865:1460–76. [DOI] [PubMed] [Google Scholar]
- 57. Leber A, Hontecillas R, Tubau‐Juni N, Zoccoli‐Rodriguez V, Hulver M, McMillan R, et al. NLRX1 regulates effector and metabolic functions of CD4+ T cells. J Immunol. 2017;198:2260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leber A, Hontecillas R, Tubau‐Juni N, Zoccoli‐Rodriguez V, Abedi V, Bassaganya‐Riera J. nlrX1 Modulates immunometabolic Mechanisms controlling the host–gut Microbiota interactions during inflammatory Bowel Disease. Front Immunol 2018;9:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leber A, Hontecillas R, Zoccoli‐Rodriguez V, Bienert C, Chauhan J, Bassaganya‐Riera J. Activation of NLRX1 by NX‐13 alleviates inflammatory bowel disease through immunometabolic mechanisms in CD4+ T cells. J Immunol. 2019;203:3407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leber A, Hontecillas R, Zoccoli‐Rodriguez V, Chauhan J, Bassaganya‐Riera J. Su1818–preclinical efficacy and safety of Nx‐13: a novel Nlrx1‐targeting immunometabolic therapeutic for Crohn’s disease and ulcerative colitis. Gastroenterology. 2019;156:S‐623. [Google Scholar]
- 61. Leber A, Hontecillas R, Zoccoli‐Rodriguez V, Ehrich M, Chauhan J, Bassaganya‐Riera J. Exploratory studies with NX‐13: oral toxicity and pharmacokinetics in rodents of an orally active, gut‐restricted first‐in‐class therapeutic for IBD that targets NLRX1. Drug Chem Toxicol. 2019;3:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Theus MH, Brickler T, Meza AL, Coutermarsh‐Ott S, Hazy A, Gris D, et al. Loss of NLRX1 exacerbates neural tissue damage and NF‐κB signaling following brain injury. J Immunol. 2017;199:3547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Landeghem FKV, Weiss T, Oehmichen M, Deimling AV. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. 2006;23:1518–28. [DOI] [PubMed] [Google Scholar]
- 64. Wang Y, Qin Z‐h. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–402. [DOI] [PubMed] [Google Scholar]
- 65. Mahmoud S, Gharagozloo M, Simard C, Amrani A, Gris D. NLRX1 enhances glutamate uptake and inhibits glutamate release by astrocytes. Cells. 2019;8:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. [DOI] [PubMed] [Google Scholar]
- 67. Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein‐coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sriram K, Insel PA. G protein‐coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol Pharmacol. 2018;93:251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gad AA, Balenga N. The emerging role of adhesion GPCRs in cancer. ACS Pharmacol Transl Sci. 2020;3:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite‐sensing receptors GPR43 and GPR109A facilitate dietary fibre‐induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6:6734. [DOI] [PubMed] [Google Scholar]
- 71. Xu M, Jiang Z, Wang C, Li N, Bo L, Zha Y, et al. Acetate attenuates inflammasome activation through GPR43‐mediated Ca 2+‐dependent NLRP3 ubiquitination. Exp Mol Med. 2019;51:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zoccal KF, Sorgi CA, Hori JI, Paula‐Silva FW, Arantes EC, Serezani CH, et al. Opposing roles of LTB 4 and PGE 2 in regulating the inflammasome‐dependent scorpion venom‐induced mortality. Nat Commun. 2016;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gong Z, Zhou J, Zhao S, Tian C, Wang P, Xu C, et al. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget. 2016;7:83951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Williams‐Bey Y, Boularan C, Vural A, Huang N‐N, Hwang I‐Y, Shan‐Shi C, et al. Omega‐3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF‐κB activation and enhancing autophagy. PLoS One. 2014;9:e97957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shang S, Wang L, Zhang Y, Lu H, Lu X. The beta‐hydroxybutyrate suppresses the migration of glioma cells by inhibition of NLRP3 inflammasome. Cell Mol Neurobiol. 2018;38:1479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman‐Schapira G, Mahdi JA, et al. Microbiota‐modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163:1428–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Henao‐Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome‐mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schulz MD, Atay Ç, Heringer J, Romrig FK, Schwitalla S, Aydin B, et al. High‐fat‐diet‐mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature. 2014;514:508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non‐alcoholic fatty liver disease. Sci Rep. 2015;5:8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wlodarska M, Thaiss CA, Nowarski R, Henao‐Mejia J, Zhang J‐P, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host‐microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang G, Wang Q, Bai J, Zhao N, Wang Y, Zhou R, et al. Upregulation of intestinal NLRP6 inflammasomes after roux‐en‐Y gastric bypass promotes gut immune homeostasis. Obes Surg. 2020;30:327–35. [DOI] [PubMed] [Google Scholar]
- 82. Lemire P, Robertson SJ, Maughan H, Tattoli I, Streutker CJ, Platnich JM, et al. The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep. 2017;21:3653–61. [DOI] [PubMed] [Google Scholar]
- 83. Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira‐Silva S, Yilmaz B, Martens L, et al. Nlrp6‐and ASC‐dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity. 2017;47:339–48.e4. [DOI] [PubMed] [Google Scholar]
- 84. Radulovic K, Normand S, Rehman A, Delanoye‐Crespin A, Chatagnon J, Delacre M, et al. A dietary flavone confers communicable protection against colitis through NLRP6 signaling independently of inflammasome activation. Mucosal Immunol. 2018;11:811–9. [DOI] [PubMed] [Google Scholar]
- 85. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T‐D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192–9. [DOI] [PubMed] [Google Scholar]
- 87. Siegmund B, Lehr H‐A, Fantuzzi G, Dinarello CA. IL‐1β‐converting enzyme (caspase‐1) in intestinal inflammation. Proc Natl Acad Sci. 2001;98:13249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Qin M, Long F, Wu W, Yang D, Huang M, Xiao C, et al. Hydrogen sulfide protects against DSS‐induced colitis by inhibiting NLRP3 inflammasome. Free Radic Biol Med. 2019;137:99–109. [DOI] [PubMed] [Google Scholar]
- 89. Elinav E, Strowig T, Kau AL, Henao‐Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Alimov I, Menon S, Cochran N, Maher R, Wang Q, Alford J, et al. Bile acid analogues are activators of pyrin inflammasome. J Biol Chem. 2019;294:3359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen L, Wilson JE, Koenigsknecht MJ, Chou W‐C, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. 2017;18:541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Arbore G, Kemper C. A novel “complement–metabolism–inflammasome axis” as a key regulator of immune cell effector function. Eur J Immunol. 2016;46:1563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ratajczak MZ, Mack A, Bujko K, Domingues A, Pedziwiatr D, Kucia M, et al. ATP‐Nlrp3 inflammasome‐complement cascade axis in sterile brain inflammation in psychiatric patients and its impact on stem cell trafficking. Stem Cell Rev Rep. 2019;15:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement‐dependent inflammasome activation and cytokine release. J Immunol. 2014;192:2837–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. An LL, Mehta P, Xu L, Turman S, Reimer T, Naiman B, et al. Complement C5a potentiates uric acid crystal‐induced IL‐1β production. Eur J Immunol. 2014;44:3669–79. [DOI] [PubMed] [Google Scholar]
- 97. Cumpelik A, Ankli B, Zecher D, Schifferli JA. Neutrophil microvesicles resolve gout by inhibiting C5a‐mediated priming of the inflammasome. Ann Rheum Dis. 2016;75:1236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Phieler J, Garcia‐Martin R, Lambris JD, Chavakis T. editors. The role of the complement system in metabolic organs and metabolic diseases. In: Seminars in immunology, Vol 25. Amsterdam, The Netherlands: Elsevier; 2013:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity. 2015;42:1033–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Arbore G, West EE, Spolski R, Robertson AA, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement‐driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352:aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46‐mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Investig. 2006;116:3252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cardone J, Le Friec G, Vantourout P, Roberts A, Fuchs A, Jackson I, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol. 2010;11:862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Moreno‐Navarrete JM, Fernández‐Real JM. editors. The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. In: Seminars in cell & developmental biology, Vol. 85. Amsterdam, The Netherlands: Elsevier; 2019:164–172. [DOI] [PubMed] [Google Scholar]
- 104. Arbore G, West EE, Rahman J, Le Friec G, Niyonzima N, Pirooznia M, et al. Complement receptor CD46 co‐stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. Nat Commun. 2018;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: The NLRP3 inflammasome links complement‐mediated inflammation and IL‐1β release. J Immunol. 2013;191(3):1006–1010. 10.4049/jimmunol.1300489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. 2013;126:2903–13. [DOI] [PubMed] [Google Scholar]
- 107. Narasimhan R, Coras R, Rosenthal SB, Sweeney SR, Lodi A, Tiziani S, et al. Serum metabolomic profiling predicts synovial gene expression in rheumatoid arthritis. Arthritis Res Ther. 2018;20(1). 10.1186/s13075-018-1655-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Neumann E, Barnum SR, Tarner IH, Echols J, Fleck M, Judex M, et al. Local production of complement proteins in rheumatoid arthritis synovium. Arthritis Rheum. 2002;46(4):934–945. 10.1002/art.10183 [DOI] [PubMed] [Google Scholar]
- 109. Pellegrini C, Antonioli L, Calderone V, Colucci R, Fornai M, Blandizzi C. Microbiota‐gut‐brain axis in health and disease: Is NLRP3 inflammasome at the crossroads of microbiota‐gut‐brain communications? Prog Neurogibol. 2020;191:101806. [DOI] [PubMed] [Google Scholar]
- 110. Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, et al. Association of brain amyloidosis with pro‐inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–8. [DOI] [PubMed] [Google Scholar]
- 111. Shen H, Guan Q, Zhang X, Yuan C, Tan Z, Zhai L, et al. New mechanism of neuroinflammation in Alzheimer's disease: the activation of NLRP3 inflammasome mediated by gut microbiota. Prog Neuropsychopharmacol Biol Psychiatry. 2020;100:109884. [DOI] [PubMed] [Google Scholar]
- 112. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wang C, Tan Z, Niu B, Tsang KY, Tai A, Chan WC, et al. Inhibiting the integrated stress response pathway prevents aberrant chondrocyte differentiation thereby alleviating chondrodysplasia. Elife. 2018;7:e37673. [DOI] [PMC free article] [PubMed] [Google Scholar]