

Summary
The impact of treatment on the risk of lymphoma in patients with rheumatoid arthritis (RA) is unclear. Here, we aimed to assess if the risk of lymphoma differs according to the type of tumor necrosis factor inhibitor (TNFi), comparing monoclonal anti‐TNF antibodies to the soluble TNF receptor. We used B cell activating factor belonging to the TNF family (BAFF)‐transgenic (Tg) mice as a model of autoimmunity‐associated lymphoma. Six‐month‐old BAFF‐Tg mice were treated with TNFi for 12 months. Histological examination of the spleen, assessment of the cellular composition of the spleen by flow cytometry and assessment of B cell clonality were performed at euthanasia. Crude mortality and incidence of lymphoma were significantly higher in mice treated with monoclonal anti‐TNF antibodies compared to both controls and mice treated with the soluble TNF receptor, even at a high dose. Flow cytometry analysis revealed decreased splenic macrophage infiltration in mice treated with monoclonal anti‐TNF antibodies. Overall, this study demonstrates, for the first time, that a very prolonged treatment with monoclonal anti‐TNF antibodies increase the risk of lymphoma in B cell‐driven autoimmunity. These data suggest a closer monitoring for lymphoma development in patients suffering from B cell‐driven autoimmune disease with long‐term exposure to monoclonal anti‐TNF antibodies.
Keywords: anti‐TNF, autoimmunity, BAFF, lymphoma, macrophages
Long‐term inhibition of TNF with monoclonal anti‐TNF increases the risk of lymphoma in the autoimmune BAFF transgenic mice. These data suggest the need for continued monitoring of lymphoma occurrence in patients with B cell‐driven autoimmune disease with long‐term exposure to monoclonal anti‐TNF Ab.

INTRODUCTION
Autoimmunity is associated with an increased risk of lymphoma. This risk is well demonstrated in patients with rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and primary Sjogren’s syndrome (pSS) [1]. Two major risk factors have been identified. The first is the activity of the autoimmune disease, as demonstrated in patients with RA [2] and with pSS [3]. The second factor is the use of immunosuppressive drugs. It is, indeed, well known that immunosuppression may induce specific types of lymphoma, particularly Epstein–Barr virus‐associated lymphomas as described in post‐transplant lymphoproliferation disorders [4] or in Crohn’s disease treated with thiopurines [5]. In SLE patients, the risk of non‐Hodgkin lymphoma (NHL) is likely to be associated with exposure to cyclophosphamide and high cumulative steroids [6]. In this context, the role of immunosuppressive drugs in promoting lymphoma is difficult to assess, as patients who are the most exposed to immunosuppression are also those with the most active disease.
Tumor necrosis factor (TNF) inhibitors (TNFi) therapy has revolutionized the management of patients with RA as well as of other autoimmune and inflammatory diseases. It is the cornerstone of treatment of methotrexate (MTX)‐resistant RA. To date, epidemiological studies have not disclosed any increased risk of lymphoma in RA patients treated with TNFi [7, 8]. However, in these studies, the median duration of treatment is approximately 4 years and the potential differential risk according to the type of TNFi remains an ongoing matter of debate. Indeed, there are two types of licensed TNFi: a dimeric soluble form of p75/TNF receptor 2 [the TNF‐R2‐Fc, etanercept (ETA)] and monoclonal anti‐TNF antibodies [infliximab, adalimumab (ADA), golimumab and certolizumab]. Differences in terms of efficacy and safety profile between these two types of TNFi are already established. Etanercept is not effective in inflammatory bowel disease [9], and is probably less effective than monoclonal anti‐TNF antibodies in both uveitis [10] and psoriasis [11]. Infectious safety profile also differs. The risk of opportunistic infections and of reactivation of latent tuberculosis has been shown to be higher with monoclonal anti‐TNF antibodies than with ETA [12]. These data underline the notion of differences in the mechanism of action of TNFi that might differentially impact the risk of lymphoma. In 2010, the French registry Research Axed on Tolerance of Biotherapies (RATIO) found that the risk of lymphoma was higher than the risk found in the general population in patients treated with infliximab or ADA [standardized incidence ratio (SIR) = 3.7 (2.5–5.4)], although remaining in the range of what is expected in patients with long‐term active RA. Moreover, in the same study the risk of lymphoma in patients treated with ETA reached that of the general population [SIR = 0.9 (0.4–1.8)] [13]. In the Japanese RA cohort SafEty of biologics in Clinical Use in Japanese patients with Rheumatoid arthritis (SECURE), patients exposed to infliximab had a significantly higher risk of lymphoma than patients exposed to etanercept with an unadjusted incidence rates of 3.38 (2.57–4.38) with infliximab compared to 1.30 (0.87–1.87) with etanercept (p < 6.6 × 10−4) [14]. Of note, in the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis (BSRBR‐RA) registry, RA patients exposed to etanercept had the lowest crude incidence rate of lymphoma compared to those with infliximab or ADA, but the difference did not reach statistical significance after adjustment [15]. More recently, it has been demonstrated that patients with inflammatory bowel diseases (IBD) treated with TNFi (only monoclonal anti‐TNF antibodies) are exposed to an increased risk of lymphoma [16].
To decipher the impact of the different TNFi on the risk of lymphoma associated with autoimmune diseases, we used B cell activating factor belonging to the TNF family (BAFF)‐transgenic (Tg) mice, which is a model of lymphoma complicating B cell autoimmunity. These mice develop a B cell‐driven autoimmune disease with clinical and biological symptoms reminiscent of RA [rheumatoid factor (RF), polyarthritis], SLE [anti‐double strand (ds)DNA, hypergammaglobulinemia, glomerulonephritis, lymphadenopathies, splenomegaly] and SS (sialadenitis) [16, 17, 18]. Three to 5% of BAFF‐Tg mice spontaneously develop B cell lymphoproliferative disease during aging. Notably, development of B cell lymphoproliferative diseases in these mice seems to be linked to the action of TNF, as introduction of TNF deficiency into a BAFF‐Tg background (TNF−/− BAFF‐Tg mice) leads to a surprisingly high incidence of B cell lymphoma (38% of mice at 12 months of age) [19]. To extend these previous findings, we intended to evaluate the risk of lymphoma induced by a long‐term treatment with the two types of TNFi in these BAFF‐Tg autoimmune mice.
MATERIAL AND METHODS
Mice
BAFF‐Tg mice, with a C57BL/6 genetic background, were provided by F. Mackay (Monash University, Melbourne, Australia) [17]. Animals chosen for experimental procedure were homozygous for the BAFF transgene. The genotype of BAFF‐Tg mice was determined using polymerase chain reaction (PCR) on genomic DNA obtained from 5‐mm tail snips. Because spontaneous lymphomas have been shown to occur in BAFF‐Tg mice aged 12–18 months [19], we decided to begin treating 6‐month‐old mice for 12 months to assess the impact of TNFi on the risk of lymphoma associated with B cell autoimmunity. Euthanasia was performed after 12 months of treatment or before, in cases with criteria of imminent death, to allow the collection of terminal tissue [20].
Treatment of BAFF‐Tg mice with TNFi
The number of mice to include in the experimental design was calculated based on data reported in BAFF‐Tg.TNF−/− mice [19]. In this study, 38% of the BAFF‐Tg.TNF−/− mice developed lymphoma compared to 5% usually described in the BAFF‐Tg mice [17, 18, 19]. Given that TNFi treatment would be less potent than TNF−/−, we have based the hypothesis on the incidence of 30% of lymphoma in BAFF‐Tg mice treated with TNFi compared to 5% in the control group. With a risk alpha of 5% and a power of 80%, a minimum of 12 mice were needed in each group. Mice were randomized into four treatment groups, as follows:
anti‐mouse TNF monoclonal antibody (clone TN3 19.12 (referred to as TN3) + MTX (n = 15)
humanized monoclonal anti‐TNF antibody, adalimumab (referred to as ADA) + MTX (n = 13)
human recombinant TNF‐R2‐Fc fragment, etanercept (referred to as ETA) + MTX (n = 15)
controls (n = 22), consisting of mice treated with water for injection + MTX (n = 8) and immunized mice treated with the three anti‐TNF without MTX leading to undetectable drug levels as soon as week 8 (n = 14)
An additional group of BAFF Tg mice was treated with ETA at high‐dose + MTX (referred to as ETA high dose). To prevent immunization against TNFi, MTX (5 mg/kg) was administered intraperitoneally within minutes before the first TNFi injection and repeated only twice at 24 and 4 h as previously described [22].
All the drugs were administrated by intraperitoneal (i.p.) injection. Details of the doses and frequency of administration of the drugs are presented in the Supporting information, Table S3.
TNFi drug monitoring and anti‐drug antibody detection
Blood samples were drawn every 4 weeks starting at week 8 of treatment. Serum drug concentration (ADA and ETA) and detection of anti‐drug antibodies were simultaneously measured using the Lisa Tracker® (Theradiag, Croissy‐Beaubourg, France) duo enzyme‐linked immunosorbent assay (ELISA), according to the manufacturer’s instructions [17]. An in‐house sandwich ELISA was used to measure serum TN3 drug concentration.
L929 cytotoxicity assay
TNF‐sensitive L929 cell line (Sigma, St Louis, MO, USA) was cultured in Dulbecco’s modified Eagle’s medium (DMEM) 2 mM + glutamine + 10% fetal bovine serum. Recombinant murine TNF (R&D Systems, Minneapolis, MN, USA) was used throughout this study. TNF bioactivity was assessed in vitro using an L929 bioassay, as previously described [23]. We employed a fixed concentration of TNF (20 ng/ml) that was incubated with increasing concentrations of ADA and ETA ranging from 1 to 500 µg/ml. For experiments with sera, we used TNF at 0.1 ng/ml and added 50 µl of sera of BAFF‐Tg mice treated with ADA, 50 µl of sera of BAFF‐Tg mice treated with TN3, 10 µl of sera of BAFF‐Tg mice treated with ETA and 50 µl of sera of untreated mice. Apoptosis of TNF‐exposed L929 cells was assessed by annexin V/7‐aminoactinomycin D (7AAD) staining.
Lymphoma assessment
Spleens were divided into three parts: one for flow cytometry analysis (single‐cell suspension), one for clonality assessment and one for histological assessment. As BAFF‐Tg mice are characterized by splenomegaly and increased in the B cell compartment, assessment of the presence of a B cell lymphoma versus B lymphoid hyperplasia is difficult. Thus, to detect lymphoma with high specificity in our mice, we used the three following parameters: (1) the presence of tumor‐like masses clinically assessed (lymphadenopathies, tumoral spleen); (2) histological examination of hematoxylin and eosin slides based on a specific composite score that we have developed (see Supporting information methods) in the absence of a pre‐existing one; and (3) assessment of the clonality (see Supporting information methods). Diagnosis of lymphoma was made when two of these three criteria were present.
Inducible nitric oxide synthase (iNOS) and macrophage receptor with collagenous structure (MARCO) quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from the spleen with the gene JET kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. The cDNA synthesis was performed using the enhanced avian first‐strand synthesis kit (Sigma). Quantification of mRNA expression was determined by Taqman real‐time PCR, according to the manufacturer’s instructions. Primers used were from Thermo Fisher Scientific [nitric oxide synthase 2 (Nos2) = INOS Mm 00440502_m1; MARCO = Mm 00440265_m1; glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) = Mm 99999915_g1]. Relative mRNA expression was determined from normalized Ct values with GAPDH as housekeeping gene and the 2‐ ΔCt method.
Statistical analysis
All analyses were performed using GraphPad Prism version 7.04 software. Continuous variables were summarized as mean ± standard deviation (s.d.). Non‐parametric tests were used to compare differences in continuous variables between groups (two groups: Mann–Whitney U–test; more than two groups: Kruskal–Wallis test followed by Dunn’s multiple comparison test). Categorical variables were presented as frequencies (%) and differences in proportions were analysed using Fisher’s exact tests. Crude mortality and progression‐free survival (PFS) without lymphoma were compared among the different groups using the log‐rank test.
RESULTS
BAFF Tg mice treated with monoclonal anti‐TNF have a reduced overall survival rate
Overall survival rate of the four groups of mice was analyzed during the 12 months of treatment with TNFi. The median (range) duration of survival was 221 (56–374) days in the TN3 group, 269 (10–374) days in the ADA group, 368 (119–374) days in the ETA group and 374 (212–374) days in the control group. We observed a significantly reduced survival rate for the BAFF‐Tg mice treated with the two monoclonal anti‐TNF antibodies compared to the control group (Figure 1). Conversely, survival of mice treated with ETA was not significantly different from controls. Of note, among the controls, no difference was observed between immunized mice without detectable drug levels and mice treated with water + MTX (Supporting information, Table S1). We aimed to determine the causes of this increased mortality.
FIGURE 1.
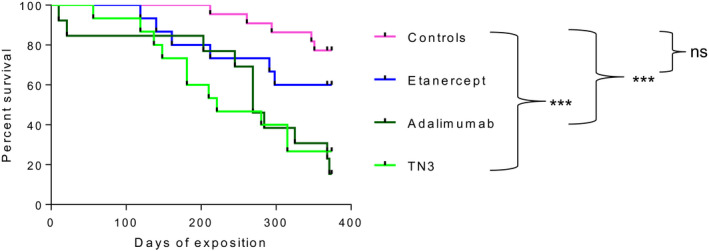
Survival rate of B cell activating factor belonging to the tumor necrosis factor (TNF) family‐transgenic (BAFF‐Tg) mice is significantly reduced in the two monoclonal anti‐TNF antibody groups compared to controls. Representation of the Kaplan–Meier curve of survival is shown. Mice were randomized into four groups of treatment: anti‐mouse TNF monoclonal antibody [clone TN3 19.12 (referred to as TN3) = MTX (n = 15] humanized monoclonal anti‐TNF antibody, adalimumab (referred to as ADA) = MTX (n = 13), human recombinant TNF‐R2‐Fc fragment, etanercept (referred to as ETA) = MTX (n = 15) and controls (n = 22). Survival is compared using log‐rank (Mantel–Cox) test
Immunogenicity and pharmacokinetics of TNFi in BAFF‐tg mice
First, we wanted to carefully check for pharmacokinetics of TNFi in our mice. B cell lymphomagenesis linked to activity of autoimmune diseases or to immunosuppression is thought to be a long‐term process. Thus, to test the impact of TNFi on the risk of lymphoma in BAFF‐Tg mice, we performed chronic exposure to the drugs for 1 year (half the life of mice). However, treatment of mice with TNFi, murine or humanized, leads to immunization occurring as soon as 2 weeks after starting treatment [24]. To prevent such immunization, we performed a short course of i.p. administration of MTX at a dose of 5 mg/kg at days 0, 1 and 2, as previously reported [22]. This led to a complete prevention of immunization and to the maintenance of significant drug concentration during the 12 months of the experiment, as we have previously published [22]. Taking advantage of this strategy, we obtained the following mean ± s.d. serum TNFi concentrations over 1 year: anti‐mouse TNF monoclonal antibody (clone TN3) referred as to TN3 (69 ± 50 µg/ml), ADA (105 ± 67 µg/ml) and ETA (7 ± 9 µg/ml). While serum concentrations of TNFi differed among these three groups, reaching a higher concentration of ADA was an objective in our study design, given its lowest capacity to inhibit murine TNF compared to ETA [25]. This observation was confirmed by performing a cytotoxicity assay using TNF‐sensitive L929 cells as targets. Inhibitory concentration leading to a 50% decrease in TNF‐induced L929 cell death was 300 µg/ml and 25 µg/ml for ADA and ETA, respectively (Figure 2a). To assess the bioactivity of the different anti‐TNF in vivo, we performed the L929 assay using sera from the mice treated by anti‐TNF [ETA, n = 6; ADA, n = 4; TN3, n = 8; or untreated mice as controls (n = 5)] and confirmed the bioactivity of the two types of TNF blockers used in the mice (Figure 2b).
FIGURE 2.
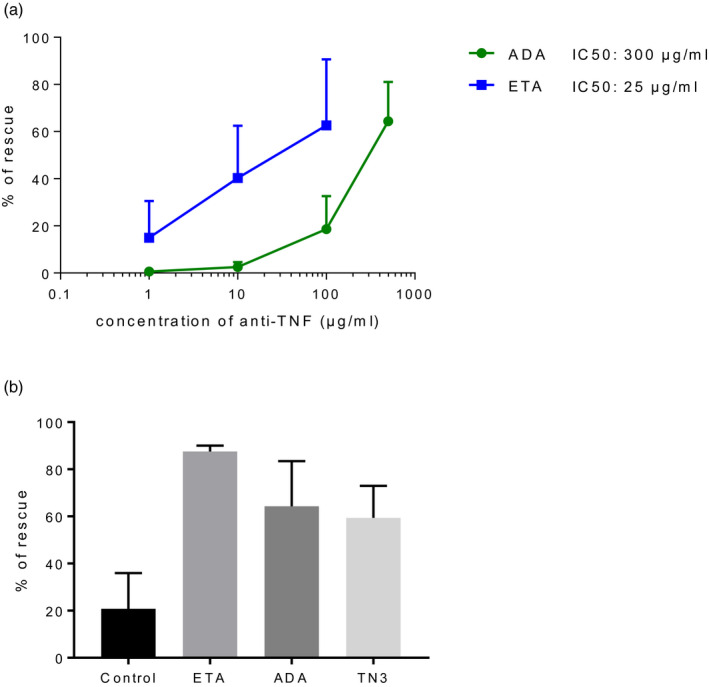
L929 tumor necrosis factor (TNF) cytotoxicity assay using adalimumab (ADA) and eternacept (ETA). (a) Comparison of the concentrations of ETA and ADA allowing rescue of L929 cell lines exposed to murine TNF. Results are mean and standard deviation (s.d.) of five experiments. (b) L929 assay using serum from untreated mouse (control, n = 5), sera from mice treated with ETA (n = 6), sera from mice treated with ADA (n = 4) and sera from mice treated with TN3 (n = 8) to inhibit TNF. Results are shown as mean and s.d
Exposure to the different types of TNFi does not impact manifestations of autoimmunity in BAFF Tg mice
We then assessed if signs of autoimmunity differed in the different groups of mice (ADA, ETA, controls) which thus might explain the difference in term of overall survival. We did not observe any difference between groups regarding the types of the B cells infiltrating the spleen, except for T1 and T2, that were increased in mice treated with ADA compared to controls and with no differences between the ETA and ADA groups (Figure 3a–d). There was no difference concerning the titer of autoantibodies (anti‐DNA and RF), the immunoglobulin (Ig)M and IgG serum level (Figure 3e–h), the renal involvement by studying the presence of glomerular (Figure 4a–c) and tubule‐interstitial involvement (Figure 4b,c). Lastly, as sialadenitis has been described in BAFF‐Tg mice, we also assessed submandibular salivary gland involvement in our mice. We confirmed the presence of sialadenitis with lymphocytic infiltrate of the serous gland and ductal atrophy (Figure 4d–f). Again, we did not detect differences in terms of severity among the different treatment groups (Figure 4d,e). Altogether, we were unable to detect any impact of the different TNFi treatments on the signs of autoimmunity in the BAFF‐Tg mice.
FIGURE 3.
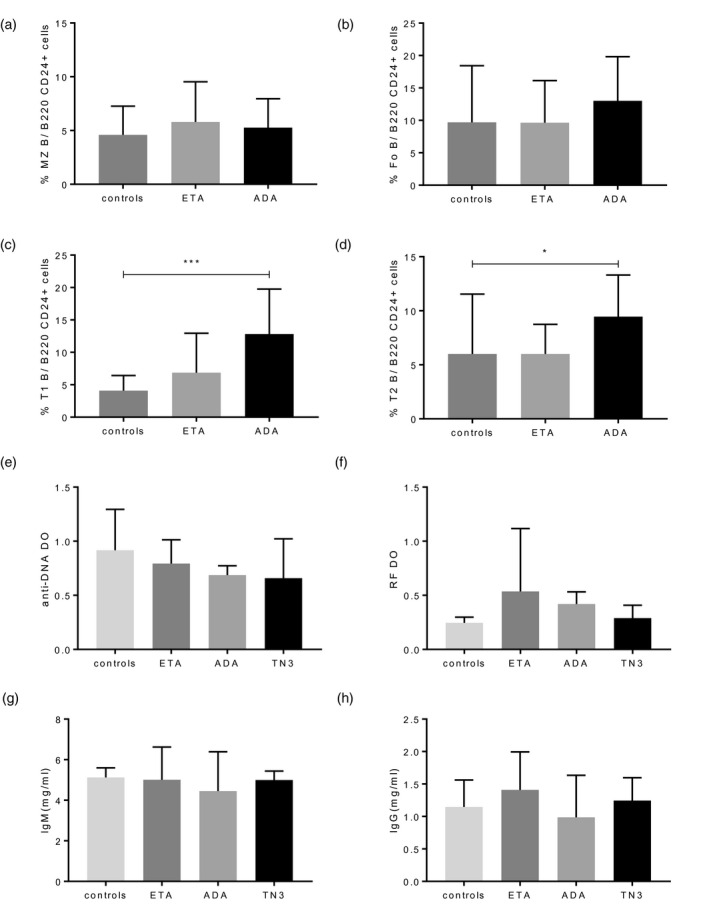
Assessment of autoimmunity in the tumor necrosis factor (TNF) family‐transgenic (BAFF‐Tg) mice treated with TNF inhibitor (TNFi). Assessment of the cellular composition of the spleen by flow cytometry performed at euthanasia in 10 mice treated with control, 14 treated with eternacept (ETA) and 11 mice treated with adalimumab (ADA) and focused on (a) marginal zone (MZ) B cells, (b) follicular (Fo) B cells, (c) T1 B cells and (d) T2 B cells. (e) Anti‐dsDNA antibodies, (f) rheumatoid factor (RF) titers and immunoglobulin (Ig)M (g) and IgG (h) concentrations measured in the sera of the mice at euthanasia (18 in the control group, 12 in the group treated with eternacept (ETA), six in the group treated with ADA and 12 in the group treated with TN3). Results are shown as mean and standard deviation (s.d.). Kruskal–Wallis test followed by Dunn’s multiple comparison test. *p < 0.05, **p < 0.01
FIGURE 4.
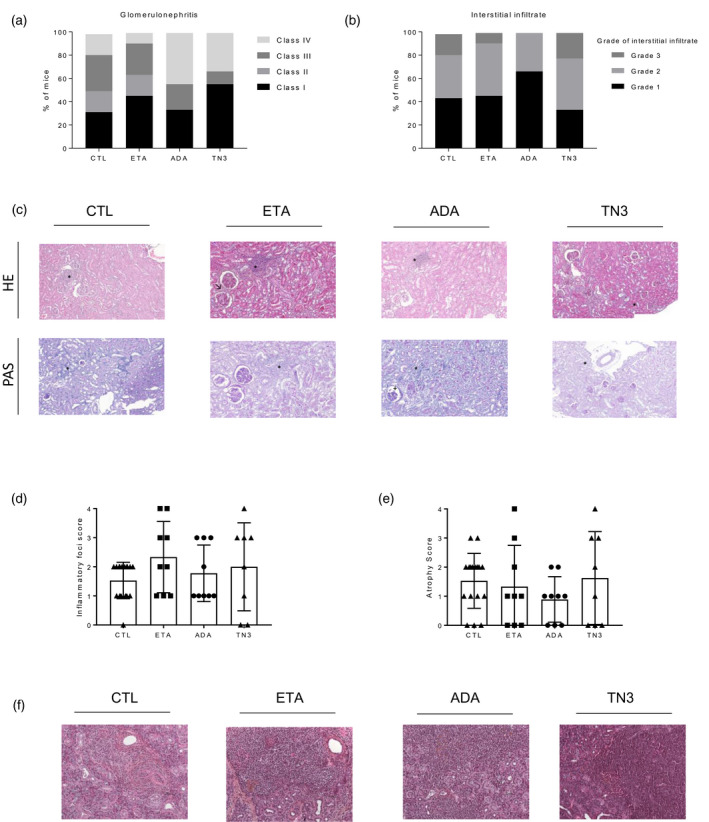
Assessment of renal and salivary glands involvement in tumor necrosis factor (TNF) family‐transgenic (BAFF‐Tg) mice treated with TNF inhibitor (TNFi). Assessment of renal involvement in 19 mice from the control group, 11 mice treated with eternacept (ETA), nine treated with adalimumab (ADA) and nine treated with TN3: proportion and severity of glomerular (a) and tubule‐interstitial (b) lesions in mice. (c) Representative kidney tissues [hematoxylin and eosin (H&E) staining upper panel, periodic acid‐Schiff (PAS) staining lower staining, original magnification ×20] in mice from the four groups. Kidneys amongs the four groups were characterized mild mesangial cell proliferation (↗), interstitial infiltration (*) with presence of plasma cells and rare presence of thrombi (+). Assessment of salivary gland involvement in 17 mice from the control group, nine mice treated with eternacept (ETA), nine treated with ADA and eight treated with TN3 with evaluation of the lymphocytic infiltrate (focus score, d) and ductal atrophy (e). (f) Representative submandibular salivary gland tissues (H&E staining, original magnification ×20) in mice from the four groups showing sialadenitis associated with ductal atrophy
Chronic exposure and high dose of monoclonal anti‐TNF antibodies are associated with an increased prevalence of lymphoma in BAFF Tg mice
Based on our stringent definition of lymphoma (see Methods: the presence of two criteria among macroscopic abnormality, histological composite score and assessment of B cell clonality), we observed a statistically significantly higher incidence of lymphoma in the BAFF‐Tg mice treated by monoclonal anti‐TNF antibodies compared to controls (Table 1). For more details concerning TNFi‐related B cell lymphomas, see Supporting information, Table S2. Interestingly, conversely to monoclonal anti‐TNF, etanercept is able to inhibit lymphotoxin‐α (LTα) that is known to participate in germinal center formation. Thus, we examined the size of the follicular compartment between the different groups of mice and did not detect a significant difference in this specific item of the histological composite score (Supporting information, Figure S1). This suggests that the difference in terms of lymphoma incidence was not linked to a protective effect of etanercept via inhibition of LT. As no mouse presented lymph node enlargement without splenomegaly, histological examination and assessment of B cell clonality were performed only on spleens. Assessment of survival rate without lymphoma confirmed the higher incidence of lymphoma in mice treated with monoclonal anti‐TNF antibodies (Figure 5).
TABLE 1.
Incidence of lymphoma in the four groups of mice
| Mice (–) | Lymphoma (n, %) | |
|---|---|---|
| TN3 | 15 | 5 (33) |
| ETA | 15 | 0 (0) |
| ADA | 12 | 4 (33) |
| Controls | 22 | 1 (5) |
Frequencies (percentage) are presented.
Abbreviations: ADA, adalimumab; ETA, etanercept; TN3, anti‐mouse tumor necrosis factor monoclonal antibody.
FIGURE 5.
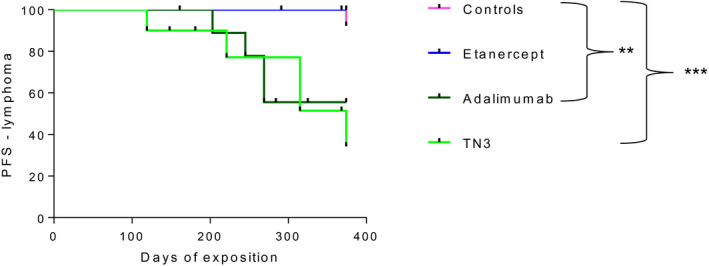
Survival rate without lymphoma of tumor necrosis factor (TNF) family‐transgenic (BAFF‐Tg) mice treated with TNF inhibitor (TNFi). Representation of the Kaplan–Meier curve of survival. Mice were randomized into four groups of treatment: anti‐mouse TNF monoclonal antibody [clone TN3 19.12 (referred to as TN3) = MTX (n = 15], humanized monoclonal anti‐TNF antibody, adalimumab [referred to as ADA = MTX (n = 13)], human recombinant TNF‐R2‐Fc fragment, etanercept [referred to as ETA) = MTX (n = 15] and controls (n = 22). Survival rate is compared using log‐rank (Mantel–Cox) test
Notwithstanding that our objective was to reach a 10‐fold higher concentration of ADA than ETA (because of the substantial lower efficacy of ADA than ETA on murine TNF), we decided to test if treatment with higher concentration of ETA might modify the occurrence of lymphoma. This new group of mice was treated for 12 months at a concentration of 40 mg/kg (Supporting information, Table S1) three times a week. The obtained mean concentration of ETA ± s.d. was 84.5 M 67 µg/ml. Only one lymphoma was observed among the 11 mice treated with a high ETA dosage, leading to a similar low incidence of lymphoma (9%) observed in control mice. Thus, the difference in drug concentrations did not explain the observed difference in incidence of lymphoma in these mice.
Chronic exposure to monoclonal anti‐TNF antibodies is associated with a decreased macrophage infiltration
To gain more insight into the mechanisms associate with different lymphomagenesis incidence associated with TNFi administration in mice, we explored the effect of the tested TNFi on the local microenvironment surrounding lymphoma. We particularly focused our attention upon three cellular subsets known to be strongly involved in lymphoma immunosurveillance and which express membrane TNF, i.e. natural killer cells (NK), forkhead box protein 3 (FoxP3+) regulatory T cells (Treg) and macrophages. Unexpectedly, we did not detect any significant splenic NK cell infiltration in these mice (data not shown). In addition, we did not detect any significant difference between splenic pool of Treg between groups (Figure 6a). Conversely, flow cytometry analysis using an exclusion panel (Supporting information, Figure S3) revealed that splenic macrophage infiltration was significantly reduced in the ADA group compared to ETA (Figure 6b). The decrease in macrophage infiltration in the ADA group compared to ETA was confirmed in immunohistochemistry (IHC) using CD68 staining (Figure 6c–e). We aimed to further assess the origin of this observed decrease splenic macrophage infiltration in mice treated with ADA. We did not detect an impact of in‐vivo exposure to TNFi on the capacity of migration of monocytes in response to monocyte chemoattractant protein‐1/chemokine ligand 2 (MCP‐1/CCL2) stimulation (Supporting information, Figure S2).
FIGURE 6.
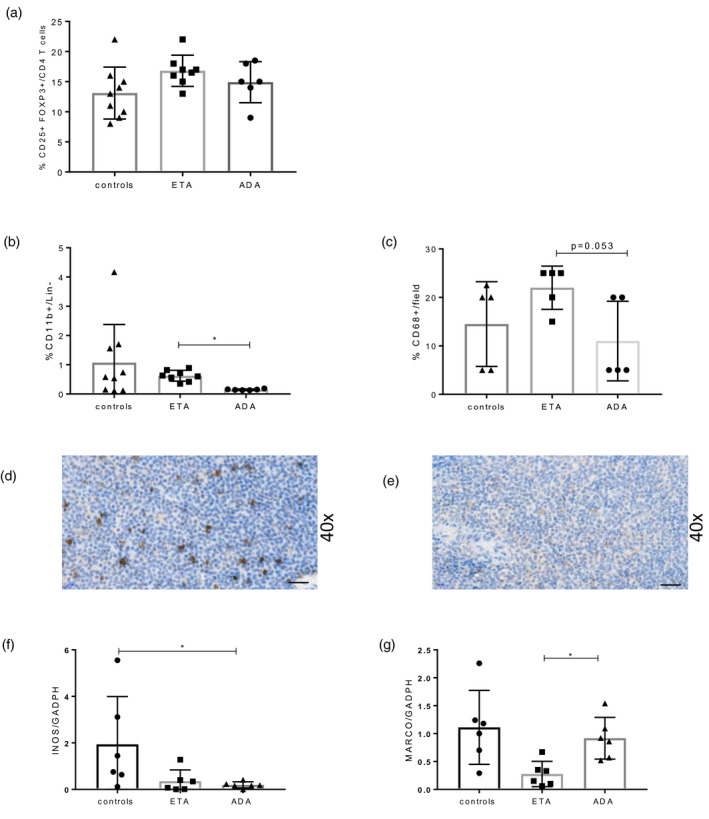
Assessment of cellular infiltrate involved in lymphoma immunosurveillance in tumor necrosis factor (TNF) family‐transgenic (BAFF‐Tg) mice treated with TNF inhibitor (TNFi). Flow cytometry analysis was performed on spleen of BAFF‐Tg mice treated by control (n = 9), eternacept (ETA) (n = 8) or adalimumab (ADA) (n = 6). (a) Splenic regulatory T cells (Treg) and (b) macrophages were assessed. (c) Macrophage infiltrate was also assessed by CD68 staining in immunohistochemistry (IHC) on spleen sections (n = 5 for each group). Panels (d) and (e) are representative of CD68 staining in IHC, with ×40 magnification in mice treated with (d) ETA and (e) ADA. Inducible nitric oxide synthase (INOS) (f) and MARCO (g) in ADA (n = 6), ETA (n = 6) or control immunoglobulin (Ig) mice (n = 6) quantified by quantitative polymerase chain reaction (qPCR). Results are shown as mean and standard deviation (s.d.). Kruskal–Wallis test followed by Dunn’s multiple comparison test. *p < 0.05, **p < 0.01
Finally, we investigated the nature of the macrophages infiltrating the spleen. We measured INOS in as an M1‐like marker and MARCO as a M2‐like marker [26] by qPCR. Our results showed that M1‐like markers were more decreased with ADA than with ETA and that M2‐like markers were spared by ADA but not by ETA (Figure 6f,g).
DISCUSSION
In this study, we showed that long‐term exposure to monoclonal anti‐TNF antibodies but not to the soluble TNF‐R2‐Ig in BAFF‐Tg mice, a mouse model of B cell autoimmunity, were predisposed to the development of lymphoma. We found that this higher risk of lymphoma in mice treated with monoclonal anti‐TNF antibodies compared with ETA could be linked in part to a decrease in macrophage infiltration of lymphoid organ that might be involved in lymphoma immunosurveillance.
Difference in terms of survival between BAFF‐Tg mice treated or not by TNFi could have been linked to a negative impact of TNFi on autoimmunity. In fact, TNFi have been shown to be inefficient in the two major autoimmune diseases driven by BAFF, i.e. SLE [27] and SS [28]. Moreover, several studies support an aberrant immune‐regulatory effect of TNF in SLE [29]. The interferon (IFN) signature is well established in SLE and SS, and cross‐regulation between TNF and IFN type I has been described. It has been demonstrated that TNF regulates generation of plasmacytoid dendritic cells (pDC), the main producers of IFN‐α and IFN‐α release by pDC [30]. In patients, the occurrence of autoantibodies to nuclear antigens as well as occasional, transient lupus‐like syndromes in patients exposed to TNFi could suggest a protective role of TNF in SLE [31]. However, this hypothesis remains controversial. Some reports, including open‐label studies, have suggested that TNFi might be efficient in some SLE patients [32]. In the present study, we did not observe any impact of TNFi treatment on manifestations of autoimmunity confirming this balanced effect, and further suggest that the observed increased mortality in mice treated by TNFi was not linked to flare of the autoimmune disease.
We observed an increased incidence of lymphoma in BAFF‐Tg mice treated by monoclonal anti‐TNF antibodies that could explain much of the difference in terms of mortality. However, other causes of death not linked to autoimmunity or to lymphoma in some animals cannot be formally excluded. We paid great attention to the definition of lymphoma given the basal Bcell hyperactivity observed in BAFF‐Tg mice [17]. Batten et al. defined the presence of lymphoproliferation by macroscopic abnormalities and histological examination [19]. In our study, we also added assessment of B cell clonality to be more stringent. However, given the strict definition of lymphoma that we had (two of three items among macroscopic tumor, histological score and clonality), lymphoma incidence might have been underestimated. Nevertheless, the percentage of lymphoma observed in these experiments was in line with previously described rates in both BAFF‐Tg and BAFF‐Tg.TNF−/− mice [19].
As mentioned above our data suggest that, in the context of chronic autoimmune B cell stimulation, long‐term inhibition of TNF by monoclonal anti‐TNF antibodies could increase the risk of lymphoma. In this study, we used two monoclonal anti‐TNF: TN3, which is the reference anti‐TNF in mice recognizing mouse TNF and, due to supply difficulties with TN3, ADA, an anti‐human TNF that has the advantage of being used in human clinics. However, a limitation of using ADA is its lower affinity to mouse TNF than to human TNF. To overcome this difficulty, we adapted the concentration of ADA to induce the same apoptosis rate of the TNF‐sensitive L929 cell line than with ETA that recognizes mouse TNF. We could determine that a concentration of ADA approximately 10 times higher than that of ETA induced the same biological effect on the TNF‐sensitive L929 cell line (Figure 1).
Even if the impact of inhibition of TNF on the risk of lymphoma has already been demonstrated by the study of Batten et al. [19], this previous model was extreme, as these mice were totally TNF‐deficient. Here, we demonstrate that inhibition by therapeutic antibodies targeting TNF could have the same effect. Two parameters need to be considered to interpret the data. First, mice were treated for 12 months, which represents the half‐life of the mice, hence a very long‐term exposure that has no parallel in humans. Secondly, as indicated above, the mean blood concentration of ADA was very high (105 µg/ml), while that of ETA was 7 µg/ml in order to have the same anti‐TNF effect in mice with the two drugs (see above). Of note, higher doses of ETA did not increase the risk of lymphoma, and thus may suggest that the difference of effect between drugs could be due to a different mechanism of action, possibly on membrane TNF.
If the differential risk of lymphoma between TNFi drugs is not associated with their dose, it could be due to a different mechanism of action between TNFi [33]. From our results, one could speculate the presence of (1) a deleterious impact of monoclonal anti‐TNF antibodies on lymphoma immunosurveillance; and/or (2) a protective role of ETA. This second hypothesis could be linked to the inhibition of LTα by ETA but not by monoclonal anti‐TNF antibodies [34]. It is well demonstrated that, in mice, LTα is critical for the development and maintenance of splenic and lymph node microarchitecture [35]. In addition, LTα participates in the formation of germinal centers (GC) [36] and to generation of memory B cells [37]. Lastly, it has been demonstrated that LTα participates in the establishment of a permissive niche for lymphoma [38]. However, in our study we did not observe a difference in term of size of the follicular compartment between the three groups of treatments, suggesting that ETA did not inhibit GC formation in these BAFF Tg mice. In addition, treatment with ETA did not decrease the incidence of lymphoma compared to the control group, even at a higher dose.
Immune surveillance plays an important role in B cell lymphomagenesis, as demonstrated by the development of immune checkpoint inhibitors in the treatment of Hodgkin lymphoma and NHL [39, 40]. Clearly, several cellular actors of immune surveillance might be affected by TNFi. First, we previously found that NK cells were negatively impacted by TNFi, but with no difference between ETA and monoclonal anti‐TNF antibodies [41]. In the current study, we did not detect any significant NK cell infiltrate within the spleen of BAFF‐Tg mice. Secondly, Treg might be involved. The impact of Treg in the microenvironment of lymphoma is controversial [42]. Some studies suggest that the presence of Treg within the microenvironment could be associated with better prognosis [43]. Conversely, other studies suggest that Treg, through a strong immunosuppressive effect on anti‐tumor response, are a major deleterious factor for tumor control [44]. Interestingly, recent report demonstrated that ADA but not ETA was able to induce expansion of Treg through the membrane TNF–TNF‐R2 pathway [45]. However, in our study we did not detect any difference in splenic Treg infiltration in mice treated with ADA, ETA or controls. Thirdly, the role of macrophages in anti‐lymphoma immunosurveillance is now strongly established [46]. Very recently, the development of an anti‐CD47 immunotherapy aiming to reverse inhibition of macrophages has shown very promising results in NHL [47]. This highlights the role of macrophages in controlling lymphoma. In this study, we obtained data suggesting that exposure to ADA may alter macrophage infiltration with data suggesting that M1‐like were more decreased with ADA than with ETA and that ADA spared M2‐like conversely to ETA. This could be deleterious in the context of cancer control. Interestingly, it has been demonstrated that adalimumab could promote M2‐like macrophages while ETA could not. The mechanism leading to this effect remains unclear, and could involve interaction between the anti‐TNF Fc region and Fcγ receptors [48]. An acknowledged difference in the mode of action between monoclonal anti‐TNF antibodies and ETA is a better stability of monoclonal anti‐TNF/membrane TNF complexes, possibly leading to reverse signaling and modulating macrophages [49]. It may explain the differential effect in IBD patients and the differential risk of latent tuberculosis reactivation observed with different TNFi. Moreover, our group has demonstrated the presence of a specific defect in RA of differentiation of monocytes into CD206+ M2‐like macrophages that can be corrected by ADA and not by ETA [50]. As well as macrophages, other actors of immunosurveillance including cytotoxic T CD8+ could be modulated by anti‐TNF and may also play a role.
Collectively, our study demonstrates for the first time, to our knowledge, that that very long‐term inhibition of TNF, especially with monoclonal anti‐TNF antibody, in the context of chronic autoimmune B cell stimulation increases the risk of lymphoma in mice. This increase in lymphoma incidence might be driven by a specific mechanism of action of monoclonal anti‐TNF antibodies for decreasing immune surveillance by macrophages both quantitatively and qualitatively. Further research aiming to more clearly understand how TNFi impair the phenotype and function of macrophages involved in immune surveillance is needed. These data suggest the need for a closer monitoring of lymphoma occurrence in patients with B cell‐driven autoimmune disease with long‐term exposure to monoclonal anti‐TNF. These data also underline the need to decrease the dose of TNFi when the target of treatment is achieved (i.e. remission or low disease activity), as advised in the EULAR recommendations for treatment of RA patients [51].
CONFLICT OF INTERESTCONFLICT OF INTEREST
G. N. received honorarium from Novartis, Lilly and UCB. R. S. received honoraria from Pfizer, BMS and UCB. X. M. received honoraria from BMS, Gilead, Pfizer and UCB.
ETHICS STATEMENT
This study was approved under number CEEA34.CN.089.12 by the Regional Animal Care and Ethics Committee. Animal care and use was in accordance with the EU Directive 2010/63/EEU. The institute was approved as compliant with ETS123 recommendations for animal breeding and with Standards for Human Care and Use of Laboratory Animals.
AUTHOR CONTRIBUTIONS
G. N., B. L., A. P., J. P. and C. N., performed experiments; G. N., R. S., F. M., F. V., T. L., S. F, L. S, S. R., R. K., S. H. B and X. M. analysed results; G. N. and X. M. designed the research and wrote the paper.
Supporting information
Figure S1
Figure S2
Figure S3
Supplementary Material
ACKNOWLEDGEMENTS
Funding was obtained from the French Ministry of Research: ANR‐10‐LABX‐LERMIT, from Société Française de Rhumatologie and Fondation pour la Recherche Médicale DEQ20150934719: Sjögren’s syndrome and Autoimmunity‐associated Lymphomas (SAIL). An unrestricted grant was also obtained from Pfizer. The authors thank Christiane Chéreau, Marine, Stephane Bloquet and Axel Perrot for their help in mouse housing and Katia Posseme and Nathalie Ba for their support for tissue processing for histopathology. The authors also thank Christopher Mueller for his help and advice concerning macrophages experiments and Manon Zala for her help in the V(D)J rearrangement analysis.
DATA AVAILABILITY STATEMENT
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Smedby KE, Hjalgrim H, Askling J, Chang ET, Gregersen H, Porwit‐MacDonald A, et al. Autoimmune and chronic inflammatory disorders and risk of non‐Hodgkin lymphoma by subtype. J Natl Cancer Inst. 2006;98:51–60. [DOI] [PubMed] [Google Scholar]
- 2. Baecklund E, Iliadou A, Askling J, Ekbom A, Backlin C, Granath F, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54:692–701. [DOI] [PubMed] [Google Scholar]
- 3. Nocturne G, Virone A, Ng W‐F, Le Guern V, Hachulla E, Cornec D, et al. Rheumatoid factor and disease activity are independent predictors of lymphoma in primary Sjögren’s syndrome. Arthritis Rheumatol. 2016;68:977–85. [DOI] [PubMed] [Google Scholar]
- 4. Dierickx D, Habermann TM. Post‐transplantation lymphoproliferative disorders in adults. N Engl J Med. 2018;378(6):549–62. [DOI] [PubMed] [Google Scholar]
- 5. Beaugerie L, Brousse N, Bouvier AM, Colombel JF, Lémann M, Cosnes J, et al. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet. 2009;374(9701):1617–25. [DOI] [PubMed] [Google Scholar]
- 6. Bernatsky S, Ramsey‐Goldman R, Joseph L, Boivin J‐F, Costenbader KH, Urowitz MB, et al. Lymphoma risk in systemic lupus: effects of disease activity versus treatment. Ann Rheum Dis. 2014;73:138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mercer LK, Regierer AC, Mariette X, Dixon WG, Baecklund E, Hellgren K, et al. Spectrum of lymphomas across different drug treatment groups in rheumatoid arthritis: a European registries collaborative project. Ann Rheum Dis. 2017;76:2025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mariette X, Matucci‐Cerinic M, Pavelka K, Taylor P, van Vollenhoven R, Heatley R, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta‐analysis. Ann Rheum Dis. 2011;70:1895–904. [DOI] [PubMed] [Google Scholar]
- 9. Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, et al. Etanercept for active Crohn’s disease: a randomized, double‐blind, placebo‐controlled trial. Gastroenterology. 2001;121:1088–94. [DOI] [PubMed] [Google Scholar]
- 10. Guignard S, Gossec L, Salliot C, Ruyssen‐Witrand A, Luc M, Duclos M, et al. Efficacy of tumour necrosis factor blockers in reducing uveitis flares in patients with spondylarthropathy: a retrospective study. Ann Rheum Dis. 2006;65:1631–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zweegers J, Groenewoud J, van den Reek J, Otero ME, van de Kerkhof P, Driessen R, et al. Comparison of the 1‐ and 5‐year effectiveness of adalimumab, etanercept and ustekinumab in patients with psoriasis in daily clinical practice: results from the prospective BioCAPTURE registry. Br J Dermatol. 2017;176:1001–9. [DOI] [PubMed] [Google Scholar]
- 12. Salmon‐Ceron D, Tubach F, Lortholary O, Chosidow O, Bretagne S, Nicolas N, et al. Drug‐specific risk of non‐tuberculosis opportunistic infections in patients receiving anti‐TNF therapy reported to the 3‐year prospective French RATIO registry. Ann Rheum Dis. 2011;70:616–23. [DOI] [PubMed] [Google Scholar]
- 13. Mariette X, Tubach F, Bagheri H, Bardet M, Berthelot JM, Gaudin P, et al. Lymphoma in patients treated with anti‐TNF: results of the 3‐year prospective French RATIO registry. Ann Rheum Dis. 2010;69:400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harigai M, Nanki T, Koike R, Tanaka M, Watanabe‐Imai K, Komano Y, et al. Risk for malignancy in rheumatoid arthritis patients treated with biological disease‐modifying antirheumatic drugs compared to the general population: a nationwide cohort study in Japan. Mod Rheumatol. 2016;26:642–50. [DOI] [PubMed] [Google Scholar]
- 15. Mercer LK, Galloway JB, Lunt M, Davies R, Low ALS, Dixon WG, et al. Risk of lymphoma in patients exposed to antitumour necrosis factor therapy: results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Ann Rheum Dis. 2017;76:497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lemaitre M, Kirchgesner J, Rudnichi A, Carrat F, Zureik M, Carbonnel F, et al. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. JAMA. 2017;318:1679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190 :697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B‐cell autoimmune disease. Nature 2000;404(6781):995–9. [DOI] [PubMed] [Google Scholar]
- 19. Batten M, Fletcher C, Ng LG, Groom J, Wheway J, Laâbi Y, et al. TNF deficiency fails to protect BAFF transgenic mice against autoimmunity and reveals a predisposition to B cell lymphoma. J Immunol. 2004;172(2):812–22. [DOI] [PubMed] [Google Scholar]
- 20. Khare SD, Sarosi I, Xia XZ, McCabe S, Miner K, Solovyev I, et al. Severe B cell hyperplasia and autoimmune disease in TALL‐1 transgenic mice. Proc Natl Acad Sci USA. 2000;97(7):3370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ray MA, Johnston NA, Verhulst S, Trammell RA, Toth LA. Identification of markers for imminent death in mice used in longevity and aging research. J Am Assoc Lab Anim Sci. 2010;49:282–8. [PMC free article] [PubMed] [Google Scholar]
- 22. Bitoun S, Nocturne G, Ly B, Krzysiek R, Roques P, Pruvost A, et al. Methotrexate and BAFF interaction prevents immunization against TNF inhibitors. Ann Rheum Dis. 2018;77:1463–70. [DOI] [PubMed] [Google Scholar]
- 23. Shiau MY, Chiou HL, Lee YL, Kuo TM, Chang YH. Establishment of a consistent L929 bioassay system for TNF‐alpha quantitation to evaluate the effect of lipopolysaccharide, phytomitogens and cytodifferentiation agents on cytotoxicity of TNF‐alpha secreted by adherent human mononuclear cells. Mediat Inflamm. 2001;10:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams RO, Mason LJ, Feldmann M, Maini RN. Synergy between anti‐CD4 and anti‐tumor necrosis factor in the amelioration of established collagen‐induced arthritis. Proc Natl Acad Sci USA. 1994;91:2762–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. humira‐epar‐scientific‐discussion_en.pdf [Internet]. [cité 3 juill 2019]. Disponible sur: https://www.ema.europa.eu/en/documents/scientific‐discussion/humira‐epar‐scientific‐discussion_en.pdf
- 26. Georgoudaki A‐M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor‐associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15:2000–11. [DOI] [PubMed] [Google Scholar]
- 27. Aringer M, Houssiau F, Gordon C, Graninger WB, Voll RE, Rath E, et al. Adverse events and efficacy of TNF‐alpha blockade with infliximab in patients with systemic lupus erythematosus: long‐term follow‐up of 13 patients. Rheumatology 2009;48:1451–4. [DOI] [PubMed] [Google Scholar]
- 28. Nocturne G, Cornec D, Seror R, Mariette X. New biological therapies in Sjögren’s syndrome. Best Pract Res Clin Rheumatol. 2015;29:783–93. [DOI] [PubMed] [Google Scholar]
- 29. Zhu L‐J, Yang X, Yu X‐Q. Anti‐TNF‐α therapies in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:465898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palucka AK, Blanck J‐P, Bennett L, Pascual V, Banchereau J. Cross‐regulation of TNF and IFN‐α in autoimmune diseases. Proc Natl Acad Sci USA. 2005;102(9):3372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aringer M, Smolen JS. Therapeutic blockade of TNF in patients with SLE‐promising or crazy? Autoimmun Rev. 2012;11:321–5. [DOI] [PubMed] [Google Scholar]
- 32. Aringer M, Graninger WB, Steiner G, Smolen JS. Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open‐label study. Arthritis Rheum. 2004;50:3161–9. [DOI] [PubMed] [Google Scholar]
- 33. Smolen JS, Kay J, Doyle M, Landewé R, Matteson EL, Gaylis N, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor α inhibitors: findings with up to five years of treatment in the multicenter, randomized, double‐blind, placebo‐controlled, phase 3 GO‐AFTER study. Arthritis Res Ther. 2015;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–79. [DOI] [PubMed] [Google Scholar]
- 35. Fu YX, Huang G, Wang Y, Chaplin DD. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin alpha‐dependent fashion. J Exp Med. 1998;187(7):1009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsumoto M, Fu YX, Molina H, Chaplin DD. Lymphotoxin‐alpha‐deficient and TNF receptor‐I‐deficient mice define developmental and functional characteristics of germinal centers. Immunol Rev. 1997;156:137–44. [DOI] [PubMed] [Google Scholar]
- 37. Fu YX, Huang G, Wang Y, Chaplin DD. Lymphotoxin‐alpha‐dependent spleen microenvironment supports the generation of memory B cells and is required for their subsequent antigen‐induced activation. J Immunol. 2000;164(5):2508–14. [DOI] [PubMed] [Google Scholar]
- 38. Rehm A, Mensen A, Schradi K, Gerlach K, Wittstock S, Winter S, et al. Cooperative function of CCR7 and lymphotoxin in the formation of a lymphoma‐permissive niche within murine secondary lymphoid organs. Blood. 2011;118(4):1020–33. [DOI] [PubMed] [Google Scholar]
- 39. Witkowska M, Smolewski P. Immune checkpoint inhibitors to treat malignant lymphomas. J Immunol Res. 2018. 10.3389/fonc.2018.00288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar D, Xu ML. Microenvironment cell contribution to lymphoma immunity. Front Oncol. 2018;8:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nocturne G, Boudaoud S, Ly B, Pascaud J, Paoletti A, Mariette X. Impact of anti‐TNF therapy on NK cells function and on immunosurveillance against B‐cell lymphomas. J Autoimmun. 2017;80:56–64. [DOI] [PubMed] [Google Scholar]
- 42. Wang J, Ke X‐Y. The four types of Tregs in malignant lymphomas. J Hematol Oncol. 2011;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tzankov A, Meier C, Hirschmann P, Went P, Pileri SA, Dirnhofer S. Correlation of high numbers of intratumoral FOXP3+ regulatory T cells with improved survival in germinal center‐like diffuse large B‐cell lymphoma, follicular lymphoma and classical Hodgkin’s lymphoma. Haematologica 2008;93:193–200. [DOI] [PubMed] [Google Scholar]
- 44. Yang Z‐Z, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Attenuation of CD8(+) T‐cell function by CD4(+)CD25(+) regulatory T cells in B‐cell non‐Hodgkin’s lymphoma. Cancer Res. 2006;66(20):10145‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nguyen DX, Ehrenstein MR. Anti‐TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF‐TNF‐RII binding in rheumatoid arthritis. J Exp Med. 2016;213(7):1241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pham LV, Pogue E, Ford RJ. The role of macrophage/B‐cell interactions in the pathophysiology of B‐cell lymphomas. Front Oncol. 2018;8:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by Hu5F9‐G4 and rituximab in non‐Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bloemendaal FM, Levin AD, Wildenberg ME, Koelink PJ, McRae BL, Salfeld J, et al. Anti‐tumor necrosis factor with a glyco‐engineered Fc‐region has increased efficacy in mice with colitis. Gastroenterology 2017;153:1351–62.e4. [DOI] [PubMed] [Google Scholar]
- 49. Scallon B, Cai A, Solowski N, Rosenberg A, Song X‐Y, Shealy D, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–26. [DOI] [PubMed] [Google Scholar]
- 50. Paoletti A, Rohmer J, Ly B, Pascaud J, Rivière E, Seror R, et al. Monocyte/macrophage abnormalities specific to rheumatoid arthritis are linked to miR‐155 and are differentially modulated by different TNF inhibitors. J Immunol. 2019;203(7):1766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Supplementary Material
Data Availability Statement
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.