

Abstract
CD4+ regulatory T (Treg) cells, dependent upon the transcription factor Foxp3, contribute to tumour immunosuppression but are also required for immune homeostasis. There is interest in developing therapies that selectively target the immunosuppressive function of Treg cells within tumours without disrupting their systemic anti‐inflammatory function. High levels of expression of chemokine (C‐C motif) receptor 8 (CCR8) discriminate Treg cells within tumours from those found in systemic lymphoid tissues. It has recently been proposed that disruption of CCR8 function using blocking anti‐CCR8 antibodies results in reduced accumulation of Treg cells within tumours and disruption of their immunosuppressive function. Here, using Ccr8 −/− mice, we show that CCR8 function is not required for Treg cell accumulation or immunosuppression in the context of syngeneic MC38 colorectal adenocarcinoma and B16 melanoma tumours. We observed high levels of CCR8 expression on tumour‐infiltrating Treg cells which were abolished in Ccr8 −/− mice. High levels of CCR8 marked cells with high levels of suppressive function. However, whereas systemic ablation of Treg cells resulted in strikingly diminished tumour burden, growth of subcutaneously implanted tumours was unaffected by systemic CCR8 loss. Consistently, we observed minimal impact of systemic CCR8 ablation on the frequency, phenotype and function of tumour‐infiltrating Treg cells and conventional T (Tconv) function. These findings suggest that CCR8 is not required for Treg cell accumulation and immunosuppressive function within tumours and that depletion of CCR8+ Treg cells rather than blockade of CCR8 function is a more promising avenue for selective immunotherapy.
Keywords: Treg, CCR8, Foxp3, CD4+ T cells, CD8+ T cells, cancer, immunotherapy
There is interest in developing therapies that selectively target the immunosuppressive function of Treg cells within tumours without disrupting their systemic anti‐inflammatory function. It has recently been proposed that disruption of CCR8 function using blocking anti‐CCR8 antibodies results in reduced accumulation of Treg cells within tumours and disruption of their immunosuppressive function.We show that CCR8 marks highly suppressive Treg cells within tumours but is not required for Treg cell accumulation and immunosuppressive function within tumours, suggesting that depletion of CCR8+ Treg cells rather than blockade of CCR8 function is a more promising avenue for selective immunotherapy.

Abbreviations
- ADCC
antibody‐dependent cellular cytotoxicity
- APCs
antigen‐presenting cells
- Areg
amphiregulin
- C‐C motif
chemokine
- CCR8
receptor 8
- CTV
CellTrace Violet™
- DTR
diphtheria toxin receptor
- DTx
diphtheria toxin
- EGFP
enhanced green fluorescent protein
- NK
natural killer
- (PPAR)‐γ
peroxisome proliferator‐activated receptor
- Tconv
conventional T
- Th
T helper
- Treg
regulatory T
- WT
wild‐type
INTRODUCTION
Tumours grow in immunocompetent hosts despite the ability of cells of the adaptive immune system to recognise and kill cancer cells. In part, this phenomenon is attributable to the process of immunosuppression. Tumour immunosuppression is dependent upon a number of peripheral tolerance mechanisms normally employed to prevent unwanted inflammation and autoimmune responses. Therapeutic approaches aimed at disrupting tumour immunosuppression would ideally do so without disturbing systemic peripheral tolerance. CD4+ regulatory T (Treg) cells are a suppressive T‐cell subset required to prevent autoimmune and allergic inflammation [1, 2, 3]. Treg cells are often found at high relative frequencies within tumours where they limit immune‐mediated rejection of disease [4, 5, 6]. Consistent with their suppressive function, low Treg to conventional T (Tconv) cell ratios are associated with favourable survival in several types of cancer including ovarian cancer [7, 8], breast cancer [9], non‐small‐cell lung cancer [10], hepatocellular carcinoma [11], renal cell cancer [12], pancreatic cancer [13], gastric cancer [14], cervical cancer [15] and colorectal cancer [16]. Thus, Treg cells diminish both autoimmune and allergic inflammation, but also hinder effective immune responses against tumours. There is a need to develop therapies that selectively target the immunosuppressive function of Treg cells within tumours without disrupting their systemic anti‐inflammatory function.
Recent evidence suggests that Treg cells within different tissues exhibit distinct molecular profiles and functional characteristics. For instance, adipose tissue Treg cells, which express the transcription factor peroxisome proliferator‐activated receptor (PPAR)‐γ, are critical regulators of tissue metabolism and insulin sensitivity [17, 18]. Treg cells in skeletal muscle expressing IL‐33 contribute to muscle repair through expression of molecules such as the epidermal growth factor receptor ligand amphiregulin (AREG) [19, 20]. Recent studies have now shown that Treg cells infiltrating a variety of human tumours, including breast, colorectal and non‐small‐cell lung cancers, exhibit an altered transcriptional profile [21, 22]. These studies showed that Treg cells within tumours are highly activated when compared to systemic and/or normal tissue Treg cells. Among the identified differences was highly increased expression of the chemokine (C‐C motif) receptor 8 (Ccr8) gene, encoding CCR8, within tumour‐infiltrating Treg cells as compared with Treg cells found in other tissues.
Chemokines are small (∼8–14 kDa) secreted proteins, structurally similar to cytokines, which regulate cell signalling and trafficking through interactions with a subset of seven transmembrane G protein‐coupled receptors called chemokine receptors [23]. The chemokine receptor CCR8 is a receptor for CCL1 (in humans and mice) and CCL18 (in humans) [24, 25]. CCR8 is expressed on Treg cells, a subset of type helper (Th)‐2 cells, monocytic cells and natural killer (NK) cells [24, 26, 27, 28, 29]. CCR8 has been proposed to play a role in allergic inflammation through loss‐of‐function studies in mice [28], although the extent of its involvement is unclear, with contradictory results in the literature [30, 31]. CCR8 signalling is thought to contribute to Treg cell suppressive function and has been found to promote donor Treg cell survival in a murine model of graft‐versus‐host disease [32]. Moreover, CCR8 signalling by CCL1 has been proposed to potentiate Treg cell proliferation and suppressive function in the context of inflammation of the central nervous system [33].
A recent study has suggested that CCR8 function is required for Treg cell‐mediated tumour immunosuppression [34]. Antibodies with proposed blocking activity were shown to reduce Treg cell accumulation within tumours, drive Tconv cell activation and reduce tumour growth. In addition to blockade, antibodies can induce cell depletion through antibody‐dependent cellular cytotoxicity (ADCC) and fixation of complement via the classical pathway of complement activation. Indeed, surface plasmon resonance experiments have shown that rat IgG2b antibodies of different specificity bind all mouse fixed chain receptors with relatively high affinity, such that their biological effect was reduced in Fcer1g‐deficient mice [35]. These findings raise the untested possibility that the IgG2b antibody used in prior experiments to determine the function of CCR8 in tumour immunity depleted Treg cells via ADCC in addition to blocking CCR8 function [34]. Therefore, the function of CCR8 in tumour immunity remains unclear.
In this study, we examined the function of CCR8 in tumour immunosuppression using mice in which CCR8 expression has been genetically ablated. We confirmed high levels of CCR8 expression on tumour‐infiltrating Treg cells, which was abolished on cells from Ccr8 −/− mice. Whereas systemic ablation of Treg cells resulted in strikingly diminished tumour growth, growth of subcutaneously implanted tumours was unaffected by systemic CCR8 loss. Consistently, we observed minimal impact of systemic CCR8 ablation on the frequency, phenotype and function of tumour‐infiltrating Treg cells. These findings suggest that CCR8 is not required for Treg cell accumulation and immunosuppressive function within tumours and that depletion of CCR8+ Treg cells rather than blockade of CCR8 function may provide a means of selective immunotherapy.
METHODS
Mice
Foxp3 EGFP‐DTR mice, originally described by Kim et al [36], were obtained from Jackson Laboratories. Ccr8 −/− mice were a kind gift from Sergio Lira [26] and Frank Tacke. Animals were genotyped using a custom genotyping service provided by Transnetyx® Inc. All mice were housed at the Babraham Institute Biological Services Unit or the Cambridge University Biomedical Services Gurdon Institute animal facilities. Experiments were performed using mice 8–14 weeks of age, with male and female mice equally distributed into experiment and control groups. Tumour measurements were completed by an independent investigator who was not aware of treatment groups or genotypes. Experiments were repeated 2–4 times using 3–8 mice per group. All animal experiments were conducted in accordance with UK Home Office guidelines and were approved by the Babraham Institute and/or University of Cambridge Animal Welfare and Ethics Review Board.
Depletion of Treg cells with diphtheria toxin
Diphtheria toxin (DTx) from Corynebacterium diphtheriae (Sigma‐Aldrich) was obtained in lyophilized powder form and reconstituted in sterile double‐distilled water according to the manufacturer's instructions. Solutions for injection were made up in sterile PBS to a dose of 25 μg/kg. To achieve transient depletion of Treg cells in Foxp3 EGFP‐DTR mice, DTx was administered via intraperitoneal injection in 100 μl on days 7, 9, 11 and 14 after tumour implantation.
MC38 and B16‐F10 heterotopic subcutaneous tumour implantation model
MC38 colon carcinoma cells were purchased from Kerafast. B16‐F10 melanoma cells were purchased from ATCC. Cell lines were passaged in DMEM (Invitrogen) supplemented with 10% FCS and antibiotics. 3·5 × 105 − 2 × 106 MC38 cells in 100 µl PBS or 1·25 × 105 B16‐F10 cells in 100 µl PBS were injected subcutaneously into the right flanks of mice, and tumours were measured with digital callipers at serial time‐points after implantation as previously described [37].
Suppression of Tconv cells by Tregs
The suppressive capacity of Treg cells was tested as previously described [38]. CCR8+ and CCR8− Treg cells were FACS sorted from MC38 tumours of Foxp3 EGFP ‐ DTR mice. Naïve CD4+ Tconv cells (CD25− CD44− CD62L+) were obtained from the spleens of WT CD45.1 mice via florescence‐activated cell sorting (FACS) and stained with CellTrace Violet™ (CTV) according to the manufacturer's protocol (Thermo Fisher Scientific). Treg cells and Tconv cells were plated in a 1:4 ratio in the presence of anti‐CD3 (BioLegend 1 µg/ml) and Rag2 −/− antigen‐presenting cells (APCs). Naïve Tconv cells cultured without Treg cells were used as the proliferating control. Cell division was evaluated after 4 days of culture.
Flow cytometry analysis
Tumour samples were digested using collagenase and DNase for 30 min at 37°C, and Lympholyte® (Cedarlane) was used to isolate lymphocytes from tumours. Cell suspensions were filtered using 40µm cell strainers (BD Biosciences). Spleens were mechanically dissociated over a 40µm cell strainer. Red blood cells were lysed using ACK Lysing Buffer (Gibco). Cells were stained with the Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) to discriminate between live and dead cells and then incubated with the following surface antibodies for 30 min on ice: anti‐TCRβ PE (H57‐597), anti‐CD8 PE‐Cy7 (53–6·7), anti‐CD25 APC (PC61.5), anti‐CD44 PerCP‐Cyanine5.5 (IM7), anti‐CD45.1 APC (A20) from eBioscience; anti‐CD4 BUV395 (GK1.5), anti‐CD62L BUV737 (MEL‐14) from BD Biosciences and anti‐CCR8 BV421 (SA214G2), anti‐Thy1.2 BV605 (53‐2.1) from BioLegend. Cells were stimulated with phorbol 12‐myristate 13‐acetate (PMA) and ionomycin and blocked with brefeldin A (BFA) for 4 h in RPMI 1640 complete medium. Intracellular antibodies anti‐Foxp3 APC (FJK‐16S), anti‐IFN‐γ FITC (XMG1.2) and anti‐TNF PE‐Cy7 (MP6‐XT22) were purchased from eBioscience and used with the eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol. Samples were analysed using BD Fortessa and Beckman Coulter CytoFLEX analysers. After analysis, data were analysed using FlowJo software (Tree Star, Inc.).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Two‐tailed Student's t tests were used to calculate statistical significance of the difference in sample means. P values of less than 0·05 were considered statistically significant. In all figures, data represent the mean ± the standard error of the mean (SEM). P values correlate with symbols as follows: ns = not significant, *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001, ****P ≤ 0·0001.
RESULTS
CCR8 is highly expressed by tumour‐infiltrating Treg cells and a subset of Tconv cells within tumours
To examine the expression of CCR8 on tumour‐infiltrating T cells, we subcutaneously implanted syngeneic MC38 colorectal adenocarcinoma cells into wild‐type (WT) C57BL/6 animals. Flow cytometry analysis of tumours revealed high levels of CCR8 expression in a substantial fraction of Foxp3+ CD4+ T cells and a smaller fraction of CD4+ and CD8+ Tconv cells within tumours of tumour‐bearing animals, whereas CCR8 expression within corresponding T cell populations in systemic lymphoid tissues was substantially lower (Figure 1a). To test the specificity of this signal and of the antibody used in these experiments, we examined anti‐CCR8 antibody staining on the surface of cells within MC38 tumours implanted in WT and Ccr8 −/− animals. Whereas a substantial proportion of Foxp3+ Treg cells within tumours of WT animals were positive for anti‐CCR8 antibody staining, this signal was abolished upon cells infiltrating tumours of Ccr8 −/− animals, confirming both the target and specificity of the antibody used (Figure 1b). CCR8+ Treg cells have been previously described as having enhanced suppressive potential compared to their negative counterparts [34, 39]. In order to test in our model whether CCR8 expression marks Treg cells with enhanced suppressive function, we sorted CCR8− and CCR8+ intratumoral Treg cells by FACS and tested their capacity to suppress proliferation of autologous naïve CD4+ Tconv cells in vitro (Figure 2). Both CCR8− and CCR8+ Treg cells were capable of suppressing proliferation at 1:4 Treg cell:Tconv cell ratio. However, CCR8+ Treg cells had higher suppressive capacity.
FIGURE 1.
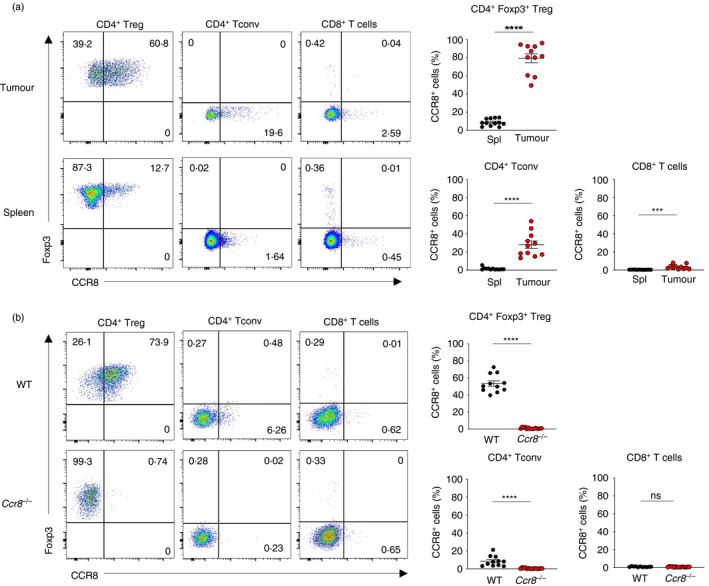
High levels of CCR8 expression discriminate Foxp3+ Treg cells within subcutaneously implanted syngeneic MC38 colorectal adenocarcinoma tumours. (a) Representative flow cytometry (left) and replicate measurements (right) of CCR8 expression on indicated CD4+ and CD8+ Tcell subsets within tumours and spleens of MC38 tumour‐bearing animals at day 21 following tumour implantation. (b) Representative flow cytometry (left) and replicate measurements (right) of CCR8 antibody staining on Treg and CD4+ Tconv and CD8+T cells within MC38 tumours of WT and Ccr8 −/− animals at day 21 following tumour implantation. Data are representative of 2 independently repeated experiments. Bars and error represent mean and SEM. Student's t test; ***P < 0·001; ****P < 0·0001; ns, not significant
FIGURE 2.
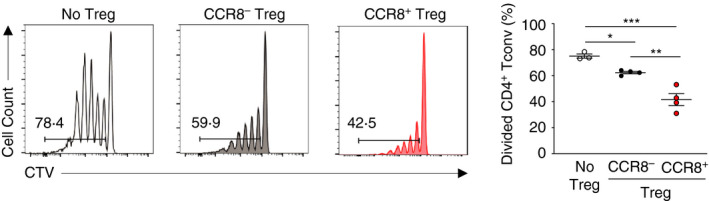
CCR8 marks highly suppressive Foxp3+ Treg cells within MC38 colorectal adenocarcinoma tumours. Representative flow cytometry (left) of CTV‐labelled naïve CD4+ Tconv cells incubated with no Treg cells, or at a 4:1 ratio with intratumoral CCR8− Treg cells or CCR8+ Treg cells from Foxp3 EGFP‐DTR mice after 4 days incubation, and replicate measurements of Tconv cell division (right). Data are representative of 2 independently repeated experiments. Bars and error represent mean and SEM. ordinary one‐way ANOVA; *P < 0·05;**P < 0·01 ***P < 0·001
Loss of CCR8 expression does not affect the growth of subcutaneously implanted syngeneic tumours
To test the function of CCR8 in anti‐tumour immunity, we measured the growth of subcutaneously implanted MC38 tumours in littermate WT and Ccr8 −/− animals and compared this to the effect of systemic experimental ablation of Treg cells using Foxp3 EGFP‐DTR mice, which express human diphtheria toxin receptor (DTR) and enhanced green fluorescent protein (EGFP) under the transcriptional control of the endogenous Foxp3 gene, enabling selective depletion of Foxp3+ Treg cells through administration of diphtheria toxin (DTx) [36]. Whereas systemic ablation of Treg cells resulted in substantially reduced growth of MC38 tumours (Figure 3a), systemic loss of CCR8 expression had no significant effect on tumour growth (Figure 3b). Importantly, we had similar observations using the syngeneic B16‐F10 melanoma tumour model, growth of which was highly sensitive to Treg cell depletion (Figure 3c) but not to germline ablation of Ccr8 (Figure 3d). These findings suggest that CCR8 function does not have a measurable effect on tumour growth using a syngeneic tumour model highly sensitive to the suppressive function of Treg cells.
FIGURE 3.
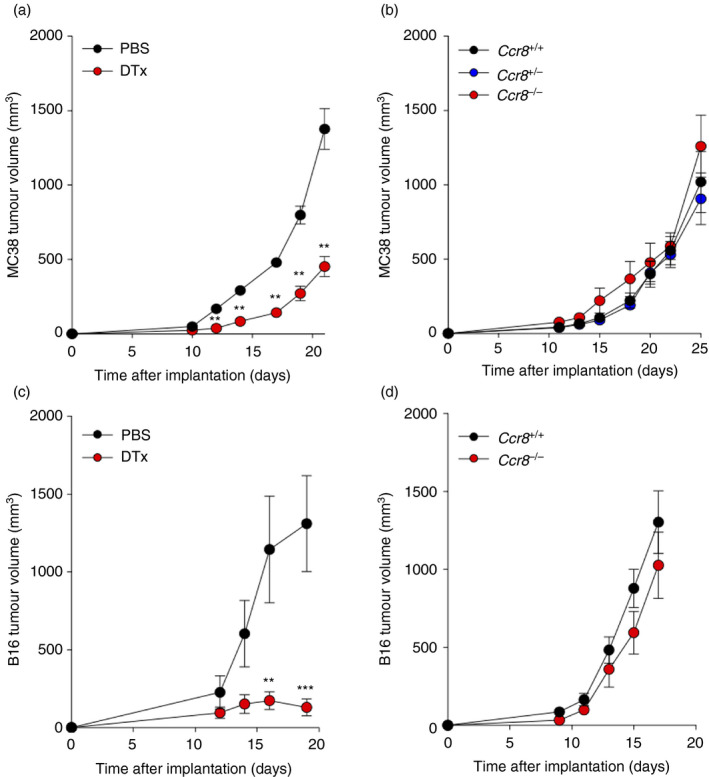
Systemic loss of CCR8 does not affect growth of subcutaneously implanted MC38 or B16‐F10 tumours in contrast to total Treg cell ablation. (a) Volume of heterotopic MC38 colorectal adenocarcinoma tumours at indicated time‐points following implantation into Foxp3 EGFP‐DTR animals which were administered with phosphate‐buffered saline (PBS) or diphtheria toxin (DTx) on days 7, 9, 11 and 14. (b) Volume of heterotopic MC38 colorectal adenocarcinoma tumours at indicated time‐points following implantation into animals of the indicated genotypes. (c) Volume of heterotopic B16‐F10 melanoma tumours at indicated time‐points following implantation into Foxp3 EGFP‐DTR animals which were administered with PBS or DTx on days 7, 9, 11 and 14. (d) Volume of heterotopic B16‐F10 melanoma tumours at indicated time‐points following implantation into animals of the indicated genotypes. n = 5–9 animals per genotype. Data are representative of 2 independently repeated experiments. Bars and error represent mean and SEM. Student's t test; **P < 0·01; ***P < 0·001
Loss of CCR8 expression does not affect Treg cell accumulation or activation of CD4+ or CD8+ Tconv cells within tumours
To formally test the function of CCR8 in Treg cell accumulation within tumours, we examined the frequency and number of Foxp3+ Treg cells within MC38 tumours implanted in WT and Ccr8 −/− animals (Figure 4a). This analysis revealed that loss of CCR8 function does not affect the frequency or total number of Treg cells within tumours and spleens. No increase was observed in the number of CD4+ or CD8+ Tconv cells within tumours of Ccr8 −/− animals (Figure 4b). Consistently, we did not observe increased production of the type I cytokines IFN‐γ and TNF among CD4+ Tconv (Figure 5a) and CD8+ (Figure 5b) T cells within tumours or spleens. Collectively, these findings suggest that CCR8 function does not substantially affect anti‐tumour immune responses in the syngeneic MC38 colorectal adenocarcinoma model, despite its sensitivity to Treg cell ablation.
FIGURE 4.
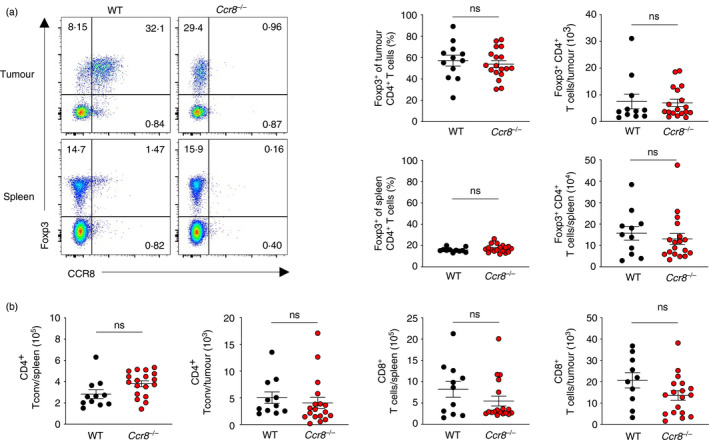
CCR8 expression is dispensable for Foxp3+ Treg cell accumulation within tumours. (a) Representative flow cytometry (left) and replicate measurements (right) of Foxp3+ Treg cells within spleens and MC38 tumours of WT and Ccr8 −/− animals at day 21 following tumour implantation. (b) Representative flow cytometry (left) and replicate measurements (right) of CD4+ and CD8+ Tconv cells within spleens and MC38 tumours of WT and Ccr8 −/− animals at day 21 following tumour implantation. n = 11–18 mice per genotype. Data are representative of 4 independently repeated experiments. Bars and error represent mean and SEM. Student's t test; ns, not significant
FIGURE 5.
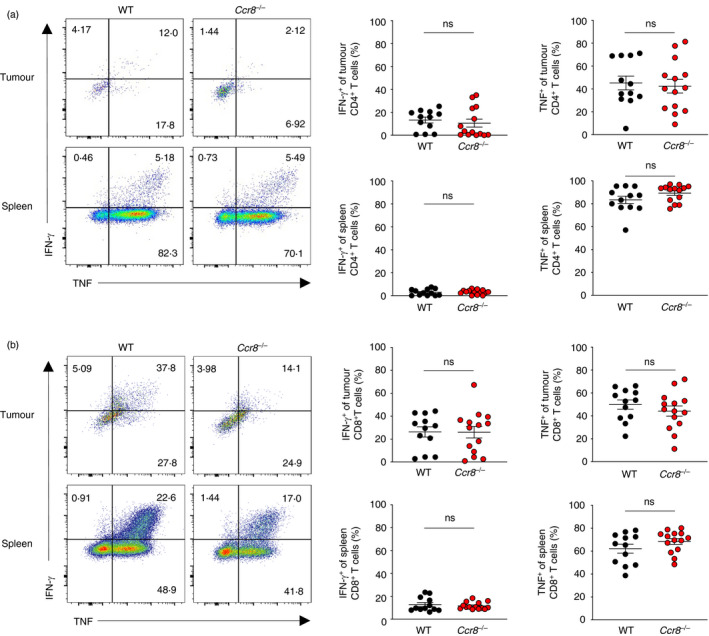
CCR8 expression does not impact suppression of CD4+ or CD8+ Tconv activation within tumours. (a) Representative flow cytometry (left) and replicate measurements (right) of IFN‐γ and TNF expression as detected by intracellular cytokine staining of CD4+ Tconv cells from spleens and MC38 tumours of WT and Ccr8 −/− animals at day 21 following tumour implantation. (b) Representative flow cytometry (left) and replicate measurements (right) of IFN‐γ and TNF expression as detected by intracellular cytokine staining of CD8+ T cells from spleens and MC38 tumours of WT and Ccr8‐KO animals at day 21 following tumour implantation. n = 11–18 mice per genotype. Data are representative of 4 independently repeated experiments. Bars and error represent mean and SEM. Student's t test; ns, not significant
DISCUSSION
The immunosuppressive function of Treg cells is an important therapeutic target in the immunotherapy of cancer. However, Treg‐targeted therapies should ideally spare the systemic anti‐inflammatory function of Treg cells in other tissues. There is consequently considerable interest in understanding whether Treg cells within tumours possess unique molecular characteristics enabling their selective targeting, either through functional disruption or cellular depletion. Recent studies have identified high levels of CCR8 expression as a distinguishing feature of Treg cells within tumours. It has also been proposed, through experiments where anti‐CCR8 antibodies have been systemically administered, that blockade of CCR8 function impairs the ability of Treg cells to suppress anti‐tumour immunity [34]. Here, we formally tested the contribution of CCR8 to anti‐tumour immunity using genetic loss‐of‐function experiments in mice. We found that CCR8 expression was dispensable both for Treg cell accumulation within tumours and for their immunosuppressive function. CCR8 is also reported to be expressed by Th2 cells, monocytic cells and NK cells. We observed no changes in the frequency of total CD4+ Tconv cells in the spleens or tumours of Ccr8 −/− mice compared to Ccr8 +/+ animals but did not in this study examine whether there were differences in the composition of the CD4+ Tconv compartment. In addition, the contribution of CCR8 to the function of NK cells and monocytes within tumours was not resolved. Thus, while we observed no overall difference in Treg cell infiltration and tumour immunity in the absence of CCR8, it will be important to examine its functions in greater cellular and molecular resolution in future studies.
We would like to emphasise that our observations are not inconsistent with the recently reported ability of anti‐CCR8 antibodies to reduce tumour growth in syngeneic tumour models in mice, but suggest a re‐interpretation of the mechanism underlying these observations [34]. In particular, while anti‐CCR8 antibodies may have blocking activity, it is possible that the isotypes used also caused some extent of cellular depletion through ADCC. Indeed, mouse fixed chain receptors can cross‐react with antibodies of the rat IgG2b isotype [35]. Whether the anti‐CCR8 antibodies used functioned in part through induction of Treg cell depletion has yet to be formally tested. The hypothesis that therapeutic depletion of CCR8+ cells rather than blockade of CCR8 function leads to induction of anti‐tumour immunity is indeed consistent with recently reported findings that administration of anti‐CCR8 nanobodies with blocking function does not augment tumour immunity, but does so when provided the capability for ADCC [40].
Using Ccr8‐deficient mice to confirm the specificity of anti‐CCR8 staining, our findings validate prior conclusions that Treg cells infiltrating MC38 colorectal adenocarcinoma tumours express high levels of CCR8 on their cell surface. Thus, depletion of CCR8‐expressing cells remains a potentially important therapeutic approach. Our findings therefore do not lessen the importance of CCR8 as a potential target in therapies aimed at selectively targeting tumour‐associated Treg cells, but suggest that therapeutic depletion of CCR8+ Treg cells rather than blockade of CCR8 function is likely to be more efficacious.
CONFLICT OF INTERESTS
The authors have no competing interests to declare.
ACKNOWLEDGEMENTS
We thank Bernhard Moser of Cardiff University and David Withers of the University of Birmingham for ideas and discussion. The research was supported by the Wellcome Trust/Royal Society grant 105663/Z/14/Z, the UK Biotechnology and Biological Sciences Research Council grant BB/N007794/1, Cancer Research UK grant C52623/A22597, MRC grant MR/S024468/1 and Hungarian National Scientific Research Fund grant (NKFIH‐OTKA FK132971). We thank members of the Babraham Institute Biological Services Facility and University of Cambridge UBS Gurdon Facility for technical support with animal experiments and the members of the Flow Cytometry Core from the Babraham Institute and the Department of Pathology at the University of Cambridge. We thank members of the Roychoudhuri laboratory for sharing of reagents, protocols, ideas and discussion.
Funding information
The research was supported by Wellcome Trust–Royal Society Fellowship 105663/Z/14/Z (R. Roychoudhuri); Biotechnology and Biological Sciences Research Council grants BB/N007794/1, BBS/E/B/000C0427 and BBS/E/B/000C0428; Cancer Research UK grant C52623/A22597; Medical Research Council grant MR/S024468/1; and Hungarian National Research, Development and Innovation Office Fund (NKFIH‐OTKA Grant No. FK132971 to DSG).
Contributor Information
Sarah K. Whiteside, Email: sw925@cam.ac.uk, Email: rr257@cam.ac.uk.
Rahul Roychoudhuri, Email: sw925@cam.ac.uk, Email: rr257@cam.ac.uk.
REFERENCES
- 1. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. [DOI] [PubMed] [Google Scholar]
- 3. Benoist C, Mathis D. Treg cells, life history, and diversity. Cold Spring Harb Perspect Biol. 2012;4:a007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roychoudhuri R, Eil RL, Restifo NP. The interplay of effector and regulatory T cells in cancer. Curr Opin Immunol. 2015;33:101–11. [DOI] [PubMed] [Google Scholar]
- 5. Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241:104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stockis J, Roychoudhuri R, Halim TYF. Regulation of regulatory T cells in cancer. Immunology. 2019;157:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. [DOI] [PubMed] [Google Scholar]
- 8. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high‐risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24:5373–80. [DOI] [PubMed] [Google Scholar]
- 10. Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi MB, Harpole DH Jr, et al. Tumor infiltrating Foxp3+ regulatory T‐cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107:2866–72. [DOI] [PubMed] [Google Scholar]
- 11. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93. [DOI] [PubMed] [Google Scholar]
- 12. Griffiths RW, Elkord E, Gilham DE, Ramani V, Clarke N, Stern PL, et al. Frequency of regulatory T cells in renal cell carcinoma patients and investigation of correlation with survival. Cancer Immunol Immunother. 2007;56:1743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–34. [DOI] [PubMed] [Google Scholar]
- 14. Perrone G, Ruffini PA, Catalano V, Spino C, Santini D, Muretto P, et al. Intratumoural FOXP3‐positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer. 2008;44:1875–82. [DOI] [PubMed] [Google Scholar]
- 15. Jordanova ES, Gorter A, Ayachi O, Prins F, Durrant LG, Kenter GG, et al. Human leukocyte antigen class I, MHC class I chain‐related molecule A, and CD8+/regulatory T‐cell ratio: which variable determines survival of cervical cancer patients? Clin Cancer Res. 2008;14:2028–35. [DOI] [PubMed] [Google Scholar]
- 16. Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T‐cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology. 2009;137:1270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR‐gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. 2015;162:1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45:1122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor‐infiltrating T regulatory cells. Immunity. 2016;45:1135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. [DOI] [PubMed] [Google Scholar]
- 24. Roos RS, Loetscher M, Legler DF, Clark‐Lewis I, Baggiolini M, Moser B. Identification of CCR8, the receptor for the human CC chemokine I‐309. J Biol Chem. 1997;272:17251–4. [DOI] [PubMed] [Google Scholar]
- 25. Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J Exp Med. 2013;210:1889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Freeman CM, Chiu BC, Stolberg VR, Hu J, Zeibecoglou K, Lukacs NW, et al. CCR8 is expressed by antigen‐elicited, IL‐10‐producing CD4+CD25+ T cells, which regulate Th2‐mediated granuloma formation in mice. J Immunol. 2005;174:1962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inngjerdingen M, Damaj B, Maghazachi AA. Human NK cells express CC chemokine receptors 4 and 8 and respond to thymus and activation‐regulated chemokine, macrophage‐derived chemokine, and I‐309. J Immunol. 2000;164:4048–54. [DOI] [PubMed] [Google Scholar]
- 28. Chensue SW, Lukacs NW, Yang TY, Shang X, Frait KA, Kunkel SL, et al. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J Exp Med. 2001;193:573–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soler D, Chapman TR, Poisson LR, Wang L, Cote‐Sierra J, Ryan M, et al. CCR8 expression identifies CD4 memory T cells enriched for FOXP3+ regulatory and Th2 effector lymphocytes. J Immunol. 2006;177:6940–51. [DOI] [PubMed] [Google Scholar]
- 30. Chung CD, Kuo F, Kumer J, Motani AS, Lawrence CE, Henderson WR Jr, et al. CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J Immunol. 2003;170:581–7. [DOI] [PubMed] [Google Scholar]
- 31. Goya I, Villares R, Zaballos A, Gutierrez J, Kremer L, Gonzalo JA, et al. Absence of CCR8 does not impair the response to ovalbumin‐induced allergic airway disease. J Immunol. 2003;170:2138–46. [DOI] [PubMed] [Google Scholar]
- 32. Coghill JM, Fowler KA, West ML, Fulton LM, van Deventer H, McKinnon KP, et al. CC chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft‐versus‐host disease. Blood. 2013;122:825–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, et al. CCR8(+)FOXp3(+) Treg cells as master drivers of immune regulation. Proc Natl Acad Sci USA. 2017;114:6086–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Villarreal DO, L'Huillier A, Armington S, Mottershead C, Filippova EV, Coder BD, et al. Targeting CCR8 induces protective antitumor immunity and enhances vaccine‐induced responses in colon cancer. Cancer Res. 2018;78:5340–8. [DOI] [PubMed] [Google Scholar]
- 35. Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcgammaRs modulate the anti‐tumor activity of antibodies targeting the PD‐1/PD‐L1 Axis. Cancer Cell. 2015;28:285–95. [DOI] [PubMed] [Google Scholar]
- 36. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–7. [DOI] [PubMed] [Google Scholar]
- 37. Gyori D, Lim EL, Grant FM, Spensberger D, Roychoudhuri R, Shuttleworth SJ, et al. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI. Insight. 2018;3:e120631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collison LW, Vignali DA. In vitro Treg suppression assays. Methods Mol Biol. 2011;707:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alvisi G, Brummelman J, Puccio S, Mazza EM, Tomada EP, Losurdo A, et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J Clin Invest. 2020;130:3137–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, et al. Therapeutic depletion of CCR8(+) tumor‐infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti‐PD‐1 therapy. J Immunother Cancer. 2021;9:e001749. [DOI] [PMC free article] [PubMed] [Google Scholar]