

Summary
Tofacitinib is an oral small molecule targeting the intracellular Janus kinase–signal transducer and activator of transcription (JAK‐STAT) pathways approved for the treatment of active rheumatoid arthritis (RA). We investigated the effects of tofacitinib on the response of RA lymphocytes to B and T cell collagen epitopes in their native and post‐translationally modified forms. In particular, peripheral blood mononuclear cells (PBMCs) from patients with RA and healthy subjects were cultured with type II collagen peptides (T261‐273, B359‐369, carT261‐273, citB359‐369) or with phorbol myristate acetate (PMA)/ionomycin/CD40L in the presence or absence of 100 nM tofacitinib for 20 h and analyzed by fluorescence activated cell sorter (FACS). Cultures without brefeldin A were used for cytokine supernatant enzyme‐linked immunosorbent assay (ELISA) analysis. Tofacitinib down‐regulated inflammatory cytokines by stimulated B [interleukin (IL)‐6 and tumor necrosis factor (TNF)‐α] and T [interferon (IFN)‐γ, IL‐17 or TNF‐α] cells in the short term, while a significant reduction of IL‐17 and IL‐6 levels in peripheral blood mononuclear cell (PBMC) supernatant was also observed. IL‐10 was significantly reduced in collagen‐stimulated B cells from patients with RA and increased in controls, thus mirroring an altered response to collagen self‐epitopes in RA. Tofacitinib partially prevented the IL‐10 down‐modulation in RA B cells stimulated with collagen epitopes. In conclusion, the use of tofacitinib exerts a rapid regulatory effect on B cells from patients with RA following stimulation with collagen epitopes while not reducing inflammatory cytokine production by lymphocytes.
Keywords: acquired immunity, collagen epitope, Janus kinase, post‐translational modifications, regulatory effect, targeted synthetic DMARDs
To mimic RA synovial inflammation we investigated the response to tofacitinib in lymphocytes stimulated with the B‐ and T‐cell collagen epitopes in their native and post‐translationally modified forms. Tofacitinib treatment partially prevented IL‐10 down‐modulation in RA B cells stimulated with collagen self‐epitopes while weakly increasing the IL‐6 expression in RA stimulated B cells.

INTRODUCTION
Rheumatoid arthritis (RA) is a systemic disease and its autoimmune pathogenesis includes the dysregulation of both the innate and adaptive arms of the immune system, ultimately causing chronic inflammation and erosive synovitis mediated by inflammatory cytokines (1). Tofacitinib represents the first oral small molecule approved for RA and targets the intracellular Janus kinase–signal transducer and activator of transcription (JAK‐STAT) pathways that mediate cytokine effects; it significantly inhibits JAK‐1 and JAK‐3, and to a lesser extent JAK‐2 and TYK‐2, indirectly targeting interleukin (IL‐6), IL‐10, IL‐12, IL‐21 and IL‐23, among others (2). Tofacitinib reduces STAT‐1 and STAT‐3 phosphorylation and consequently inhibits the T helper type 1 (Th1) and Th17 differentiation (2) while inducing a persistent, dose‐dependent increase in B cell count (3), similar to that observed in hereditary JAK‐3 deficiency (4), and a slight reduction of immunoglobulin (Ig) levels, probably a consequence of the IL‐21 impaired signaling (3). Of note, IL‐21 also promotes the transition from naive B cells to plasmablasts when combined with CD40–CD40L co‐stimulation (5); i.e. in the case of antigen activated T cell–B cell interaction, otherwise resulting in B cell apoptosis as a rescue mechanism to eliminate non‐antigen‐activated B cells (6).
We have previously reported that human collagen epitopes differentially modulate the T and B cell cytokine response in RA by altering the balance between inflammatory and regulatory cytokines (7). In the present work, we have investigated the lymphocyte response to tofacitinib after the stimulation with the B and T cell collagen epitopes in their native and post‐translationally modified forms to mimic RA synovial inflammation.
METHODS
Subjects
Peripheral blood was obtained from eight patients with RA attending the outpatient clinics of Rheumatology Department at the Humanitas Clinical and Research Center. Seven of eight patients were seropositive for both anti‐citrullinated peptides and rheumatoid factors; two of eight patients were human leukocyte antigen (HLA)‐DR4‐positive; median C‐reactive protein (CRP) was 0.48 mg/dl (range = 0.12–1.44); median disease activity score in 28 joints (DAS28) was 2.8 (range = 0.96–5.75); five of eight were women, the median age was 55 years (range = 31–71). Peripheral blood samples from four healthy subjects (two women, median age = 47 years, range = 31–66) were used as controls. The experimental protocol of this study was approved by the Ethical Committee of the Humanitas Clinical and Research Center and written informed consent was obtained from study participants. All patient data and samples were handled according to Helsinki and Health Insurance Portability and Accountability acts.
Experimental design
Peripheral blood mononuclear cells (PBMCs) were isolated by Lympholyte® Cell Separation density gradient centrifugation media for humans (Cedarlane, Burlington, ON, Canada). PBMCs were subsequently washed twice with phosphate‐buffered saline (PBS), counted in trypan blue excluding dead cells, and re‐suspended in freezing medium containing 90% fetal calf serum (FCS) and 10% dimethyl sulfoxide (DMSO), aliquoted into cryovials, kept at −80°C for 24 h and ultimately stored in liquid nitrogen tanks until used. From each subject, 2 × 106 cells/well in duplicate were cultured into 96‐well plates with 200 μl of complete RPMI [10% of FCS, 1% of penicillin and streptomycin, 1% of glutamine, 1% of 4‐(2‐hydroxyethyl)‐1‐piperazine etan‐sulfonic acid (HEPES)] for 20 h with the RA‐specific activation stimulus with type II collagen peptides (1 μg/ml of T261‐273 + 1 μg/ml of B359‐369 + 1 μg/ml of carT261‐273 + 1 μg/ml of citB359‐369), as previously described (7) or with the non‐specific lymphocyte stimulators phorbol myristate acetate (PMA) + ionomycin (50 ng/ml + 1 μg/ml) for T cells and CD40L (1 μg/ml) for B cells in the presence or absence of 100 nM tofacitinib (Pfizer Inc., Peaback, NJ, USA) (8). After 2 h culture, brefeldin A (BFA) 10 μl/ml was added, except for cultures whose supernatant has been used for enzyme‐linked immunosorbent assay (ELISA) cytokine detection. Supernatants were collected and stored at −80°C until analyzed.
Lymphocyte phenotypes
After stimulation, cells were collected and stained with extracellular and intracellular markers using fluorochrome‐conjugated monoclonal antibodies (mAbs) titrated to determine the optimal concentrations. The mAbs used included CD19 BV650 (Biolegend, San Diego, CA, USA), CD138 peridinin chlorophyl‐cyanin 5.5 (PerCp‐Cy 5.5) (BD Biosciences, San Jose, CA, USA), CD24 phycoerythrin (PE)‐Cy 7 (BD Biosciences), CD27 BV605 (BD Biosciences), CD38 Alexa Fluor 700 (BD Biosciences), CD3 BV650 (Biolegend), CD4 BV570 (Biolegend), CD45RO PerCP‐Cy 5.5 (Biolegend), CCR7 PE‐CF494 (BD Biosciences), CD69 BV650 (Biolegend), CD25 APC Alexa Tandem (BD Biosciences), CD154 APC (BD Biosciences), IgD allophycocyanin (APC)‐h7am (BD Biosciences), IL‐10 BV786 (BD Biosciences) in the B cell panel or IL‐10 BV421 (BD Biosciences) in the T cell panel, transforming growth factor (TGF)‐β PerCF594 (BD Biosciences), IL‐6 BV421 (BD Biosciences), tumor necrosis factor (TNF)‐α FITC (BD Biosciences), IFN‐γ FITC (BD Biosciences) and IL‐17 PE (Thermo Fisher, Fremont, CA, USA). Briefly, cells were stained with Zombie Aqua fixable viability dye (catalog 423102; Biolegend) for 15 min at room temperature in PBS, surface markers were marked by incubating cells with related mAbs for 20 min at room temperature, except for chemokine receptors that were detected by incubating cells at 37°C for 20 min in Hanks’ balanced salt solution (HBSS) in the presence of 2% FCS. The Cytofix/Cytoperm kit (catalog 554722/554723; BD Biosciences) was used to detect intracellular cytokines, according to the manufacturer’s instructions.
Lymphocytes were gated based on the FSC/SSC profile, excluding doublets/dead cells, and then based on specific markers. Flow cytometry analysis was performed on an LSR Fortessa Cell Analyzer (BD Biosciences) with applied automatic compensation, and flow cytometry data were analyzed using FlowJo software (Treestar Inc., Ashland, OR, USA).
Lymphocyte subpopulations were gated as follows: activated B lymphocytes as CD19+CD69+ cells; regulatory B cells as CD19+IL‐10+; naive B cells as CD19+CD27−IGD+; transitional B cells as CD19+CD24+CD38+; memory B cells as CD19+CD27+IGD−; plasmablasts as CD19+CD24−CD38+; plasma cells as CD19+CD138+; activated T cells as CD3+CD69+; antigen‐activated T cells as CD3+CD40L+; naive T cells as CD3+CD45RA+CD45RO−CCR7+; T central memory cells as CD3+CD45RO+CCR7+; T effector memory cells as CD3+CD4+CD45RA−CD45RO+CCR7−; and regulatory T cells as CD3+CD25highIL‐10+.
Supernatants from each experimental condition were analyzed for IL‐17, IL‐10, TNF‐α, IL‐6 and IFN‐γ levels using commercially available ELISA (Raybitech, Norcross, GA, USA), based on the manufacturer’s protocols; all analyses were performed in duplicate.
Statistical analysis
Results are presented as median cell percentages and mean fluorescence intensity (MFI) with range. Data were analyzed with Mann Whitney U‐ or paired t‐tests, as appropriate (GraphPad Prism version 8.1). P < 0.05 was considered statistically significant.
RESULTS
B cell activation and inflammatory cytokine production
Changes in B cell activation and cytokine production in different experimental conditions are illustrated in Figure 1 and Supporting information, Table S1.
FIGURE 1.
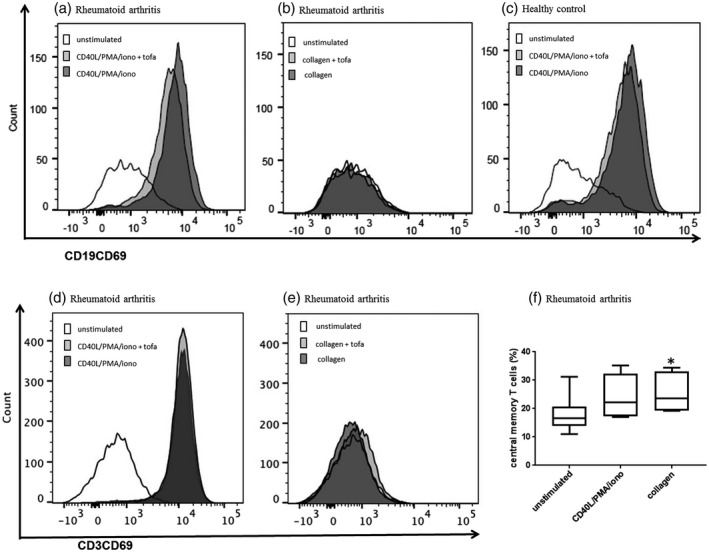
Activation of B and T cells. The percentages of B cells expressing CD69 activation marker significantly increased after CD40L/phorbol myristate acetate (PMA)/ionomycin stimulation in both rheumatoid arthritis (a, P < 0.0001 versus unstimulated) and in healthy subjects (c, P = 0.004 versus unstimulated) but not after collagen epitope stimulation (b). Tofacitinib treatment weakly reduced CD69+ B cells. CD69 significantly increased in rheumatoid arthritis (RA) T cells after non‐specific lymphocyte stimulation (d, P = 0.006) but not after collagen epitope stimulation (e). Tofacitinib treatment did not significantly decrease T cell activation mirrored by CD69 surface expression. Collagen epitope stimulation increased significantly the proportion of RA central memory T cells (f, P = 0.049)
B cell activation, i.e. the percentage of CD69‐expressing B cells and CD69 MFI, was higher in RA compared to controls at baseline conditions and increased after non‐specific lymphocyte stimulation with CD40L/PMA/ionomycin. Collagen epitope stimulation of both RA and control cells led to a weak B cell activation compared to unstimulated cultures (Figure 1). In tofacitinib‐treated cells, after the different stimulation conditions, B cell activation was slightly reduced in both RA and control cells compared to untreated cultures (Figure 1a–c). TNF‐α expression increased after stimulation with CD40L/PMA/ionomycin in both RA and control B cells and after stimulation with collagen epitopes only weakly in RA B cells. Tofacitinib did not reduce B cell TNF‐α expression induced by CD40L/PMA/ionomycin or by collagen epitopes.
B lymphocyte IL‐6 production was slightly greater in RA compared to controls at baseline. CD40L/PMA/ionomycin stimulation slightly increased IL‐6 B cell expression, while collagen epitope‐stimulated B cells were similar to unstimulated B cells. Of note, tofacitinib treatment weakly increased IL‐6 B cell expression in RA cells (Figure 2a,b).
FIGURE 2.
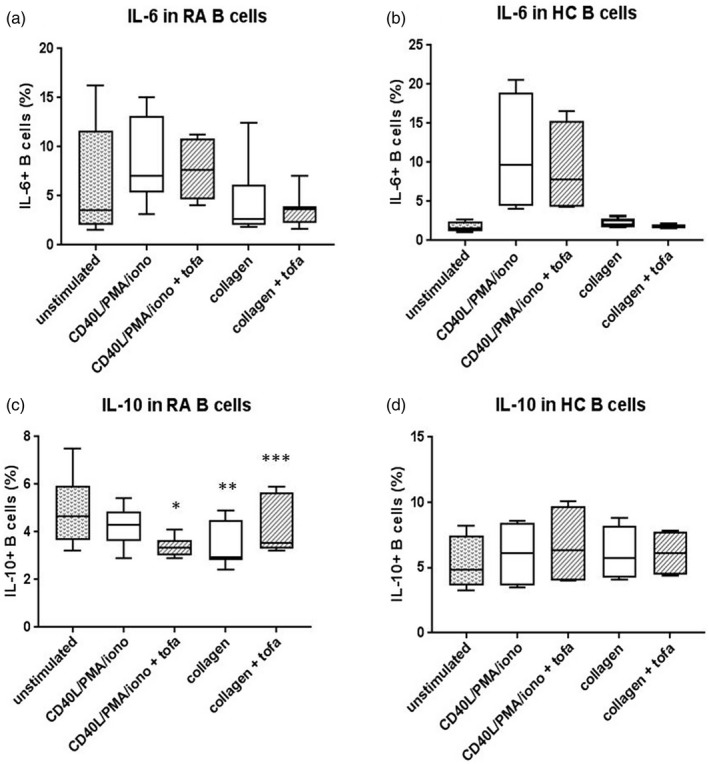
Interleukin (IL)‐6 and IL‐10 in B cells. (a,b) Percentages of IL‐6+ B cells increased after CD40L/phorbol myristate acetate (PMA)/ionomycin stimulation in rheumatoid arthritis (RA) and in healthy subjects (HC), but not after collagen epitope stimulation. In RA, tofacitinib treatment increased IL‐6+ B cells. (c,d) Percentages of IL‐10+ B cells in RA were significantly reduced after collagen epitope stimulation (**P = 0.003 versus unstimulated B cells), but not in HC. IL10+ B cells significantly increased in tofacitinib‐treated RA B cell stimulated with collagen epitopes (***P = 0.01 versus RA B cells stimulated with collagen epitopes not treated with tofacitinib), while significantly decreased in the CD40L/PMA/ionomycin setting (*P = 0.005 RA B cells stimulated with CD40L/PMA/ionomycin not treated with tofacitinib versus RA B cells stimulated with CD40L/PMA/ionomycin + tofacitinib)
Stimulation with collagen epitopes did not induce changes in the proportion of memory B lymphocytes in the RA samples, suggesting that patients did not have collagen‐specific B memory cells and explaining the weak response to collagen epitope stimulation. Data on plasmablasts and plasma cells were not reported due to the low number of these subpopulations (data not shown).
T cell activation and inflammatory cytokine production
Changes in T cell activation and cytokine production in different experimental conditions are detailed in Supporting information, Tables S2, S3 and S4.
T cell activation, i.e. CD69 and CD154 expression, was higher in RA compared to controls at baseline; CD69 significantly increased after non‐specific lymphocyte stimulation, as expected, while CD154, i.e. surface CD40L induced by antigen‐specific activation in CD4 T cells, only weakly increased after collagen epitope stimulation in RA (Figure 1d,e), suggesting the absence of collagen‐specific T helper cells in the majority of the selected patients. Tofacitinib treatment did not significantly decrease T cell activation mirrored by CD69 and CD154 (Figure 1d,e).
Collagen epitope stimulation significantly increased the proportion of RA central memory T cells (Figure 1f) and also, weakly, that of RA effector memory T cells. This observation suggested an altered self‐epitope recognition by CD8 T cells.
The percentages of T cells expressing IFN‐γ, IL‐17 or TNF‐α at baseline did not differ significantly between RA and controls, while inflammatory cytokines increased with CD40L/PMA/ionomycin stimulation, but not with collagen epitope stimulation, either considering all T cells or analyzing separately effector memory T cells and central memory T cells. The effect of treatment with tofacitinib in down‐regulating inflammatory cytokines in T cells was limited.
Regulatory B and T lymphocytes
Baseline IL‐10 production by B cells was lower in RA than in healthy subjects, especially considering transitional B cells. Of note, IL‐10 was significantly reduced in RA B cells, while increased in controls when stimulated using collagen epitopes, either considering all IL‐10+ B cells or only transitional IL‐10+ B cells, confirming an altered response to collagen self‐epitopes in RA. The treatment with tofacitinib partially prevented IL‐10 down‐modulation in RA B cells stimulated with collagen epitopes (Figure 2c,d and Supporting information, Table S1).
Baseline IL‐10 production by T cells (CD3IL‐10+) was higher in healthy subjects than in patients with RA (P = 0.006); however, IL‐10 production by regulatory T cells (CD3, CD4, CD25highIL‐10+) was higher in RA compared to healthy subjects (P = 0.006). A significant increase of RA IL‐10+ T cells (P = 0.003) and RA regulatory T cells (P > 0.05) was observed with non‐specific stimulation. Of interest, tofacitinib increased the percentages of RA IL‐10+ T cells and IL‐10 MFI of both RA IL‐10+ T cells and of RA regulatory T cells while significantly decreasing the percentage of RA regulatory T cells. Collagen stimulation did not decrease IL‐10 production in RA T cells and tofacitinib treatment did not change IL‐10 production in this setting (Supporting information, Table S4).
Cytokine modulation by tofacitinib
Cytokine levels produced by PBMCs under different experimental conditions were measured in the supernatants (Supporting information, Table S5). Non‐specific lymphocyte stimulators induced a potent cytokine release from immune cells. Tofacitinib treatment significantly reduced IL‐17, IL‐6 and IL‐10 in RA (Figure 3a).
FIGURE 3.
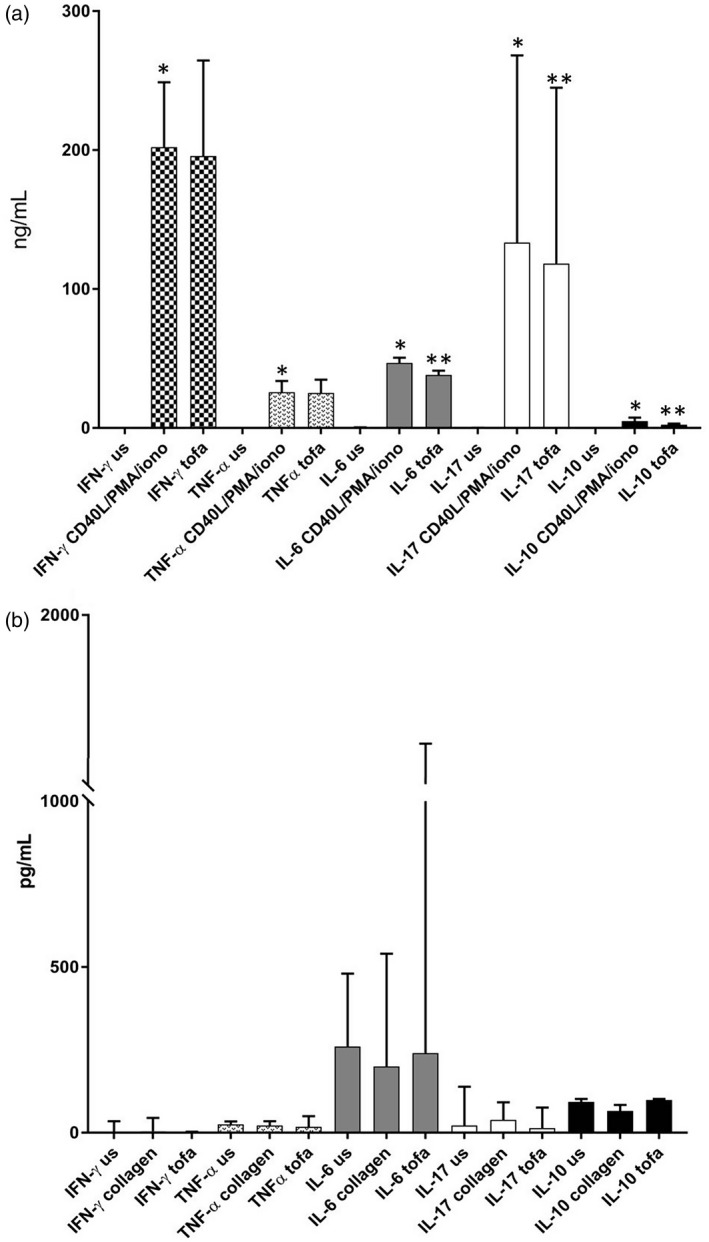
Stimulation‐induced RA peripheral blood mononuclear cell (PBMC) cytokines. (a) CD40L/phorbol myristate acetate (PMA)/ionomycin stimulation significantly increased interferon (IFN)‐γ (P = 0.003), tumor necrosis factor (TNF)‐α (P = 0.001), interleukin (IL)‐6 (P = 0.0002), IL‐17 (P = 0.007) and IL‐10 (P = 0.002) in rheumatoid arthritis (RA) peripheral blood mononuclear cell (PBMC) cultures. Tofacitinib treatment significantly reduced IL‐6 (P = 0.002), IL‐17 (P = 0.03) and IL‐10 (P = 0.0007) levels. (b) Collagen stimulation reduced IL‐10 levels, while tofacitinib treatment increased IL‐10 and IL‐6 levels. Results are presented as median and interquartile range
Collagen epitope stimulation did not increase IFN‐γ in RA cultures, as we could expect in case of collagen‐specific memory T cell activation; however, an altered self‐epitope recognition seems to be supported by the increase in IL‐17 and the reduced IL‐10 levels in RA. Of interest, tofacitinib increased IL‐6 and IL‐10 in RA, as we observed in collagen‐stimulated B cells, while reducing IL‐17 (Figure 3b).
DISCUSSION
The available treatment options for RA have significantly changed disease management but understanding of the fine immunological effects is largely incomplete, especially for more recently developed drugs with complex mechanisms of action. In this study, we investigated the impact of tofacitinib on B and T cells from patients with RA and controls exposed ex vivo to non‐specific stimulators or collagen RA‐specific antigens, i.e. native and post‐translationally modified epitopes of collagen type II (7). In the short term, tofacitinib did not significantly reduce the production of inflammatory cytokines, such as IFN‐γ, TNF‐α or IL‐17, by lymphocytes challenged with different agents, but the overall levels of these cytokines were reduced in tofacitinib‐treated PBMCs, including monocyte‐derived macrophages, the most relevant producers of inflammatory cytokines (9, 10).
Surprisingly, tofacitinib increased IL‐6 production by B cells and in culture supernatants after collagen stimulation. While the mechanisms regulating IL‐6 expression in B cells remain unknown, with the exception of the role of BAFF (11, 12), consistent data have been reported on macrophages and T cells. Tofacitinib decreased IL‐6 gene expression in unstimulated RA synovial macrophages (13), but this did not correspond to lower IL‐6 protein levels in culture supernatant of RA synovial macrophages treated with tofacitinib in the short term (8), similar to that observed in our experimental setting. IL‐6 reduction was not demonstrated also in tofacitinib‐treated RA CD4+ T cells (8). However, in longer culture conditions (1415) and in vivo (16), tofacitinib significantly reduced IL‐6, suggesting that a longer time may be necessary to modulate this cytokine. However, even at earlier time‐points, tofacitinib inhibits IL‐6‐mediated Th17 differentiation, as also confirmed by our data on IL‐17.
Cytokine production is controlled by numerous positive and negative feedback mechanisms, particularly in macrophages (10). The inflammatory cytokine production by macrophages is negatively controlled by the action of anti‐inflammatory cytokines, such as IL‐10 and IL‐1 receptor antagonist (17), to control excessive or prolonged inflammation (18). IL‐10 promotes the activation of JAK‐1 and tyrosine kinase 1 (TYK‐2), resulting in the phosphorylation of STAT‐3, which is required for the repressive effects of IL‐10 on Toll‐like receptor (TLR)‐mediated cytokine production in macrophages (19). These data suggest that, in the early phases of tofacitinib treatment, increased IL‐6 levels in B cells could be explained by the lack of IL‐10 repressive action on IL‐6 due to JAK‐1 inhibition by tofacitinib, as previously demonstrated (20).
We further observed that IL‐10 levels were reduced by collage epitope stimulation in B cells, while tofacitinib partially inhibited this effect in treated cultures. This is of particular interest, as previous data showed that higher concentrations and longer‐term culture conditions are required to observe an IL‐10 reduction induced by tofacitinib in macrophages in vitro (20), while increased IL‐10 mRNA was observed in stimulated B cells after 3 days of tofacitinib treatment (14).
The use of B and T cell‐specific collagen epitopes did not induce an antigen‐specific activation of B or CD4+ T lymphocytes in our cohort of patients with RA, and we speculate that it could be related to the absence of autoreactive lymphocytes for collagen, probably linked to the low prevalence of HLA‐DR4 in our series, being the largely HLA‐DR4‐restricted used collagen epitopes (21). This view is supported by the lack of proliferation of memory B cells and of expression of the antigen‐specific CD154 co‐stimulatory molecule on CD4 T lymphocytes after stimulation with collagen epitopes in our experiment. However, an altered self‐epitope recognition seems to occur based on the increase of IL‐17 and on the diminished IL‐10 levels after collagen epitope stimulation and the increase of central memory and effector memory CD8 T cells. These observations could be explained by two non‐exclusive mechanisms. We may envision that the adaptive immune system does not recognize type II collagen peptides as self‐epitopes through class I HLA, because these are normally hidden from the immune system (22) or that the adaptive immune system recognizes the neo‐epitopes created with post‐translational modifications (carbamylation and citrullination) as non‐self, as supported by the majority of enrolled RA cases being serum anti‐CCP‐positive.
In conclusion, the short‐term use of tofacitinib does not reduces inflammatory cytokine production by lymphocytes, probably as a result of the altered feedback mechanisms regulating cytokine expression, but the data suggest that longer drug exposures may be associated with a different cytokine modulation.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
M. D. S. and C. S. designed conception of the work, supervised experiments, manuscript revision, editing and final approval; N. I. and G. C. designed experiments and carried out experiments and analysis; A. C., M. V., G. G., M. C., N. L. and F. M. helped with patient enrollment. All authors helped equally in writing and editing the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
Financial support for this research was unconditionally provided by Pfizer Inc as part of CREARE 2016 research grant. The authors thank all our patients for signing consent to participate to this study.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and Supporting information.
REFERENCES
- 1. Winthrop KL, Weinblatt ME, Crow MK, Burmester GR, Mease PJ, So AK, et al. Unmet need in rheumatology: reports from the Targeted Therapies meeting 2018. Ann Rheum Dis. 2019;78:872–8. [DOI] [PubMed] [Google Scholar]
- 2. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34:318–28. [PubMed] [Google Scholar]
- 3. Rizzi M, Lorenzetti R, Fischer K, Staniek J, Janowska I, Troilo A, et al. Impact of tofacitinib treatment on human B‐cells in vitro and in vivo . J Autoimmun. 2017;77:55–66. [DOI] [PubMed] [Google Scholar]
- 4. Roberts JL, Lengi A, Brown SM, Chen M, Zhou Y‐J, O'Shea JJ, et al. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood 2004;103:2009–18. [DOI] [PubMed] [Google Scholar]
- 5. Konforte D, Simard N, Paige CJ. IL‐21: an executor of B cell fate. J Immunol. 2009;182:1781–7. [DOI] [PubMed] [Google Scholar]
- 6. Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL‐21. J Immunol. 2006;177:5236–47. [DOI] [PubMed] [Google Scholar]
- 7. De Santis M, Ceribelli A, Cavaciocchi F, Generali E, Massarotti M, Isailovic N, et al. Effects of type II collagen epitope carbamylation and citrullination in human leucocyte antigen (HLA)‐DR4(+) monozygotic twins discordant for rheumatoid arthritis. Clin Exp Immunol. 2016;185:309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maeshima K, Yamaoka K, Kubo S, Nakano K, Iwata S, Saito K, et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon‐gamma and interleukin‐17 production by human CD4+ T cells. Arthritis Rheum. 2012;64:1790–8. [DOI] [PubMed] [Google Scholar]
- 9. Hamilton JA, Tak Paul P. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009;60:1210–21. [DOI] [PubMed] [Google Scholar]
- 10. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140:805–20. [DOI] [PubMed] [Google Scholar]
- 11. Matsushita T. Regulatory and effector B cells: friends or foes? J Dermatol Sci. 2019;93:2–7. [DOI] [PubMed] [Google Scholar]
- 12. Shen P, Fillatreau S. Antibody‐independent functions of B cells: a focus on cytokines. Nat Rev Immunol. 2015;15:441–51. [DOI] [PubMed] [Google Scholar]
- 13. Yarilina A, Xu K, Chan C, Ivashkiv LB. Regulation of inflammatory responses in tumor necrosis factor‐activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheum. 2012;64:3856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang SP, Iwata S, Nakayamada S, Sakata K, Yamaoka K, Tanaka Y. Tofacitinib, a JAK inhibitor, inhibits human B cell activation in vitro . Ann Rheum Dis. 2014;73:2213–5. [DOI] [PubMed] [Google Scholar]
- 15. Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP‐690,550). J Immunol. 2011;186:4234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Migita K, Izumi Y, Jiuchi Y, Kozuru H, Kawahara C, Izumi M, et al. Effects of Janus kinase inhibitor tofacitinib on circulating serum amyloid A and interleukin‐6 during treatment for rheumatoid arthritis. Clin Exp Immunol. 2014;175:208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. IL‐10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–22. [PubMed] [Google Scholar]
- 18. Ananieva O, Darragh J, Johansen C, Carr JM, McIlrath J, Park JM, et al. The kinases MSK1 and MSK2 act as negative regulators of Toll‐like receptor signaling. Nat Immunol. 2008;9:1028–36. [DOI] [PubMed] [Google Scholar]
- 19. Murray PJ. Understanding and exploiting the endogenous interleukin‐10/STAT3‐mediated anti‐inflammatory response. Curr Opin Pharmacol. 2006;6:379–86. [DOI] [PubMed] [Google Scholar]
- 20. Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide‐induced cytokine production by blocking IL‐10‐mediated feedback. J Immunol. 2012;189:2784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ria F, Penitente R, De Santis M, Nicolò C, Di Sante G, Orsini M, et al. Collagen‐specific T‐cell repertoire in blood and synovial fluid varies with disease activity in early rheumatoid arthritis. Arthritis Res Ther. 2008;10:R135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tong D, Lönnblom E, Yau ACY, Nandakumar KS, Liang B, Ge C, et al. A shared epitope of collagen type XI and type II is recognized by pathogenic antibodies in mice and humans with arthritis. Front Immunol. 2018;9:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
All data generated or analyzed during this study are included in this published article and Supporting information.