

Summary
Tuberculosis (TB) is the leading cause of death from a single bacterial infectious agent and is one of the most relevant issues of public health. Another pandemic disease is type II diabetes mellitus (T2D) that is estimated to affect half a billion people in the world. T2D is directly associated with obesity and a sedentary lifestyle and is frequently associated with immunosuppression. Immune dysfunction induced by hyperglycemia increases infection frequency and severity. Thus, in developing countries the T2D/TB co‐morbidity is frequent and represents one of the most significant challenges for the health‐care systems. Several immunoendocrine abnormalities are occurring during the chronic phase of both diseases, such as high extra‐adrenal production of active glucocorticoids (GCs) by the activity of 11‐β‐hydroxysteroid dehydrogenase type 1 (11‐βHSD1). 11‐βHSD1 catalyzes the conversion of inactive cortisone to active cortisol or corticosterone in lungs and liver, while 11‐β‐hydroxysteroid dehydrogenase type 2 (11‐βHSD2) has the opposite effect. Active GCs have been related to insulin resistance and suppression of Th1 responses, which are deleterious factors in both T2D and TB. The anabolic adrenal hormone dehydroepiandrosterone (DHEA) exerts antagonistic effects on GC signaling in immune cells and metabolic tissues; however, its anabolic effects prohibit its use to treat immunoendocrine diseases. 16α‐bromoepiandrosterone (BEA) is a water miscible synthetic sterol related to DHEA that lacks an anabolic effect while amplifying the immune and metabolic properties with important potential therapeutic uses. In this work, we compared the expression of 11‐βHSD1 and the therapeutic efficacy of BEA in diabetic mice infected with tuberculosis (TB) (T2D/TB) with respect to non‐diabetic TB‐infected mice (TB). T2D was induced by feeding mice with a high‐fat diet and administering a single low‐dose of streptozotocin. After 4 weeks of T2D establishment, mice were infected intratracheally with a high‐dose of Mycobacterium tuberculosis strain H37Rv. Then, mice were treated with BEA three times a week by subcutaneous and intratracheal routes. Infection with TB increased the expression of 11‐βHSD1 and corticosterone in the lungs and liver of both T2D/TB and TB mice; however, T2D/TB mice developed a more severe lung disease than TB mice. In comparison with untreated animals, BEA decreased GC and 11‐βHSD1 expression while increasing 11‐βHSD2 expression. These molecular effects of BEA were associated with a reduction in hyperglycemia and liver steatosis, lower lung bacillary loads and pneumonia. These results uphold BEA as a promising effective therapy for the T2D/TB co‐morbidity.
Keywords: 11‐βHSD1, active glucocorticoids, BEA, central nervous system, colony‐forming units, diabetes–tuberculosis co‐morbidity, immunotherapy
Study of immune‐endocrine relation between diabetes and their relationship in tuberculosis susceptibility.

INTRODUCTION
Type II diabetes (T2D) is a metabolic disease with high incidence and prevalence in low‐ and middle‐income countries, and is highly associated with immune dysfunction. Hyperglycemia impairs overall immunity, increasing infection frequency and severity. Therefore, T2D is a critical susceptibility factor for tuberculosis (TB) (1). In immunocompetent individuals, Mycobacterium tuberculosis (Mtb) infection is controlled by immune mechanisms that contribute to granuloma development and control of bacterial growth (2). However, in subjects with T2D/TB co‐morbidity the innate and adaptive immunity are impaired and is associated with poor prognosis (3).
A type 1 T helper (Th1) cell response mediates the protective immune response to TB through macrophage activation by interferon (IFN)‐γ and tumor necrosis factor (TNF)‐α (4), while type 2 T helper (Th2) immune response has been shown that is a susceptibility factor, because the Th2 cytokines IL‐4 and IL‐13 favor microbial growth by reducing TNF‐α and inducible nitric oxide synthase (iNOS) (5). T2D patients show an impaired response to Mtb infection (6), consisting of alterations in bacterial recognition, phagocytosis and a deficient immune cellular response with low cytokine and chemokine production (7). These immune response dysfunctions in T2D individuals result in severe TB manifestations (8). The advance in understanding of the metabolic factors and neuroimmune–endocrine regulation in T2D has been an important aspect for clarifying the higher susceptibility of T2D to develop TB.
Glucocorticoids (GCs) are steroid hormones with critical homeostatic functions. GCs exert their effects by binding to glucocorticoid receptors (GR), endocrine nuclear receptors present in all cell types. GR signaling plays an essential role in the modulation of many biological functions in metabolic organs, including liver, lung, adipose tissue and muscle, and in immune tissues and cells such as thymus, macrophages and lymphocytes (9). Cortisol suppresses the expression of proinflammatory and adhesion molecules, thus preventing the extravasation of neutrophils to the site of inflammation (9). Chronic exposure induces an anti‐inflammatory gene expression profile of resident macrophages and decreases their phagocytic activity. Furthermore, active GCs suppress Th1 cytokine production and induce cell death (10), promoting a Th2 environment that impairs granuloma formation (2). GCs inhibit the proliferation of effector T cells and induce apoptosis of neutrophils, basophils and eosinophils, resulting in reduced inflammation (11). In homeostatic conditions and under acute stress, the principal source of GCs are the adrenal glands, which are regulated by the negative feedback loop of the hypothalamus–pituitary–adrenal (HPA) axis (12). However, diverse environmental insults and metabolic or endocrine alterations prompt GCs over‐production, leading to negative effects on metabolism and the immune response (13).
GCs have an important role in the physiopathology of TB. It has been reported in an experimental model of progressive TB that at the time of maximal protective activity mediated by IFN‐γ, TNF‐α and nitric oxide (NO) production the HPA axis is activated, thereby stimulating the adrenal glands to secrete GCs (corticosterone) with the apparent aim of avoiding tissue damage produced by excessive lung inflammation. However, the excess of corticosterone also inhibits the activity of Th1 lymphocytes and induces differentiation of Th2 lymphocytes, favoring bacterial survival and proliferation, ultimately causing death (10).
GCs also play a pivotal role in metabolic diseases and is determinant in the progression of obesity and metabolic syndrome to T2D. Although circulating GCs in T2D individuals are not different to healthy subjects (14), there is a local conversion of inactive to active GCs in metabolic organs such as the liver and adipose tissue (15). Non‐adrenal glucocorticoids synthesis is mediated by 11‐β‐hydroxysteroid dehydrogenase type 1 (11‐βHSD1), an oxo‐reductase enzyme that uses nicotinamide adenine dinucleotide phosphate [NADP(H)] as a co‐factor. This enzyme converts inactive cortisone to active cortisol in humans or corticosterone in rodents (15). Interestingly, isozyme 11‐βHSD2 exerts the opposite effect. This NAD‐dependent dehydrogenase converts active cortisol or active corticosterone to inactive cortisone (16). The higher activity of 11‐βHSD1 in organs such as lung, liver and adipose tissues in T2D results in increased local GC production with deleterious consequences. Increased conversion of active glucocorticoids in visceral adipose tissue increases lipolysis and circulating free fatty acid content (17). Experimental studies with transgenic mice over‐expressing 11‐βHSD1 selectively in adipose tissue have demonstrated that an increase in local levels of corticosterone induces visceral obesity, insulin resistance, diabetes and hyperlipidemia (14).
Moreover, up‐regulation of 11‐βHSD1 expression in liver of T2D patients increases local synthesis of GCs leading to augmented glycogenolysis. Hepatic GCs also stimulate glucose output by activating phosphoenolpyruvate carboxykinase (PEPCK) gene expression (18), resulting in unrestrained glucose release. Sustained hyperglycemia induces glucotoxic damage and immune dysfunction, deteriorating the condition of diabetic patients (19). As the increased activity of 11‐βHSD1 in liver and adipose tissues is implicated in the development of T2D (20) it is plausible that the same mechanism occurs in the lung during TB. However, as far as we know, there are no published studies concerning the pulmonary production of active GCs mediated by 11‐βHSD1 in TB. Accordingly, novel therapeutic approaches for T2D or TB should be aimed to prevent abnormal GCs signaling and 11‐βHSD1 expression in non‐adrenal organs.
Dehydroepiandrosterone (DHEA) is an anabolic hormone of the adrenal cortex involved in the conversion of sexual steroids. This hormone displays antagonistic activity to cortisol, counteracting the deleterious effect of cortisol on immune response (21) and glucose metabolism. The cortisol–DHEA ratio is modified in T2D/TB co‐morbidity and represents a deleterious factor for both diseases (12). However, the anabolic effects of DHEA prevents its pharmacological use in the treatment of these diseases. 16α‐bromoepiandrosterone (BEA) is a synthetic analog of DHEA that modulates immune and metabolic responses (22, 23). Compared to DHEA, BEA does not display anabolic activities, which makes it a feasible candidate drug for T2D and TB.
In this study, we studied the expression of 11‐βHSD1 and 11‐βHS2 in the lung and liver, as well as the concentrations of GCs in a murine model of T2D/TB co‐morbidity, and evaluated the therapeutic effect of BEA administration during experimental late TB and T2D/TB.
MATERIALS AND METHODS
Ethics statements
All the animal work was performed according to the guidelines of the Mexican law NOM 061‐Z00‐1999 and approved by the Internal Committee for the Care and Use of Laboratory Animals (CICUAL) of the National Institute of Medical Sciences and Nutrition in México.
Experimental model of T2D and progressive pulmonary TB in BALB/c mice
To induce T2D, we used a non‐genetic mouse model of diet‐induced insulin resistance and pancreatic beta cell insufficiency, as previously reported (24, 25). Briefly, male BALB/c 3‐week‐old mice were fed with a high‐fat diet (HFD) containing 45% of calories from fat during the whole course of the experiment. Control animals were fed with rodent chow. After 5 weeks, animals received a single low dose (100 mg/kg) of streptozotocin (STZ) (ChemCruz; Santa Cruz Biotechnology, Dallas, TX, USA) by intraperitoneal (i.p.) injection. This low dose of STZ protocol does not cause diabetes in chow‐fed mice (24). We assessed the combined effect of HFD and STZ in fasting blood glucose levels before and 1 month after STZ administration with glucose test strips (Accu‐chek Active; Roche Diabetes Care Mannheim, Germany) in blood obtained from the tail vein. Serum cholesterol and triglyceride concentrations were measured in blood obtained from the tail vein with test strips (Accutrend Plus System; Roche) 1 month after STZ administration.
After 1 month of STZ administration, animals were treated with either metformin (250 mg/kg) or glibenclamide (15 mg/kg) intragastrically and fasting glucose was determined 8 and 24 h after administration. Animals confirmed with T2D were infected by the intratracheal route with a high dose of the Mtb reference strain H37Rv (ATCC no. 25618), in order to induce progressive pulmonary TB. Briefly, Mtb strain H37Rv was grown in Middlebrook 7H9 broth (Difco, Detroit, MI, USA) supplemented with 0.2% glycerol, 10% oleic acid–albumin–dextrose–catalase (OADC) enrichment and 0.02% Tween‐80 and maintained at 37°C in agitation. Mid‐log‐phase cultures were used for infection. Mycobacteria were counted and stored at −80°C until use. Bacterial aliquots were thawed and pulse‐sonicated to remove clumps. For the infection protocol, groups of T2D and control animals were anesthetized in a gas chamber using Sevoflurane and infected intratracheally with 2.5 Å ~ 105 live bacilli using a cannula in a biosafety level III cabinet and maintained in a vertical position until spontaneous recovery. Mice were maintained in groups of five throughout the study in cages fitted with micro‐isolators connected to negative pressure in an animal biosafety level III facility.
In order to compare the evolution of TB in T2D and control mice, groups of five mice were euthanized by exsanguination under pentobarbital anesthesia after 14, 21, 28 and 120 days after infection. Left lungs were immediately excised, cleared from hilar lymph nodes and thymic tissues, frozen by immersion in liquid nitrogen and kept to −80°C for bacillary loads determination by colony‐forming unit (CFU) quantification and TNF‐α expression determined by reverse transcription–polymerase chain reaction (RT–PCR), as described below. Right lungs were intratracheally perfused with absolute ethanol and after 24 h the tissue was paraffin‐embedded for histology and automated morphometry studies. Ten animals per group were left untouched and their survival was recorded to construct survival curves.
Expression and cellular source of 11‐βHSD1, 11‐βHSD2 and glucocorticoids in the lungs and liver during late active pulmonary TB
Groups of five mice with T2D, TB and T2D/TB were euthanized by exsanguination under pentobarbital anesthesia after 60 and 120 days after infection in two independent experiments. In this model the late active disease is well established by this time, as characterized by extensive pneumonia, high bacillary loads and low expression of IFN‐γ and TNF‐α. The lungs were processed as described above. Left lungs were used for total RNA isolation and mRNA determination of 11‐βHSD1 and 11‐βHSD2 by RT–PCR, while right lungs were used for histology and immunohistochemistry studies, including the determination of the cellular source of corticosterone and cortisone and their concentrations by digital pathology. Tissue fragments from the liver were processed in the same way.
BEA treatment in infected animals with and without T2D
Two months after infection, groups of mice with TB or T2D/TB were administered every other day with 0.02 mg/kg BEA either by the intratracheal route to evaluate its direct effect on the infected lung or by subcutaneous route to determine the effect of BEA in the liver. After 30 and 60 days of treatment, groups of six animals were euthanized by exsanguination under anesthesia with 210 mg/kg of i.p. pentobarbital inside in a biosecurity‐level III cabinet. Three left lungs per time were perfused with absolute ethanol, fixed and prepared for histopathological studies, as mentioned above. Five right lungs were frozen for bacilli load quantification and three lungs were used to determine mRNA abundance by RT–PCR. Two independent experiments were performed. Animals were monitored daily and humanely euthanized if they exhibited respiratory insufficiency or substantial weight loss.
Determination of colony‐forming units in lung
Left lungs and spleen from four mice at each time‐point, in two different experiments, were used for bacillary load determination by CFU quantification. Lungs were homogenized in 1 ml of 0.05% Tween‐80 in a sample homogenizer (FastPrep‐24; MP Biomedicals, Santa Ana, CA, USA). Four consecutive logarithmic dilutions were made from this homogenate. Ten microliters of each dilution were plated in duplicate on Bacto Middlebrook 7H10 agar (Difco, Franklin Lakes, NJ, USA) enriched with OADC. Plates were incubated at 37ºC and 5% CO2 during 21 days for CFU quantification.
Preparation of lung tissue for histological and immunohistochemistry analysis
Right lungs of mice were perfused with absolute ethanol by the endotracheal route, fixed for 24 h and then embedded in paraffin blocks. Sections of 4 μm width were mounted on glass slides, deparaffinized and stained with hematoxylin and eosin. For quantification and morphometric analysis for determining the surface area affected by pneumonia, three different mouse lungs per time‐point in two independent experiments were evaluated.
For immunohistochemistry, lung tissue was sectioned and mounted on charged glass slides and then deparaffinized. Slides were first blocked for unspecific peroxidase activity with 3% methanol peroxide for 1 h. For tissue 11‐βHSD1 and 11‐βHSD2 abundance detection, rabbit anti‐mouse monoclonal antibodies (Biorbyt, Cambridge, UK) were used at a concentration of 1 : 250, incubated overnight in agitation, followed by incubation with secondary anti‐rabbit immunoglobulin (Ig)G labeled with peroxidase. For cortisone and corticosterone immunostaining, rabbit anti‐mouse monoclonal antibodies (LsBio, Seattle, WA, USA) were used at a final concentration of 1 : 250. In both cases, bound antibodies were detected with diaminobenzidine and counterstained with hematoxylin.
Immunostained sections were analyzed by digital pathology. Briefly, stained sections were digitalized at ×200 magnification using an Aperio ScanScope CS (Aperio, Sausalito, CA, USA), with a spatial resolution of 0.45 μm/pixels. The images were analyzed using ImageScope software (Aperio). All the lung tissue sections were circumscribed and sent for automated image analysis using Spectrum Software version 11.1.2.752 (Aperio). For immunostaining GC intensity quantification, an algorithm was developed to determine the total lung expression of cortisone and corticosterone in the whole tissue section (26, 27, 28).
Gene expression determination of 11‐βHSD1, 11‐βHSD2 and cytokines in lung and liver by RT–PCR
The right lungs of three mice per time‐point were rapidly excised, collected in 1.5‐ml cryotubes, immediately frozen in liquid nitrogen and maintained at −80°C until processing. For homogenization, lungs were slowly defrosted and homogenized in the FastPrepR‐24 (MP Biomedicals) in the presence of zirconium flint beads. Total RNA extraction was performed with the RNeasy Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
Total RNA was quantified by spectrophotometry (A260/280) and 100 ng of RNA from each lung was used for the production of complementary DNA (cDNA) by retrotranscription using a Omniscript kit (Qiagen, Valencia, CA, USA), following the manufacturer’s instructions. An end‐point polymerase chain reaction (PCR) was then run to amplify the constitutive housekeeping gene [ribosomal protein large P0 (RPLP0) Gen ID:11837, GenBank; NCBI, Bethesda, MD, USA] and its integrity was analyzed by running at 2% agarose gel stained with SYBR green.
The cDNA obtained from each sample was analyzed by real‐time quantitative (RT–qPCR) using the RT–PCR system 7500 (Applied Biosystems, Foster City, CA, USA) and the Quantitech SYBR Green Mastermix kit (Qiagen) with specific primers designed with the first BLAST (ncbi.nlm.nih.gov) for 11‐βHSD1, 11‐βHSD2, IFN‐γ and TNF‐α. Cycling conditions used were as follows: initial denaturation at 95°C for 15 min, followed by 40 cycles at 95°C for 20 s, 60°C for 20 s and 72°C for 34 s. Relative 11‐βHSD1, 11‐βHSD2 mRNA abundance was calculated by the 2−△△Ct method (29). For absolute quantification of IFN‐γ, TNF‐α and 11‐βHSD1 and 11‐βHSD2 mRNA in liver, the number of copies of each target gene was normalized to 1 million amplicons of the housekeeping gene RPLP0 mRNA, including the standard curves and a negative control.
Statistical analysis
All the statistical analyses were performed using GraphPad Prism Software version 6.0 (La Jolla, CA, USA). The data were analyzed using the paired t‐test and one‐ and two‐tailed analyses of variance (anova) with Bonferroni correction for multiple comparisons. p‐values < 0.05 were considered significant.
RESULTS
Biochemical characterization of the T2D mice
To compare the progression and severity of TB in mice with T2D with respect to non‐diabetic mice, we induced diet‐induced insulin resistance and pancreatic beta cell insufficiency in mice by feeding them for 5 weeks with a high‐fat diet containing 45% of calories from fat. Afterwards, animals received a single low dose (100 mg/kg) of STZ via i.p. (Figure 1a). Mice fed with the high‐fat diet showed significant weight gain with respect to control mice fed rodent chow (Figure 1b). Two months after SZT administration, T2D mice showed significantly higher serum glucose, triglycerides, cholesterol and insulin concentrations (Figure 1c–f), confirming that these mice developed the metabolic abnormalities of T2D. Moreover, treatment of these mice with the hypoglycemic drugs metformin or glibenclamide reduced blood glucose to control levels (Figure 1g), indicating that this model of diabetes is responsive to pharmacological anti‐hyperglycemic treatments. In accordance with the biochemical characterization, histological analysis of the liver showed extensive lipid cytoplasmic vacuolization of hepatocytes (steatosis) in T2D mice (Figure 1h).
FIGURE 1.
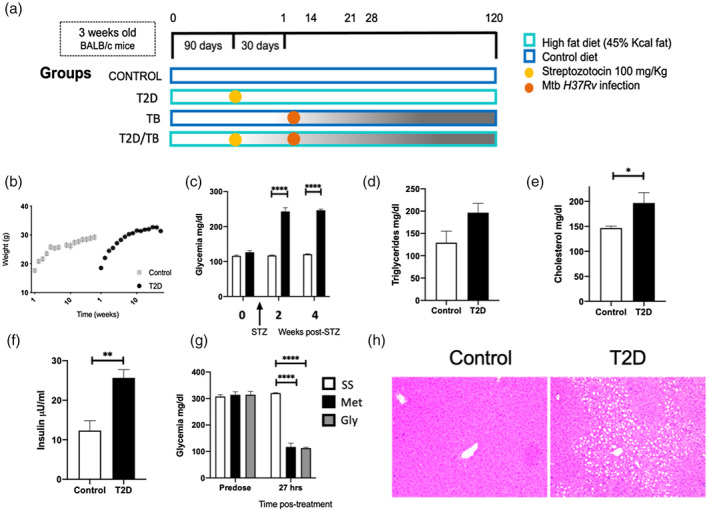
Metabolic abnormalities and liver pathology of mice with type II diabetes mellitus in comparison with non‐diabetic animals (sham). (a) General description of the experimental groups. (b) Mice were weighed weekly for weight gain monitoring. Data are presented as mean and standard deviation of five mice per group for 16 weeks. (c) Blood glucose levels were assessed at 2 and 4 weeks after administration of streptozotocin (STZ) intraperitoneally in animals fed with a high‐fat diet (HFD) and in control animals not treated with STZ and fed with rodent chow. Serum concentrations of (d) triglycerides, (e) cholesterol and (f) insulin in control (white bars) and T2D (black bars) mice after 4 months of HFD‐STZ treatment. (g) Effect of hypoglycemic drugs on serum glucose levels in T2D animals, before and after 27 h of oral administration of metformin (Met) or glibenclamide (Gly). Asterisk represents statistical significance [one‐way anlysis of variance (anova), p < 0.05 and two‐way anova for graph 1B, 1C and 1G). (h) Representative micrograph of liver tissue from a control mouse (left) and T2D mouse (right). Extensive cytoplasmic vacuolization of hepatocytes corresponding to steatosis is shown in the liver of T2D mouse
Comparative evolution of pulmonary disease in TB and T2D/TB mice
In comparison with chow‐fed mice, T2D mice showed significant higher pulmonary bacilli burdens after 28 or 120 days after infection with Mtb (Figure 2a). T2D/TB mice also showed a decreased survival rate and lower expression of TNF‐α in lungs at 120 days after infection than TB mice (Figure 2b,c). Thus, T2D/TB mice developed a more severe disease than TB mice.
FIGURE 2.
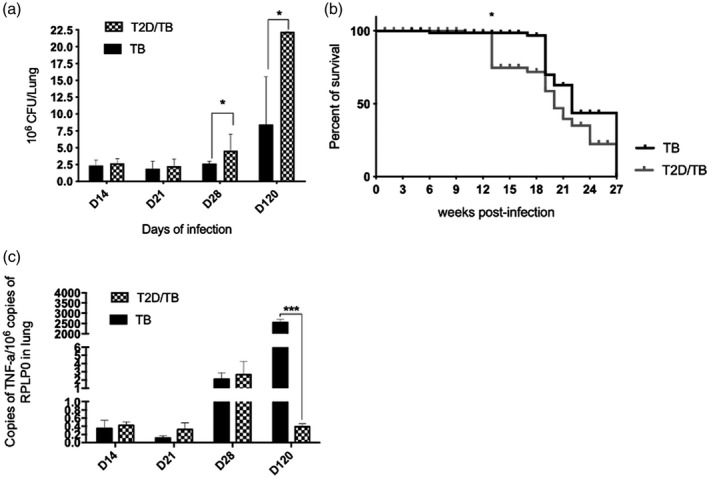
Comparative course of pulmonary tuberculosis in diabetic mice infected with Mycobacterium tuberculosis (Mtb) (T2D/TB) and non‐diabetic mice infected with Mtb (TB). (a) Five mice per group were euthanized at the indicated time‐points post‐infection and the right lungs were used to determine the bacterial loads by counting colony‐forming units (CFU). A significant increase in the bacillary load of the T2D/TB group was observed after 1 and 4 months of infection in comparison with the control group
Comparative expression of GCs and GCs converting enzymes in the late phase of TB and T2D/TB lungs
To determine the role of locally synthesized GCs in the lung in the progression of lung damage, we evaluated corticosterone (active GC) and cortisone (inactive GC) content in the lungs of non‐infected controls and T2D mice, as well as TB and T2D/TB mice at day 120 of infection by immunohistochemistry. As shown in Figure 3a, the lungs of control non‐infected mice showed scarce bronchial epithelial cells with corticosterone‐positive immunostaining, while numerous bronchial cells are strongly positive in T2D mice. In the TB and T2D/TB groups the bronchial epithelium was negative for corticosterone. However, the pneumonic areas showed intense corticosterone immunostaining in some lymphocytes and numerous macrophages, many of them vacuolated or foamy macrophages. Gene expression of the GC converting enzyme 11‐βHSD1 was significantly higher in T2D mice in comparison with the control non‐infected group (Figure 3b). TB and T2D/TB groups also presented a significant increase in 11‐βHSD1 mRNA content with respect to the control group. Immunostaining revealed that, in T2D mice, the 11‐βHSD1 was limited to bronchial epithelial cells, whereas in TB and T2D/TB this enzyme displayed strong immunostaining in foamy vacuolated macrophages of the pneumonic areas.
FIGURE 3.
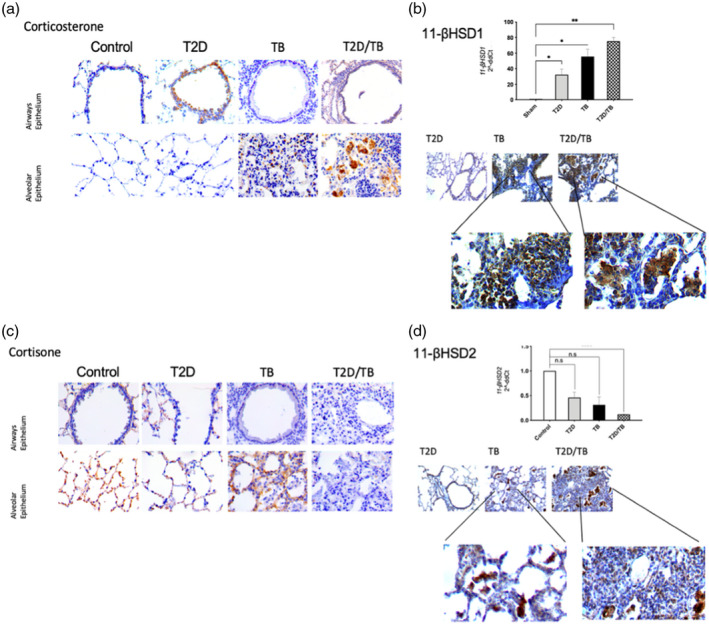
Gene expression and cellular localization of glucocorticoids converting enzymes and glucocorticoid content in late tuberculosis (TB) (day 120). (a) Representative micrographs of immunohistochemical staining of corticosterone in the epithelium of the lung airways and the alveolar epithelium. (b) Comparative gene expression of 11‐β‐hydroxysteroid dehydrogenase type 1 (11‐βHSD1) determined by quantitative polymerase chain reaction (qPCR) in the different experimental groups at day 120 (top panel). Immunohistochemical detection of 11‐βHSD1 (bottom panel). (c) Representative micrographs of immunohistochemical detection of cortisone in bronchial epithelial cells and alveoli. (d) Comparative 11‐βHSD2 gene expression and immunohistochemical staining. Asterisks represent statistical significance [****p < 0.0001, two‐ way analysis of variance (anova) and n.s. = not significant]
Immunohistochemical detection of the inactive GC cortisone revealed positive immunostaining in alveolar epithelium of control mice, while bronchial cells were negative in all conditions. TB mice showed strong immunostaining in alveolar epithelium and in some inflammatory cells, while T2D and T2D/TB were negative to this hormone (Figure 3c). The immunohistochemical results were well correlated with the 11‐βHSD2 mRNA abundance, which showed significantly higher expression in non‐infected lungs, while it was lower in all other groups, where the T2D/TB group showed the lowest content (Figure 3d). These results revealed that at a late stage of the disease there is high expression of 11‐βHSD1 and corticosterone production in the inflammatory cells of pulmonary TB, which is even higher in mice with T2D/TB. Conversely, in TB and T2D/TB mice the 11‐βHSD2 that converts active cortisol or active corticosterone to inactive cortisone is repressed, increasing TB severity.
Effect of BEA in lung and spleen bacillary burdens, pulmonary cytokine expression and tissue damage in TB and T2D/TB mice
To evaluate the therapeutic effect of BEA on Mtb‐infected mice, TB and T2D/TB mice were administered with BEA or vehicle to evaluate bacillary loads (CFUs) in the lung and spleen, extent of tissue damage (percentage of lung surface affected by pneumonia) and gene expression of the protective cytokines TNF‐α and IFN‐γ by RT–PCR. In comparison with mice receiving vehicle, both TB and T2D/TB groups showed significant reduction of pulmonary bacillary burdens after 30 or 60 days of treatment with BEA (Figure 4a). BEA administration reduced the percentage of lung affected by pneumonia in the T2D/TB mice at 60 days (Figure 3b). The administration of BEA increased IFN‐γ and TNF‐α mRNA content in both groups after 30 days of treatment and IFN‐γ in TB group after 60 days of treatment (c), as well as reduction of pneumonia. Interestingly, 60 days of BEA treatment induced a significant reduction of CFUs and weight increase in spleen of the T2D/TB group (Figure 4e,f). These results indicate that BEA administration to T2D/TB mice allows spleen hyperplasia, leading to a more effective control of bacillary dissemination in this experimental group.
FIGURE 4.
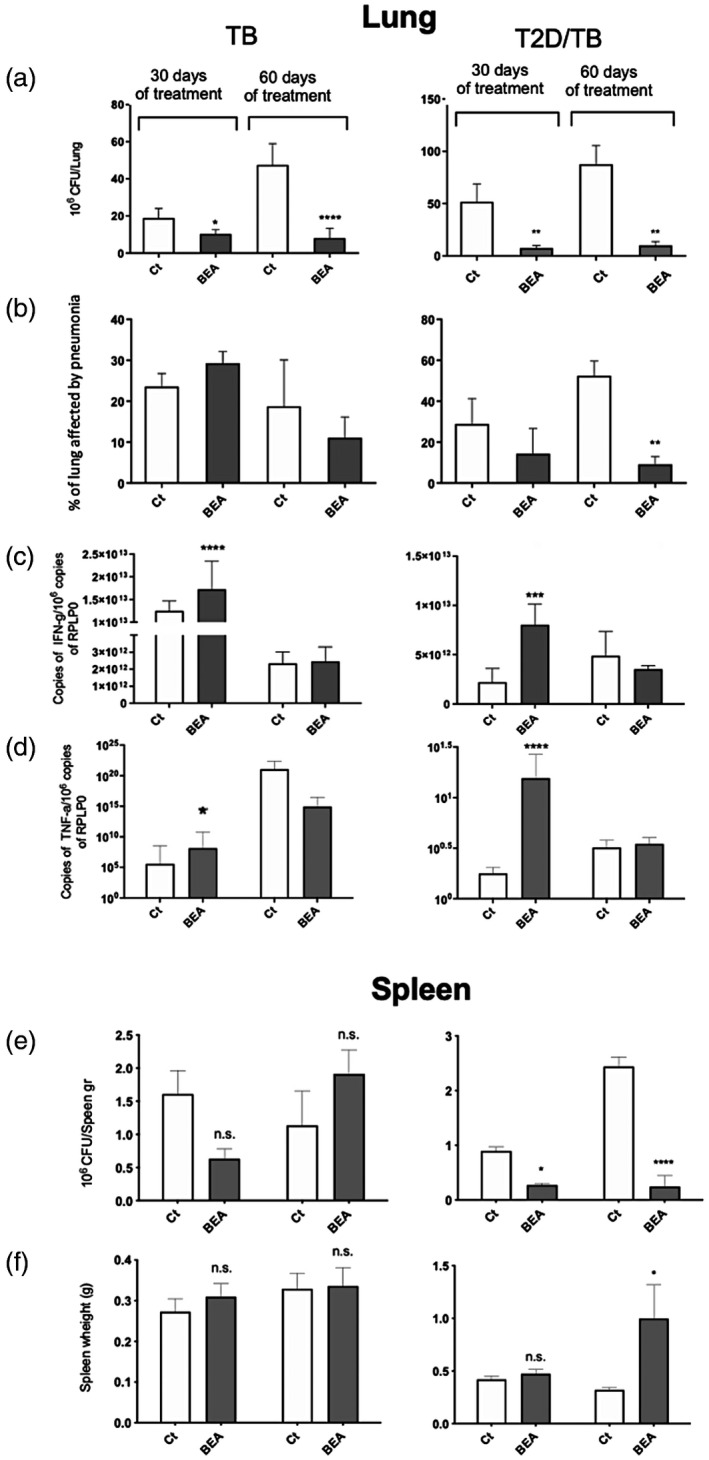
Therapeutic effect of 16α‐bromoepiandrosterone (BEA) in tuberculosis (TB) and type 2 diabetes mellitus (T2D)/TB mice. Groups of TB and T2D/TB mice after 60 days of intratracheal infection with high‐dose Mycobacterium tuberculosis (Mtb) strain H37Rv were treated during 1 or 2 months with BEA, while the control TB or T2D/TB groups received only the vehicle. Lungs were processed for the indicated determination. In comparison with the control TB or T2D/TB mice, BEA induced a significant decrease of pulmonary bacillary loads and tissue damage (pneumonia) in both TB and T2D/TB groups, particularly after 60 days of treatment. After 1 month of treatment, BEA induced higher expression of the protective cytokines interferon (IFN)‐γ and tumor necrosis factor (TNF)‐α. In the spleen, treatment with BEA did not produced significant changes in the weight and colony‐forming units (CFUs) in TB mice, while it induced significant decrease of bacillary burdens and higher weight in T2D/TB mice after 2 months of treatment. Asterisks represent statistical significance [****p < 0.0001, two‐way analysis of variance (anova); n.s. = not significant]
Effect of BEA on the expression of 11‐βHSD1 and 11‐βHSD2, corticosterone and cortisone in the lungs and liver
In the lung, treatment of TB and T2D/TB groups with BEA reduced the expression of 11‐βHSD1 after 30 or 60 days of treatment when compared with the control group (Figure 5a). This effect was even more evident in the liver of T2D/TB group, which also showed complete remission of steatosis and hyperglycemia (Figure 6).
FIGURE 5.
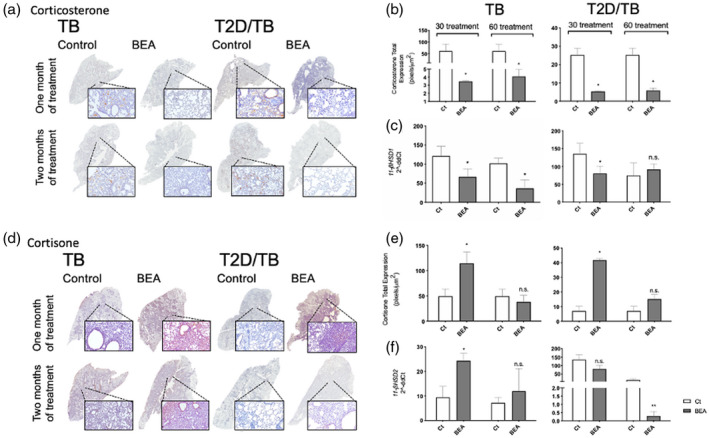
Effect of 16α‐bromoepiandrosterone (BEA) on lung glucocorticoids (GCs) production and gene expression of their converting enzymes in tuberculosis (TB) and type 2 diabetes mellitus (T2D)/TB mice. (a) Representative low‐power and mild‐power micrographs of immunohistochemistry detection of corticosterone in both groups after 1 and 2 months of BEA treatment. (b) In comparison with the TB or T2D/TB groups, the digital pathology analysis showed significant lower immunostaining of corticosterone induced by BEA treatment after 1 and 2 months in TB mice and T2D/TB mice. (c) BEA treatment induced lower 11‐β‐hydroxysteroid dehydrogenase type 1 (11β‐HSD1) gene expression in TB and T2D/TB in comparison with TB or T2D/TB mice treated with vehicle. (d) Representative micrographs of cortisone detection by immunohistochemistry in the lungs of TB and T2D mice treated with BEA at 1 and 2 months of infection. (e) The digital pathology study shows higher expression of cortisone after 1 month of treatment in the TB and T2D/TB groups. (f) Gene expression of 11b‐HSD2 corticosterone converting enzyme after BEA treatment in TB and T2D/TB animals. Asterisk represents statistical significance (p < 0.05)
FIGURE 6.
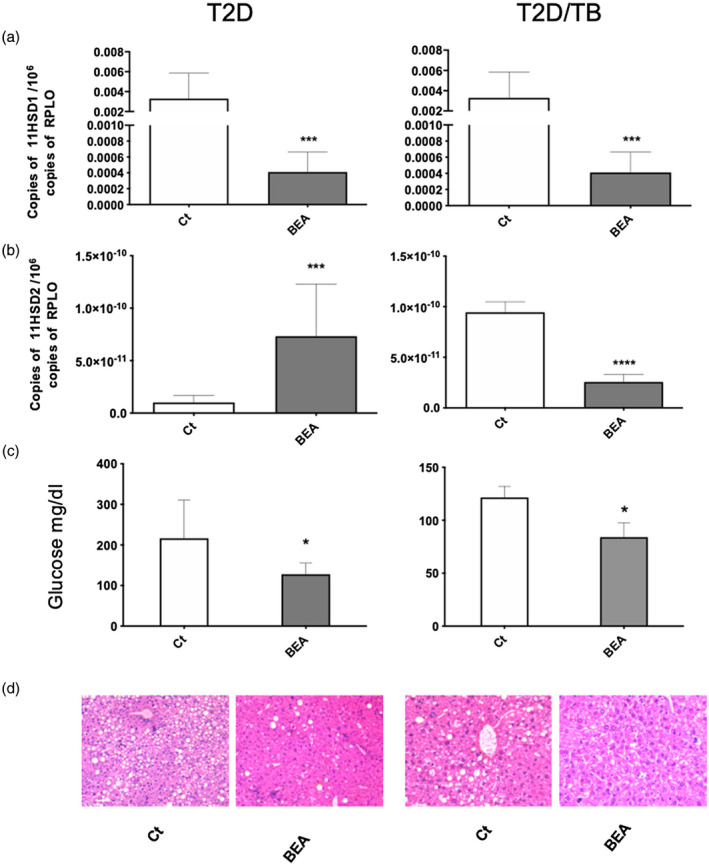
Effect of 16α‐bromoepiandrosterone (BEA) in the liver of tuberculosis (TB) and type 2 diabetes mellitus (T2D)/TB mice. Liver samples from groups of five T2D and T2D/TB mice treated with BEA for 1 month and their respective control mice that received only the vehicle were used to isolate total RNA and 11‐β‐hydroxysteroid dehydrogenase type 1 (1‐βHSD1) (a) and 11‐βHSD2) (b) mRNA determination by reverse transcription–polymerase chain reaction (RT–PCR). (c) Treatment with BEA corrected hyperglycemia in T2D and T2D/TB mice. Asterisks represent statistical significance [***p < 0.0001, ****p < 0.0001, two‐way analysis of variance (anova), n.s. = without significance]. D) Liver tissue sections stained with hematoxylin and eosin (H&E) revealed steatosis in T2D and T2D/TB mice, which was reverted by treatment with BEA
The effects of BEA on 11‐βHSD1 gene expression in the infected lung were in concordance with corticosterone immunostaining and its quantitative evaluation by digital pathology (Figure 5a–c). BEA significantly decreased corticosterone content and 11‐βHSD1 mRNA abundance in TB and TB/T2D mice, being more pronounced in the TB group (Figure 5b,c). In contrast, BEA administration increased cortisone content and 11‐βHSD2 expression in TB or T2D/TB mice after 30 and 60 days (Figure 5d–f).
In the liver, treatment of TB and T2D/TB groups with BEA also reduced the expression of 11‐βHSD1 and increased that of 11‐βHSD2 when compared with the control group (Figure 6a,b). This effect was even more evident in the liver of the T2D/TB group, which also showed complete remission of steatosis and hyperglycemia (Figure 6c,d).
These results indicate that BEA acts as a regulator of 11‐βHSD1 and 11‐βHSD2 transcription in the lung and liver, repressing 11‐βHSD1 and up‐regulating 11‐βHSD2 gene expression. In consequence, treatment of Mtb‐infected mice with BEA induced a lower production of active corticosterone and a higher production of inactive cortisone which, in turn, contribute to the increased expression of the proinflammatory cytokines TNF‐α and IFN‐γ, decreasing the pulmonary bacillary loads. BEA administration improved lipid and glucose metabolism in the liver and normalized liver histology and glucose blood concentrations.
Thus, BEA administration improved the immune response and metabolism in T2D/TB co‐morbidity, suggesting that it could potentially be used as a novel immune therapeutic agent with beneficial immune, endocrine and metabolic activities.
DISCUSSION
T2D/TB co‐morbidity is an emerging area of interest due to its alarming increase in incidence worldwide. T2D is a metabolic and endocrine disease characterized by a progressive whole‐body glucose intolerance that leads to chronic hyperglycemia, affecting multiple organs. T2D compromises host immune responses and impairs host ability to control microbial infections (6, 7). Indeed, T2D impairs the immune response against Mtb, worsening the course of the disease and requiring longer courses of chemotherapy (8, 9). Our results confirm that T2D/TB co‐morbidity leads to a more aggressive course of TB in a murine model of T2D/TB and, for the first time, demonstrates that one significant pathogenic factor for increased TB severity is the higher expression of 11‐βHSD1 and corticosterone production during late advanced disease in the lungs and liver. Moreover, treatment with BEA efficiently decreases the expression of 11‐βHSD1, reducing local corticosterone synthesis. This effect of BEA significantly reduced bacillary burdens and hyperglycemia; these results suggest that this synthetic hormone could be considered a feasible candidate drug for T2D and TB co‐morbidity.
Mtb affects mainly the lungs, producing chronic and excessive inflammation in which innate and adaptive immunity are severely affected (3, 4). Mtb infection initiates by inhalation of saliva droplets with Mtb that are engulfed by alveolar macrophages. Macrophages are key cells in bacilli elimination, and together with dendritic cells process Mtb antigens that are presented to T lymphocytes in regional lymph nodes. Lymphocytes migrate to the lung and, together with macrophages and other cells, form containment structures known as granulomas, which are the histopathological hallmark of TB (6). During early stages of active infection Th1 cellular immune responses are protective, as IFN‐γ, TNF‐α and other cytokines such as IL‐12 induce macrophage activation, allowing containment of bacterial growth. Nevertheless, during late active disease, extensive inflammation produces a shift towards anti‐inflammatory responses such as the Th2 cytokine pattern, in which IL‐4, IL‐13 and other anti‐inflammatory cytokines such as IL‐10 and TGF‐β. This shift in immune response induces a local immunosuppressive/anti‐inflammatory effect, resulting in poor containment of infection and progression of tissue damage (30). Moreover, besides the derangement in the immunological response, there is a heightened neuroendocrine response involving a complex network of cytokines, hormones and neurotransmitters that exacerbates TB pathogenesis (10, 31). These neuroendocrine responses include the cytokines released during the immune response to Mtb (proinflammatory mediators), abnormal production of several hormones and central nervous system (CNS) responses, prompting the activation of the two major stress systems, the HPA axis (32) and the sympathetic nervous system (SNS) (7, 33).
The HPA and GCs have an important contribution to the physiopathology of TB (34, 35). It was reported in the experimental model of progressive TB in BALB/c mice that at the time of maximal protective activity mediated by IFN‐γ, TNF‐α, IL‐1β and NO production (day 21 after infection), proinflammatory cytokines such as TNF‐α and IL‐1β strongly activate the HPA axis, producing high expression of corticotropin‐releasing factor (CRF) in the hypothalamus and adrenal hyperplasia with high serum concentrations of corticosterone (32). Then, during the chronic or late phase after 28 days of infection, there is progressive adrenal atrophy and a decrease in circulating corticosterone associated with extensive pneumonia (32). Thus, there is high GC adrenal production apparently with the aim of avoiding tissue damage produced by excessive lung inflammation. During the late phase of the disease, when adrenal glands atrophy, peripheral macrophages become an alternative. It seems that there is another source of corticosterone available by local production in unidentified tissues or cells.
Our results show for the first time, to our knowledge, that this extra‐adrenal source of GCs are macrophages, particularly foamy cells which strongly express the enzyme 11‐βHSD type 1, and serve as an alternative source of corticosterone in the inflamed tuberculous lung. This local source of corticosteroids acts in an autocrine and paracrine manner. However, local excess of corticosterone also inhibits Th1 lymphocyte activity and induces differentiation of Th2 lymphocytes that favors bacterial survival and proliferation causing the animals’ death (15). Thus, this local anti‐inflammatory activity mediated by high production of corticosterone in affected organs during active late TB has a deleterious effect. Antagonistic hormones such as DHEA can restore balance to the system.
DHEA is the most abundant product of the human adrenal gland after puberty, with maximum levels occurring in the late 20s. Its production then falls steadily with advanced age. DHEA has glucocorticoid counter‐regulatory effects in several systems. For example, in obesity DHEA shows opposite effects to GCs on enzyme expression in the liver (17) and in the immune system (36). In a previous study, treatment with DHEA starting on day 60 after Mtb infection in BALB/c mice reduced the pulmonary bacillary burden and decreased tissue damage by reactivating the Th1 cytokine response (37). In humans, a deficit in DHEA relative to cortisol can be striking in severe TB (12). However, DHEA is suboptimal for human use, partly because it is metabolized into sex steroids.
BEA (16a‐bromo‐5a‐androstan‐3b‐ol‐17‐one) is a synthetic adrenal steroid derivative that does not enter sex steroid pathways and has been demonstrated in tuberculous BALB/c mice treated with BEA every other day, beginning on day 60, that it produces a significant inhibition of bacterial proliferation and higher expression of protective cytokines (TNF‐α, IFN‐γ). Moreover, when given as an adjunct to conventional chemotherapy, BEA enhances bacterial clearance (38). Interestingly, this synthetic hormone showed therapeutic benefit in patients with TB, malaria (23, 39) and in the acquired immune deficiency syndrome (AIDS) (40).
In the present study we used a new water‐miscible formulation of BEA. Its ability to form a stable suspension in water is derived from a new method of formulating the molecule. Water miscibility avoids the problems associated with the organic solvents required to administer the former lipid soluble formulation. Mice treated with this new BEA formulation showed similar results to the previous DHEA synthetic analog, producing a significant increase of IFN‐γ and TNF‐α. This effect was associated with pulmonary bacillary load reduction and increased survival (38). In the present study, we demonstrated that BEA administration also reduced pulmonary expression of 11‐βHSD1 and corticosterone. Conversely, BEA increased 11‐βHSD2 which shifts the GCs equilibrium to inactive cortisone. Thus, BEA can induce CD4 Th1 cells and macrophages activation by direct activity and, as shown in the present work, by the suppression of the local production of corticosterone in the lungs.
Obesity is commonly associated with T2D and other manifestations of the metabolic syndrome (41). The metabolic syndrome is characterized by insulin resistance in adipose tissue, skeletal muscle and liver leading to hyperlipidemia, hypertension, hepatic steatohepatitis and T2D. GCs activity antagonize insulin action and are important causative factors in the development of metabolic syndrome and T2D. GCs impair glucose uptake, enhance lipolysis and hepatic gluconeogenesis and promote proteolysis. Moreover, GCs directly inhibit pancreatic secretion of insulin, enhance glucose secretion by inhibiting gluconeogenesis in the liver and oppose other metabolic actions, such as insulin signaling and glucose uptake, by inhibiting the translocation of the glucose transporter GLUT4 to the plasma membrane (20).
Our results show that, in comparison with healthy control animals, mice with T2D without Mtb infection exhibit higher 11‐βHSD1 expression and strong corticosterone immunostaining in bronchial epithelial cells and in hepatocytes. BEA treatment decreased the expression of both enzymes and hormones in lung and liver and corrected glucose concentrations in blood. Thus, BEA significantly improved metabolic abnormalities by down‐regulating 11‐βHSD1 gene expression as one of its therapeutic mechanisms. These results are in agreement with previous publications and contribute new information, such as the increase of corticosterone production in the airways epithelium of T2D individuals, which could be a factor for higher severity of respiratory infections, such as TB.
Our results demonstrate that T2D/TB mice present a higher pulmonary bacillary load than TB mice without T2D, as well as lower expression of IFN‐γ and TNF‐α. These alterations were associated with higher expression of 11‐βHSD1 and strong immunostaining to corticosterone in the pulmonary inflammatory intra‐alveolar infiltrate, particularly in foamy macrophages. Thus, it seems that the higher pulmonary production of corticosterone mediated by 11‐βHSD1 is a factor that worsens the course of TB in T2D, and thus regulation of 11‐βHSD1 expression in the lung could be a new therapeutic target.
In this regard, our results suggest that BEA can block 11‐βHSD1 in the lung and in the liver, making it an effective novel treatment for this co‐morbidity. Trials for the use of BEA in the treatment of human TB, T2D and the co‐morbidity T2D/TB are now justified.
CONCLUSION
The study of T2D/TB co‐morbidity from the immunoendocrine point of view is of utmost importance to understand the background of this binomial, as well as to develop novel strategies for the control of this co‐morbidity. Active GCs represent a negative factor in the development of both entities, with a negative effect on glucose metabolism and the development of a non‐protective immune response. Treatment with BEA represents a novel strategy as an adjuvant in the treatment of TB alone and co‐morbidity with T2D. For these reasons, it is of utmost importance to continue with the study of the BEA through its escalation to clinical studies to be used in patients.
CONFLICT of INTEREST
We declare no conflicts of interest during the course of this work.
AUTHOR CONTRIBUTIONS
L. T.‐M. O., A. C. and H. P.‐R. contributed to the background work and conceived the experiments. L. T.‐M. O. and R. E.‐O. performed, organized and analyzed the results. L. T.‐M. O., B.‐E., E. B. and M. C.‐B. experiments design and execution. B. P.‐J. performed animal infections and drug administration. M. E.‐D. performed the culture and preparation of the Mycobacterium tuberculosis H37Rv strain for infection and CFU experiments, I. T.‐V., N. T. and A. T. designed the hypercaloric diet. C.‐W. and G.‐Y. prepared and supplied BEA for the animal treatment. L. T.‐M. O., I. T.‐V. and H. P.‐R. wrote the paper.
ACKNOWLEDGEMENTS
L. T.‐M. O. is a PhD student and received fellowship 433346 from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowship 433346 from CONACyT. This work was sponsored by Consejo Nacional de Ciencia y Tecnología, Grant/Award Number: FC2015‐1/115. LT‐MO from Consejo Nacional de Ciencia y Tecnología, México (CONACyT).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ayelign B, Negash M, Genetu M, Wondmagegn T, Shibabaw T. Immunological impacts of diabetes on the susceptibility of Mycobacterium tuberculosis . J Immunol Res. 2019;2019:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host–pathogen collusion. Front Immunol. 2012;3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meenakshi P, Ramya S, Lavanya J, Vijayalakshmi V, Sumanlatha G. Effect of IFN‐c, IL‐12 and IL‐10 cytokine production and mRNA expression in tuberculosis patients with diabetes mellitus and their household contacts. Cytokine 2016;81:127–36. https://doi.org/10.1016/j.cyto.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 4. Cooper AM, Mayer‐Barber KD, Sher A. Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 2011;4:252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rook WG. Th2 cytokines in susceptibility to tuberculosis. Curr Mol Med. 2007;7:327–37. [DOI] [PubMed] [Google Scholar]
- 6. Boillat‐Blanco N, Tumbo AMN, Perreau M, Amelio P, Ramaiya KL, Mganga M, et al. Hyperglycaemia is inversely correlated with live M. bovis BCG‐specific CD4+ T cell responses in Tanzanian adults with latent or active tuberculosis. Immun Inflamm Dis. 2018;6:345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology 2015;144:171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rey AD, Mahuad CV, Bozza VV, Bogue C, Farroni MA, Bay ML, et al. Endocrine and cytokine responses in humans with pulmonary tuberculosis. Brain Behav Immun. 2007;21:171–9. [DOI] [PubMed] [Google Scholar]
- 9. Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21:259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pandolfi J, Baz P, Fernández P, Discianni Lupi A, Payaslián F, Billordo LA, et al. Regulatory and effector T‐cells are differentially modulated by dexamethasone. Clin Immunol. 2013;149:400–10. [DOI] [PubMed] [Google Scholar]
- 11. Sorrels SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Bone 2012;23:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernández R, Díaz A, D’Attilio L, Bongiovanni B, Santucci N, Bertola D, et al. An adverse immune–endocrine profile in patients with tuberculosis and type 2 diabetes. Tuberculosis 2016;101:95–101. [DOI] [PubMed] [Google Scholar]
- 13. Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11β‐hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol Cell Endocrinol. 2009;301:123–31. [DOI] [PubMed] [Google Scholar]
- 14. Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001;294:2166–70. [DOI] [PubMed] [Google Scholar]
- 15. Talabér G, Jondal M, Okret S. Extra‐adrenal glucocorticoid synthesis: Immune regulation and aspects on local organ homeostasis. Mol Cell Endocrinol. 2013;380:89–98. [DOI] [PubMed] [Google Scholar]
- 16. Gomez‐Sanchez EP, Ganjam V, Chen YJ, Liu Y, Clark SA, Gomez‐Sanchez CE. The 11β hydroxysteroid dehydrogenase 2 exists as an inactive dimer. Steroids 2001;66:845–8. [DOI] [PubMed] [Google Scholar]
- 17. Apostolova G, Schweizer RAS, Balazs Z, Kostadinova RM, Odermatt A. Dehydroepiandrosterone inhibits the amplification of glucocorticoid action in adipose tissue. Am J Physiol Endocrinol Metab. 2005;288;E957–64. [DOI] [PubMed] [Google Scholar]
- 18. Shukla R, Basu AK, Mandal B, Mukhopadhyay P, Maity A, Chakraborty S, et al. 11β Hydroxysteroid dehydrogenase‐1 activity in type 2 diabetes mellitus: a comparative study. BMC Endocr Disord. 2019;19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sävendahl L. The effect of acute and chronic stress on growth. Sci Signal. 2012;5:9–11. [DOI] [PubMed] [Google Scholar]
- 20. Chiodini I, Adda G, Scillitani A, Coletti F, Morelli V, Di Lembo S, et al. Cortisol secretion in patients with type 2 diabetes: relationship with chronic complications. Diabetes Care. 2007;30:83–8. [DOI] [PubMed] [Google Scholar]
- 21. Buford TW, Willoughby DS. Impact of DHEA(S) and cortisol on immune function in aging: a brief review. Appl Physiol Nutr Metab. 2008;33:429–33. [DOI] [PubMed] [Google Scholar]
- 22. Nicoletti F, Conrad D, Wang A, Pieters R, Mangano K, van Heeckeren A, et al. 16α‐Bromoepiandrosterone (HE2000) limits non‐productive inflammation and stimulates immunity in lungs. Clin Exp Immunol. 2009;158:308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramos‐Espinosa O, Islas‐Weinstein L, Peralta‐Álvarez MP, López‐Torres MO, Hernández‐Pando R. The use of immunotherapy for the treatment of tuberculosis. Exp Rev Respir Med. 2018;12:427–40. 10.1080/17476348.2018.1457439. [DOI] [PubMed] [Google Scholar]
- 24. Luo J, Quan J, Tsai J, Hobensack CK, Sullivan C, Hector R, et al. Nongenetic mouse models of non‐insulin‐dependent diabetes mellitus. Metabolism 1998;47:663–8. [DOI] [PubMed] [Google Scholar]
- 25. Segura‐Cerda CA, Marquina‐Castillo B, Lozano‐Ordaz V, Mata‐Espinosa D, Barrios‐Payán JA, López‐Torres MO, et al. BCG and BCGΔBCG1419c protect type 2 diabetic mice against tuberculosis via different participation of T and B lymphocytes, dendritic cells and pro‐inflammatory cytokines. NPJ Vaccines. 2020;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rangel‐Santiago JF, Baay‐Guzman GJ, Duran‐Padilla MA, Lopez‐Bochm KA, Garcia‐Romero BL, Hernandez‐Cueto DD, et al. A novel role of Yin‐Yang‐1 in pulmonary tuberculosis through the regulation of the chemokine CCL4. Tuberculosis 2016;96:87–95. [DOI] [PubMed] [Google Scholar]
- 27. Baay‐Guzman GJ, Duran‐Padilla MA, Rangel‐Santiago J, Tirado‐Rodriguez B, Antonio‐Andres G, Barrios‐Payan J, et al. Dual role of hypoxia‐inducible factor 1 α in experimental pulmonary tuberculosis: its implication as a new therapeutic target. Future Microbiol. 2018;13:785–98. [DOI] [PubMed] [Google Scholar]
- 28. Zapata‐Tarres M, Juarez‐Villegas LE, Maldonado‐Valenzuela A, Baay‐Guzman GJ, Lopez‐Perez TV, Cabrera‐Muñoz L, et al. Expression of YY1 in Wilms tumors with favorable histology is a risk factor for adverse outcomes. Futur Oncol. 2019;15:1231–41. [DOI] [PubMed] [Google Scholar]
- 29. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–8. [DOI] [PubMed] [Google Scholar]
- 30. Hernandez‐Pando R, Orozco H, Aguilar D. Factors that deregulate the protective immune response in tuberculosis. Arch Immunol Ther Exp. 2009;57:355–67. [DOI] [PubMed] [Google Scholar]
- 31. Bellavance M‐A, Rivest S. The HPA–immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front Immunol. 2014;5:136. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3978367&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hernandez‐Pando R, Orozco H, Honour J, Silva P, Leyva R, Rook GAW. Adrenal changes in murine pulmonary tuberculosis; a clue to pathogenesis? FEMS Immunol Med Microbiol. 1995;12:63–72. [DOI] [PubMed] [Google Scholar]
- 33. Barrios‐Payán J, Revuelta A, Mata‐Espinosa D, Marquina‐Castillo B, Villanueva EB, Gutiérrez MEH, et al. The contribution of the sympathetic nervous system to the immunopathology of experimental pulmonary tuberculosis. J Neuroimmunol. 2016;298:98–105. [DOI] [PubMed] [Google Scholar]
- 34. Irwin MR. Human psychoneuroimmunology: 20 years of discovery. Brain Behav Immun. 2008;22:129–39. [DOI] [PubMed] [Google Scholar]
- 35. Rook GA, Hernandez‐Pando R. Pathogenic role, in human and murine tuberculosis, of changes in the peripheral metabolism of glucocorticoids and antiglucocorticoids. Psychoneuroendocrinology 1997;22(Suppl 1):1–5. [DOI] [PubMed] [Google Scholar]
- 36. Chen CCG, Parker CR. Adrenal androgens and the immune system. Semin Reprod Med. 2004;22:369–77. [DOI] [PubMed] [Google Scholar]
- 37. Streber M, de la Luz M, Orozco H, Arriaga K, Pavon L, Al‐Nakhli SA, et al. The effects of androstenediol and dehydroepiandrosterone on the course and cytokine profile of tuberculosis in BALB/c mice. Immunology 1998;95:234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hernández‐Pando R, Aguilar‐Leon D, Orozco H, Serrano A, Ahlem C, Trauger R, et al. 16alpha‐Bromoepiandrosterone restores T helper cell type 1 activity and accelerates chemotherapy‐induced bacterial clearance in a model of progressive pulmonary tuberculosis. J Infect Dis. 2005;191:299–306. http://doi.org/10.1086/426453. [DOI] [PubMed] [Google Scholar]
- 39. Leenstra T, ter Kuile F, Kariuki S, Nixon C, Oloo A, Kager P, et al. Dehydroepiandrosterone sulfate levels associated with decreased malaria parasite density and increased hemoglobin concentration in pubertal girls from western Kenya. J Infect Dis. 2003;188:297–304. [DOI] [PubMed] [Google Scholar]
- 40. Stickney DR, Noveljic Z, Garsd A, Destiche DA, Frincke JM. Safety and activity of the immune modulator HE2000 on the incidence of tuberculosis and other opportunistic infections in AIDS patients. Antimicrob Agents Chemother. 2007;51:2639–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Zyl S, van der Merwe LJ, van Rooyen FC, Joubert G, Walsh CM. The relationship between obesity, leptin, adiponectin and the components of metabolic syndrome in urban African women, Free State, South Africa. South African J Clin Nutr. 2017;30:68–73. 10.1080/16070658.2017.1267380 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.