

Abstract
Osteoporosis is a common skeletal disorder characterized by deterioration of bone tissue in later life. The set of genetic factors contributing to osteoporosis is not completely specified. Whole exome sequencing was performed on affected-relative-pairs, approximately cousins, from 10 high-risk osteoporosis pedigrees to identify rare, highly penetrant candidate predisposition variants. Variants were filtered on population frequency and concordance between pairs of cousins. We identified 3 candidate variants that were tested for segregation in 74 additional relatives of the index carriers. A rare variant in MEGF6 (rs755467862) showed the strongest evidence of segregation, with 6 affected pedigree members carrying the variant (RVsharing probability=0.0011). Functional impact of the MEGF6 variant was analyzed via in-silico prediction of protein folding disruption using I-Tasser software that predicted substantial protein folding differences between wild type and variant MEGF6 proteins. Predicted protein folding showed that the variant (Cys200Tyr) may disrupt a disulfide bond between residues 187 and 200 in the second EGF-like calcium-binding domain of MEGF6, likely eliminating a calcium binding site. Furthermore, functional analyses in zebrafish demonstrate that the paralogous genes megf6a and megf6b contribute in a redundant fashion to cartilage and bone formation. Segregation analyses, in-silico protein structure modeling, and functional assays support a role for MEGF6 in predisposition to osteoporosis.
INTRODUCTION
Osteoporosis is a common skeletal disorder characterized by deterioration of bone mineral density (BMD) and increased risk of fracture (1). Identification of genetic factors that have a strong influence on disease could enhance screening and knowledge concerning biological pathways involved. Genome-wide association studies (GWAS) have identified over 500 common single nucleotide polymorphisms (SNPs) associated with BMD (2), revealing several relevant biological pathways including genes involved in WNT signaling, the RANK/RANKL/OPG pathway, and genes involved in endochondral ossification. GWAS studies have revealed that osteoporosis is highly polygenic. SNPs identified thus far are estimated to account for about 20% of trait variation (3), whereas overall heritability estimates for BMD are higher, ranging from 50-90% (4-5), which may in part be due to rare genetic variations that are not typically detectable in GWAS. Family based designs present a complimentary approach that may provide opportunities to discover rare, highly penetrant variants (6). Using a multi-generational pedigree resource, we performed exome sequencing on affected-relative-pairs with a deficiency in BMD from 10 high-risk osteoporosis pedigrees to identify rare, coding variants that may be contributing to osteoporosis risk in these families. Analysis of this resource identified a rare coding variant in MEGF6 (multiple epidermal growth factor like protein 6) gene (Cys200Tyr) that segregates to other family members with osteoporosis. Computational and functional analyses indicate that the MEGF6 variant likely disrupts wildtype (WT) protein function and that zebrafish paralogues contribute to bone and cartilage formation in vivo.
RESULTS
Variant prioritization
Exome sequencing of the 20 affected individuals in 10 pedigrees identified a total of ~45K variants. Among these, 548 coding variants in 480 genes were rare (MAF<0.005 in EXAC browser (29) and concordant between a sequenced pair of affected cousins. Restricting candidates to IBD regions for sequenced pairs as identified by SGS analysis further limited the selection to 125 rare, shared candidate variants in 111 genes occurring in regions labeled as IBD. A search of PubMed for each of the 111 genes to identify previous reported association to osteoporosis further limited selection to variants in 12 candidate genes. In the fourth filtering step, variants that were scored as ‘damaging’ by at least 8 of 10 in-silico pathogenicity prediction algorithms included in ANNOVAR variant annotations were retained, resulting in 3 candidate genes (MEGF6 (rs755467862), PAX8 (rs1269341622), and UMPS (rs777350640)).
Confirmation of candidate variants via segregation
Eight carriers of the MEGF6 variant rs755467862 (NC_000001.11:g.3524129C>T, NC_000001.11(MEGF6_v001):c.599G>A, NC_000001.11(MEGF6_i001):p.Cys200Tyr) were identified among 14 sampled relatives of the index cousins that were assayed for the variant. The sequenced index cousins as well as one relative had a physician confirmed diagnosis of osteoporosis, four carriers had a physician confirmed diagnosis of osteopenia, and one carrier was unaffected (screened in the 50th decade of life). Four of the eight carriers of the MEGF6 variant had a medical history of bone fractures, and four had been previously treated for osteoporosis. Of the remaining 6 relatives that did not carry the variant, 2 had a physician confirmed diagnosis of unaffected and 4 were considered undeterminable. The de-identified pedigree segregating rs755467862 is depicted in Figure 1 and had significant evidence of segregation to affected relatives (osteoporosis or osteopenia, RVsharing p=0.0011) after correcting for 3 tests of hypothesis (α=0.05/3=0.017).
Figure 1.
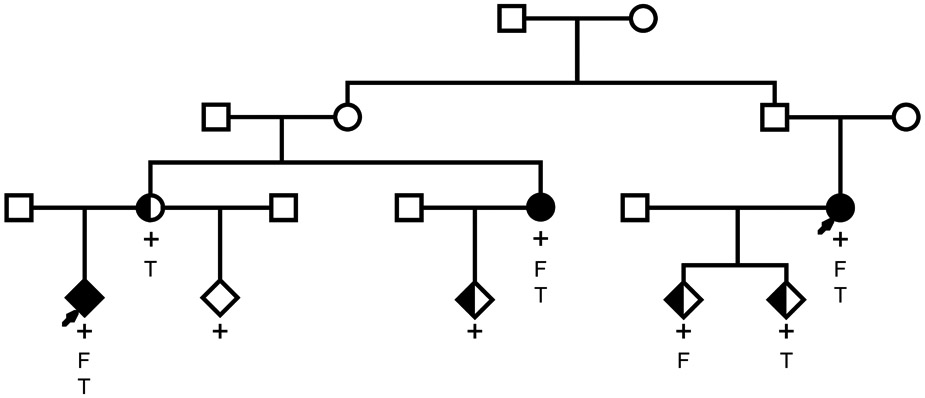
Osteoporosis pedigree segregating MEGF6 variant. Physician confirmed diagnosis of osteoporosis is denoted by black fill and physician confirmed diagnosis of osteopenia is denoted by half-shading. Arrows indicate index sequenced subjects, ‘+’ indicates confirmed carriage of the variant, ‘F’ indicates multiple fractures in the patient’s medical history, and ‘T’ indicates previous treatment for osteoporosis.
In-silico functional evaluation of rs755467862
Variant rs755467862 is a non-synonymous variant substituting base T for C (reference allele) in exon 5 that codes a Cysteine > Tyrosine amino acid substitution. Population frequency has been estimated (MAF/number of alleles tested) as T=4e-5/243,578 (30). Variant rs755467862 was predicted to be damaging by almost all of the functional prediction software included in Annovar (12), including Polyphen (31), LRT_pred (32), MutationTaster (33), FATHMM (34), Radial_SVM (35), LR score (35), but not SIFT (36). The GERP score for the variant is 5.0, indicating strong evidence for conservation of the reference allele across species (37).
Protein interaction network
Analysis with the STRING database tool (18) showed that this protein is related to pathways involved in various biological processes (Figure 2A) including carcinogenesis (GULP1, a known tumor suppressor gene (38)) angiogenesis (EMILIN1, a protein involved in vessel assembly (39), and most notably, osteoporosis (ZBTB40 (40-41)). Figure 2B contains a STRING graph re-centered on ZBTB40 that shows connections with WNT16, which has been previously associated with BMD (42).
Figure 2.

Protein interaction network from STRING database centered on MEGF6 (A) and ZBTB40 (B). Red nodes indicate the query protein and first shell of interactors, white nodes indicate second shell of interactors. Empty nodes indicate proteins of unknown 3-dimensional structure and filled nodes indicate 3-dimensional structure is known or predicted. Teal connectors indicate known interactions from curated databases, purple connectors indicate experimentally determined interactions, black connectors indicate co-expression, and yellow connectors indicate that interactions originate from text-mining.
Protein structure prediction
MEGF6 is a very large protein with 1541 amino acids and is not amenable to existing 3D structure prediction methods. The candidate variant rs755467862 produces the amino acid substitution Cys200Tyr that occurs in the second EGF-like calcium binding domains. I-TASSER software predicted the structures of the second domain of MEGF6 (residues 161-201) for both wild type and variant amino acid sequences, resulting in two distinct candidate structures for the wild type and five structures for variant proteins. From the ten possible comparisons, five (50%) showed substantial differences between the wild type and variant predicted structures. An example of this is presented in Figure 3 where it is apparent that the amino acid substitution disrupts a disulfide bond between residues 187 and 200 in the second EGF-like calcium-binding domain, where a helix is formed instead of an open loop. Similar behavior was observed in the other comparisons. The main structure of EGF-like domains is a two-stranded β-sheet followed by a loop where the calcium ion binds (43). The collapse of this loop may eliminate this calcium binding site in MEGF6, which may decrease, or even eliminate, the functionality of the protein.
Figure 3.

Comparison of the structure of MEGF6 wild type (tan) and variant (blue) in the region of the Cys200Tyr. The disulfide bond between residues 187 and 200 have been highlighted in the wild type and mutant structures in red and yellow, respectively. The elimination of a loop can be visually observed, which is likely to adversely affect the calcium binding function of the first cbEGF-like domain in MEGF6.
Analysis of Zebrafish megf6a and megf6b
The zebrafish genome contains two megf6 paralogues, megf6a and megf6b, which are orthologous to the single human MEGF6 gene. Each orthologue has similar degree of identity (megf6a - 56% and megf6b – 54%) and similarity (megf6a - 68% and megf6b – 64%) to the human orthologue. The expression patterns of megf6a and megf6b were determined by whole-mount in situ hybridization (WISH) at 24, 36, and 48 hpf. At 24 hpf megf6a is broadly expressed in the head and localized to the fin fold (Supplemental Figure 1A). At 36 hpf it is localized to the apical ectodermal ridge (AER) of the pectoral fin (Supplemental Figure 1B), the atrioventricular canal (Supplemental Figure 1C), the notochord (Supplemental Figure 1D), and broadly expressed in the head, while the expression in the fin fold is waning. megf6b is expressed in the same domains as megf6a at 24 hpf, but is also expressed in the developing somites (Supplemental Figure 1E). The expression pattern of megf6b is maintained in the fin fold and head at 36 hpf and is also expressed in the AER of the pectoral fin and in the notochord (Supplemental Figure 1F). Both megf6a and megf6b continue to be expressed in the notochord of 48 hpf larvae (data not shown). In the head of 48 hpf larvae, megf6b is expressed in the mouth and pharyngeal region (Supplemental Figure 1G). These data indicate that megf6a and megf6b have mostly overlapping expression domains during the first 48 hpf of development and are expressed in regions of the embryo that give rise to cartilage and bone.
Functional Analyses of Zebrafish megf6a and megf6b
Computational analysis suggested that the MEGF6 candidate osteoporosis variant allele (Cys200Tyr) might be deleterious or damaging to WT protein function. To determine the function of these paralogues, we produced embryos lacking both gene functions. To generate zebrafish lacking both megf6a and megf6b gene function, we first generated a germline deletion of megf6b and then utilized injection of CRISPR-Cas9 RNPs to disrupt megf6a. Both megf6a and megf6b are large genes containing 34 and 38 exons, respectively. To make a large deletion in megf6b, we injected RNPs targeting exons 6 and 35 into one-cell stage embryos. We identified one allele, z48 - a 51860 bp deletion with a 53 bp insertion, which stably transmitted through the germline (Supplemental Figure 2). megf6b mutants are homozygous viable and the only overt phenotype is ectopic migration of a small number of melanocytes in the trunk and tail of 48 hpf larvae (Figure 4A-B’).
Figure 4.

Larvae lacking megf6a and megf6b gene function have fin fold defects at 48 hpf. WT or megf6b−/+ larvae have no overt phenotypes (A and A’), while megf6b−/− larvae have a few aberrant melanocytes (B’, arrow) present in the caudal fin area (B and B’). A’ and B’ are high magnification views of dashed boxes in A and B. (C-F) Injection of megf6a RNPs into larvae from an intercross between megf6b+/− adults. Loss of megf6a gene function has no effect in a WT or megf6b+/− background (C), while loss of megf6a gene function in megf6b−/− larve disrupted fin fold development and led to aberrant migration of melanocytes into the caudal fin area (D-F). All abnormal larvae were genotyped and determined to be megf6b−/−, indicating complete loss of megf6b function is required for the phenotype observed in the megf6a RNP-injected larvae (p < 0.0056, Fisher’s exact test with Bonferroni correction). Panels D-F depict the spectrum of aberrant phenotypes observed. Lateral views with anterior to the left.
Paralogous genes in zebrafish can have redundant biological functions (44). Since megf6a and megf6b are both expressed in the fin fold, head, and notochord, we hypothesized they might have partially redundant functions. Therefore, we attempted to eliminate megf6a function in megf6b null mutants using CRISPR-Cas9 RNP reagents that efficiently eliminate target gene function (45). We injected CRISPR-Cas9 RNPs targeting exons 6 and 28 of megf6a into one-cell stage embryos generated from an intercross between megf6b+/− adults. Whereas three-quarters of the embryos appeared phenotypically WT following CRIPSR-Cas9 mutagenesis of megf6a (Figure 4C), roughly one-quarter of the larvae (21.7%, (n =115)) exhibited a severe disruption of the fin fold with aberrant migration of melanocytes into the fin fold area (Figure 4 D-F). The aberrant embryos were genotyped, and all were found to be homozygous for the megf6b mutation, indicating that mutagenesis of megf6a only has measurable effect on fin fold development in the absence of megf6b function (p = 0.0056). These results indicate that megf6a and megf6b have redundant functions during embryogenesis: loss of either gene had minimal effect on development whereas larvae lacking both megf6a and megf6b gene functions had severe defects on fin development.
In the zebrafish larvae, cartilage and bone forms in an anterioposterior wave of development, and at any single time point posterior structures are developmentally younger than anterior ones. Therefore, we examined cartilage and bone formation at two developmentally distinct time points, 10 and 14 day post fertilization (dpf). To determine if megf6a and megf6b have a function in cartilage and bone development in the head, we analyzed 10 dpf larvae that developed from megf6b+/− intercross eggs injected with megf6a RNPs. To visualize cartilage and bone formation, larvae were stained with Alcian Blue (cartilage) and Alizarin Red (bone). Injection of megf6a RNPs into WT or megf6b+/− larvae had no measurable effect on bone and cartilage formation (Figure 5A and 5A’). However, twenty percent of the injected intercross larvae (n = 110) exhibited consistent delays in jaw development, including formation of the pharyngeal arches and development of both bones (5th ceratobranchyal (5CB) or cleithrum (CL)) and cartilaginous structures of the jaw (Figure 5B and 5B’). Genotyping revealed that all of embryos with jaw defects were megf6b−/−, indicating that loss of both megf6 genes was required (p = 0.0062) to produce significant delays in jaw development.
Figure 5.

Cartilage and bone formation is delayed in larvae lacking both megf6a and megf6b genes. Progeny of an megf6b−/+ intercross were injected with megf6a RNPs. (A-B’) Cartilage and bone formation in the heads of RNP-injected WT, megf6b−/+ (A and A’), or megf6b−/− (B and B’) larvae were analyzed at 10 dpf. Arrowhead indicates the 5th ceratobranchyal (5CB) and arrow indicates the cleithrum (CL). Twenty-two of 110 larvae exhibited delayed formation of jaw bones and pharyngeal arches, and all of these were megf6b−/−, indicating loss of both genes is required for the developmental defect (p < 0.0062, Fisher’s exact test with Bonferroni correction). A and B, lateral view and A’ and B’, ventral view. Asterisk indicates the notochord. (C-F) Cartilage and bone formation in in the tails of RNP-injected WT, megf6b−/+, or megf6−/− larvae were analyzed at14 dpf. Mineralization in the vertebra and formation of cartilaginous structures of the caudal fin are significantly delayed in larvae lacking megf6a and megf6b function (17.5%, n=80) (D-F) compared to WT or megf6b+/− (C) larvae injected with megf6a RNPs. All abnormal larvae were genotyped and determined to be megf6b−/− indicating loss of both genes is required for the delayed phenotpye (p < 0.007, Fisher’s exact test with Bonferroni correction). Panels D-F depict the spectrum of phenotypes observed when megf6a RNPs are injected into megf6b−/− embryos. E- H, lateral views with anterior to the left.
Given that megf6a and megf6b have overlapping expression domains in the notochord (which gives rise to the vertebral column) and loss of both megf6a and megf6b function affects the fin fold (Figure 4D-F), we analyzed the roles of the genes in bone and cartilage formation in the vertebral column and caudal fin at 14 dpf. Progeny of a megf6b+/− intercross mating were injected with megf6a RNPs. Most larvae exhibited normal bone and cartilage formation in the vertebral column and caudal fin (Figure 5C). However, 17.5% of the larvae (n=80) exhibited a severe delay in the ossification of the posterior vertebrae and a delay in the formation of the cartilaginous structures and bony rays of the caudal fin (Figure 5D-F). As all the abnormal larvae proved to be megf6b−/− upon genotyping, only embryos lacking both megf6b and megf6a gene functions had an altered phenotype (p < 0.007). The functions of both genes are required for proper formation of both cartilaginous and bony structures in the zebrafish larvae.
To determine if zebrafish lacking megf6a and megf6b function have an adult phenotype, we separated 48 hpf larvae that developed from megf6b+/− intercross eggs injected with megf6a RNPs (Figure 4) into two groups of 25 animals, those with normal fin folds (megf6b−/?) and those with fin fold defects (megf6b−/−) and raised them to 4 months of age. We measured the weight (g) and length (cm) of individual male and female adult zebrafish. The males in the abnormal fin fold group weighed less (mean ± SEM - 0.4098g ± 0.014g) than the males with normal fin folds (0.4556g ± 0.014g), while there was no significant difference in length between the two groups (abnormal fin folds – 3.671cm ± 0.032cm vs normal fin folds – 3.721cm ± 0.035cm). In contrast to the males, females in the abnormal fin fold group weighed more (abnormal fin folds - 0.6616g ± 0.021g vs normal fin folds 0.5332g and ± 0.020g) and were longer (abnormal fin folds – 3.971cm ± 0.035cm vs normal fin folds – 3.809cm ± 0.034cm) than females with normal fin folds (Supplemental Figure 3).
DISCUSSION
Whole exome sequence data from pairs of related osteoporosis-affected individuals from 10 extended high-risk pedigrees were analyzed to identify a small set of rare, IBD-shared variants representing excellent candidates responsible for osteoporosis predisposition in the pedigrees studied. A rare MEGF6 variant segregated with osteoporosis in the pedigree in which it was identified. A combination of in silico modeling and functional analyses in zebrafish suggests that this very rare variant in MEGF6 may predispose to osteoporosis.
The design of the current study focused on exome sequencing of pairs of related affected individuals selected from extended high-risk pedigrees. Selection of subjects focused on affected cousins, which has been shown to be ideal when selecting subjects to represent dense pedigrees (9). The main benefit of the affected-relative-pairs strategy is efficiency with respect to data capture, but at the potential cost of missing evidence of disease variants that may have been detectable in a design that sequences all pedigree members. Here, the strategy was sufficiently robust to lead to identification of one variant in ten pedigrees. The reliance on SGS analysis to delineate regions of long-range sharing between target pair members provided a productive filter of potential candidate variants. The use of well-sampled high-risk pedigrees has the added advantage of being able to invoke a test of segregation among additional affected family members to gain evidence of linkage to disease, which was shown here to be particularly useful strategy for confirmation of the very rare variant rs755467862. A test of association in an independent set of cases and controls was hampered by the rarity of the variant (2 carriers per 10,000 individuals).
Previous linkage of low BMD to chromosome band 1p36.32, which contains rs755467862, has been reported; this provides further supporting evidence for the results obtained here. A LOD score of +2.74 to 1p36.32 was reported in a resource of 1,270 subjects in 324 osteoporosis pedigrees (46). Independently, a LOD score of +3.07 to 1p36.3 was reported in a single Belgian pedigree with 34 subjects (47); exon sequencing of MEGF6 did not identify any coding variants among pedigree members in the Belgian study.
In silico modeling of the WT and variant MEGF6 indicate that the Cys200Tyr may result in collapse of a loop that likely eliminates a calcium biding site in MEGF6, which may decrease, or even eliminate, the functionality of the protein. Similar arguments have been made by observation of mutations of EGF-like domains in other proteins. For instance, more than 60% of the mutations causing Marfan syndrome occur within fibrillin-1 cbEGF-like domains, emphasizing that correct folding of these domains is critical for molecular function and that mutations in cbEGF-like domains that affect cysteine residues are likely to alter disulfide bond formation, thereby disrupting the correct fold, while mutations affecting residues in the calcium binding consensus sequence reduce calcium binding affinity, leading to structural destabilization (48).
MEGF6 is a calcium binding protein that regulates cell adhesion that accelerates cell migration via TGF-β/SMAD signaling mediated epithelial-mesenchymal tissue transition (49) and showed that depletion of MEGF6 caused reduction in TGF-β1 and phosphorylation of SMAD2 and SMAD3. Furthermore, MEGF6 expression was positively correlated with that of TGF-β1, which indicates a role for MEGF6 in TGF-β/SMAD signaling (49). TGF-β and its proposed role in osteoporosis is due to its regulation of osteoclastogenesis through activation of the osteoprotegerin (OPG) promoter (50). OPG has been shown to inhibit the activity and survival of osteoclasts in vitro and bone resorption in vivo (41). It is plausible that increased TGF-β expression caused by loss of MEGF6 function could lead to disinhibition of osteoclastogenesis that could lead to osteoporosis. Furthermore, we identified another potential link between MEGF6 and osteoporosis through a ZBTB40 -WNT16 interaction using the STRING database. Variants in ZBTB40 and WNT16 have been previously associated with osteoporosis (42,51) and osteoblast-derived WNT16 inhibits osteoclastogenesis to prevent bone fragility (52). Further studies are needed to determine if MEGF6 interacts with ZBTB40 or WNT16.
Our functional analyses demonstrate that the zebrafish orthologues of human MEGF6, megf6a and megf6b, have redundant activities needed for proper development and mineralization of both cartilage and bone in the head, vertebrae, and caudal fin. In the head of 10 dpf larvae, we observed a delay in the formation of the cartilaginous pharyngeal arches and a delay in mineralization of the 5th ceratobranchyal (5CB) and the cleithrum (CL) bones, while there was no defect in the initial mineralization of the notochord (Figure 5A-B’). At 14 dpf there was a delay in the mineralization of the vertebral column and bones of the caudal fin, and a delay in the formation of cartilaginous structures of the caudal fin (Figure 5C-F). The mineralization of the 5CB, CL, and notochord (vertebral column) represent distinct modes of bone ossification: endochondral, membranous, and perichordal bone ossification, respectively. Given there is a delay in mineralization of all of these structures, megf6a and megf6b have important functions in the distinct modes of bone ossification.
The bone phenotype observed in larvae lacking megf6a and megf6b function is strikingly similar to several previously reported zebrafish mutant phenotypes. A mutation in the macrophage-stimulating protein (msp) gene or morpholino knockdown of its receptor, ron-2, display similar defects in the 5CB bone (53). The msp mutant phenotype can be rescued by exogenous calcium supplementation to the media, indicating that Ron-2 and Msp are necessary for bone mineralization and calcium homeostasis (53). The zebrafish Chihuahua (Chi) mutant disrupts Col1a1a (G574D) and is a model for classical osteogenesis imperfecta. Heterozygous Chi mutants have defects in the 5CB and CL bones and a delay in vertebral mineralization that are conspicuous of larvae with disrupted mefg6a and megf6b function (54). The zebrafish frilly fins (frf) mutant disrupts the bmp1a gene and is phenotypically similar to the microwaved mutant, which carries a mutation in Col1a1a (Q888K) (55). BMP1 has a known function in the proteolytic processing of procollagen I C-propeptide to generate mature collagen type I (56) and may account for the phenotype similarities between the mutants. The frf mutant has a delay in the mineralization of the vertebrae reminiscent of larvae without megf6a and megf6b gene function (55). Given the similarity between these mutant phenotypes and that MEGF6 is a calcium binding protein, MEGF6 may have a role in regulating calcium homeostasis and collagen processing during embryonic development.
A MEGF6-deficient mouse was recently reported with no discernable phenotype (57). The apparent discrepancy between the mouse and zebrafish mutants may lay in the phenotypic analyses performed. There was no detailed histological analysis of embryonic skeletal development, which may have overlooked subtle defects, and BMD was not analyzed in the adult. Skeletal phenotypes may be uncovered upon a more detailed analysis, since Megf6 is expressed in the mouse musculoskeletal system during development (58), during osteoblast differentiation, and in mature bone (59). Furthermore, Wang et. al., used CRISPR-Cas9 technology to produce deletions in exon 5, which resulted in no detectable Megf6 transcript 57). The mutant transcript likely undergoes non-sense mediated decay, which may trigger genetic compensation that upregulates related genes with sequence similarity to mask the null phenotype (60-61).
Despite the dramatic developmental phenotype of megf6b−/− larvae injected with megf6a RNPS (zebrafish with abnormal fin folds), adult zebrafish do not exhibit severe phenotypes (e.g., kyphosis), although there is a significant difference in the weight and length of these zebrafish. The adult males with abnormal fin folds weigh less than controls with normal fin folds without a reduction in overall length, which may reflect a loss in bone density. In contrast to the adult males, adult females with abnormal fin folds weigh more and are longer than controls with normal fin folds (Supplemental Figure 3). This phenotype may be due to factors unique to the teleost lineage, including sex differences and compensatory growth mechanisms. Zebrafish grow in size throughout their lifetime and have prodigious regenerative capacities, which may contribute to the mild adult phenotype and the observed phenotypic differences between males and females. Further characterization of the zebrafish megf6a;6b mutant phenotype in aged adults and generation of zebrafish with the human disease allele (25) will provide insight into the genetic mechanisms of osteoporosis and may yield new targets for therapeutic intervention. Evidence of segregation, protein folding prediction, and in vivo functional evaluation support a role for MEGF6 in predisposition to development of osteoporosis.
METHODS
Utah osteoporosis pedigrees
Osteoporosis cases with a self-reported family history were ascertained and recruited from clinics associated with Intermountain Healthcare, the largest Utah health care provider, and local advertising in health care clinics; relatives of these cases were also recruited. All recruited individuals were offered a BMD scan for determination of osteoporosis phenotype. BMD was measured on a Hologic QDR 4500A fan-beam dual X-ray absorptiometry (DXA) bone densitometer by a single technician (8). Quality control for the DXA was performed daily on a spine phantom and all scans were reviewed and evaluated by a single experienced physician. BMD was measured at the hip and spine (or at the wrist if hip or spine was not possible); in addition to the anteroposterior (AP) view of the spine, lateral measurements were taken. Scans were excluded if their interpretation was confounded by poor positioning or abnormal anatomy.
A total of 1,871 individuals were consented, phenotyped, and sampled in 276 multi-generational pedigrees with 4–46 sampled individuals per pedigree. In addition to BMD measurements, a single physician reviewed a subject medical history questionnaire, and available medical records to assign a phenotype of osteoporosis, osteopenia, unaffected, or unknown. Ten pedigrees were selected for sequencing based on a physician diagnosis of osteoporosis in a pair of (approximately) cousins (9), and having additional sampled affected relatives for tests for segregation This study was approved by the Institutional Review Boards of Intermountain Healthcare and the University of Utah.
Utah genomic data
Whole exome sequencing was performed on the 10 pairs of affected cousins at the Huntsman Cancer Institute’s Genomics Core facility. DNA libraries were prepared from 3 micrograms of DNA using the Agilent SureSelect XT Human All Exon + UTR (v5) capture kit. Samples were run on the Illumina HiSeq 2000 instrument. Reads were mapped to the human genome GRCh37 reference using BWA-mem (10) for alignment and variants were called using Genome Analysis Toolkit (GATK) (11) software following Broad Institute Best Practices Guidelines. Exome capture resulted in an average of 85% of target bases being covered by greater than 10x coverage across the genome. Variants occurring outside the exon capture kit intended area of coverage were removed. Variants were annotated with Annovar, which contains predicted pathogenicity scores from 10 in-silico functional prediction algorithms (12). Samples were also genotyped on the Illumina OmniExpress high density SNP array (720,000 SNPs).
Candidate variant prioritization
Variants were filtered in four phases to achieve a small set of highly probable candidates. In the first phase, variants with population minor allele frequency (MAF)<0.005 that were observed in both index cases from a pedigree (affected cousins) were retained. In the second phase of variant filtration, the genomes of index pairs were restricted to regions exhibiting evidence of identity-by-descent (IBD) transmission, or shared common ancestry. Focus on these IBD regions between sequenced cousin-pairs provides a useful filter that can rule out variants that are shared but were not inherited from a common ancestor and are inconsistent with a dominant mode of inheritance. SNP genotypes were used to estimate regions of long shared haplotypes between related sequenced cases using Shared Genomic Segments analysis (SGS) (13). SGS analysis identifies the set of contiguous markers at which genotyped individuals could share alleles; long runs of such markers indicate likely regions of IBD sharing from a common ancestor (14). Genomic regions were considered as IBD for a sequenced relative pair if the normalized shared-segment length was ≥20 standard deviations from the mean shared-segment length (15). This selection criteria was based on the expected amount of IBD sharing that should occur for a pair of cousins (~12.5%), and provided an equivalent reduction to the genomic search space. The third phase of variant filtration required that the gene had at least one published study connecting the gene with osteoporosis through some genetic study design, identified via PubMed search (16) with search terms ‘osteoporosis’ and gene name or alias provided by OMIM (Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), January 17, 2018: https://omim.org/). In the final filtering step, variants that were scored as ‘damaging’ by at least 8 of 10 in-silico pathogenicity prediction algorithms included in ANNOVAR variant annotations were retained.
Confirmation of candidate variants via segregation to affected relatives
The 10 pedigrees of the sequenced relative pairs had a total of 68 additional sampled relatives, ranging from 4 to 15 per family. To confirm segregation of the candidate predisposition variants in the pedigrees, each variant was assayed in the sampled relatives of sequenced cases using custom Taqman assays (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s specifications on a Bio-Rad CFX96 Real-Time PCR instrument. Evidence of segregation to additional affected relatives was evaluated with RVsharing software (17) that provides the probability of the observed configuration of sharing in a pedigree, expressed as an exact probability, assuming the shared variant is rare (<1%) and entered the pedigree only once.
Pathway analysis and protein structure prediction
The STRING database tool (18) with default settings was used to query a large number of known protein interactions concerning the candidate gene. Protein structure prediction software I-TASSER using full homology modeling was used to model the structure of critical domains assuming either wild-type or variant amino acid sequences (19-22). Predicted structures were visualized and analyzed using Chimera software (23).
Zebrafish experiments
Zebrafish -
Danio rerio were maintained in accordance with approved institutional protocols at the University of Utah. Adult zebrafish were maintained under standard conditions (24) and kept on a light-dark cycle of 14 hours in light and 10 hours in dark at 27°C. The Tu strain was used in all experiments.
Megf6a and Megf6b Mutant Zebrafish Generation –
Mutations were generated with CRISPR-Cas9 reagents as described in (25). gRNA target sequences are as follows: megf6a exon 6 - CGCTGTCAGCATGGTGTTCT(TGG) and exon 28 - GCTCGCAGTGACCCTCTCAC(AGG); and megf6b exon 6 - AGCACACATGTGTGAACACT(AGG) and exon 35 - TCCTGTATCCGGATGACAGG(AGG). The PAM sequence is indicated in parentheses. Target-specific Alt-R® crRNA and common Alt-R® tracrRNA were synthesized by IDT and dissolved in duplex buffer (IDT) as a 100μM stock solution. Equal volumes of the Alt-R® crRNA and Alt-R® tracrRNA stock solutions were mixed together and annealed in PCR machine using the following settings: 95°C, 5 min; cool at 0.1°C/sec to 25°C; 25°C, 5 min; 4°C. Cas9 protein (Alt-R® S.p. Cas9 nuclease, v.3, IDT, dissolved in 20mM HEPES-NaOH (pH 7.5), 350mM KCl, 20% glycerol) and crRNA:tracrRNA duplex mixed to generate a 5μM gRNA:Cas9 RNP complex (referred to as RNPs). Prior to microinjection, the RNP complex solution was incubated at 37°C, 5 min and then placed at room temperature. Approximately one nanoliter of 5μM RNP complex was injected into the cytoplasm of one-cell stage embryos.
Genomic DNA extraction, HRMA, and PCR genotyping -
Genomic DNA was extracted from individual embryos at 24 hours post fertilization (hpf). Dechorionated embryos were incubated in 50 ul 50 mM NaOH at 95°C, 20 min. 1/10 volume of 1 M Tris-HCl (pH 8.0) was added to neutralize. Genome sequences containing CRISPR/Cas9 target sites were amplified with pairs of primers: megf6a ex6 HRMA F1 – GGAGATTGTCAATACCTGTGACT and megf6a ex6 HRMA R1 – AGGTCTTCGGCGAGCTGATA; megf6a ex28 HRMA F1 – GGTCAGGACTGTGCTGGAGTG and megf6a ex28 HRMA R1 – CCCACAGTCCCGTCCACGC; megf6b ex6 HRMA F1 – ATATTGACGAGTGCCAGGTTCAT and megf6b ex6 HRMA R1 – ATGCAGTCGAGAGCCGGCGTTA; megf6b ex35 HRMA F1 – GCTCTGGGTGTCAGCAGCAGT and megf6b ex35 HRMA R1 – GCTGACAGCGCGGGCCGT. To determine if individual gRNA:Cas9 RNPs produced mutations at the desired target sites, HMRA was performed on DNA isolated from 8 individual 24 hpf gRNA:Cas9 RNP-injected embryos using KAPA HRM FAST PCR Master Mix (26). To generate the megf6b deletion allele, a mixture of gRNA:Cas9 RNPs targeting exons 6 and 35 was injected into the cytoplasm of one-cell stage embryos. To detect deletion events, PCR was performed with megf6b ex6 HRMA F1 and megf6b ex35 HRMA R1 primers on DNA isolated from 8 individual 24 hpf G0 gRNA:Cas9 RNP injected embryos using KAPA 2G FAST PCR Master Mix. To remove megf6a gene function in G0 embryos, a mixture of gRNA:Cas9 RNPs targeting exons 6 and 28 was injected into the cytoplasm of one-cell stage embryos.
In Situ Hybridization –
Whole mount in situ hybridization (WISH) was performed on embryos as described in (27). cDNA fragments used to generate riboprobes probes were amplified by RT-PCR using the following primers: megf6a ISH F – TGGCACCTGCAGCTGCCC, and megf6a ISH R – TCCAGCCGTTCAGACACGTGCA, or megf6b ISH F – TGAACAGACGTGTCCGCAGGG, and megf6b ISH R – TCACACTCGCACAGCAGAGAGC using KAPA 2G FAST Master Mix, subcloned into pGEM-T Easy, and subjected to Sanger sequencing for verification.
Cartilage and Bone Staining –
Ten and 14 dpf zebrafish larvae were anesthetized with Tricaine methanesulfonate (ethyl 3-aminobenzoate methanesulfonate) and processed as described in (28) and https://wiki.zfin.org/pages/viewpage.action?pageId=13107375 with the following modifications. Larvae were fixed in 2% paraformaldehyde for 1 hour, washed for 10 minutes in 50% EtOH, and then transferred to a solution containing 0.01% Alizarin Red and 0 .04% Alcian Blue for 24 hours. Larvae were washed in 80% EtOH/10mM MgCl2 for 60 minutes, 50% EtOH for 30 minutes, 25% EtOH for 30 minutes, bleached in 3% H2O2/0.5% KOH for 15 minutes, washed in 2X 25% glycerol/0.1% KOH and then transferred to 50% glycerol/0.1% KOH for imaging.
Length and Weight Measurements –
One-cell stage embryos from an intercross between megf6b+/− adults were injected with megf6a RNPs and larvae were sorted at 48 hpf into two groups of 25 animals, those with normal fin folds (megf6b−/?) and those with fin fold defects (megf6b−/−). Animals were raised to 4 months of age. At 4 months of age adults were anesthetized using Tricaine methanesulfonate and the length (cm) of each zebrafish was determined by measuring from the anterior most portion of the head to the tip of the tail. Zebrafish were then weighed and allowed to recover from anesthesia.
Statistical analysis –
One-cell stage embryos from an intercross between megf6b+/− adults were injected with megf6a RNPs and larvae were sorted at 48 hpf based on whether or not they displayed the mutant fin fold phenotype. Embryos were then genotyped as describe above. For the cartilage and bone analysis, embryos were stained, sorted based on phenotype, and then genotyped. To determine whether phenotypically defective larvae were significantly enriched for the megf6b−/− genotype, Fisher’s exact test with Bonferroni correction was used to determine statistical significance. A Student’s t-test was used to determine statistical significance for the weight and length comparisons.
Supplementary Material
megf6a and megf6b have both overlapping and unique expression domains. Whole mount in situ hybridization of megf6a (A-D) and megf6b (E-F) at 24 hpf (A and E), 36 hpf (B, C, D, and F), and 48 hpf (G). megf6a and megf6b are expressed in the fin fold (arrow in A) and in the head of 24 hpf embryos (A and E), while only megf6b is expressed in somitic tissue (arrow in E). At 36 hpf megf6a and megf6b are expressed in the fin fold (D and F), AER (arrowhead in B and F), atrioventricular canal (arrowhead in C), and notochord (arrowhead in D and F), and head. megf6b is expressed in the mouth (arrow in G) and pharyngeal region (arrowheads in B) in 48 hpf larvae. Lateral views with anterior to the left.
Generation of a heritable megf6b deletion mutant. (A) RNPs were designed to target sites (lightning bolts) within exon 6 (ex 6 gRNA) and exon 35 (ex 35 gRNA) of megf6b. Primer pairs used to amplify junction fragments joining target sites are indicated (red arrows). (B) Detailed view of the ex 6 gRNA and ex 35 gRNA sites with gRNA target sequence in read, PAM site in blue, and Cas9 cleavage site indicated by lightning bolts. megf6bΔ ex 6/35 represents a conceptual precise joining of sequences flanking the presumed cleavage sites in target sites ex 6 and ex 35. megf6bz48 represents the deletion allele recovered in the germline. This allele contains a 52,960 bp deletion with a 53 bp insertion.
Weight (g) and length (cm) of adult zebrafish lacking megf6a and megf6b function. The weight (A) and length (B) of individual male and female 4 month old zebrafish generated from megf6b+/− intercross eggs injected with megf6a RNPs. Circles indicate embryos identified at 48 hpf with normal fin folds (megf6b−/?) and squares indicate those with fin fold defects (megf6b−/−). Males with abnormal fin folds weighed less than controls with normal fin folds (A), while there was no difference in length between the two groups (B). Females with abnormal fin folds weighed more and were longer when compared to controls with normal fin folds (A and B). n = 14 males and n = 11 females with normal fin folds and n= 14 males and n = 7 females with abnormal fin folds. A Student’s t-test was used to determine statistical significance. Data are represented as means ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
ACKNOWLEDGEMENTS
The samples used for this project originated from a collaboration between Myriad Genetics, Intermountain Healthcare, and the University of Utah. We are especially grateful to the families who participated in this study.
FUNDING
This work was supported by a pilot award from the University of Utah Center on Aging to C.C.T.; the Utah Genome Project to M.J.J.; and the National Institutes of Health [1R01HD081950 to D.J.G., T15LM00712418 to J.C.F., 1S10OD02164401A1 to J.C.F., 1ULTR002538 to J.C.F., P30 CA42014 to L.A.C.A].
Footnotes
CONFLICT OF INTEREST STATEMENT
No conflicts of interest are reported by the authors.
REFERENCES
- 1.Rocha-Braz MGM, and Ferraz-de-Souza B (2016) Genetics of osteoporosis: searching for candidate genes for bone fragility. Arch. Endocrinol. Metab , 60 , 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trajanoska K, and Rivadeneira F (2019) The genetic architecture of osteoporosis and fracture risk. Bone. , S8756-3282 , 30133–30134. [DOI] [PubMed] [Google Scholar]
- 3.Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, Vulpescu NA, Forgetta V, Kleinman A, and Sindhu T (2019) An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet , 51 , 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.François S, Benmalek A, Guaydier-Souquières G, Sabatier JP, and Marcelli C (1999) Heritability of bone mineral density. Rev. Rhum. Engl. Ed , 66 , 146–151. [PubMed] [Google Scholar]
- 5.Duncan EL, Cardon LR, Sinsheimer JS, Wass JA, and Brown MA (2003) Site and gender specificity of inheritance of bone mineral density. J. Bone. Miner. Res , 18 , 1531–1538. [DOI] [PubMed] [Google Scholar]
- 6.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. (2009) Finding the missing heritability of complex diseases. Nature. , 461 , 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bansal A, Hughes DC, Farnham JM, Bagi CM, O'Neil G, Rowe K, Shakib JH, Wood GC, Wyckoff JA, and Cannon-Albright LA (2000) Impact of correlated factors on bone density in individuals with a family history of osteoporosis. J. Clin. Densitometry , 3 , 333–338. [DOI] [PubMed] [Google Scholar]
- 8.Kanis JA, McCloskey EV, Johansson H, Oden A, Melton LJ, and Khaltaev N (2008) A reference standard for the description of osteoporosis. Bone. , 42 , 467–475. [DOI] [PubMed] [Google Scholar]
- 9.Feng BJ, Tavtigian SV, Southey MC, and Goldgar DE (2012) Design considerations for massively parallel sequencing studies of common familial cancers. Hered. Cancer. Clin. Prac , 10 , A38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, and Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics. , 26 , 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Gen. Res , 20 , 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K, Li M, and Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequence data. Nucleic Acids Res. , 38 , 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas A, Camp NJ, Farnham JM, Allen-Brady K, and Cannon-Albright LA (2008) Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. Ann. Hum. Genet , 72 , 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas A, Skolnick MH, and Lewis CM (1994) Genomic mismatch scanning in pedigrees. I.M.A.J. Math. Appl. Med. Biol , 11 , 1–16. [DOI] [PubMed] [Google Scholar]
- 15.Huff CD, Witherspoon DJ, Simonson TS, Xing J, Watkins WS, Zhang Y, Tuohy TM, Neklason DW, Burt RW, Guthery SL, et al. (2011) Maximum-likelihood estimation of recent shared ancestry (ERSA). Gen. Res , 21 , 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.N.C.B.I. Resource Coordinators. (2016) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. , 44 , D7–D19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bureau A, Younkin SG, Parker MM, Bailey-Wilson JE, Marazita ML, Murray JC, Mangold E, Albacha-Hejazi H, Beaty TH, and Ruczinski I (2014) Inferring rare disease risk variants based on exact probabilities of sharing by multiple affected relatives. Bioinformatics. , 30 , 2189–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Research. , 45 , 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y (2008) I-TASSER server for protein 3D structure prediction. B.M.C. Bioinf , 9 , 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y (2009) I-TASSER: Fully automated protein structure prediction in CASP8. Prot. Struct. Func. & Bioinf , 77 , 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy A, Kucukural A, and Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protocols , 5 , 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods , 12 , 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J. Comp. Chem , 25 , 1605–1612. [DOI] [PubMed] [Google Scholar]
- 24.Westerfield M (2010) The Zebrafish Book. Univ. of Oregon Press, Eugene, OR. [Google Scholar]
- 25.Hoshijima K, Jurynec MJ, and Grunwald DJ (2016) Precise editing of the zebrafish genome made simple and efficient. Dev. Cell , 36 , 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dahlem T, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, and Grunwald DJ (2012) Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. , 8 , 1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurynec MJ, Xia R, Mackrill JJ, Gunther D, Crawford T, Flanigan KM, Abramson JJ, Howard MT, and Grunwald DJ (2008) Selenoprotein N is required for ryanodine receptor calcium release channel activity in human and zebrafish muscle. Proc. Nat. Acad. Sci. U.S.A , 105 , 12485–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker MB, and Kimmel CB (2007) A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem , 82 , 23–28. [DOI] [PubMed] [Google Scholar]
- 29.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature. , 536 , 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, and McVean GA (2012) An integrated map of genetic variation from 1,092 human genomes. Nature. , 491 , 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat. Methods , 7 , 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Jian X, and Boerwinkle E (2013) dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutation , 34 , E2393–E2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwarz JM, Cooper DN, Schuelke M, and Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods , 11 , 361–362. [DOI] [PubMed] [Google Scholar]
- 34.Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR, and Campbell C (2015) An integrative approach to predicting the functional consequences of non-coding and coding sequence variation. Bioinformatics. , 31 , 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, and Gaunt TR (2013) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutation , 34 , 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng PC, and Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. , 31 , 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, and Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comp. Biol , 6 , e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayashi M, Reis LO, Baras A, and Maldonado L (2015) Engulfment gene GULP1 is a functional tumor suppressor through influencing TGF-β pathway and is silenced by promoter methylation in urothelial carcinoma. A.A.C.R , Abstract 4943. [Google Scholar]
- 39.Munjal C, Opoka AM, Osinska H, James JF, Bressan GM, and Hinton RB (2014) TGF-β mediates early angiogenesis and latent fibrosis in an Emilin1-deficient mouse model of aortic valve disease. Dis. Mod. & Mech , 7 , 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chao TH, Yu HN, Huang CC, Liu WS, and Lu KH (2012) Opposite associations of osteoprotegerin and ZBTB40 polymorphisms with bone mineral density of the hip in postmenopausal Taiwanese women. J. Chin. Med. Assoc , 75 , 335–340. [DOI] [PubMed] [Google Scholar]
- 41.Boyle WJ, Simonet WS, and Lacey DL (2003) Osteoclast differentiation and activation. Nature. , 423 , 337. [DOI] [PubMed] [Google Scholar]
- 42.García-Ibarbia C, Delgado-Calle J, Casafont I, Velasco J, Arozamena J, Pérez-Núñez MI, Alonso MA, Berciano MT, Ortiz F, Pérez-Castrillón JL, et al. (2013) Contribution of genetic and epigenetic mechanisms to Wnt pathway activity in prevalent skeletal disorders. Gene. , 532 , 165–172. [DOI] [PubMed] [Google Scholar]
- 43.Rao Z, Handford P, Mayhew M, Knott V, Brownlee GG, and Stuart ZD (1995) The structure of a Ca2+-binding epidermal growth factor-like domain: Its role in protein-protein interactions. Cell. , 82 , 131–141. [DOI] [PubMed] [Google Scholar]
- 44.Prince VE and Pickett FB (2002) Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet , 3 , 827–837. [DOI] [PubMed] [Google Scholar]
- 45.Hoshijima K, Jurynec MJ, Klatt-Shaw D, Jacobi AM, Behlke AM, and Grunwald DJ (2019) Generation of heritable deletion mutations and F0 embryos that lack gene function in zebrafish. Dev. Cell, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karasik D, Myers RH, Hannan MT, Gagnon D, McLean RR, Cupples LA, and Kiel DP (2002) Mapping of quantitative ultrasound of the calcaneus bone to chromosome 1 by genomewide linkage analysis. Osteoporos. Int , 13 , 796–802. [DOI] [PubMed] [Google Scholar]
- 47.Willaert A, Van Pottelbergh I, Zmierczak H, Goemaere S, Kaufman JM, De Paepe A, and Coucke P (2008) A genome-wide linkage scan for low spinal bone mineral density in a single extended family confirms linkage to 1p36. Eur. J. Hum. Genet , 16 , 970–976. [DOI] [PubMed] [Google Scholar]
- 48.Kielty CM, Sherratt MJ, Marson A, and Baldock C (2005) Fibrillin Microfibrils. Adv. Prot. Chem , 70 , 405–436. [DOI] [PubMed] [Google Scholar]
- 49.Hu H, Wang M, Wang H, Liu Z, Guan X, Yang R, Huang R, Tang Q, Zou C, Wang G, et al. (2018) MEGF6 promotes the epithelial-to-mesenchymal transition via the TGFβ/SMAD signaling pathway in colorectal cancer metastasis. Cell. Physiol. Biochem , 46 , 1895–1906. [DOI] [PubMed] [Google Scholar]
- 50.Thirunavukkarasu K, Miles RR, Halladay DL, Yang X, Galvin RJ, Chandrasekhar S, Martin TJ, and Onyia JE (2001) Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-β (TGF-β) mapping of the OPG promoter region that mediates TGF-β effects. J. Biol. Chemistry , 276 , 36241–36250. [DOI] [PubMed] [Google Scholar]
- 51.Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Center JR, Nguyen TV, et al. (2008) Multiple genetic loci for bone mineral density and fractures. N. Engl. J. Med , 358 , 2355–2365. [DOI] [PubMed] [Google Scholar]
- 52.Movérare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Börjesson AE, Sjögren K, Windahl SH, Farman H, Kindlund B, et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med 2014;20(11):1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huitema LF, Renn J, Logister I, Gray JK, Waltz SE, Flik G, and Schulte-Merker S (2012) Macrophage-stimulating protein and calcium homeostasis in zebrafish. F.A.S.E.B , 26 , 4092–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gioia R, Tonelli F, Ceppi I, Biggiogera M, Leikin S, Fisher S, Tenedini E, Yorgan TA, Schinke T, Tian K, et al. (2017) The chaperone activity of 4PBA ameliorates the skeletal phenotype of Chihuahua, a zebrafish model for dominant osteogenesis imperfecta. Hum. Mol. Genet , 26 , 2897–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asharani PV, Keupp K, Semler O, Wang W, Li Y, Thiele H, Yigit G, Pohl E, Becker J, Frommolt P, et al. (2012) Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. Am. J. Hum. Genet , 90 , 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge G, and Greenspan DS (2006) Developmental roles of the BMP1/TLD metalloproteinases. Birth Defects Res. C. Embryo Today , 78 , 47–68. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Song H, Wang W, and Zhang Z (2019) Generation and characterization of Megf6 null and Cre knock-in alleles. Genesis. , 57 , 23262. [DOI] [PubMed] [Google Scholar]
- 58.Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I, et al. (2011) A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. , 9 , 1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chim SM, Qin A, Tickner J, Pavlos N, Davey T, Wang H, Guo Y, Zheng MH, and Xu J (2011) EGFL6 promotes endothelial cell migration and angiogenesis through the activation of extracellular signal-regulated kinase. J. Biol. Chem , 286 , 22035–22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Günther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai SL, et al. (2019) Genetic compensation triggered by mutant mRNA degradation. Nature. , 568 , 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma Z, Zhu P, Shi H, Guo L, Zhang Q, Chen Y, Chen S, Zhang Z, Peng J, and Chen J (2019) PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature. , 568 , 259–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
megf6a and megf6b have both overlapping and unique expression domains. Whole mount in situ hybridization of megf6a (A-D) and megf6b (E-F) at 24 hpf (A and E), 36 hpf (B, C, D, and F), and 48 hpf (G). megf6a and megf6b are expressed in the fin fold (arrow in A) and in the head of 24 hpf embryos (A and E), while only megf6b is expressed in somitic tissue (arrow in E). At 36 hpf megf6a and megf6b are expressed in the fin fold (D and F), AER (arrowhead in B and F), atrioventricular canal (arrowhead in C), and notochord (arrowhead in D and F), and head. megf6b is expressed in the mouth (arrow in G) and pharyngeal region (arrowheads in B) in 48 hpf larvae. Lateral views with anterior to the left.
Generation of a heritable megf6b deletion mutant. (A) RNPs were designed to target sites (lightning bolts) within exon 6 (ex 6 gRNA) and exon 35 (ex 35 gRNA) of megf6b. Primer pairs used to amplify junction fragments joining target sites are indicated (red arrows). (B) Detailed view of the ex 6 gRNA and ex 35 gRNA sites with gRNA target sequence in read, PAM site in blue, and Cas9 cleavage site indicated by lightning bolts. megf6bΔ ex 6/35 represents a conceptual precise joining of sequences flanking the presumed cleavage sites in target sites ex 6 and ex 35. megf6bz48 represents the deletion allele recovered in the germline. This allele contains a 52,960 bp deletion with a 53 bp insertion.
Weight (g) and length (cm) of adult zebrafish lacking megf6a and megf6b function. The weight (A) and length (B) of individual male and female 4 month old zebrafish generated from megf6b+/− intercross eggs injected with megf6a RNPs. Circles indicate embryos identified at 48 hpf with normal fin folds (megf6b−/?) and squares indicate those with fin fold defects (megf6b−/−). Males with abnormal fin folds weighed less than controls with normal fin folds (A), while there was no difference in length between the two groups (B). Females with abnormal fin folds weighed more and were longer when compared to controls with normal fin folds (A and B). n = 14 males and n = 11 females with normal fin folds and n= 14 males and n = 7 females with abnormal fin folds. A Student’s t-test was used to determine statistical significance. Data are represented as means ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.