

Abstract
Brain function requires connecting neuronal networks to empower movement, sensation, behavior, and cognition. Studies published early this year provide evidence that in humans, Netrin receptor, Deleted in Colorectal Cancer (DCC), is a master regulator of axonal crossing throughout the neuraxis.
Introduction
The molecular rules governing axonal midline crossing have been elegantly investigated using cell culture, invertebrate, and vertebrate animal models (reviewed in Nugent et al., 2012). Yet interspecies evolutionary studies indicate that the particulars of those rules differ significantly among organisms (reviewed in Beamish and Kennedy, 2015). Understanding these details is critical to the potential application of cell-based and reparative therapies in the treatment of neurological disease. Modern high-resolution, non-invasive imaging tools have accelerated the detection of human disorders affecting axonal guidance. Coupled with advanced clinical genetics, these tools are generating new insights into the molecules, their functional domains, and interactions that regulate the crossing of axons in commissures to connect our right side of the nervous system with our left to enable information processing in three and four dimensions. Two human genetic studies published earlier this year by Marsh and colleagues (Marsh et al., 2017) and Jamuar et al. (Jamuar et al., 2017) provide a fascinating look into DCC-mediated axonal guidance in ourselves.
The major axonal crossings or decussations in mammalian brain encompass the corpus callosum, anterior and posterior commissures, corticospinal tract, and various hindbrain and brainstem commissures that, for example, yoke the eye muscles for conjugate gaze or coordinate a smooth gait. Several genes have been implicated in human axon projection disorders (reviewed in Nugent et al., 2012). Heterozygous missense mutations in neuron-specific tubulin subunit, TUBB3, have been linked to congenital fibrosis of extraocular muscles type 3 (CFEOM3) defined by blepharoptosis (drooping eyelids) and ophthalmoplegia (impaired eye movement). CFEOM3 results from reduced axon outgrowth and miswiring of corpus callosum, anterior commissure, and oculomotor nerves. Biallelic loss-of-function mutations in the axon-guidance receptor Roundabout 3 (ROBO3) cause horizontal gaze palsy with progressive scoliosis (HGPPS), characterized by abnormal conjugate eye movements and early-onset scoliosis associated with disruption of midbrain, pontine, and corticospinal commissural tracts, but normal corpus callosum. Finally, heterozygous mutations in DCC cause congenital mirror movements (CMM), defined by involuntary movement of one side of the body occurring in concert with voluntary movement of the other side of the body. Partial decussation of the corticospinal tract (CST) or brainstem motor axons is thought necessary to produce CMM, so that normally crossed neuronal fibers can make either ipsilateral or contralateral projections (Srour et al., 2010).
Adding to this molecular constellation, Jamuar et al. (2017) reported a new human syndrome caused by biallelic mutation in DCC that combines features of HGPPS and agenesis of the corpus callosum (ACC), resulting in complete disruption of corpus callosum, anterior, posterior, and hippocampal commissures. Around the same time, Marsh et al. (2017) identified several families and sporadic individuals manifesting isolated ACC with or without CMM, with incomplete penetrance, as a result of heterozygous mutations in DCC. These findings implicate DCC in novel human phenotypes and shed new light on its expansive role in axon guidance.
Mutation Loci and Associated Phenotypes
First identified in Drosophila and C. elegans as a cell-surface receptor for the guidance signaling protein Netrin, DCC contains four immunoglobulin-like domains, six fibronectin type 3-like domains, a transmembrane domain, and an intracellular C terminus. Heterozygous mutations in DCC cause CMM in an autosomal-dominant manner (Srour et al., 2010). Marsh et al. (2017) expanded the DCC-linked phenotypic spectrum, finding heterozygous DCC mutations in four families having members with ACC (complete or partial), CMM, or both, and varying intellectual ability. DCC mutations differed in type and location between families (Figure 1A). For example, heterozygous truncating mutations occurred in the immunoglobulin-like domains proximal to the N terminus, likely yielding a functionally null protein and haploinsufficiency. Within the same family, individuals exhibited partial or complete ACC with or without CMM, while others harboring the same mutations were healthy. Additional families had missense DCC mutations located in fibronectin-like extracellular repeats of the Netrin binding domain or in the cytoplasmic domain, thought to be important for DCC dimerization (Stein et al., 2001). Among all heterozygous missense mutations, 12 were associated with partial or complete ACC with or without CMM and two carriers were healthy.
Figure 1. Summary of Reported Mutations in DCC and ROBO3 Associated with Disruptions of Commissure and Axonal Decussations in Humans.
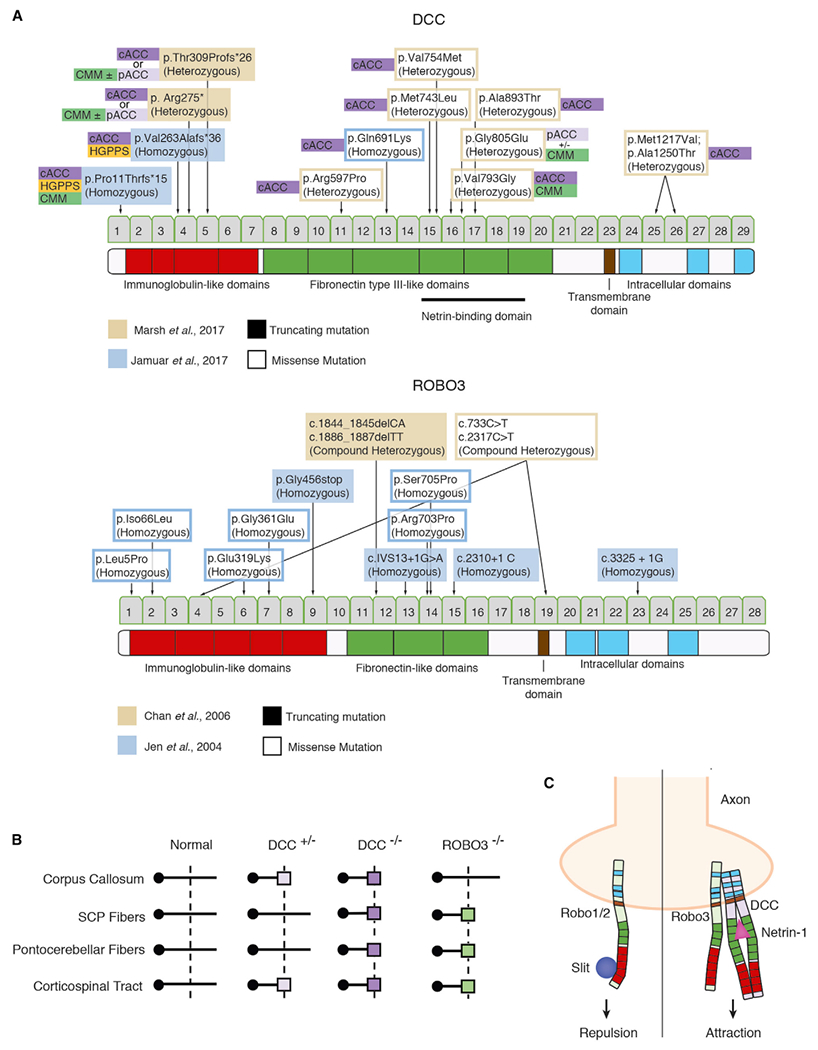
(A) Mutations and their location in encoded proteins DCC (taken from Jamuar et al., 2017 and Marsh et al., 2017) and ROBO3 (from Chan et al., 2006 and Jen et al., 2004). Whether mutation is homozygous/compound heterozygous or heterozygous is indicated, as is the associated commissural defect. ACC, agenesis of corpus callosum (p, partial ACC; c, complete ACC); CMM, congenital mirror movements; HGPPS, horizontal gaze palsy with progressive scoliosis.
(B) Summary of mutation gene and type associated with rostro-caudal level of crossing defects. Modified from Jamuar et al. (2017).
(C) Relationship in mammals among ROBO1/2, ROBO3, and DCC in generating attractive versus repulsive guidance signals from Netrin and Slit ligands. Adapted from Beamish and Kennedy (2015).
Jamuar et al. (2017) surveyed seven individuals from three separate families, four of whom harbored homozygous loss-of-function mutations for DCC. Two affected brothers shared a 7 kb deletion with resultant skipping of exon 1 that encodes the membrane localization signal crucial for its axonal guidance function. The single affected female in another family had a homozygous 7 bp deletion in exon 4, causing a premature termination of DCC consistent with a functional null. All three affected individuals from families 1 and 2 exhibited horizontal gaze palsy, intellectual disability, and scoliosis yet, interestingly, only the two males in family 1 displayed CMM. A fourth individual previously reported to have ACC, interhemispheric cysts, and mild intellectual delays (Griebel et al., 1995) was found by Jamuar and colleagues to have a homozygous DCC missense mutation in the third fibronectin-like repeat, in close proximity to the Netrin-binding site. No information about eye movement, scoliosis, or mirror movement was available. This change was strongly predicted to be deleterious by in silico algorithms. However, the missense mutant protein may still be partially functional, suggesting that the potency of the mutation dictates the severity of clinical phenotypes observed.
Neuroimaging
MRI and diffusion tractography of surveyed individuals in these studies documented complete or partial loss of corpus callosum and hippocampal commissures (summarized in Figure 1). Interestingly, those with CMM (with or without ACC) revealed reduced, but not absent, CST crossing at the brainstem/spinal cord junction (Marsh et al., 2017). The remaining commissural tracts, including anterior and posterior commissures, as well as decussation of superior cerebellar peduncles (SCP), remained intact. This is in stark contrast to the DCC−/− individuals reported by Jamuar et al. (2017) who exhibited a complete loss of all commissural tracts (callosal, anterior, posterior, and hippocampal), along with fewer and disorganized cortical-cortical and descending subcortical projections. Decussations of SCP and CST axons were fully impaired in these individuals. These combined results suggest a widespread role for DCC as a regulator of axon guidance for all cortical projections. They also emphasize the increased sensitivity of the callosal and subcerebral pyramidal projections to DCC loss of function, as compared to the remaining commissures and tracts.
Disease Penetrance
All three DCC−/− individuals surveyed in two families by Jamuar et al. (2017) displayed a “split-brain” syndrome, unifying features of HGPPS and ACC. Though a larger sample size would be needed to draw definitive conclusions, it seems that complete DCC loss of function reliably causes full loss of commissural tracts and corticospinal decussations. However, DCC+/− individuals in the Marsh et al. (2017) study displayed varying levels of impairments, ranging from healthy to complete ACC, with or without CMM. The sample size in these new studies combined with previous published reports enabled estimation of CMM penetrance at 42% and an ACC penetrance of 26% of DCC mutations. Surprisingly, males exhibited CMM almost twice as often as females (male/female ratio 1.8). Both DCC−/− males reported by Jamuar et al. (2017) exhibited CMM while the DCC−/− female did not. In contrast, more females than males with truncating DCC mutations exhibited ACC (male/female ratio = 0.2). Marsh et al. (2017) postulate that sex could play a role in the penetrance of these diseases since DCC expression level was shown by the authors to be dependent on testosterone levels in vitro. Other genetic modifiers such as ethnicity or epigenetic marks may also play a role in modulating phenotypic expression.
Regulation of Axons Crossing the Midline in Mammals
The newly reported DCC syndromes (Marsh et al., 2017; Jamuar et al., 2017) share features with HGPPS, the cause of which was previously identified as biallelic mutations in the axon-guidance receptor ROBO3 (reviewed in Nugent et al., 2012). The authors emphasize the importance of this connection as they explain that ROBO3 is a known interactor of DCC and their interaction is required for proper midline crossing of axons (Figure 1C). Unlike its cousins ROBO1/2, ROBO3 does not bind Slit to mediate axonal repulsion but instead directly interacts with DCC and enhances binding to Netrin-1, thus mediating attraction toward the midline (reviewed in Beamish and Kennedy, 2015). Yet, recent data by Marsh et al. (2017) and Jamuar et al. (2017) suggest that global axonal midline crossing is intimately dependent on DCC and less so on ROBO3. For example, whereas ROBO3 is expressed only in hindbrain and spinal cord (Jen et al., 2004), the regulatory role of DCC is more pervasive, mediating axonal guidance in all commissural tracts, including SCP and pontocerebellar decussations (Figure 1B). Furthermore, only biallelic loss-of-function of ROBO3 has been associated with disruption of the corresponding midbrain, hindbrain, and corticospinal commissural tracts, while heterozygous DCC mutation is sufficient to impair midline crossing, specifically at callosal and corticospinal tracts (Figure 1B).
Conclusion
Together, these papers suggest that isolated ACC or CMM can result from DCC loss of function, either through protein truncation or missense mutations disrupting Netrin- or intracytoplasmic- protein interactions, in an autosomal-dominant fashion. Biallelic DCC loss-of-function results in a new syndrome that combines features of HGPPS, ACC, and CMM. These insights indicate that DCC is a master regulator of connectivity across the midline throughout the human neuraxis. An intriguing question becomes what compensatory signals cause the incomplete penetrance and differential sensitivity of the commissures to DCC deficiencies. Moreover, if TUBB3 is a downstream target of Netrin/Slit signaling to modulate dynamics of axon guidance, what is the mechanism and is TUBB3 special among tubulins. Currently, it appears that axon guidance of callosal and CST fibers is dependent on Netrin binding to the extracellular domain of DCC, and they are additionally reliant on DCC intracellular interaction(s), perhaps on dimerization of the receptor, and/or, especially in the hindbrain and spinal cord, on interaction with ROBO3.
ACKNOWLEDGMENTS
Supported by P01HD067244 and T32HD060600.
REFERENCES
- Beamish IV, and Kennedy TE (2015). Dev. Cell 32, 3–4. [DOI] [PubMed] [Google Scholar]
- Chan WM, Traboulsi EI, Arthur B, Friedman N, Andrews C, and Engle EC (2006). J. Med. Genet 43, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel ML, Williams JP, Russell SS, Spence GT, and Glasier CM (1995). Pediatr. Neurol 13, 119–124. [DOI] [PubMed] [Google Scholar]
- Jamuar SS, Schmitz-Abe K, D’Gama AM, Drottar M, Chan WM, Peeva M, Servattalab S, Lam AN, Delgado MR, Clegg NJ, et al. (2017). Nat. Genet 49, 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen JC, Chan WM, Bosley TM, Wan J, Carr JR, Rüb U, Shattuck D, Salamon G, Kudo LC, Ou J, et al. (2004). Science 304,1509–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh AP, Heron D, Edwards TJ, Quartier A, Galea C, Nava C, Rastetter A, Moutard ML, Anderson V, Bitoun P, et al. (2017). Nat. Genet 49, 511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, Kolpak AL, and Engle EC (2012). Curr. Opin. Neurobiol 22, 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srour M, Rivière JB, Pham JM, Dubé MP, Girard S, Morin S, Dion PA, Asselin G, Rochefort D, Hince P, et al. (2010). Science 328, 592. [DOI] [PubMed] [Google Scholar]
- Stein E, Zou Y, Poo M, and Tessier-Lavigne M (2001). Science 291, 1976–1982. [DOI] [PubMed] [Google Scholar]