

Abstract
Objective:
Skin inflammation heralds systemic disease in juvenile myositis (JM), yet we lack an understanding of pathogenic mechanisms driving skin inflammation in JM. The goal of this study is to define cutaneous gene expression signatures in JM and identify key genes and pathways that differentiate skin disease in JM from childhood-onset systemic lupus erythematosus (cSLE).
Methods:
We utilized formalin-fixed paraffin-embedded (FFPE) skin biopsy samples from 15 JM (9 lesional, 6 non-lesional), 5 cSLE, and 8 control patients to perform transcriptomic analysis and identify significantly differentially expressed genes (DEGs; q-value ≤ 5%) between patient groups. We utilized Ingenuity Pathway Analysis (IPA) to highlight enriched biological pathways and validated DEGs using immunohistochemistry and quantitative real-time PCR.
Results:
Comparison of lesional JM to control revealed 221 DEGs, with the majority of upregulated genes representing interferon-stimulated genes. CXCL10, CXCL9 and IFI44L represented the top three DEGs (fold-change respectively = 23.2, 13.3, 13.0, q-value < 0.0001). IPA revealed interferon (IFN) signaling as the top canonical pathway. When compared to cSLE, JM lesional skin shared a similar gene expression pattern, with only 28 unique DEGs, including FBLN2, CHKA and SLURP1. Notably, JM patients with NXP2 autoantibodies exhibited the strongest IFN signature and also demonstrated the most extensive MX1 immunostaining, both in keratinocytes and perivascular regions.
Conclusion:
JM lesional skin demonstrates a striking IFN signature similar to that previously reported in muscle and peripheral blood. Further investigation into the association of a higher IFN score with NXP2 autoantibodies may lend insight into disease endotypes and pathogenesis.
Introduction
Juvenile myositis (JM) is a potentially life-threatening idiopathic inflammatory myopathy of childhood, often presenting with skin inflammation and following a highly heterogeneous disease course. Skin inflammation frequently persists in the absence of active muscle disease and prevents complete disease remission(1), yet there also exists uncertainty as to the role of skin disease in directing a change in systemic therapy. Multiple prior studies have highlighted the importance of skin inflammation as an indicator of ongoing disease activity, leading to disease chronicity and damage over time(2, 3); however, skin disease in JM is understudied.
Interferons (IFNs) are likely important in JM pathophysiology. A striking upregulation of interferon-stimulated genes (ISGs) has been noted in adult dermatomyositis skin, similar to that seen in systemic lupus erythematosus (SLE)(4). IFN signaling is also known to be upregulated in JM muscle and peripheral blood(5, 6). Yet, the role of interferons in disease pathogenesis is less clear. While the IFN signature in peripheral blood for both JM and DM has been shown to correlate with disease activity(7, 8), it does not differ with disease duration in JM(9). Higher IFN scores in muscle are associated with increased disease severity based on muscle biopsy histopathology, and type II IFN scores may predict a longer time to clinically inactive disease(10). In DM skin disease, type I IFNs have been purported to lead to recruitment of lymphocytes, macrophages, and plasmacytoid dendritic cells. Further similar to lupus(11, 12), non-lesional JM skin may also be abnormal with increased numbers of plasmacytoid dendritic cells and mast cells(13).
Here, we investigated the transcriptional changes in JM lesional and non-lesional skin and compared these with childhood-onset SLE (cSLE) skin disease. This was examined in the context of patient data, including myositis-specific autoantibodies (MSAs). This work thus lays the foundation for understanding JM skin lesions and identifies IFN-targeting therapies as appropriate for trials in JM.
Materials and Methods
Sample acquisition and clinical data collection
JM and cSLE patient biopsies at either the University of Michigan or Ann & Robert H. Lurie Children’s Hospital of Chicago were identified under University of Michigan IRBMED approval with a waiver of consent. Diagnosis was verified through chart review of current and historical clinical findings, labs, imaging, histopathology and specified diagnosis of juvenile dermatomyositis (JDM) or cSLE by a Pediatric Rheumatologist. All but four JDM patients met Bohan and Peter criteria for definite or probable JDM. Of the four patients not meeting Bohan and Peter criteria, two had amyopathic disease and two lacked typical JDM rash at diagnosis. We therefore chose to use the more general term JM for our patient cohort. All cSLE patients met the 1997 American College of Rheumatology classification criteria for SLE at time of skin biopsy with the exception of one patient with isolated cutaneous lupus at diagnosis, who later developed systemic disease features. Overall, we identified a total of 25 formalin-fixed, paraffin-embedded (FFPE) skin biopsies, including 17 JM (9 lesional, 8 non-lesional) and 8 cSLE (all lesional, two patients with 2 separate biopsies from different sites at individual time points). Lesional JM skin (JM_L) was from varied locations, including the elbow (n = 3), finger (n = 2), arm, knee, leg and thigh (all n = 1). Lesional cSLE skin was also from multiple locations, including the upper arm, toe, cheek, palm, scalp, finger, anterior lateral proximal thigh and elbow. All non-lesional JM skin (JM_NL) was from the thigh or lower back. We additionally obtained FFPE skin from 8 pediatric control (CTL) patients (uninvolved skin removed with nevi excision). Summary reports from patient biopsies done for clinical care are listed in Supplemental Table 1.
Clinical data was collected retrospectively by chart review from JM and cSLE patients both at time of diagnosis and within one month of skin biopsy (Table 1, patients passing quality controls, see methods below for details). MSAs were measured by either the myositis autoantibody profile at the Oklahoma Medical Research Foundation (OMRF) Clinical Immunology Laboratory (all non-lesional and 1 lesional sample) or the Myomarker Panel 3 at Mayo Clinic Laboratories (5 lesional samples). These clinically available MSA testing methodologies utilize differing techniques for MSA detection, with the OMRF profile being determined predominantly by immunoprecipitation and immunoblotting and the Myomarker Panel 3 by enzyme immunoassay (EIA). While a direct comparison of test performance characteristics between these two methodologies has not been published for reference, it has been noted that EIA methodology has a lower sensitivity for detection of some autoantibodies such as anti-TIF1γ while also potentially leading to more false positive results(14–17).
Table 1.
Summary characteristics of controls, patients with juvenile myositis and patients with childhood-onset systemic lupus erythematosus included in the study. Comparisons between patients with lesional and non-lesional skin were made using two-sided unpaired Students’ t-test.
| Controls (n=8) | JM non-lesional (JM_NL) (n=6) | JM lesional (JM_L) (n=9) | cSLE lesional (n=5)**** | P value (JM_L vs. JM_NL) | P value (cSLE vs. JM_L) | |
|---|---|---|---|---|---|---|
| Mean age at diagnosis, years (±SEM) | - | 7.2 (1.6) | 9.1 (1.7) | 12.8 (0.8) | 0.4560 | 0.1598 |
| Mean age at time of biopsy (±SEM) | 12.3 (1.7) | 13.0 (2.0) | 10.9 (1.7) | 13.2 (0.5) | 0.4428 | 0.3561 |
| Sex, n (% of female) | 6 (75) | 6 (100) | 8 (88.9) | 5 (83) | 0.4346 | 0.6785 |
| Race, n (%) | ||||||
| White | 5 (62.5) | 6 (100) | 7 (77.8) | 3 (60) | 0.2445 | 0.5185 |
| African American | 0 (0) | 0 (0) | 2 (22.2) | 1 (20) | 0.2445 | 0.9298 |
| Other | 2 (25) | 0 (0) | 0 (0) | 1 (20) | - | 0.1902 |
| Unknown | 1 (12.5) | 0 (0) | 0 (0) | 0 (0) | - | - |
| Ethnicity, n (%) | ||||||
| Non-hispanic | 7 (87.5) | 4 (66.7) | 8 (88.9) | 5 (100) | 0.0888 | - |
| Hispanic | 0 (0) | 2 (33.3) | 0 (0) | 0 (0) | 0.0888 | - |
| Unknown | 1 (12.5) | 0 (0) | 1 (11.1) | 0 (0) | 0.4346 | 0.4783 |
| Disease duration at biopsy, years (±SEM) | - | 5.8 (1.4) | 1.8 (0.9) | 0.4 (0.2) | 0.0213 | 0.2861 |
| Duration of untreated disease prior to diagnosis, months (±SEM) | - | 8.8 (2.9) | 5.9 (1.6) | - | 0.3687 | - |
| Dysphagia, n (% of patients at time of biopsy) | - | 0 (0) | 3 (33.3) | - | 0.1309 | - |
| Childhood Myositis Assessment Scale (CMAS) score at time of biopsy (0–52) (±SEM)* | - | 44 (4.8) | 39 (11.5) | - | 0.6416 | - |
| Amyopathic disease, n (% of patients) | - | 0 (0) | 2 (22.2) | - | 0.2445 | - |
| Lupus nephritis (% of patients) | - | - | - | 2 (40) | - | - |
| Skin manifestations, n (% of presence at time of biopsy) | ||||||
| Cutaneous lupus erythematosus only (% of patients) | - | - | - | 1 (20) | - | - |
| Discoid lupus (% of samples) | - | - | - | 1 (14.2) | - | - |
| Heliotrope rash | - | 2 (33.3) | 4 (44.4) | - | 0.6934 | - |
| Gottron’s sign/papules | - | 4 (66.7) | 7 (77.8) | - | 0.6621 | - |
| Nailfold capillary changes | - | 2 (33.3) | 7 (77.8) | - | 0.0970 | - |
| Calcinosis | - | 2 (33.3) | 0 (0) | - | 0.0699 | - |
| Skin ulceration | - | 0 (0) | 0 (0) | - | N/A | - |
| Laboratory tests (±SEM) | ||||||
| Positive anti-dsDNA (% of patients) | - | - | - | 2 (40.0) | - | - |
| C3 (mg/dL) | - | - | - | 115.8 (10.9) | - | - |
| C4 (mg/dL) | - | - | - | 16.6 (2.1) | - | - |
| Myositis associated autoantibody (MAA), n (% of presence)** | - | 3 (50.0) | 1 (12.5) | - | 0.0476 | - |
| Myositis specific autoantibody (MSA), n (% of presence)*** | - | 4 (66.7) | 3 (60.0) | - | 0.8402 | - |
| NXP2+ | - | 1 (16.7) | 2 (40.0) | - | 0.4385 | - |
| TIF1γ+ | - | 3 (50.0) | 1 (20.0) | - | 0.3527 | - |
| MSA negative | - | 2 (33.3) | 2 (40.0) | - | 0.8402 | - |
| MSA unknown | - | 0 (0) | 4 (44.4) | - | 0.0623 | - |
| Serum muscle enzymes at time of biopsy (± SEM) | ||||||
| CK (U/L) | - | 48.2 (8.5) | 1,474.4 (1,341.0) | - | 0.4514 | - |
| Aldolase (U/dL) | - | 4.4 (1.0) | 12.8 (5.5) | - | 0.2856 | - |
| LDH (U/L) | - | 189.0 (13.0) | 386.9 (109.4) | - | 0.1885 | - |
| AST (U/L) | - | 31.6 (2.7) | 68.8 (32.0) | - | 0.4126 | - |
| ALT (U/L) | - | 35.0 (6.0) | 41.3 (17.7) | - | 0.8009 | - |
| Medications, n (% of patients on drug at time of biopsy) | ||||||
| None | - | 0 (0) | 4 (44.4) | 1 (20) | 0.0623 | 0.3997 |
| Oral steroids | - | 6 (100) | 3 (33.3) | 4 (80) | 0.0066 | 0.1089 |
| IV steroids | - | 4 (66.7) | 2 (22.2) | 1 (20) | 0.0970 | 0.9298 |
| Mycophenolate Mofetil | - | 5 (83.3) | 1 (11.0) | 3 (60) | 0.0024 | 0.0575 |
| Cyclosporine | - | 3 (50.0) | 1 (11.0) | 0 (0) | 0.1089 | 0.4783 |
| Hydroxychloroquine | - | 5 (83.3) | 0 (0) | 2 (40) | <0.0001 | 0.0426 |
| Methotrexate | - | 2 (33.3) | 4 (44.4) | 0 (0) | 0.6934 | 0.0888 |
| IVIG | - | 3 (50.0) | 0 (0) | 0 (0) | 0.0152 | - |
Information was missing for 6 patients with lesional skin.
Information was missing for 1 patient with lesional skin.
Information was missing for 4 patients with lesional skin.
Two cSLE patients had 2 separate biopsies from different sites at individual time points. Nailfold capillary changes include nailfold capillary dilatation or dropout. Myositis-specific autoantibodies were measured by either the myositis autoantibody profile at the OMRF Clinical Immunology Laboratory (all non-lesional and 1 lesional sample) or the Myomarker Panel 3 at Mayo Clinic Laboratories (5 lesional samples).
RNA isolation and microarray procedures
We obtained 10 10μm sections per FFPE skin block and isolated RNA using an Omega Bio-tek FFPE RNA kit (Norcross, GA). Library prep and microarray were completed by the Advanced Genomics Core at the University of Michigan. Complementary DNA (cDNA) was prepared (NuGEN, Ovation PicoSL WTA System V2 Manual, P/N M01226 v4) from approximately 30 ng total RNA. 2.5 ug cDNA was biotinylated using the NuGEN Encore Biotin Module (Encore Biotin Module Manual, P/N M01111 v6). A Poly-A RNA Control Kit was used. Affymetrix Human Gene ST 2.1 array plates were run using the Affymetrix GeneTitan system (software version 3.2.4.1515). Quality control and RMA (Robust Multi-array Average) normalization of CEL files were performed in R software version 3.5.1 using custom CDF version 23 and the associated modified Affymetrix package from BrainArray http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/CDF_download.asp)(18).
All samples underwent Normalized Unscaled Standard Error (NUSE), Relative Log Expression (RLE) and principal component analysis (PCA) quality controls. Two JM_NL and one cSLE sample were excluded from further analysis, as one patient was an extreme outlier on PCA and two had atypical histopathology. The final cohort consisted of 15 JM (9 lesional, 6 non-lesional), 7 cSLE (from 5 patients) and 8 CTL biopsy samples. The baseline Log2 expression value was defined as minimum plus one standard deviation of the median of all genes. A variance filter of 80% was then applied. Of the 29,635 unique genes represented on the Human ST2.1 chip, a total of 23,698 genes passed the defined criteria. Data from the microarrays are available through GEO GSE148810.
Canonical pathways and literature-based network analyses, hierarchical clustering and heatmap generation
Canonical pathways (well-established signaling and metabolic pathways) were identified using Ingenuity Pathway Analysis software (IPA) (www.ingenuity.com). Biological literature-based networks were built using Genomatix Pathway System software (GePS) (www.genomatix.de) with the function-word level as minimum evidence level parameter. Heatmaps were generated using the gene expression values as input for the HeatmapViewer module in GenePattern (https://cloud.genepattern.org).
Cell types enrichment analysis
Cell types enrichment analysis was performed on the normalized dataset of 23,698 genes using the xCell webtool (http://xcell.ucsf.edu/)(19).
Calculation of IFN and JM-specific disease signature scores
IFN scores were calculated using 6 IFN-stimulated genes (IFIT1, IRF7, MX1, EIF2AK2, OASL, IFI44) with the algorithm described by Feng et al. (20) and as previously published(21). Except EIF2AK2, these genes were used by Feng et al. and include 2 of the 5 recommended ISGs (OASL and MX1). EIF2AK2 has been shown to be an ISG in lupus(22) and is also specifically upregulated in keratinocytes upon IFNa stimulation(23). Our 6 gene IFN score strongly correlated with the IFN score calculated using the 5 ISGs from Feng et al. (LY6E, OAS1, OASL, MX1, ISG15) (r=0.9828, p<0.0001). A skin-directed IFN score was also calculated based on this algorithm with 18 genes specifically upregulated in keratinocytes upon IFNα stimulation: EIF2AK2, IFI16, IFI27, IFI44, IFIH1, IFIT5, IRF9, ISG15, NMI, OAS3, PARP12, PARP14, PARP9, PLSCR1, SP100, STAT1, TNFSF10, ZNFX1(23). Finally, the same algorithm was applied to a JM-specific signature consisting of 23 genes (28 genes minus 2 miR, 2 LOC and one C-orf genes) derived from comparison of cSLE and JM_L. The 3 gene JM-specific score was calculated using the 3 most regulated genes (smallest q-value) with a fold-change (FC)≥2 (FBNL2 (q-value=0.0137, FC=2.15), CHKA (q-value=0.0137, FC=2.14), SLURP1 (q-value=0.0239, FC=2.13) in our study samples and also skin disease array datasets available from GEO(23). Association between the JM-specific signature and the skin-directed IFN score was performed using Pearson correlation with GraphPad Prism software version 8.0.0.
Immunohistochemistry
Four micron sections were cut from FFPE skin blocks. Skin tissue was deparaffinized in histoclear and rehydrated in graded ethanol. Heat-mediated antigen retrieval was performed in sodium citrate buffer, followed by incubation steps with BLOXALL and 1.5% goat serum (VECTOR Laboratories, Burlingame, CA). Slides were incubated overnight at 4°C with anti-MX1 at 1:500 (ab97921, Abcam, Cambridge, MA), rabbit IgG isotype control, or PBS. Slides were developed with biotinylated secondary antibody and HRP/Vectastain Elite ABC Reagent, followed by ImmPACT DAB Peroxidase (HRP) Substrate for 90 seconds before quenching in water (VECTOR Laboratories, Burlingame, CA) and counterstaining with hematoxylin.
Quantitative real-time PCR
cDNA was prepared from FFPE-isolated RNA. Expression of MX1, IFI44, CXCL10 and SLURP1 were measured by real-time PCR (RT-PCR) on an ABI Prism 7900HT (Applied Biosystems) using SYBR green (Life Technologies). FC expression was calculated relative to GAPDH using 2−ΔΔCT. Primers were as follows (5’ to 3’): MX1 - TACCAGGACTACGAGATTG (forward), TGCCAGGAAGGTCTATTAG (reverse), IFI44 - GGTGGGCACTAATACAACTGG (forward), CACACAGAATAAACGGCAGGTA (reverse), CXCL10 - GTGGCATTCAAGGAGTACCTC (forward), TGATGGCCTTCGATTCTGGATT (reverse), SLURP1 – CTGCAAGCCAGAGGACACA (forward), CACACAGGAGCTGGAGCAG (reverse), GAPDH – CTGGGCTACACTGAGCACC (forward), AAGTGGTCGTTGAGGGCAATG (reverse).
Statistical analyses
DEGs were compared between JM_L, JM_NL, cSLE and CTL biopsies using the Significance Analysis of Microarrays (SAM) method implemented in the TIGR MultiExperiment Viewer application version 4.9.0 (unpaired analysis)(24). Genes regulated with the False Discovery Rate (FDR) q-value<0.05 were considered significant and used for further transcriptional and pathway analyses. Statistical analysis of clinical data and gene score comparisons were generated using an unpaired parametric t-test with GraphPad Prism software version 8.0.0; p-values<0.05 were considered significant and reported in all Figures. All comparisons across all groups were performed and for clarity, only the most relevant were reported if significant.
Results
Clinical cohort characteristics
In the JM cohort, JM_L patients had shorter disease duration at time of skin biopsy (Table 1). Overall skin manifestations and serum muscle enzyme levels were similar between JM_L and JM_NL. Two JM_L patients had amyopathic disease. Of both JM_L and JM_NL patients tested for presence of a myositis-specific autoantibody (MSA), the majority were MSA positive. JM patients in our cohort exclusively demonstrated NXP2 and TIF1γ MSAs. Only four JM_L patients were treatment naïve at time of skin biopsy. JM_NL patients were more likely to be on oral steroids, mycophenolate mofetil, hydroxychloroquine and IVIG. In the cSLE cohort, only one patient had isolated cutaneous lupus at time of skin biopsy. The majority of biopsies were subacute cutaneous lupus, with only one discoid lupus (Table 1; Supplemental Table 1). Overall treatment was similar between JM_L and cSLE patients, with the only difference being more cSLE patients on hydroxychloroquine at time of skin biopsy.
Comparison of JM lesional to control skin
A total of 221 genes were differentially regulated in JM_L compared to CTL, with all but one upregulated in JM_L (q-value<0.05) (Supplemental Table 2). The majority of upregulated genes in JM_L were IFN-sensitive, with CXCL10, CXCL9 and IFI44L representing the top three upregulated genes (FC=23.2, 13.3, 13.0 respectively, q-value<0.0001). Figure 1 highlights the top upregulated canonical pathways of the DEGs in JM_L relative to CTL (left panel) (Supplemental Table 3) and a heatmap of the genes regulated in each pathway (right panel). Canonical pathway analysis revealed IFN signaling as the top upregulated pathway and showed activation of pathways involving antigen presentation, pattern recognition receptors, communication between innate and adaptive immune cells, T cell signaling, complement system and dendritic cell maturation (Figure 1, Supplemental Table 3). Literature-based network analysis of all 221 DEGs identified upregulation of STAT1 as a central node linking dysregulated genes (FC=5.16, q-value<0.0001). The top predicted upstream regulator was IFNα (p-value=7.91×10−88) (Supplemental Table 4).
Figure 1.
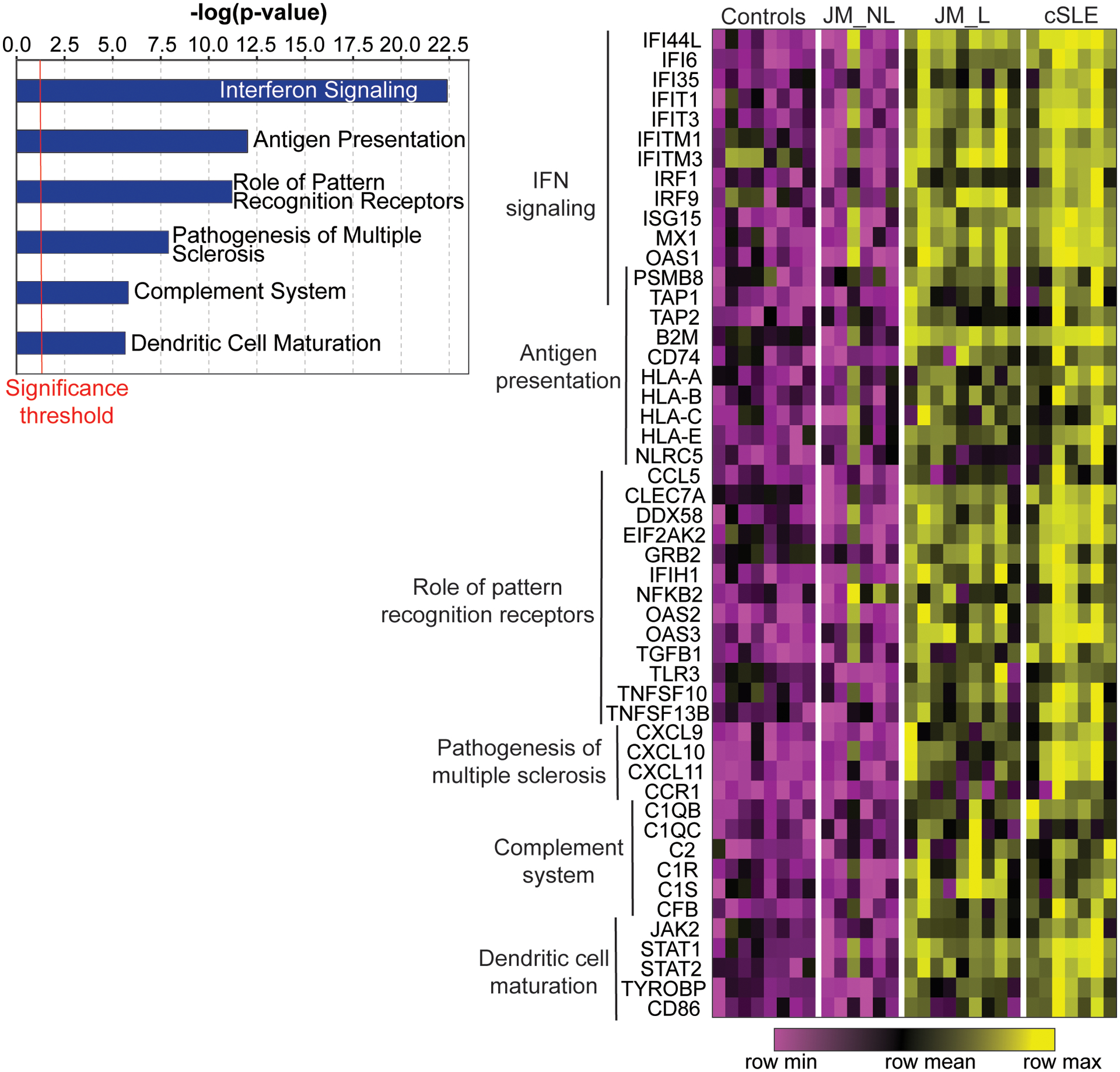
Genome-wide expression analysis of CTL, JM and cSLE skin biopsies. Selected top canonical pathways (p-value<0.05) from the 221 genes regulated in lesional skin biopsies compared to controls (q-value<0.05) (left panel). Heatmap of selected genes from those top pathways in control (CTL), non-lesional JM (JM_NL), lesional JM (JM_L) and cSLE skin biopsies. Each column represents an individual patient sample while each row represents a differentially expressed gene in JM_L relative to CTL. Gene expression values are depicted using the color scale shown with purple to yellow indicating increasing expression. Genes overlapping between pathways are represented only once.
Comparison of JM lesional to non-lesional skin
JM_NL skin had a strikingly different gene expression signature compared to JM_L biopsies, most notably lack of a prominent IFN signature (Figure 1). Multiple genes and pathways were downregulated in JM_NL compared to JM_L, including pathways in protein ubiquitination, glucocorticoid receptor signaling, IFN signaling and oxidative phosphorylation (Supplemental Tables 5 and 6). We applied a cell type enrichment webtool on the gene expression data (xCell) to characterize potential immune cell types in JM_L and JM_NL skin, identifying increased macrophages and CD4+ memory T-cells in JM_L skin (Supplemental Figure 1).
Comparison of JM to cSLE lesional skin
JM_L skin shared a highly similar gene expression pattern with cSLE (Figures 1 and 2). Notably, lesional skin from both JM and cSLE demonstrated a prominent type I interferon signature. There were only 28 unique DEGs in JM_L compared to cSLE (Figure 2). The top most significant unique DEGs in JM_L included FBLN2, CHKA and SLURP1, genes with diverse roles in extracellular matrix structure, keratinocyte proliferation and differentiation, calcium signaling and phospholipid metabolism. In contrast, cSLE skin had 722 unique DEGs compared to JM_L (Supplemental Table 7). Figure 2 shows that cSLE skin uniquely exhibits increased expression of IFNγ relative to CTL, illustrating a more pronounced type II interferon signature in addition to the type I interferon signature in common with JM_L. We confirmed a predominant type I IFN signature in JM_L skin by utilizing RNAseq data from control keratinocytes treated with either IFNα or IFNγ (Supplemental Figure 2). While both cSLE and JM_L skin have upregulation of genes stimulated by IFNα, cSLE demonstrates a more robust upregulation of genes regulated by IFNγ stimulation. With the xCell cell type enrichment analysis, cSLE skin exhibited an overall higher inflammatory cell signature when compared to JM_L, with increased T-cells, B-cells, macrophages and plasmacytoid dendritic cells (Supplemental Figure 1).
Figure 2.

Transcriptomic comparison of cSLE skin with lesional JM skin (q-value<0.05). Literature-based networks (GePS) obtained from the genes regulated in cSLE and JM vs. control biopsies. The pictures display the 100 best connected genes co-cited in PubMed abstracts in the same sentence linked to a function word (most relevant genes/interactions). Orange represents the genes that are upregulated and green represent the genes that are downregulated in lesional compared to control skin.
Skin-directed interferon scores are similar in JM and cSLE
JM_L patients had higher skin-directed IFN scores than JM_NL (p = 0.0001, Supplemental Figure 3). JM_L and cSLE patients had similar skin-directed IFN scores. The findings from our skin-directed interferon scoring of patient samples were validated with a more standard six-gene IFN score (Supplemental Figure 3). We also evaluated expression of three candidate ISGs (MX1, IFI44 and CXCL10) using RT-PCR, confirming higher MX1, IFI44 and CXCL10 expression in JM_L compared to CTL skin (Supplemental Figure 4). MX1 and CXCL10 expression were similar in JM_L and cSLE, while IFI44 expression was slightly higher in cSLE.
Derivation and evaluation of JM disease signature
Using the top three unique DEGs in JM_L skin relative to cSLE (FBLN2, CHKA and SLURP1), a JM-specific skin score was developed (details in methods) and evaluated relative to expression data from independent adult skin microarray datasets, including patients with dermatomyositis, cutaneous lupus, psoriasis, lichen planus, or graft versus host disease. Figure 3 illustrates that the JM disease signature is much higher in dermatomyositis (both pediatric and adult) as compared to other skin diseases, including pediatric and adult lupus. We confirmed that SLURP1 expression is higher in JM_L as compared to cSLE in our study samples (Supplemental Figure 5). We also evaluated a 23-gene JM-specific skin score, obtaining similar results (Supplemental Figure 6). The 23-gene JM-specific skin score was strongly associated with the skin-directed IFN score in JM_L samples (r=0.8713, p-value=0.0022) (Supplemental Figure 6).
Figure 3.
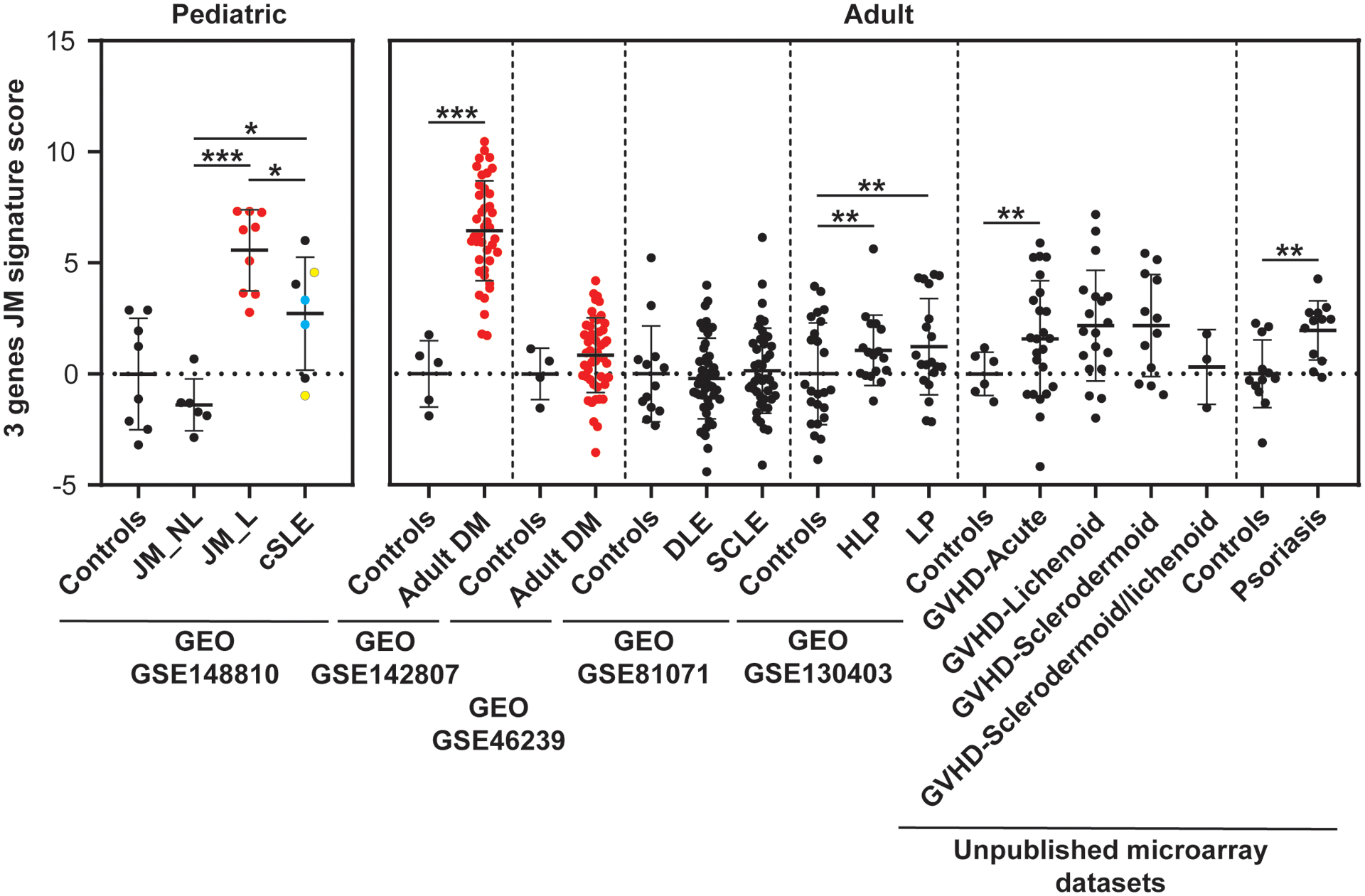
JM disease signature: comparison with transcriptomic datasets of skin lesions from adult DM and other inflammatory skin diseases. The 3 gene JM transcriptomic signature identified is the highest in juvenile and adult DM compared to other skin disease lesions (GEO GSE142807, GSE46239, GSE81071, GSE130403, and unpublished microarray datasets, courtesy of Dr. Johann Gudjonsson, Department of Dermatology, University of Michigan)(21, 23, 40). DM lesional samples are represented in red. Vertical dashed lines separate the studied datasets. Each dataset had its control sample set. DLE: discoid lupus erythematosus, SCLE: subacute cutaneous lupus erythematosus, HLP: Hypertrophic lichen planus, LP: lichen planus, GVHD: graft versus host disease. cSLE patients who had two biopsies are colored in blue and yellow. * p-value<0.05; ** p-value<0.01; *** p-value<0.0001.
NXP2+ JM patients demonstrate higher interferon scores and increased MX1 immunostaining in skin
Upon evaluation of skin-directed interferon scores in JM patients based on clinical features, we found that skin-directed IFN scores did not differ based on individual skin disease manifestations (Figure 4A; Supplemental Figure 7A). Specifically, skin-directed IFN scores did not differentiate between patients with amyopathic disease, nailfold capillary changes or calcinosis. There was no difference in skin-directed IFN scores based on treatment status (Figure 4B; Supplemental Figure 7B). While the skin-directed IFN scores did not differ between patients with MSA presence versus absence, the skin-directed IFN scores did differ by MSA subtype when lesional and non-lesional skin were analyzed together (Figure 4C; Supplemental Figure 7C). Of note, we only had three NXP2+ patients for this comparison. Anti-NXP2+ patients demonstrated higher skin-directed IFN scores than NXP2− patients (FC=8, p-value=0.034). JM patients with elevation of multiple serum muscle enzymes (levels of CK, aldolase, AST, ALT, LDH considered) also had higher skin-directed IFN scores when lesional and non-lesional skin were analyzed together (Figure 4D; Supplemental Figure 7D). We additionally evaluated expression of individual ISGs in JM patients. NXP2+ versus NXP2− JM patients had higher cutaneous expression levels of MX1, IFI44 and USP18 when lesional and non-lesional skin were analyzed together (Supplemental Figure 8). In JM_L skin biopsies, MX1 immunostaining localizes to keratinocytes, inflammatory cells and also the perivascular region (Figure 5). MX1 staining is more pronounced in NXP2+ patients compared to TIF1γ+ and MSA negative patients (Figure 5), in concordance with gene expression data (Supplemental Figure 9).
Figure 4.
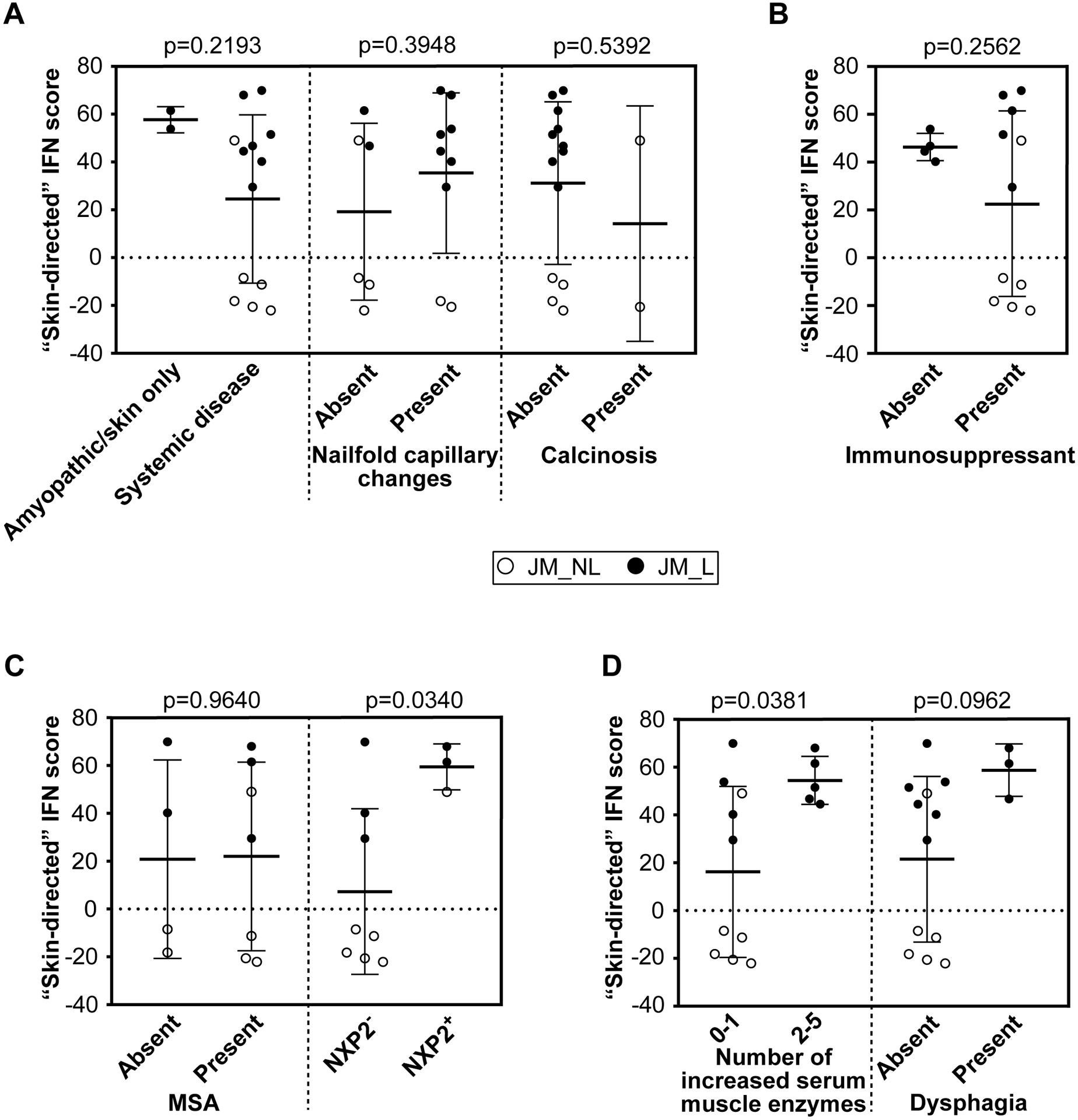
JM skin-directed IFN score with clinical variables. A. The skin-directed IFN score is not significantly modified by the presence of systemic disease, nailfold capillary changes or calcinosis. B. The skin-directed IFN score is not significantly changed by treatment status. C. The presence alone of any myositis-specific autoantibody (MSA) does not significantly alter the skin-directed IFN score; however, NXP2+ JM patients have a significantly higher skin-directed IFN score when lesional and non-lesional skin are analyzed together. D. An increased overall number of serum muscle enzymes was associated with a higher skin-directed IFN score (p-value=0.0381) when lesional and non-lesional skin are analyzed together. A higher skin-directed IFN score demonstrated a trend toward presence of dysphagia.
Figure 5.
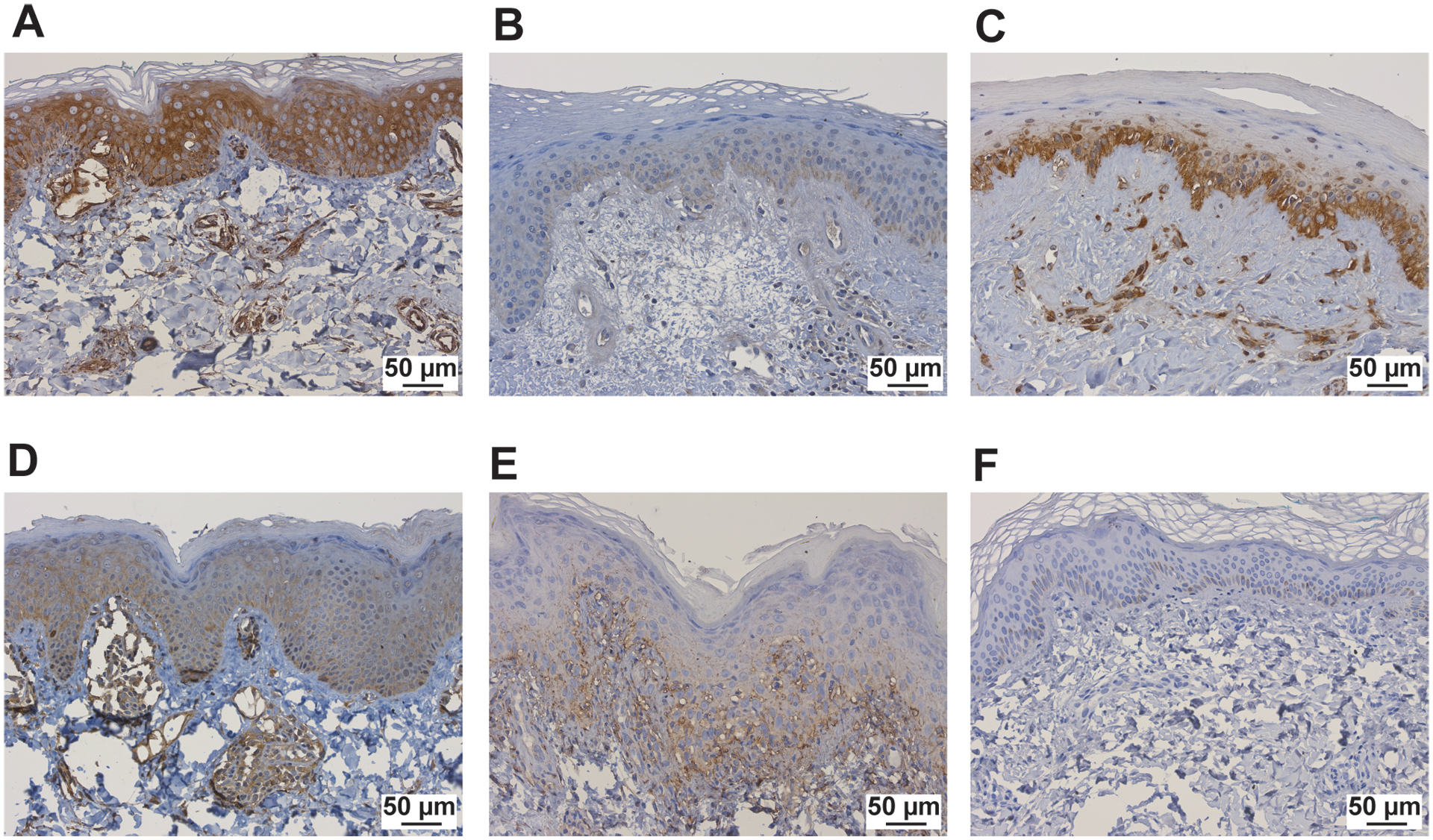
MX1 immunostaining in study samples, including JM patients A. NXP2+, B. TIF1γ+, C. Ku+, D. MSA neg, and E. cSLE patients, F. control patients. The scale bar represents 50μm.
Discussion
In this study, we provide the first characterization of cutaneous gene expression signatures in a juvenile myositis cohort. Similar to previous gene expression studies in JM muscle and peripheral blood(5, 6, 9, 25), we identified a striking type I interferon signature in JM lesional skin. Interestingly, JM lesional skin was found to have a predominant type I interferon signature whereas cSLE exhibited upregulation of both type I and II IFNs. A candidate JM-specific skin signature was derived using FBLN2, CHKA and SLURP1, all genes not typically considered to have immunomodulatory roles but instead functions in cellular structure and metabolism. While a skin-directed IFN score did not distinguish JM patients by cutaneous features or treatment status, the three NXP2+ JM patients in our study demonstrated a higher IFN score and stronger MX1 immunostaining in lesional skin.
Our study suggests interferons play a role in JM skin disease pathogenesis, in line with what has previously been described through gene expression and immunohistochemistry studies of adult and juvenile dermatomyositis skin(4, 13, 26). The specific mechanisms by which IFNs contribute to JM skin disease pathophysiology are not well understood. In dermatomyositis skin lesions, the number of CXCR3+ lymphocytes correlates with strength of MX1 immunostaining(26, 27), suggesting a role for IFN-inducible chemokines that are also CXCR3 ligands in recruitment of inflammatory cells. Indeed, we identified CXCL9 and CXCL10 within the top three DEGs in JM lesional skin, suggesting that these chemokines may play a role in cutaneous disease pathogenesis. CXCL10 has also been evaluated as a serum biomarker in JM and was demonstrated to outperform creatine kinase as disease activity marker(28). Using cell type enrichment analysis, we identified CD4+ memory T-cells and macrophages as increased in JM_L compared to JM_NL and CTL skin, in line with prior IHC studies of DM skin(27, 29).
In our study, we also demonstrated that NXP2+ patients exhibit a stronger IFN signature in skin, suggesting a potential role for NXP2 in contributing to the IFN signature. Given that we identified an elevated IFN signature even in a non-lesional skin sample from an NXP2+ patient, it is possible that the IFN signature is reflective of overall higher levels of systemic inflammation versus skin-specific inflammation. NXP2, also known as MORC3, is an understudied protein with RNA-binding activity that functions as an epigenetic regulator(30) and has also been described as both an antiviral factor(31) and a positive regulator of influenza virus transcription(32). Further studies are needed to understand the relationship between interferons, the autoantigen NXP2 and how NXP2 autoantibodies influence disease phenotype.
When comparing JM and cSLE lesional skin, we noted an overwhelming similarity in gene expression profiles. In particular, JM and cSLE shared a common type I IFN signature, with a major difference being that cSLE lesions had a central IFNγ node on network analysis. While this finding might be reflective of the cSLE samples in our study and influenced in part by presence of a discoid lesion(21), it is also possible that lack of a strong type II IFN signature distinguishes JM from cSLE skin, with implications for disease pathogenesis and treatment. We found that a molecular score incorporating expression of the top three DEGs, FBLN2, CHKA and SLURP1, was higher in both JM and DM, even when considering other autoimmune skin diseases. SLURP1 is expressed in differentiated keratinocytes(33) whereas FBLN2 is an extracellular matrix protein involved in basement membrane stability(34) and CHKA serves as a catalyst in phospholipid biosynthesis. Future validation of this JM-specific skin score and how these genes contribute to pathogenesis will be needed.
A major limitation of our study was our small sample size, including only three NXP2+ JM patients, and the retrospective nature of this study. JM patients in our study had variable disease duration at biopsy, which may have limited our ability to detect evolving clinical features such as calcinosis in our lesional JM cohort and also influenced gene expression profiles, with JM_NL samples skewed more toward chronic versus acute inflammatory changes. Given that we had no treatment naïve patients with non-lesional samples and JM_NL patients were on more immunosuppression; this may have dampened inflammatory pathway signatures that might otherwise have been represented in our gene expression data. JM_L skin samples were also all predominantly from sun-exposed areas whereas JM_NL samples were from non-sun-exposed skin, which may have contributed to the difference in gene expression signatures between JM_L and JM_NL skin. Typically, the difference in gene expression identified with FFPE tissue tends to be the more pronounced changes, as sensitivity of detection is diminished. This likely explains why we may not have seen as many DEGs as might be anticipated. However, it has been shown that gene expression from fresh vs. frozen vs. FFPE tissue can yield comparable findings(35–37), and the genes that we did identify are in fact more likely to be truly DEGs since we are likely under-detecting differences. JM clinical phenotypes were also quite heterogeneous, as we did have two clinically amyopathic JM patients enrolled in our study. We did not have all described MSAs represented in our cohort and lacked MSA data in four patients; therefore, we could only compare TIF1γ+ and MSA negative patients to NXP2+ patients. Notably, we lacked MDA5+ patients, and higher type I IFN signatures have been reported in skin and peripheral blood of adult DM patients(38). MSAs were also tested by two separate methodologies based on center of enrollment, which may have influenced testing results and patient categorization. It is also not clear what cell types were contributing to the IFN signature and how large a role skin resident versus infiltrating immune cells might play in disease pathogenesis given analysis of bulk tissue. While we did try and characterize potential immune cell types present in JM skin using xCell, we did not directly quantify cell types and lacked histopathology reports for non-lesional skin biopsies. Further work is ongoing to characterize the cellular origin of the type I IFN signature in JM skin and how this relates to MSA subtype.
In conclusion, our work provides the largest study of genome-wide expression analysis of JM and cSLE skin disease to date, serving to begin characterization of dysregulated genes and pathways specific to skin inflammation in these multisystem diseases. We identify a link between NXP2 autoantibodies and strength of the interferon signature in JM skin, which could lead to a better understanding of disease heterogeneity and pave the way for individualized treatment in juvenile myositis.
Supplementary Material
Supplemental Figure 2. Gene expression analysis of JM_L and cSLE in the context of type I or type II IFN signatures. Keratinocytes were treated with either IFNα or IFNγ. The gene expression signatures unique to these cytokines in keratinocytes (as assessed by RNAseq) were compared with the DEGs in JM_L and cSLE lesions. This eliminates the genes that are upregulated by both cytokines. Both cSLE and JM_L have upregulation of genes stimulated by IFNα, cSLE demonstrates a more robust upregulation of genes regulated by IFNγ stimulation.
Supplemental Figure 1. Cell type enrichment analysis using xCell webtool. A. Heatmap of relevant cell types representing average xCell enrichment score for HC, JM_NL, JM_L and cSLE. B. Graph of the macrophage xCell score, showing a significantly higher score in JM_L compared to JM_NL and controls and in cSLE compared to JM_L. C. Graph of the CD4+ memory T-cells xCell score, showing a significantly higher score in JM_L compared to JM_NL and in cSLE compared to JM_L. D. Graph of the B-cells xCell score showing a significantly higher score in cSLE compared to JM_L. E. Graph of the plasmacytoid dendritic cells xCell score, showing a significantly higher score in cSLE compared to JM_L. For (B-E), comparisons were made via unpaired Students’ t-test. * p-value<0.05; ** p-value<0.01; *** p-value<0.0001.
Supplemental Figure 3. IFN scores in controls, JM_NL, JM_L and cSLE skin. A. Skin-directed IFN score. B. Literature-derived IFN score.
Supplemental Figure 4. Validation of the expression of three candidate ISGs (MX1, IFI44 and CXCL10) using real-time PCR, which confirmed higher MX1, IFI44 and CXCL10 expression in lesional JM skin compared control skin. IFI44 expression was higher in cSLE skin when compared to JM_L skin. Comparisons were made via unpaired Students’ t-test.* p-value<0.05; ** p-value<0.01; *** p-value<0.0001.
Supplemental Figure 5. Validation of the expression of one JM candidate marker (SLURP1) using real-time PCR, which confirmed higher SLURP1 mRNA expression in lesional JM skin compared to cSLE skin. Comparison was made via Mann-Whitney test.
Supplemental Figure 6. JM disease signature. A. Comparison with transcriptomic datasets of skin lesions from adult DM and other inflammatory skin diseases. The 23 gene JM transcriptomic signature identified is the highest in JM (pediatric and adult) compared to other skin disease lesions. DM lesional samples are represented in red. Vertical dashed lines separate the studied datasets. Each dataset had its control sample set. DLE: discoid lupus erythematosus, SCLE: subacute cutaneous lupus erythematosus, HLP: Hypertrophic lichen planus, LP: lichen planus, GVHD: graft versus host disease. * p-value<0.05; ** p-value<0.01; *** p-value<0.0001. B. Correlation of skin-directed IFN score with JM disease signature scores in JM_L.
Supplemental Figure 7. Categorized JM skin-directed IFN score with clinical variables. A. The skin-directed IFN score in JM_L is not significantly modified by the presence of systemic disease, nailfold capillary changes or calcinosis. B. The skin-directed IFN score in JM_L is not significantly changed by treatment status. C. The presence of a myositis-specific autoantibody (MSA) and MSA subtype do not significantly alter the skin-directed IFN score in JM_L. D. An increased overall number of serum muscle enzymes and dysphagia are not associated with a higher skin-directed IFN score in JM_L.
Supplemental Figure 9. MX1 microarray expression data in controls and JM patients (NXP2− and NXP2+).
Supplemental Figure 8. Transcriptional expression levels of selected ISGs from skin microarray data in NXP2+ vs. NXP2− patients. A. Analysis from combined lesional and non-lesional skin biopsies. NXP2+ JM patients had higher cutaneous expression levels of MX1, IFI44 and USP18. B. Analysis categorizing JM samples based on non-lesional and lesional skin samples. NXP2+ JM patients had higher cutaneous expression levels of USP18.
Supplemental Table 1. Histological reports from patients with juvenile myositis and childhood-onset systemic lupus erythematosus included in microarray study.
Supplemental Table 2. List of the 221 genes regulated in JM_L compared to controls.
Supplemental Table 3. List of Canonical Pathways (IPA) regulated in JM_L compared to controls.
Supplemental Table 4. List of Top upstream regulators (IPA) in JM_L compared to controls.
Supplemental Table 6. List of Canonical Pathways (IPA) regulated in JM_L compared to JM_NL.
Supplemental Table 5. List of the 7,990 genes regulated in JM_L compared to JM_NL.
Supplemental Table 7. List of the genes regulated in the comparison of JM_L with sCLE (relative to controls).
Acknowledgements
We would like to thank the University of Michigan Advanced Genomics and Research Histology Core for their support and equipment for processing and sequencing of skin samples. We express our many thanks to the myositis and lupus patients for generously sharing their samples for our work. We also thank Matthew Manninen for his expertise in R programming and assistance with calculation of interferon scores and Tammi Reed for assisting with the FFPE skin biopsy processing protocol. This work was presented in part at both the 2019 and 2020 American College of Rheumatology Annual Meetings(39).
Funding
This work was supported by a Cure JM Foundation Research Grant (PI: JLT), a K12 Child Health Research Center Career Development Award to the University of Michigan Department of Pediatrics (K12 HD028820-28), a Rheumatology Research Foundation Investigator Award (PI: JLT), a MICHR Pathway to First Grant Award under award number UL1TR002240 (PI: JLT), and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award numbers R01-AR071384 (JMK), K24-AR076975 (JMK), P30-AR075043 (JEG) and K01-AR072129 (LCT).
Competing Interests
Dr. Kahlenberg reports personal fees from AstraZeneca, Aurinia, Boehringer Ingleheim, Bristol Myers Squibb (BMS), Eli Lilly, and grants from BMS/Celgene and Q32 bio all outside the submitted work. Dr. Gudjonsson reports research grant funding from Almirall, Novartis, Pfizer, Eli Lilly, Celgene/BMS Advisory board Almirall, Novartis, Eli Lilly, AnaptysBio, also outside the submitted work.
References
- 1.Almeida B, Campanilho-Marques R, Arnold K, Pilkington CA, Wedderburn LR, Nistala K, et al. Analysis of Published Criteria for Clinically Inactive Disease in a Large Juvenile Dermatomyositis Cohort Shows That Skin Disease Is Underestimated. Arthritis Rheumatol. 2015;67(9):2495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christen-Zaech S, Seshadri R, Sundberg J, Paller AS, Pachman LM. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty-six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;58(2):571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stringer E, Singh-Grewal D, Feldman BM. Predicting the course of juvenile dermatomyositis: significance of early clinical and laboratory features. Arthritis Rheum. 2008;58(11):3585–92. [DOI] [PubMed] [Google Scholar]
- 4.Wong D, Kea B, Pesich R, Higgs BW, Zhu W, Brown P, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLoS One. 2012;7(1):e29161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168(8):4154–63. [DOI] [PubMed] [Google Scholar]
- 6.O’Connor KA, Abbott KA, Sabin B, Kuroda M, Pachman LM. MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement. Clin Immunol. 2006;120(3):319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg SA, Higgs BW, Morehouse C, Walsh RJ, Kong SW, Brohawn P, et al. Relationship between disease activity and type 1 interferon- and other cytokine-inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun. 2012;13(3):207–13. [DOI] [PubMed] [Google Scholar]
- 8.Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 2009;60(11):3436–46. [DOI] [PubMed] [Google Scholar]
- 9.Chen YW, Shi R, Geraci N, Shrestha S, Gordish-Dressman H, Pachman LM. Duration of chronic inflammation alters gene expression in muscle from untreated girls with juvenile dermatomyositis. BMC Immunol. 2008;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moneta GM, Pires Marafon D, Marasco E, Rosina S, Verardo M, Fiorillo C, et al. Muscle Expression of Type I and Type II Interferons Is Increased in Juvenile Dermatomyositis and Related to Clinical and Histologic Features. Arthritis Rheumatol. 2019;71(6):1011–21. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. 2018;77(11):1653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, et al. Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI Insight. 2017;2(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum. 2010;62(9):2813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tansley SL, Li D, Betteridge ZE, McHugh NJ. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatology (Oxford). 2020;59(8):2109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tansley SL, Snowball J, Pauling JD, Lissina A, Kuwana M, Rider LG, et al. The promise, perceptions, and pitfalls of immunoassays for autoantibody testing in myositis. Arthritis Res Ther. 2020;22(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mecoli CA, Albayda J, Tiniakou E, Paik JJ, Zahid U, Danoff SK, et al. Myositis Autoantibodies: A Comparison of Results From the Oklahoma Medical Research Foundation Myositis Panel to the Euroimmun Research Line Blot. Arthritis Rheumatol. 2020;72(1):192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Espinosa-Ortega F, Holmqvist M, Alexanderson H, Storfors H, Mimori T, Lundberg IE, et al. Comparison of autoantibody specificities tested by a line blot assay and immunoprecipitation-based algorithm in patients with idiopathic inflammatory myopathies. Ann Rheum Dis. 2019;78(6):858–60. [DOI] [PubMed] [Google Scholar]
- 18.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. [DOI] [PubMed] [Google Scholar]
- 19.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2951–62. [DOI] [PubMed] [Google Scholar]
- 21.Berthier CC, Tsoi LC, Reed TJ, Stannard JN, Myers EM, Namas R, et al. Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. J Clin Med. 2019;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiche L, Jourde-Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014;66(6):1583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsoi L, Gharaee-Kermani M, Berthier CC, Nault T, Hile G, Estadt SN, et al. IL-18-containing five-gene signature distinguishes histologically identical dermatomyositis and lupus erythematosus skin lesions. JCI Insight. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–93. [DOI] [PubMed] [Google Scholar]
- 25.Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. 2007;13(1–2):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wenzel J, Schmidt R, Proelss J, Zahn S, Bieber T, Tuting T. Type I interferon-associated skin recruitment of CXCR3+ lymphocytes in dermatomyositis. Clin Exp Dermatol. 2006;31(4):576–82. [DOI] [PubMed] [Google Scholar]
- 27.Caproni M, Torchia D, Cardinali C, Volpi W, Del Bianco E, D’Agata A, et al. Infiltrating cells, related cytokines and chemokine receptors in lesional skin of patients with dermatomyositis. Br J Dermatol. 2004;151(4):784–91. [DOI] [PubMed] [Google Scholar]
- 28.Wienke J, Bellutti Enders F, Lim J, Mertens JS, van den Hoogen LL, Wijngaarde CA, et al. Galectin-9 and CXCL10 as Biomarkers for Disease Activity in Juvenile Dermatomyositis: A Longitudinal Cohort Study and Multicohort Validation. Arthritis Rheumatol. 2019;71(8):1377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hausmann G, Herrero C, Cid MC, Casademont J, Lecha M, Mascaro JM. Immunopathologic study of skin lesions in dermatomyositis. J Am Acad Dermatol. 1991;25(2 Pt 1):225–30. [DOI] [PubMed] [Google Scholar]
- 30.Li DQ, Nair SS, Kumar R. The MORC family: new epigenetic regulators of transcription and DNA damage response. Epigenetics. 2013;8(7):685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sloan E, Orr A, Everett RD. MORC3, a Component of PML Nuclear Bodies, Has a Role in Restricting Herpes Simplex Virus 1 and Human Cytomegalovirus. J Virol. 2016;90(19):8621–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ver LS, Marcos-Villar L, Landeras-Bueno S, Nieto A, Ortin J. The Cellular Factor NXP2/MORC3 Is a Positive Regulator of Influenza Virus Multiplication. J Virol. 2015;89(19):10023–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favre B, Plantard L, Aeschbach L, Brakch N, Christen-Zaech S, de Viragh PA, et al. SLURP1 is a late marker of epidermal differentiation and is absent in Mal de Meleda. J Invest Dermatol. 2007;127(2):301–8. [DOI] [PubMed] [Google Scholar]
- 34.Ibrahim AM, Sabet S, El-Ghor AA, Kamel N, Anis SE, Morris JS, et al. Fibulin-2 is required for basement membrane integrity of mammary epithelium. Sci Rep. 2018;8(1):14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodgin JB, Borczuk AC, Nasr SH, Markowitz GS, Nair V, Martini S, et al. A molecular profile of focal segmental glomerulosclerosis from formalin-fixed, paraffin-embedded tissue. Am J Pathol. 2010;177(4):1674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bossel Ben-Moshe N, Gilad S, Perry G, Benjamin S, Balint-Lahat N, Pavlovsky A, et al. mRNA-seq whole transcriptome profiling of fresh frozen versus archived fixed tissues. BMC Genomics. 2018;19(1):419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wimmer I, Troscher AR, Brunner F, Rubino SJ, Bien CG, Weiner HL, et al. Systematic evaluation of RNA quality, microarray data reliability and pathway analysis in fresh, fresh frozen and formalin-fixed paraffin-embedded tissue samples. Sci Rep. 2018;8(1):6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ono N, Kai K, Maruyama A, Sakai M, Sadanaga Y, Koarada S, et al. The relationship between type 1 IFN and vasculopathy in anti-MDA5 antibody-positive dermatomyositis patients. Rheumatology (Oxford). 2020;59(4):918. [DOI] [PubMed] [Google Scholar]
- 39.Turnier JBC, Tsoi L, Lowe L, Morgan G, Gudjonsson J, Pachman L, Kahlenberg J. . Cutaneous Gene Expression Signatures in Juvenile Myositis Reveal a Prominent IFN Signature in Lesional Skin [abstract]. Arthritis Rheumatol. 2019;71. [Google Scholar]
- 40.Shao S, Tsoi LC, Sarkar MK, Xing X, Xue K, Uppala R, et al. IFN-gamma enhances cell-mediated cytotoxicity against keratinocytes via JAK2/STAT1 in lichen planus. Sci Transl Med. 2019;11(511). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 2. Gene expression analysis of JM_L and cSLE in the context of type I or type II IFN signatures. Keratinocytes were treated with either IFNα or IFNγ. The gene expression signatures unique to these cytokines in keratinocytes (as assessed by RNAseq) were compared with the DEGs in JM_L and cSLE lesions. This eliminates the genes that are upregulated by both cytokines. Both cSLE and JM_L have upregulation of genes stimulated by IFNα, cSLE demonstrates a more robust upregulation of genes regulated by IFNγ stimulation.
Supplemental Figure 1. Cell type enrichment analysis using xCell webtool. A. Heatmap of relevant cell types representing average xCell enrichment score for HC, JM_NL, JM_L and cSLE. B. Graph of the macrophage xCell score, showing a significantly higher score in JM_L compared to JM_NL and controls and in cSLE compared to JM_L. C. Graph of the CD4+ memory T-cells xCell score, showing a significantly higher score in JM_L compared to JM_NL and in cSLE compared to JM_L. D. Graph of the B-cells xCell score showing a significantly higher score in cSLE compared to JM_L. E. Graph of the plasmacytoid dendritic cells xCell score, showing a significantly higher score in cSLE compared to JM_L. For (B-E), comparisons were made via unpaired Students’ t-test. * p-value<0.05; ** p-value<0.01; *** p-value<0.0001.
Supplemental Figure 3. IFN scores in controls, JM_NL, JM_L and cSLE skin. A. Skin-directed IFN score. B. Literature-derived IFN score.
Supplemental Figure 4. Validation of the expression of three candidate ISGs (MX1, IFI44 and CXCL10) using real-time PCR, which confirmed higher MX1, IFI44 and CXCL10 expression in lesional JM skin compared control skin. IFI44 expression was higher in cSLE skin when compared to JM_L skin. Comparisons were made via unpaired Students’ t-test.* p-value<0.05; ** p-value<0.01; *** p-value<0.0001.
Supplemental Figure 5. Validation of the expression of one JM candidate marker (SLURP1) using real-time PCR, which confirmed higher SLURP1 mRNA expression in lesional JM skin compared to cSLE skin. Comparison was made via Mann-Whitney test.
Supplemental Figure 6. JM disease signature. A. Comparison with transcriptomic datasets of skin lesions from adult DM and other inflammatory skin diseases. The 23 gene JM transcriptomic signature identified is the highest in JM (pediatric and adult) compared to other skin disease lesions. DM lesional samples are represented in red. Vertical dashed lines separate the studied datasets. Each dataset had its control sample set. DLE: discoid lupus erythematosus, SCLE: subacute cutaneous lupus erythematosus, HLP: Hypertrophic lichen planus, LP: lichen planus, GVHD: graft versus host disease. * p-value<0.05; ** p-value<0.01; *** p-value<0.0001. B. Correlation of skin-directed IFN score with JM disease signature scores in JM_L.
Supplemental Figure 7. Categorized JM skin-directed IFN score with clinical variables. A. The skin-directed IFN score in JM_L is not significantly modified by the presence of systemic disease, nailfold capillary changes or calcinosis. B. The skin-directed IFN score in JM_L is not significantly changed by treatment status. C. The presence of a myositis-specific autoantibody (MSA) and MSA subtype do not significantly alter the skin-directed IFN score in JM_L. D. An increased overall number of serum muscle enzymes and dysphagia are not associated with a higher skin-directed IFN score in JM_L.
Supplemental Figure 9. MX1 microarray expression data in controls and JM patients (NXP2− and NXP2+).
Supplemental Figure 8. Transcriptional expression levels of selected ISGs from skin microarray data in NXP2+ vs. NXP2− patients. A. Analysis from combined lesional and non-lesional skin biopsies. NXP2+ JM patients had higher cutaneous expression levels of MX1, IFI44 and USP18. B. Analysis categorizing JM samples based on non-lesional and lesional skin samples. NXP2+ JM patients had higher cutaneous expression levels of USP18.
Supplemental Table 1. Histological reports from patients with juvenile myositis and childhood-onset systemic lupus erythematosus included in microarray study.
Supplemental Table 2. List of the 221 genes regulated in JM_L compared to controls.
Supplemental Table 3. List of Canonical Pathways (IPA) regulated in JM_L compared to controls.
Supplemental Table 4. List of Top upstream regulators (IPA) in JM_L compared to controls.
Supplemental Table 6. List of Canonical Pathways (IPA) regulated in JM_L compared to JM_NL.
Supplemental Table 5. List of the 7,990 genes regulated in JM_L compared to JM_NL.
Supplemental Table 7. List of the genes regulated in the comparison of JM_L with sCLE (relative to controls).