

Abstract
Photoacoustic (PA) imaging has emerged as a reliable in vivo technique for diverse biomedical applications ranging from disease screening to analyte sensing. Most contemporary PA imaging agents employ NIR-I light (650–900 nm) to generate an ultrasound signal; however, there is significant interference from endogenous biomolecules such as hemoglobin that are PA active in this window. Transitioning to longer excitation wavelengths (i.e., NIR-II) reduces the background and facilitates the detection of low abundance targets (e.g., nitric oxide, NO). In this study, we employed a two-phase tuning approach to develop APNO-1080, a NIR-II NO-responsive probe for deep-tissue PA imaging. First, we performed Hammett and Brønsted analyses to identify a highly reactive and selective aniline-based trigger that reacts with NO via N-nitrosation chemistry. Next, we screened a panel of NIR-II platforms to identify chemical structures that have a low propensity to aggregate since this can diminish the PA signal. In a head-to-head comparison with a NIR-I analogue, APNO-1080 was 17.7-fold more sensitive in an in vitro tissue phantom assay. To evaluate the deep-tissue imaging capabilities of APNO-1080 in vivo, we performed PA imaging in an orthotopic breast cancer model and a heterotopic lung cancer model. Relative to control mice not bearing tumors, the normalized turn-on response was 1.3 ± 0.12 and 1.65 ± 0.07, respectively.
Graphical Abstract
INTRODUCTION
Photoacoustic (PA) imaging is a powerful in vivo technique that leverages the PA effect to convert absorbed light into an ultrasound readout.1 Because sound at clinically relevant frequencies (2–18 MHz) can readily propagate through tissue with minimal perturbation,2, 3 PA imaging has emerged as a versatile approach for various biomedical applications.4 For instance, label-free PA imaging has been used to detect breast cancer,5 thyroid cancer,6 inflammatory arthritis,7 and scleroderma8 in human patients. Moreover, in the context of analyte sensing, a variety of design strategies have been employed to develop acoustogenic probes (also known as activatable PA probes) to selectively visualize and monitor diverse imaging targets in vivo.9–11 Examples include acoustogenic probes for hypoxia,12–15 redox status,16 enzymatic activity,17–20 metal ions,21–24 biological thiols,25,26 and various reactive oxygen and nitrogen species (ROS/RNS).10,27,28 Most small-molecule PA probes have been designed on NIR-I dye platforms which have absorbance maxima between 650 and 900 nm. Unfortunately, several endogenous biomolecules (i.e., hemoglobin, oxy-hemoglobin, lipids, and melanin) absorb within this region, which results in high background. In addition to undesirable signal, these biological chromophores attenuate the incident light, thereby limiting sensitivity and imaging depth. Notable NIR-I examples include our first-generation probe (APNO-5) and the second-generation congener (SR-APNO-3) that facilitated the detection of nitric oxide (NO) in live mice but were confined to subcutaneous models of inflammation9 and breast cancer, respectively.10 Red-shifted PA probes that absorb within the NIR-II region (>1000 nm) can potentially overcome these limitations and afford improved signal-to-noise ratios, enhanced sensitivity, and deeper tissue penetration.29–31 Toward this end, we undertook a two-phase tuning approach, which yielded APNO-1080, the first NIR-II probe for deep-tissue NO PA imaging.
RESULTS AND DISCUSSION
Design and Synthesis of APNO-1080.
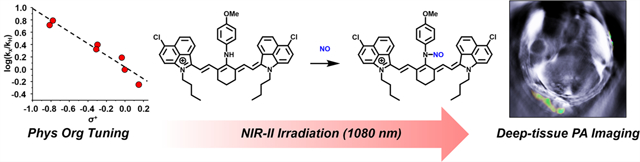
First, we sought to translate our previous N-nitrosation-based detection strategy to the cyanine dye platform. This was accomplished by substituting the meso-chloro of Cy7-Cl (a readily accessible NIR-I cyanine) with a panel of anilines bearing different substitutions at the para position (−Br, −H, −CH2OH, −Et, −Me, −OEt, and −OMe) to afford probes 1–7 (Figure 1a). Of note, we selected the para position, over our previous ortho substitution pattern, to limit steric effects. The corresponding Hammett plot revealed a ρ value of −0.94 which is consistent with a modest positive charge character accumulating at the transition state (e.g., N-nitrosation prior to deprotonation) that is promoted by increased electron density (Figure 1b). This was corroborated in our Brønsted plot, in which we observed a strong correlation between the pKa of the aniline’s conjugate acid and the initial rate of the reaction (R2 = 0.87, Figure 1c). The p-anisidine trigger facilitated near-complete conversion with NO (100 equiv at 25 °C) in less than 30 s and was selected for further probe development.
Figure 1.
(a) Schematic showing the reaction between the various NIR-I APNOs (probes 1–7) and NO to form an N-nitroso product. (b) Hammett plot for the N-nitrosation reaction between para-substituted APNOs and NO (introduced as MAHMA-NONOate) at 25 °C. Dotted line represents the best linear fit. R2 = 0.94. (c) Brønsted plots indicating the linear relationship between log(k) and the pKa value of the conjugate acid form of each aniline. Dotted line represents the best linear fit. R2 = 0.87. (d) Chemical structures of NIR-II APNOs (probes 8–12) derived from IR-26, IR-1061, IR-1048, Et-1080, and Flav7.
Specifically, we installed the trigger onto three commercial NIR-II cyanines (IR-26, IR-1061, and IR-1048), as well as Et-108032 and Flav733 to afford probes 8–12, respectively (Figure 1d). The cyanine scaffold was selected, as opposed to our previous work on the aza-BODIPY dyes, because its intrinsic lipophilic and cationic character can be exploited to target cancer cells.34 The goal of this phase was to identify probes that satisfy the following criteria to maximize performance in deep tissue. First, the extinction coefficient must be large (>104 M−1 cm−1) to generate a strong PA output upon irradiation. Second, there must be a low propensity to form insoluble aggregates in aqueous media that commonly result in broad absorbance bands and lower PA signals. Moreover, most probes in an aggregated form do not react with the target analyte. Third, the probe must respond rapidly to NO to afford a large spectral separation between the wavelength of maximum absorbance (λabs) of the probe and the NO-reacted product. Fourth, an ideal probe must exhibit good photostability for continuous monitoring over several minutes.
In Vitro Characterization and Biocompatibility Assays.
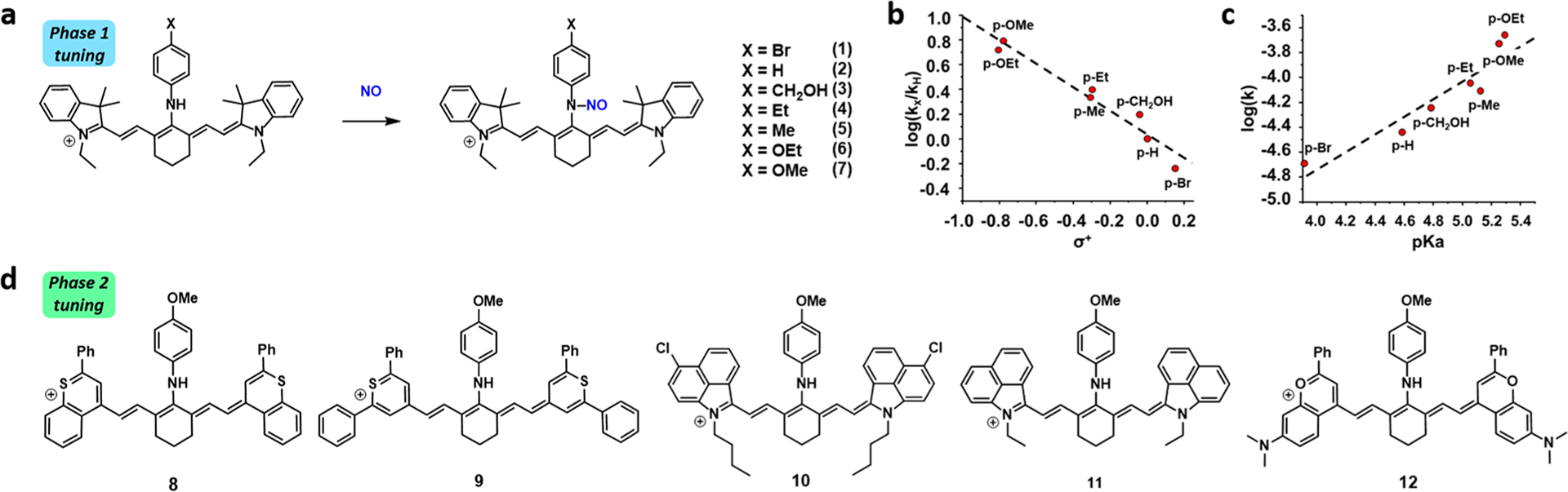
Each member of the series had a large extinction coefficient (∼105 M−1cm−1) with an absorbance maximum in the NIR II window (Table S1). However, this was not the only factor that influences the PA “brightness”. For example, probes 8, 9, and 12 were poorly soluble in all the aqueous buffer systems that we evaluated. This resulted in broad spectral properties and a weaker PA signal. Probes 10 and 11, on the other hand, had sharp, well-defined spectral properties in aqueous media and a significantly stronger PA signal upon irradiation. Only two of the probes from the series (10 and 11) reacted completely when they were treated with an excess of NO. In contrast, probes 8 and 12 did not react, and while probe 9 did respond, the conversion to product did not go to completion even after an extended incubation period of 1 h. This is consistent with our previous work that indicated that solubility impacts the rate of N-nitrosation in vitro, likely because the trigger is inaccessible in this aggregated state. Probe 10 (herein referred to as APNO-1080) underwent rapid conversion to the N-nitrosated product with a 206 nm shift in the absorbance maximum from 874 to 1080 nm (Figure 2a). Of note, this version was selected over probe 11 because the λabs of the N-nitrosated product is more red-shifted by nearly 40 nm. Lastly, continuous irradiation of APNO-1080 and its N-nitrosated product did not result in any photo-oxidation, or photodenitrosylation, which we have observed in previous generations (Figure 2b).15,35 Notably, 97% of activated APNO-1080 remained after 5 min, whereas only 41% was present for an NIR-I congener (vide infra, Figure S1).
Figure 2.
(a) Normalized absorbance spectra of APNO-1080 with (red) and without (blue) NO treatment. (b) Photostability assay in which APNO-1080 or the N-nitrosated product (50 μM) is continuously irradiated with a pulsed laser at their respective λabs for 300 s. (c) Reaction of APNO-1080 (5 μM) with biologically relevant metal ions (1 mM), GSH (1 mM), Cys (500 μM), H2S (100 μM), reactive oxygen species (1 mM), reactive carbonyl species (1 mM), reactive nitrogen species (1 mM), ONOO− (50 μM), and NO (100 μM) after 1 h incubation. (d) MTT viability assay in A549 cells after 24 h incubation at 37 °C.
Having identified APNO-1080 as our optimized NIR-II acoustogenic probe, we first examined its selectivity for NO. This was accomplished by treating APNO-1080 with a panel of biologically relevant analytes for 1 h and measuring the absorbance at 1080 nm. For instance, glutathione (GSH) was among the tested analytes because it has the potential to displace the trigger and generate a red-shifted GSH-cyanine adduct.36 Likewise, reactive carbonyl species, such as formaldehyde, could presumably form red-shifted iminium products and lead to false positive results. Under no circumstance did we observe any undesirable activation of our probe (Figure 2c). In addition, to determine suitability for potential cellular and in vivo applications, we performed a standard MTT viability assay in A549 cells in which no cytotoxicity was observed at concentrations up to 25 μM for 24 h (Figure 2d). Lastly, we examined the metabolic stability of APNO-1080 by incubating it with rat liver microsomes (RLM) rich in CYP450 enzymes at 37 °C for 1 h. NIR spectroscopic analysis revealed that there was no increase in absorbance at 1080 nm, suggesting APNO-1080 was stable in the presence of RLMs. To corroborate these results, we subjected a RLM reaction to ESI-MS analysis and found that the probe was indeed stable for 24 h under these conditions (Figure S2).
NIR-I and NIR-II PA Imaging of Tissue Phantoms.
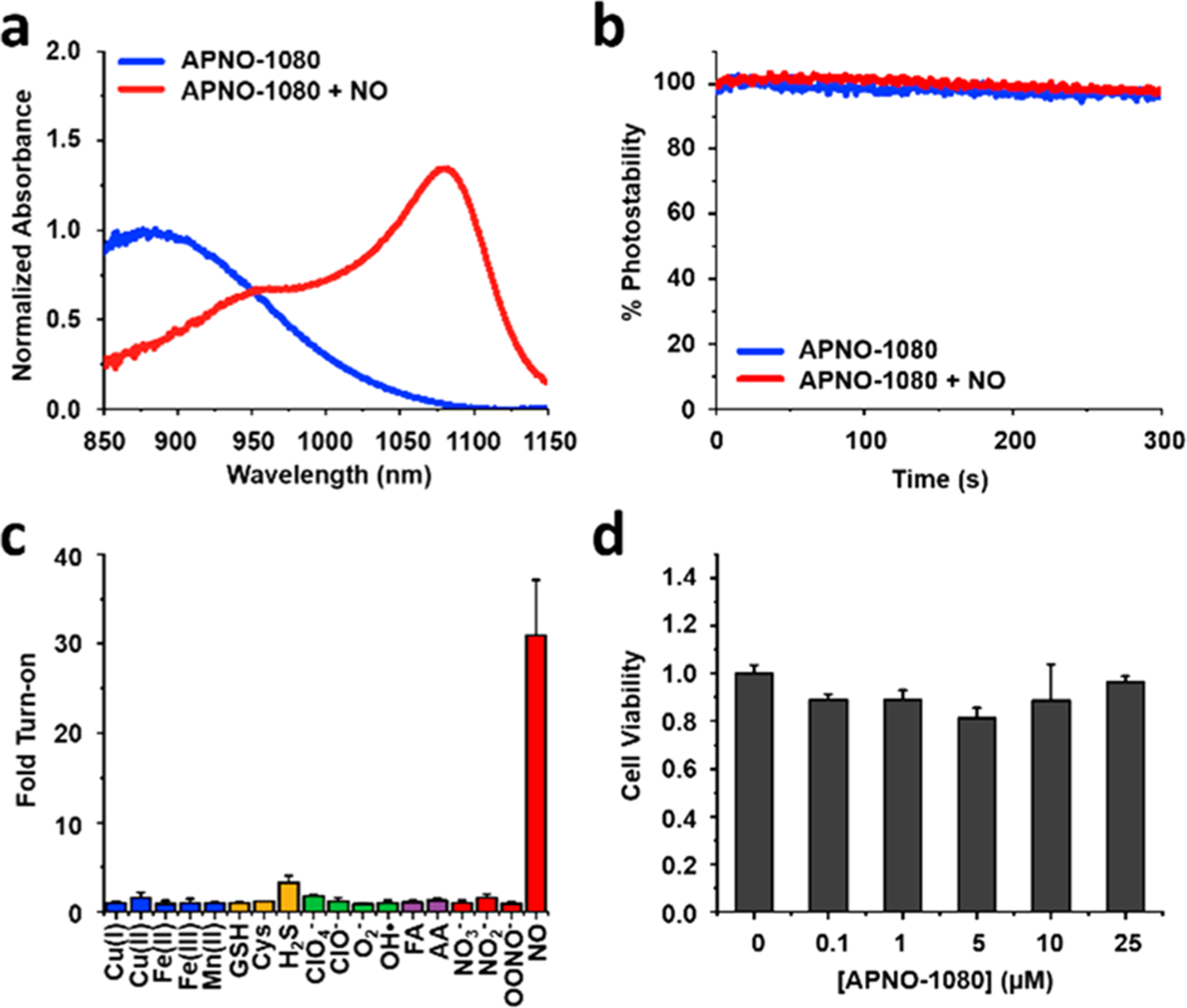
Next, we devised a head-to-head experiment to directly compare the deep-tissue capabilities of APNO-1080 and its NIR-I analogue, probe 7 (herein referred to as APNO-780). Solutions of each probe were treated with an NO-donor (MAHMA-NONOate) to ensure full activation. The resulting products were then embedded in a 3 cm thick tissue mimicking phantom composed of agarose and 2% milk (six times the size of a standard phantom used with our MSOT InVision 128 system) and imaged at their absorbance maxima. APNO-1080 can be clearly observed, while APNO-780 was not discernible from the background at these imaging depths (Figure 3b). This corresponds to a 17.7-fold increase in sensitivity when corrected for wavelength-dependent differences in fluence (Figure 3c). It is noteworthy that the PA brightness of both probes was similar (less than 2-fold difference), suggesting that the increased penetration of the excitation light is driving the difference in sensitivity.15 It is reasonable to assume that this is an underapproximation of the effect because the tissue-mimicking phantoms we employed lack hemoglobin and oxy-hemoglobin, which attenuates NIR-I light more significantly than NIR-II light (Figure 3a).
Figure 3.
(a) Schematic illustrating that NIR-II light can penetrate deeper into tissue compared to NIR-I light. (b) Representative PA images of APNO-780 and APNO-1080 (10 μM) treated with NO and overlaid with a 3 cm thick tissue imaging phantom. (c) Quantified data from (b). Error bars = SD (n = 3) Statistical analysis was performed using a two-tailed Student’s t test (α = 0.05). ****: p < 0.0001.
Evaluation of APNO-1080 in Breast and Liver Cancer Models.
The detection of endogenous NO associated with cancer is critical to understanding its role in modulating the tumor microenvironment.37 For instance, it is believed that tumor-associated macrophages produce low steady-state concentrations of NO (<200 nM) and this is responsible for creating a chronic inflammatory state that drives tumor progression, leading to poorer clinical prognosis.38 To this end, APNO-1080 was first applied in an orthotopic 4T1-Luc breast cancer model. In comparison to the subcutaneous breast cancer model we previously employed, the resulting orthotopic tumors can grow deeper into the body, and thus, this is an ideal model for us to evaluate the deep tissue imaging capabilities of APNO-1080 in vivo. A luciferase-expressing cell line was employed to confirm implantation and to track tumor growth. Once the tumors had grown to a volume of ∼400 mm3, APNO-1080 was administered systemically, the animals were imaged, and the signal from the probe was isolated using spectral unmixing. We observed a 1.3 ± 0.1-fold turn-on response (defined as PAFinal/PAInitial) in the tumor-bearing mice as compared to 1.0 ± 0.2-fold in the tumor-free control (Figure 4a,b,e). Next, we surgically implanted A549-Luc2 lung cancer cells onto the liver of Nu/J mice. This model simulates the liver metastases found in lung cancer patients and provides an opportunity to image cancer-derived NO within significantly deeper tissue. Tumors were detectable several weeks post implantation using bioluminescence imaging, at which point APNO-1080 was administered via retroorbital injection for PA imaging. After 30 min, we performed real-time PA monitoring over 5 min and observed a marked signal increase in the tumor region (Supplementary Video 1). Overall, we observed a 1.65 ± 0.07-fold turn-on response in tumor-bearing mice relative to 1.00 ± 0.07-fold in mice that received a sham surgery (Figure 4c,d,f).
Figure 4.
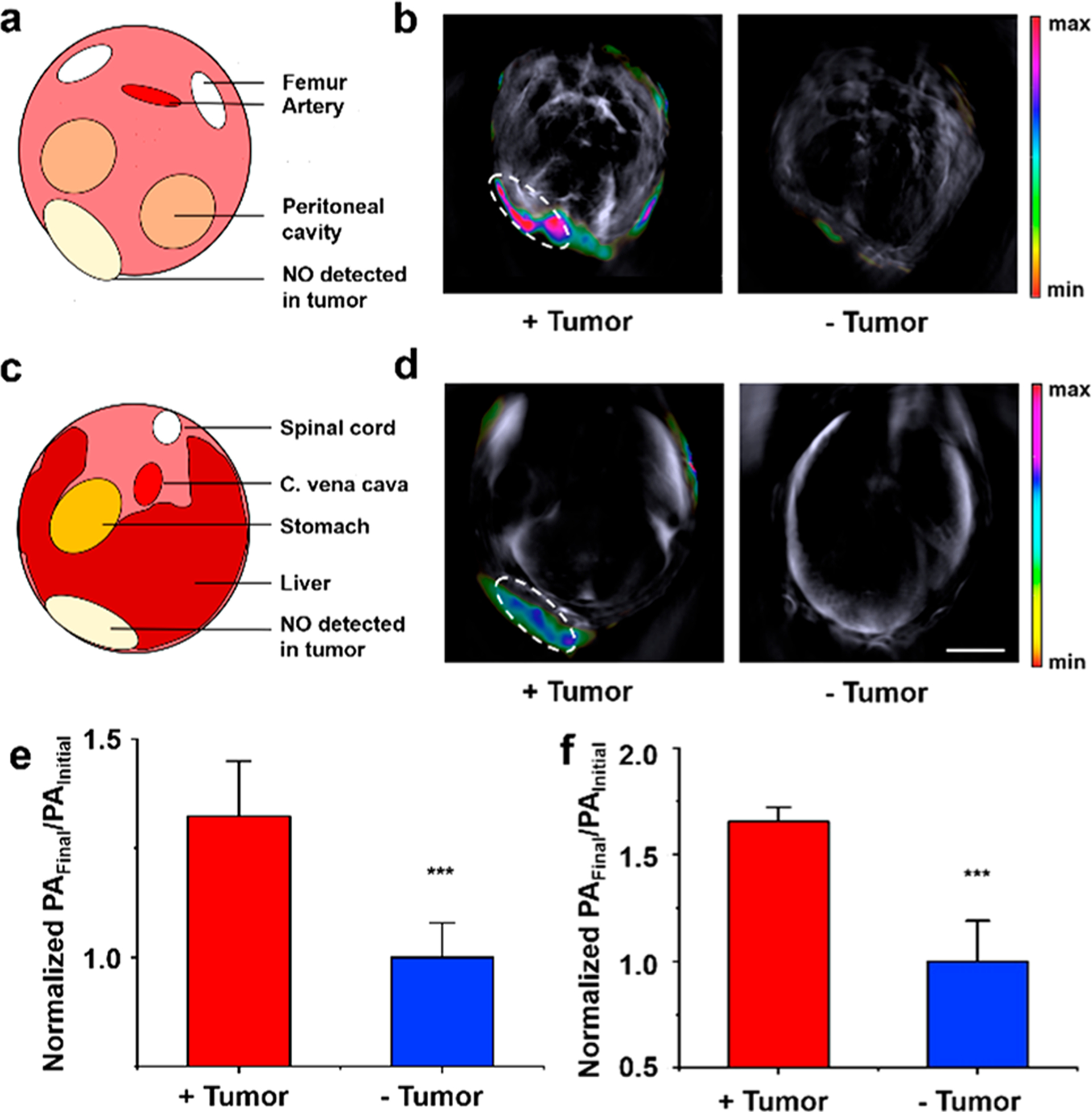
(a) Cartoon schematic depicting the cross-sectional anatomy of the images in (b). (b) Representative PA images of a 4T1-Luc tumor, as well as the tumor-less control after treatment with APNO-1080 (50 μM). Scale bar = 5 mm. (c) Cartoon schematic depicting the cross-sectional anatomy of images in (d). (d) Representative PA images of a heterotopic A549-Luc2 tumor, as well as the tumor-less control after treatment with APNO-1080. Scale bar = 5 mm. (e) Normalized PA fold turn-on in 4T1-Luc tumors after treatment with APNO-1080. Error bars = SD (n = 6). (f) Normalized PA fold turn-on in A549 tumors (n = 6) and the tumor-less control (n = 3) after treatment with APNO-1080. Error bars = SD. Statistical analysis was performed using a two-tailed Student’s t test (α = 0.05). ***: p < 0.001
CONCLUSIONS
In conclusion, we successfully employed a two-phase tuning approach to develop the first NIR-II acoustogenic probe, APNO-1080, for the detection of endogenous, cancer-derived NO within deep-tissue. It is noteworthy that an organic semiconducting nanoprobe was recently developed for NIR-II fluorescence imaging of NO; however, the λabs value was only at 680 nm.39 The design of NIR PA probes (regardless if they are small-molecule- or nanoparticle-based) is distinctly different from that of the more abundant NIR-II fluorescence designs where the λabs does not have to be NIR-II to generate a fluorescent readout. APNO-1080 features an optimized p-anisidine trigger on a NIR-II cyanine platform (IR-1048). Upon reaction with NO, the N-nitrosated product exhibits a larger extinction coefficient relative to the probe (Figure S3) and absorbs maximally at 1080 nm with no spectral overlap with the probe. While there are many NO probes, most examples are only suitable for cellular studies. Indeed, very few are applicable for in vivo applications and fewer yet, are sensitive enough to detect NO in cancer where the steady-state concentrations are in the low nM range. Prior to this work, our most responsive probe could only detect NO in shallow, subcutaneous models using NIR-I PA imaging. This study highlights the value of shifting the λabs of PA contrast agents and acoustogenic probes into the NIR-II window to achieve greater imaging depths and superior sensitivity. Without these improvements, it would not have been possible to detect endogenous NO in the two deep-tissue murine models of cancer we generated in this study. Current investigations are focused on applying APNO-1080 to study how external factors, such as diet, aging, and oxygenation, can alter NO concentration and/or activity.
EXPERIMENTAL DETAILS
Synthetic Procedures.
Thin-layer chromatography (TLC) was performed on glass-backed TLC plates precoated with silica gel containing an UV254 fluorescent indicator (Macherey-Nagel). TLC’s were visualized with a 254/365 nm UV hand-held lamp (UVP). Flash silica gel chromatography was performed using 0.04–0.063 mm 60 M silica (Macherey-Nagel). All glassware used under anhydrous reaction conditions were flame-dried under vacuum and cooled immediately before use.
Method A.
Cy7-Cl (1 equiv) and the corresponding p-aniline (5 equiv) were dissolved in anhydrous DMSO and then treated with triethylamine (5 equiv). The reaction was heated at 55 °C for 8 h before cooling to room temperature. The DMSO was blown off using N2, resuspended in CH2Cl2, and washed with 1 M HCl. The organic solution was washed 3× with an aqueous potassi-um iodide solution (saturated, 3× 25 mL), dried (Na2SO4) and the solvent was removed under reduced pressure. The product was purified twice via silica gel chromatography (5% MeOH/CH2Cl2 and then 10%/45%/45% MeOH/toluene/CH2Cl2).
Method B.
Cy7-Cl (1 equiv) and the corresponding p-aniline (3 equiv) were dissolved in anhydrous DMF and then treated with triethylamine (3 equiv). The reaction was heated at 55 °C for 8 h before cooling to room temperature. The reaction was diluted in brine and extracted with CH2Cl2 (3×). The organic solution was washed 3× with an aqueous potassi-um iodide solution (saturated, 3× 25 mL), dried (Na2SO4) and the solvent was removed under reduced pressure. The product was purified via silica gel chromatography (5% MeOH/CH2Cl2).
Compound 1.
Method A was used and afforded a purple solid (8.2 mg, 0.011 mmol, 1.4% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), p-bromoaniline (673 mg, 4.0 mmol, 5 equiv), DMSO (13 mL), and triethylamine (545 μL, 4.0 mmol, 5 equiv) were used. 1H NMR (500 MHz, CD3OD) δ 8.11 (d, J = 13.9 Hz, 2H), 7.36 (dt, J = 12.2, 5.7 Hz, 6H), 7.20 (d, J = 8.3 Hz, 2H), 7.16 (dd, J = 12.8, 7.1 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 6.10 (d, J = 13.9 Hz, 2H), 4.10 (q, J = 7.2 Hz, 4H), 2.67 (t, J = 6.3 Hz, 4H), 1.98 (t, J = 6.2 Hz, 2H), 1.40 (s, 12H), 1.35 (t, J = 7.2 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ 170.91, 162.17, 143.00, 141.94, 132.30, 128.53, 128.25, 127.82, 124.87, 124.18, 121.93, 117.43, 111.29, 109.77, 98.08, 48.54, 38.34, 36.17, 29.38, 26.88, 24.16, 20.07, 10.77. MS (ES+): m/z calcd for C40H45BrN3 646.2791 and 648.2771, found 646.2766 and 648.2747.
Compound 2.
Method A was used and afforded a blue solid (54.3 mg, 0.078 mmol, 10.0% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), aniline (684 mg, 4.0 mmol, 5 equiv), DMSO (13 mL), and triethylamine (4.0 mmol, 5 equiv) were used. 1H NMR (500 MHz, CD3OD): δ 8.17 (d, J = 13.8 Hz, 2H), 7.36 (ddt, J = 6.1, 4.7, 1.9 Hz, 4H), 7.27 (dd, J = 8.5, 7.3 Hz, 2H), 7.21−7.12 (m, 5H), 7.05−6.99 (m, 2H), 6.82 (t, J = 7.4 Hz, 1H), 6.07 (d, J = 13.7 Hz, 2H), 4.10 (q, J = 7.2 Hz, 4H), 2.69 (t, J = 6.3 Hz, 4H), 2.00 (p, J = 6.4 Hz, 2H), 1.39 (s, 12H), 1.36 (t, J = 7.2 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ 170.56, 159.23, 146.93, 143.07, 142.01, 140.85, 129.56, 128.53, 128.19, 127.82, 124.35, 123.93, 121.85, 120.21, 116.47, 109.56, 97.51, 78.08, 38.22, 26.99, 24.19, 21.57, 10.71. MS (ES+): m/z calcd for C40H46N3 568.3686, found 568.3681.
Compound 3.
Method B was used and afforded a blue solid (67.3 mg, 0.112 mmol, 11.5% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), p-amino-benzyl alcohol (360.7 mg, 2.3 mmol, 3 equiv), DMF (8 mL), and triethylamine (327 μL, 2.3 mmol, 3 equiv) were used. 1H NMR (500 MHz, CD3OD): δ 8.13 (d, J = 13.8 Hz, 2H), 7.34 (dt, J = 7.7, 3.9 Hz, 5H), 7.25 (d, J = 8.2 Hz, 2H), 7.21−7.11 (m, 4H), 7.00 (d, J = 8.4 Hz, 2H), 6.05 (d, J = 13.8 Hz, 2H), 4.46 (s, 2H), 4.07 (q, J = 7.2 Hz, 4H), 2.66 (t, J = 6.3 Hz, 4H), 1.98 (t, J = 6.3 Hz, 2H), 1.37 (s, 12H), 1.34 (t, J = 7.2 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ 170.50, 159.35, 152.70, 149.85, 142.98, 142.02, 140.83, 138.62, 138.00, 137.69, 133.50, 128.61, 128.18, 124.17, 123.90, 121.85, 116.58, 109.53, 97.42, 63.45, 38.19, 27.00, 24.18, 23.40, 21.58, 19.33, 12.53, 10.70. MS (ES+): m/z calcd for C41H48N3O 598.3792, found 598.3784.
Compound 4.
Method B was used and afforded a blue solid (110.2 mg, 0.152 mmol, 19.4% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), p-ethyl aniline (284.4 mg, 2.3 mmol, 3 equiv), DMF (8 mL) and triethylamine (327 μL, 2.3 mmol, 3 equiv) were used. 1H NMR (500 MHz, CDCl3): δ 8.02 (d, J = 13.7 Hz, 2H), 7.27−7.16 (m, 4H), 7.13−6.97 (m, 6H), 6.95−6.85 (m, 2H), 5.90 (d, J = 13.7 Hz, 2H), 3.95 (q, J = 7.2 Hz, 4H), 2.54 (t, J = 6.3 Hz, 5H), 2.42 (q, J = 7.6 Hz, 2H), 1.86 (q, J = 6.33, 2H), 1.26 (s, 12H), 1.23 (t, J = 7.2 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ 170.13, 156.30, 148.58, 148.20, 147.78, 142.81, 142.07, 140.74, 129.00, 128.16, 123.73, 123.47, 121.81, 117.56, 109.38, 96.94, 48.10, 38.07, 27.75, 27.06, 24.18, 21.66, 14.98, 10.63. MS (ES+): m/z calcd for C42H50N3 596.3999, found 596.3979.
Compound 5.
Method B was used and afforded a blue solid (66.4 mg, 0.094 mmol, 12.0% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), p-toluidine (360.7 mg, 2.3 mmol, 3 equiv), DMF (8 mL), and triethylamine (408 μL, 2.3 mmol, 3 equiv) were used. 1H NMR (500 MHz, CD3OD): δ 8.11 (d, J = 13.7 Hz, 2H), 7.33 (t, J = 7.1 Hz, 4H), 7.18−7.06 (m, 6H), 6.97 (d, J = 8.2 Hz, 2H), 6.01 (d, J = 13.7 Hz, 2H), 4.06 (q, J = 7.2 Hz, 4H), 2.64 (t, J = 6.3 Hz, 4H), 2.21 (s, 3H), 1.96 (t, J = 6.3 Hz, 2H), 1.37 (s, 12H), 1.33 (t, J = 7.2 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ 170.13, 160.21, 144.08, 142.81, 142.08, 140.74, 131.32, 130.37, 130.07, 128.16, 123.73, 123.63, 121.83, 117.44, 109.41, 97.03, 38.13, 27.00, 24.23, 21.66, 19.24, 10.69. MS (ES+): m/z calcd for C41H48N3 582.3843, found 582.3832.
Compound 6.
Method B was used and afforded a blue solid (182.5 mg, 0.247 mmol, 31.5% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), p-ethoxyaniline (322.0 mg, 2.3 mmol, 3 equiv), DMF (8 mL), and triethylamine (327 μL, 2.3 mmol, 3 equiv) were used. 1H NMR (500 MHz, CD3OD): δ 8.12 (d, J = 13.6 Hz, 2H), 7.34 (td, J = 7.3, 1.3 Hz, 4H), 7.19−7.12 (m, 5H), 7.11−7.05 (m, 2H), 6.90 (d, J = 8.9 Hz, 2H), 5.99 (d, J = 13.6 Hz, 2H), 4.06 (q, J = 7.2 Hz, 4H), 3.98 (q, J = 7.0 Hz, 2H), 2.64 (t, J = 6.3 Hz, 4H), 2.03−1.93 (m, 2H), 1.40 (s, 12H), 1.34 (td, J = 7.1, 3.2 Hz, 9H). 13C NMR (125 MHz, CD3OD): δ 169.74, 142.46, 142.13, 140.61, 139.59, 129.36, 128.13, 123.57, 122.92, 121.79, 119.51, 115.88, 109.23, 96.51, 63.58, 27.05, 24.22, 21.71, 13.79, 13.79, 10.58. MS (ES+): m/z calcd for C42H50N3O 612.3948, found 612.3936.
Compound 7.
Method B was used and afforded a blue solid (101.9 mg, 0.140 mmol, 18.0% yield). Cy7-Cl (500 mg, 0.80 mmol, 1 equiv), p-anisidine (289.1 mg, 2.3 mmol, 3 equiv), DMF (8 mL), and triethylamine (408 μL, 2.3 mmol, 3 equiv) were used. 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 13.6 Hz, 2H), 7.27 (ddd, J = 10.9, 5.4, 3.1 Hz, 5H), 7.24−7.18 (m, 2H), 7.08 (t, J = 7.4 Hz, 2H), 7.04 (d, J = 8.1 Hz, 2H), 6.94 (d, J = 7.9 Hz, 2H), 5.79 (d, J = 13.6 Hz, 2H), 3.97 (q, J = 7.2 Hz, 4H), 2.56 (t, J = 6.4 Hz, 4H), 1.98−1.86 (m, 2H), 1.37 (s, 12H), 1.37−1.33 (m, 6H). 13C NMR (125 MHz, CDCl3): δ 169.43, 160.69, 143.50, 142.90, 142.24, 140.74, 131.88, 130.84, 130.18, 129.03, 128.77, 128.29, 128.23, 125.29, 123.64, 123.59, 122.07, 118.91, 109.07, 96.87, 48.40, 28.34, 24.77, 21.70, 20.62, 11.88. MS (ES+): m/z calcd for C41H48N3O 598.3792, found 598.3779.
Compound 8.
p-Anisidine (14 mg, 0.11 mmol) was added to a solution of IR-26 (40 mg, 0.056 mmol) in DMF (1 mL), and the reaction mixture was stirred for 0.5 h at room temperature. The solvent was removed under reduced pressure, and the resulting solid was dissolved in CH2Cl2 (25 mL). The organic solution was washed 3× with an aqueous potassium iodide solution (saturated, 3 × 25 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude mixture was purified via trituration (cold acetone) to afford the title compound as a shiny red solid (15 mg, 0.021 mmol, 38%). 1H NMR (400 MHz, DMSO-d6): δ 11.66 (s, 1H), 8.31 (s, 2H), 7.95 (m, 2H), 7.66 (m, 6H), 7.61−7.49 (m, 10H), 7.39 (m, 4H), 7.13 (d, J = 12.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 3.42 (s, 3H), 2.81 (br. s, 4H), 1.88 (s, 2H). The product was too insoluble to obtain a 13C NMR spectrum. MS (ES+): m/z calcd for C47H38OS2 696.2395, found 696.2385.
Compound 9.
p-Anisidine (13 mg, 0.11 mmol) was added to a solution of IR-1061 (40 mg, 0.053 mmol) in DMF (1 mL), and the reaction mixture was stirred for 1 h at room temperature. The solvent was removed under reduced pressure, and the resulting solid was dissolved in CH2Cl2 (25 mL). The organic solution was washed 3× with an aqueous potassium iodide solution (saturated, 3× 25 mL) and dried (Na2SO4), and the solvent was removed under reduced pressure. The crude mixture was purified via silica column chromatography (5% MeOH/CH2Cl2) followed by trituration (Et2O) to afford the title compound as a shiny red solid (22 mg, 0.029 mmol, 55%). 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 13.2 Hz, 2H), 7.69−7.64 (m, 4H), 7.52 (m, 4H), 7.40−7.29 (m, 17H), 7.05 (s, 2H), 6.60 (d, J = 8.5 Hz, 2H), 6.21 (d, J = 13.2 Hz, 2H), 3.39 (s, 3H), 2.52 (t, J = 6.3 Hz, 4H), 1.78 (q, J = 6.4 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 162.35, 156.26, 145.98, 145.78, 144.59, 140.06, 137.31, 136.50, 136.45, 130.32, 130.21, 129.19, 127.16, 126.31, 123.86, 121.00, 119.84, 114.64, 55.25, 28.84, 25.49, 21.78. MS (ES+): m/z calcd for C51H42NOS2 748.2708, found 748.2703.
Compound 10 (APNO-1080).
p-Anisidine (27 mg, 0.10 mmol) was added to a solution of IR-1048 (80 mg, 0.11 mmol) in DMF (1 mL), and the reaction mixture was stirred for 4 h at room temperature. The reaction mixture was partitioned with CH2Cl2 (50 mL) and brine (50 mL). The organic layer was further washed with brine (6× 25 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude mixture was purified via silica column chromatography (5% MeOH/CH2Cl2) followed by trituration (Et2O) to afford the title compound as a dark solid (21 mg, 0.029 mmol, 25%). 1H NMR (500 MHz, CD3OD/CDCl3): δ 8.35 (d, J = 12.8 Hz, 2H), 8.11 (d, J = 8.2 Hz, 2H), 7.64 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.8 Hz, 2H), 7.34 (br. s, 2H), 7.27 (d, J = 8.4 Hz, 2H), 6.95 (m, 4H), 6.36 (d, J = 13.4 Hz, 2H), 4.08 (m, 4H), 3.76 (s, 3H), 2.77 (m, 4H), 2.11−2.00 (m, 2H), 1.83 (m, 4H), 1.48 (m, 4H), 1.01 (m, 6H). 13C NMR (125 MHz, CDCl3): δ 161.24, 157.18, 151.36, 140.85, 140.12, 136.77, 129.84, 128.37, 128.31, 126.52, 126.32, 125.02, 122.17, 114.97, 106.04, 102.96, 30.36, 25.07, 21.52, 20.26, 13.65. MS (ES+): m/z calcd for C47H38OS2 738.3018, found 738.3021.
Compound 11.
p-Anisidine (100 mg, 0.81 mmol) was added to a solution of Et-10802 (50 mg, 0.08 mmol) in DMF (2 mL), and the reaction mixture was heated to 40 °C for 24 h. The solvent was removed under reduced pressure, and the resulting solid was dissolved in CH2Cl2 (25 mL). The organic solution was washed 3× with an aqueous potassium iodide solution (saturated, 3× 25 mL) and dried (Na2SO4), and the solvent was removed under reduced pressure. The crude mixture was purified via silica column chromatography (5% MeOH/CH2Cl2) followed by trituration (Et2O followed by MeOH) to afford the title compound as a dark solid (18 mg, 0.027 mmol, 34%). 1H NMR (500 MHz, DMSO-d6): δ 10.10 (s, 1H), 8.27 (d, J = 13.2 Hz, 2H), 8.07 (d, J = 8.1 Hz, 2H), 7.66 (t, J = 7.7 Hz, 2H), 7.58 (m, 4H), 7.37 (m, 2H), 7.31 (m, 4H), 7.02 (d, J = 8.4 Hz, 2H), 6.54 (d, J = 13.3 Hz, 2H), 4.27 (m, 4H), 3.70 (s, 3H), 2.80 (m, 4H), 1.97−1.91 (m, 2H), 1.31 (m, 6H). 13C NMR (125 MHz, DMSO-d6): δ 161.11, 156.96, 150.90, 141.54, 140.12, 137.62, 130.29, 130.15, 129.98, 129.65, 126.78, 126.08, 125.51, 122.58, 120.75, 115.41, 107.29, 103.57, 55.91, 38.29, 25.24, 21.68, 13.81. MS (ES+): m/z calcd for C43H40N3O 614.3171, found 614.3181.
Compound 12.
p-Anisidine (76 mg, 0.62 mmol) was added to a solution of Flav73 (50 mg, 0.075 mmol) in DMF (2 mL), and the reaction mixture was heated to 40 °C for 24 h. The reaction mixture was partitioned with CH2Cl2 (50 mL) and brine (50 mL). The organic layer was further washed with brine (6 × 25 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude mixture was purified via silica column chromatography (5% MeOH/CH2Cl2) followed by trituration (Et2O) to afford the title compound as a dark red solid (10 mg, 0.013 mmol, 18%). 1H NMR (500 MHz, CDCl3): δ 8.26 (m, 1H), 7.98 (m, 3H), 7.68 (m, 2H), 7.47−7.42 (m, 6H), 7.39 (d, J = 8.5 Hz, 2H), 7.23 (s, 2H), 6.71 (d, J = 9.0 Hz, 2H), 6.65 (d, J = 8.5 Hz, 2H), 6.51 (d, J = 13.0 Hz, 2H), 6.40−6.36 (m, 2H), 3.44 (s, 3H), 3.03 (s, 12H), 2.64 (s, 4H), 1.90 (s, 2H). The product was too insoluble to obtain a 13C NMR spectrum. MS (ES+): m/z calcd for C51H48N3O3 750.3696, found 750.3674.
Supplementary Material
ACKNOWLEDGMENTS
M.Y.L. thanks the Alfred P. Sloan Foundation for financial support. A.K.E. acknowledges the Beckman Institute Graduate Fellowship for financial support. C.J.R. thanks the Chemistry-Biology Interface Training Grant (T32 GM070421) and the Seemon Pines Graduate Fellowship for support. M.C.L. thanks the National Science Foundation for a Graduate Fellowship (Grant No. 1746047). Major funding for the 500 MHz Bruker CryoProbe was provided by the Roy J. Carver Charitable Trust (Muscatine, Iowa; Grant No. 15-4521) to the School of Chemical Sciences NMR Lab. The Q-Tof Ultima mass spectrometer was purchased in part with a grant from the National Science Foundation, Division of Biological Infrastructure (DBI-0100085). We also acknowledge Dr. Iwona Dobrucka and the Molecular Imaging Laboratory at the Beckman Institute for use of the IVIS imaging system, Drs. Nicole Herndon and Jessica Xu for help generating the liver metastasis model, and Prof. Hee-Sun Han for access to the Cary 5000 UV-vis-NIR spectrophotometer.
Funding
This work was supported the National Institutes of Health (R35GM133581).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03004.
Further experimental details, including synthetic procedures, spectral data, and supplemental in vitro and in vivo procedures and data (PDF)
Deep tissue imaging (MOV)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c03004
The authors declare no competing financial interest.
REFERENCES
- (1).Wang LV; Hu S Photoacoustic Tomography: In Vivo Imaging from Organelles to Organs. Science 2012, 335, 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Carovac A; Smajlovic F; Junuzovic D Application of ultrasound in medicine. Acta Inform Med. 2011, 19, 168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yadav AK; Hernandez S; Su S; Chan J Acoustic-based chemical tools for profiling the tumor microenvironment. Curr. Opin. Chem. Biol 2020, 57, 114–121. [DOI] [PubMed] [Google Scholar]
- (4).Yao J; Wang LV Recent progress in photoacoustic molecular imaging. Curr. Opin. Chem. Biol 2018, 45, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Heijblom M; Piras D; Brinkhuis M; van Hespen JCG; van den Engh FM; van der Schaaf M; Klaase JM; van Leeuwen TG; Steenbergen W; Manohar S Photoacoustic image patterns of breast carcinoma and comparisons with Magnetic Resonance Imaging and vascular stained histopathology. Sci. Rep 2015, 5, 11778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yang M; Zhao L; He X; Su N; Zhao C; Tang H; Hong T; Li W; Yang F; Lin L; Zhang B; Zhang R; Jiang Y; Li C Photoacoustic/ultrasound dual imaging of human thyroid cancers: an initial clinical study. Biomed. Opt. Express 2017, 8, 3449–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jo J; Xu G; Cao M; Marquardt A; Francis S; Gandikota G; Wang X A Functional Study of Human Inflammatory Arthritis Using Photoacoustic Imaging. Sci. Rep 2017, 7, 15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu Y; Zhang L; Li S; Han X; Yuan Z Imaging molecular signatures for clinical detection of scleroderma in the hand by multispectral photoacoustic elastic tomography. Journal of Biophotonics 2018, 11, e201700267. [DOI] [PubMed] [Google Scholar]
- (9).Reinhardt CJ; Chan J Development of Photoacoustic Probes for in Vivo Molecular Imaging. Biochemistry 2018, 57, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Reinhardt CJ; Xu R; Chan J Nitric oxide imaging in cancer enabled by steric relaxation of a photoacoustic probe platform. Chemical Science 2020, 11, 1587–1592. [Google Scholar]
- (11).East AK; Lucero MY; Chan J New directions of activity-based sensing for in vivo NIR imaging. Chemical Science 2021, 12, 3393–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Knox HJ; Hedhli J; Kim TW; Khalili K; Dobrucki LW; Chan J A bioreducible N-oxide-based probe for photoacoustic imaging of hypoxia. Nat. Commun 2017, 8, 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Knox HJ; Kim TW; Zhu Z; Chan J Photophysical Tuning of N-Oxide-Based Probes Enables Ratiometric Photoacoustic Imaging of Tumor Hypoxia. ACS Chem. Biol 2018, 13, 1838–1843. [DOI] [PubMed] [Google Scholar]
- (14).Chen M; Knox HJ; Tang Y; Liu W; Nie L; Chan J; Yao J Simultaneous photoacoustic imaging of intravascular and tissue oxygenation. Opt. Lett 2019, 44, 3773–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhou EY; Knox HJ; Liu C; Zhao W; Chan J A Conformationally Restricted Aza-BODIPY Platform for Stimulus-Responsive Probes with Enhanced Photoacoustic Properties. J. Am. Chem. Soc 2019, 141, 17601–17609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zheng J; Zeng Q; Zhang R; Xing D; Zhang T Dynamic-Reversible Photoacoustic Probe for Continuous Ratiometric Sensing and Imaging of Redox Status in Vivo. J. Am. Chem. Soc 2019, 141, 19226–19230. [DOI] [PubMed] [Google Scholar]
- (17).Wu Y; Huang S; Wang J; Sun L; Zeng F; Wu S Activatable probes for diagnosing and positioning liver injury and metastatic tumors by multispectral optoacoustic tomography. Nat. Commun 2018, 9, 3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yin L; Sun H; Zhang H; He L; Qiu L; Lin J; Xia H; Zhang Y; Ji S; Shi H; Gao M Quantitatively Visualizing Tumor-Related Protease Activity in Vivo Using a Ratiometric Photoacoustic Probe. J. Am. Chem. Soc 2019, 141, 3265–3273. [DOI] [PubMed] [Google Scholar]
- (19).Wang Y; Hu X; Weng J; Li J; Fan Q; Zhang Y; Ye D A Photoacoustic Probe for the Imaging of Tumor Apoptosis by Caspase-Mediated Macrocyclization and Self-Assembly. Angew. Chem., Int. Ed 2019, 58, 4886–4890. [DOI] [PubMed] [Google Scholar]
- (20).Cheng P; Chen W; Li S; He S; Miao Q; Pu K Fluoro-Photoacoustic Polymeric Renal Reporter for Real-Time Dual Imaging of Acute Kidney Injury. Adv. Mater 2020, 32, 1908530. [DOI] [PubMed] [Google Scholar]
- (21).Li H; Zhang P; Smaga LP; Hoffman RA; Chan J Photoacoustic Probes for Ratiometric Imaging of Copper(II). J. Am. Chem. Soc 2015, 137, 15628–15631. [DOI] [PubMed] [Google Scholar]
- (22).Wang S; Sheng Z; Yang Z; Hu D; Long X; Feng G; Liu Y; Yuan Z; Zhang J; Zheng H; Zhang X Activatable Small-Molecule Photoacoustic Probes that Cross the Blood–Brain Barrier for Visualization of Copper(II) in Mice with Alzheimer‘s Disease. Angew. Chem., Int. Ed 2019, 58, 12415–12419. [DOI] [PubMed] [Google Scholar]
- (23).Roberts S; Seeger M; Jiang Y; Mishra A; Sigmund F; Stelzl A; Lauri A; Symvoulidis P; Rolbieski H; Preller M; Deán-Ben XL; Razansky D; Orschmann T; Desbordes SC; Vetschera P; Bach T; Ntziachristos V; Westmeyer GG Calcium Sensor for Photoacoustic Imaging. J. Am. Chem. Soc 2018, 140, 2718–2721. [DOI] [PubMed] [Google Scholar]
- (24).Zhang C; Gao R; Zhang L; Liu C; Yang Z; Zhao S Design and Synthesis of a Ratiometric Photoacoustic Probe for In Situ Imaging of Zinc Ions in Deep Tissue In Vivo. Anal. Chem 2020, 92, 6382–6390. [DOI] [PubMed] [Google Scholar]
- (25).Chen Z; Mu X; Han Z; Yang S; Zhang C; Guo Z; Bai Y; He W An Optical/Photoacoustic Dual-Modality Probe: Ratiometric in/ex Vivo Imaging for Stimulated H2S Upregulation in Mice. J. Am. Chem. Soc 2019, 141, 17973–17977. [DOI] [PubMed] [Google Scholar]
- (26).Lucero MY, Chan J Towards Personalized Medicine: Photoacoustic Imaging Enables Companion Diagnosis and Targeted Treatment of Lung Cancer ChemRxiv, 2020, DOI: 10.26434/chemrxiv.11888214.v2 (accessed 2021-04-15). [DOI] [Google Scholar]
- (27).Reinhardt CJ; Zhou EY; Jorgensen MD; Partipilo G; Chan J A Ratiometric Acoustogenic Probe for in Vivo Imaging of Endogenous Nitric Oxide. J. Am. Chem. Soc 2018, 140, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ikeno T; Hanaoka K; Iwaki S; Myochin T; Murayama Y; Ohde H; Komatsu T; Ueno T; Nagano T; Urano Y Design and Synthesis of an Activatable Photoacoustic Probe for Hypochlorous Acid. Anal. Chem 2019, 91, 9086–9092. [DOI] [PubMed] [Google Scholar]
- (29).Upputuri PK; Pramanik M Photoacoustic imaging in the second near-infrared window: a review. J. Biomed. Opt 2019, 24, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sun A; Guo H; Gan Q; Yang L; Liu Q; Xi L Evaluation of visible NIR-I and NIR-II light penetration for photoacoustic imaging in rat organs. Opt. Express 2020, 28, 9002–9013. [DOI] [PubMed] [Google Scholar]
- (31).Zhang J; Ning L; Zeng Z; Pu K Development of Second Near-Infrared Photoacoustic Imaging Agents. Trends in Chemistry 2021, 3, 305–317. [Google Scholar]
- (32).Feng W; Zhang Y; Li Z; Zhai S; Lv W; Liu Z Lighting Up NIR-II Fluorescence in Vivo: An Activable Probe for Noninvasive Hydroxyl Radical Imaging. Anal. Chem 2019, 91, 15757–15762. [DOI] [PubMed] [Google Scholar]
- (33).Cosco ED; Caram JR; Bruns OT; Franke D; Day RA; Farr EP; Bawendi MG; Sletten EM Flavylium Polymethine Fluorophores for Near- and Shortwave Infrared Imaging. Angew. Chem., Int. Ed 2017, 56, 13126–13129. [DOI] [PubMed] [Google Scholar]
- (34).Usama SM; Lin C-M; Burgess K On the Mechanisms of Uptake of Tumor-Seeking Cyanine Dyes. Bioconjugate Chem. 2018, 29, 3886–3895. [DOI] [PubMed] [Google Scholar]
- (35).Zhou EY; Knox HJ; Reinhardt CJ; Partipilo G; Nilges MJ; Chan J Near-Infrared Photoactivatable Nitric Oxide Donors with Integrated Photoacoustic Monitoring. J. Am. Chem. Soc 2018, 140, 11686–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang X; Lv J; Yao X; Li Y; Huang F; Li M; Yang J; Ruan X; Tang B Screening and investigation of a cyanine fluorescent probe for simultaneous sensing of glutathione and cysteine under single excitation. Chem. Commun 2014, 50, 15439–15442. [DOI] [PubMed] [Google Scholar]
- (37).Yadav AK, Lee MC, Lucero MY, Reinhardt CJ, Su S, Chan J NIR Bioluminescence Probe Enables Discovery of Diet-Induced Modulation of the Tumor Microenvironment via Nitric Oxide ChemRxiv, 2021, DOI: 10.26434/chemrxiv.14176835.v1 (accessed 2021-04-15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lin Y; Xu J; Lan H Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J. Hematol. Oncol 2019, 12, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tang Y; Li Y; Wang Z; Pei F; Hu X; Ji Y; Li X; Zhao H; Hu W; Lu X; Fan Q; Huang W Organic semiconducting nanoprobe with redox-activatable NIR-II fluorescence for in vivo real-time monitoring of drug toxicity. Chem. Commun 2019, 55, 27–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.