

Abstract
Background:
Early cancer detection could identify tumors at a time when outcomes are superior and treatment is less morbid. This prospective case-control sub-study (from NCT02889978 and NCT03085888) assessed the performance of targeted methylation analysis of circulating cell-free DNA (cfDNA) to detect and localize multiple cancer types across all stages at high specificity.
Participants and methods:
The 6689 participants [2482 cancer (>50 cancer types), 4207 non-cancer] were divided into training and validation sets. Plasma cfDNA underwent bisulfite sequencing targeting a panel of >100 000 informative methylation regions. A classifier was developed and validated for cancer detection and tissue of origin (TOO) localization.
Results:
Performance was consistent in training and validation sets. In validation, specificity was 99.3% [95% confidence interval (CI): 98.3% to 99.8%; 0.7% false-positive rate (FPR)]. Stage I–III sensitivity was 67.3% (CI: 60.7% to 73.3%) in a pre-specified set of 12 cancer types (anus, bladder, colon/rectum, esophagus, head and neck, liver/bile-duct, lung, lymphoma, ovary, pancreas, plasma cell neoplasm, stomach), which account for ~63% of US cancer deaths annually, and was 43.9% (CI: 39.4% to 48.5%) in all cancer types. Detection increased with increasing stage: in the pre-specified cancer types sensitivity was 39% (CI: 27% to 52%) in stage I, 69% (CI: 56% to 80%) in stage II, 83% (CI: 75% to 90%) in stage III, and 92% (CI: 86% to 96%) in stage IV. In all cancer types sensitivity was 18% (CI: 13% to 25%) in stage I, 43% (CI: 35% to 51%) in stage II, 81% (CI: 73% to 87%) in stage III, and 93% (CI: 87% to 96%) in stage IV. TOO was predicted in 96% of samples with cancer-like signal; of those, the TOO localization was accurate in 93%.
Conclusions:
cfDNA sequencing leveraging informative methylation patterns detected more than 50 cancer types across stages. Considering the potential value of early detection in deadly malignancies, further evaluation of this test is justified in prospective population-level studies.
Keywords: cell-free DNA, next-generation sequencing, methylation, cancer
INTRODUCTION
Earlier cancer detection offers the opportunity to identify tumors when cures are more achievable, outcomes are superior, and treatment can be less morbid.1,2 Effective screening paradigms exist only for a small subset of cancers, are focused on single cancer types, and have variable adoption and compliance.3–6 Thus, diagnoses are often prompted by symptoms and are made at later stages. Utilization of blood-based circulating tumor cell-free DNA (cfDNA) to simultaneously detect and localize multiple cancer types7,8 may address this large unmet need. In large-scale population screening, such a multi-cancer detection approach would require high specificity, clinically useful sensitivity, and highly accurate tissue of origin (TOO) identification to limit the scope, cost, and complexity of evaluating asymptomatic patients.
There are few studies interrogating simultaneous detection and localization of multiple cancer types using cfDNA or other analytes.9–11 These studies generally analyzed a handful of cancer types in geographically-restricted cohorts9–11 or interrogated a single cfDNA-based molecular approach.11 Current commercially available cfDNA-based approaches interrogating single-nucleotide variants (SNVs/indels) that focus on key alterations associated with specific tumor types or treatment options12 may be hampered by confounding signals from white blood cells (WBCs) or other tissue.13,14 Similarly, approaches based on detecting somatic copy number alterations may be limited by smaller relative differences between cases and controls resulting in a need for increased sequencing depth as well as technical variation restricting the signal-to-noise ratio.15,16 These approaches as well as others such as protein biomarkers have not yet demonstrated robust TOO assignment across a broad range of tumor types to direct a diagnostic evaluation. To date there have been no population-scale studies of cancer cfDNA signatures nor have there been studies designed to ensure consistent performance in a representative screening population.
The Circulating Cell-free Genome Atlas (CCGA; NCT02889978) study was designed to determine whether genome-wide cfDNA sequencing in combination with machine learning could detect and localize a large number of cancer types at sufficiently high specificity to be considered for a general population-based cancer screening program. During discovery work in the first CCGA sub-study, whole-genome bisulfite sequencing (WGBS) interrogating genome-wide methylation patterns outperformed whole-genome sequencing (WGS) and targeted sequencing approaches interrogating copy-number variants (CNVs) and single-nucleotide variants (SNVs)/small insertions and deletions, respectively.7,17 Additionally, targeted sequencing with SNV-based classification was significantly confounded by clonal hematopoiesis of indeterminate potential (CHIP)18; such a test would thus require concurrent sequencing of WBCs to return accurate results. Herein, we report results from the second case-control sub-study designed to develop, train, and validate a methylation-based assay for simultaneous multi-cancer detection across stages as well as TOO localization in preparation for clinical validation and utility studies (NCT03085888, NCT03934866) and for a study in which results will be returned to health care providers and patients (NCT04241796).
METHODS
Study design and participants
CCGA (NCT02889978) is a prospective, multicenter, case-control, observational study with longitudinal follow-up. De-identified biospecimens were collected from 15 254 participants with (n = 8584; 56%) and without (n = 6670; 44%) cancer from 142 sites in North America. Up to 80 ml whole blood was collected from all participants as part of the research study; only two tubes of plasma were processed separately per participant. Pre-treatment tumor tissue when available was submitted from those with cancer (supplementary information, available at Annals of Oncology online). The CCGA study was divided into three pre-specified sub-studies: (1) discovery,17 (2) further analysis (training/validation) with the selected assay (reported herein), and (3) further validation (forthcoming) (Figure 1A). The first sub-study aimed to identify the highest performing assay(s) for further development and included three independent, comprehensive sequencing approaches.17,19 A methylation-based assay was selected for further development in this second sub-study based on the previous finding that WGBS outperformed targeted sequencing and WGS approaches targeting SNVs/small insertions and deletions and CNVs, respectively.7,17 The primary objective was to train and validate a classifier for cancer versus non-cancer and TOO identification utilizing an updated targeted methylation assay (Figure 1B and C, Figure 2). Pre-specified analysis groups included all cancer types (more than 50 cancer types20; cancers grouped for reporting purposes, see supplementary information, available at Annals of Oncology online) and a subset of 12 high-signal cancers (supplementary information, available at Annals of Oncology online) based on results from the first sub-study (>50% sensitivity on at least one of three prototype sequencing assays) and Surveillance, Epidemiology, and End Results (SEER) mortality data (anus, bladder, colon/rectum, esophagus, head and neck, liver/bile-duct, lung, lymphoma, ovary, pancreas, plasma cell neoplasm, stomach).17,21 The third sub-study was designed to further validate the classifier in a large population and is ongoing.
Figure 1. The CCGA study for development and validation of a cfDNA-based assay for multi-cancer detection.
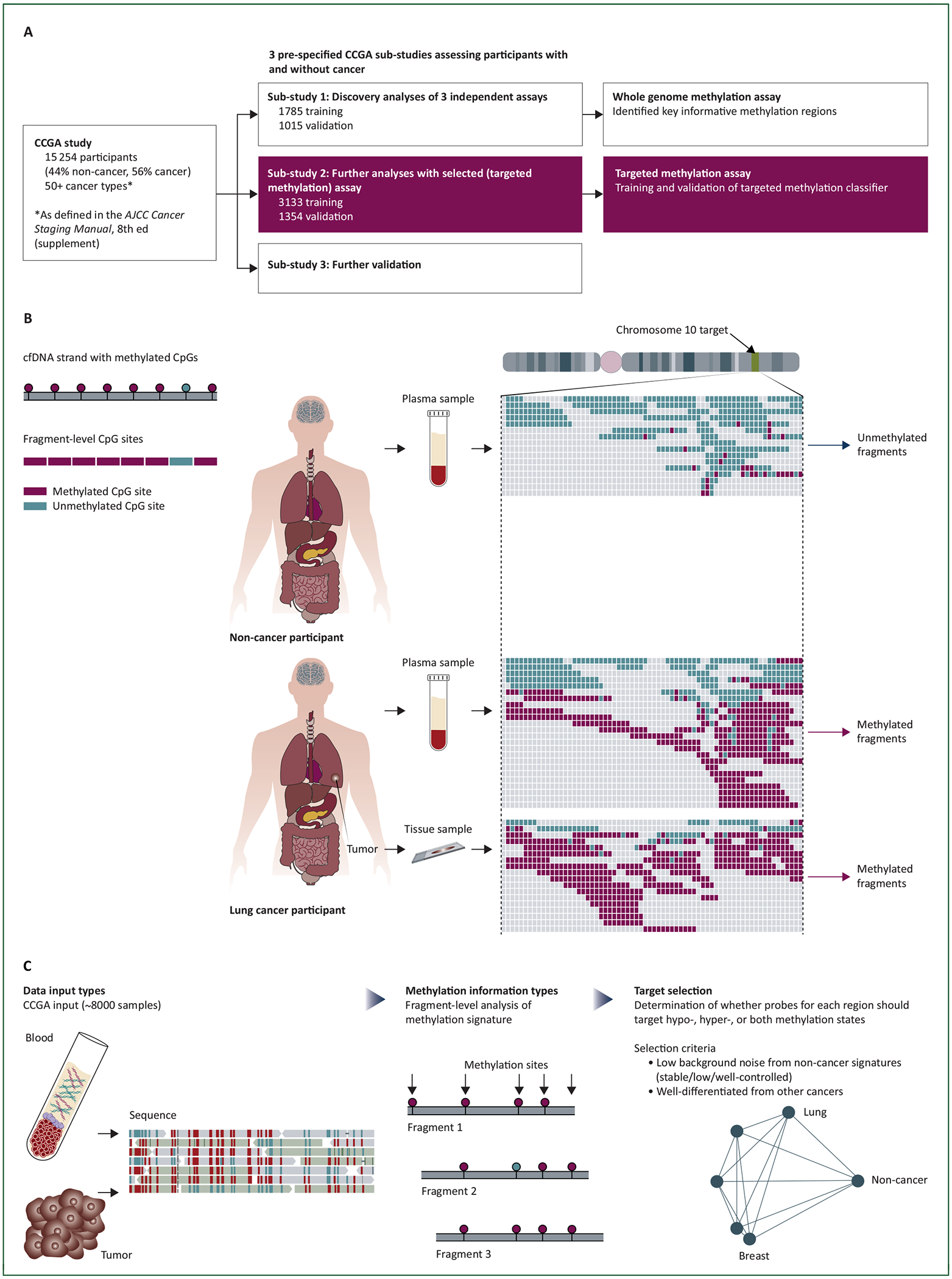
(A) CCGA study design. The CCGA study included three pre-specified sub-studies designed to discover, train, and validate an assay for multi-cancer detection and localization. The burgundy, shaded boxes highlight the second sub-study, which is the focus of this report. (B) Methylation biology discriminates cancer from non-cancer. One circulating cfDNA fragment is represented on the top left; individual CpGs are indicated as burgundy (methylated) or teal (unmethylated) circles. This assay interrogated fragment-level methylation patterns as indicated on the bottom left (‘Fragment-Level CpG Sites’). In non-cancer participants (top right), cfDNA is shed from cells across the body including WBCs13 and is present in plasma. These DNA fragments retain methylation marks from the originating cells as indicated in this example from a region on chromosome 10. Individual cfDNA fragment sequencing reads are indicated as horizontal lines of differing sizes and are aligned vertically. In non-cancer participants, these fragments are largely unmethylated as indicated by the almost uniformly teal fragments. In a participant with lung cancer (bottom right), the plasma contains a mix of methylated (burgundy) and unmethylated (teal) fragments as the circulating cfDNA is a mixture of tumor cfDNA and cfDNA from other cells in the body. Sequencing of the tumor tissue sample confirms that this region is almost entirely methylated as indicated. Note that tumor tissue is not a requirement for this assay but is illustrative. (C) Target selection. A large database of methylation patterns (‘data input types’) was constructed from WGBS analysis of cfDNA and tissue samples from the CCGA study as well as WGBS analysis of a set of commercially sourced tissue samples. Systematic examination of the fragment-level methylation signature (‘methylation information type’) from these samples allowed the identification of a large number of genomic regions (‘target selection’) containing informative biological signatures of cancer and TOO.
Figure 2. Pre-classifier sample preparation and preprocessing overview.
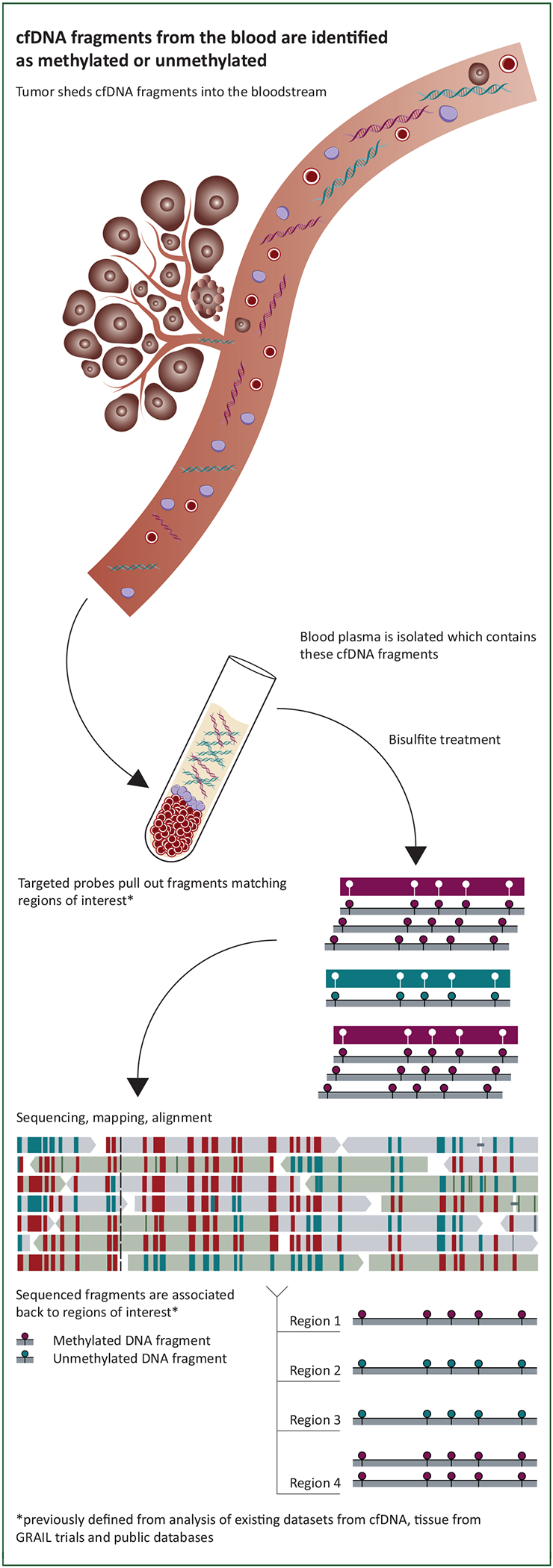
Illustration of how cfDNA fragments from the blood are processed: cfDNA was extracted from plasma, subjected to bisulfite treatment, and regions of interest were pulled down, followed by sequencing and alignment. In this way the methylation state of fragments was obtained.
This second sub-study reported herein included 4841 participants from CCGA divided into a training set (n = 3133: 1742 cancer and 1391 non-cancer; Figure 3) and an independent validation set (n = 1354: 740 cancer and 614 non-cancer); 354 samples were reserved for a tumor biopsy reference set. Samples for each sub-study were selected to ensure a pre-specified distribution of cancer types and non-cancers across sites in each cohort (supplementary information, available at Annals of Oncology online).
Figure 3. Participant disposition.
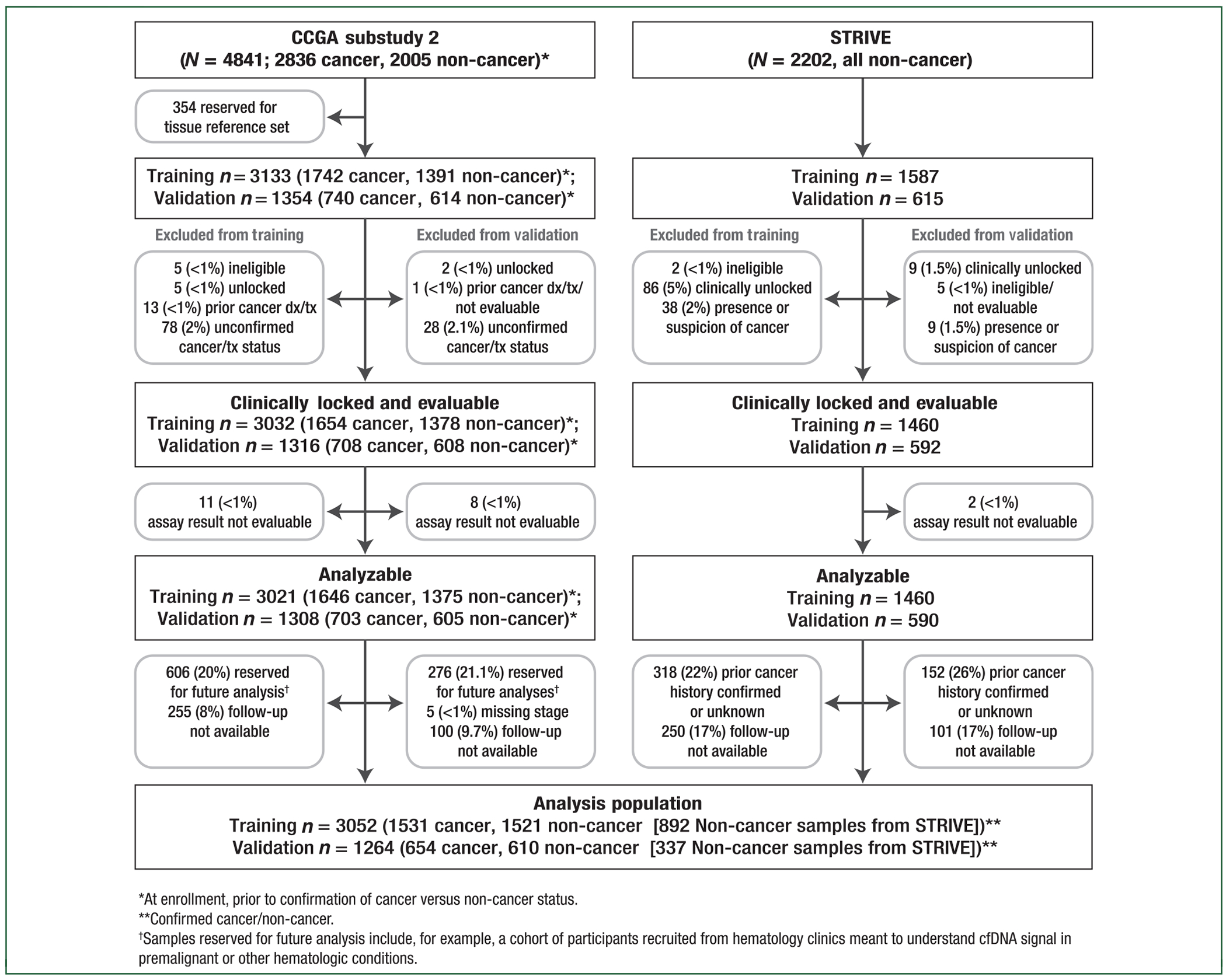
A total of 4841 participants (2836 cancer, 2005 non-cancer) from the CCGA study and 2202 non-cancer participants from the STRIVE study were included in this pre-specified analysis. Of these, 3133 samples from CCGA were allocated to training (1742 cancer, 1391 non-cancer) and 1354 were allocated to validation (740 cancer, 614 non-cancer); 1587 samples from STRIVE were allocated to training and 615 to validation. STRIVE non-cancer samples were used to train the classifier and to ensure >99% specificity was achieved with >90% confidence (see Methods, supplementary information, available at Annals of Oncology online). Participant disposition is indicated. Overall, 3052 samples in training (1531 cancer, 1521 non-cancer) and 1264 samples in validation (654 cancer, 610 non-cancer) were analyzable and in the pre-specified primary analysis population. Samples reserved for pre-specified future analyses (as indicated) included, for example, samples lacking 1-year follow-up and samples from participants with carcinoma in situ (CIS) (see Methods).
To operate at high specificity, large numbers of well-characterized controls were required both to train classification and to measure specificity accurately due to sampling variability (i.e. to ensure >99% specificity was achieved with >90% confidence). These additional non-cancer blood samples were obtained from the independent STRIVE study (NCT03085888), a prospective, multicenter, case-cohort, observational study with longitudinal follow-up. STRIVE was designed to independently validate the ability of this multi-cancer early detection test to detect and localize multiple invasive cancers including breast in a population of women undergoing screening mammography (supplementary information, available at Annals of Oncology online). De-identified biospecimens were collected from 99 286 participants from 35 sites. Samples from women without a known history of cancer (non-cancer) were selected from a single STRIVE site and incorporated into the training (n = 1587) and independent validation sets (n = 615). As noted above, these STRIVE non-cancer samples were used to train the classifier and to ensure >99% specificity was achieved with >90% confidence. Thus, 6689 total samples were analyzed in this second sub-study: 4720 in the training set (1742 cancer; 2978 non-cancer) and 1969 in the independent validation set (740 cancer; 1229 non-cancer; Figure 3).
All participants were required to provide informed consent; eligibility and exclusion criteria for each study are described in the supplementary information, available at Annals of Oncology online. Institutional Review Board or independent ethics committee approval was obtained at each participating site and the study was conducted in accordance with the Good Clinical Practice Guidelines of the International Conference on Harmonization.
Sample collection, accessioning, storage, and processing
Details of plasma and tumor tissue sample collection, accessioning, storage, and processing are described in the supplementary information, available at Annals of Oncology online.
WGBS
WGBS resulted in 3508 analyzable samples: cfDNA [n = 2628 (1493 cancer; 1135 non-cancer)], formalin fixed paraffin embedded (FFPE) tumor biopsies (n = 242), and WBCs (n = 70) from the first CCGA sub-study; commercial tissue or cells [n = 227; Discovery Life Sciences (formerly Conversant Biologics, Inc.); Huntsville, AL]; non-cancer cells (n = 1; from Yuval Dor, Hebrew University, Jerusalem, Israel); and FFPE tumor biopsies from the second CCGA sub-study (n = 340; these participants were not used in subsequent evaluation of the classifier). WGBS is described in the supplementary information, available at Annals of Oncology online.
Targeted methylation panel design, sample processing, and sequencing
Based on the WGBS results as noted above17,19 and methylation array data from The Cancer Genome Atlas,21,22 regions of the hg19 genome22 predicted to contain cancer and/or tissue-specific methylation patterns in cfDNA relative to non-cancer controls were identified and the most informative targets were combined into a custom hybridization capture panel (Twist Bioscience, San Francisco, CA) using a custom algorithm (supplementary information, available at Annals of Oncology online). The final targeted methylation panel covered 103 456 distinct regions (17.2 Mb) and 1 116 720 cytosine-guanine dinucleotides (CpGs).
Plasma cfDNA (up to 75 ng) was subjected to bisulfite conversion (EZ-96 DNA Methylation Kit; Zymo Research, Irvine, CA), prepared as a dual indexed sequencing library, and enriched using standard hybridization capture conditions, followed by 150-bp paired-end sequencing on Illumina NovaSeq (supplementary information, available at Annals of Oncology online). Individual libraries were sequenced to a median depth of 113 million fragments (median unique on-target depth: 139X).
Classification of cancer versus non-cancer and TOO
Custom software was built to classify samples using source models that recognized methylation patterns per region as similar to those derived from a particular cancer type, followed by a pair of ensemble logistic regressions: one to determine cancer/non-cancer status and the other to resolve the TOO to one of the listed sites (supplementary Figure S1C, available at Annals of Oncology online). Both levels of model (source models, logistic regression) as well as any thresholding parameters were trained on 11 154 samples from 5854 participants (supplementary information, available at Annals of Oncology online) using cross-validation so that no data in held-out folds could be accessed during training.
Tumor fraction
Tumor fraction analyses are described in the supplementary information, available at Annals of Oncology online.
Blinding
All analyses in training and validation were double-blinded; classifiers developed on the training set were locked and the final classifier was selected before the validation dataset was released and before release of the validation dataset a data integrity team blinded to classifier development, analysis, and clinical/assay evaluability reviewed merged data to ensure completeness (supplementary information, available at Annals of Oncology online). Researchers developing classifiers were also blinded to cancer status in the validation set.
RESULTS
This second pre-specified CCGA sub-study included 6689 participants with previously untreated cancer (n = 2482) or without cancer (n = 4207) (Figure 3). More than 50 primary cancer types20 across all clinical stages were represented. Samples were divided into training (n = 4720) and independent validation sets (n = 1969). A total of 4316 participants [training: 3052 (1531 cancer: stage I: 28%; stage II: 25%; stage III: 20%; stage IV: 24%; missing/not expected: 3%; 1521 non-cancer); validation: 1264 (654 cancer: stage I: 28%; stage II: 25%; stage III: 21%; stage IV: 23%; missing/not expected: 3%; 610 non-cancer)] were analyzable and included in the primary analysis population (supplementary Table S1 available at Annals of Oncology online). Training and validation sets were generally comparable with respect to age, sex, race/ethnicity, and body mass index in the cancer and non-cancer groups; as expected, fewer never-smokers were in the cancer group in the training and validation sets (supplementary Table S1 available at Annals of Oncology online).
The classifier achieved consistently high specificity between the cross-validated training and independent validation sets [99.8% (95% CI: 99.4% to 99.9%) versus 99.3% (95% CI: 98.3% to 99.8%), respectively; P = 0.095] (Figure 4A); this reflected a single, consistent, false-positive rate (FPR) of less than 1% across the more than 50 cancer types. The FPR in the validation set was similar for the CCGA and STRIVE non-cancer samples [0.7% (95% CI: 0.1% to 2.6%) versus 0.6% (95% CI: 0.1% to 2.1%), respectively; P = 0.830]; supporting that performance was not biased by sites or selected samples.
Figure 4. Targeted methylation cfDNA test performance.
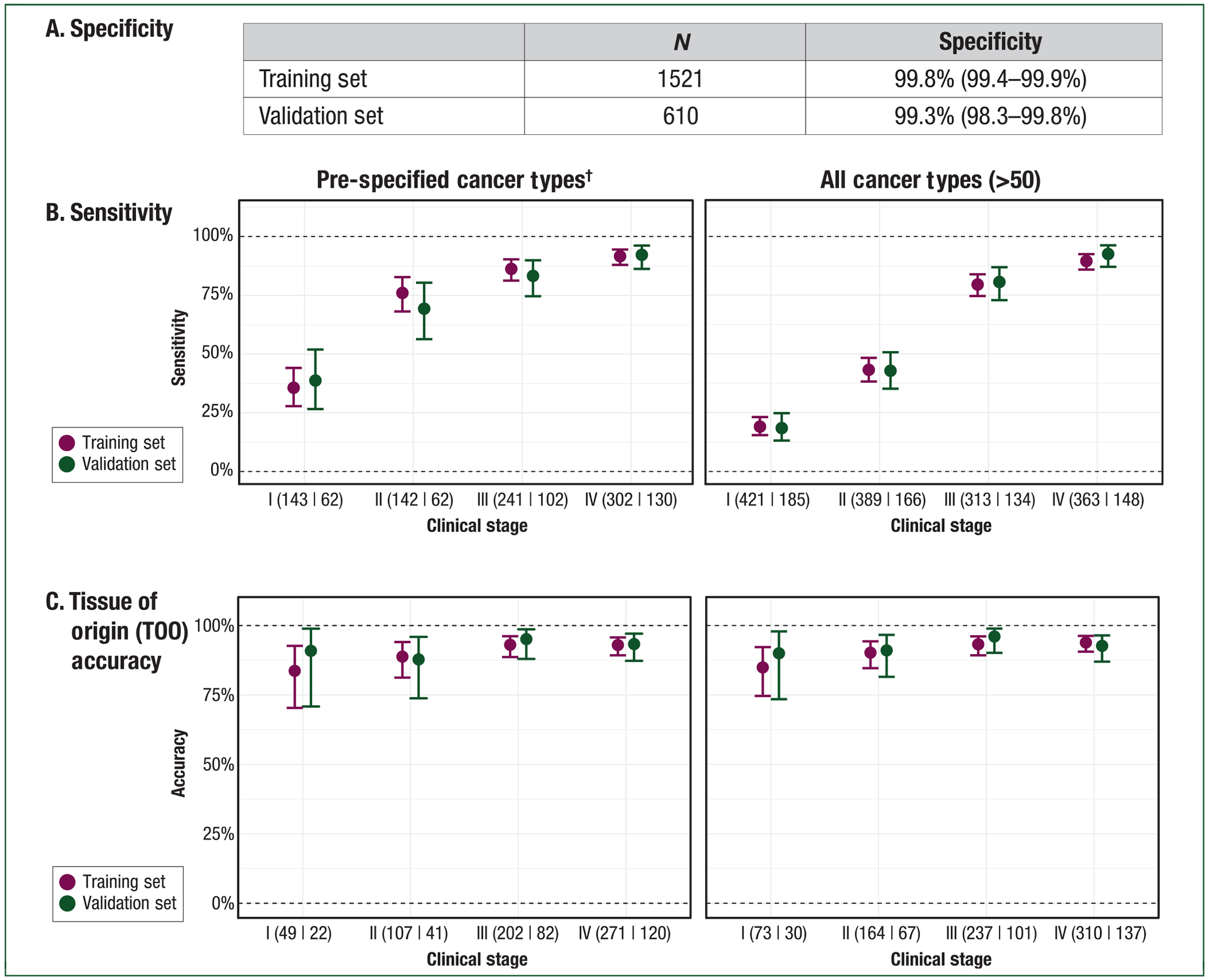
(A) Specificity. Specificity was >99% in the training and validation sets. Importantly, this represents a consistent, single false-positive rate (FPR) across the >50 cancer types in this study. (B) Sensitivity. Sensitivity (y-axis) is reported by clinical stage (x-axis) in the pre-specified cancer types (left panel) and in all cancer types (right panel) for training and validation. Numbers indicate samples in training|validation sets. It excludes 45 samples in training and 21 samples in validation without stage information (e.g. leukemias). (C) Tissue of origin. Tissue of origin (TOO) accuracy (y-axis) is reported by clinical stage (x-axis) in the pre-specified cancer types (left panel) and in all cancer types (right panel) for training and validation. Numbers indicate samples in training|validation sets.
Sensitivity was consistent in the training and validation sets. In all cancers, stage I–III sensitivity was 44.2% (95% CI: 41.3% to 47.2%) versus 43.9% (95% CI: 39.4% to 48.5%) (P = 1.000), respectively. For the pre-specified set of 12 high-signal cancers, stage I–III sensitivity was 69.8% (95% CI: 65.6% to 73.7%) versus 67.3% (95% CI: 60.7% to 73.3%), respectively (P = 0.988). Similarly, stage I–IV sensitivity across all cancer types was 55.2% (95% CI: 52.7% to 57.7%) versus 54.9% (95% CI: 51.0% to 58.8%), respectively (P = 0.897), and in the pre-specified cancers was 77.9% (95% CI: 75.0% to 80.7%) versus 76.4% (95% CI: 71.6% to 80.7%), respectively (P = 0.573). Clinical models incorporating baseline demographic information and blood sample quality metrics alone resulted in <10% sensitivity in the training set and as such were not evaluated in the validation set (supplementary information, available at Annals of Oncology online).
Sensitivity increased with increasing stage of disease (Figure 4B). In validation, sensitivity in pre-specified cancer types was 39% (95% CI: 27% to 52%) in stage I (n = 62), 69% (95% CI: 56% to 80%) in stage II (n = 62), 83% (95% CI: 75% to 90%) in stage III (n = 102), and 92% (95% CI: 86% to 96%) in stage IV (n = 130). Among all cancer types, sensitivity was 18% (95% CI: 13% to 25%) in stage I (n = 185), 43% (95% CI: 35% to 51%) in stage II (n = 166), 81% (95% CI: 73% to 87%) in stage III (n = 134), and 93% (95% CI: 87% to 96%) in stage IV (n = 148). Performance in individual tumor types is depicted in Figure 5. These included numerous deadly cancer types without screening paradigms; for example in pancreatic cancer sensitivity was 63% (95% CI: 24% to 91%) in stage I, 83% (95% CI: 36% to 100%) in stage II, 75% (95% CI: 35% to 97%) in stage III, and 100% (80% to 100%) in stage IV. Tumor fraction (supplementary Figure S2, available at Annals of Oncology online) as measured by the frequency of abnormal tumor methylation patterns in plasma correlated with tumor fraction based on tumor mutation variant allele frequencies in plasma confirming the tumor-derived nature of the methylation signal.
Figure 5. Sensitivity in individual tumors by stage.
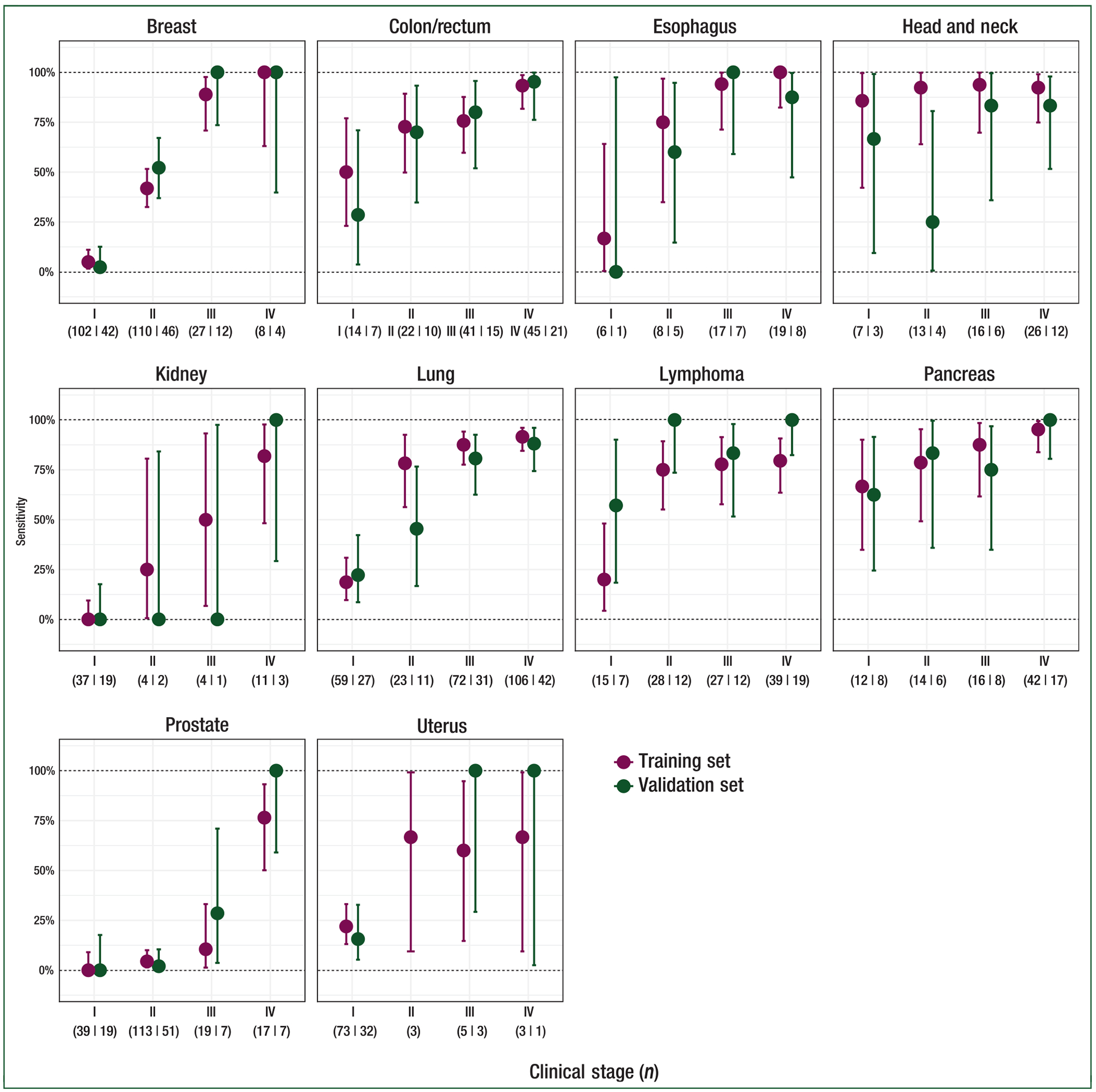
Sensitivity at 99.8% specificity (training) or 99.3% specificity (validation) with 95% confidence intervals is reported for individual cancer types with at least 50 samples. Clinical stage is indicated below the plots as is the number of samples in training and validation (separated by a vertical line).
To ensure consistent performance in centers that did not contribute to training and to ensure that single sites did not over-contribute, a post hoc site balancing analysis interrogated performance in a subset of centers dropped from the analysis (supplementary information, available at Annals of Oncology online). A limited shift in sensitivity consistent with variability in the training set was observed when omitting those sites from training [sensitivity for included versus excluded sites: 53.6% (95% CI: 51.8% to 55.4%) versus 50.0% (95% CI: 48.2% to 51.8%), respectively]; specificity was also within the expected range of variation [FPR of 0.5% (95% CI: 0.2% to 1%) versus 0.4% (95% CI: 0.2% to 0.8%), respectively].
A critical attribute of a blood-based multi-cancer detection test is the ability to localize the TOO to direct the diagnostic workup. A pre-specified analysis of TOO accuracy (the fraction of all TOO predictions that were correct) found that TOO was predicted in 96% (344/359) of samples with a cancer-like signal in the validation set; among these, accuracy was 93% (321/344). Accuracy was consistent between the training and validation sets and across stages (Figure 4C). The classifier distinguished from among the numerous cancer types included in the study with consistent performance in individual cancer types (Figure 6).
Figure 6. Tissue of origin accuracy by individual cancer type in the training and validation sets.
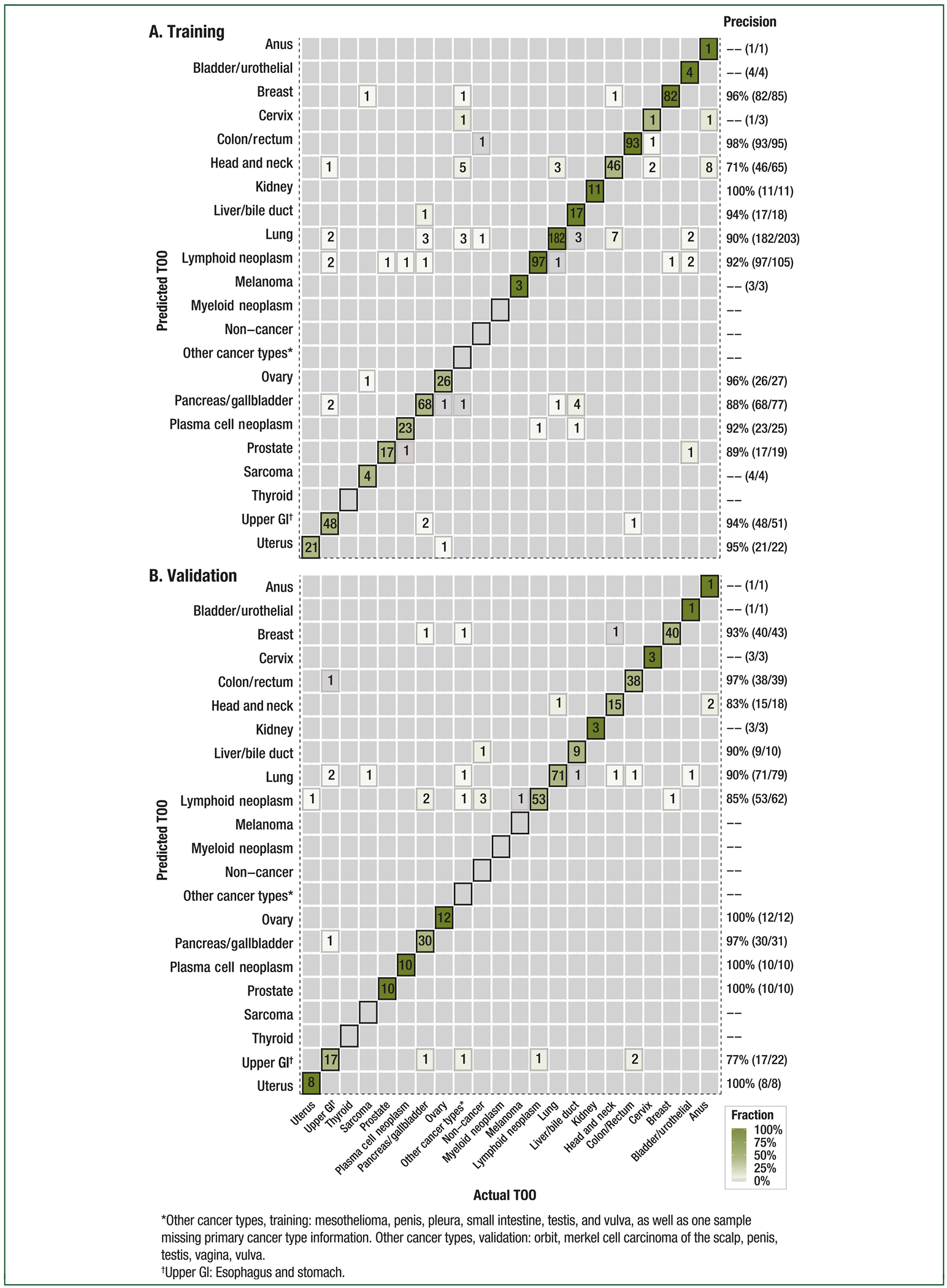
Confusion matrices representing the accuracy of tissue of origin (TOO) localization in the (A) training and (B) validation sets. Agreement between the actual (x-axis) and predicted (y-axis) TOO per sample using the targeted methylation classifier is depicted. Color corresponds to the proportion of predicted TOO calls. Included participants (training: n = 844, validation: n = 359) are those with cancer predicted as having cancer at 99.8% specificity (training) or 99.3% specificity (validation). The TOO calls were assigned in 95% (806/844) of cases in training and in 96% (344/359) of cases in validation; calls were correct in 92% (744/806) of cases in training and in 93% (321/344) of cases in validation.
DISCUSSION
Herein we report on the largest clinical genomics programs, to our knowledge, on participants with and without cancer, to develop and validate a blood-based test for multi-cancer early detection. The CCGA study was designed such that results may be generalizable as well as to minimize bias, a problem that has plagued the early detection field. This was accomplished by pre-specifying analyses, controlling for pre-analytic factors (e.g., age, sex, site location) and ensuring that demographics were comparable between the cancer and non-cancer groups, ensuring that stage distribution and method of diagnosis were consistent in independent training and validation sets (supplementary Table S1 available at Annals of Oncology online), ensuring that multiple cancer types at all stages (including early stages) were represented such that resultant cancer classifiers would not be confounded by inappropriate comparison cohorts, and ensuring that there were no site-specific effects on classifier performance. Inclusion of an independent validation set confirmed that the classifier was not over-fitted. Lastly, the inclusion of a large non-cancer cohort enriched in potentially confounding conditions demonstrated with confidence a high specificity (i.e. safety) that may be appropriate for population-level screening, minimizing potential harm from false positives. Together, these data provide compelling evidence that targeted methylation analysis of cfDNA can detect and localize a broad range of non-metastatic and metastatic cancer types including many common and deadly cancers that lack effective screening strategies.
Methylation outperformed WGS and targeted mutation panels in cancer detection and TOO localization7,8 for a number of reasons. Methylation is more pervasive compared with canonical mutation sites23 typically interrogated in traditional liquid biopsy approaches. Indeed, this targeted methylation approach interrogated approximately 1 million informative CpG sites out of the roughly 30 million CpGs across the genome that can be methylated or unmethylated.24 This allowed deeper sequencing of those informative regions compared with WGBS and may overcome expected cost and efficiency limitations of WGS or WGBS approaches. Although WGS detected cancer at high tumor fractions, it had a worse limit of detection than a methylation-based approach.7 Targeted mutation detection also suffered a worse limit of detection7 and was subject to highly prevalent mutations present in individuals due to biological processes such as CHIP.18 As such, unlike methylation, targeted sequencing required concurrent WBC sequencing to achieve strong performance. Finally, epigenetic signals inherently reflect tissue differentiation and malignant cancer states; this likely contributed to the strong cancer detection and TOO classification.
Other recently published studies also reported the feasibility of blood-based multi-cancer detection. These studies combined mutation detection with serum protein biomarkers,25 leveraged differences in cfDNA fragment lengths between cancer and non-cancer participants,10 or utilized a methylation-based immunoprecipitation approach to avoid perceived issues with bisulfite sequencing degradation.11 To overcome potential concerns about applicability to broad populations and less prevalent cancers, here we reported on >50 cancer types (supplementary information, available at Annals of Oncology online20) covering sites with >95% of the SEER program cancer incidence and included more than 4300 cancer and non-cancer participants in the primary analysis population. Additionally, this approach was developed after exhaustive preclinical analyses into three complementary and comprehensive sequencing approaches,17,19 ensuring that the highest performing assay was further developed.
A blood-based multi-cancer detection test should demonstrate certain fundamental performance characteristics to be useful in a general screening population. These include a sufficiently high specificity to ensure a low rate of false positives as well as accuracy in determining TOO. We reported a <1% FPR, a single, fixed FPR across all cancer types, such that inclusion of additional cancer types to the test would increase the number of tumors detected but not the number of false positives. By contrast, single-cancer early detection tests used in combination would generally have a cumulative FPR higher than the individual tests,26 potentially increasing unnecessary diagnostic work-ups. Accurate TOO localization is critical to direct the diagnostic workup; in its absence, patients with a positive test may be subjected to a diagnostic odyssey. This also applies to blood-based single-cancer detection, which would still require accurate TOO to avoid a diagnostic odyssey from other potential cancers not being tested for. The ability to discriminate from among so many cancer types may also be useful in cases of diagnostic uncertainty such as cancer of unknown primary origin. Finally, there would be little-to-no benefit from artificially increasing sensitivity by excluding cancer types in a multi-cancer test; the higher overall prevalence of cancer versus the prevalence of any single-cancer type means that a multi-cancer test with moderate sensitivity may result in a higher yield of detected cancers than a single-cancer test with very high sensitivity. As such, sensitivity must be considered in light of the number of interrogated cancer types.
Screening tests are subject to FPRs and the subsequent morbidity and psychological, physical, and financial costs associated with secondary screening or diagnostic tests. Minimizing these risks and costs requires high positive predictive value (PPV) in the target population, especially in asymptomatic populations. PPV is more significantly impacted by specificity and disease prevalence than by sensitivity. As noted above, multi-cancer detection would thus benefit from aggregate cancer incidence compared with single-cancer screening given that most cancer types have low prevalence in a screening population. While precise PPV calculations require measurement in a prospective trial of asymptomatic individuals, preliminary calculations can be carried out based on available cancer statistics. Specifically, assuming test performance replicates in an asymptomatic population, a multi-cancer test with a stage I–IV sensitivity of 55% and a specificity of 99.3%, both from representative populations as reported here, applied to a similar population with a 1.3% incidence rate per year of cancer1,27 would detect 715 cancers per 100 000 screened persons in a long-term screening program and would necessitate diagnostic work-ups in 691 FPRs, yielding a PPV of 51%. By contrast, the PPVs for United States Preventive Services Task Force (USPSTF) recommended screening for breast, colorectal (stool-based), and lung cancer (in the USPSTF-recommended high-risk population) range from 3.7% to 4.4%28–30 for every one person with cancer correctly detected, there would be between 22 and 27 people incorrectly identified as having cancer.
Despite the scale of and care in developing and validating this targeted methylation approach, the study has limitations. Participants with cancer were not all asymptomatic; to understand performance in an asymptomatic screening population will require additional studies, which are ongoing. Establishing a mortality benefit will also require additional studies as the CCGA study was not designed to examine all-cause mortality outcomes. Until such longer-term studies are completed, a multi-cancer test that shifts detection to earlier stages may function as a proxy for mortality, given that cancer-specific mortality is improved when cancer is diagnosed at earlier stages. Whether cancers detected at later stages can be intercepted at earlier stages using this cfDNA-based multi-cancer early detection test will require additional studies in intended use populations. At the time of analysis, complete 1-year follow-up was not available on all non-cancer participants to ensure their ascribed non-cancer status was accurate, thus potentially overestimating the FPR and underestimating PPV. Indeed, prior results from the first sub-study identified cancer signal up to 15 months before clinical diagnosis in participants enrolled without a cancer diagnosis.31 Follow-up of non-cancer participants in this sub-study is ongoing. Aggregate sensitivities were likely affected by stage and cancer distribution; reporting by individual cancer type and by stage as in this report is thus critical to contextualizing aggregate performance metrics. Confusion in TOO identification often occurred among HPV-driven cancers (e.g. cervix, anus, head and neck cancers); analyses are ongoing to further improve accuracy by leveraging this information. Finally, despite the broad range of cancer types captured in this study, for some cancer types the sample size was small, precluding a full representation of heterogeneity within some cancer types.
In summary, cfDNA sequencing of informative methylation patterns detected a broad range of cancer types at metastatic and non-metastatic stages with specificity and sensitivity performance approaching the goal for population-level screening. The pre-specified cancer types identified here account for ~63% of all estimated cancer deaths.21,27 Clinical validation in intended use populations is ongoing (NCT03085888, NCT03934866) and a study has been initiated that is returning results to health care providers and patients (NCT04241796). These results support the feasibility of employing this targeted methylation analysis of cfDNA in ongoing clinical trials in the intended use population for early cancer detection.
Supplementary Material
ACKNOWLEDGEMENTS
Editorial support provided by Kristi Whitfield, PhD from PosterDocs (Oakland, CA) as well as Steve Brunn and Ray Hunziker from ProEd Communications, Inc. (Beachwood, OH).
FUNDING
This work was supported by GRAIL, Inc. (Menlo Park, CA; no grant number). This publication was also partially supported by Princess Margaret Cancer Centre’s McCain GU BioBank in the Department of Surgical Oncology (grant number REB # 08-0124); its contents are solely the responsibility of the authors and do not necessarily represent the official views of the University Health Network. CS is supported by the Francis Crick Institute that receives its core funding from Cancer Research UK [grant numbers FC001169, FC001202]; the UK Medical Research Council [grant numbers FC001169, FC001202]; and the Wellcome Trust [grant numbers FC001169, FC001202]. CS is funded by Cancer Research UK (TRACERx; PEACE; and CRUK Cancer Immunotherapy Catalyst Network, no grant number), the CRUK Lung Cancer Centre of Excellence (no grant number); the Rosetrees Trust (no grant number); and the Breast Cancer Research Foundation (BCRF, no grant number).
DISCLOSURES
The Mayo Clinic was compensated for MCL’s advisory board activities for GRAIL, Inc. GRO reports personal fees from GRAIL, Inc. during the conduct of the study as well as personal fees from Inivata, Sysmex, AstraZeneca, Janssen, Illumina, and Foundation Medicine outside the submitted work. EAK reports personal fees from GRAIL, Inc. during the conduct of the study. MVS reports personal fees and other from McKesson and personal fees from GRAIL, Inc. during the conduct of the study as well as other from Merck and Bristol-Myers Squibb outside the submitted work. CS reports grants from Pfizer, AstraZeneca, BMS, Roche-Ventana, and Boehringer-Ingelheim; has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, BMS, Celgene, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Inc., Medicxi, and the Sarah Cannon Research Institute; has stock options of Apogen Biotechnologies, Epic Bioscience, GRAIL, Inc., and has stock options in and is co-founder of Achilles Therapeutics. CS is Royal Society Napier Research Professor. HA reports personal fees from GRAIL, Inc., during the conduct of the study as well as other from Illumina Inc. outside the submitted work; in addition, HA has patents pending to GRAIL, Inc. DAB reports grants from the National Cancer Institute, other from Berry Consultants, LLC, outside the submitted work. TCC reports personal fees and other from Illumina, Inc., outside the submitted work. CC reports personal fees and other (stock options) from GRAIL, Inc., during the conduct of the study as well as personal fees from Genentech outside the submitted work; in addition, CC has a patent pending outside the submitted work. KD reports personal fees from GRAIL, Inc. during the conduct of the study and other from Alphabet outside the submitted work. FJC reports research support from GRAIL, Inc. MD reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, MD has patents pending to GRAIL, Inc. SF reports personal fees and other from GRAIL, Inc., during the conduct of the study as well as personal fees and other from 23andMe and other from Illumina outside the submitted work. APF reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, APF has patents pending to GRAIL, Inc. DF reports personal fees from GRAIL, Inc. during the conduct of the study as well as personal fees from Roche Sequencing Solutions outside the submitted work; in addition, DF has patents pending to GRAIL, Inc. SG reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as other from Illumina outside the submitted work. S. Gross reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, S. Gross has patents pending to GRAIL, Inc. MPH reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as other from Jazz Pharmaceuticals and Natera outside the submitted work. SAP reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as other from Natera, Inc. outside of the submitted work. EH reports personal fees and other from GRAIL, Inc. during the conduct of the study; in addition, EH has patents pending to GRAIL, Inc. NH reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, NH has patents pending to GRAIL, Inc. CH reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as other from Illumina outside the submitted work. QL reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, QL has patents pending to GRAIL, Inc. AJ reports personal fees from GRAIL, Inc. during the conduct of the study and personal fees from Illumina outside the submitted work; in addition, AJ has patents pending to GRAIL, Inc. and a patent (differential tagging of RNA for preparation of a cell-free DNA/RNA sequencing library) issued to GRAIL, Inc. RK reports personal fees from GRAIL, Inc. during the conduct of the study as well as personal fees from Mind-strong Health, personal fees from Lyell Immunopharma, personal fees from LifeMine, personal fees from Wisdo, personal fees from Medical Creations/Extremity, and personal fees from FOG Pharma all outside the submitted work. KNK reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as other from Illumina outside the submitted work. ML reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as personal fees and other from Genentech, Inc., personal fees and other from Google Life Sciences, personal fees and other from Boreal Genomics, and personal fees and other from Genomic Health, Inc. outside the submitted work; in addition, ML has a patent arising from the CCGA work pending to GRAIL, Inc. MCM reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, MCM has patents pending to GRAIL, Inc. CM reports personal fees and other from GRAIL, Inc. during the conduct of the study; in addition, CM has a patent pending to GRAIL, Inc. VD reports personal fees and other from GRAIL, Inc. during the conduct of the study. JN reports personal fees from GRAIL Inc. during the conduct of the study as well as personal fees from Verily Life Sciences (formerly part of Google) outside the submitted work. Joshua N. reports personal fees and other from GRAIL, Inc. during the conduct of the study. VN reports personal fees from GRAIL Inc. during the conduct of the study; in addition, VN has patents pending to GRAIL, Inc. RVS reports personal fees from GRAIL, Inc. during the conduct of the study as well as personal fees from Guardant Health outside the submitted work; in addition, RVS has a patent pending to GRAIL, Inc. AS reports personal fees from GRAIL, Inc. during the conduct of the study and is the owner of Illumina stock. LS reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, LS has a patent pending to GRAIL, Inc. MS reports personal fees from Celgene, personal fees from Millenium/Takeda, and personal fees from Syros outside the submitted work. DS reports other from US Oncology during the conduct of the study. AV reports personal fees and other from GRAIL, Inc. during the conduct of the study as well as other from Illumina outside the submitted work. OV reports personal fees from GRAIL, Inc. during the conduct of the study; in addition, OV has patents pending to GRAIL, Inc. SRC reports research support from GRAIL, Inc. JY reports personal fees and other from GRAIL Inc. during the conduct of the study as well as personal fees and other from Acerta Pharma B.V., personal fees from Forty Seven Inc., other from BeiGene, Ltd., other from Celgene Corporation, other from Loxo Oncology, Inc., other from Nektar Therapeutics, other from Corvus Pharmaceuticals, Inc., and other from Illumina, Inc., all outside the submitted work. AB reports a financial interest in GRAIL, Inc. via Foresite Capital’s funds and personal equity. AMA is a founder, employee, and shareholder at GRAIL, Inc. and a paid advisor to Foresite Capital and Myst Therapeutics. JB reports personal fees from GRAIL, Inc. as well as patents pending to GRAIL, Inc. during the conduct of the study; JB also has patents issued to Roche and Philips Medical Systems outside of this work. PF reports personal fees from GRAIL, Inc. as well as patents pending to GRAIL, Inc. during the conduct of the study. WL reports personal fees and other from GRAIL, Inc. during the conduct of the study and personal fees from Genentech outside of this work. TM reports personal fees and other from GRAIL, Inc. and a patent pending to GRAIL, Inc. during the conduct of the study; TM also reports personal fees and other from Lexent Bio, HTG Molecular, and NDA Partners, as well as other from Genomic Health and personal fees from Terumo Medical outside of this work. RS, RL, TW, AS, ON, LZ, RC, CY, PS, NR, CC, AY, A. Shanmugam, JS, GA, AM, JZ, HC, GC, KCS, XC, BA, JL, JY, FA, LB, J. Berman, JC, TK, SB, JFB, CB, TCC, DC, ZD, ETF, A-RH, RJ, BJ, QL, SN, CN, SP, RR, OS, ES, AJS, SS, KKS, ST, JMT, RTW, XY, JY, and NZ report personal fees from GRAIL, Inc. during the conduct of the study. The remaining authors have declared no conflict of interest.
APPENDIX A. AUTHORS FOR THE CCGA CONSORTIUM
CCGA investigators
Minetta C. Liu1†, Geoffrey R. Oxnard2†, Eric A. Klein3, David Smith4, Donald Richards5, Timothy J. Yeatman6,a, Allen L. Cohn7, Rosanna Lapham8, Jessica Clement9, Alexander S. Parker10,b, Mohan K. Tummala11, Kristi McIntyre12, Mikkael A. Sekeres13, Alan H. Bryce14, Robert Siegel15, Xuezhong Wang15, David P. Cosgrove16, Nadeem R. Abu-Rustum17, Jonathan Trent18, David D. Thiel19, Carlos Becerra20, Manish Agrawal21, Lawrence E. Garbo22, Jeffrey K. Giguere23, Ross M. Michels23,c, Ronald P. Harris24, Stephen L. Richey25, Timothy A. McCarthy26, David M. Waterhouse27, Fergus J. Couch28, Sharon T. Wilks29, Amy K. Krie30, Rama Balaraman31, Alvaro Restrepo32, Michael W. Meshad33, Kimberly Rieger-Christ34, Travis Sullivan34, Christine M. Lee35, Daniel R. Greenwald36, William Oh37, Che-Kai Tsao37, Neil Fleshner38, Hagen F. Kennecke39, Maged F. Khalil40, David R. Spigel41, Atisha P. Manhas42, Brian K. Ulrich43, Philip A. Kovoor44, Christopher Stokoe45, Jay G. Courtright46, Habte A. Yimer47, Timothy G. Larson48, Charles Swanton49,50, Michael V. Seiden51†*
STRIVE investigators
Steven R. Cummings52
GRAIL, Inc. (alphabetical)
Farnaz Absalan53, Gregory Alexander53, Brian Allen53, Hamed Amini53, Alexander M. Aravanis53, Siddhartha Bagaria53, Leila Bazargan53, John F. Beausang53, Jennifer Berman53, Craig Betts53, Alexander Blocker53,d, Joerg Bredno53, Robert Calef53, Gordon Cann53, Jeremy Carter53, Christopher Chang53, Hemanshi Chawla53, Xiaoji Chen53, Tom C. Chien53, Daniel Civello53, Konstantin Davydov53, Vasiliki Demas53, Mohini Desai53, Zhao Dong53, Saniya Fayzullina53, Alexander P. Fields53, Darya Filippova53, Peter Freese53, Eric T. Fung53, Sante Gnerre53,e, Samuel Gross53, Meredith Halks-Miller53,f, Megan P. Hall53, Anne-Renee Hartman53,g, Chenlu Hou53, Earl Hubbell53, Nathan Hunkapiller53, Karthik Jagadeesh53,h, Arash Jamshidi53, Roger Jiang53, Byoungsok Jung53, TaeHyung Kim53,i, Richard D. Klausner53, Kathryn N. Kurtzman53, Mark Lee53,j, Wendy Lin53,k, Jafi Lipson53, Hai Liu53, Qinwen Liu53, Margarita Lopatin53, Tara Maddala53,l, M. Cyrus Maher53, Collin Melton53, Andrea Mich53, Shivani Nautiyal53,f, Jonathan Newman53, Joshua Newman53, Virgil Nicula53, Cosmos Nicolaou53,m, Ongjen Nikolic53,n, Wenying Pan53, Shilpen Patel53,o, Sarah A. Prins53, Richard Rava53,f, Neda Ronaghi53, Onur Sakarya53, Ravi Vijaya Satya53,e, Jan Schellenberger53, Eric Scott53, Amy J. Sehnert53,p, Rita Shaknovich53, Avinash Shanmugam53, K.C. Shashidhar53, Ling Shen53,q, Archana Shenoy53, Seyedmehdi Shojaee53, Pranav Singh53, Kristan K. Steffen53, Susan Tang53, Jonathan M. Toung53, Anton Valouev53,r, Oliver Venn53, Richard T. Williams53,s, Tony Wu53, Hui H. Xu53,q, Christopher Yakym53, Xiao Yang53, Jessica Yecies53, Alexander S. Yip53, Jack Youngren53, Jeanne Yue53, Jingyang Zhang53, Lily Zhang53, Lori (Quan) Zhang53, Nan Zhang53
Advisors
Christina Curtis54, Donald A. Berry55
*Corresponding author.
†Contributed equally.
Affiliations
1. Division of Medical Oncology, Department of Oncology, Mayo Clinic, Rochester, USA.
2. Lowe Center for Thoracic Oncology, Dana Farber Cancer Institute, Boston, USA.
3. Glickman Urological and Kidney Institute, Cleveland Clinic, Cleveland, USA.
4. Compass Oncology, Vancouver, USA.
5. Texas Oncology, Tyler, USA.
6. Gibbs Cancer Center and Research Institute, Spartanburg, USA.
7. Rocky Mountain Cancer Center, Denver, USA.
8. Spartanburg Regional Healthcare System, Spartanburg, USA.
9. Hartford HealthCare Cancer Institute, Hartford, USA.
10. Mayo Clinic, Jacksonville, USA.
11. Mercy Clinic Cancer Center, Springfield, USA.
12. TOPA Dallas Presbyterian, Dallas, USA.
13. Hematology and Medical Oncology, Cleveland Clinic, Cleveland, USA.
14. Genomic Oncology Clinic, Mayo Clinic, Phoenix, USA.
15. Bon Secours St. Francis Cancer Center, Greenville, USA.
16. Compass Oncology, Vancouver Cancer Center, Vancouver, USA.
17. Department of Surgery, Memorial Sloan Kettering Cancer Center, New York, USA.
18. Department of Hematology/Oncology, University of Miami Health System, Miami, USA.
19. Department of Urology, Mayo Clinic, Jacksonville, USA.
20. Department of Medical Oncology, Texas Oncology-Baylor Charles A. Sammons Cancer Center, Irving, USA.
21. Department of Medical Oncology, Maryland Oncology Hematology, Rockville, USA.
22. New York Oncology Hematology, Albany, USA.
23. Greenville Health System Cancer Institute, Seneca, USA.
24. Department of Internal Medicine, Broome Oncology, Binghamton, USA.
25. Texas Oncology, Fort Worth, USA.
26. Virginia Cancer Specialists, Fairfax, USA.
27. Oncology Hematology Care, Cincinnati, USA.
28. Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, USA.
29. Texas Oncology, San Antonio, USA.
30. Avera Medical Group Hematology and Oncology, Sioux Falls, USA.
31. Florida Cancer Affiliates, Ocala, USA.
32. Texas Oncology, McAllen, USA.
33. Southern Cancer Center, Daphne, USA.
34. Translational Research, Lahey Hospital and Medical Center, Burlington, USA.
35. Texas Oncology, The Woodlands, USA.
36. Sansom Clinic, Ridley-Tree Cancer Center, Santa Barbara, USA.
37. Department of Medicine, Icahn School of Medicine, The Mount Sinai Hospital, New York, USA.
38. Princess Margaret Cancer Centre’s McCain GU BioBank, in the Department of Surgical Oncology, University of Toronto, Princess Margaret Cancer Centre, Toronto, Canada.
39. Department of Medical Oncology, Virginia Mason Benaroya Research Institute, Seattle, USA.
40. Hematology Oncology, Lehigh Valley Health Network, Allentown, USA.
41. Sarah Cannon Research Institute/Tennessee Oncology, PLLC, Nashville, USA.
42. Department of Hematology/OncologyTexas Oncology-Methodist Dallas Cancer Center, Dallas, USA.
43. Department of Medical Oncology, Texoma Cancer Center, Wichita Falls, USA.
44. Department of Medical Oncology, Texas Oncology, Plano West, Plano, USA.
45. Department of Medical Oncology, Texas Oncology, Plano East, Plano, USA.
46. Department of Medical Oncology, Texas Oncology, Medical City Dallas, Dallas, USA.
47. Department of Medical Oncology, Texas Oncology, Tyler, Tyler, USA.
48. Department of Medical Oncology, Minnesota Oncology, Minneapolis, USA.
49. Cancer Evolution and Genome Instability Laboratory, The Francis Crick Institute, London, UK.
50. Cancer Evolution and Genome Instability Laboratory, University College London Cancer Institute, London, UK.
51. Gynecologic Medical Oncology, US Oncology Research, The Woodlands, USA.
52. San Francisco Coordinating Center, Sutter Health Research, San Francisco, USA.
53. Research and Development, GRAIL, Inc., Menlo Park, USA.
54. School of Medicine, Departments of Medicine and Genetics, Stanford University, Stanford, USA.
55. Department of Biostatistics, Division of Basic Sciences, MD Anderson Cancer Center, Houston, USA.
Current affiliations:
aCurrent affiliation: Department of General Surgery, Intermountain Healthcare, Murray, USA.
bCurrent affiliation: University of Florida College of Medicine, Jacksonville, USA.
cCurrent affiliation: Prisma Health Cancer Institute, Seneca, USA.
dCurrent affiliation: Foresite Labs, San Francisco, USA.
eCurrent affiliation: Research and Development, Guardant Health, Redwood City, USA.
fFormerly of GRAIL, Inc., Menlo Park, USA.
gCurrent affiliation: Operator Collective, San Francisco, USA.
hCurrent affiliation: Department of Computer Science, Stanford University, Stanford, USA.
iCurrent affiliation: Department of Computer Science, University of Toronto, Toronto, Canada.
jCurrent affiliation: Personalized Healthcare, Product Development, Genentech/Roche, San Francisco, USA.
kCurrent affiliation: Companion Diagnostics, Genentech, San Francisco, USA.
lCurrent affiliation: TMBiostats, LLC, Sunnyvale, USA.
mCurrent affiliation: Research and Development, CloudEng LLC, Palo Alto, USA.
nCurrent affiliation: Data Science Department, Better-Omics.com, San Francisco, USA.
oCurrent affiliation: Medical Affairs, Genentech, San Francisco, USA.
pCurrent affiliation: Clinical Development, MyoKardia, San Francisco, USA.
qCurrent affiliation: Research and Development, Inter-Venn Biosciences, Redwood City, USA.
rCurrent affiliation: Research and Development Department, ArsenalBio, San Francisco, USA.
sCurrent affiliation: WuXi NextCODE, Cambridge, USA.
REFERENCES
- 1.NIH National Cancer Insitute. Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER 18 Regs Research Data, Nov 2017 Sub (1973-2015) National Cancer Institute, DCCPS, Surveillance Research Program, released April 2018, based on the November 2017 submission. Statistic based on all invasive cancers, ages 50+ at diagnosis. [www.seer.cancer.gov].
- 2.Noone A, Howlander N, Krapcho M, et al. , eds. SEER Cancer Statistics Review, 1975-2015, National Cancer Institute, software version 8.3.6. Bethesda. Available at: https://seer.cancer.gov/csr/1975_2015/. [Google Scholar]
- 3.Cossu G, Saba L, Minerba L, Mascalchi M. Colorectal cancer screening: the role of psychological, social and background factors in decision-making process. Clin Pract Epidemiol Ment Health. 2018;14(1):63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayan A, Fischer A, Zhang Z, et al. Nationwide cross-sectional adherence to mammography screening guidelines: national behavioral risk factor surveillance system survey results. Breast Cancer Res Treat. 2017;164(3):719–725. [DOI] [PubMed] [Google Scholar]
- 5.Brasher P, Tanner N, Yeager D, Silvestri G. Adherence to annual lung cancer screening within the Veterans Health Administration lung cancer screening demonstration project. Chest. 2018;154(4):636A–637A. [DOI] [PubMed] [Google Scholar]
- 6.Limmer K, LoBiondo-Wood G, Dains J. Predictors of cervical cancer screening adherence in the United States: a systematic review. J Adv Pract Oncol. 2014;5(1):31–41. [PMC free article] [PubMed] [Google Scholar]
- 7.Oxnard GR, Klein EA, Seiden MV, et al. Simultaneous multi-cancer detection and tissue of origin (TOO) localization using targeted bisulfite sequencing of plasma cell-free DNA (cfDNA). Ann Oncol. 2019;30(suppl 5):LBA77. [Google Scholar]
- 8.Liu MC, Jamshidi A, Venn O, et al. Genome-wide cell-free DNA (cfDNA) methylation signatures and effect on tissue of origin (TOO) performance. J Clin Oncol. 2019;37(suppl 15):3049. [Google Scholar]
- 9.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563(7732):579–583. [DOI] [PubMed] [Google Scholar]
- 12.Merker JD, Oxnard GR, Compton C, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. J Clin Oncol. 2018;36(16):1631–1641. [DOI] [PubMed] [Google Scholar]
- 13.Razavi P, Li BT, Brown DN, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25(12):1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Ulrich BC, Supplee J, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24(18):4437–4443. [DOI] [PubMed] [Google Scholar]
- 15.Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4(162):162ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan KCA, Jiang P, Chan CWM, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci. 2013;110(47):18761–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu MC, Klein E, Hubbell E, et al. Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: the circulating cell-free genome atlas (CCGA) study. Ann Oncol. 2018;29(suppl 8):500. [Google Scholar]
- 18.Swanton C, Venn O, Aravanis A, et al. Prevalence of clonal hematopoiesis of indeterminate potential (CHIP) measured by an ultra-sensitive sequencing assay: exploratory analysis of the Circulating Cancer Genome Atlas (CCGA) study. J Clin Oncol. 2018;36(suppl 15):12003. [Google Scholar]
- 19.Klein E, Hubbell E, Maddala T, et al. Development of a comprehensive cell-free DNA (cfDNA) assay for early detection of multiple tumor types: the Circulating Cell-free Genome Atlas (CCGA) study. J Clin Oncol. 2018;36(suppl 15):12021. [Google Scholar]
- 20.Amin MB, Greene FL, Edge SB, et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more ‘personalized’ approach to cancer staging. CA Cancer J Clin. 2017;67(2):93–99. [DOI] [PubMed] [Google Scholar]
- 21.Surveillance, Epidemiology, and End Results (SEER) Program SEER*Stat Database: Mortality - All COD, Aggregated With State, Total U.S. (1969-2016) <Katrina/Rita Population Adjustment>, National Cancer Institute, DCCPS, Surveillance Research Program, released December 2018. Underlying mortality data provided by NCHS (www.cdc.gov/nchs). Statistic based on 2015-2016 data, all ages. [www.seer.cancer.gov].
- 22.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. [DOI] [PubMed] [Google Scholar]
- 23.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Research. 2002;6:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croswell JM, Kramer BS, Kreimer AR, et al. Cumulative incidence of false-positive results in repeated, multimodal cancer screening. Ann Fam Med. 2009;7(3):212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surveillance, Epidemiology, and End Results SEER*Stat software. Available at: www.seer.cancer.gov/seerstat.
- 28.Lehman CD, Arao RF, Sprague BL, et al. National performance benchmarks for modern screening digital mammography: update from the Breast Cancer Surveillance Consortium. Radiology. 2017;283(1):49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.U. S. Food and Drug Administration. Cologuard Summary of Safety and Effectiveness Data (Premarket Approval Application P130017). 2014. Available at https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130017B.pdf.
- 30.The National Lung Screening Trial Research Team. Results of initial low-dose computed tomographic screening for lung cancer. N Engl J Med. 2013;368(21):1980–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohn AL, Seiden MV, Kurtzman KN, et al. The Circulating Cell-free Genome Atlas (CCGA) study: follow-up (F/U) on non-cancer participants with cancer-like cell-free DNA signals. J Clin Oncol. 2019;37(suppl 15):5574. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.