

Abstract
Lymphocyte-specific protein tyrosine kinase (Lck), a non-receptor Src family kinase, has a vital role in various cellular processes such as cell cycle control, cell adhesion, motility, proliferation, and differentiation. Lck is reported as a key factor regulating the functions of T-cell including the initiation of TCR signalling, T-cell development, in addition to T-cell homeostasis. Alteration in expression and activity of Lck results in numerous disorders such as cancer, asthma, diabetes, rheumatoid arthritis, atherosclerosis, and neuronal diseases. Accordingly, Lck has emerged as a novel target against different diseases. Herein, we amass the research efforts in literature and pharmaceutical patents during the last decade to develop new Lck inhibitors. Additionally, structure-activity relationship studies (SAR) and docking models of these new inhibitors within the active site of Lck were demonstrated offering deep insights into their different binding modes in a step towards the identification of more potent, selective, and safe Lck inhibitors.
Keywords: Lck inhibitors, structure-activity relationship (SAR), Src family kinase, lymphocyte-specific protein tyrosine kinase (Lck), molecular modelling
1. Introduction
Lymphocyte-specific protein tyrosine kinase (Lck), a 56 KDa protein, is a member of the Src family of non-receptor protein kinases. Lck is involved in the phosphorylation process of a number of intracellular signalling molecules such as IL-2-inducible T-cell kinase (ITK), protein kinase C, Phosphoinositide 3-kinase (PI3K), and Zeta-chain-associated protein kinase 70 (ZAP-70). Accordingly, it regulates numerous cellular processes including cell cycle control, cell adhesion, motility, proliferation, and differentiation. The function of Lck has been extensively studied and various reports revealed different mechanistic insights into the regulation of its activity including its major role as a key activator of T cells via T cell antigen receptors (TCR) signalling1–5. In addition to T cells, Lck is expressed in natural killer (NK) cells, NK T cells, CD5+ B-1 B cells, germinal centre and to a lesser extent in mantle zone B cells, aryl hydrocarbon receptor-activated primary human B cells, and brain including the hippocampus, cerebellum and retina6–10. In addition to leukaemia, Lck expression was also detected in a number of solid cancers including colon cancer, lung carcinoma, and breast cancer11–16, which led to the hypothesis that Lck may also have cancer promoting functions and hence may act as a potential therapeutic target for solid cancers.
Accordingly, Lck inhibitors were found to be promising not only for the treatment of leukaemia but also in various solid cancers. In this review, we focus on presenting the newly discovered Lck inhibitors during the last decade, discussing their structure-activity relationship (SAR), in addition to performing docking simulation models of the most promising candidates into the binding site(s) of Lck in an attempt to get insights for further investigations towards more selective, potent and safe Lck inhibitors.
2. Lck (structure, regulation, and physiological roles)
The strcuture of Lck has the typical backbone found in all members of the Src kinase family (Figure 1); an N-terminal site (SH4 domain), SH3 and SH2 domains, a catalytic domain at the carboxy terminal (SH1 domain), and a short C-terminal tail17–19. The C-terminal lobe contains the activation loop (alpha-helix) which forms the phosphorylation site. Both SH2 and SH3 domains are folded to be involved in protein-protein interactions responsible for the regulation of Lck activity and signal transmission; while the main function of SH2 domain is to regulate interactions with phosphotyrosine containing elements, the SH3 domain regulates interactions with proline rich elements. The SH4 domain contains a glycine and two cysteine residues, which are myristoylated and palmitoylated, respectively, to target Lck to the plasma membrane.
Figure 1.

(A) Structure of Lck kinase domains; (B) Schematic structure of Lck: SH4, unique region (UR), SH3, SH2, SH2-kinase domain linker region (LR), kinase domain, and the C-terminal negative regulatory tail (NR), Reprinted from Ref.4; (C) Lck conformations and regulation of Lck activation, Reprinted from Ref.4.
The regulation of Lck activity occurs via phosphorylation/dephosphorylation of crucial tyrosine residues, and by some conformational changes; Phosphorylation of Tyr505 residue by the C-terminal Src kinase (Csk) leads to Lck closure through an intramolecular interaction with the SH2 domain. The closed conformation of Lck is further stabilised by the interaction between the SH3 domain and a proline-reach region located in the SH2-kinase domain linker. On the other side, Lck opening depends on dephosphorylation of Tyr505 catalysed by the protein tyrosine phosphatase CD45. The open conformation of Lck auto- and transphosphorylates Tyr394 residue located in the activation loop within the catalytic domain resulting in Lck activation. In addition to Tyr505 and Tyr394, there are other amino acid residues regulate Lck activity; a recent study by Courtney et al. on a phosphomimetic Lck mutant found that phosphorylation of Tyr192 located in the SH2 domain may restrict the interaction between Lck and CD45, leading to hyperphosphorylation of Tyr505 and accordingly in Lck inactivation20. Another study proposed that the phosphorylation of this site is Zap-70-dependent, in addition, Tyr192 residue was found to be a part of an inhibitory feedback loop, which controls the regulation of the amount of active Lck and the strength/duration of TCR signalling21. Moreover, Lck activity was found to be also regulated by phosphorylation of Ser59 (another feedback circuit required for the regulation of TCR signalling)22–24. Accordingly, a number of biochemical modifications, conformational dynamics, and signalling circuits were found to regulate the activity of Lck.
At physiological level, Lck is a key factor for development of T cells in the thymus and for the function of mature T cells. It also has a major role in the activation of TCR linked signal transduction pathways (Figure 2)25–28. Plus, Lck is involved in regulation of neurite outgrowth since it plays an important role in maintaining long-term synaptic plasticity in neurons in addition to other roles related to spatial learning and memory29,30. As mentioned earlier, Lck is also expressed in NK T cells, NK cells, and B cells. Although the function of Lck in B-cell remains unclear, Lck was suggested to regulate B Cell Receptor Signalling (BCR) signalling31,32.
Figure 2.

The pathway of Lck signalling. Reprinted from Ref. 5.
3. Lck-related diseases
The human genome contains more than 500 protein kinases transfer a γ-phosphate group from ATP to serine, threonine, or tyrosine residues. Several kinases were found to be associated with different human disorders including cancer initiation and progression. Also, the recent medicinal chemistry research targeting development of small molecule kinase inhibitors for the treatment of various diseases including cancer has been proven to be a successful strategy33–46. Among these cancer-related kinases, Lck was reported to be the promotor of BCR signals in chronic lymphocytic leukaemia (CLL) via catalysis of the proximal phosphorylation of CD79a and the induction of distal signalling events involving phosphorylation of Syk, activation of MAPK, NF-kB, ERK, and PI3K/Akt signalling pathways that are responsible for CLL cell survival following BCR cross-linking. The treatment of CLL cells with Lck inhibitors suppressed BCR dependent cell survival leading to apoptosis suggesting the potential role of Lck inhibitors in the treatment of CLL47–49. Lck was also found to be overexpressed and hyperactivated in patients with B-cell precursor acute lymphoblastic leukaemia (BCP-ALL)50,51. Low levels of Lck were also detected in thymoma and suggested to be responsible of the abnormal proliferation of immature thymocytes causing thymic tumorigenesis. Co-expression of Lck-Fyn has been reported in the development of thymomas52. Other studies showed that Lck functions as a therapeutic target in acute myeloid leukaemia (AML)53–55. A recent study showed that Lck was expressed at a high level in primary central nervous system lymphoma (PCNSL) patients56. Lck expression was also detected in cholangiocarcinoma57,58, breast cancer12,13,59,60, colon cancer14,15,61,62, and lung carcinoma16,62,63. It was also found that Lck seems to play a role in cancer stem cells (CSC) in endometrioid cancer models and cisplatin resistance of glioma cancer stem cells64,65. An additional function of Lck in glioma cells has been recently described66. Moreover, Lck overexpression was reported in several small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) cell lines and lung cancer specimens from patients67.
The success of small molecule kinase inhibitors in the treatment of cancer, coupled with a greater understanding of inflammatory signalling cascades, has led to kinase inhibitors coming to the fore in the pursuit for new anti-inflammatory agents for the treatment of inflammatory and immune-mediated diseases68. Non-receptor tyrosine kinases of the Jak, Src, Syk, and Btk families play major roles in various inflammatory and immune-mediated disorders69. Lck was found to be a key player in the early allergic immune response. Antigen activation of TCR results in the activation of Lck and further downstream signalling, resulting in T cell differentiation as well as cytokine secretion. It was also reported that Lck mediates Th2 differentiation. The chronic inflammatory disease of the bronchial airways (asthma) was associated with activation of a Th2 type of T cell in the airway70,71. A study by Pernis et al. found that mice overexpressing or lacking Lck gene showed altered lung function suggesting their involvement in pathogenesis of asthma72. Histological assessment of mice lung tissues by Zhang et al. revealed that Lck specific siRNA attenuated the pulmonary inflammation in asthma mice proposing Lck as a potential therapeutic target for asthma73. Accordingly, since Lck was proved to be involved in the pathogenesis of asthma, a novel therapy for treatment of asthma can be developed based on Lck novel specific inhibitors.
Different studies reported the relation between Lck and other diseases rather than cancer and inflammation; the expression of Lck with Type I diabetes suggesting Lck as one of the main targets for diabetes treatment74. A recent study reviewed the interplay of protein tyrosine phosphatases with Src kinases including Lck establishing their role in auto-immune mediated diabetes75. The concept of Lck inhibition for the management of Type 1 diabetes was supported by another report suggested that βig-h3 represses T cell activation in Type 1 diabetes via inhibition of Lck76. Moreover, blocking of Lck may provide a novel therapeutic target to manage atherosclerosis. A recent study showed T-cells in atherosclerosis patients to be cytotoxic towards vascular smooth muscle cells as well as endothelial cells, leading to vascular injury and plaque destabilisation. Lck might inhibit heat shock protein 65–mediated Reverse Cholesterol Transport in T cells which has been well established as one of the causes involved for atherosclerosis77. Lck was also reported as a potential therapeutic target for acute rejection after kidney transplantation78. Organ graft rejection occurs when the tissue transplanted in the recipient’s body is rejected by his immune system79. Thus, inhibition of Lck has been established as a potential target to prevent organ graft rejection80,81.
4. Early discovery of Lck kinase inhibitors (selected examples)
By the year 2010, a large number of small molecules incorporating various chemical scaffolds were already reported to inhibit Lck82,83. In this section, we demonstrate some examples of the most promising candidates; the earliest members of this family are the ones possessing the pyrazolopyrimidine chemical scaffold; PP1 (I) and PP2 (II) (Figure 3), reported by Pfizer in 199684. Despite the low nanomolar Lck IC50 range of these two compounds (0.005 and 0.004 µM for PP1 and PP2, respectively), they showed lack of selectivity within Src kinase family. Further extended studies offered a direct descendant of PP1 (A-770041, III, Figure 3) which demonstrated a specific inhibition over Lck with an IC50 value of 0.147 µM. The final structure of this molecule is a result of both strategic modification and extensive SAR exploration aiming to improve the activity towards Lck and reduce activity against other members of the Src family while offering compounds with suitable pharmacokinetic properties80,81,85–88.
Figure 3.

Chemical structures of compounds I–IV.
For different reasons such as the discovery of a chemical space available in the hydrophobic pocket and the solvent exposed binding region of Lck80,81, the difficulty of generating N7 variants of A-770041, and the hope to discover a highly selective Lck inhibitor via making productive contacts with the side chains of Tyr318 and the unique Glu320 in the extended hinge region of Lck, the pyrazolopyrimidine core was replaced with a thienopyridine scaffold offering compound IV with Lck IC50 value of 0.21 µM (Figure 3)89. The further SAR exploration confirmed that specificity could be generated through interactions with the hinge region. Analysis of compound IV against a larger kinase set showed improved selectivity within the Src family with significant decreases in activity against Src and Fyn relative to A-770041 (III). However, upon administration to mice, compound IV inhibited TCR stimulated IL-2 production with an ED50 of 5 mg/kg; the pharmacokinetic analysis demonstrated poor performance regarding clearance and oral bioavailability.
The benzothiazole compound BMS-243117 (V, Figure 4) was then reported following SAR exploration of a thiazole compound initially obtained via high throughput screening. Although compound V demonstrated a highly potent nanomolar activity over Lck (IC50 = 4 nM) and a promising inhibitory activity over T Cell proliferation with an IC50 value of 1.1 µM, it showed high inhibitory activity against other isoforms of Src family (Src IC50 = 632 nM, Fyn IC50 = 128 nM, Hck IC50 = 3.84 µM, Blk IC50 = 336 nM, Lyn IC50 = 1.32 µM, and Fgr IC50 = 240 nM), in addition, no in vivo data is reported for this promising candidate to date90. Another aminoquinazoline-based highly potent Lck inhibitor (VI, Figure 4), possessing IC50 of 0.2 nM was identified via a high-throughput screening (HTS)91. Extended SAR studies of compound VI offered a series of novel aminoquinazolines possessing in vitro mechanism-based potency. Orally bioavailable optimised analogs of compound VI exhibited a promising anti-inflammatory activity over the anti-CD3-induced production of interleukin-2 (IL-2) in mice. Although the selectivity of compound VI within the Src family was not studied during these initial SAR studies, some analogs showed potent nanomolar activity against other Src family isoforms91. Screening of some pyrimidopyridazine-based small molecules against Lck led to the discovery of a novel 1,2-dihyrdropyrimido[4,5-c]pyridazine derivative (VII, Figure 4) with low micromolar activity towards Lck. Optimisation of this compound revealed the most promising analog of this series (VIII, Figure 4) which demonstrated good solubility and activity towards Lck (IC50 = 2 nM), although still with strong activity towards Src (IC50 = 3 nM).
Figure 4.

Chemical structures of compounds V–VIII.
A novel 4-amino-5,6-biaryl-furo[2,3-d]pyrimidine lead (IX, Figure 5) was discovered by DiMauro et al. as potent, non-selective inhibitor of Lck (IC50 = 0.081 µM) via HTS92. The study further offered novel and expeditious synthetic route allowed for rapid diversification of the core scaffold and identification of compounds (X and XI, Figure 5) possessing higher potency over Lck with IC50 values of 0.009 and 0.036 µM, respectively. However, lack of selectivity was found; X and XI showed Src IC50 values of 0.045 and 0.914 µM, and Ack1 IC50 values of 0.098 and 0.078 µM, respectively. Further exploration of new 2,3-diarylfuro[2,3-b]pyridin-4-amines by Martin et al. offered some derivatives with promising potency but the lack of selectivity and the non-optimal pharmacokinetic properties limited the research efforts in this area93. Martin et al. reported another series of 2-aminopyrimidine carbamates as a new class of compounds with potent and selective inhibition of Lck. The most promising compound of this series (XII, Figure 5) exhibits good activity when evaluated in an in vivo model of T cell activation. It showed an IC50 value of 0.0006 µM over Lck with an interesting selectivity profile (Src IC50 = 0.001 µM, Kdr IC50 = 0.14 µM, Syk IC50 = 0.20 µM, Zap-70 IC50 = 0.37 µM, and Btk IC50 = 0.10 µM)94.
Figure 5.

Chemical structures of compounds IX–XII.
5. New horizons in drug discovery of Lck inhibitors (2011–2020)
In the last decade, novel small molecules related to new chemical scaffolds were reported to inhibit Lck offering new horizons of drug discovery in this research area. By searching literature and pharmaceutical patents, we amass these efforts in this section. In addition, SAR studies and docking models of the most promising inhibitors within Lck active site were carried out to offer deep insights of their different binding modes in a step towards development of more potent, selective and safe Lck inhibitors as promising therapy for Lck-related human diseases.
The molecular docking study of the following discussed Lck inhibitors was performed in an attempt to assist in defining and categorising the functional groups of each series (which are involved in the ligand binding and which are not detrimental in binding). Classifying these groups will determine which must be excised and which should be preserved or modified, which in turn will pave the way for the development of more potent and selective inhibitors. Guided by co-crystal structures of different ligands to their corresponding Lck domains, the key interactions in ATP pocket are determined as follow: (1) The native ligands anchored in the hinge binding adenine pocket by hydrogen bond interactions with either the NH or the carbonyl groups of the main chain of Met319 amino acid; however, some co-crystal structures showed additional hydrogen bond interaction in the adenine region with the carbonyl oxygen of Glu317 backbone, (2) The hydrophobic pocket of Lck is occupied by the ligand via Van der Waals interaction with Asp382 residue, (3) Amongst the employed crystal structures, staurosporine-Lck complex revealed deep embedding of the methylamino nitrogen of the glycoside ring in ATP ribose pocket via participation in hydrogen bond interaction with Ser323 residue, 4) Finally, the ligand is positioned in Lck gatekeeper residue via hydrogen acceptor bond with the γ–OH of Thr316 residue95,96.
The molecular docking studies was performed using Molecular Operating Environment (MOE, 2014). The X-ray crystal structures of Lck domain were downloaded from the Protein Data Bank (PDB IDs: 1QPC, 1QPJ, 2OF2, 2OFU, 2PL0, 3BYM, 3BYO, 3LCK, and 6PDJ). Amino acid sequences of all protein were protonated and their energies were minimised. The employed crystal structures were docked with their native ligands, and their RMSD values were calculated. Only four PDB IDs: 2PL0, 3BYM, 3BYO, and 6PDJ of the lowest RMSD values were selected for operating docking protocol to the discussed inhibitors 1–38 (Figures 6(A), 7(A), 8, 10, 12, 14(A), 15) aiming at evaluation of their binding scores, and determination of their crucial binding interactions within Lck active site, comparable to the native ligand of the corresponding PDB file (Table 1). As depicted in Table 1, most of the docked compounds preserved the key interaction in the hinge binding site by hydrogen bond formation with Met319; while, the hydrophobic pocket was occupied by some compounds via Van der Waals interaction with Asp382; however, the gatekeeper Thr316 H-bonded with majority of the compounds, that in turn hypothesised their selectivity to Lck kinase among Src-family kinases. The correlation between the docking findings and the variable inhibitory activities is discussed in more details for each class.
Figure 6.

(A) Chemical structures of halogenated alkaloids 1–3; (B) 3D molecular interaction docking model of compound 1 in Lck kinase domain active site (PDB ID: 3BYO) (C) 3D molecular interaction docking model of compound 3 in Lck kinase domain active site (PDB ID: 3BYO).
Figure 7.

(A) Chemical strucutre of compound 4; (B) 3D molecular interaction docking model of compound 4 in Lck kinase domain active site (PDB ID: 2PL0).
Figure 8.

Chemical structures of pyrrolopyrimidine-based Lck inhibitors 5–23.
Figure 10.

Chemical structures of triazole-based compounds 24–29.
Figure 12.

Chemical structure of Dasatinib (30) and its derivative 31.
Figure 14.

(A) Chemical structure of compound 32; (B) 3D molecular interaction docking model of compound 32 in Lck kinase domain active site (PDB ID: 3BYM).
Figure 15.

Chemical structures of phenoxypyrimidine scaffold-based Lck inhibitors 33–35.
Table 1.
Molecular docking study of compounds 1–38 in Lck kinase domain represented in 2D diagrams.
| Cpd. ID | PDB ID | Energy Score (Kcal/mol) |
2D diagram | Amino acids | Binding group | Molecular interactions |
|---|---|---|---|---|---|---|
| (Native ligand95) 6-(2,6-dimethylphenyl)-2-((4-(4-methyl-1-piperazinyl)phenyl)amino)pyrimido[5′,4′:5,6] pyrimido-[1,2-a]benzimidazol-5(6H)-one |
3BYO | −8.29 | 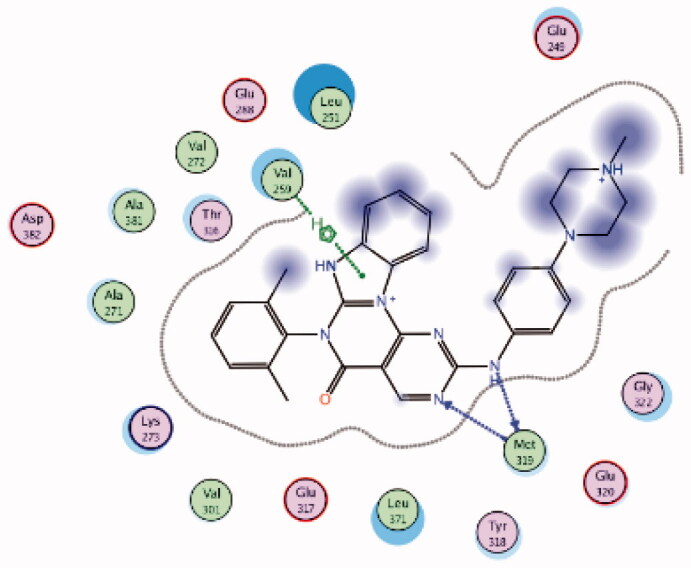 |
Met319 | Pyrimidine (N)-NH | H-bond |
| Val259 | Imidazole ring | Arene-H | ||||
| 1 | 3BYO | −5.57 | 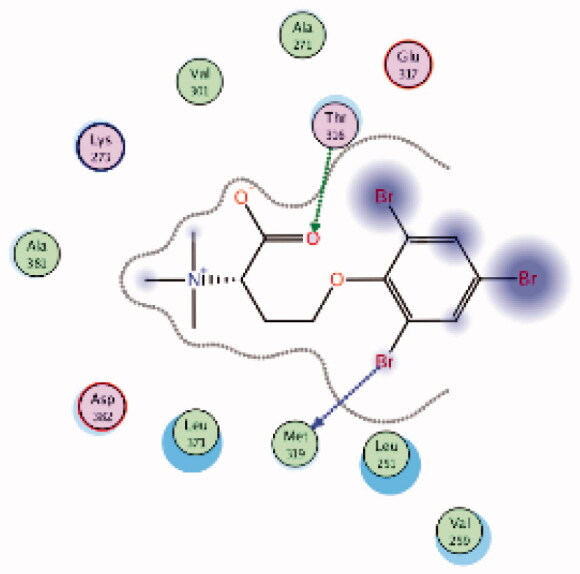 |
Met319 | Phenoxy (Br) | H-bond |
| Thr316 | COOH (C = O) | H-bond | ||||
| 2 | 3BYO | −5.69 | 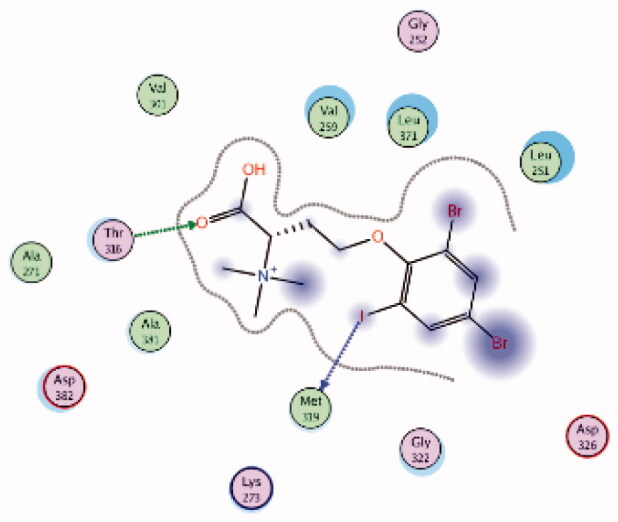 |
Met319 | Phenoxy (I) | H-bond |
| Thr316 | COOH (C = O) | H-bond | ||||
| 3 | 3BYO | −5.75 | 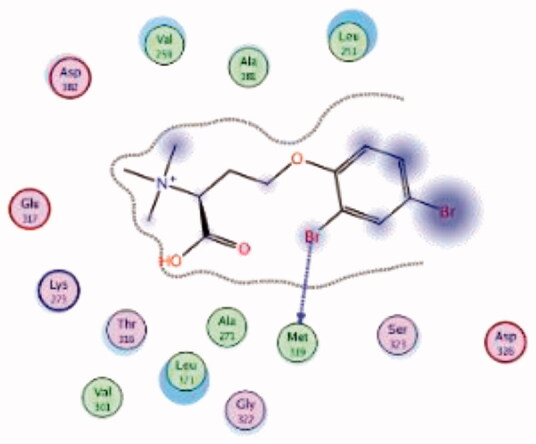 |
Met319 | Phenoxy (Br) | H-bond |
| (Native ligand97) Imatinib |
2PL0 | −10.66 | 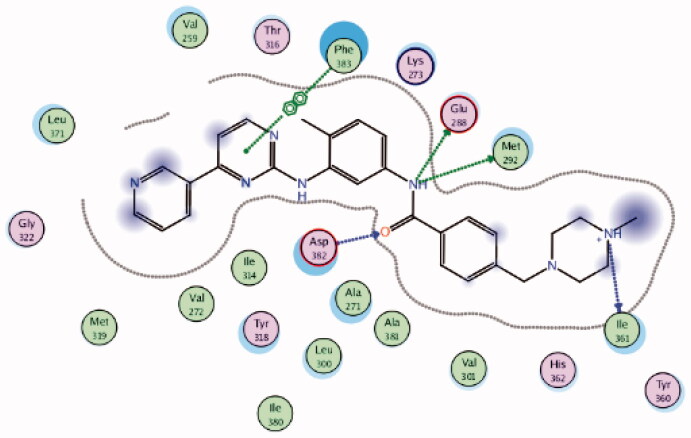 |
Asp382 | Amide-C = O | H-bond |
| Glu288 | Amide-NH | H-bond | ||||
| Ile361 | Piperazine-NH | H-bond | ||||
| Met292 | Amide-NH | H-bond | ||||
| Phe383 | Pyrimidine ring | Ar-Ar | ||||
| 4 | 2PL0 | −8.36 | 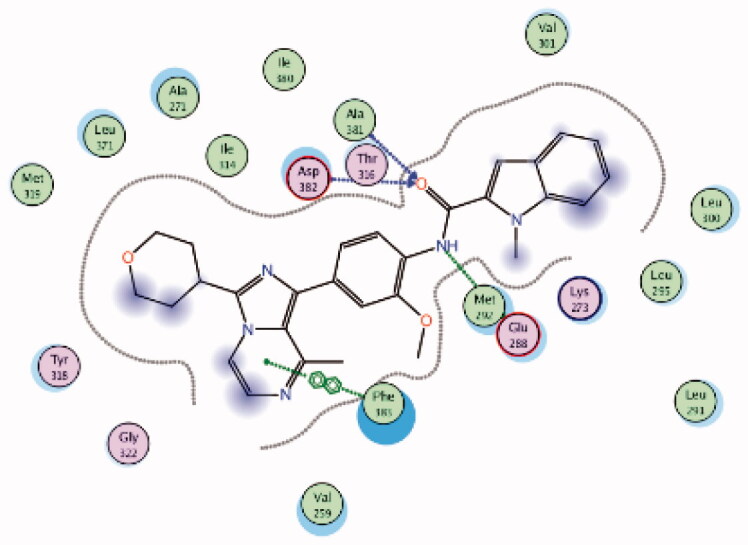 |
Ala381 | Amide-C = O | H-bond |
| Asp382 | Amide-C = O | H-bond | ||||
| Met292 | Amide-NH | H-bond | ||||
| Phe383 | Pyrimidine ring | Ar-Ar | ||||
| (Native ligand95) N-phenyl-1-(4-((3,4,5-trimethoxyphenyl)amino)-1,3,5-triazin-2-yl)-1H-benzo[d]imidazol-2-amine |
3BYM | −8.55 | 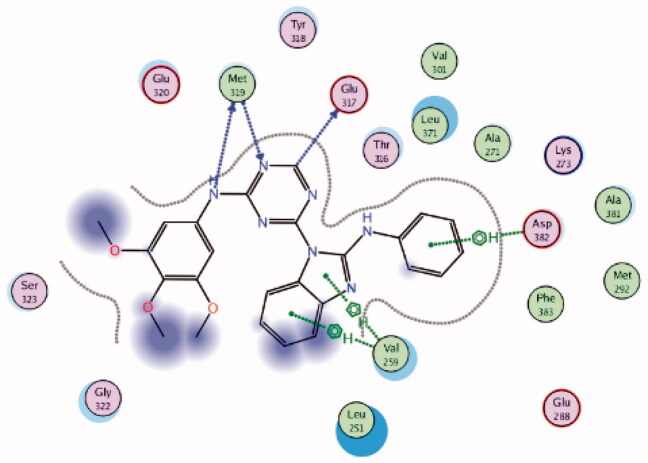 |
Asp382 | Phenyl ring | Arene-H |
| Glu317 | Triazine-CH | H-bond | ||||
| Met319 | Triazine (N)-NH | H-bond | ||||
| Val259 | benzo[d]imidazole | Arene-H | ||||
| 5 | 3BYM | −6.63 | 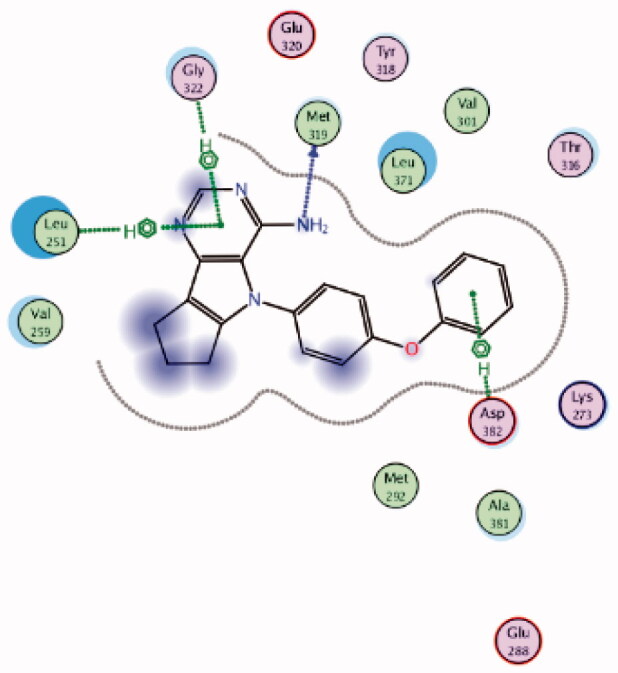 |
Asp382 | Phenoxy ring | Arene-H |
| Gly322 | Pyrimidine ring | Arene-H | ||||
| Leu251 | Pyrimidine ring | Arene-H | ||||
| Met319 | NH2 | H-bond | ||||
| 6 | 3BYM | −5.98 | 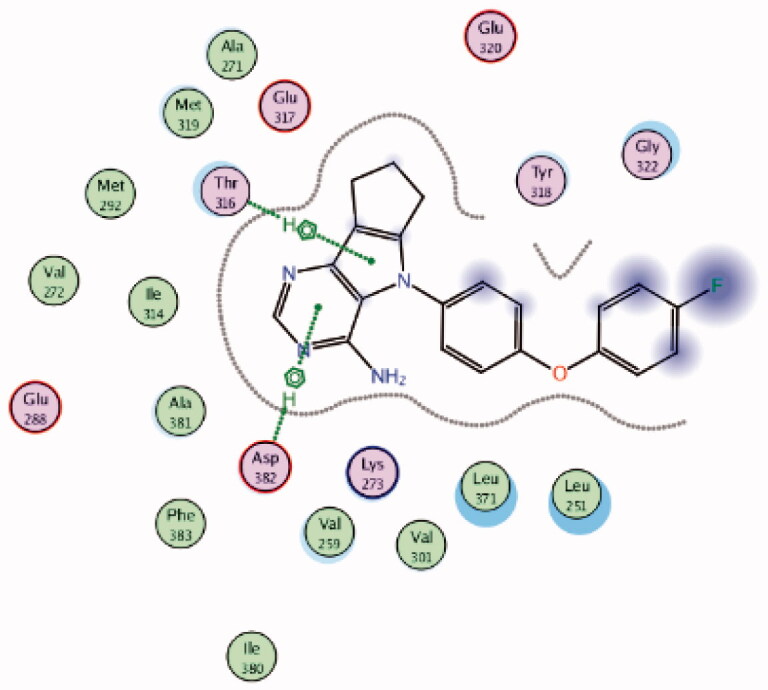 |
Asp382 | Pyrimidine ring | Arene-H |
| Thr316 | Pyrrole ring | Arene-H | ||||
| 7 | 3BYM | −6.57 | 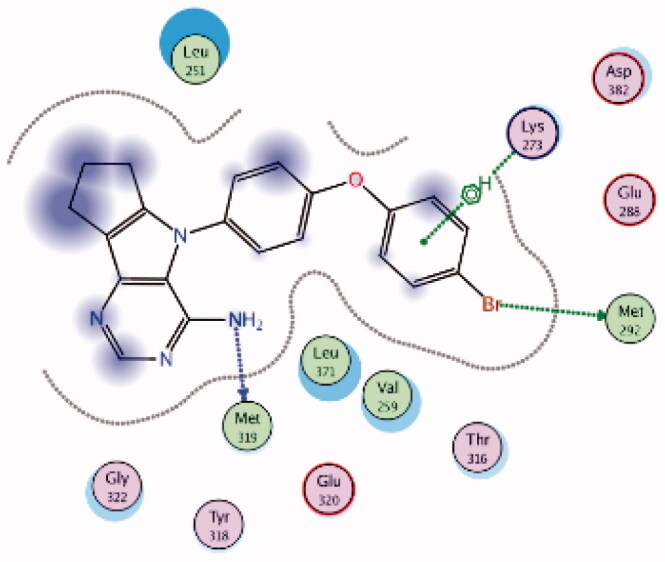 |
Lys273 | 4-Br Phenoxy ring | Arene-H |
| Met292 | 4-Br Phenoxy (Br) | H-bond | ||||
| Met319 | NH2 | H-bond | ||||
| 8 | 3BYM | −6.46 | 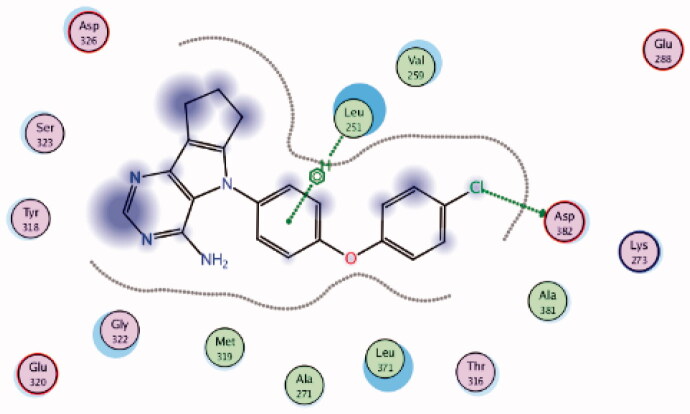 |
Asp382 | 4-Cl Phenoxy (Cl) | H-bond |
| Leu251 | Phenyl ring | Arene-H | ||||
| 9 | 3BYM | −6.66 | 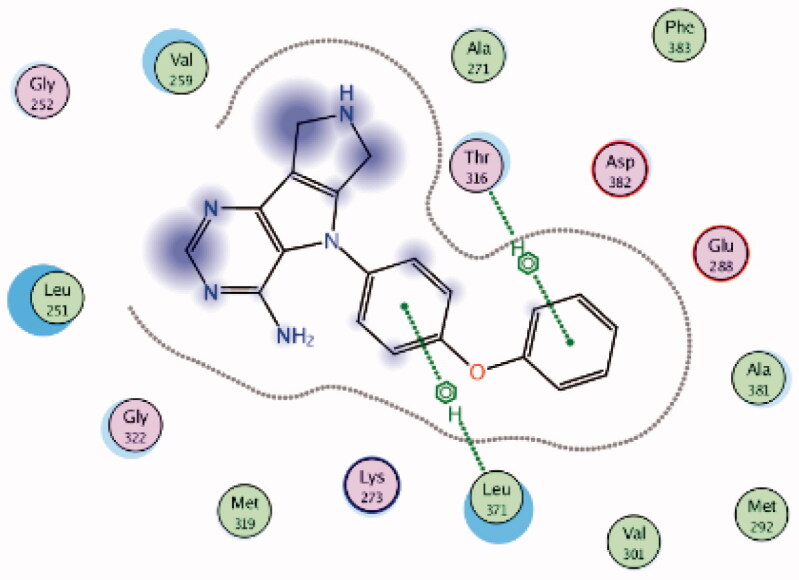 |
Leu371 | Phenyl ring | Arene-H |
| Thr316 | Phenoxy ring | Arene-H | ||||
| 10 | 3BYM | −7.54 | 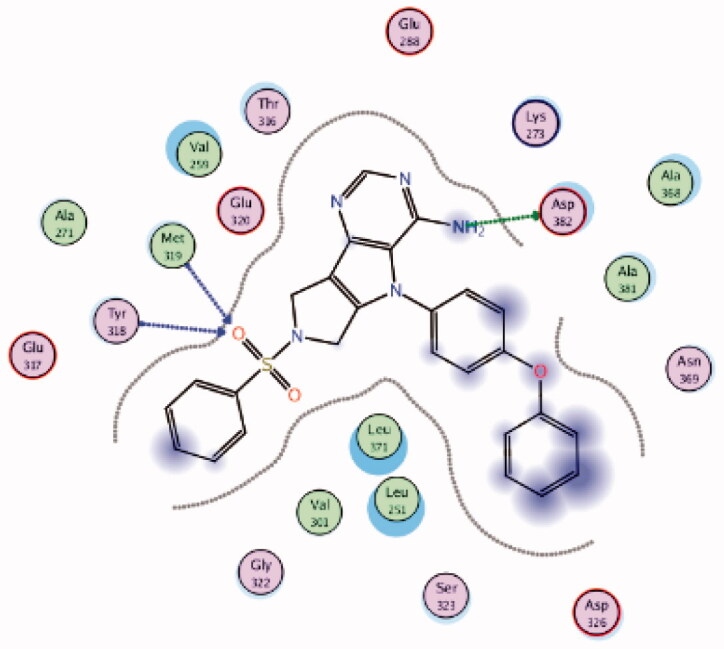 |
Asp382 | NH2 | H-bond |
| Met319 | SO2 | H-bond | ||||
| Tyr318 | SO2 | H-bond | ||||
| 11 | 3BYM | −6.89 | 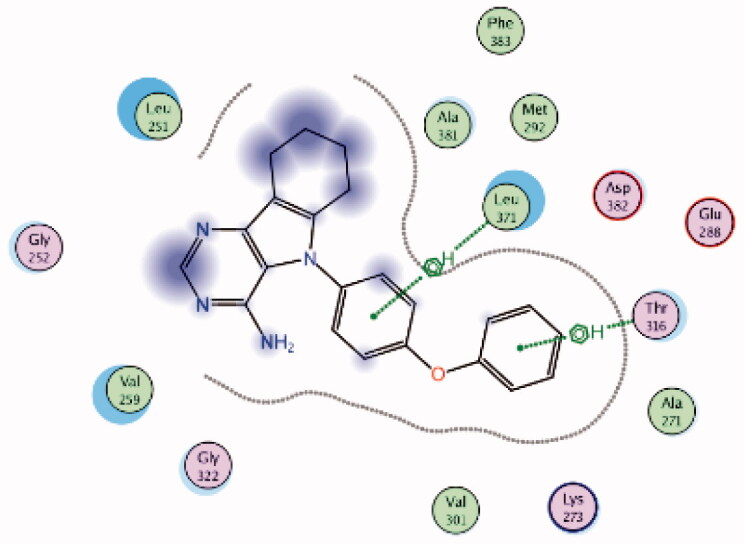 |
Leu371 | Phenyl ring | Arene-H |
| Thr316 | Phenoxy ring | Arene-H | ||||
| 12 | 3BYM | −6.66 | 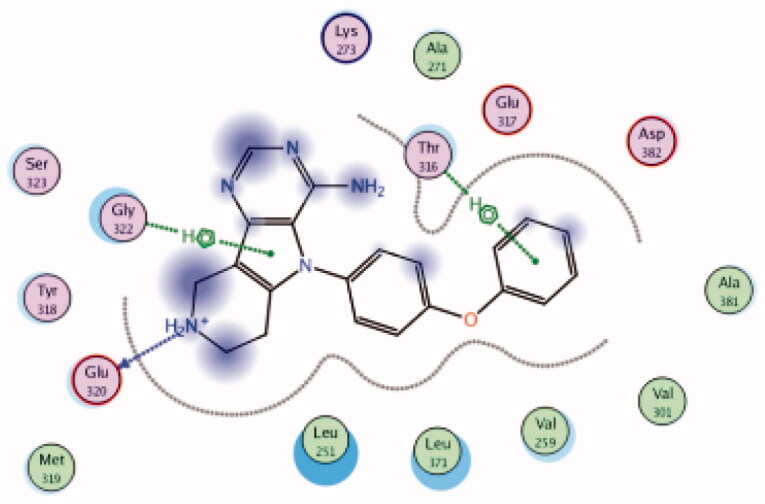 |
Glu320 | Piperidine (NH) | H-bond |
| Gly322 | Pyrrole ring | Arene-H | ||||
| Thr316 | Phenoxy ring | Arene-H | ||||
| 13 | 3BYM | −7.71 | 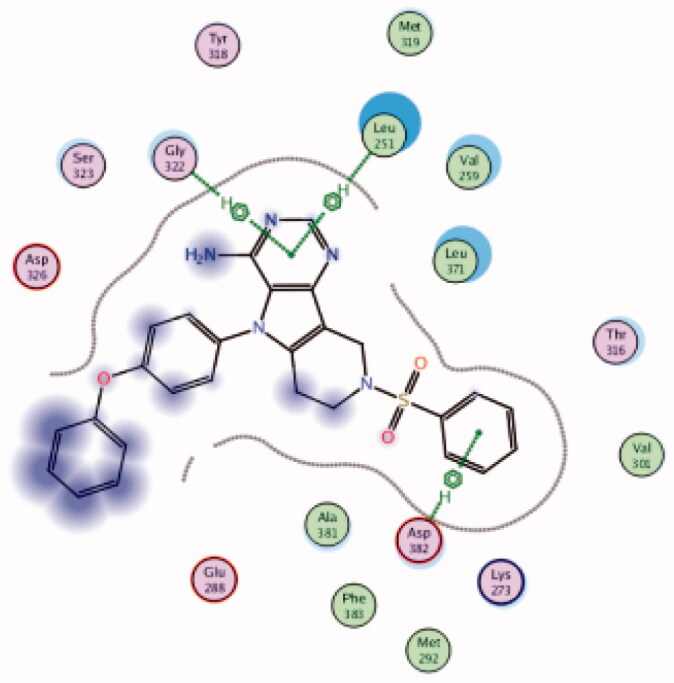 |
Asp382 | Phenyl ring | Arene-H |
| Gly322 | Pyrimidine ring | Arene-H | ||||
| Leu251 | Pyrimidine ring | Arene-H | ||||
| 14 | 3BYM | −5.69 | 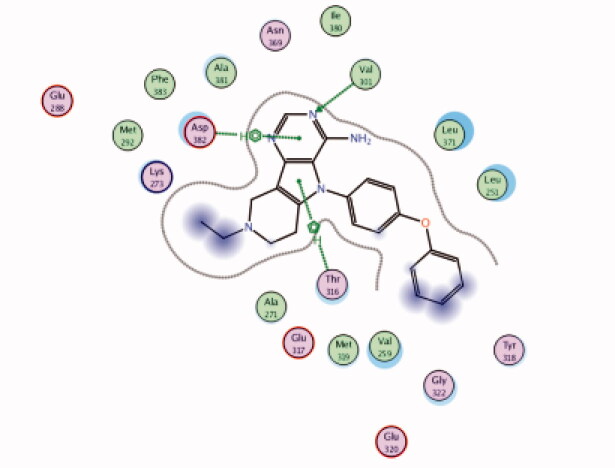 |
Asp382 | Pyrimidine ring | Arene-H |
| Thr316 | Pyrrole ring | Arene-H | ||||
| Val301 | Pyrimidine (N) | H-bond | ||||
| 15 | 3BYM | −6.38 | 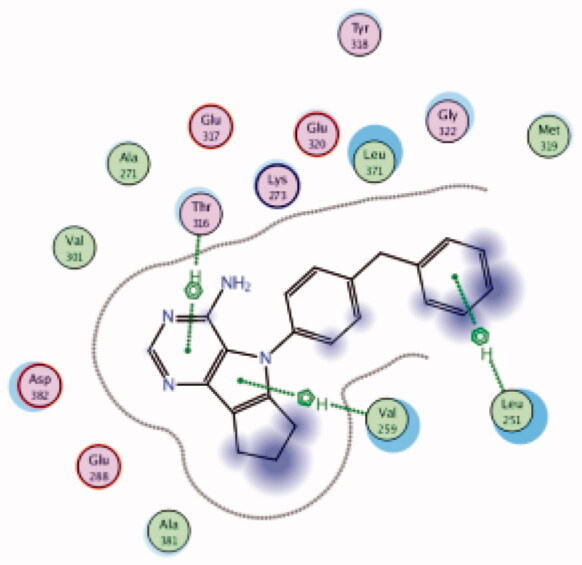 |
Leu251 | Benzyl group | Arene-H |
| Thr316 | Pyrimidine ring | Arene-H | ||||
| Val259 | Pyrrole | Arene-H | ||||
| 16 | 3BYM | −6.64 | 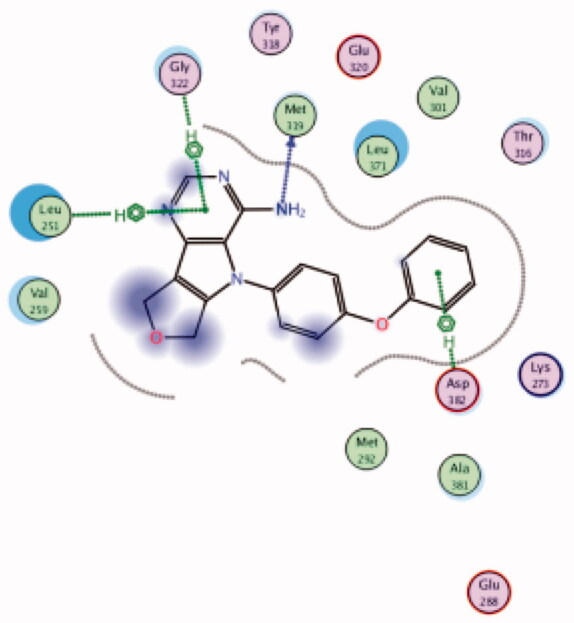 |
Asp382 | Phenyl ring | Arene-H |
| Gly322 | Pyrimidine ring | Arene-H | ||||
| Leu251 | Pyrimidine ring | Arene-H | ||||
| Met319 | NH2 | H-bond | ||||
| 17 | 3BYM | −6.44 | 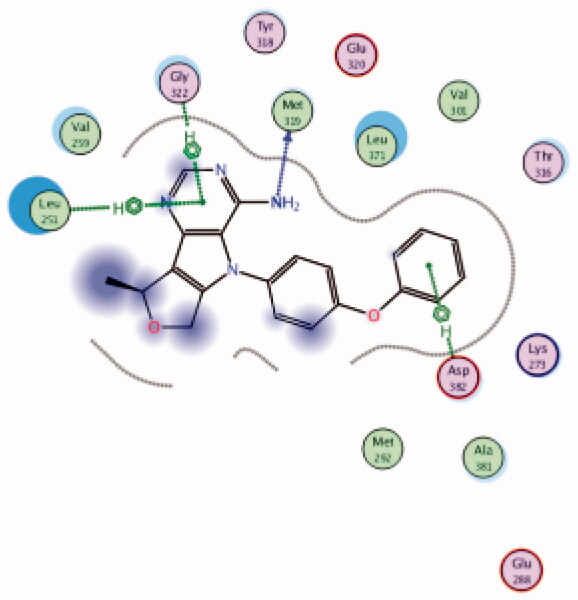 |
Asp382 | Phenoxy ring | Arene-H |
| Gly322 | Pyrimidine ring | Arene-H | ||||
| Leu251 | Pyrimidine ring | Arene-H | ||||
| Met319 | NH2 | H-bond | ||||
| 18 | 3BYM | −6.97 | 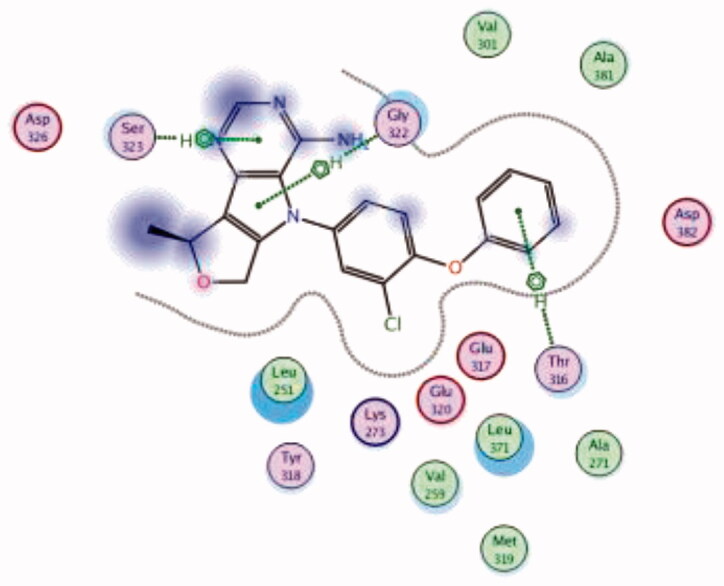 |
Gly322 | Pyrrole ring | Arene-H |
| Ser323 | Pyrimidine ring | Arene-H | ||||
| Thr316 | Phenoxy ring | Arene-H | ||||
| 19 | 3BYM | −6.61 | 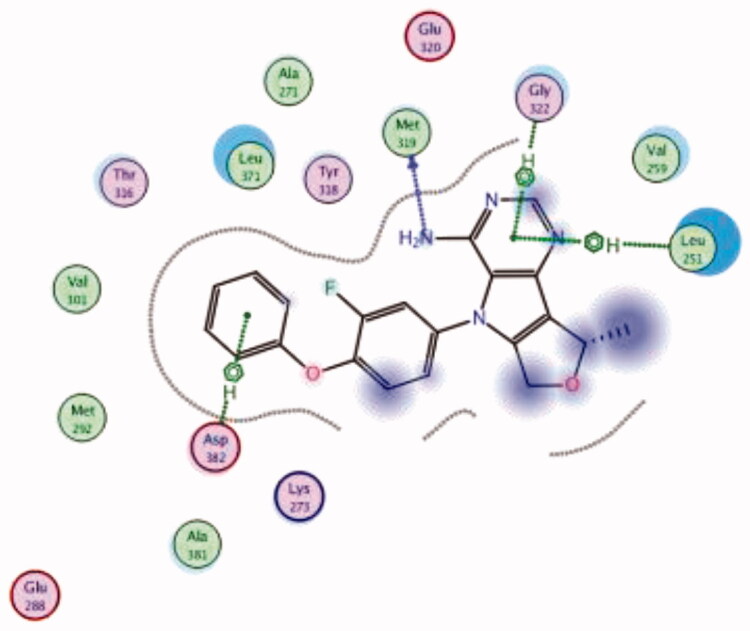 |
Asp382 | Phenoxy ring | Arene-H |
| Gly322 | Pyrimidine ring | Arene-H | ||||
| Leu251 | Pyrimidine ring | Arene-H | ||||
| Met319 | NH2 | H-bond | ||||
| 20 | 3BYM | −6.62 | 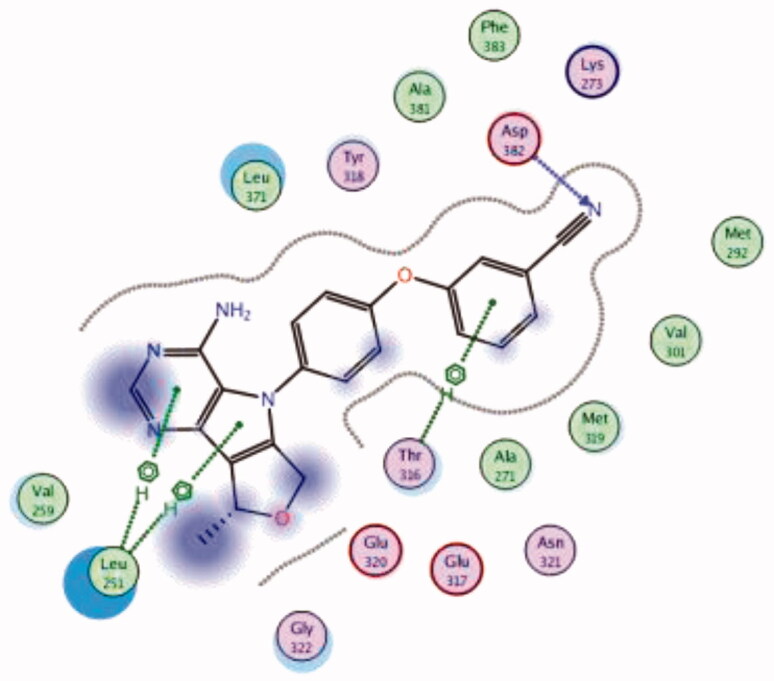 |
Asp382 | 3-CN phenoxy (CN) | H-bond |
| Leu251 | Pyrrolo pyrimidine | Arene-H | ||||
| Thr316 | Phenyl ring | Arene-H | ||||
| 21 | 3BYM | −6.50 | 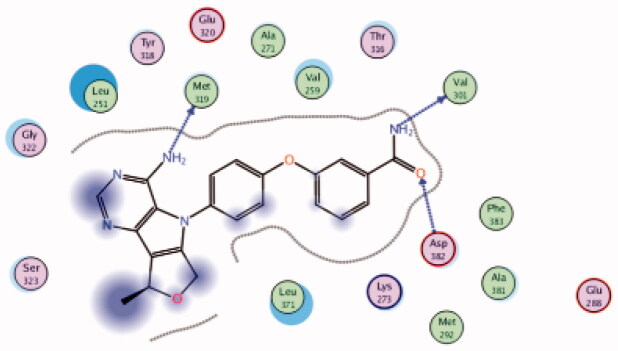 |
Asp382 | Amide-(C = O) | H-bond |
| Met319 | NH2 | H-bond | ||||
| Val301 | Amide-(NH2) | H-bond | ||||
| 22 | 3BYM | −6.70 | 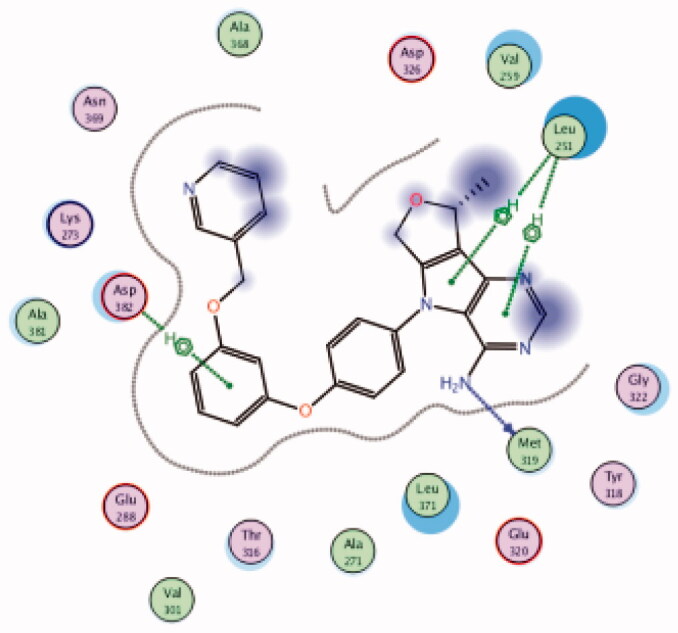 |
Asp382 | Phenyl ring | Arene-H |
| Leu251 | Pyrrolo pyrimidine | Arene-H | ||||
| Met319 | NH2 | H-bond | ||||
| 23 | 3BYM | −6.81 | 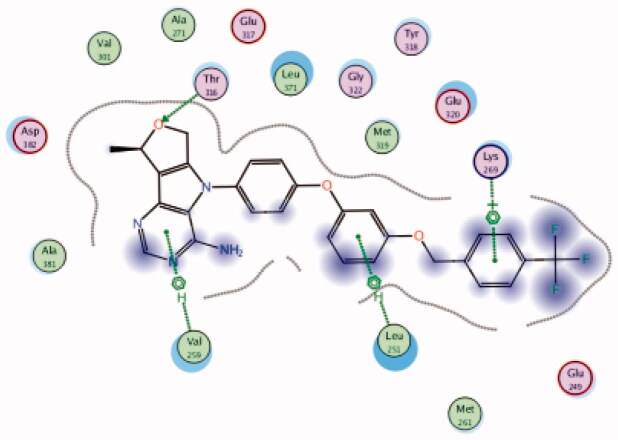 |
Leu251 | Phenoxy ring | Arene-H |
| Lys269 | 4-CF3 phenyl group | Arene-H | ||||
| Thr316 | Furan (O) | H-bond | ||||
| Val259 | Pyrimidine ring | Arene-H | ||||
| 24 | 3BYM | −5.86 | 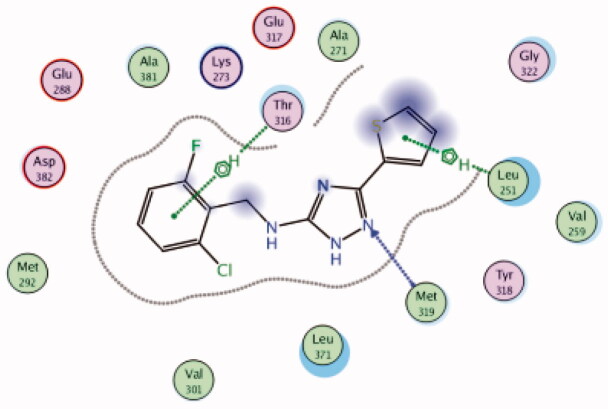 |
Leu251 | Thiophene ring | Arene-H |
| Met319 | Triazole (N) | H-bond | ||||
| Thr316 | 2-Cl,6-F-phenyl group | Arene-H | ||||
| 25 | 3BYM | −6.17 | 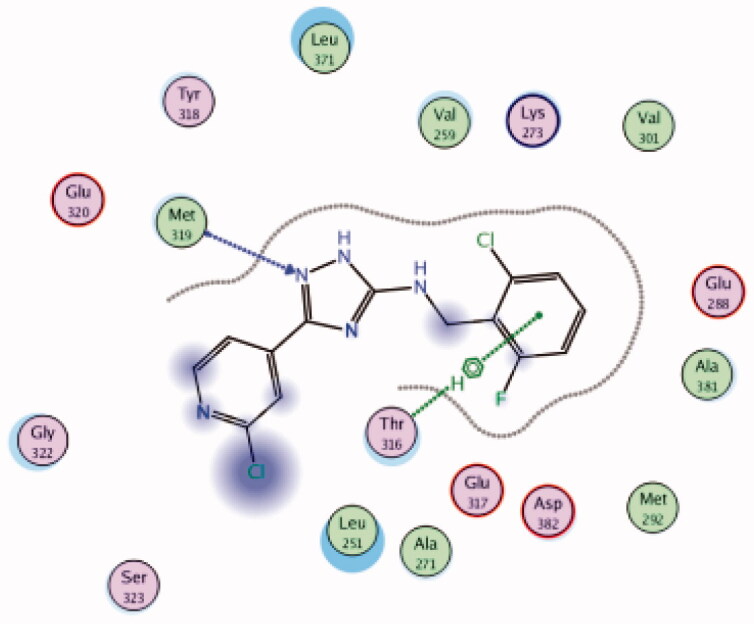 |
Met319 | Triazole (N) | H-bond |
| Thr316 | 2-Cl,6-F-phenyl group | Arene-H | ||||
| 26 | 3BYM | −6.57 | 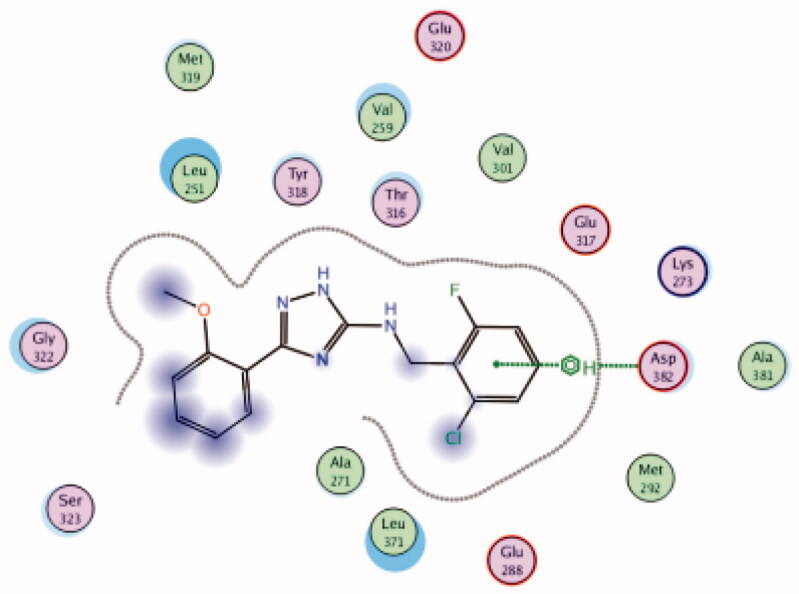 |
Asp382 | 2-Cl,6-F-phenyl group | Arene-H |
| 27 | 3BYM | −6.87 | 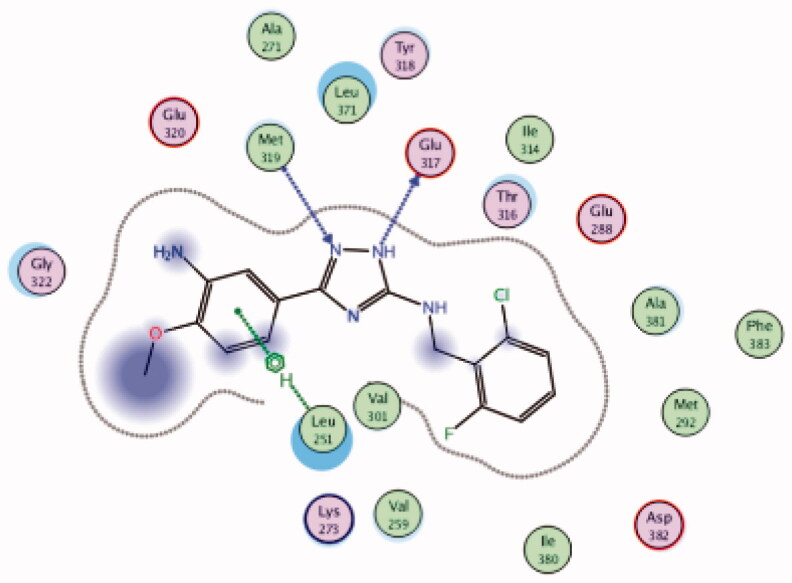 |
Glu317 | Triazole (NH) | H-bond |
| Leu251 | 3-NH2, 4-OMe phenyl group | Arene-H | ||||
| Met319 | Triazole (N) | H-bond | ||||
| 28 | 3BYM | −6.34 | 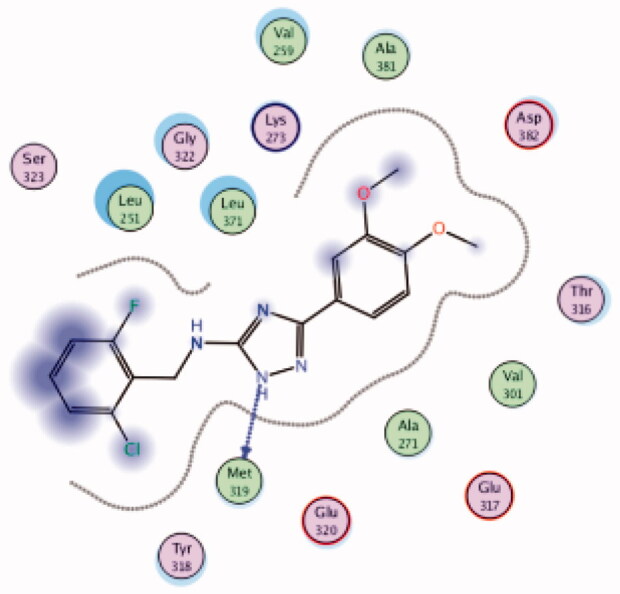 |
Met319 | Triazole (N) | H-bond |
| 29 | 3BYM | −6.16 | 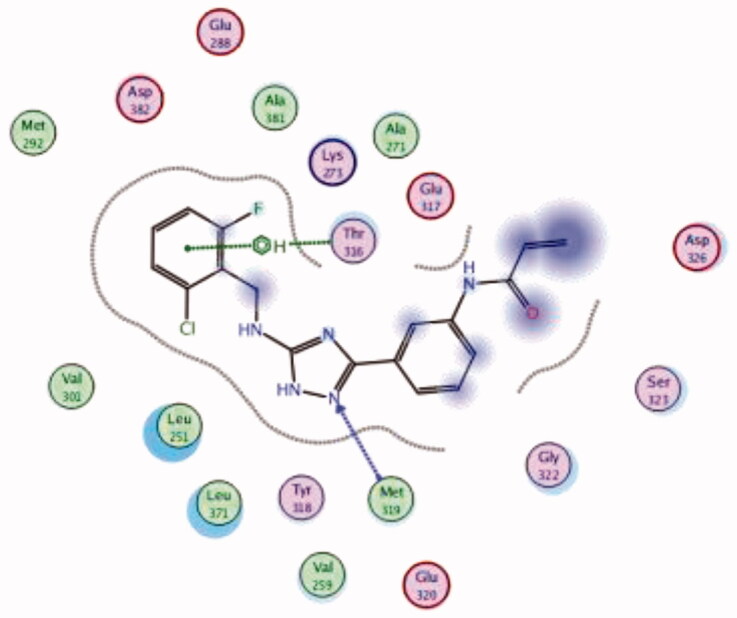 |
Met319 | Triazole (N) | H-bond |
| Thr316 | 2-Cl,6-F-phenyl group | Arene-H | ||||
| 30 | 3BYM | −7.00 | 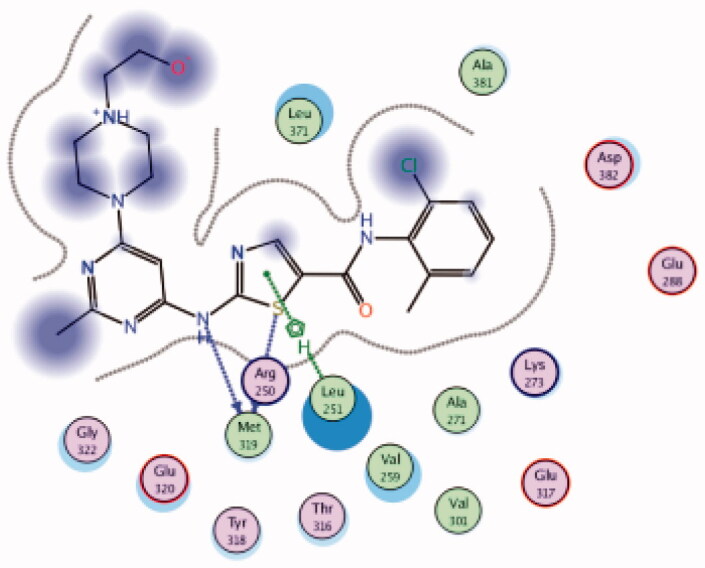 |
Leu251 | Thiazole ring | Arene-H |
| Met319 | Thiazole (S) | H-bond | ||||
| NH | H-bond | |||||
| 31 | 3BYM | −7.63 | 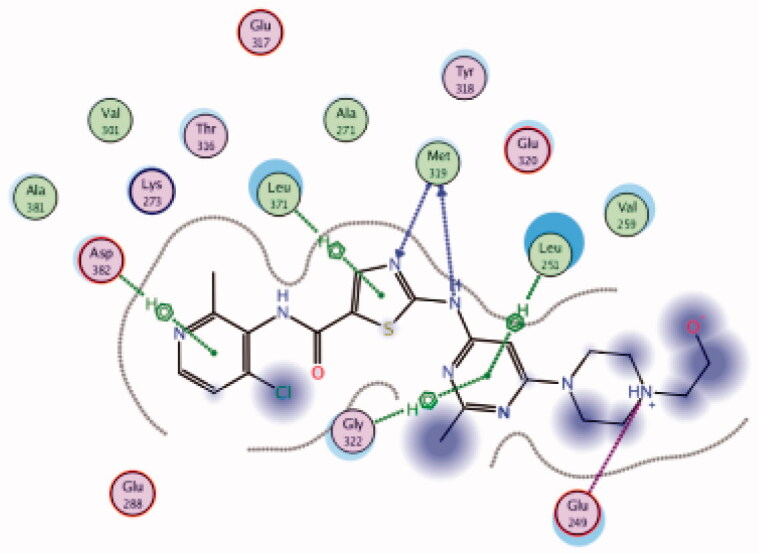 |
Asp382 | Pyridine ring | Arene-H |
| Glu249 | Piperazine (NH) | Metal/Ione | ||||
| Gly322 | Pyrimidine ring | Arene-H | ||||
| Leu251 | Pyrimidine ring | Arene-H | ||||
| Leu371 | Thiazole ring | Arene-H | ||||
| Met319 | Thiazole (N) | H-bond | ||||
| NH | H-bond | |||||
| 32 | 3BYM | −6.98 | 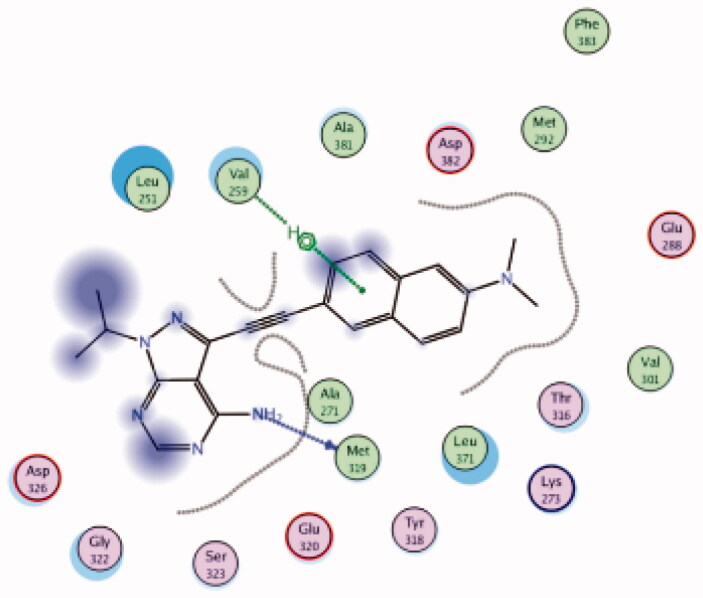 |
Met319 | NH2 | H-bond |
| Val259 | Naphthyl group | Arene-H | ||||
| (Native ligand98) N-(4-(6-methoxypyrazolo[1,5-a]pyridine-3-carboxamido)-3-methylphenyl)-1-methyl-1H-indazole-3-carboxamide (37) |
6PDJ | −11.39 | 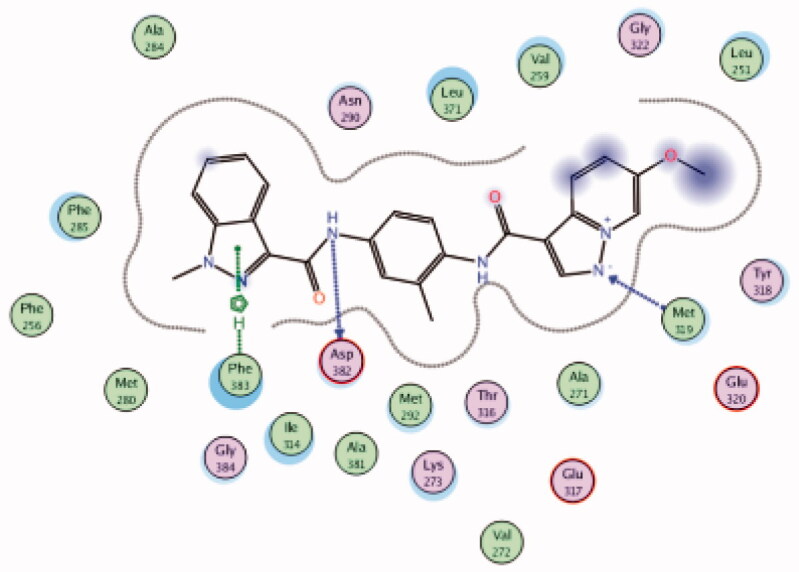 |
Asp382 | Amide-C = O | H-bond |
| Met319 | Pyrazole (N) | H-bond | ||||
| Phe283 | Pyrazole ring | Arene-H | ||||
| 33 | 6PDJ | −7.59 | 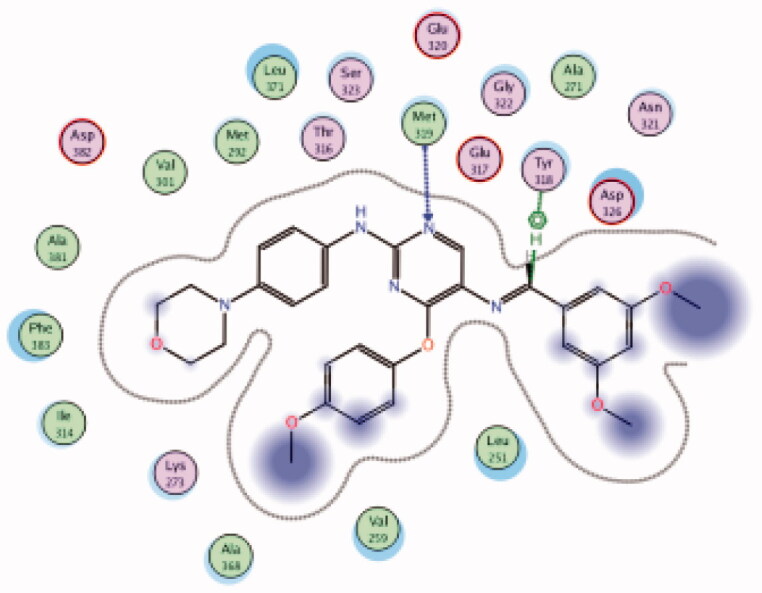 |
Met319 | Pyrimidine (N) | H-bond |
| Tyr318 | CH | Arene-H | ||||
| 34 | 6PDJ | −7.95 | 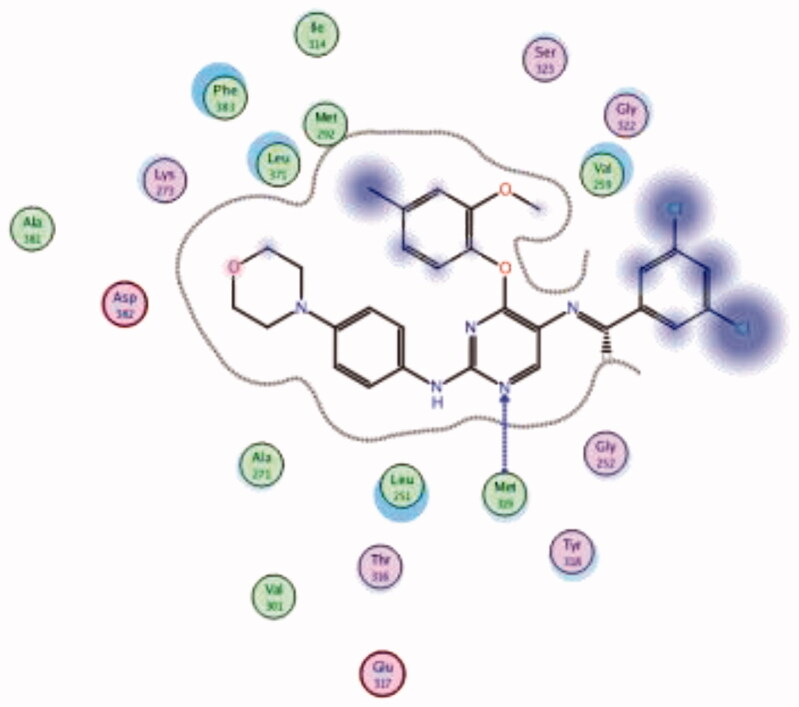 |
Met319 | Pyrimidine (N) | H-bond |
| 35 | 6PDJ | −7.17 | 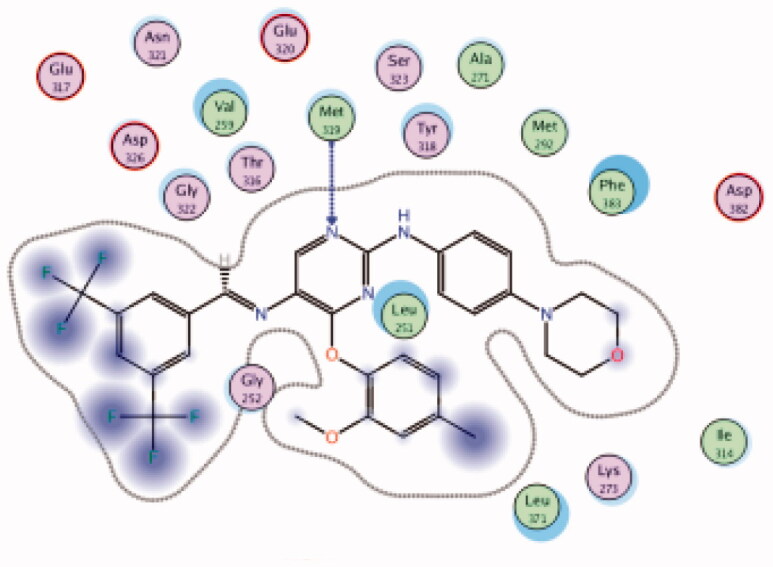 |
Met319 | Pyrimidine (N) | H-bond |
| 36 | 6PDJ | −6.52 | 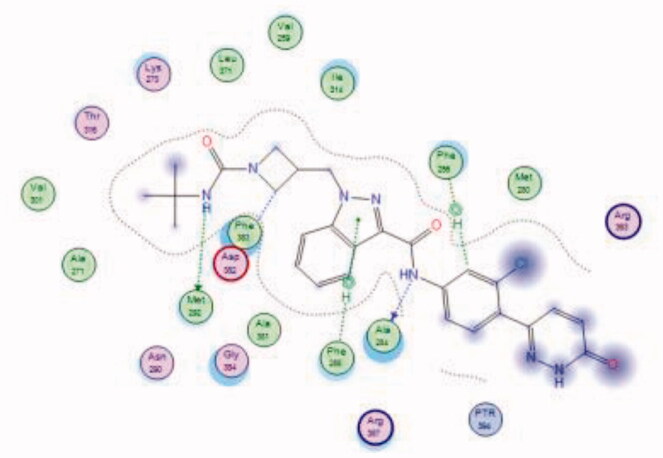 |
Ala284 | Amide (NH) | H-bond |
| Asp382 | Azetidine (CH) | Arene-H | ||||
| Met292 | Urea (NH) | H-bond | ||||
| Phe285 | Indazole (Pyrazole ring) | Arene-H | ||||
| Phe256 | 3-Cl phenyl group | Arene-H | ||||
| 38 | 6PDJ | −9.50 | 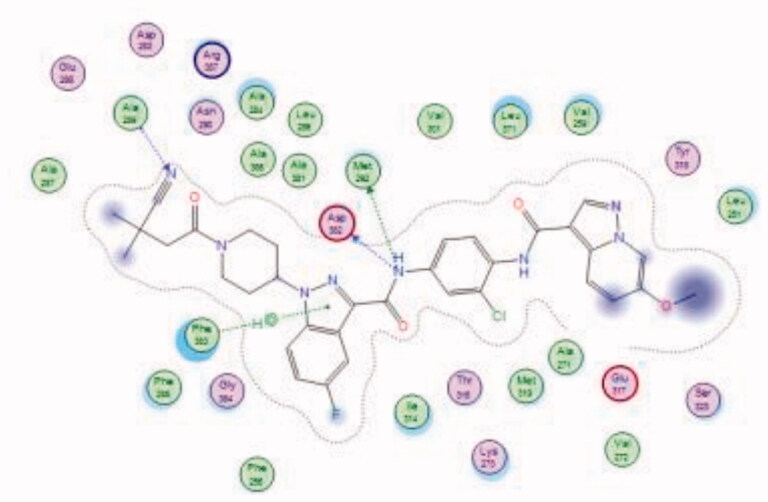 |
Ala289 | CN group | H-bond |
| Asp382 | Amide (NH) | H-bond | ||||
| Met292 | Indazole (Pyrazole ring) | H-bond | ||||
| Phe383 | Amide (NH) | Arene-H |
5.1. Halogenated alkaloids
HPLC-ESIMS (High-performance liquid chromatography combined with electrospray mass spectrometry) guiding fractionation of the sponge I. purpurea resulted in the isolation of ten polyhalogenated alkaloids (Purpuroine A–J)99. The newly isolated purpuroines were assayed for their antibiotic and kinase inhibition activities. Although the initial assays were limited to a small panel of three different kinases including Lck, cyclin-dependent kinase 2 (CDK2), and polo-like kinase 1 (PLK1), purpuroines A (1) and D (2) (Figure 5(A)) showed potent inhibitory activity against Lck kinase with IC50 values of 2.35 and 0.94 µg/mL, respectively. Purpuroine D was also found to inhibit PLK1 with an IC50 value of 1.45 µg/mL. As a reference, staurosporine (a broad-spectrum protein kinase inhibitor) exhibited IC50 values of 3.73 and 0.92 µg/mL over Lck and PLK1, respectively. All purpuroines displayed weak inhibition to CDK2 (IC50 > 50 µg/mL). The primary SAR analysis of the trihalogen substituted analogs including the most potent compound (purpuroine D) presented their ability to show more inhibitory activity against Lck than the dihalogentaed analogs as in purpuroine B (3, Figure 6(A)). A molecular docking simulation was performed to get more insights about the different binding modes of this series within the Lck active site and to understand the possible reason(s) behind the difference in their biological activities. The docking study indicated that compounds 1 (Figure 6(B)) and 2 possessing tri-halogenated phenoxy group were deeply embedded in the hinge binding region via formation of H-bond with Met319 residue, leading to orientation of molecule’s lateral carboxylic acid group towards H-bonding with Thr316. On the other hand, the di-halogenated phenoxy in compound 3 (Figure 6(C)) is H-bonded through the bromo group with Met319, even though, the molecule didn’t show any additional H-bonds with amino acid residues in the adenine binding area.
5.2. 8-Methyl-1-phenyl-imidazo[l,5-a]pyrazines
Using Lck IMAP assay, design of a new series of 8-methyl-1-phenyl-imidazo[l,5-a]pyrazines as Lck inhibitors resulted in the discovery of novel Lck inhibitory derivatives with a wide range of pIC50 values against Lck (≥6 – ≥8)100. Compound 4 (Figure 7(A)) is reported as one among many derivatives exhibited a potent inhibitory activity over Lck with pIC50 value ≥8. Docking of compound 4 (Figure 7(B)) in the active site of Lck (PDB ID: 2PL0) illustrated a similar binding behaviour of imatinib (selective inhibitor of Lck among Src-family kinases); the amide linker of compound 4 conserved the essential hinge binding interactions with the back bone of Met292 and Glu288 amino acids via H-bond formation with NH, while the carbonyl part H-bonds with the NH of both Ala381and Asp381 residues. Moreover, the imidazopyrimidine moiety occupied the hydrophobic pocket and involved in Van der Waals interactions with Phe383.
5.3. Pyrrolopyrimidines
Novel pyrrolopyrimidine-based Lck inhibitors were patented by Laurent et al. from the Canadian pharmaceutical company Pharmascience Inc.101 At the molecular level, the kinase inhibitory activity (expressed as Ki values) of the newly synthesised compounds was assessed against Lck and Bruton’s tyrosine kinase (Btk). Using splenic cell proliferation assay, EC50 values (50% proliferation in the presence of compound as compared to vehicle treated controls) were also determined at the cellular level. As illustrated in Figure 8, nineteen compounds belonging to five different general structures were selected to elucidate the SAR of this new series (Table 2).
Table 2.
Biological activity of compounds 5–23 over Lck.
| Cpd | Ki Lck (nM) |
|---|---|
| 5, 14, 15, 16, 17, 18, 19, 22, and 23 | <100 |
| 7, 8, 9, 10, 11, 12, and 13 | >100 – <1000 |
| 6, 20, and 21 | >1000 |
It was noted that compounds 5–8 possessing cyclopentene ring exhibited a wide range of Lck inhibition; while compound 5 possessing unsubstituted phenoxy moiety exhibited a potent inhibition constant (Ki value < 100 nM), compounds 7 and 8 with bromo and chloro substituted phenoxy, respectively, exhibited higher Ki values (> 100 – < 1000 nM). A total loss of the nanomolar activity was found in case of compound 6 possessing p-fluorophenoxy moiety (Ki value > 1000 nM). Alteration of the cyclopentene ring into the 5-membered (un)substituted 2,5-dihydro-1H-pyrrole (9 and 10) and ring expansion into the 6-membered cyclohexene (11) and 1,2,3,6-tetrahydropyridine (12) retrieved the modest activity (Ki value > 100 – < 1000 nM). While substitution of the free NH in 1,2,3,6-tetrahydropyridine ring with benzenesulphonyl moiety (13) did not improve this modest activity, substitution with a small size ethyl group (14) greatly increased the inhibitory activity against Lck (Ki value < 100 nM). Retrieving the 5-membered cyclopentene moiety along with replacement of the phenoxy moiety with benzyl resulted in compound 15 which also demonstrated a potent Lck inhibition (Ki value < 100 nM). Although an introduction of the isostere 5-membered 2,5-dihydrofuran (16) or 2-methyl-2,5-dihydrofuran (17–19) instead of the cyclopentene ring, along with keeping the phenoxy moiety, maintained the high potency, substitution of the phenoxy moiety in the meta position with cyano (20) or carboxamide group (21) resulted in loss of the nanomolar activity. Interestingly, the high potency was retrieved when the phenoxy moiety was substituted in the meta position with pyridin-3-ylmethoxy group (22) and 4-(trifluoromethyl)benzyloxy group (23). Molecular modelling studies in the active site of Lck (PDB ID: 3BYM) were carried out to understand the superiority in activity of the 5-membered rings (cyclopentene, 2,5-dihydrofuran, and 2-methyl-2,5-dihydrofuran ring) over the 6-membered ring 1,2,3,6-tetrahydropyridine, in addition, to figure out the role of the meta position substitution of the phenoxy moiety in the biological activity over Lck.
As demonstrated in Table 1, the docked derivatives exhibited variable interaction modes; however, the most potent inhibitors exhibited the highest affinity to the enzyme active site. For instance, compound 5 (Figure 9(A)) embedded deeply via multiple interactions within the pocket residues. Compound 5 anchored to the adenine area by H-bond with Met319 residue, also, the un-substitution on the phenoxy moiety allowed its deep interaction into the hydrophobic pocket via Arene-H bond with Asp382 back chain, while, the pyrrolopyrimidine scaffold contributed in holding the compound in this position by hydrophobic interaction with Gly322 and Leu251 amino acid residues. In contrary, p-fluoro substitution on the phenoxy group in compound 6 (Figure 9(B)) flipped the compound in the active site and resulted in moving the amino group away from the hinge binder which is supposed to badly affect the compound stability in the enzyme active site and reduce its activity. However, the observed moderate activity upon replacement of the cyclopentene ring into substituted 2,5-dihydro-1H-pyrrole in compound 10 (Figure 9(C)) could be explained due to the contribution of the substituted sulphonyl (SO2) group in two H-bonds with Met319 in the hinge binder and Thr316 in the hydrophobic pocket.
Figure 9.

3D molecular interaction docking models of compound 5 (A), compound 6 (B), and compound 10 (C) in Lck kinase domain active site (PDB ID: 3BYM).
5.4. Substituted triazoles
A series of substituted triazole-based compounds were designed and synthesised as new kinase inhibitors from the national institute of biological sciences in Beijing102,103. Upon screening of forty-two compounds over a panel of seven autoimmune disease-related kinases including Lck, Btk, P38a, Fyn, Lyn, BMX, and Blk, only two compounds (24 and 25, Figure 10) exhibited highly potent and selective activities over Lck with IC50 values less than 0.1 µM.
While most of other compounds exhibited moderate activities against Lck with an IC50 range of 0.1–10 µM, it was noted that only compound 26 (Figure 10) was totally inactive over Lck (IC50 > 10 µM)102. It was also found that compounds 27, 28, and 29 (Figure 10) belonging to the same series were able to inhibit Lck in a high nanomolar IC50 range (0.077 ± 0.022, 0.018 ± 0.007, and 0.044 ± 0.02, respectively) despite their high activity against other kinases. A molecular docking study of this groupoffered insights into their different binding modes in the active site of Lck and proposed an explanation for their variable activities. The highly potent derivatives 24 (Figure 11(A)) and 25 were able to fit into the active site, where the triazole nitrogen atom is conserving H-bonding interaction with Met319 amino acid backbone in the hinge binding region, while, the lateral substituted benzylamine moiety was oriented towards the gatekeeper pocket through Arene-H interaction with Thr316 residue. The conformation of the moderately active non selective inhibitors 27–29 preserved the central triazole ring held in the adenine binding region, but hindered the benzyl moiety interaction in the hydrophobic pocket (Figure 11(C)). Compound 26 did not exhibit the fundamental binding interactions in the hinge region (Figure 11(B)).
Figure 11.

3D molecular interaction docking models of compound 24 (A), compound 26 (B), and compound 27 (C) in Lck kinase domain active site (PDB ID: 3BYM).
5.5. Dasatinib-derived Lck inhibitor
Dasatinib (30, Figure 12) is one of the tyrosine kinase inhibitors (TKIs) which has transformed the treatment of Chronic Myeloid Leukaemia (CML), with chronic-phase CML now considered a manageable chronic disease. It is an orally administered small molecule inhibitor of many tyrosine kinases at nanomolar concentrations, including BCR-ABL1, c-Kit, EphA2, platelet-derived growth factor receptor-b and the Src family of kinases (e.g. Src, Lck, Yes, Fyn)104–107. However, dasatinib which is metabolised in humans primarily by the cytochrome P450 enzyme 3A4 (CYP3A4) is also a time-dependent inhibitor of CYP3A4, accordingly, the dosage of dasatinib must be significantly decreased if the patient is concomitantly medicated with a strong CYP3A4 inhibitor such as ketoconazole, clarithromycin, and indinavir, since these drugs may increase the plasma concentration of dasatinib to unsafe levels. The administration of dasatinib should be stopped upon occurrence of myelosuppression. In addition, dasatinib causes inhibition of hERG (the human “Ether-a-go-go-Related Gene”) which is an ion channel involved in the electrical activity of the heart and the coordination of heart beating.Dasatinib also suffers from an extremely short half-life, with an overall mean terminal half-life of only 3–5 h. Accordingly, in a recent trial to develop a dasatinib-derived new inhibitor with better pharmacological and safety profile, compound 31 (Figure 12) was reported by Sennthenn et al. and found to inhibit multiple kinases including Lck with an IC50 value of 1.5 nM108.
Docking the structurally modified derivative 31 (Figure 13(B)) revealed significant changes in the compound conformation in the active site of Lck (PDB ID: 3BYM), compared to the lead compound (dasatinib, Figure 13(A)) . While the main hinge binder interaction with Met319 via the 2-aminothiazole central scaffold was conserved in the modified compound, such a small change in the terminal aromatic amide in dasatinib by the pyridinyl amide in compound 31 oriented the compound to bind deeply in the hydrophobic pocket via Arene-H interaction with Asp382. Additional molecular interactions were observed as a result of these conformational changes, the pyrimidine ring contributed by a pair of Arene-H interactions with Gly322 and Leu 251 amino acids. Also, the nitrogen atom of the lateral piperazine participated in Metal/Ione interaction with Glu249.
Figure 13.

3D molecular interaction docking models of dasatinib (30) (A) and compound 31 (B) in Lck kinase domain active site (PDB ID: 3BYM).
5.6. Prodan-derived Lck inhibitor
In an attempt to find a prodan-derived Lck inhibitor which could serve as a molecular tool for real-time intracellular studies of Lck signalling, a small ATP-competitive Lck inhibitor (32, IC50 = 124 nM, Figure 14(A)) with innate fluorescent properties has been discovered by Fleming et al. through the integration of a prodan-derived fluorophore into the pharmacophore of the kinase inhibitor109. Docking of compound 32 in the Lck active site (PDB ID: 3BYM, Figure 14(B)) revealed the pyrazolopyrimidine main scaffold to be buried in the adenine binding site by H-bonding between the amino group in the compound and Met319 backbone. While, the hydrophobic pocket was occupied by the naphthyl moiety which participated in Arene-H interaction with Val259.
5.7. Phenoxypyrimidines
A novel series of phenoxypyrimidine scaffold-based inhibitors was recently reported from Korea Institute of Science and Technology (KIST) targeting Lck and FMS kinases for inflammatory disorders110. While this study concluded the discovery of a new Lck/FMS dual inhibitor (33, Figure 15) with highly potent nanomolar IC50 values of 22.0 ± 10.0 and 4.6 ± 0.05 nM against Lck and FMS kinases, respectively, in addition to its ability to demonstrate a promising anti-inflammatory effect, compounds 34 and 35 (Figure 15) with Lck IC50 value of 0.0065 ± 0.002 and 0.006 ± 0.0005 µM, respectively, were found to be the most potent Lck inhibitors in this series.
Molecular docking of the synthesised phenoxypyrimidine derivatives 33–35 in the Lck active site (PDB ID: 6PDJ, Figure 16), revealed the fundamental role of 2-aminopyrimidine core in stabilising the inhibitors in the active site of the enzyme. The nitrogen atom of the pyrimidine acted as a H-bond acceptor and kept the molecules in the hinge binding region by forming a H-bond interaction with Met319. Furthermore, the substituted phenoxy moiety was oriented towards the hydrophobic pocket, even though it didn’t show remarkable interactions with the amino acid residues in this area.
Figure 16.

3D molecular interaction diagrams of compound 33 (A), compound 34 (B), and compound 35 (C) in Lck kinase domain active site (PDB ID: 6PDJ).
5.8. Pyrazolo[1,5-a]pyridines
Bristol-Myers Squibb screened an internal kinase inhibitor collection which led to identify a pyridazinone lead compound (36, Figure 17) as a starting point for development of novel inhibitors of C-terminal Src Kinase to evaluate the potential of this target for an immuno-oncology therapy98. Upon a series of modifications included switching from a pyridazinone to pyrazolopyridine hinge binder, the optimised analog 37 (Figure 17) showed a promising ability to increase T cell proliferation induced by T cell receptor signalling and an excellent potential to reduce Lck phosphorylation in vivo upon oral dosing with Lck IC50 = 260 nM. The most potent compound in this series over Lck was compound 38 (Figure 17, IC50 = 26 nM) which showed 10-folds of potency compared to compound 37.
Figure 17.

Chemical structures of pyrazolo[1,5-a]pyridine-based Lck inhibitors 36–38.
The molecular interactions of the developed inhibitors were elaborated by their docking in the Lck active site (PDB ID: 6PDJ). As illustrated in Figure 18, the native ligand (37) showed fit binding in the enzyme pocket; where the pyrazolo[1,5-a]pyridine’s N1 formed a H-bond with Met319 in the hinge binding area, while, the hydrophobic pocket was occupied by the lateral indazole-3-carboxamide moiety via H-bonding with Asp382 and Arene-H interaction with Phe383. On the other hand, the pyridazinone moiety of the initial identified lead 36 was positioned away from the hinge region with no observed interactions. In addition, the indazole-3-carboxamide moiety was anchored in the hydrophobic pocket through H-bonding with Met292, and a couple of Arene-H interactions between the azetidine ring and the indazole moiety with Asp382 and Phe285, respectively. The modified potent inhibitor 38 showed the highest affinity to the binding site; the pyrazolo[1,5-a]pyridine carboxamide moiety was positioned to the hinge region, while, the indazole-3-carboxamide moiety was bound in the hydrophobic pocket where the carboxamide-NH group H-bound with both Met292 and Asp382. Also, the indazole moiety formed an Arene-H interaction with Phe285. Moreover, the long chain substitution on the indazole N1 of 38 allowed the compound to extend deeply in the pocket via formation of a H-bond between the terminal CN group and Ala289 residue.
Figure 18.

3D molecular interaction diagrams of compound 36 (A), compound 37 (B), and compound 38 (C) in Lck kinase domain active site (PDB ID: 6PDJ).
6. Conclusion
Fuelled by the recent development of kinase inhibitor small molecules as an area of intense research, Lck is well established as a promising target for the next generation of kinase inhibitors. However, due to the high homology of Lck with other members of the Src family isoforms, complications in the development of Lck inhibitors are still found. There is no doubt that off target inhibition of other Src family members has the potential to inhibit numerous essential cellular functions. Accordingly, the successful Lck inhibitory chemical scaffold must show high activity towards Lck, relatively little activity towards other Src kinases as well as a promising in vitro cell-based and in vivo data to support its further consideration as promising clinical candidate. Despite the extensive research efforts to optimise a promising Lck inhibitor possessing the above-mentioned criteria, the development of Lck specific inhibitors with good bioavailability and pharmacokinetics is still elusive. The efforts thrown in developing potent selective inhibitors are focussing on deep analysis of the targeted enzyme active site and defining its specific key interactions. Among Src kinases family, Lck has an advantage of sequence differences where Lck gatekeeper is characterised by hydrogen bonding between the γ OH of Thr316 and a H-bond acceptor group in the corresponding inhibitors. Considering this specific hydrogen bonding might help in designing Lck selective inhibitors by introducing well-positioned groups to accept H-bond from Thr316.
Molecular docking of the presented inhibitors showed variable interaction modes in Lck active site; however, following their reported SAR revealed that the key point for improving the activity is conserving the essential interactions in the Lck active site including H-bond interaction with Met319 in the hinge binder, Van der Waals interaction with Asp382 in the hydrophobic pocket, and binding to Lck gatekeeper with Thr316. Moreover, additional molecular interactions were detected by some inhibitors, which in turn boosted the inhibitors affinity and stability in Lck active site and explained their improved activity. In summary, deep understanding of the different structural interactions of inhibitor molecules with multiple closely related enzymes has the potential to provide data useful in the rational design of kinase inhibitors and the development of novel Lck inhibitors. Moreover, small‐molecule allosteric kinase inhibitors possessing the significant advantages over ATP‐competitive kinase inhibitors such as greater selectivity and lower off‐target toxicity could be the next generation of specific Lck inhibitors that can be optimised for clinical use. Thus, the efficient rational approaches for rapid discovery of new allosteric hits for Lck, as well as systematic biological assay technologies, are urgently needed.
CRediT authorship contribution statement
Ahmed Elkamhawy: Conceptualisation, Methodology, Writing-original draft, and Data curation.
Eslam M.H. Ali: Visualisation, Software, Writing-modelling section.
Kyeong Lee: Supervision, Funding acquisition, Review and editing.
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) [No. NRF-2018R1A5A2023127].
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Voronova AF, Sefton BM.. Expression of a new tyrosine protein kinase is stimulated by retrovirus promoter insertion. Nature 1986;319:682–5. [DOI] [PubMed] [Google Scholar]
- 2.Rohrs JA, Wang P, Finley SD.. Predictive model of lymphocyte-specific protein tyrosine kinase (LCK) autoregulation. Cell Mol Bioeng 2016;9:351–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marth JD, Peet R, Krebs EG, Perlmutter RM.. A lymphocyte-specific protein-tyrosine kinase gene is rearranged and overexpressed in the murine T cell lymphoma LSTRA. Cell 1985;43:393–404. [DOI] [PubMed] [Google Scholar]
- 4.Bommhardt U, Schraven B, Simeoni L.. Beyond TCR signaling: emerging functions of Lck in cancer and immunotherapy. Int J Mol Sci 2019;20:3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh PK, Kashyap A, Silakari O.. Exploration of the therapeutic aspects of Lck: a kinase target in inflammatory mediated pathological conditions. Biomed Pharmacother 2018;108:1565–71. [DOI] [PubMed] [Google Scholar]
- 6.Ta HQ, Jackson SR, Whitworth H, et al. Identification of a novel long noncoding RNA within the LCK gene locus that regulates prostate cancer cell growth. AACR 107th Annual Meeting 2016; 2016. Apr 16–20; New Orleans, LA. [Google Scholar]
- 7.Weiss A. T cell antigen receptor signal transduction: a tale of tails and cytoplasmic protein-tyrosine kinases. Cell 1993;73:209–12. [DOI] [PubMed] [Google Scholar]
- 8.Omri B, Crisanti P, Marty M-C, et al. The Lck tyrosine kinase is expressed in brain neurons. J Neurochem. 1996;67:1360–4. [DOI] [PubMed] [Google Scholar]
- 9.Van Tan H, Allée G, Benes C, et al. Expression of a novel form of the p56lck protooncogene in rat cerebellar granular neurons. J Neurochem 1996;67:2306–15. [DOI] [PubMed] [Google Scholar]
- 10.Omri B, Blancher C, Neron B, et al. Retinal dysplasia in mice lacking p56lck. Oncogene 1998;16:2351–6. [DOI] [PubMed] [Google Scholar]
- 11.Chen R, Chen B.. The role of dasatinib in the management of chronic myeloid leukemia. Drug Des Devel Ther 2015;9:773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu T, Wang X, Li J, et al. Identification of personalized chemoresistance genes in subtypes of basal-like breast cancer based on functional differences using pathway analysis. PloS One 2015;10:e0131183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakraborty G, Rangaswami H, Jain S, Kundu G.. Hypoxia regulates cross-talk between Syk and Lck leading to breast cancer progression and angiogenesis. J Biol Chem 2006;281:11322–31. [DOI] [PubMed] [Google Scholar]
- 14.Clarke CN, Lee MS, Wei W, et al. Proteomic features of colorectal cancer identify tumor subtypes independent of oncogenic mutations and independently predict relapse-free survival. Ann Surg Oncol 2017;24:4051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janikowska G, Janikowski T, Pyka-Pająk A, et al. Potential biomarkers for the early diagnosis of colorectal adenocarcinoma–transcriptomic analysis of four clinical stages. Cancer Biomark 2018;22:89–99. [DOI] [PubMed] [Google Scholar]
- 16.Mahabeleshwar GH, Kundu G.. Tyrosine kinase p56lck regulates cell motility and nuclear factor κB-mediated secretion of urokinase type plasminogen activator through tyrosine phosphorylation of IκBα following hypoxia/reoxygenation. J Biol Chem 2003;278:52598–612. [DOI] [PubMed] [Google Scholar]
- 17.Carrera AC, Paradis H, Borlado LR, et al. Lck unique domain influences Lck specificity and biological function. J Biol Chem 1995;270:3385–91. [DOI] [PubMed] [Google Scholar]
- 18.Ventimiglia LN, Alonso MA.. The role of membrane rafts in Lck transport, regulation and signalling in T-cells. Biochem J 2013;454:169–79. [DOI] [PubMed] [Google Scholar]
- 19.Sicheri F, Kuriyan J.. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol 1997;7:777–85. [DOI] [PubMed] [Google Scholar]
- 20.Courtney AH, Amacher JF, Kadlecek TA, et al. A phosphosite within the SH2 domain of Lck regulates its activation by CD45. Mol Cell 2017;67:498–511.e496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sjölin-Goodfellow H, Frushicheva MP, Ji Q, et al. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci Signal 2015;8:ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watts JD, Sanghera JS, Pelech S, Aebersold R.. Phosphorylation of serine 59 of p56lck in activated T cells. J Biol Chem 1993;268:23275–82. [PubMed] [Google Scholar]
- 23.Dutta D, Barr VA, Akpan I, et al. Recruitment of calcineurin to the TCR positively regulates T cell activation. Nat Immunol 2017;18:196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler DG, Park I, Kim T, et al. Phosphorylation of Ser-42 and Ser-59 in the N-terminal region of the tyrosine kinase p56lck. Proc Natl Acad Sci USA 1993;90:5176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguilera-Montilla N, Pérez-Blas M, Valeri AP, et al. Higher proliferative capacity of T lymphocytes from patients with Crohn disease than from ulcerative colitis is disclosed by use of Herpesvirus saimiri-transformed T-cell lines. Scand J Gastroenterol 2004;39:1236–42. [DOI] [PubMed] [Google Scholar]
- 26.Yan Q, Barros T, Visperas PR, et al. Structural basis for activation of ZAP-70 by phosphorylation of the SH2-kinase linker. Mol Cell Biol 2013;33:2188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thill PA, Weiss A, Chakraborty AKJM, biology c.. Phosphorylation of a tyrosine residue on Zap70 by Lck and its subsequent binding via an SH2 domain may be a key gatekeeper of T cell receptor signaling in vivo. Mol Cell Biol 2016;36:2396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo W-L, Shah NH, Ahsan N, et al. Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat Immunol 2018;19:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishii T, Warabi E, Siow RCM, Mann GE.. Sequestosome1/p62: a regulator of redox-sensitive voltage-activated potassium channels, arterial remodeling, inflammation, and neurite outgrowth. Free Radic Biol Med 2013;65:102–16. [DOI] [PubMed] [Google Scholar]
- 30.Kim E-J, Monje FJ, Li L, et al. Alzheimer’s disease risk factor lymphocyte-specific protein tyrosine kinase regulates long-term synaptic strengthening, spatial learning and memory. Cell Mol Life Sci 2013;70:743–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulivieri C, Valensin S, Majolini MB, et al. Normal B-1 cell development but defective BCR signaling in LCK–/– mice. Eur J Immunol 2003;33:441–5. [DOI] [PubMed] [Google Scholar]
- 32.Dal Porto JM, Burke K, Cambier JC.. Regulation of BCR signal transduction in B-1 cells requires the expression of the Src family kinase Lck. Immunity 2004;21:443–53. [DOI] [PubMed] [Google Scholar]
- 33.Bhullar KS, Lagarón NO, McGowan EM, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018;17:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heffron TP. Small molecule kinase inhibitors for the treatment of brain cancer. J Med Chem 2016;59:10030–10066. [DOI] [PubMed] [Google Scholar]
- 35.Chahrour O, Cairns D, Omran Z.. Small molecule kinase inhibitors as anti-cancer therapeutics. Mini Rev Med Chem. 2012;12:399–411. [DOI] [PubMed] [Google Scholar]
- 36.Nada H, Elkamhawy A, Lee K.. Structure activity relationship of key heterocyclic anti-angiogenic leads of promising potential in the fight against cancer. Molecules. 2021;26:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Sanea MM, Elkamhawy A, Paik S, et al. Abdelgawad, Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: synthesis, biological evaluation and in silico insights. Bioorg Med Chem 2020;28:115525. [DOI] [PubMed] [Google Scholar]
- 38.Elkamhawy A, Paik S, Hassan AHE, et al. Hit discovery of 4-amino-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: a novel EGFR inhibitor from a designed small library. Bioorgan Chem 2017;75:393–405. [DOI] [PubMed] [Google Scholar]
- 39.Elkamhawy A, Park JE, Cho NC, et al. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: cell line-based assay, kinase panel, molecular docking, and toxicity studies. J Enzyme Inhib Med Chem 2016;31:158–166. [DOI] [PubMed] [Google Scholar]
- 40.Elkamhawy A, Al-Sanea MM, Song C, et al. Design and synthesis of new [1,2,3]triazolo[4,5-d]pyrimidine derivatives as potential antiproliferative agents. Bull Kor Chem Soc 2015;36:1863–73. [Google Scholar]
- 41.Elkamhawy A, Farag AK, Viswanath AN, et al. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg Med Chem Lett 2015;25:5147–54. [DOI] [PubMed] [Google Scholar]
- 42.Elkamhawy A, Kim Ny, Hassan AH, et al. . Thiazolidine-2,4-dione-based irreversible allosteric IKK-β kinase inhibitors: optimization into in vivo active anti-inflammatory agents. European Journal of Medicinal Chemistry 2020;188:111955. [DOI] [PubMed] [Google Scholar]
- 43.Elkamhawy A, youn Kim N, Hassan AH, et al. . Optimization study towards more potent thiazolidine-2,4-dione IKK-β modulator: synthesis, biological evaluation and in silico docking simulation. Bioorganic Chemistry 2019;92:103261. [DOI] [PubMed] [Google Scholar]
- 44.Elkamhawy A, Hassan AH, Paik S, et al. . EGFR inhibitors from cancer to inflammation: discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorganic Chemistry 2019;86:112–8. [DOI] [PubMed] [Google Scholar]
- 45.Elkamhawy A, Kim Ny, Hassan AH, et al. . Design, synthesis and biological evaluation of novel thiazolidinedione derivatives as irreversible allosteric IKK-β modulators. European Journal of Medicinal Chemistry 2018;157:691–704. [DOI] [PubMed] [Google Scholar]
- 46.Al-Sanea M, Elkamhawy A, Zakaria A, et al. . Synthesis and in vitro screening of phenylbipyridinylpyrazole derivatives as potential antiproliferative agents. Molecules 2015;20:1031–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Till KJ, Allen JC, Talab F, et al. Lck is a relevant target in chronic lymphocytic leukaemia cells whose expression variance is unrelated to disease outcome. Sci Rep 2017;7:16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Talab F, Allen JC, Thompson V, et al. LCK is an important mediator of B-cell receptor signaling in chronic lymphocytic leukemia cells. Mol Cancer Res MCR 2013;11:541–54. [DOI] [PubMed] [Google Scholar]
- 49.Harr MW, Caimi PF, McColl KS, et al. Inhibition of Lck enhances glucocorticoid sensitivity and apoptosis in lymphoid cell lines and in chronic lymphocytic leukemia. Cell Death Differ 2010;17:1381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cazzaniga V, Bugarin C, Bardini M, et al. LCK over-expression drives STAT5 oncogenic signaling in PAX5 translocated BCP-ALL patients. Oncotarget 2015;6:1569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Accordi B, Espina V, Giordan M, et al. Functional protein network activation mapping reveals new potential molecular drug targets for poor prognosis pediatric BCP-ALL. PloS One 2010;5:e13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salmond RJ, Filby A, Pirinen N, et al. Mislocalization of Lck impairs thymocyte differentiation and can promote development of thymomas. Blood 2011;117:108–117. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Zhang X, Zhao Y, et al. Identification of potential therapeutic target genes, key miRNAs and mechanisms in acute myeloid leukemia based on bioinformatics analysis. Med Oncol 2015;32:152. [DOI] [PubMed] [Google Scholar]
- 54.Li L, Cui Y, Shen J, et al. Evidence for activated Lck protein tyrosine kinase as the driver of proliferation in acute myeloid leukemia cell, CTV-1. Leuk Res 2019;78:12–20. [DOI] [PubMed] [Google Scholar]
- 55.Marhäll A, Kazi JU, Rönnstrand L.. The Src family kinase LCK cooperates with oncogenic FLT3/ITD in cellular transformation. Sci Rep 2017;7:13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge L, Xu L, Lu S, Yan H.. LCK expression is a potential biomarker for distinguishing primary central nervous system lymphoma from glioblastoma multiforme. FEBS Open Bio 2020;10:904–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sugihara T, Werneburg NW, Hernandez MC, et al. YAP tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells are regulated by LCK and independent of LATS activity. Mol Cancer Res 2018;16:1556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pei T, Li Y, Wang J, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015;6:17206–17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Santpere G, Alcaráz-Sanabria A, Corrales-Sánchez V, et al. Transcriptome evolution from breast epithelial cells to basal-like tumors. Oncotarget 2017;9:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Köster A, Landgraf S, Leipold A, et al. Expression of oncogenes in human breast cancer specimens. Anticancer Res 1991;11:193–201. [PubMed] [Google Scholar]
- 61.Matsueda S, Shichijo S, Nagata S, et al. Identification of novel Lck-derived T helper epitope long peptides applicable for HLA-A2(+) cancer patients as cancer vaccine. Cancer Sci 2015;106:1493–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Veillette A, Foss FM, Sausville EA, et al. Expression of the lck tyrosine kinase gene in human colon carcinoma and other non-lymphoid human tumor cell lines. Oncogene Res 1987;1:357–74. [PubMed] [Google Scholar]
- 63.Krystal GW, DeBerry CS, Linnekin D, Litz J.. Lck associates with and is activated by Kit in a small cell lung cancer cell line: inhibition of SCF-mediated growth by the Src family kinase inhibitor PP1. Cancer Res 1998;58:4660–6. [PubMed] [Google Scholar]
- 64.Saygin C, Wiechert A, Rao VS, et al. CD55 regulates self-renewal and cisplatin resistance in endometrioid tumors. J Exp Med 2017;214:2715–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim RK, Yoon CH, Hyun KH, et al. Role of lymphocyte-specific protein tyrosine kinase (LCK) in the expansion of glioma-initiating cells by fractionated radiation. Biochem Biophys Res Commun 2010;402:631–36. [DOI] [PubMed] [Google Scholar]
- 66.Zepecki JP, Snyder KM, Moreno MM, et al. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a Lck targeted inhibitor. Oncogene 2019;38:1734–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rupniewska E, Roy R, Mauri FA, et al. Targeting autophagy sensitises lung cancer cells to Src family kinase inhibitors. Oncotarget 2018;9:27346–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patterson H, Nibbs R, McInnes I, Siebert S.. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin Exp Immunol 2014;176:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szilveszter KP, Németh T, Mócsai A.. Tyrosine kinases in autoimmune and inflammatory skin diseases. Front Immunol 2019;10:1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kemp KL, Levin SD, Bryce PJ, Stein PL.. Lck mediates Th2 differentiation through effects on T-bet and GATA-3. J Immunol 2010;184:4178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinson DS. The role of the T cell in asthma. J Allergy Clin Immunol 2010;126:1081–91. [DOI] [PubMed] [Google Scholar]
- 72.Pernis AB, Rothman PB.. JAK-STAT signaling in asthma. J Clin Invest 2002;109:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang S, Yang R, Zheng Y.. The effect of siRNA-mediated lymphocyte-specific protein tyrosine kinase (Lck) inhibition on pulmonary inflammation in a mouse model of asthma. Int J Clin Exp Med 2015;8:15146–54. [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J, Zhuang S.. Src family kinases in chronic kidney disease. Am J Physiol Renal Physiol 2017;313:F721–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gurzov EN, Stanley WJ, Brodnicki TC, Thomas HE.. Protein tyrosine phosphatases: molecular switches in metabolism and diabetes. Trends Endocrinol Metabol 2015;26:30–39. [DOI] [PubMed] [Google Scholar]
- 76.Patry M, Teinturier R, Goehrig D, et al. βig-h3 Represses T-cell activation in type 1 diabetes. Diabetes 2015;64:4212–19. [DOI] [PubMed] [Google Scholar]
- 77.Luo T, Hu J, Xi D, et al. Lck inhibits heat shock protein 65-mediated reverse cholesterol transport in T cells. J Immunol 2016;197:3861–70. [DOI] [PubMed] [Google Scholar]
- 78.Jia L, Jia R, Li Y, et al. LCK as a potential therapeutic target for acute rejection after kidney transplantation: a bioinformatics clue. J Immunol Res 2018;2018:6451298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Womer KL, Kaplan B.. Recent developments in kidney transplantation–a critical assessment. Am J Transplant 2009;9:1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Borhani DW, Calderwood DJ, Friedman MM, et al. A-420983: a potent, orally active inhibitor of lck with efficacy in a model of transplant rejection. Bioorg Med Chem Lett 2004;14:2613–16. [DOI] [PubMed] [Google Scholar]
- 81.Burchat A, Borhani DW, Calderwood DJ, et al. Discovery of A-770041, a src-family selective orally active lck inhibitor that prevents organ allograft rejection. Bioorg Med Chem Lett 2006;16:118–22. [DOI] [PubMed] [Google Scholar]
- 82.Khatik R, Pathak AK.. LcK inhibitors and its analogues: a review. Der Pharma Chem 2011;3:310–20. [Google Scholar]
- 83.Meyn MA, 3rd, Smithgall TE.. Small molecule inhibitors of Lck: the search for specificity within a kinase family. Mini Rev Med Chem 2008;8:628–37. [DOI] [PubMed] [Google Scholar]
- 84.Hanke JH, Gardner JP, Dow RL, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem 1996;271:695–701. [DOI] [PubMed] [Google Scholar]
- 85.Arnold LD, Calderwood DJ, Dixon RW, et al. Pyrrolo[2,3-d]pyrimidines containing an extended 5-substituent as potent and selective inhibitors of lck I. Bioorg Med Chem Lett 2000;10:2167–70. [DOI] [PubMed] [Google Scholar]
- 86.Burchat AF, Calderwood DJ, Hirst GC, et al. Pyrrolo[2,3-d]pyrimidines containing an extended 5-substituent as potent and selective inhibitors of lck II. Bioorg Med Chem Lett 2000;10:2171–4. [DOI] [PubMed] [Google Scholar]
- 87.Calderwood DJ, Johnston DN, Munschauer R, Rafferty P.. Pyrrolo[2,3-d]pyrimidines containing diverse N-7 substituents as potent inhibitors of Lck. Bioorg Med Chem Lett 2002;12:1683–6. [DOI] [PubMed] [Google Scholar]
- 88.Stachlewitz RF, Hart MA, Bettencourt B, et al. A-770041, a novel and selective small-molecule inhibitor of Lck, prevents heart allograft rejection. J Pharmacol Exp Therap 2005;315:36–41. [DOI] [PubMed] [Google Scholar]
- 89.Abbott L, Betschmann P, Burchat A, et al. Discovery of thienopyridines as Src-family selective Lck inhibitors. Bioorg Med Chem Lett 2007;17:1167–71. [DOI] [PubMed] [Google Scholar]
- 90.Das J, Lin J, Moquin RV, et al. Molecular design, synthesis, and structure-activity relationships leading to the potent and selective p56(lck) inhibitor BMS-243117. Bioorg Med Chem Lett 2003;13:2145–49. [DOI] [PubMed] [Google Scholar]
- 91.DiMauro EF, Newcomb J, Nunes JJ, et al. Discovery of aminoquinazolines as potent, orally bioavailable inhibitors of Lck: synthesis, SAR, and in vivo anti-inflammatory activity. J Med Chem 2006;49:5671–86. [DOI] [PubMed] [Google Scholar]
- 92.DiMauro EF, Newcomb J, Nunes JJ, et al. Discovery of 4-amino-5,6-biaryl-furo[2,3-d]pyrimidines as inhibitors of Lck: development of an expedient and divergent synthetic route and preliminary SAR. Bioorg Med Chem Lett 2007;17:2305–9. [DOI] [PubMed] [Google Scholar]
- 93.Martin MW, Newcomb J, Nunes JJ, et al. Discovery of novel 2,3-diarylfuro[2,3-b]pyridin-4-amines as potent and selective inhibitors of Lck: synthesis, SAR, and pharmacokinetic properties. Bioorg Med Chem Lett 2007;17:2299–304. [DOI] [PubMed] [Google Scholar]
- 94.Martin MW, Newcomb J, Nunes JJ, et al. Novel 2-aminopyrimidine carbamates as potent and orally active inhibitors of Lck: synthesis, SAR, and in vivo antiinflammatory activity. J Med Chem 2006;49:4981–91. [DOI] [PubMed] [Google Scholar]
- 95.Martin MW, Newcomb J, Nunes JJ, et al. Structure-based design of novel 2-amino-6-phenyl-pyrimido[5',4':5,6]pyrimido[1,2-a]benzimidazol-5(6H)-ones as potent and orally active inhibitors of lymphocyte specific kinase (Lck): synthesis, SAR, and in vivo anti-inflammatory activity. J Med Chem 2008;51:1637–48. [DOI] [PubMed] [Google Scholar]
- 96.Zhu X, Kim JL, Newcomb JR, et al. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure 1999;7:651–61. [DOI] [PubMed] [Google Scholar]
- 97.Jacobs MD, Caron PR, Hare BJ.. Classifying protein kinase structures guides use of ligand-selectivity profiles to predict inactive conformations: structure of lck/imatinib complex. Proteins 2008;70:1451–60. [DOI] [PubMed] [Google Scholar]
- 98.O’Malley DP, Ahuja V, Fink B, et al. Discovery of pyridazinone and pyrazolo[1,5-a]pyridine inhibitors of C-terminal Src kinase. ACS Med Chem Lett 2019;10:1486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shen S, Liu D, Wei C, et al. Purpuroines A-J, halogenated alkaloids from the sponge Iotrochota purpurea with antibiotic activity and regulation of tyrosine kinases. Bioorg Med Chem 2012;20:6924–28. [DOI] [PubMed] [Google Scholar]
- 100.De Man A, Rewinkel J, Jans C, et al. Preparation of 8-methyl-1-phenyl-imidazo[1,5-a]pyrazine compounds as Lck kinase inhibitors. Netherlands: N.V. Organon. 2011:156. [Google Scholar]
- 101.Laurent A, Rose Y, Morris S, Jaquith J.. Preparation of pyrrolopyrimidine derivatives as protein kinase inhibitors. Montreal, Canada: Pharmascience Inc. 2012:227. [Google Scholar]
- 102.Wang Y, Sun Y, Cao R, et al. In silico identification of a novel Hinge-Binding Scaffold for kinase inhibitor discovery. J Med Chem 2017;60:8552–64. [DOI] [PubMed] [Google Scholar]
- 103.Huang N, Qi X, Wang Y, Sun Y.. Preparation of substituted triazoles as kinase inhibitors. Beijing: National Institute of Biological Sciences; 2017. [Google Scholar]
- 104.Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013;122:872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-Chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a Dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 2004;47:6658–61. [DOI] [PubMed] [Google Scholar]
- 106.Rix U, Hantschel O, Dürnberger G, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007;110:4055–63. [DOI] [PubMed] [Google Scholar]
- 107.Huang F, Reeves K, Han X, et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Res 2007;67:2226–38. [DOI] [PubMed] [Google Scholar]
- 108.Sennhenn P, Meier-Ewert S, Khandelwal N, Bancroft D. Heterocyclic kinase inhibitors and uses thereof for treatment of proliferative disorders. Planegg, Germany: iOmx Therapeutics AG. 2020:191. [Google Scholar]
- 109.Fleming CL, Sandoz PA, Inghardt T, et al. A fluorescent kinase inhibitor that exhibits diagnostic changes in emission upon binding. Angew Chem Int Ed Engl 2019;58:15000–15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Farag AK, Elkamhawy A, Londhe AM, et al. Novel LCK/FMS inhibitors based on phenoxypyrimidine scaffold as potential treatment for inflammatory disorders. Eur J Med Chem 2017;141:657–675. [DOI] [PubMed] [Google Scholar]