

Abstract
Osteocytes are an ancient cell, appearing in fossilized skeletal remains of early fish and dinosaurs. Despite its relative high abundance, even in the context of nonskeletal cells, the osteocyte is perhaps among the least studied cells in all of vertebrate biology. Osteocytes are cells embedded in bone, able to modify their surrounding extracellular matrix via specialized molecular remodeling mechanisms that are independent of the bone forming osteoblasts and bone-resorbing osteoclasts. Osteocytes communicate with osteoclasts and osteoblasts via distinct signaling molecules that include the RankL/OPG axis and the Sost/Dkk1/Wnt axis, among others. Osteocytes also extend their influence beyond the local bone environment by functioning as an endocrine cell that controls phosphate reabsorption in the kidney, insulin secretion in the pancreas, and skeletal muscle function. These cells are also finely tuned sensors of mechanical stimulation to coordinate with effector cells to adjust bone mass, size, and shape to conform to mechanical demands.
Keywords: osteocytes, perilacunar remodeling, mechanosensation, FGF23, RANKL, sclerostin
1. HISTORY OF THE FIELD
Osteocytes have an ancient history, a recent past history, and a very modern history. The earliest evidence for the existence of osteocytes within bone comes from jawless fish that lived during the Ordovician period (1). The remains of osteocytes have been discovered within dinosaur bone, implicating osteocytes as an ancient cell (2) that may yield biological information (3). However, almost nothing was known about the function of these cells until the last decade. Prior to that, most hypotheses regarding the function of osteocytes were based on microscopic images of bone. One of the earliest (more than 100 years ago) published descriptions of osteocytes was by von Recklinghausen (4), who described the delicate extensions of osteocytes in bone and proposed that osteocytes might be capable of removing their surrounding mineralized matrix. The field then became dormant until the early modern osteocyte pioneers of the 1960–1970s revived the concept of osteocytic osteolysis (5) and also proposed an additional function for these cells—as mechanosensors in bone tissue (6). The field became dormant once again until the age of molecular biology and new biotechnology were ushered in. As a consequence of incorporating modern molecular biology techniques to the study of osteocytes, our knowledge of the biology and function of these cells has exploded. In this review, we trace the history of osteocyte-focused research and summarize what is now known about these unusual and unique cells.
The main reason for our delayed understanding of and lack of progress in osteocyte biology was simple—it was extremely difficult to access cells embedded within a hard mineralized matrix. While great strides were being made in studying circulating cells and cells from soft tissue (immunology, hematology, cancer, etc.), little progress was being made in the study of osteocytes. Researchers in the bone field focused on cells they could retrieve from the bone surface, the osteoblasts and osteoclasts. Although Nijweide and colleagues (7) were the first to isolate avian chick osteocytes, Mikuni-Takagaki (8, 9) was one of the first to remove primary osteocytes from mammalian bone using a series of collagenase digestions and calcium chelation. This advance in the field opened a means to study primary cells, but the yields were low and, though enriched, the populations were heterogeneous. The generation of osteocyte cell lines provided opportunity to perform molecular manipulations and to test for function. The identification of osteocyte markers and their subsequent use to drive Cre transgene expression in engineered mouse models provided a means to delete or overexpress key genes in osteocytes (see below). These tools have helped to expand the field of osteocyte biology and function in numerous ways.
2. OSTEOCYTE BIOLOGY
2.1. The Origin of Osteocytes
The early pioneers questioned the origin of osteocytes. Did they descend from osteoblasts, osteoclasts, or another cell type? We now know that osteocytes are descended from mature, matrix-producing osteoblasts (see Figure 1). Manolagas (10) showed that osteoblasts have one of three fates: They can become osteocytes or bone lining cells, or they can undergo programmed cell death, apoptosis. The mechanisms responsible for cell fate of the mature osteoblasts are not known. Embedding cells become surrounded by collagen, made by their own matrix-producing machinery or that of neighboring cells. As these cells begin to embed, they generate cellular extensions—the future dendritic processes of mature osteocytes—that continuously extend and contract until a proper contact has been made with a previously embedded cell (11). The dendrites then appear to anchor to the existing cell and the cell begins the mineralization process, surrounding itself within a hydroxyapatite cave of lacunae. Once the process is completed, the embedded cells are functionally part of the osteocyte lacunocanalicular network (Figure 2).
Figure 1.

(a) The graph shows the relative temporal expression of keratocan (KTN), a marker for osteoblasts; E11/gp38 (E11), a marker of early embedding osteocytes; dentin matrix protein 1 (DMP1), an early osteocyte marker responsible for mineralization; neuropeptide Y (NPY), a neurotransmitter expressed in maturing osteocytes; and MEPE and SOST, both markers for late, mature osteocytes. (b) The Goldner-stained bone section below shows marrow, the surface osteoblasts (①), the embedding osteoid osteocyte (②), early mineralizing osteocytes (③), and mature osteocytes (④). The time scale for the expression graph in panel a corresponds temporally to the differentiation process represented histologically in panel b.
Figure 2.

(a) Three-dimensional reconstruction image of the avian osteocyte network by IMARIS software. Note the ordered array of the osteocytes in chick bone. (b) Field emission scanning electron microscope images of chick osteocyte. Adapted with permission from Reference 12. Copyright 1998, John Wiley & Sons.
A number of markers have been identified that are differentially expressed during this process. Whereas numerous markers for osteoblasts are known (e.g., Cbfa1/Runx2, Osx, Alp, Col1a1, Bglap), only within the past decade or so have markers been identified for osteocytes. Osteocytes share some markers with their progenitors, the osteoblasts, but also express unique markers based on their morphology and potential function (Figure 1). Markers for osteocytes include phosphate regulating neutral endopeptidase on the chromosome X (PHEX), dentin matrix protein 1 (DMP1), and E11/gp38 for early osteocytes; and receptor activator of nuclear factor-κB ligand (RANKL), SOST/sclerostin, fibroblast growth factor 23 (FGF23), and matrix extracellular phosphoglycoprotein (MEPE) for mature osteocytes. Several of these markers, such as PHEX, DMP1, MEPE, and FGF23, play a role in mineralization and phosphate homeostasis. As a regulator of skeletal remodeling, osteocytes secrete sclerostin, a negative regulator of osteoblastic bone formation, and RANKL, an activator of osteoclast formation and function. The function of these biomarkers is reviewed in more detail below.
2.2. Osteocyte Viability and Death
Very few cells in the body can live as long as the osteocyte. Unlike osteoblasts and osteoclasts that can live days to weeks, osteocytes can live for years or even decades (10). As osteocytes are multifunctional, critically essential cells, a major goal in healthcare research has been to keep osteocytes alive and healthy, especially with aging. With aging, osteocyte death is accelerated, mainly through apoptosis. Osteocyte death leaves behind empty lacunae that can fill in with mineral, a process called micropetrosis. Micropetrosis may function as a compensatory mechanism in aged bone by removing the empty lacunae that can function as a stress concentrator if left open. Osteocytes can also undergo programmed cell death, especially in the presence of microdamage, which stimulates the release of chemical signals for osteoclasts to remodel the damaged bone (13). A major regulatory signal sent to osteoclasts by osteocytes is RankL. Deletion of RankL in osteocytes results in increased bone mass (14, 15) because resorption is impaired. Osteocytes can also undergo the process of autophagy (16). Autophagy is a cellular state that promotes survival especially under stressful conditions, and autophagic cells remove unnecessary organelles until they can return to a more functional, healthy state. Should the stress causing autophagy not cease, then the cells can undergo apoptosis, leaving dead or osteonecrotic bone that does not heal or respond to mechanical loading.
Aging is accompanied by a decline in osteocyte connectivity and viability in the bone of osteoporotic patients (17) and aged mice (17). This is most likely due to osteocyte cell death, apoptosis, and autophagy (18). As osteocytes are master regulators of both osteoblasts and osteoclasts, disruption of osteoblast and osteoclast coupling activity is observed in aged bone, with bone resorption far exceeding bone formation (19). A recent study by Tiede-Lewis et al. (20) showed that the dendrite number was greatly reduced in aged mice, where reduced connectivity preceded osteocyte death. Deletion of superoxide dismutase 2 in osteocytes resulted in an osteoporosis-like bone phenotype due to increased generation of reactive oxygen species (ROS) (21). Osteocytes in aged mice also have increased expression of markers of senescence that may contribute to increased osteoclast activity and bone resorption (22).
3. TOOLS FOR STUDYING OSTEOCYTES
3.1. In Vitro Cell Lines
As stated above, it has only been within the past 10–15 years that significant tools have been developed to study osteocytes, including cell lines, transgenic mouse models, and instrumentation. The first osteocyte-like cell line was the MLO-Y4, a dendritic, highly mechanosensitive cell (23). Subsequent lines were developed that possessed the feature of mature osteoblasts that then differentiated into cells with the characteristics of osteocytes. To date, six cell line models have been generated: HOB-01-C1, MLO-Y4, MLO-A5, IDG-SW3, OCY454, and OMGFP66. The HOB-01-C1 human bone cell line was generated to serve as a preosteocyte or early osteocyte, but there are few published studies of this cell line (24). In contrast, the MLO-Y4 murine osteocyte-like cell line has been used extensively to investigate osteocyte function (23). A transgenic mouse in which the immortalizing T antigen was expressed under control of the osteocalcin promoter was used to derive this cell line. Numerous laboratories (over 270 publications as of the writing of this review) have used the MLO-Y4 cell line to investigate osteocyte cell function, especially gap junctions, hemichannels, mechanosensitivity, factor secretion, dendrite formation, regulation of osteoblasts and osteoclasts, and other functions.
The MLO-A5 is postosteoblast/preosteocyte-like cell line, established from the same animals used to derive the MLO-Y4 cells (25). Within one week of culture, these cells will rapidly mineralize in sheets, not nodules. Mineralization is accelerated by the addition of an external source of phosphate. This cell line reproduces primary mineralization as shown by similar spectra compared to primary bone using Fourier transform infrared spectroscopy (25). These cells mineralize their collagen matrix by generating nanospherulites that mineralize while budding from developing cellular processes to become associated with and initiate collagen-mediated mineralization (26). These cells have been useful for studies that require a mineralizing cell (50 studies to date).
The IDG-SW3 cell line differentiates in culture from the late osteoblast to the late osteocyte, closely replicating the phenotype of primary cells (27). Both the IDG-SW3 and Ocy 454 (28) were created by crossing the 8-kb Dmp1-GFP (green fluorescent protein) transgenic mouse line (29) with the Immortomouse. The Immortomouse has an interferon-gamma (IFN-γ)-inducible promoter driving expression of a thermolabile large T antigen (H-2Kb-tsA58) that enables conditional immortalization of cells derived from this mouse. Therefore, both the IDG-SW3 and the Ocy 454 express the SV40 T antigen when cultured at 33°C in the presence of IFN-γ and proliferate rapidly. In the absence of IFN-γ at 37°C, they no longer express T antigen and therefore behave like primary cells. Like normal primary cells, Fgf23 mRNA expression in response to treatment with 1,25-dihydroxyvitamin D3 is increased. Also like primary cells, Sost/sclerostin expression with parathyroid hormone (PTH) treatment is downregulated. Therefore, in these responses and others, the IDG-SW3 cell line reproduces the osteoblasts to late osteocyte differentiation process, as observed in vivo (27). (More than 20 publications have reported using IDG-SW3 cells.) OCY 454 cells are reported to express Sost at an earlier time in culture than the IDG-SW3 cells (9 publications).
A new exciting cell line is OMGFP66, which spontaneously forms bone-like structures containing cells that appear very similar to osteocytes in vivo (30). This cell line was generated from a mouse model where membrane GFP was driven by the Dmp1 promoter (Figure 3). We predict that the OMGFP66 cell line will be extremely useful for the study of osteocytes.
Figure 3.
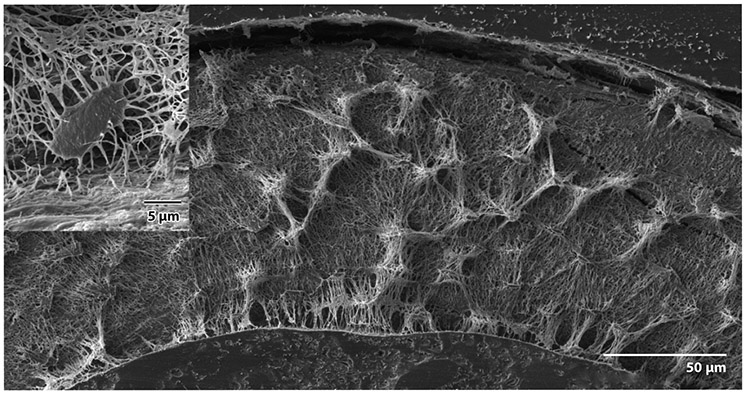
Backscatter scanning electron microscopy of a resin-filled, acid-etched cross section of murine long bone. Note the extensive connections between lacunae. (Inset) Note the dendrite-filled canalicular connections with the bone surface.
3.2. In Vivo Imaging
Since the invention of microscopy, scientists have been able to view cells and tissues under static conditions. These pioneers used their intellect, knowledge, and imagination to hypothesize functions for cells and their components. Generally, their hypotheses have been validated. However, the capacity to track cells and their components in real time and in the live animal has led to tremendous new insight into function. For example, osteocytes were thought to be static cells, but dynamic imaging has shown them to have the capacity to extend and retract their dendritic processes (11). Knowledge gained from these approaches is reviewed below.
3.2.1. Lineage tracing.
Transgenic mouse models using osteoblast and osteocyte promoters have been useful for studying osteocytes. The first Cre line to be generated to study osteocytes and odontoblasts was the 10-kb Dmp1-Cre model (31). This Cre model was the first to target osteocytes, and it opened the field with regard to studying osteocytes in vivo. Subsequently, Kalajzic and coworkers (29) used a slightly shorter version of the 10-kb Dmp1 promoter—the 8-kb Dmp1 promoter—to drive GFP expression selectively in osteocytes. This mouse model has proved useful to examine osteocyte ontogeny (lineage tracing) and to determine osteocyte function, especially when used in conjunction with other fluorophore-linked transgenes (e.g., osteoblast promoters such as osteocalcin and collagen type 1 driving topaz or GFP) to examine transgene expression during osteoblast differentiation (32).
3.2.2. Cre mice for studying osteocytes.
The 10-kb Dmp1 promoter was also used to generate an inducible model, 10-kb Dmp1-Cre-ERT2 (33), which showed highly specific expression in osteocytes. The next model generated was the 8-kb Dmp1-Cre, but no significant differences in Cre activation in embedding cells and early osteocytes were shown between the 10-kb and 8-kb Dmp1-Cre (34). However, it has been found that approximately half of the 10-kb Dmp1-Cre animals also have Cre activation in muscle, an event thought to occur during embryogenesis. The 8-kb Dmp1-Cre mice also exhibit expression of the transgene in muscle, similar to the 10-kb version.
More recently, mice have been made using the Sost promoter to drive Cre expression (34), and reports indicate that only osteocytes are targeted, not osteoblasts or lining cells. However, hematopoietic cells in the marrow and some osteoclasts were also shown to be positive in this model. Recently, an inducible Sost ERT2 has been generated with very specific expression in osteocytes with little off target Cre expression, but a muscle phenotype was also observed in these mice (35). Thus, to date, there is no perfect Cre model that only targets osteocytes. When performing experiments with Cre models, the pros and cons of each model must be taken into account and, ideally, more than one Cre model should be utilized (34).
3.2.3. Confocal multiplex imaging.
Instrumentation to image structures and cells within a mineralized matrix can now help to visualize the osteocyte lacunocanalicular network and the mineral matrix surrounding the cells. Originally, early osteocyte pioneers used light microscopy to examine stained bone sections. Later, scanning electron microscopy, especially of acid-etched resin-embedded bone, was used to image the osteocyte lacunar morphology (Figure 4). Currently, serial focused ion beam and serial block-face scanning electron microscopy have been developed for osteocyte imaging, which increases the field of view. Confocal microscopy, several forms of tomography such as X-ray computed tomography, microcomputed tomography, transmission electron computed tomography, and synchrotron radiation band have subsequently been developed to better understand osteocyte morphology (see 36 for a comprehensive review). Recently, label-free third-harmonic generation microscopy has been shown to provide a great deal of information, similar to confocal fluorescence (37). These studies have mainly visualized the mineralized matrix surrounding the osteocytes, providing structural information.
Figure 4.

OmGFP66 cells form bone-like structures with a lacunar and osteocyte morphology resembling in vivo bone. Maximal Z-projected confocal images of (a) an OmGFP66 bone-like structure from a day-28 culture and (b) a 7-day-old Dmp1-mGFP transgenic mouse calvarium. Green denotes Dmp1-mGFP and red the alizarin reed staining for mineral. Note the similarities in morphology and spacing of the Dmp1-mGFP-positive osteocytes and similar appearance of the mineralized lacunae by alizarin red fluorescence. Adapted with permission from Reference 30. Copyright 2019, John Wiley & Sons.
Two- and three-dimensional confocal multiplex imaging was developed using bones from mice injected with fixable dyes or from novel reporter mice (38). For example, a recently generated 10-kb Dmp1 promoter-driving membrane GFP mouse model provides increased clarity and resolution of canaliculi and vesicles within the matrix. Another key finding from these studies was the observation of multiple collagen rings around osteocytes, suggesting successive waves of perilacunar remodeling. But clearly the most exciting imaging has been of live cells either in culture or in the live animal (intravital imaging). Ishihara and colleagues (39) have performed time-lapse imaging of osteocytes in live animals and shown that Ca2+ oscillations are modulated by gap junctions. Dallas & Veno (11) have documented the motion of dendrites in an embedding cell and shown real-time mineralization dynamics of the cell line MLO-A5 (40) and primary cells (11). Tanaka and colleagues (41) performed Ca2+ imaging in live bone and found that mature osteocytes are more mechanosensitive (in terms of calcium oscillations) than early osteocytes. Intravital imaging will open new gateways to the discovery and understanding of osteocyte function.
4. FUNCTIONAL ASPECTS OF OSTEOCYTES
4.1. Primary Direct Functions
Osteocytes are embedded within a hard mineralized environment for life (exceptions being when released by fracture or during remodeling). Within this hard tissue the cell is bathed by a bone fluid that travels over the dendrites and cell bodies creating shear stresses. It is thought that these stresses are sensed by the cell to induce signals to osteoblasts and osteoclasts to initiate remodeling. Interestingly, under certain conditions, such as calcium-demanding conditions, the osteocyte can actually remove its perilacunar matrix (mimicking the osteoclast) and also can replace that matrix (mimicking the osteoblast); this is known as osteocyte perilacunar remodeling. These are the interactions of the osteocyte with its environment.
4.1.1. Remodeling matrix.
Osteocytes have the capacity to remove and replace their perilacunar and pericanalicular matrix. This process occurs under healthy calcium-demanding conditions, such as lactation and hibernation. It is logical to assume that the osteocyte can remember being an osteoblast and therefore be able to reexpress the genes necessary for making matrix, but surprisingly, these cells are also capable of expressing genes that were once thought to be osteoclast specific (42). It is estimated that there are up to 42 × 109 osteocytes in the adult human skeleton, with a total lacunocanalicular surface area of 215 m2 (43). Therefore, the ability of osteocytes to release even a minute or trace amount of calcium from their perilacunocanalicular matrix would have a significant effect on the overall serum calcium level. Rapid release of calcium by osteocytes may replace the need for osteoclastic bone resorption, thereby protecting skeletal integrity.
Lactating mice with large litters creating a high calcium demand have larger lacunae in their long bones and vertebrae (loaded bone) but not in their calvaria (unloaded bone) compared to virgin mice (42). Osteocytes from lactating mice express genes normally associated with osteoclasts, such as cathepsin K, tartrate-resistant acid phosphatase (Trap), carbonic anhydrase 2, and the v-Atpase subunit Atp6v0d2. PTHrP, which is highly elevated during lactation, was shown to induce a decrease in osteocyte extracellular pH in vitro, and in vivo lactating mice showed a decreased pH within their osteocyte network (44). This same study also reported that osteocytes are more resistant to acidification of their extracellular environment than osteoblastic cells, suggesting that they can remain viable in low-pH environments.
In addition to lactation, hibernation can lead to enlarged osteocyte lacunae (45). Similarly, mechanical unloading due to microgravity during spaceflight induces perilacunar remodeling in monkeys (46) and mice (47). Egg-laying hens fed a low-calcium diet showed tetracycline labeling around the osteocyte lacunae when a normal calcium diet was reintroduced (48). It has been suggested that sclerostin, a negative regulator of Wnt signaling that is increased by mechanical unloading, could partially be responsible for lacunar enlargement. Adding sclerostin to MLO-Y4 cells or to primary human osteocytes induces the expression of perilacunar remodeling genes such as cathepsin K, carbonic anhydrase 2, and matrix metalloproteinase 13 (49). In addition, osteocyte lacunar area was increased in human and bovine trabecular bone explants cultured with sclerostin. The molecular mechanisms responsible for the removal and the replacement of their perilacunar matrix remain to be determined.
Osteocytic osteolysis refers to the pathologic removal of the perilacunar matrix as occurs with diseases such as hyperparathyroidism, hypophosphatemic rickets, and osteoporosis (50). Glucocorticoid administration, a common cause of secondary osteoporosis, increases lacunar area in mice (51). X-linked hypophosphatemia is a disorder characterized by elevated Fgf23 levels, decreased 1,25(OH)2D3 levels, and osteomalacia. Significantly enlarged lacunae were found in the mouse model of X-linked hypophosphatemia, Hyp, compared to controls. Osteocytes from these mice had increased expression of cathepsin K, Trap, matrix metalloproteinase 13, and the v-Atpase subunit Atp6v0d2 (52). These effects could be partially rescued by treating the mice with either daily 1,25(OH)2D3 or an Fgf23-neutralizing antibody. Many questions still remain regarding osteocytic osteolysis and perilacunocanalicular remodeling: (a) Is the matrix also removed in addition to the mineral? (b) Are growth factors removed and put into local or systemic circulation during the process? (c) If so, what are their effects locally and systemically? (d) Does an enlargement of the lacunocanalicular system change the load-sensing apparatus of the resident osteocyte?
4.1.2. Mechanosensing.
The notion that bone tissue is responsive to mechanical influences has been known for well over 100 years (53). Decades ago, it was postulated that the osteocyte was the most likely candidate for the primary sensor cell type in bone (reviewed in 6). There were several observational reasons for this premise that predated any experiments on the issue: (a) Osteocytes are regularly distributed throughout cortical and trabecular bone, even in areas of mineralized matrix devoid of vasculature (54), which could serve as a broad load-monitoring net that infiltrates every cubic millimeter of bone tissue; (b) osteocytes are intricately connected to one another (and to surface cells such as bone lining cells, osteoblasts, and vascular cells) via a large number (~50/cell) of long cellular processes that join one another and transmit information intercellularly via gap junctions. As such, their propensity for communication—a key attribute for cellular networks that integrate information over a large area—is ideal; and (c) as osteocytes are not effector cells (capable of changing bone size and shape, as osteoblasts and osteoclasts are), they have been thought of historically, perhaps somewhat by default, as sensor cells. However, as discussed earlier, osteocytes can remodel the small rim of bone lining their lacunae, which collectively might have profound effects on plasma calcium levels.
Moving beyond philosophical arguments addressing why osteocytes might be the primary mechanosensory cell type in bone, accumulating experimental evidence shows that these cells are robust responders to and translators of mechanical energy applied to bone. Osteocytes are more responsive to mechanical stimulation than osteoblasts. They release more nitric oxide (NO) (55) and prostaglandin (PGE2) (56, 57), trigger a greater calcium influx (58), and exhibit more rapid β-catenin-mediated transcription (57) than osteoblasts (see Figure 5). Interestingly, within the osteocyte, the cell processes are more mechanosensitive than the cell body (59). This has been demonstrated using a Transwell filter culture system that permitted mechanical stimulation of either the osteocyte cell body or the cell processes by fluid flow (59), or by glass microneedle manipulation of the membrane surrounding the cell body or cell process (60). The connections between osteocytes are also important for mechanotransduction. The presence of gap junctions between cell processes of osteocytes has been demonstrated by electron microscopy (61), but their role in osteocyte mechanotransduction has only recently come to light. Deletion of the most highly expressed gap junction subunit in bone—Cx43—using Dmp1-Cre-mediated recombination of Cx43-floxed alleles resulted in a gain in mechanosensitivity for periosteal bone formation. This result is consistent with other skeletal Cre drivers used to delete Cx43 earlier in the lineage of Dermo1 (62) and Bglap (63) and suggests the presence of an inhibitory signal transmitted through gap junctions that is disabled in the mutants. It is important to consider that Cx43, beyond gap junction formation, can also assemble into hemichannels on osteocytes, which has been implicated in osteocyte PGE2 release into the extracellular space upon mechanical stimulation (64).
Figure 5.

(Top) Osteocytes function as mechanosensory cells that mediate loading effects in bone through enhanced release or inhibition of regulatory molecules. (Bottom) Bone homeostasis cells regulate the normal process of bone remodeling via activation of osteoclasts (left) and osteoblasts (right) by distinct pathways. (Inset) Endocrine cells regulate such diverse and distant processes as phosphate handling in the kidney and muscle maintenance in the limbs and trunk. Abbreviations: ATP, adenosine triphosphate; DKK1, Dickkopf gene codes for the protein that is an inhibitor of WNT signaling; FGF23, fibroblast growth factor 23; IGF-1, insulin-like growth factor 1; M-CSF, macrophage colony stimulating factor; NO, nitric oxide; OPG, osteoprotegerin; PGE2, prostaglandin E2; RANKL, receptor activator of NF-κB ligand; SOST, gene coding for the protein sclerostin, a specific inhibitor of WNT signaling.
As a mechanosensitive cell, osteocytes must be equipped with a collection of proteins that sense changes in the mechanical environment. The identity of the primary mechanosensory apparatus in osteocytes has been of great interest in the field for a long time, but a consensus on the exact mechanism(s) is lacking. Integrin complexes and ion channels are considered likely candidates for osteocyte mechanosensors, based on a wealth of studies. For example, the β1 integrin subunit appears to be crucial for osteocyte mechanotransduction (65-67), as does the Trpv4 channel (68, 69). Although the signal reception mechanism in the osteocyte is unclear, downstream signaling is better characterized (though still incomplete). Mechanical stimulation of the osteocyte induces the release of ATP (70), PGE2 (71-73), NO (74, 75), and growth factors such as insulin-like growth factor 1 (IGF-1) (76, 77). A later cascade that has received considerable attention in osteocyte mechanotransduction is the activation of Wnt signaling. Osteocytic deletion of Lrp5 (78) or β-catenin (79, 80), or overexpression of Sost in osteocytes (81), all severely impair mechanotransduction in bone. Conversely, disuse mechanotransduction (i.e., bone wasting) is impaired with osteocyte-selective stabilization of β-catenin (82) or overexpression of a gain-of-function Lrp5 mutation (83). In summary, both the initial receptor(s) and subsequent downstream signaling cascades involved in osteocyte mechanotransduction are incompletely known, but new biological models and technological advances are fostering progress in delineating the exact mechanisms.
The physical stimulus to which osteocytes respond is also a matter of ongoing research. Osteocytes are mechanically stimulated by bone loading, but a number of possible mechanisms for precise stimulation have been advanced. Fluid flow through the lacunocanalicular (spelled as elsewhere) system is one possible mechanism, whereby pressure gradients from bone bending (physical activity) induce fluid movement and subsequent shear stress on the cell membranes; osteocytes are very sensitive to fluid shear stress in vitro (84, 85). Another mechanism related to fluid movement in the canaliculi is the strain amplification phenomenon, which has two different submechanisms. One purports that the fluid movement between the osteocyte cell process membrane and the canalicular wall creates drag forces on the tethering structures, which deflect and consequently create a hoop strain on the cell process (86). The resulting hoop strain far exceeds the macroscopic global strains measured at the bone surface. The other purports that the stress risers in the bone tissue created by the lacunocanalicular voids generate large local strains immediately adjacent to osteocytes, which also far exceed the macroscopic global strains measured at the bone surface (87-89). Rather than relying on just one particular mechanical input, it is likely that osteocytes integrate several of these physical stimuli to achieve the appropriate signaling cues in order to maintain proper whole bone adaptation.
4.2. Primary Indirect Function of Osteocytes
Whereas the earliest functions proposed for osteocytes were mechanosensing and removal of their perilacunar matrix, a total and unanticipated function was the osteocyte-producing factors that could regulate not only bone cells but also distant organs, such as the kidney. This latter function is the definition of an endocrine cell. Even though these cells have much less cytoplasm and/or organelles than osteoblasts or other cell types, they still have the capacity to produce and secrete potent local and soluble factors.
4.2.1. Regulation of osteoclasts.
In addition to orchestrating mechanical adaptation of bone structure, osteocytes also specifically coordinate the activity of osteoclasts. They accomplish this task through a variety of mechanisms. To begin with, the most widely studied role for osteocyte control of osteoclast biology is the RANKL/OPG mechanism. Osteocytes are a major source of RANKL for osteoclastogenesis. Two comprehensive independent studies (15, 60) demonstrated that selective deletion of the gene for RankL, Tnfsf11, in the osteocyte population (but not osteoblasts or precursors) of engineered mice accounts for the deficient osteoclastogenesis phenotype seen in global mutants (90). Osteocytes are also a significant source for OPG, which, as discussed earlier, functions as a soluble decoy receptor for RANK and inhibits osteoclast formation. Thus, osteocytes control osteoclastogenesis in a positive direction by increasing the expression/availability of RANKL and decreasing the expression/availability of OPG, or conversely, the proportions can be reversed to decrease resorptive activity.
As mentioned earlier, physical contact is required for transmission of the RANKL signal from osteocytes to osteoclasts. How then does this happen, given that osteocytes are buried in the bone matrix? Recent experiments show that osteocytic RankL is provided as a membrane-bound form to osteoclast precursors through osteocyte dendritic processes (91) that reach beyond the bone surface and into the marrow and periosteal compartments. Here, newly synthesized RankL molecules are transferred from the Golgi apparatus to lysosomes in a complex that involves OPG. Experiments using cells from Opg knockout mice revealed that Opg is required for proper transfer of RankL from the Golgi apparatus to the secretory lysosomes and, ultimately, osteoclastogenesis (92). Thus, these observations establish two important points about osteocytic control of osteoclasts: (a) Osteocyte-derived Opg is necessary for proper function of RankL trafficking in osteocytes and subsequent osteoclast effects, since the Opg-mediated effect on trafficking process occurs intracellularly; and (b) secreted OPG, while important for serving as a decoy receptor for RANKL, is not the only mechanism of action for OPG’s modulatory effect on the RANK/RANKL axis.
Taking a broader view of spatial control of osteoclastogenesis by osteocytes, earlier observational studies had suggested that osteocytes somehow chemically attract remodeling units into bone that was in need of replacement. For example, new remodeling basic multicellular units (teams of osteoclast and osteoblasts that function in a coordinated arrangement as they move through tissue space) are 4 to 6 times more likely to be associated with fatigue-induced microcracks than by chance alone in canine bone samples (93, 94). The experimental model that most easily facilitates a functionally compromised moiety of bone is the introduction of microdamage by fatigue loading in rodents. Mouse and rat cortical bone do not typically undergo osteonal remodeling, but induction of microdamage via fatigue loading results in osteon-like structures, which appear selectively in fatigue-riddled locales (95). If severe enough, cortical bone microdamage causes osteocyte apoptosis, which initiates the remodeling response (96). Schaffler’s group (97) has shown that apoptotic osteocytes in fatigue damaged regions signal neighboring, healthy osteocytes residing ~200 μm away to release RankL, which ultimately serves to attract remodeling units. The communication from dying osteocytes to trigger nearby bystander cells to release RankL involves ATP signaling via Panx1 and P2X7 activation.
Other osteocyte-derived factors that contribute to osteoclast differentiation and function include M-CSF (98), interleukin 6 (IL-6) (99), tumor necrosis factor alpha (TNF-α) (100) (perhaps through osteocyte-derived apoptotic bodies) (101), and HMGB1 (102). While the field of osteocyte control of osteoclastogenesis has made great strides in recent years, several RANKL/OPG-independent mechanisms are currently under investigation (e.g., 103, 104).
4.2.2. Regulation of osteoblasts.
Osteocytes have both indirect and direct effects on osteoblasts. Many of the indirect effects on osteoblasts can be attributed to the coupling phenomenon in bone remodeling, where osteoblastic activity (indirect effect) follows osteoclastic activity (direct effect from osteocytes) during bone turnover (105). However, osteocytes also have direct effects on osteoblasts that occur through the production of both stimulatory and inhibitory factors. Regarding stimulatory factors, osteocytes are a rich source of signaling lipids (e.g., PGE2) (106), growth factors (e.g., IGF-1) (77), glycoproteins (e.g., Wnts) (107), free radicals (e.g., NO) (55), and nucleotides (e.g., ATP) (108) that have potent effects on osteoblastogenesis and matrix formation. One of the most potent signals generated by osteocytes that control osteoblast biology is the secreted inhibitor of Wnt signaling. Osteocytes are a major source of the Lrp5/6 antagonists sclerostin and Dkk1. Although sclerostin expression can be found in several tissues other than bone (109, 110), the osteocyte is a major source. Dkk1 is also highly enriched in the osteocyte population. A body-wide screen of Dkk1 expression in young adult mice found that Dkk1 expression was restricted largely to bone (i.e., osteocyte-enriched fraction), where very strong expression was detected (111). Moreover, both fluorescence-activated cell sorting (112) and laser capture microdissection (113) approaches revealed that Dkk1 is highly upregulated in the osteocyte compared with the osteoblast. Sclerostin and Dkk1 are strong antagonists of Wnt-mediated activity in osteoblasts. Numerous anabolic stimuli [e.g., loading (114), PTH (115), and PGE2 (116)] reduce Sost/sclerostin expression in osteocytes, which ultimately facilitates osteoblast-mediated anabolism through Wnt. Another emerging osteocyte-derived key player in osteoblast activity is neuropeptide Y, which is highly expressed in osteocytes and exerts inhibitory actions on osteoblasts (117). Much of the work on the osteocyte-to-osteoblast communication pathway has focused on the regulation of osteocyte-derived osteoblast inhibitors. Osteocyte-derived molecular activators of osteoblast function are also an important part of the cross talk and are receiving increasing attention.
4.2.3. Communication with and regulation of distant organs.
The human skeleton hanging in the classroom frequently leads the observer to the conclusion that the skeleton is more like an inanimate object that only provides structure to the body necessary for movement. However, bone is a dynamic organ, constantly remodeling itself. Therefore, it was a novel and unexpected observation to find that osteocytes embedded in bone can secrete factors that can target distant organs (Figure 5). The first description of this phenomenon was based on the observation that Fgf23—a growth factor that is highly expressed in osteocytes—regulates phosphate homeostasis in the kidney. This was the first description of osteocytes in bone as endocrine cells; as a consequence, our view of osteocytes changed and now includes the view that bone is an endocrine organ (118). Following the observation that Fgf23 is a bone endocrine factor produced by osteocytes, osteocalcin was described as a bone hormone having effects on distant organs, targeting male fertility, and affecting both cognition and energy metabolism and recently shown to target muscle (see 119 for a review). Osteocalcin is produced by both osteoblasts and osteocytes.
As discussed above, osteocytes regulate plasma calcium by releasing mineral from their surrounding matrix, especially in response to PTH and PTHrP, but osteocytes regulate phosphate by targeting the kidney. The induction of the phosphaturic hormone, FGF23 in osteocytes by 1,25(OH)2D3 in both animal models and in humans is believed to be an essential part of the kidney/bone phosphate-regulatory axis (120) and is highly elevated in hypophosphatemia. Fgf23 decreases the expression of the sodium/phosphate cotransporters NaPi-IIa and NaPi-IIc in the renal proximal tubule, leading to increased phosphate excretion. In addition, it inhibits the conversion of 25(OH)2D into the active metabolite 1,25(OH)2D3 by decreasing the expression of 1-α-hydroxylase (121). Thus, Fgf23 promotes phosphate excretion and inhibits phosphate uptake, leading to phosphate wasting. Fgf23 signals through the FGF receptor (FGFR) family and requires the presence of a coreceptor, Klotho (120).
Fgf23 expression in osteocytes is regulated by PHEX and DMP1, which are promoters of bone mineralization. PHEX inhibits Fgf23 transcription, and mice with inactivating Phex mutations (Hyp mice) have highly elevated bone Fgf23 mRNA levels (122). Mice lacking Dmp1 expression have osteomalacia and abnormally high serum Fgf23 levels (118). In addition to inhibitors of Fgf23, osteocytes also express Mepe, which acts as an inhibitor of matrix mineralization. While the precise function of Mepe is not yet known, it is thought to inhibit mineralization by promoting renal phosphate excretion (123). Full-length Mepe protein can be cleaved to release a 19 amino acid fragment known as the ASARM peptide (acidic serine aspartate-rich MEPE-associated motif). The ASARM peptide can bind to Phex and inhibit its activity (124), but it does not regulate Fgf23. Therefore, the role of Mepe in the regulation of Fgf23 remains controversial.
Recently, studies have been performed examining communication between bone and muscle and vice versa. Bone cells produce prostaglandins, Wnt1, Wnt3a, and Fgf9 that can have effects on the proliferation of muscle progenitors and differentiation into myotubes. PGE2 (125) and the Wnts (126) are elevated in response to fluid flow shear stress in osteocytes and Fgf9 is expressed as the late osteoblast is embedding and becoming an osteocyte. Whereas PGE2 and the Wnts stimulate myogenesis, Fgf9 inhibits myogenesis but stimulates proliferation of muscle progenitors. RANKL produced by osteocytes also has negative effects on muscle (127). Therefore, osteocytes make both positive and negative regulators of muscle. The balance between these factors under conditions such as exercise or disuse-induced osteoporosis may be key for coupling bone and muscle mass. In addition, osteocytes and osteoblasts are known to secrete osteocalcin, which can signal to the pancreas to increase insulin synthesis and increase insulin sensitivity in skeletal muscle and adipose tissue (128).
Conversely, it has now been shown that muscle can produce both negative regulators of bone such as myostatin and positive regulators of bone such as β-aminoisobutyric acid, BAIBA. Myostatin has negative effects not only on muscle but also on bone mass (129). The muscle metabolite, BAIBA, protects osteocytes against ROS-induced cell death and protects hindlimb unloaded mice against bone and muscle loss (130). A muscle protein, irisin, has been shown to have both positive and negative effects on bone. Colaianni and colleagues (131, 132) found that small injections of irisin increased bone mass, but Kim et al. (129) found that mice with global deletion of the precursor for irisin, FNDC5, were protected from bone loss during ovariectomy. It will be important to understand how each factor is being regulated in order to develop therapeutics. In addition to irisin and L-BAIBA, several other myokines are known to act on bone in addition to myostatin, including IL-6, IGF-1, and basic fibroblast growth factor 2 (bFGF-2) (133), but the direct effect of most of these myokines on osteocytes is unknown.
5. TRANSLATIONAL IMPLICATIONS
5.1. Anti-Sclerostin Antibodies to Treat Osteoporosis
In the late 1990s, identification of the SOST gene as the cause for the bone sclerosing disorder sclerosteosis represented a major milestone in the history of harnessing the osteocyte to generate anabolic action in the skeleton (134). The sclerostin protein represented the ideal target to improve bone mass in patients because, (a) despite being an antagonist for the Wnt signaling pathway (a fairly ubiquitous pathway), sclerostin expression was relatively restricted to the osteocyte population, (b) sclerostin is a secreted protein thus it is amenable to antibody targeting, (c) sclerostin deprivation had clear and well-described high bone mass–causing effects on the skeleton with few nonskeletal side effects (e.g., cancer concerns), and (d) its inhibition stimulated anabolic action in the skeleton. By the mid-2000s, at least three large pharmaceutical companies had active sclerostin programs in a race to bring to market the first non-PTHR1-based anabolic therapy. Recently, the sclerostin antibody Evenity (romosozumab) was approved for clinical use in the United States as a first-in-class therapy for the treatment of postmenopausal women with osteoporosis at high risk of fracture. In preclinical studies, treatment with sclerostin monoclonal antibodies resulted in potent osteoanabolic responses in mice, rats, and monkeys (reviewed in 135). Phase III studies designed to evaluate the reduction in fracture risk in a large international population of women (~7,000 in the FRAME trial and ~4,100 in the ARCH trial) showed significant reductions in vertebral fractures at 1 and 2 years (reviewed in 136). Despite its clinical efficacy and selectivity to bone, romosozumab therapy comes with a black box warning to patients at increased risk for cardiovascular disease, due to a slight increase in MACE—defined as cardiovascular death, myocardial infarction, and stroke—observed in the ARCH trial. As romosozumab has only been on the market for several months at the time of this writing, it is too early to tell how commercially successful sclerostin targeting will be. But the antibody-based study and elucidation of this pathway will have lasting effects on our understanding of osteocyte biology.
5.2. Anti-RANKL, Anti-FGF23, and Others
In addition to the anti-sclerostin antibody that targets and neutralizes the osteocyte factor sclerostin, there are other therapeutic antibodies available that target other osteocyte factors. These include anti-FGF23, burosumab, and anti-RANKL, denosumab. Treatment with anti-FGF23 antibody increases serum phosphate in X-linked hypophosphatemic rickets (see 137 for a review). As described above, FGF23 is highly elevated in osteocytes and in the circulation in hypophosphatemic rickets caused by deletion or mutation of PHEX or DMP1; autosomal dominant hypophosphatemic rickets caused by gain-of-function mutations in FGF23 that prevent proteolytic cleavage (see 138 for a review); in chronic kidney disease (139); and hypophosphatemic osteomalacia caused by homozygous mutation in FAM20, a regulatory molecule of FGF23 (140). Studies have linked high levels of circulating FGF23 to an increased risk of heart disease, vascular calcification, and increased fat mass (141). In addition to anti-FGF-23 antibody, other potential therapeutics include FGF receptor inhibitors. The mechanisms for regulating FGF23 production are complex (137).
In 2002, researchers reported that the osteocyte-like cell line MLO-Y4 expressed high levels of RankL and supported osteoclast formation (142). In spite of studies showing that primary osteocytes will support osteoclast formation and activation (143), this concept was not generally accepted until two groups showed that deletion of RankL in osteocytes in vivo using the Dmp1-Cre model resulted in increased bone mass (14, 15). Denosumab, a human anti-RANKL antibody, is now approved for the treatment of osteoporosis (144).
It appears that other therapeutics such as bisphosphonates and hormones also target osteocytes in addition to affecting osteoclast and osteoblast function. Bisphosphonates have been shown to reduce osteocyte apoptosis (145) and therefore may maintain osteocyte viability and function. Although they are thought to target osteoblasts, hormone replacement therapy, selective estrogen receptor modulators (Evista), and PTH peptides (Forteo) could also have significant effects on osteocytes. There may exist other osteocyte factors, unknown at this time, that could be targets for development of future therapeutics.
6. SUMMARY AND CHALLENGES
The early pioneers hypothesized two main functions for osteocytes: one as a mechanosensor and the other as having the capacity to remove their surrounding matrix. Novel functions, never before hypothesized, have been discovered, such as phosphate regulators, regulators of resorption, and regulators of formation, among others. Although they are not covered here, other functions have been attributed to osteocytes, such as controlling hemopoietic stem cell mobilization and lymphopoiesis (146). The role of osteocytes in cancer metastasis and burden is being examined (147), as is the interaction between the immune system and osteocytes (148). One question continuously arises: How can one cell type have so many functions? Most cells have one major function and a few minor functions. Are there subpopulations of osteocytes that specialize in specific regulatory processes or do all osteocytes have multiple functions? Has the full iceberg been uncovered or just the tip? We propose that we have only revealed the tip and anticipate more exciting discoveries in the future.
FUTURE ISSUES.
What is the role of the osteocyte in glucose and energy metabolism?
Is there a role for osteocytes in bone repair and fracture healing?
What are the effects of fatty acid overload (obesity) on osteocyte function?
What is the role of osteocytes in disease and aging and as targets for therapeutics?
ACKNOWLEDGMENTS
The authors’ work is funded by the US National Institutes of Health (grant R01AR053237 to A.G.R.), PO1AG039355 (to L.F.B.), and R61AR073551 (to A.G.R. and L.F.B.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Smith MM, Hall BK. 1990. Development and evolutionary origins of vertebrate skeletogenic and odontogenic tissues. Biol. Rev. Camb. Philos. Soc 65:277–373 [DOI] [PubMed] [Google Scholar]
- 2.Pawlicki R 1975. Studies of the fossil dinosaur bone in the scanning electron microscope. Z. Mikrosk. Anat. Forsch 89:393–98 [PubMed] [Google Scholar]
- 3.Schweitzer MH, Zheng W, Cleland TP, Bern M. 2013. Molecular analyses of dinosaur osteocytes support the presence of endogenous molecules. Bone 52:414–23 [DOI] [PubMed] [Google Scholar]
- 4.von Recklinghausen F 1910. Untersuchungen über Rachitis und Osteomalacia. Jena, Ger.: Gustav Fischer [Google Scholar]
- 5.Bélanger LF. 1969. Osteocytic osteolysis. Calcif. Tissue Res 4:1–12 [DOI] [PubMed] [Google Scholar]
- 6.Lanyon LE. 1993. Osteocytes, strain detection, bone modeling and remodeling. Calcif. Tissue Int 53:S102–6 [DOI] [PubMed] [Google Scholar]
- 7.Van Der Plas A, Aarden EM, Feijen JH, de Boer AH, Wiltink A, et al. 1994. Characteristics and properties of osteocytes in culture. J. Bone Miner. Res 9:1697–704 [DOI] [PubMed] [Google Scholar]
- 8.Mikuni-Takagaki Y, Kakai Y, Satoyoshi M, Kawano E, Suzuki Y, et al. 1995. Matrix mineralization and the differentiation of osteocyte-like cells in culture. J. Bone Miner. Res 10:231–42 [DOI] [PubMed] [Google Scholar]
- 9.Mikuni-Takagaki Y, Suzuki Y, Kawase T, Saito S. 1996. Distinct responses of different populations of bone cells to mechanical stress. Endocrinology 137:2028–35 [DOI] [PubMed] [Google Scholar]
- 10.Manolagas SC. 2000. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev 21:115–37 [DOI] [PubMed] [Google Scholar]
- 11.Dallas SL, Veno PA. 2012. Live imaging of bone cell and organ cultures. Methods Mol. Biol 816:425–57 [DOI] [PubMed] [Google Scholar]
- 12.Tanaka-Kamioka K, Kamioka H, Ris H. 1998. Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J. Bone Miner. Res 10:1555–68 [DOI] [PubMed] [Google Scholar]
- 13.Verborgt O, Gibson GJ, Schaffler MB. 2000. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J. Bone Miner. Res 15:60–67 [DOI] [PubMed] [Google Scholar]
- 14.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. 2011. Matrix-embedded cells control osteoclast formation. Nat. Med 17:1235–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, et al. 2011. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med 17:1231–34 [DOI] [PubMed] [Google Scholar]
- 16.Yao W, Dai W, Jiang JX, Lane NE. 2013. Glucocorticoids and osteocyte autophagy. Bone 54:279–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tate MLK, Adamson JR, Tami AE, Bauer TW. 2004. The osteocyte. Int. J. Biochem. Cell Biol 36:1–8 [DOI] [PubMed] [Google Scholar]
- 18.Jilka RL, O’Brien CA. 2016. The role of osteocytes in age-related bone loss. Curr. Osteoporos. Rep 14:16–25 [DOI] [PubMed] [Google Scholar]
- 19.Boskey AL, Coleman R. 2010. Aging and bone. J. Dent. Res 89:1333–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, et al. 2017. Degeneration of the osteocyte network in the C57BL/6 mouse model of aging. Aging 9:2190–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi K, Nojiri H, Saita Y, Morikawa D, Ozawa Y, et al. 2015. Mitochondrial superoxide in osteocytes perturbs canalicular networks in the setting of age-related osteoporosis. Sci. Rep 5:9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, et al. 2016. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res 31:1920–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato Y, Windle JJ, Koop BA, Mundy GR, Bonewald LF. 1997. Establishment of an osteocyte-like cell line, MLO-Y4. J. Bone Miner. Res 12:2014–23 [DOI] [PubMed] [Google Scholar]
- 24.Bodine PV, Vernon SK, Komm BS. 1996. Establishment and hormonal regulation of a conditionally transformed preosteocytic cell line from adult human bone. Endocrinology 137:4592–604 [DOI] [PubMed] [Google Scholar]
- 25.Kato Y, Boskey A, Spevak L, Dallas M, Hori M, Bonewald LF. 2001. Establishment of an osteoid preosteocyte-like cell MLO-A5 that spontaneously mineralizes in culture. J. Bone Miner. Res 16:1622–33 [DOI] [PubMed] [Google Scholar]
- 26.Barragan-Adjemian C, Nicolella D, Dusevich V, Dallas MR, Eick JD, Bonewald LF. 2006. Mechanism by which MLO-A5 late osteoblasts/early osteocytes mineralize in culture: similarities with mineralization of lamellar bone. Calcif. Tissue Int 79:340–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF. 2011. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J. Bone Miner. Res 26:2634–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spatz JM, Wein MN, Gooi JH, Qu Y, Garr JL, et al. 2015. The Wnt inhibitor sclerostin is up-regulated by mechanical unloading in osteocytes in vitro. J. Biol. Chem 290:16744–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalajzic I, Braut A, Guo D, Jiang X, Kronenberg MS, et al. 2004. Dentin matrix protein 1 expression during osteoblastic differentiation, generation of an osteocyte GFP-transgene. Bone 35:74–82 [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Le L, Chun BM, Tiede-Lewis LAM, Shiflett LA, et al. 2019. A novel osteogenic cell line that differentiates into GFP-tagged osteocytes and forms mineral with a bone-like lacunocanalicular structure. J. Bone Miner. Res 34:979–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. 2007. DMP1-targeted Cre expression in odontoblasts and osteocytes. J. Dent. Res 86:320–25 [DOI] [PubMed] [Google Scholar]
- 32.Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, et al. 2002. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J. Bone Miner. Res 17:15–25 [DOI] [PubMed] [Google Scholar]
- 33.Powell WF Jr., Barry KJ, Tulum I, Kobayashi T, Harris SE, et al. 2011. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol 209:21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, et al. 2015. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLOS ONE 10:e0138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurel DB, Matsumoto T, Vallejo JA, Johnson ML, Dallas SL, et al. 2019. Characterization of a novel murine Sost ERT2 Cre model targeting osteocytes. Bone Res. 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster DJ, Schneider P, Dallas SL, Muller R. 2013. Studying osteocytes within their environment. Bone 54:285–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genthial R, Beaurepaire E, Schanne-Klein MC, Peyrin F, Farlay D, et al. 2017. Label-free imaging of bone multiscale porosity and interfaces using third-harmonic generation microscopy. Sci. Rep 7:3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamel-ElSayed SA, Tiede-Lewis LM, Lu Y, Veno PA, Dallas SL. 2015. Novel approaches for two and three dimensional multiplexed imaging of osteocytes. Bone 76:129–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishihara Y, Sugawara Y, Kamioka H, Kawanabe N, Kurosaka H, et al. 2012. In situ imaging of the autonomous intracellular Ca2+ oscillations of osteoblasts and osteocytes in bone. Bone 50:842–52 [DOI] [PubMed] [Google Scholar]
- 40.Dallas SL, Veno PA, Rosser JL, Barragan-Adjemian C, Rowe DW, et al. 2009. Time lapse imaging techniques for comparison of mineralization dynamics in primary murine osteoblasts and the late osteoblast/early osteocyte-like cell line MLO-A5. Cells Tissues Organs 189:6–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, Hoshijima M, Sunaga J, Nishida T, Hashimoto M, et al. 2018. Analysis of Ca2+ response of osteocyte network by three-dimensional time-lapse imaging in living bone. J. Bone Miner. Metab 36:519–28 [DOI] [PubMed] [Google Scholar]
- 42.Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, et al. 2012. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res 27:1018–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buenzli PR, Sims NA. 2015. Quantifying the osteocyte network in the human skeleton. Bone 75:144–50 [DOI] [PubMed] [Google Scholar]
- 44.Jahn K, Kelkar S, Zhao H, Xie Y, Tiede-Lewis LM, et al. 2017. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J. Bone Miner. Res 32:1761–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wysolmerski JJ. 2013. Osteocytes remove and replace perilacunar mineral during reproductive cycles. Bone 54:230–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodionova NV, Oganov VS, Zolotova NV. 2002. Ultrastructural changes in osteocytes in microgravity conditions. Adv. Space Res 30:765–70 [DOI] [PubMed] [Google Scholar]
- 47.Blaber EA, Dvorochkin N, Lee C, Alwood JS, Yousuf R, et al. 2013. Microgravity induces pelvic bone loss through osteoclastic activity, osteocytic osteolysis, and osteoblastic cell cycle inhibition by CDKN1a/p21. PLOS ONE 8:e61372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zallone AZ, Teti A, Primavera MV, Pace G. 1983. Mature osteocytes behaviour in a repletion period: the occurrence of osteoplastic activity. Basic Appl. Histochem 27:191–204 [PubMed] [Google Scholar]
- 49.Kogawa M, Wijenayaka AR, Ormsby RT, Thomas GP, Anderson PH, et al. 2013. Sclerostin regulates release of bone mineral by osteocytes by induction of carbonic anhydrase 2. J. Bone Miner. Res 28:2436–48 [DOI] [PubMed] [Google Scholar]
- 50.Tsourdi E, Jahn K, Rauner M, Busse B, Bonewald LF. 2018. Physiological and pathological osteocytic osteolysis. J. Musculoskelet. Neuronal Interact 18:292–303 [PMC free article] [PubMed] [Google Scholar]
- 51.Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, et al. 2006. Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J. Bone Miner. Res 21:466–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tokarz D, Martins JS, Petit ET, Lin CP, Demay MB, Liu ES. 2018. Hormonal regulation of osteocyte perilacunar and canalicular remodeling in the Hyp mouse model of X-linked hypophosphatemia. J. Bone Miner. Res 33:499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolff J 1892. Das Gesetz der Transformen der Knochen. Berlin: A. Hirschwald [Google Scholar]
- 54.Frost HM. 1960. Measurement of osteocytes per unit volume and volume components of osteocytes and canaliculae in man. Henry Ford Hosp. Med. Bull 8:208–11 [PubMed] [Google Scholar]
- 55.Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH. 1995. Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts—correlation with prostaglandin upregulation. Biochem. Biophys. Res. Commun 217:640–48 [DOI] [PubMed] [Google Scholar]
- 56.Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA,et al. 1995. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 9:441–45 [DOI] [PubMed] [Google Scholar]
- 57.Kamel MA, Picconi JL, Lara-Castillo N, Johnson ML. 2010. Activation of β-catenin signaling in MLO-Y4 osteocytic cells versus 2T3 osteoblastic cells by fluid flow shear stress and PGE2: implications for the study of mechanosensation in bone. Bone 47:872–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu XL, Huo B, Chiang V, Guo XE. 2012. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. J. Bone Miner. Res 27:563–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burra S, Nicolella DP, Francis WL, Freitas CJ, Mueschke NJ, et al. 2010. Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. PNAS 107:13648–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adachi T, Aonuma Y, Tanaka M, Hojo M, Takano-Yamamoto T, Kamioka H. 2009. Calcium response in single osteocytes to locally applied mechanical stimulus: differences in cell process and cell body. J. Biomech 42:1989–95 [DOI] [PubMed] [Google Scholar]
- 61.Palumbo C, Ferretti M, Marotti G. 2004. Osteocyte dendrogenesis in static and dynamic bone formation: an ultrastructural study. Anat. Rec. A Discov. Mol. Cell. Evol. Biol 278:474–80 [DOI] [PubMed] [Google Scholar]
- 62.Grimston SK, Watkins MP, Brodt MD, Silva MJ, Civitelli R 2012. Enhanced periosteal and endocortical responses to axial tibial compression loading in conditional connexin43 deficient mice. PLOS ONE 7:e44222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimston SK, Screen J, Haskell JH, Chung DJ, Brodt MD, et al. 2006. Role of connexin43 in osteoblast response to physical load. Ann. N. Y. Acad. Sci 1068:214–24 [DOI] [PubMed] [Google Scholar]
- 64.Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, et al. 2008. Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J. Biol. Chem 283:26374–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Litzenberger JB, Kim JB, Tummala P, Jacobs CR. 2010. β1 Integrins mediate mechanosensitive signaling pathways in osteocytes. Calcif. Tissue Int 86:325–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Watabe H, Furuhama T, Tani-Ishii N, Mikuni-Takagaki Y. 2011. Mechanotransduction activates α5β1 integrin and PI3K/Akt signaling pathways in mandibular osteoblasts. Exp. Cell Res 317:2642–49 [DOI] [PubMed] [Google Scholar]
- 67.Phillips JA, Almeida EA, Hill EL, Aguirre JI, Rivera MF, et al. 2008. Role for β1 integrins in cortical osteocytes during acute musculoskeletal disuse. Matrix Biol. 27:609–18 [DOI] [PubMed] [Google Scholar]
- 68.Lyons JS, Joca HC, Law RA, Williams KM, Kerr JP, et al. 2017. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci. Signal 10:eaan5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mizoguchi F, Mizuno A, Hayata T, Nakashima K, Heller S, et al. 2008. Transient receptor potential vanilloid 4 deficiency suppresses unloading-induced bone loss. J. Cell. Physiol 216:47–53 [DOI] [PubMed] [Google Scholar]
- 70.Genetos DC, Kephart CJ, Zhang Y, Yellowley CE, Donahue HJ. 2007. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J. Cell. Physiol 212:207–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang JN, Zhao Y, Liu C, Han ES, Yu X, et al. 2015. The role of the sphingosine-1-phosphate signaling pathway in osteocyte mechanotransduction. Bone 79:71–78 [DOI] [PubMed] [Google Scholar]
- 72.McGarry JG, Klein-Nulend J, Prendergast PJ. 2005. The effect of cytoskeletal disruption on pulsatile fluid flow-induced nitric oxide and prostaglandin E2 release in osteocytes and osteoblasts. Biochem. Biophys. Res. Commun 330:341–48 [DOI] [PubMed] [Google Scholar]
- 73.Klein-Nulend J, van Oers RF, Bakker AD, Bacabac RG. 2014. Nitric oxide signaling in mechanical adaptation of bone. Osteoporos. Int 25:1427–37 [DOI] [PubMed] [Google Scholar]
- 74.Bakker AD, Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, et al. 2009. Tumor necrosis factor α and interleukin-1β modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum. 60:3336–45 [DOI] [PubMed] [Google Scholar]
- 75.Vatsa A, Smit TH, Klein-Nulend J. 2007. Extracellular NO signalling from a mechanically stimulated osteocyte. J Biomech. 40(Suppl. 1):S89–95 [DOI] [PubMed] [Google Scholar]
- 76.Inoue M, Ono T, Kameo Y, Sasaki F, Ono T, et al. 2019. Forceful mastication activates osteocytes and builds a stout jawbone. Sci. Rep 9:4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tian F, Wang Y, Bikle DD. 2018. IGF-1 signaling mediated cell-specific skeletal mechano-transduction. J. Orthop. Res 36:576–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao L, Shim JW, Dodge TR, Robling AG, Yokota H. 2013. Inactivation of Lrp5 in osteocytes reduces young’s modulus and responsiveness to the mechanical loading. Bone 54:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Javaheri B, Stern AR, Lara N, Dallas M, Zhao H, et al. 2014. Deletion of a single β-catenin allele in osteocytes abolishes the bone anabolic response to loading. J. Bone Miner. Res 29:705–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kang KS, Hong JM, Robling AG. 2016. Postnatal β-catenin deletion from Dmp1-expressing osteocytes/osteoblasts reduces structural adaptation to loading, but not periosteal load-induced bone formation. Bone 88:138–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, et al. 2012. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50:209–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Broudy VC, Kaushansky K, Shoemaker SG, Aggarwal BB, Adamson JW. 1990. Muramyl dipeptide induces production of hemopoietic growth factors in vivo by a mechanism independent of tumor necrosis factor. J. Immunol 144:3789–94 [PubMed] [Google Scholar]
- 83.Saxon LK, Jackson BF, Sugiyama T, Lanyon LE, Price JS. 2011. Analysis of multiple bone responses to graded strains above functional levels, and to disuse, in mice in vivo show that the human Lrp5 G171V High Bone Mass mutation increases the osteogenic response to loading but that lack of Lrp5 activity reduces it. Bone 49:184–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smalt R, Mitchell FT, Howard RL, Chambers TJ. 1997. Induction of NO and prostaglandin E2 in osteoblasts by wall-shear stress but not mechanical strain. Am. J. Physiol 273:E751–58 [DOI] [PubMed] [Google Scholar]
- 85.Smalt R, Mitchell FT, Howard RL, Chambers TJ. 1997. Mechanotransduction in bone cells: induction of nitric oxide and prostaglandin synthesis by fluid shear stress, but not by mechanical strain. Adv. Exp. Med. Biol 433:311–14 [DOI] [PubMed] [Google Scholar]
- 86.Han Y, Cowin SC, Schaffler MB, Weinbaum S. 2004. Mechanotransduction and strain amplification in osteocyte cell processes. PNAS 101:16689–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Verbruggen SW, McGarrigle MJ, Haugh MG, Voisin MC, McNamara LM. 2015. Altered mechanical environment of bone cells in an animal model of short- and long-term osteoporosis. Biophys. J 108:1587–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonivtch AR, Bonewald LF, Nicolella DP. 2007. Tissue strain amplification at the osteocyte lacuna: a microstructural finite element analysis. J. Biomech 40:2199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nicolella DP, Moravits DE, Gale AM, Bonewald LF, Lankford J. 2006. Osteocyte lacunae tissue strain in cortical bone. J. Biomech 39:1735–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, et al. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–23 [DOI] [PubMed] [Google Scholar]
- 91.Honma M, Ikebuchi Y, Kariya Y, Hayashi M, Hayashi N, et al. 2013. RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J. Bone Miner. Res 28:1936–49 [DOI] [PubMed] [Google Scholar]
- 92.Aoki S, Honma M, Kariya Y, Nakamichi Y, Ninomiya T, et al. 2010. Function of OPG as a traffic regulator for RANKL is crucial for controlled osteoclastogenesis. J. Bone Miner. Res 25:1907–21 [DOI] [PubMed] [Google Scholar]
- 93.Burr DB, Martin RB. 1993. Calculating the probability that microcracks initiate resorption spaces. J. Biomech 26:613–16 [DOI] [PubMed] [Google Scholar]
- 94.Burr DB, Martin RB, Schaffler MB, Radin EL. 1985. Bone remodeling in response to in vivo fatigue microdamage. J. Biomech 18:189–200 [DOI] [PubMed] [Google Scholar]
- 95.Bentolila V, Boyce TM, Fyhrie DP, Drumb R, Skerry TM, Schaffler MB. 1998. Intracortical remodeling in adult rat long bones after fatigue loading. Bone 23:275–81 [DOI] [PubMed] [Google Scholar]
- 96.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. 2009. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J. Bone Miner. Res 24:597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheung WY, Fritton JC, Morgan SA, Seref-Ferlengez Z, Basta-Pljakic J, et al. 2016. Pannexin-1 and P2X7-receptor are required for apoptotic osteocytes in fatigued bone to trigger RANKL production in neighboring bystander osteocytes. J. Bone Miner. Res 31:890–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harris SE, MacDougall M, Horn D, Woodruff K, Zimmer SN, et al. 2012. Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone 50:42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blanchard F, Duplomb L, Baud’huin M, Brounais B. 2009. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. 20:19–28 [DOI] [PubMed] [Google Scholar]
- 100.Pathak JL, Bakker AD, Luyten FP, Verschueren P, Lems WF, et al. 2016. Systemic inflammation affects human osteocyte-specific protein and cytokine expression. Calcif. Tissue Int 98:596–608 [DOI] [PubMed] [Google Scholar]
- 101.Kogianni G, Mann V, Noble BS. 2008. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J. Bone Miner. Res 23:915–27 [DOI] [PubMed] [Google Scholar]
- 102.He F, Bai J, Wang J, Zhai J, Tong L, Zhu G. 2019. Irradiation-induced osteocyte damage promotes HMGB1-mediated osteoclastogenesis in vitro. J. Cell. Physiol 234:17314–25 [DOI] [PubMed] [Google Scholar]
- 103.Nakano M, Ikegame M, Igarashi-Migitaka J, Maruyama Y, Suzuki N, Hattori A 2019. Suppressive effect of melatonin on osteoclast function via osteocyte calcitonin. J. Endocrinol 242:13–23 [DOI] [PubMed] [Google Scholar]
- 104.O’Brien W, Fissel BM, Maeda Y, Yan J, Ge X, et al. 2016. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 68:2889–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Parfitt AM. 1982. The coupling of bone formation to bone resorption: a critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab. Bone Dis. Relat. Res 4:1–6 [DOI] [PubMed] [Google Scholar]
- 106.Ajubi NE, Klein-Nulend J, Nijweide PJ, Vrijheid-Lammers T, Alblas MJ, Burger EH. 1996. Pulsating fluid flow increases prostaglandin production by cultured chicken osteocytes—a cytoskeleton-dependent process. Biochem. Biophys. Res. Commun 225:62–68 [DOI] [PubMed] [Google Scholar]
- 107.Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, et al. 2017. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J. Clin. Investig 127:2678–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kringelbach TM, Aslan D, Novak I, Ellegaard M, Syberg S, et al. 2015. Fine-tuned ATP signals are acute mediators in osteocyte mechanotransduction. Cell Signal 27:2401–9 [DOI] [PubMed] [Google Scholar]
- 109.van Bezooijen RL, Deruiter MC, Vilain N, Monteiro RM, Visser A, et al. 2007. SOST expression is restricted to the great arteries during embryonic and neonatal cardiovascular development. Dev. Dyn 236:606–12 [DOI] [PubMed] [Google Scholar]
- 110.Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, et al. 2011. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage 19:874–85 [DOI] [PubMed] [Google Scholar]
- 111.Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, et al. 2006. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 39:754–66 [DOI] [PubMed] [Google Scholar]
- 112.Paic F, Igwe JC, Nori R, Kronenberg MS, Franceschetti T, et al. 2009. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone 45:682–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Taylor S, Ominsky MS, Hu R, Pacheco E, He YD, et al. 2016. Time-dependent cellular and transcriptional changes in the osteoblast lineage associated with sclerostin antibody treatment in ovariectomized rats. Bone 84:148–59 [DOI] [PubMed] [Google Scholar]
- 114.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, et al. 2008. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem 283:5866–75 [DOI] [PubMed] [Google Scholar]
- 115.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, et al. 2005. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146:4577–83 [DOI] [PubMed] [Google Scholar]
- 116.Genetos DC, Yellowley CE, Loots GG. 2011. Prostaglandin E2 signals through PTGER2 to regulate sclerostin expression. PLOS ONE 6:e17772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Igwe JC, Jiang X, Paic F, Ma L, Adams DJ, et al. 2009. Neuropeptide Y is expressed by osteocytes and can inhibit osteoblastic activity. J. Cell. Biochem 108:621–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, et al. 2006. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet 38:1310–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karsenty G 2017. Update on the biology of osteocalcin. Endocr. Pract 23:1270–74 [DOI] [PubMed] [Google Scholar]
- 120.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, et al. 2004. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig 113:561–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Quarles LD. 2012. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol 8:276–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. 2003. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J. Biol. Chem 278:37419–26 [DOI] [PubMed] [Google Scholar]
- 123.Rowe PS. 2004. The wrickkened pathways of FGF23, MEPE and PHEX. Crit. Rev. Oral Biol. Med 15:264–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rowe PS, Garrett IR, Schwarz PM, Carnes DL, Lafer EM,et al. 2005. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: a model for impaired mineralization in X-linked rickets (HYP). Bone 36:33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cheng B, Kato Y, Zhao S, Luo J, Sprague E, et al. 2001. PGE2 is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology 142:3464–73 [DOI] [PubMed] [Google Scholar]
- 126.Huang J, Romero-Suarez S, Lara N, Mo C, Kaja S, et al. 2017. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/β-catenin pathway. JBMR Plus 1:86–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huang J, Wang K, Shiflett LA, Brotto L, Bonewald LF, et al. 2019. Fibroblast Growth Factor 9 (FGF9)—a potential osteokine with dual effects: inhibition of myogenic differentiation, and enhancement of proliferation. Cell Cycle 18:3562–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zoch ML, Clemens TL, Riddle RC. 2016. New insights into the biology of osteocalcin. Bone 82:42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, et al. 2018. Irisin mediates effects on bone and fat via αV integrin receptors. Cell 175:1756–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kitase Y, Vallejo JA, Gutheil W, Vemula H, Jähn K, et al. 2018. β-Aminoisobutyric acid, l-BAIBA, is a muscle-derived osteocyte survival factor. Cell Rep. 22:1531–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, et al. 2015. The myokine irisin increases cortical bone mass. PNAS 112:12157–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Colaianni G, Mongelli T, Cuscito C, Pignataro P, Lippo L, et al. 2017. Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Sci. Rep 7:2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Maurel DB, Jähn K, Lara-Castillo N. 2017. Muscle-bone crosstalk: emerging opportunities for novel therapeutic approaches to treat musculoskeletal pathologies. Biomedicines 5:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paszty C, Turn CH, Robinson MK. 2010. Sclerostin: a gem from the genome leads to bone building antibodies. J. Bone Miner. Res 25:1897–904 [DOI] [PubMed] [Google Scholar]
- 135.Ominsky MS, Boyce RW, Li X, Ke HZ. 2017. Effects of sclerostin antibodies in animal models of osteoporosis. Bone 96:63–75 [DOI] [PubMed] [Google Scholar]
- 136.McClung MR. 2018. Romosozumab for the treatment of osteoporosis. Osteoporos. Sarcopenia 4:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Imel E, White KE. 2018. Pharmacological management of X-linked hypophosphateaemia. Br. J. Clin. Pharmacol 85:1188–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bonewald LF, Wacker MJ. 2013. FGF23 production by osteocytes. Pediatr. Nephrol 28:563–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pereira RC, Jüppner H, Azucena-Serrano CE, Yadin O, Salusky IB, Wesseling-Perry K. 2009. Patterns of FGF-23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone 45:1161–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Takeyari S, Yamamoto T, Kinoshita Y, Glorieux FH, Michigami T, et al. 2014. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone 67:56–62 [DOI] [PubMed] [Google Scholar]
- 141.Leifheit-Nestler M, Haffner D. 2018. Paracrine effects of FGF23 on the heart. Front. Endocrinol 9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhao S, Kato Y, Zhang Y, Harris S, Ahuja SS, Bonewald LF. 2002. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res 17:2068–79 [DOI] [PubMed] [Google Scholar]
- 143.Tanaka K, Yamaguchi Y, Hakeda Y. 1995. Isolated chick osteocytes stimulate formation and bone-resorbing activity of osteoclast-like cells. J. Bone Miner. Metab 13:61–70 [Google Scholar]
- 144.Miller PD. 2011. A review of the efficacy and safety of denosumab in postmenopausal women with osteoporosis. Ther. Adv. Musculoskel. Dis 3:271–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bellido T, Plotkin LI. 2011. Novel actions of bisphosphonates in bone: preservation of osteoblast and osteocyte viability. Bone 49:50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pajevic PD, Krause DS. 2019. Osteocyte regulation of bone and blood. Bone 119:13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Atkinson EG, Delgado-Calle J. 2019. The emerging role of osteocytes in cancer in bone. JBMR Plus 3:e10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhou M, Li S, Pathak JL 2019. Pro-inflammatory cytokines and osteocytes. Curr. Osteoporos. Rep 17:97–104 [DOI] [PubMed] [Google Scholar]