

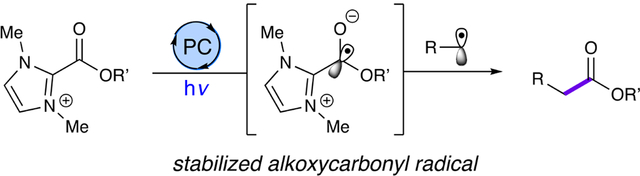
Abstract
Despite recent advancements in the selective generation and coupling of organic radical species, the alkoxycarbonyl radical remains underexplored relative to other carbon-containing radical species. Drawing inspiration from new strategies for generating acyl radical equivalents utilizing dual N-heterocyclic carbene catalysis and photocatalysis, we have prepared dimethylimidazolium esters that can function as an alkoxycarbonyl radical surrogate under photocatalytic conditions. We demonstrate the synthetic utility of these azolium-based partners through the preparation of esters arising from the coupling of this radical surrogate with an oxidatively generated alkyl radical.
Keywords: Photocatalysis, N-Heterocyclic Carbene, Alkoxycarbonyl radical, Esterification, Radical-radical coupling
Graphical Abstract
1. Introduction
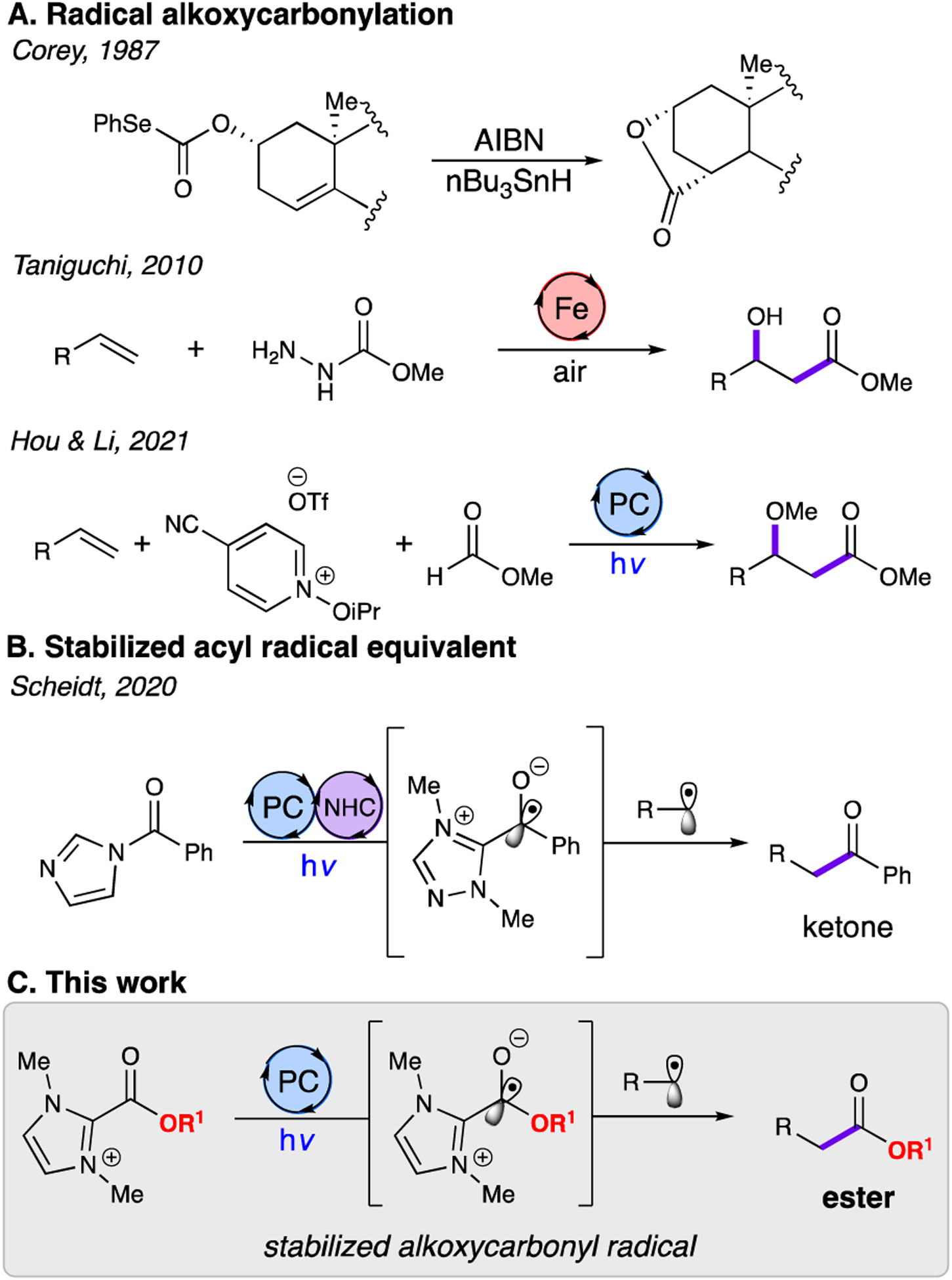
During the past few decades, organic synthetic methodology has seen a resurgence of interest in radical reactivity. Out of the various carbon-containing radical species that have recently shown novel applications, acyl radicals have attracted special interest due to their potential synthetic utility.1 A subset of acyl radicals, alkoxycarbonyl radicals, are difficult to generate and extremely short-lived due to their tendency to decarboxylate and decarbonylate, generating undesired species in situ that can then participate in competing reactions.2 Nevertheless, they have been historically employed in intramolecular reactions to construct complex lactones from acylic esters. One representative example is from Corey and coworkers’ 1987 synthesis of atractyligenin, wherein a phenylselenocarbonate, in the presence of tributyltinhydride and azobisisobutyronitrile (AIBN), fragments to form an alkoxycarbonyl radical (Fig. 1A). This alkoxycarbonyl radical is trapped by an adjacent alkene to form a bridging lactone.3 Trost and coworkers employed a similar strategy to construct the key lactone component of (−)-pseudolaric acid B.4
Figure 1:
Synthetic applications of alkoxycarbonyl radicals, development of NHC-stabilized acyl radicals, and design of a stabilized alkoxycarbonyl radical
Recent advancements in radical chemistry have enabled the selective generation of a much broader variety of useful carbon-containing radicals, but the alkoxycarbonyl radical remains comparatively unexplored due to its relative instability. In 2010, Taniguchi and coworkers demonstrated the catalytic generation of alkoxycarbonyl radicals from carbazates (Fig. 1A).5 Using an iron (II) catalyst and a superstoichiometric quantity of alkoxycarbonyl radical precursor, their group was able to difunctionalize alkenes using the alkoxycarbonyl radical. In contrast, a recent report from the groups of Hou and Li details the generation of alkoxycarbonyl radicals through hydrogen atom abstraction from formate esters by a reductively generated isopropoxy radical.6 Notably, the reaction was performed using the formate ester as the solvent which limited the scope of compatible ester substrates (Fig.1A).7
To further expand the reactivity accessible by single-electron transformations, our group has developed photochemical N-heterocyclic carbene (NHC) catalysis.8 Work from the Ohmiya group in this field demonstrated that thiazolium-derived Breslow intermediates can be spontaneously oxidized to generate a stabilized acyl radical that engages in radical-radical coupling.9 Subsequently, our group identified that acyl azoliums, both isolated8a and generated in-situ8b, can undergo single-electron reduction to generate stabilized acyl radical equivalents for coupling, providing ketones in high yield under mild conditions (Fig.1B). Studer and others have since harnessed the reaction of single-electron NHC operators to forge key C–C and C–X bonds.10
Drawing inspiration from the NHC-derived stabilization demonstrated earlier by our group and others, we sought to develop a modular alkoxycarbonyl radical surrogate for the formation of C–C bonds. An azolium ester, following single-electron transfer, could act as a stabilized alkoxycarbonyl radical to couple with an oxidatively generated alkyl radical (Fig. 1C). If successful, this reaction would be, to the best of our knowledge of the literature, the first intermolecular radical-radical coupling employing an alkoxycarbonyl radical.
2. Results and Discussion
2.1. Initial Screening and Optimization
Our initial investigation of the radical-radical coupling of stabilized alkoxycarbonyl radicals began with the selection of coupling partners. Since we planned on developing an azolium-tethered species that would be reduced by the photocatalyst, an oxidative radical precursor would be needed to turn over the photocatalyst. From a variety of oxidative alkyl radical precursors, we selected potassium benzyltrifluoroborate which was prepared on multigram scale without chromatography. Potassium alkyltrifluoroborates have multiple attractive features including bench stability, substrate diversity, and higher atom economy11 compared to other oxidative alkyl radical precursors.12
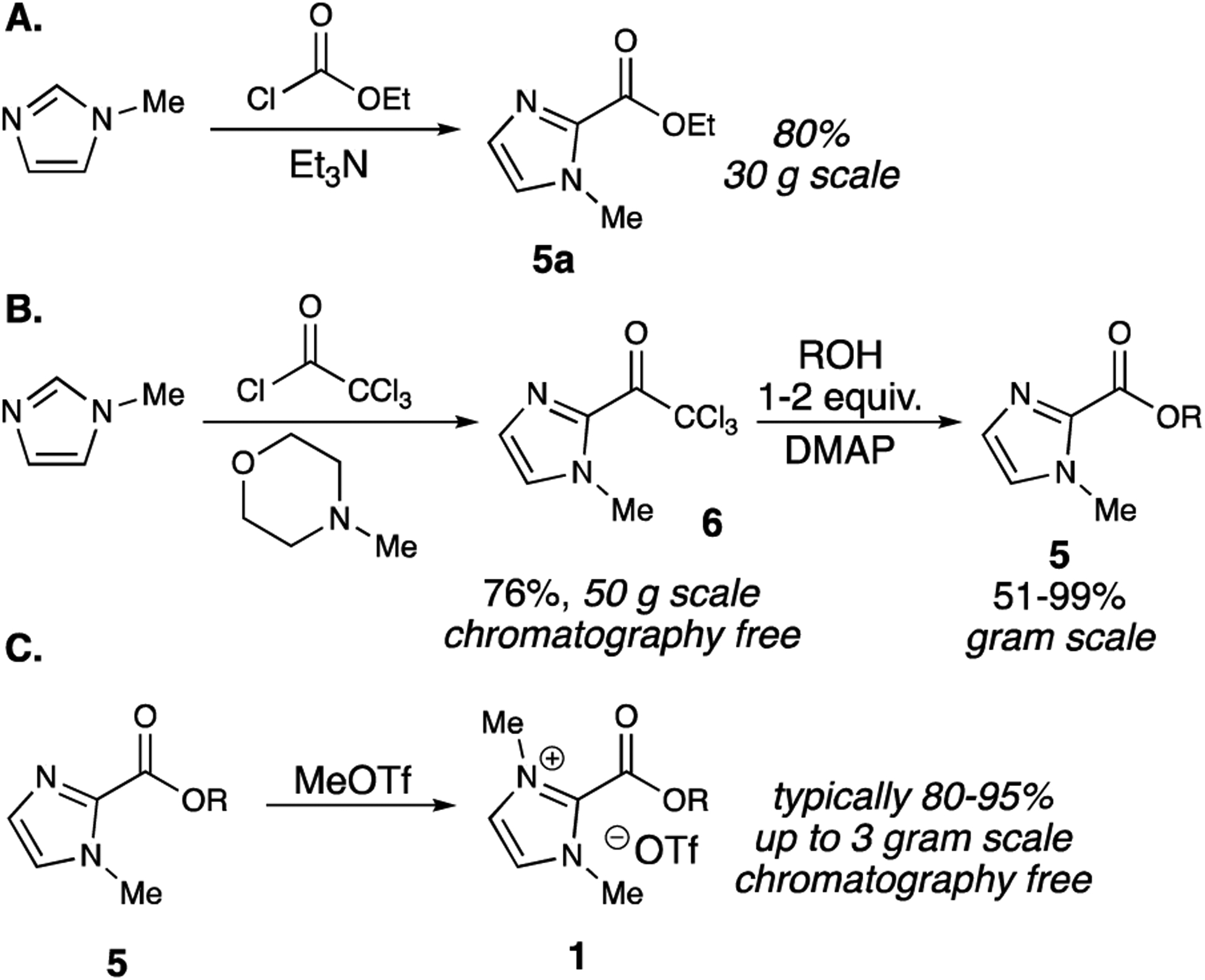
To proceed with the optimization and substrate scope, we required a streamlined route to 1a and other imidazolium esters. Fortunately, the precursor monomethylated imidazole ester 5a could be prepared on decagram scale according to a known procedure (Scheme 1A).13 To synthesize a variety of imidazolium esters, we needed a common precursor that could react directly with a variety of alcohols to yield various precursor esters. This precursor came in the form of 2-trichloroacetyl-N-methylimidazole 6, which could be prepared on 50 gram scale without chromatography.14 Furthermore, reacting 6 with a slight excess of partner alcohol and 4-dimethylaminopyridine (DMAP) gave precursor esters 5 in moderate to high yields (Scheme 1B). With this general route secured, we prepared imidazolium esters 1 in high yields without column chromatography by simple addition of methyl triflate, in up to a 3 gram scale (Scheme 1C).
Scheme 1:
Synthetic routes to imidazolium esters
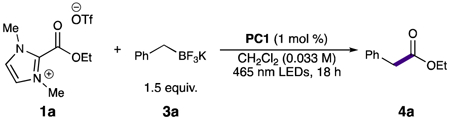
Previous work from our group demonstrated that isolated acyl imidazoliums and acyl benzimidazoliums were both effective reductive precursors to acyl radical equivalents.8a After preparing both the ethyl imidazolium and benzimidazolium esters, screening the two with a variety of solvent systems and photocatalysts provided a number of hits, with the highest-performing utilizing the imidazolium ester in dichloromethane with heteroleptic iridium photocatalyst PC1. Contrary to our group’s previously reported results that determined acyl benzimidazoliums outperform other scaffolds in terms of effiency,8a the benzimidazolium ester precursor provided lower yields than the imidazolium ester (Table 1, entries 1–2), so we decided to pursue optimization using the imidazolium scaffold.
Table 1:
Optimization for radical-radical coupling
 | ||
|---|---|---|
| entry | deviation from standard | GC yield of 4a (%)a |
| 1 | none | 39 |
| 2 | 2a instead of 1a | 21 |
| 3 | CH3CN instead of CH2CI2 | 31 |
| 4 | DMF instead of CH2CI2 | 16 |
| 5 | 1,4-dioxane instead of CH2CI2 | 17 |
| 6 | 2 equiv. 3a | 39 |
| 7 | 0.1 M | 32 |
| 8 | 0.05 M | 38 |
| g | 0.025 M | 38 |
| 10 | 3 mol % PC2 instead of PC1 | 25 |
| 11 | 390 nm LEDs | 36 |
| 12 | 427 nm LEDs | 50 |
| 13 | 1.0 equiv lutidine | 24 |
| 14 | 1.0 equiv Cs2CO3 | 30 |
| 15 | no light | n.d. |
| 16 | no photocatalyst | n.d. |
 | ||
Gas chromatography (GC) yield is based on a calibration curve using 1,3,5-trimethoxybenzene as the internal standard. n.d. = not detected
With ready access to imidazolium esters, we pursued optimization of the radical-radical coupling (Table 1). A solvent screen indicated that the reaction performed better in dichloromethane compared to other more polar solvents (entries 3–5). Further, we discovered that increasing the equivalents of trifluoroborate did not lead to an increase in yield (entry 6). Given the relative insolubility of the trifluoroborate salt in dichloromethane, it was unsurprising that lower reaction concentrations, which would allow for more trifluoroborate in solution, performed better (entries 7–9). The organophotocatalyst PC2, which has similar oxidation and reduction potentials to PC1,15 did not improve the yield (entry 10). Finally, a screen of light sources led to an increase in yield to 50% under 427 nm LED irradiation (entries 11–12). We examined Lewis and Brønsted basic additives commonly employed in reactions using potassium trifluoroborate salts16 but neither approach improved the yields in our system (entries 13–14).
2.2. Trifluoroborate Substrate Scope
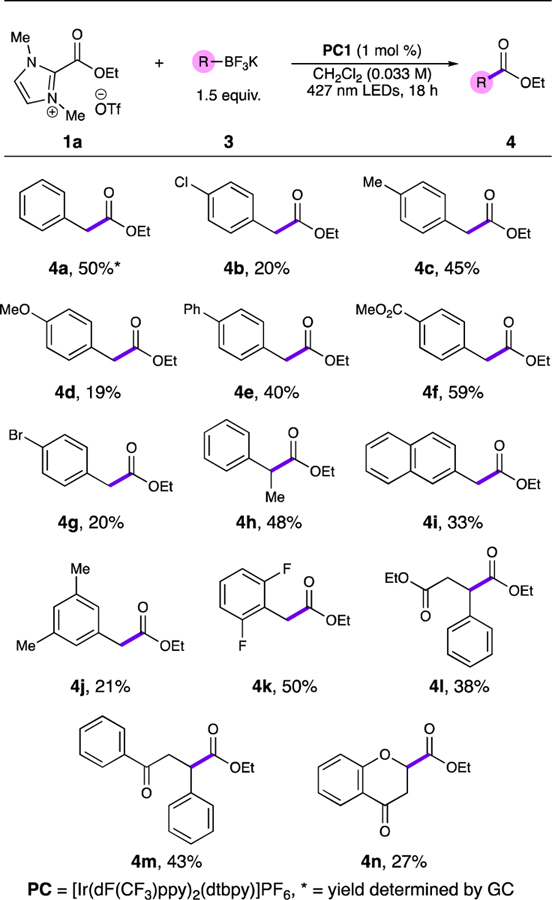
We initially investigated the scope of this radical-radical coupling, initially examining substitution on the alkyl portion of the trifluoroborate salt. A variety of functional groups were tolerated on the aryl ring of the benzyl trifluoroborate in low to moderate yields (4a-4g). The reaction also accommodated a secondary benzylic alkyl group (4h) and a naphthylic group (4i) in moderate yields. Disubstitution was also tolerated on the aryl ring, again in low to moderate yields (4j-k). More interestingly, the radical-radical coupling was accomplished with alkyltrifluoroborates derived from conjugate acceptors (4l-n). These products represent a formal umpolung addition of an ester group to a conjugate acceptor, which remains a challenging transformation currently inaccessible by two-electron NHC chemistry.
Additionally, the chromone-derived trifluoroborate product 4n is generated from a heteroatom-stabilized alkyl radical, rather than a benzylic stabilized alkyl radical, suggesting the reaction could be optimized for other heteroatom-stabilized radicals to produce glycolic and α-amino esters. This product also demonstrates a new way to access 2-carboxychromanone products such as 4n, which form the core of a large number of natural products.17 Secondary alkyl radical precursors (cyclohexyl and 2-indanyl) were also tested under the standard conditions, but provided no desired product.
2.3. Imidazolium Ester Substrate Scope
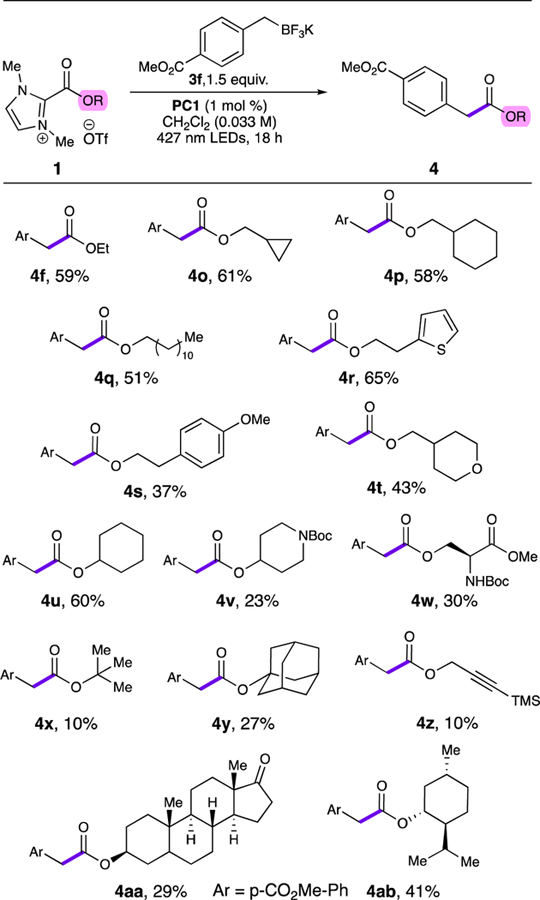
The scope with regard to imidazolium esters was investigated using the ester-containing alkyl trifluoroborate 3f (Table 3). We obtained moderate to good yields with primary and secondary alcohol-derived esters including one containing a sensitive thiophene ring (4f,4o-4u). Substrates containing carbamates proceeded in low to moderate yields, potentially due to competing hydrogen atom transfer from the substrate to the benzylic radical to generate α-amino stabilized radicals (4v,4w). Tertiary alcohol-derived esters provided products in low yields, likely due to competing β-scission of the reduced imidazolium ester resulting in a stabilized tertiary radical (4x,4y). Inclusion of a trimethylsilyl propargyl ester resulted in low yields as well (4z). Finally, esters of more complex alcohols such as epi-androsterone proceeded in low to moderate yields (4aa,4ab). It is worth noting that when starting with enantiopure imidazolium esters, the products were obtained with complete retention of stereochemistry (4w,4aa,4ab). Despite the mixed performance of the coupling, this demonstrates a readily accessible and modular method to prepare these unique di- and triesters.
Table 3:
Scope with regards to imidazolium ester
|
2.4. Mechanistic Studies and Proposed Pathway
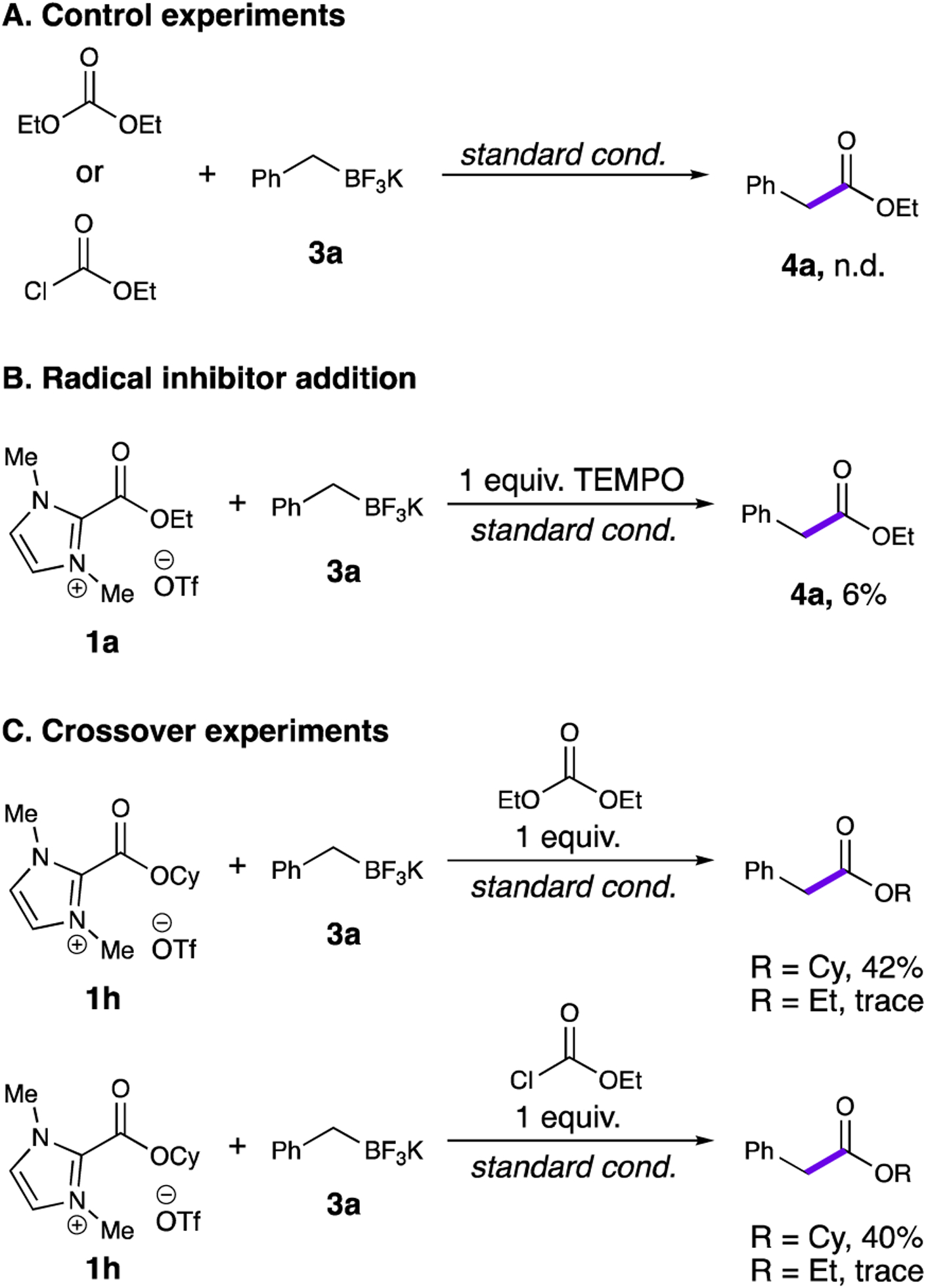
To probe the mechanism of this coupling, several control experiments were performed (Scheme 2). Both light and photocatalyst proved necessary for formation of product (Table 1, entries 15–16). Substituting ethyl chloroformate or diethyl carbonate for 1a also yielded no product, suggesting that the imidazolium ester is necessary to turn over the photocatalyst (Scheme 2A). Addition of one equivalent of the radical trapping agent (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) resulted in dramatically reduced yields, suggesting the involvement of open-shell species (Scheme 2B). In 1994, the Smith group reported the formation of asymmetric carbonates through the addition of alcohols to dimethylimidazolium esters similar to 1a.18 To ensure that the products 4 formed in this reaction were not generated from a nucleophilic addition to the imidazolium ester with elimination of the NHC, we performed a crossover experiment, combining cyclohexyl imidazolium ester 1h with an equimolar amount of diethyl carbonate or ethyl chloroformate and subjecting the mixture to the standard reaction conditions with trifluoroborate 3a (Scheme 2C). For both reactions, the cyclohexyl ester was the major product, with only trace quantities of the ethyl ester detected by gas chromatography mass spectrometry (GC-MS), indicating that the reaction likely proceeds through a direct radical-radical coupling rather than in-situ generation of an anionic carbon nucleophile.
Scheme 2:
Mechanistic studies
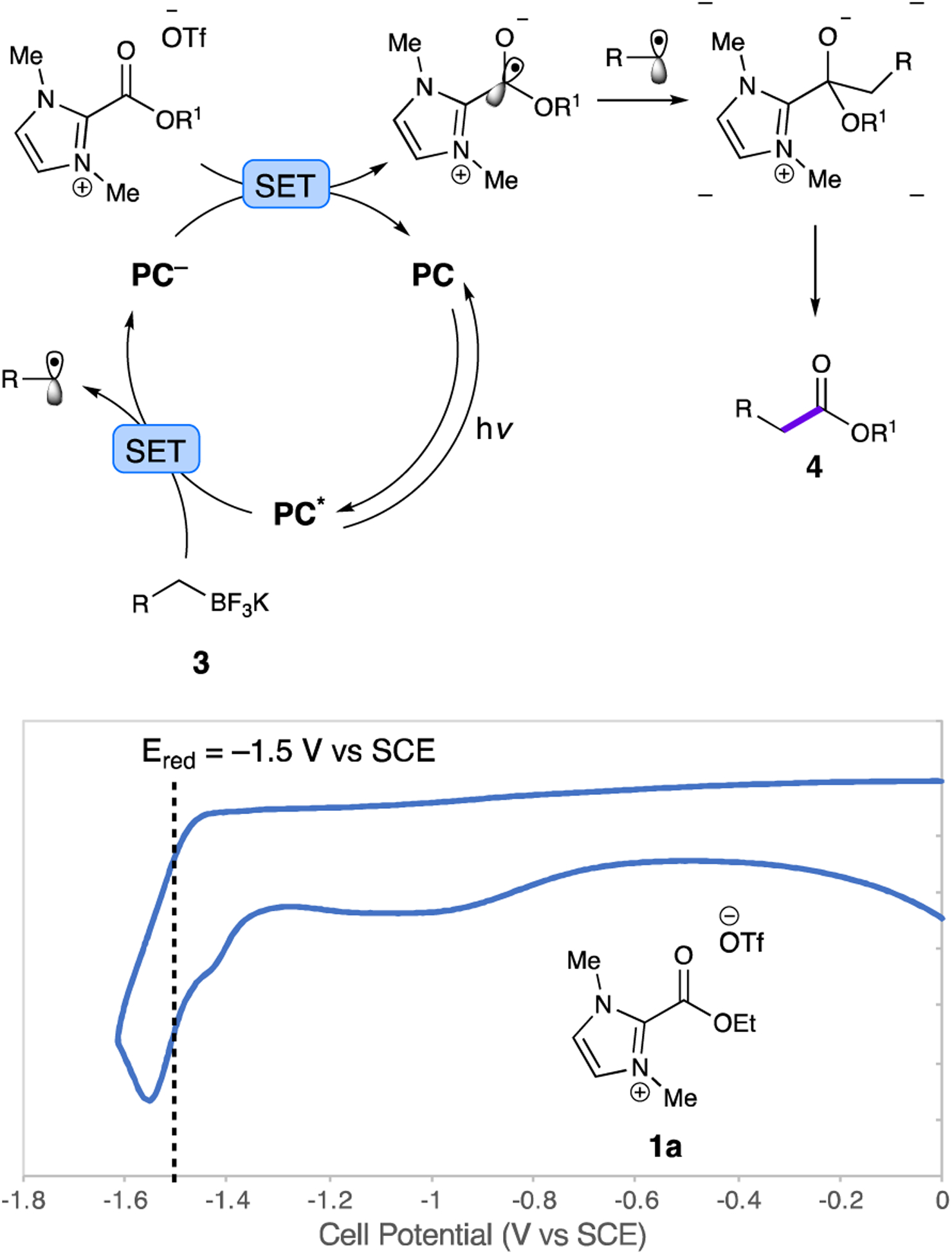
Given the oxidation and reduction potentials of the substrates and PC1,19 we propose the photocatalyst proceeds through a reductive quenching cycle (Fig. 2). The reduction potential of 1a at −1.5 V vs SCE (Fig. 2) is outside the excited state reduction potential of PC1 (E1/2 IrIII*/IrIV = −0.89 V), but more in line with the reduced state reduction potential of PC1 (E1/2 IrIII/IrII = −1.37 V). Moreover, the oxidation potential of 3a, +1.1 V vs SCE,20 is within the excited state oxidation potential of PC1, (E1/2 IrIII*/IrII = +1.21 V). In accordance with cyclic voltammetry data and mechanistic studies, we propose that the mechanism begins with the initial oxidation of the trifluoroborate salt 3 by excited state PC1. Subsequent reduction of 1a by reduced PC1 generates the stabilized alkoxycarbonyl radical. Fragmentation of the oxidized trifluoroborate to the alkyl radical, followed by radical-radical coupling and elimination of the free carbene affords the product ester 4.
Figure 2:
Proposed mechanism/pathway for alkoxycarbonyl radical-radical coupling and cyclic voltammogram of 1a
3. Conclusion
We have demonstrated a new reductive alkoxycarbonyl radical surrogate easily accessed from inexpensive commercial chemicals via two different routes. Inspired by recent developments in single-electron NHC chemistry, we demonstrate the unique potential of the dimethylimidazolium moiety to enable single-electron reduction and stabilize the highly transient alkoxycarbonyl radical. Additionally, we have developed a new radical-radical cross-coupling reaction combining diakylimidazolium esters and potassium alkyltrifluoroborates to prepare esters bearing a variety of functional groups. Current studies are underway to leverage the unique stabilizing properties of N-heterocyclic carbenes to further the scope of NHC-radical surrogates beyond acyl and alkoxycarbonyl radicals.
4. Experimental
4.1. General Information
All reactions were carried out under an argon or nitrogen atmosphere in oven-dried glassware with magnetic stirring. All solvents were purified by passing through a bed of activated alumina, dried over 3Å molecular sieves, and then degassed using the freeze-pump-thaw method (3–4 cycles). Reagents were purified prior to use unless otherwise stated following the guidelines of Perrin and Armarego. Purification of reaction products was carried out by flash chromatography on Biotage Isolera 4 systems with Ultra-grade silica cartridges. Analytical thin layer chromatography was performed on EM Reagent 0.25 mm silica gel 60-F plates. Visualization was accomplished with UV light. 1H NMR spectra were recorded on a Bruker AVANCE III 500 MHz with direct cryoprobe (500 MHz) spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm). Data are reported as (ap = apparent, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, b = broad; coupling constant(s) in Hz; integration.) Proton-decoupled 13C NMR spectra were recorded on a Bruker AVANCE III 500 MHz with direct cryoprobe (125 MHz) spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 77.16 ppm). 19F and 11B NMR spectra were acquired on a 500 MHz Bruker AVANCE III HD spectrometer equipped with a TXO prodigy probe. Reactions were monitored by LCMS or GCMS using a WATERS Acquity-H UPLC-MS with a single quad detector (ESI) or an Agilent 7890 gas chromatograph equipped with a 5975C single quadrupole EI-MS, respectively. High-resolution mass spectrometry (HRMS) was obtained using an Agilent 6201 MSLC-TOF (ESI). FTIR data were collected at room temperature on a Bruker Tensor 37 FTIR Spectrometer equipped with a Mid IR detector and KBr beam splitter. The spectrum was collected in attenuated total reflectance (ATR) mode in the range of 4000 to 600 cm−1. The data were averaged over 16 scans. The OPUS software was used for the data acquisition. Optical rotation data was obtained with an AUTOPOL VI polarimeter using the 589 nm sodium D line. All photocatalytic reactions were irradiated using a SynLED Parallel Photoreactor (465–470 nm) purchased from Sigma-Aldrich or four PR160L Kessil Lamps of the corresponding wavelength (390 or 427 nm).
See Supporting Information for complete structures of all compounds as well as relevant characterization data for new compounds.
4.2. Preparation of Trifluoroborate Salts
Trifluoroborate salts 3a,21 3b,13 3c,13 3d,22 3e,13 3f,22 3g,23 3h,24 3i,22 3j,25 3l,24 3m,26 and 3n27 were prepared according to known procedures and 1H and 11B NMR were consistent with the literature.
4.2.1. Potassium (2,6-difluorobenzyl)trifluoroborate 3k
Potassium (2,6-difluorobenzyl)trifluoroborate 3k was prepared according to a modified literature procedure.22 To an oven-dried 20 mL vial equipped with a stir bar was added 2,6-difluorobenzyl bromide (5 mmol), triphenylphosphine (0.65 mmol), and bis(pinacolato)diboron (7.5 mmol). The vial was taken into the glovebox and copper (I) iodide (0.5 mmol) and lithium methoxide (10 mmol) were added. DMF (10 mL) was added, and the reaction was sealed. The mixture was stirred vigorously for 24 hours, after which the slurry was diluted with EtOAc (10 mL) and filtered through a plug of silica, washing with additional EtOAc (50 mL). The unpurified boronic ester was concentrated to ~25 mL and diluted with methanol (25 mL), then cooled to 0 °C. Saturated aq. KHF2 (7 mL, 6 equiv.) was added dropwise, and the mixture was allowed to come to room temperature. After stirring an additional 1 hr, the solution was concentrated, and residual pinacol/water was removed by azeotropic evaporation with toluene (3×50 mL). The solid was dried under high vacuum overnight, then triturated with acetone (5×50 mL). The solution was concentrated, dissolved in a minimum volume of acetone, and diethyl ether (approx. 3x volume of acetone) was added to give a white precipitate. The solid was filtered, washed with diethyl ether and hexanes, and dried to afford 3k as a white powder (300 mg, 26%). 1H NMR (500 MHz, acetone-d6) □ 6.90 (m, 1H), 6.71 (m, 2H), 1.65 (bs, 2H). 13C NMR (126 MHz, acetone-d6) δ 162.6 (d, J = 10.9 Hz), 160.6 (d, J = 11.2 Hz), 122.8 (t, J = 10.2 Hz), 109.8 (dd, J = 19.5, 7.3 Hz). 19F NMR (470 MHz, acetone-d6) δ −115.87, −140.49 (dd, J = 112.0, 51.9 Hz). 11B NMR (160 MHz, acetone-d6) δ 3.86 (q, J = 58.1 Hz). FTIR (ATR) cm−1: 3229, 1589, 1465, 1261, 1065, 1011, 957, 776. HRMS (ESI/TOF) m/z: [M-K]− Calcd. for C7H5BF5 195.0410; Found 195.0410.
4.3. Preparation of Imidazole Esters
Imidazole ester 5a was prepared according to a known procedure, and 1H NMR was consistent with the literature.28
General procedure for the preparation of N-methyl imidazole esters 5: To an oven-dried 20 mL vial equipped with a stir bar was added 2-trichloroacetyl-N-methylimidazole (5.0 mmol), DMAP (0.5 mmol), and the corresponding alcohol (10.0 mmol). The mixture was dissolved in THF (10 mL), then stirred overnight at room temperature. The unpurified reaction was concentrated and purified by column chromatography (ethyl acetate/hexanes) to afford imidazole ester 5.
4.3.1. cyclopropylmethyl 1-methyl-1H-imidazole-2-carboxylate 5b
Prepared according to the general imidazole ester synthesis using cyclopropylmethanol. Isolated as a white solid (771 mg, 85%). 1H NMR (500 MHz, CDCl3) δ 7.05 (s, 1H), 6.95 (s, 1H), 4.08 (d, J = 7.4 Hz, 2H), 3.92 (s, 3H), 1.32 – 1.12 (m, 1H), 0.62 – 0.46 (m, 2H), 0.37 – 0.25 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.1, 136.5, 129.2, 126.0, 70.0, 35.6, 9.7, 3.4. FTIR (ATR) cm−1: 3103, 3008, 1709, 1427, 1394, 1261, 1137, 959, 789, 662. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C9H13N2O2 181.0972; Found 181.0975.
4.3.2. cyclohexylmethyl 1-methyl-1H-imidazole-2-carboxylate 5c
Prepared according to the general imidazole ester synthesis using cyclohexylmethanol. Isolated as a white solid (937 mg, 84%). 1H NMR (500 MHz, CDCl3) δ 7.06 (s, 1H), 6.95 (s, 1H), 4.07 (d, J = 6.4 Hz, 2H), 3.93 (s, 3H), 1.82 – 1.71 (m, 3H), 1.70 – 1.63 (m, 2H), 1.62 – 1.56 (m, 1H), 1.26 – 1.05 (m, 3H), 1.02 – 0.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.2, 136.6, 129.2, 126.0, 70.1, 36.8, 35.6, 29.5, 26.1, 25.4. FTIR (ATR) cm−1: 3123, 2921, 2852, 1701, 1426, 1257, 1132, 787, 664. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C12H19N2O2 223.1441; Found 223.1441.
4.3.3. dodecyl 1-methyl-1H-imidazole-2-carboxylate 5d
Prepared according to the general imidazole ester synthesis using 1-dodecanol. Isolated as a white solid (1.38 g, 94%). 1H NMR (500 MHz, CDCl3) δ 7.13 (s, 1H), 7.01 (s, 1H), 4.32 (t, J = 6.9 Hz, 2H), 4.00 (s, 3H), 1.78 (p, J = 7.1 Hz, 2H), 1.44 – 1.36 (m, 2H), 1.36 – 1.18 (m, 16H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 159.3, 136.7, 129.3, 126.1, 65.5, 35.8, 31.9, 29.6, 29.6, 29.5, 29.4, 29.3, 29.2, 28.6, 25.9, 22.6, 14.1. FTIR (ATR) cm−1: 3096, 2951, 2920, 2850, 1721, 1466, 1421, 1253, 1135, 799, 663. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C17H31N2O2 295.2380; Found 295.2378.
4.3.4. 2-(thiophen-2-yl)ethyl 1-methyl-1H-imidazole-2-carboxylate 5e
Prepared according to the general imidazole ester synthesis using 2-thiopheneethanol. Isolated as a white solid (1.02 g, 86%). 1H NMR (500 MHz, CDCl3) δ 7.14 – 7.11 (m, 2H), 7.00 (s, 1H), 6.93 – 6.86 (m, 2H), 4.53 (t, J = 7.3 Hz, 2H), 3.94 (s, 3H), 3.30 (td, J = 7.3, 0.8 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 158.8, 139.1, 136.3, 129.4, 126.9, 126.3, 125.6, 123.9, 65.2, 35.7, 29.1. FTIR (ATR) cm−1: 3092, 2953, 1715, 1411, 1258, 1129, 801, 699, 660. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C11H13N2O2S 237.0692; Found 237.0694;
4.3.5. 4-methoxyphenethyl 1-methyl-1H-imidazole-2-carboxylate 5f
Prepared according to the general imidazole ester synthesis using 4-methoxyphenethyl alcohol. Isolated as a clear oil (1.05 g, 81%). 1H NMR (500 MHz, CDCl3) δ 7.18 (d, J = 8.4 Hz, 2H), 7.14 (s, 1H), 7.01 (s, 1H), 6.83 (d, J = 8.4 Hz, 2H), 4.48 (t, J = 7.7 Hz, 2H), 3.96 (s, 3H), 3.77 (s, 3H), 3.05 (t, J = 7.6 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 159.0, 158.3, 136.6, 129.9, 129.4, 129.2, 126.2, 113.9, 65.9, 55.2, 35.8, 34.2. FTIR (ATR) cm−1: 2955, 2835, 2361, 1706, 1512, 1415, 1243, 1123, 1030, 779, 663. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C14H17N2O3 261.1234; Found 261.1235.
4.3.6. (tetrahydro-2H-pyran-4-yl)methyl 1-methyl-1H-imidazole-2-carboxylate 5g
Prepared according to the general imidazole ester synthesis using (tetrahydro-2H-pyran-4-yl)methanol. Isolated as a clear oil (896 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 7.26 (s, 1H), 7.18 (s, 1H), 4.31 (d, J = 6.9 Hz, 2H), 4.13 (s, 3H), 4.10 (dd, J = 11.5, 4.1 Hz, 2H), 3.52 (t, J = 11.1 Hz, 2H), 2.24 (ttt, J = 11.1, 7.1, 3.8 Hz, 1H), 1.86 (d, J = 12.7 Hz, 2H), 1.54 (qd, J = 12.3, 4.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 158.9, 136.2, 129.2, 126.1, 69.0, 67.0, 35.5, 34.2, 29.3. FTIR (ATR) cm−1: 2948, 2843, 1707, 1418, 1257, 1123, 1088, 988, 780, 664. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C11H17N2O3 225.1234; Found 225.1238.
4.3.7. cyclohexyl 1-methyl-1H-imidazole-2-carboxylate 5h
Prepared according to the general imidazole ester synthesis using cyclohexanol. Isolated as a clear oil (1.03 g, 99%). 1H NMR (500 MHz, CDCl3) δ 7.12 (s, 1H), 7.00 (s, 1H), 4.97 (tt, J = 9.9, 4.1 Hz, 1H), 3.99 (s, 3H), 2.08 – 1.95 (m, 2H), 1.88 – 1.78 (m, 2H), 1.68 – 1.54 (m, 3H), 1.47 – 1.32 (m, 2H), 1.32 – 1.18 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 158.8, 137.0, 129.3, 126.0, 74.3, 35.9, 31.6, 25.2, 24.1. FTIR (ATR) cm−1: 2934, 2858, 1702, 1412, 1256, 1127, 1011, 920, 781, 664. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C11H17N2O2 209.1285; Found 209.1286.
4.3.8. tert-butyl 4-((1-methyl-1H-imidazole-2-carbonyl)oxy)piperidine-1-carboxylate 5i
Prepared according to the general imidazole ester synthesis using 1-Boc-4-hydroxypiperidine. Isolated as an amorphous white solid (1.37 g, 88%). 1H NMR (500 MHz, CDCl3) δ 7.15 (s, 1H), 7.03 (s, 1H), 5.14 (tt, J = 8.6, 4.0 Hz, 1H), 4.00 (s, 3H), 3.90 (bs, 2H), 3.16 (ddd, J = 13.4, 9.6, 3.4 Hz, 2H), 2.05 – 1.90 (m, 2H), 1.80 (dtd, J = 13.2, 9.2, 4.1 Hz, 2H), 1.46 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 158.6, 154.8, 136.6, 129.5, 126.4, 79.7, 71.5, 41.6, 35.9, 30.6, 28.4. FTIR (ATR) cm−1: 2967, 2932, 2867, 1716, 1683, 1423, 1233, 1171, 1125, 1025, 795. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C15H24N3O4 310.1761; Found 310.1759.
4.3.9. (S)-2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxopropyl 1-methyl-1H-imidazole-2-carboxylate 5j
Prepared according to the general imidazole ester synthesis using N-Boc-L-serine methyl ester. Isolated as a thick clear oil (574 mg, 35%). 1H NMR (500 MHz, CDCl3) δ 7.07 (s, 1H), 6.99 (s, 1H), 5.93 (d, J = 8.7 Hz, 1H), 4.71 (dd, J = 11.2, 3.9 Hz, 1H), 4.65 (dd, J = 8.4, 4.0 Hz, 1H), 4.46 (dd, J = 11.2, 3.6 Hz, 1H), 3.92 (s, 3H), 3.71 (s, 3H), 1.37 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.1, 158.3, 155.3, 135.7, 129.3, 126.5, 80.0, 64.8, 52.7, 52.6, 35.7, 28.1. FTIR (ATR) cm−1: 2978, 1710, 1510, 1417, 1249, 1157, 1125, 1065, 916, 779, 729, 663. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C14H22N3O6 328.1503; Found 328.1500.
4.3.10. tert-butyl 1-methyl-1H-imidazole-2-carboxylate 5k
To a 25 mL round-bottom flask equipped with a stir bar and reflux condenser was added 2-trichloroacetyl-N-methylimidazole (5 mmol), followed by sodium tert-butoxide (10 mmol). The mixture was suspended in tert-butanol (15 mL) and refluxed for 24 hours, after which it was cooled and diluted with dichloromethane (15 mL). Water (30 mL) was added and the layers separated. The aqueous layer was extracted with dichloromethane (3×15 mL), and the organic layers were combined, dried with sodium sulfate, and concentrated in vacuo. The unpurified residue was purified by silica gel chromatography (ethyl acetate/hexanes) to afford 5j as a tan solid (341 mg, 37%). 1H NMR (500 MHz, CDCl3) δ 7.09 (s, 1H), 6.96 (s, 1H), 3.96 (s, 3H), 1.60 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 158.6, 137.7, 129.0, 125.8, 82.5, 69.1, 35.9, 28.2, 21.8. FTIR (ATR) cm−1: 3110, 2983, 1698, 1410, 1273, 1129, 922, 845, 782, 665. HRMS (ESI/TOF) m/z: [M+Na]+ Calcd. for C9H14N2NaO2 205.0947; Found 205.0951.
4.3.11. adamantan-1-yl 1-methyl-1H-imidazole-2-carboxylate 5l
To a 25 mL round-bottom flask equipped with a stir bar and reflux condenser was added sodium hydride (12 mmol), which was suspended in THF (15 mL). 1-adamantanol (10 mmol) was added in portions, and the mixture was stirred for 2 hours. 2-trichloroacetyl-N-methylimidazole (5 mmol) was added in portions, and the mixture was refluxed for 24 hours. The unpurified mixture was cooled to room temperature, then diluted with dichloromethane (15 mL) and water (15 mL). The layers were separataed, and the aqueous layer further extracted with dichloromethane (3×15 mL). The organic layers were combined, dried with sodium sulfate, and concentrated in vacuo. The unpurified residue was purified by silica gel chromatography (ethyl acetate/hexanes) to afford 5k as a white solid (120 mg, 9%). 1H NMR (500 MHz, CDCl3) δ 7.11 (s, 1H), 6.97 (s, 1H), 3.97 (s, 3H), 2.30 (s, 6H), 2.22 (s, 3H), 1.70 (q, J = 12.3 Hz, 6H), 1.64 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 158.3, 137.8, 129.0, 125.9, 82.8, 41.4, 36.2, 36.0, 31.0. FTIR (ATR) cm−1: 3085, 2911, 2852, 2361, 2338, 1700, 1405, 1258, 1134, 1051, 799, 664. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C15H21N2O2 261.1598; Found 261.1600.
4.3.12. 3-(trimethylsilyl)prop-2-yn-1-yl 1-methyl-1H-imidazole-2-carboxylate 5m
Prepared according to the general imidazole ester synthesis using 3-(trimethylsilyl)propargyl alcohol. Isolated as a clear oil (1.00 g, 85%). 1H NMR (500 MHz, CDCl3) δ 7.15 (s, 1H), 7.04 (s, 1H), 4.91 (s, 2H), 4.00 (s, 3H), 0.15 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 158.3, 136.0, 129.7, 126.5, 98.4, 92.5, 53.2, 35.8, −0.4. FTIR (ATR) cm−1: 2959, 2184, 1716, 1410, 1248, 1114, 1049, 1028, 839, 759, 644. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C11H17N2O2Si 237.1054; Found 237.1054.
4.3.13. (3S,8R,9S,10S,13S,14S)-10,13-dimethyl-17-oxohexadecahydro-1H-cyclopenta[a] phenanthren-3-yl 1-methyl-1H-imidazole-2-carboxylate 5n
Prepared according to the general imidazole ester synthesis using epi-androsterone. Isolated as a white solid (1.02 g, 51%). 1H NMR (500 MHz, CDCl3) δ 7.09 (s, 1H), 6.99 (s, 1H), 4.92 (tt, J = 11.2, 5.0 Hz, 1H), 3.97 (s, 3H), 2.39 (ddd, J = 19.2, 8.9, 1.1 Hz, 1H), 2.02 (dt, J = 19.2, 9.1 Hz, 1H), 1.95 – 1.85 (m, 2H), 1.82 – 1.68 (m, 5H), 1.61 (t, J = 11.8 Hz, 2H), 1.58 – 1.40 (m, 2H), 1.37 – 1.15 (m, 6H), 1.11 – 1.02 (m, 1H), 0.96 (qd, J = 12.2, 5.1 Hz, 1H), 0.84 (s, 3H), 0.82 (s, 3H), 0.70 (ddd, J = 12.2, 10.5, 4.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 158.7, 136.8, 129.2, 126.1, 74.7, 54.2, 51.3, 47.7, 44.7, 36.7, 35.8, 35.7, 35.6, 34.9, 33.8, 31.4, 30.7, 28.2, 27.2, 21.7, 20.4, 13.7, 12.1. FTIR (ATR) cm−1: 2949, 2852, 1732, 1705, 1421, 1256, 1131, 1009, 919, 792, 665. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C24H35N2O3 399.2642; Found 399.2644.
4.3.14. (1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 1-methyl-1H-imidazole-2-carboxylate 5o
Prepared according to the general imidazole ester synthesis using (−)-menthol. Isolated as a white solid (976 mg, 74%). 1H NMR (500 MHz, CDCl3) δ 7.02 (s, 1H), 6.92 (s, 1H), 4.85 (td, J = 10.9, 4.4 Hz, 1H), 3.90 (s, 3H), 2.01 – 1.88 (m, 2H), 1.67 – 1.51 (m, 3H), 1.43 (ddtd, J = 15.1, 12.0, 6.6, 3.2 Hz, 1H), 1.12 (td, J = 12.1, 10.9 Hz, 1H), 0.99 (qd, J = 13.3, 12.7, 12.7, 3.6 Hz, 1H), 0.86 – 0.76 (m, 7H), 0.68 (d, J = 7.0 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 158.7, 136.6, 129.0, 125.9, 75.0, 46.3, 40.5, 35.6, 33.9, 31.3, 25.7, 23.0, 21.7, 20.5, 15.8. FTIR (ATR) cm−1: 2925, 2870, 1702, 1410, 1257, 1126, 955, 783, 665. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C15H25N2O2 265.1911; Found 265.1905.
4.4. Preparation of dimethylimidazolium esters
General procedure for the preparation of dimethylimidazolium esters 1: To an oven-dried 20 mL scintillation vial equipped with a stir bar was added the corresponding imidazole ester 5. The ester was dissolved in diethyl ether (0.1 M), and methyl triflate (1.1 equiv.) was added dropwise with vigorous stirring. The product immediately began precipitating and the mixture was stirred an additional 30 minutes. The mixture was filtered and rinsed with diethyl ether (5 mL). The unpurified solid was then dissolved in a minimum amount of dichloromethane, and then diethyl ether was added dropwise until the product precipitated. The mixture was filtered again, and rinsed with additional diethyl ether (5 mL) and hexanes (5 mL). The product was dried under high vacuum overnight to afford dimethylimidazolium 1.
4.4.1. 2-(ethoxycarbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1a
Prepared according to the general dimethylimidazolium ester synthesis using 5a (20.0 mmol). Isolated as a white powder (5.23 g, 82%). 1H NMR was consistent with literature data.18
4.4.2. 2-((cyclopropylmethoxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1b
Prepared according to the general dimethylimidazolium ester synthesis using 5b (1.0 mmol). Isolated as a white powder (286 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 2H), 4.35 (d, J = 7.7 Hz, 2H), 4.23 (s, 6H), 1.31 (pt, J = 7.9, 4.7 Hz, 1H), 0.77 – 0.67 (m, 2H), 0.43 (dt, J = 6.2, 4.8 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 153.7, 126.7, 73.7, 39.6, 9.4, 3.8. FTIR (ATR) cm−1: 3132, 2361, 2339, 1735, 1448, 1248, 1222, 1153, 1026, 944, 795, 637. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C10H15N2O2 195.1128; Found 195.1128.
4.4.3. 2-((cyclohexylmethoxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1c
Prepared according to the general dimethylimidazolium ester synthesis using 5c (1.0 mmol). Isolated as a white powder (335 mg, 87%). 1H NMR (500 MHz, CDCl3) δ 7.73 (s, 2H), 4.27 (d, J = 5.9 Hz, 2H), 4.15 (s, 6H), 1.85 – 1.63 (m, 6H), 1.32 – 1.20 (m, 2H), 1.15 (qt, J = 12.7, 3.1 Hz, 1H), 1.02 (qd, J = 13.1, 12.1, 3.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 153.8, 132.2, 126.7, 120.6 (q, J = 320.2 Hz), 73.3, 39.4, 36.7, 29.6, 26.0, 25.4. FTIR (ATR) cm−1: 3126, 2935, 2860, 2360, 1737, 1448, 1261, 1149, 1029, 637. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C13H21N2O2 237.1598; Found 237.1602.
4.4.4. 2-((dodecyloxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1d
Prepared according to the general dimethylimidazolium ester synthesis using 5d (1.0 mmol). Isolated as a white powder (377 mg, 82%). 1H NMR (500 MHz, CDCl3) δ 7.76 (s, 2H), 4.48 (t, J = 6.9 Hz, 2H), 4.18 (s, 6H), 1.82 (p, J = 7.1 Hz, 2H), 1.51 – 1.20 (m, 18H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.8, 132.3, 126.8, 120.6 (q, J = 320.5 Hz), 68.6, 39.4, 31.9, 29.6, 29.6, 29.4, 29.3, 29.1, 28.2, 25.8, 22.7, 14.1. FTIR (ATR) cm−1: 3130, 2915, 2851, 2361, 2338, 1735, 1456, 1274, 1248, 1153, 1030, 640. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C18H33N2O2 309.2537; Found 309.2532.
4.4.5. 1,3-dimethyl-2-((2-(thiophen-2-yl)ethoxy)carbonyl)-1H-imidazol-3-ium trifluoromethanesulfonate 1e
Prepared according to the general dimethylimidazolium ester synthesis using 5e (1.0 mmol). Isolated as a white foam (272 mg, 68%). 1H NMR (500 MHz, CDCl3) δ 7.68 (s, 2H), 7.19 (dd, J = 5.1, 1.2 Hz, 1H), 6.96 (dd, J = 5.1, 3.4 Hz, 1H), 6.92 (dd, J = 3.4, 1.1 Hz, 1H), 4.75 (t, J = 6.5 Hz, 2H), 4.07 (s, 7H), 3.37 (t, J = 6.5, 0.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 153.5, 138.7, 132.1, 127.3, 126.8, 126.2, 124.5, 121.9, 119.3, 68.3, 39.4, 28.8. FTIR (ATR) cm−1: 3123, 1743, 1530, 1434, 1255, 1226, 1154, 1027, 817, 705, 629. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C12H15N2O2S 251.0849; Found 251.0841.
4.4.6. 2-((4-methoxyphenethoxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1f
Prepared according to the general dimethylimidazolium ester synthesis using 5f (1.0 mmol). Isolated as an amorphous white solid (375 mg, 88%). 1H NMR (500 MHz, CDCl3) δ 7.67 (s, 2H), 7.16 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 4.70 (t, J = 6.9 Hz, 2H), 4.03 (s, 6H), 3.78 (s, 3H), 3.07 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 158.7, 153.6, 132.2, 129.7, 128.4, 126.7, 119.4 (q, J =319.7 Hz), 114.3, 68.7, 55.3, 39.3, 33.5. FTIR (ATR) cm−1: 3126, 2954, 1742, 1439, 1245, 1158, 1024, 631. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C15H19N2O3 275.1390; Found 275.1387.
4.4.7. 1,3-dimethyl-2-(((tetrahydro-2H-pyran-4-yl)methoxy)carbonyl)-1H-imidazol-3-ium trifluoromethanesulfonate 1g
Prepared according to the general dimethylimidazolium ester synthesis using 5g (1.0 mmol). Isolated as an amorphous white solid (264 mg, 68%). 1H NMR (500 MHz, CDCl3) δ 7.72 (s, 2H), 4.35 (d, J = 6.6 Hz, 2H), 4.17 (s, 6H), 3.99 (dd, J = 11.7, 4.9 Hz, 2H), 3.42 (t, J = 11.8 Hz, 2H), 2.12 (dddd, J = 18.5, 14.4, 7.1, 3.7 Hz, 1H), 1.70 (d, J = 12.9 Hz, 2H), 1.43 (qd, J = 12.4, 4.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 153.8, 132.3, 126.7, 120.6 (q, J = 320.3 Hz), 72.3, 67.2, 39.4, 34.1, 29.4. FTIR (ATR) cm−1: 3124, 2962, 2858, 1742, 1446, 1259, 1144, 1031, 804, 639. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C12H19N2O3 239.1390; Found 239.1390.
4.4.8. 2-((cyclohexyloxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1h
Prepared according to the general dimethylimidazolium ester synthesis using 5h (1.0 mmol). Isolated as a pale yellow powder (303 mg, 81%). 1H NMR (500 MHz, CDCl3) δ 7.75 (s, 2H), 5.18 (tdd, J = 9.5, 3.8, 1.6 Hz, 1H), 4.18 (s, 6H), 2.02 (dd, J = 12.7, 5.3 Hz, 2H), 1.81 – 1.72 (m, 2H), 1.70 – 1.54 (m, 3H), 1.46 (dt, J = 13.6, 10.0 Hz, 2H), 1.40 – 1.30 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 153.1, 132.3, 126.7, 120.6 (q, J = 320.2 Hz), 78.3, 39.4, 31.2, 24.9, 23.5. FTIR (ATR) cm−1: 3128, 2930, 2860, 1740, 1530, 1446, 1253, 1151, 1030, 635. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C12H19N2O2 223.1441; Found 223.1438.
4.4.9. 2-(((1-(tert-butoxycarbonyl)piperidin-4-yl)oxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1i
Prepared according to the general dimethylimidazolium ester synthesis using 5i (1.0 mmol). Isolated as a white powder (369 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 7.68 (s, 2H), 5.35 (tt, J = 8.2, 4.0 Hz, 1H), 4.19 (s, 6H), 3.77 (dt, J = 14.3, 5.0 Hz, 2H), 3.30 (ddd, J = 13.3, 8.5, 3.6 Hz, 2H), 2.06 (ddt, J = 10.0, 7.0, 3.9 Hz, 2H), 1.82 (dtd, J = 12.8, 8.4, 3.9 Hz, 2H), 1.47 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.6, 153.1, 132.4, 126.6, 121.8, 119.3, 80.1, 75.5, 41.0, 39.6, 30.3, 28.4. FTIR (ATR) cm−1: 3128, 2976, 1739, 1680, 1443, 1425, 1248, 1153, 1027, 878, 636. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C16H26N3O4 324.1918; Found 324.1916.
4.4.10. (S)-2-((2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxopropoxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1j
Prepared according to a modified version of the general dimethylimidazolium ester synthesis using 5j (1.0 mmol). Instead of filtering and recrystallizing, the reaction mixture was concentrated, then triturated with diethyl ether (3×5 mL) and hexanes (3×5 mL). Isolated as a thick clear oil (187 mg, 38%). 1H NMR (500 MHz, CDCl3) δ 7.68 (s, 2H), 6.01 (d, J = 7.0 Hz, 1H), 4.92 (d, J = 9.4 Hz, 1H), 4.72 – 4.63 (m, 2H), 4.12 (s, 6H), 3.79 (s, 3H), 1.41 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 155.6, 153.4, 132.1, 126.7, 120.5 (q, J = 320.2 Hz), 80.4, 67.4, 53.0, 52.5, 39.4, 28.2. FTIR (ATR) cm−1: 3333, 3132, 2980, 1746, 1707, 1531, 1442, 1256, 1156, 1031, 638. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C15H24N3O6 342.1660; Found 342.1657.
4.4.11. 2-(tert-butoxycarbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1k
Prepared according to the general dimethylimidazolium ester synthesis using 5k (1.0 mmol). Isolated as a tan powder (346 mg, 95%). 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 2H), 4.16 (s, 6H), 1.66 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 152.5, 132.9, 126.4, 121.9, 119.4, 88.8, 39.5, 28.0. FTIR (ATR) cm−1: 3129, 1737, 1442, 1253, 1146, 1028, 800, 635. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C10H17N2O2 197.1285; Found 197.1289.
4.4.12. 2-((((1s,3s)-adamantan-1-yl)oxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1l
Prepared according to the general dimethylimidazolium ester synthesis using 5l (0.4 mmol). Isolated as a white powder (158 mg, 93%). 1H NMR (500 MHz, CDCl3) δ 7.73 (s, 2H), 4.16 (s, 6H), 2.28 (s, 9H), 1.72 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 152.0, 132.7, 126.5, 122.0, 119.4, 89.2, 41.4, 39.5, 35.8, 31.1. FTIR (ATR) cm−1: 2915, 2860, 2361, 2339, 1726, 1272, 1255, 1168, 1027, 636. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C16H23N2O2 275.1754; Found 275.1752.
4.4.13. 1,3-dimethyl-2-(((3-(trimethylsilyl)prop-2-yn-1-yl)oxy)carbonyl)-1H-imidazol-3-ium trifluoromethanesulfonate 1m
Prepared according to the general dimethylimidazolium ester synthesis using 5m (1.0 mmol). Isolated as a white powder (294 mg, 73%). 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 2H), 5.08 (s, 2H), 4.21 (s, 6H), 0.19 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 152.9, 131.7, 127.1, 120.6 (q, J = 321.0 Hz), 96.1, 95.2, 55.9, 39.5, −0.5. FTIR (ATR) cm−1: 3128, 2965, 2361, 2338, 1741, 1436, 1254, 1152, 1030, 941, 810, 758, 637. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C12H19N2O2Si 251.1210; Found 251.1208.
4.4.14. 2-((((3S,8R,9S,10S,13S,14S)-10,13-dimethyl-17-oxohexadecahydro-1H-cyclopenta[a] phenanthren-3-yl)oxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1n
Prepared according to the general dimethylimidazolium ester synthesis using 5n (1.0 mmol). Isolated as an amorphous white solid (365 mg, 65%). 1H NMR (500 MHz, CDCl3) δ 7.72 (s, 2H), 5.10 (tt, J = 11.0, 4.9 Hz, 1H), 4.19 (s, 6H), 2.45 (dd, J = 19.3, 8.9 Hz, 1H), 2.13 – 2.03 (m, 2H), 1.94 (ddd, J = 12.4, 8.8, 5.9 Hz, 1H), 1.83 (tt, J = 12.6, 3.8 Hz, 4H), 1.76 – 1.46 (m, 5H), 1.45 – 1.19 (m, 6H), 1.12 (td, J = 13.8, 3.4 Hz, 1H), 1.01 (qd, J = 12.4, 4.8 Hz, 1H), 0.90 (s, 3H), 0.87 (s, 3H), 0.75 (td, J = 11.2, 4.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 153.1, 132.3, 126.7, 79.2, 54.2, 51.3, 47.7, 44.7, 39.5, 36.5, 35.8, 35.6, 35.0, 33.6, 31.5, 30.7, 28.1, 27.3, 21.7, 20.5, 13.8, 12.3. FTIR (ATR) cm−1: 2931, 2854, 1733, 1444, 1260, 1153, 1031. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C25H37N2O3 413.2799; Found 413.2787.
4.4.15. 2-((((1R,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)carbonyl)-1,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate 1o
Prepared according to the general dimethylimidazolium ester synthesis using 5o (1.0 mmol). Isolated as an amorphous white solid (365 mg, 65%). 1H NMR (500 MHz, CDCl3) δ 7.82 (s, 2H), 5.12 (td, J = 11.0, 4.5 Hz, 1H), 4.19 (s, 6H), 2.17 (dtd, J = 12.0, 4.4, 4.0, 1.8 Hz, 1H), 1.87 (pd, J = 6.9, 2.6 Hz, 1H), 1.81 – 1.71 (m, 2H), 1.62 – 1.50 (m, 2H), 1.20 (q, J = 12.1, 12.0, 11.4 Hz, 1H), 1.17 – 1.07 (m, 1H), 0.99 – 0.89 (m, 7H), 0.80 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.3, 132.0, 127.0, 120.6 (q, J = 320.2 Hz), 79.9, 47.1, 40.6, 39.5, 33.7, 31.6, 26.29, 23.0, 21.8, 20.8, 15.8. FTIR (ATR) cm−1: 3127, 2957, 2924, 2867, 1736, 1442, 1254, 1145, 1030, 948, 910, 804, 635. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C16H27N2O2 279.2067; Found 279.2066.
4.4.16. 2-(ethoxycarbonyl)-1,3-dimethyl-1H-benzo[d] imidazol-3-ium trifluoromethanesulfonate 2a
Prepared according to the general dimethylimidazolium ester synthesis using ethyl 1-methyl-1H-benzo[d]imidazole-2-carboxylate (1.0 mmol). Isolated as a white powder (347 mg, 92%). 1H NMR (500 MHz, CDCl3) δ 7.83 (dt, J = 7.2, 3.6 Hz, 2H), 7.75 (dt, J = 6.5, 3.6 Hz, 2H), 4.68 (q, J = 7.2 Hz, 2H), 4.37 (s, 6H), 1.53 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 154.2, 137.3, 132.1, 129.1, 121.8, 119.2, 113.6, 65.4, 35.0, 13.8. FTIR (ATR) cm−1: 3003, 2979, 2362, 1742, 1522, 1256, 1224, 1140, 1006, 750, 637. HRMS (ESI/TOF) m/z: [M-CF3O3S]+ Calcd. for C12H15N2O2 219.1128; Found 219.1123.
4.5. General procedure for the esterification of potassium trifluoroborate salts
To an oven-dried 2-dram vial equipped with a stir bar was added the corresponding trifluoroborate salt 3 (0.3 mmol) and the corresponding dimethylimidazolium ester 1 (0.2 mmol). The vial was cycled into the glovebox, and PC1 (0.2 μmol) was added. Dichloromethane (6 mL) was added, and the vial was sealed. Parafilm was wrapped around the cap, and the mixture was irradiated with stirring using four 427 nm Kessil PR160L lamps (~5 cm away). After 18 hours, the irradiation was stopped and the reaction mixture was diluted with water (10 mL). The layers were separated and the aqueous layer was extracted with dichloromethane (2×10 mL). The organic layers were combined, dried with sodium sulfate, and concentrated. The unpurified residue was purified using silica gel chromatography (ethyl acetate/hexanes) to afford ester 4.
4.5.1. ethyl 2-phenylacetate 4a
Prepared according to the general esterification procedure with trifluoroborate 3a and ester 1a, except yield (16 mg, 50%) was determined by GC-MS without workup. Retention time and fragmentation matched a commercial analytical standard from Sigma Aldrich.
4.5.2. ethyl 2-(4-chlorophenyl)acetate 4b
Prepared according to the general esterification procedure with trifluoroborate 3b and ester 1a. Isolated as a clear oil (8 mg, 20%). 1H NMR was consistent with literature data.29
4.5.3. ethyl 2-(p-tolyl)acetate 4c
Prepared according to the general esterification procedure with trifluoroborate 3c and ester 1a. Isolated as a clear oil (16 mg, 45%). 1H NMR was consistent with literature data.29
4.5.4. ethyl 2-(4-methoxyphenyl)acetate 4d
Prepared according to the general esterification procedure with trifluoroborate 3d and ester 1a. Isolated as a clear oil (7 mg, 19%). 1H NMR was consistent with literature data.29
4.5.5. ethyl 2-([1,1′-biphenyl] −4-yl)acetate 4e
Prepared according to the general esterification procedure with trifluoroborate 3e and ester 1a. Isolated as a clear oil (19 mg, 40%). 1H NMR was consistent with literature data.29
4.5.6. methyl 4-(2-ethoxy-2-oxoethyl)benzoate 4f
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1a. Isolated as a clear oil (26 mg, 59%). 1H NMR was consistent with literature data.30
4.5.7. ethyl 2-(4-bromophenyl)acetate 4g
Prepared according to the general esterification procedure with trifluoroborate 3g and ester 1a. Isolated as a clear oil (9 mg, 20%). 1H NMR was consistent with literature data.29
4.5.8. ethyl 2-phenylpropanoate 4h
Prepared according to the general esterification procedure with trifluoroborate 3h and ester 1a. Isolated as a clear oil (17 mg, 48%). 1H NMR was consistent with literature data.29
4.5.9. ethyl 2-(naphthalen-2-yl)acetate 4i
Prepared according to the general esterification procedure with trifluoroborate 3i and ester 1a. Isolated as a clear oil (14 mg, 33%). 1H NMR was consistent with literature data.29
4.5.10. ethyl 2-(3,5-dimethylphenyl)acetate 4j
Prepared according to the general esterification procedure with trifluoroborate 3j and ester 1a. Isolated as a clear oil (8 mg, 21%). 1H NMR was consistent with literature data.29
4.5.11. ethyl 2-(2,6-difluorophenyl)acetate 4k
Prepared according to the general esterification procedure with trifluoroborate 3k and ester 1a. Isolated as a clear oil (20 mg, 50%). 1H NMR (500 MHz, CDCl3) δ 7.23 (tt, J = 8.4, 6.6 Hz, 1H), 6.94 – 6.85 (m, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.70 (s, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.7, 162.5 (d, J = 8.0 Hz), 160.5 (d, J = 8.0 Hz), 128.9 (t, J = 10.3 Hz), 111.1 (dd, J = 20.4, 5.7 Hz), 110.7 (t, J = 20.0 Hz), 61.2, 28.0 (t, J = 3.2 Hz), 14.1. 19F NMR (470 MHz, CDCl3) δ −114.6. FTIR (ATR) cm−1: 2983, 2934, 1741, 1471, 1272, 1216, 1171, 1019, 785. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C10H11F2O2 201.0722; Found 201.0722.
4.5.12. diethyl 2-phenylsuccinate 4l
Prepared according to the general esterification procedure with trifluoroborate 3l and ester 1a. Isolated as a clear oil (19 mg, 38%). 1H NMR was consistent with literature data.30
4.5.13. ethyl 4-oxo-2,4-diphenylbutanoate 4m
Prepared according to the general esterification procedure with trifluoroborate 3m and ester 1a. Isolated as a white solid (24 mg, 43%). 1H NMR was consistent with literature data.31
4.5.14. ethyl 4-oxochromane-2-carboxylate 4n
Prepared according to the general esterification procedure with trifluoroborate 3n and ester 1a. Isolated as a clear oil (12 mg, 27%). 1H NMR was consistent with literature data.32
4.5.15. methyl 4-(2-(cyclopropylmethoxy)-2-oxoethyl)benzoate 4o
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1b. Isolated a clear oil (30 mg, 61%). 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 3.93 (d, J = 7.3 Hz, 2H), 3.90 (s, 3H), 3.69 (s, 2H), 1.17 – 1.05 (m, 1H), 0.57 – 0.52 (m, 2H), 0.28 – 0.23 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.9, 166.8, 139.3, 129.8, 129.3, 129.0, 69.8, 52.0, 41.3, 9.7, 3.2. FTIR (ATR) cm−1: 3006, 2952, 1718, 1613, 1435, 1276, 1155, 1106, 1022, 984, 765, 721. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C14H17O4 249.1121; Found 249.1125.
4.5.16. methyl 4-(2-(cyclohexylmethoxy)-2-oxoethyl)benzoate 4p
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1c. Isolated as a clear oil (34 mg, 58%). 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 3.91 (s, 3H), 3.91 (d, J = 6.5 Hz, 2H), 3.67 (s, 2H), 1.74 – 1.59 (m, 6H), 1.27 – 1.08 (m, 3H), 0.97 – 0.86 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.9, 166.9, 139.3, 129.8, 129.3, 129.0, 70.2, 52.1, 41.4, 37.0, 29.6, 26.3, 25.6. FTIR (ATR) cm−1: 2924, 2852, 1720, 1435, 1275, 1149, 1106, 999, 741. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C17H23O4 291.1591; Found 291.1586.
4.5.17. methyl 4-(2-(dodecyloxy)-2-oxoethyl)benzoate 4q
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1d. Isolated as a white waxy solid (37 mg, 51%). 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.08 (t, J = 6.7 Hz, 2H), 3.90 (s, 3H), 3.66 (s, 2H), 1.60 (p, J = 6.8 Hz, 2H), 1.33 – 1.21 (m, 18H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.9, 166.8, 139.3, 129.8, 129.3, 129.0, 65.2, 52.0, 41.4, 31.9, 29.6, 29.6, 29.5, 29.5, 29.3, 29.1, 28.5, 25.8, 22.7, 14.1. FTIR (ATR) cm−1: 2952, 2918, 2850, 1725, 1275, 1223, 1178, 1108, 721. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C22H35O4 363.2530; Found 262.2528.
4.5.18. methyl 4-(2-oxo-2-(2-(thiophen-2-yl)ethoxy)ethyl)benzoate 4r
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1e. Isolated as a clear oil (40 mg, 65%). 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 6.9 Hz, 2H), 7.33 (d, J = 7.5 Hz, 2H), 7.15 (d, J = 5.1 Hz, 1H), 6.92 (ddd, J = 4.9, 3.5, 1.1 Hz, 1H), 6.78 (d, J = 3.4 Hz, 1H), 4.33 (t, J = 6.7 Hz, 2H), 3.91 (s, 3H), 3.69 (s, 3H), 3.14 (t, J = 6.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 170.6, 166.8, 139.6, 138.9, 129.8, 129.3, 129.0, 126.8, 125.6, 124.0, 65.2, 52.1, 41.3, 29.1. FTIR (ATR) cm−1: 2952, 1717, 1434, 1276, 1152, 1106, 1020, 698. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C16H17O4S 305.0842; Found 305.0846.
4.5.19. methyl 4-(2-(4-methoxyphenethoxy)-2-oxoethyl)benzoate 4s
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1f. Isolated as a clear oil (24 mg, 37%). 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 4.28 (t, J = 6.9 Hz, 2H), 3.92 (s, 3H), 3.79 (s, 3H), 3.65 (s, 3H), 2.85 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 170.7, 166.8, 158.3, 139.1, 129.8, 129.5, 129.3, 129.0, 113.9, 65.7, 55.2, 52.1, 41.4, 34.1. FTIR (ATR) cm−1: 2953, 2837, 1720, 1613, 1584, 1513, 1435, 1278, 1246, 1179, 1157, 1108, 1034, 831. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C19H21O5 329.1384; Found 329.1384.
4.5.20. methyl 4-(2-oxo-2-((tetrahydro-2H-pyran-4-yl)methoxy)ethyl)benzoate 4t
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1g. Isolated as a clear oil (25 mg, 43%). 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 3.97 – 3.92 (m, 4H), 3.90 (s, 3H), 3.67 (s, 2H), 3.35 (td, J = 11.8, 2.2 Hz, 2H), 1.92 – 1.82 (m, 1H), 1.54 (ddq, J = 13.0, 4.0, 2.0 Hz, 2H), 1.32 (dtd, J = 13.4, 11.9, 4.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 170.7, 166.8, 139.1, 129.8, 129.3, 129.0, 69.2, 67.3, 52.1, 41.3, 34.4, 29.4. FTIR (ATR) cm−1: 2950, 2844, 1718, 1613, 1435, 1419, 1276, 1147, 1107, 1017, 853. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C16H21O5 293.1384; Found 293.1387.
4.5.21. methyl 4-(2-(cyclohexyloxy)-2-oxoethyl)benzoate 4u
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1h. Isolated as a clear oil (33 mg, 60%). 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.7 Hz, 2H), 4.78 (tt, J = 8.8, 3.9 Hz, 1H), 3.91 (s, 3H), 3.65 (s, 2H), 1.88 – 1.76 (m, 2H), 1.74 – 1.62 (m, 2H), 1.54 – 1.48 (m, 1H), 1.45 – 1.29 (m, 4H), 1.29 – 1.20 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 170.3, 166.9, 139.6, 129.8, 129.3, 128.9, 73.4, 52.1, 41.8, 31.5, 25.3, 23.6. FTIR (ATR) cm−1: 2937, 2859, 1721, 1435, 1278, 1165, 1107, 1017, 742. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C16H21O4 277.1434; Found 277.1440.
4.5.22. tert-butyl 4-(2-(4-(methoxycarbonyl)phenyl)acetoxy)piperidine-1-carboxylate 4v
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1i. Isolated as an amorphous white solid (17 mg, 23%). 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H), 4.94 (tt, J = 7.8, 3.8 Hz, 1H), 3.91 (s, 3H), 3.67 (s, 2H), 3.64 – 3.55 (m, 2H), 3.22 (ddd, J = 13.6, 8.3, 3.7 Hz, 2H), 1.85 – 1.77 (m, 2H), 1.62 – 1.52 (m, 2H), 1.44 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.1, 166.8, 154.6, 139.1, 129.9, 129.2, 129.1, 79.7, 70.4, 52.1, 41.6, 40.8, 30.4, 28.4. FTIR (ATR) cm−1: 2976, 2949, 2926, 1721, 1680, 1416, 1358, 1271, 1180, 1163, 1107, 1019, 767, 726. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C20H28NO6 378.1911; Found 378.1913.
4.5.23. methyl (S)-4-(2-(2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxopropoxy)-2-oxoethyl)benzoate 4w
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1j. Isolated as a clear oil (24 mg, 30%). 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 5.20 (d, J = 8.2 Hz, 1H), 4.55 (dd, J = 8.3, 4.0 Hz, 1H), 4.46 – 4.35 (m, 2H), 3.90 (s, 3H), 3.68 (s, 3H), 3.67 (s, 2H), 1.44 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.3, 170.1, 166.8, 155.1, 138.6, 129.9, 129.4, 80.4, 64.8, 52.8, 52.7, 52.1, 49.2, 41.0, 28.3. FTIR (ATR) cm−1: 3362, 2977, 2955, 1743, 1715, 1508, 1436, 1278, 1158, 1108, 1020, 768. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C19H26NO8 396.1653; Found 396.1649. Optical data: [α]23(D) −25.0 (c, 0.90, MeOH).
4.5.24. methyl 4-(2-(tert-butoxy)-2-oxoethyl)benzoate 4x
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1k. Isolated as a clear oil (5 mg, 10%). 1H NMR was consistent with literature data.33
4.5.25. methyl 4-(2-(((3s,5s,7s)-adamantan-1-yl)oxy)-2-oxoethyl)benzoate 4y
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1l. Isolated as a clear oil (18 mg, 27%). 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 3.91 (s, 3H), 3.57 (s, 2H), 2.14 (bs, 3H), 2.07 (d, J = 3.0 Hz, 6H), 1.64 (bs, J = 3.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.8, 167.0, 140.0, 129.7, 129.3, 128.8, 81.3, 52.0, 42.8, 41.2, 36.1, 30.8. FTIR (ATR) cm−1: 2911, 2853, 1724, 1279, 1254, 1107, 1055, 970. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C20H25O4 329.1747; Found 329.1753
4.5.26. methyl 4-(2-oxo-2-((3-(trimethylsilyl)prop-2-yn-1-yl)oxy)ethyl)benzoate 4z
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1m. Isolated as a clear oil (6 mg, 10%). 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 4.71 (s, 2H), 3.91 (s, 3H), 3.73 (s, 2H), 0.18 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.0, 166.8, 138.6, 129.9, 129.3, 129.2, 98.6, 92.5, 53.3, 52.1, 40.9, −0.4. FTIR (ATR) cm−1: 2956, 1745, 1724, 1436, 1280, 1251, 1147, 1108, 845, 762. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C16H21O4Si 305.1204; Found 305.1201.
4.5.27. methyl 4-(2-(((3S,8R,9S,10S,13S,14S)-10,13-dimethyl-17-oxohexadecahydro-1H-cyclopenta[a] phenanthren-3-yl)oxy)-2-oxoethyl)benzoate 4aa
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1n. Isolated as an amorphous white solid (27 mg, 29%). 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 4.70 (tt, J = 11.4, 4.9 Hz, 1H), 3.90 (s, 3H), 3.63 (s, 2H), 2.42 (ddd, J = 19.3, 8.9, 1.1 Hz, 1H), 2.05 (dt, J = 19.2, 9.1 Hz, 1H), 1.95 – 1.88 (m, 1H), 1.82 – 1.75 (m, 3H), 1.72 (dt, J = 13.4, 3.7 Hz, 1H), 1.65 – 1.55 (m, 2H), 1.55 – 1.42 (m, 3H), 1.40 – 1.13 (m, 7H), 1.06 – 0.93 (m, 2H), 0.84 (s, 3H), 0.83, (s, 3H), 0.69 (ddd, J = 12.0, 10.4, 4.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 170.3, 166.9, 139.5, 129.8, 129.2, 128.9, 74.2, 54.2, 52.0, 51.3, 47.7, 44.6, 41.7, 36.6, 35.8, 35.6, 35.0, 33.8, 31.5, 30.7, 28.2, 27.3, 21.7, 20.4, 13.8, 12.2. FTIR (ATR) cm−1: 2932, 2851, 1734, 1710, 1276, 1167, 1106, 1012, 727. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C29H39O5 467.2792; Found 467.2786. Optical data: [α]23(D) −72.7 (c, 0.42, MeOH).
4.5.28. methyl 4-(2-(((1R,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)-2-oxoethyl)benzoate 4ab
Prepared according to the general esterification procedure with trifluoroborate 3f and ester 1o. Isolated as an amorphous white solid (27 mg, 41%). 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 4.56 (td, J = 10.9, 4.4 Hz, 1H), 3.79 (s, 3H), 3.53 (s, 2H), 1.89 – 1.78 (m, 1H), 1.65 – 1.58 (m, 1H), 1.58 – 1.50 (m, 2H), 1.34 (ddtd, J = 15.1, 12.0, 6.6, 3.1 Hz, 1H), 1.22 (ddt, J = 12.3, 10.8, 3.2 Hz, 1H), 0.91 (qd, J = 13.4, 12.8, 3.8 Hz, 1H), 0.82 (td, J = 12.1, 10.9 Hz, 1H), 0.74 (m, 7H), 0.56 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.3, 166.9, 139.5, 129.7, 129.2, 128.9, 75.0, 52.0, 47.0, 41.8, 40.7, 34.2, 31.3, 26.2, 23.4, 21.9, 20.6, 16.2. FTIR (ATR) cm−1: 2953, 2927, 2869, 1721, 1435, 1275, 1148, 1106, 985, 967, 724. HRMS (ESI/TOF) m/z: [M+H]+ Calcd. for C20H29O4 333.2060; Found 333.2054. Optical data: [α]23(D) −59.2 (c, 1.22, MeOH).
Supplementary Material
Table 2:
Scope with regards to alkyl trifluoroborate
|
Acknowledgments
We thank the NIH/NIGMS (R35 GM136440) and Northwestern University for support of this work. Use was made of the IMSERC FTIR, NMR, and MS Facilities at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experiment (SHyNE) Resource (NSF ECCS-2025633).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supporting Information contains complete structures of all compounds as well as relevant characterization data for new compounds.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (1).(a) Banerjee A; Lei Z; Ngai M-Y Synthesis 2019, 51, 303–333; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh J; Sharma S; Sharma A J. Org. Chem 2021, 86, 24–48. [DOI] [PubMed] [Google Scholar]
- (2).Bucher G; Halupka M; Kolano C; Schade O; Sander W Eur. J. Org. Chem 2001, 2001, 545–552. [Google Scholar]
- (3).Singh AK; Bakshi RK; Corey EJ J. Am. Chem. Soc 1987, 109, 6187–6189. [Google Scholar]
- (4).Trost BM; Waser J; Meyer A J. Am. Chem. Soc 2008, 130, 16424–16434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Taniguchi T; Sugiura Y; Zaimoku H; Ishibashi H Angew. Chem. Int. Ed 2010, 49, 10154–10157. [DOI] [PubMed] [Google Scholar]
- (6).Zheng M; Hou J; Zhan L-W; Huang Y; Chen L; Hua L-L; Li Y; Tang W-Y; Li B-D ACS Catal. 2021, 11, 542–553. [Google Scholar]
- (7).For related work using ketyl radicals to prepare esters see the following:; (a) Rafferty SM; Rutherford JE; Zhang L; Wang L; Nagib DA J. Am. Chem. Soc 2021, 143, 5622–5628; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang L; Lear JM; Rafferty SM; Fosu SC; Nagib DA Science 2018, 362, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Bayly AA; McDonald BR; Mrksich M; Scheidt KA Proc. Natl. Acad. Sci 2020, 117, 13261; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies AV; Fitzpatrick KP; Betori RC; Scheidt KA Angew. Chem. Int. Ed 2020, 59, 9143–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Ishii T; Kakeno Y; Nagao K; Ohmiya H J. Am. Chem. Soc 2019, 141, 3854–3858; [DOI] [PubMed] [Google Scholar]; (b) Ishii T; Ota K; Nagao K; Ohmiya H J. Am. Chem. Soc 2019, 141, 14073–14077; [DOI] [PubMed] [Google Scholar]; (c) Kakeno Y; Kusakabe M; Nagao K; Ohmiya H ACS Catal. 2020, 10, 8524–8529; [Google Scholar]; (d) Ota K; Nagao K; Ohmiya H Org. Lett 2020, 22, 3922–3925. [DOI] [PubMed] [Google Scholar]
- (10).(a) Meng Q-Y; Döben N; Studer A Angew. Chem. Int. Ed 2020, 59, 19956–19960; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hopkinson MN; Mavroskoufis A Synlett 2021, 32, 95–101; [Google Scholar]; (c) Liu K; Studer A J. Am. Chem. Soc 2021; [Google Scholar]; (d) Meng Q-Y; Lezius L; Studer A Nat. Commun 2021, 12, 2068; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ren S-C; Lv W-X; Yang X; Yan J-L; Xu J; Wang F-X; Hao L; Chai H; Jin Z; Chi YR ACS Catal. 2021, 11, 2925–2934. [Google Scholar]
- (11).Molander GA; Ellis N Acc. Chem. Res 2007, 40, 275–286. [DOI] [PubMed] [Google Scholar]
- (12).(a) Goddard J-P; Ollivier C; Fensterbank L Acc. Chem. Res 2016, 49, 1924–1936; [DOI] [PubMed] [Google Scholar]; (b) Matsui JK; Lang SB; Heitz DR; Molander GA ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Stache EE; Rovis T; Doyle AG Angew. Chem. Int. Ed 2017, 56, 3679–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Masiukiewicz E; Mrugala D; Rzeszotarska B Org. Prep. Proced. Int 2005, 37, 403–405. [Google Scholar]
- (15).Shang T-Y; Lu L-H; Cao Z; Liu Y; He W-M; Yu B Chem. Commun 2019, 55, 5408–5419. [DOI] [PubMed] [Google Scholar]
- (16).(a) DeLano TJ; Bandarage UK; Palaychuk N; Green J; Boyd MJ J. Org. Chem 2016, 81, 12525–12531; [DOI] [PubMed] [Google Scholar]; (b) Ryu D; Primer DN; Tellis JC; Molander GA Chem. Eur. J 2016, 22, 120–123; [DOI] [PubMed] [Google Scholar]; (c) Tellis JC; Kelly CB; Primer DN; Jouffroy M; Patel NR; Molander GA Acc. Chem. Res 2016, 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Zhang W; Krohn K; Zia U; Flörke U; Pescitelli G; Di Bari L; Antus S; Kurtán T; Rheinheimer J; Draeger S; Schulz B Chem. Eur. J 2008, 14, 4913–4923; [DOI] [PubMed] [Google Scholar]; (b) Kikuchi H; Isobe M; Sekiya M; Abe Y; Hoshikawa T; Ueda K; Kurata S; Katou Y; Oshima Y Org. Lett 2011, 13, 4624–4627; [DOI] [PubMed] [Google Scholar]; (c) El-Elimat T; Figueroa M; Raja HA; Graf TN; Swanson SM; Falkinham Iii JO; Wani MC; Pearce CJ; Oberlies NH Eur. J. Org. Chem 2015, 2015, 109–121; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu G; Yu G; Kurtán T; Mándi A; Peng J; Mo X; Liu M; Li H; Sun X; Li J; Zhu T; Gu Q; Li D J. Nat. Prod 2015, 78, 2691–2698; [DOI] [PubMed] [Google Scholar]; (e) Maha A; Rukachaisirikul V; Phongpaichit S; Poonsuwan W; Sakayaroj J Tetrahedron 2016, 72, 2874–2879. [Google Scholar]
- (18).Bakhtiar C; Smith EH J. Chem. Soc. Perkin Trans 1 1994, 239–243. [Google Scholar]
- (19).Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA; Malliaras GG; Bernhard S Chem. Mater 2005, 17, 5712–5719. [Google Scholar]
- (20).Yasu Y; Koike T; Akita M Adv. Synth. Catal 2012, 354, 3414–3420. [Google Scholar]
- (21).Sorin G; Martinez Mallorquin R; Contie Y; Baralle A; Malacria M; Goddard J-P; Fensterbank L Angew. Chem. Int. Ed 2010, 49, 8721–8723. [DOI] [PubMed] [Google Scholar]
- (22).Tellis JC; Primer DN; Molander GA Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li Y; Zhou K; Wen Z; Cao S; Shen X; Lei M; Gong L J. Am. Chem. Soc 2018, 140, 15850–15858. [DOI] [PubMed] [Google Scholar]
- (24).Cazorla C; Métay E; Lemaire M Tetrahedron 2011, 67, 8615–8621. [Google Scholar]
- (25).Jones MR; Fast CD; Schley ND J. Am. Chem. Soc 2020, 142, 6488–6492. [DOI] [PubMed] [Google Scholar]
- (26).Molander GA; McKee SA Org. Lett 2011, 13, 4684–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Matsui JK; Molander GA Org. Lett 2017, 19, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Baird EE; Dervan PB J. Am. Chem. Soc 1996, 118, 6141–6146. [Google Scholar]
- (29).Xie P; Xie Y; Qian B; Zhou H; Xia C; Huang H J. Am. Chem. Soc 2012, 134, 9902–9905. [DOI] [PubMed] [Google Scholar]
- (30).Wu G; Deng Y; Wu C; Zhang Y; Wang J Angew. Chem. Int. Ed 2014, 53, 10510–10514. [DOI] [PubMed] [Google Scholar]
- (31).Gu P; Wu X-P; Su Y; Xue P; Li X-Q; Gong B-L; Li R Tetrahedron Lett. 2013, 54, 4957–4959. [Google Scholar]
- (32).Ghosh S; Chandar N; Sarkar D; Ghosh M; Ganguly B; Chakraborty I Synlett 2014, 25, 2649–2653. [Google Scholar]
- (33).Abdiaj I; Huck L; Mateo JM; de la Hoz A; Gomez MV; Díaz-Ortiz A; Alcázar J Angew. Chem. Int. Ed 2018, 57, 13231–13236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.