

Abstract:
Cardiovascular disease ranks the leading cause of mortality worldwide. Prenyldiphosphate synthase subunits collectively participate in the formation and development of atherosclerosis (AS). This study aimed to investigate the role of PDSS2 in AS and its underlying mechanisms. Human coronary artery endothelial cells (HCAECs) were treated with oxidized low-density lipoprotein to establish the AS model. The gene expression levels were determined by qRT-PCR, Western blot, and ELISA. CCK-8, colony formation was applied to determine the proliferation of HCAECs. Chromatin immunoprecipitation assay and luciferase assay were applied to verify the interaction between PDSS2 and Nrf2. The results showed that the serum levels of PDSS2 and Nrf2 were decreased in patients with AS. Overexpression of PDSS2 suppressed the release of reactive oxygen species, iron content and ferroptosis of HCAECs, and promoted the proliferation of HCAECs. Moreover, PDSS2 activated antioxidant Nrf2. PDSS2 interacted with Nrf2 to alleviate the ferroptosis of HCAECs. However, knockdown of Nrf2 alleviated the effects of PDSS2 on the proliferation and ferroptosis of HCAECs. In vivo assays, overexpression of PDSS2 and Nrf2 suppressed the progression of AS. In conclusion, overexpression of PDSS2 suppressed the ferroptosis of HCAECs by promoting the activation of Nrf2 pathways. Thence PDSS2 may play a cardio-protective role in AS.
Key Words: atherosclerosis, PDSS2, Nrf2, oxidative stress, ferroptosis
INTRODUCTION
Cardiovascular disease (CVD), characterized with high morality, is a major detriment to human health.1 Atherosclerosis (AS) is a main factor of CVD.2 The pathogenesis of AS is complicated by various factors, among which oxidative stress is evidenced to be an initiator of AS.3 In the early stage of AS, accumulation of reactive oxygen species (ROS) induces endothelial dysfunction or death, which, in turn, increases abundant lipids in arterial wall and formation of atherosclerotic plaque.3 Oxidative stress–induced endothelial death is based on the following underlying mechanisms: cell ferroptosis, apoptosis, necrosis, and autophagy.4 Recently, the roles of ferroptosis in CVD attract increasingly attention. A previous study reveal that oxidized phospholipids and excess iron-induced ferroptosis contribute to the development of CVD, such as AS and thrombosis.5,6 However, the molecular mechanism of ferroptosis in endothelial cell response to oxidative stress in AS is not clear.
Prenyldiphosphate synthase subunit 2 (PDSS2) resides within chromosome 6q21.7 PDSS2 is the first key enzyme for the synthesis of coenzyme Q10 (CoQ10). PDSS2 mutation induces degradation of CoQ10, which leads to metabolic and nephritic disorders. Previous studies evidence that PDSS2 functions as a tumor suppressor in gastric cancer, melanoma, and lung cancer,8–10 and its deficiency predicts poor prognosis. In CVD, knockdown of PDSS2 contributes to structural and functional defects in congenital heart disease and heart failure.11,12 However, the possible molecular mechanisms is still unclear.
Nuclear factor erythroid 2–related factor-2 (Nrf2) participates in the development of CVD, such as AS, ischemia-reperfusion injury, and diabetic cardiomyopathy through regulating cell signaling, anabolic metabolism, and cell proliferation.13 As a redox-sensitive transcription factor, Nrf2 translocates to the nucleus and binds to the antioxidant response element or the electrophile-response element to alleviate oxidative stress damage and restore heart function. Moreover, activation of Nrf2 mimics the roles of NF-κB on oxidative stress and inhibits inflammatory response in CVD.14 Nonetheless, the potential roles of Nrf2 in AS has not been fully elucidated.
In this study, we investigated the roles of PDSS2 in AS and its underlying mechanisms. The results showed that PDSS2 was decreased in AS. Overexpression of PDSS2 decreased the release of ROS and activated Nrf2 pathways. PDSS2-induced upregulation of Nrf2 promoted the proliferation and inhibited the ferroptosis of human coronary artery endothelial cells (HCAECs). This results suggested that PDSS2/Nrf2 axis may be a promising strategy for the treatment of AS.
MATERIALS AND METHODS
Specimens
Specimens were collected from patients with AS (n = 42) and healthy volunteers (n = 30). Exclusion criteria were diabetes, cancer, or infection. This study was approved by the Ethics Committee of the First People's Hospital of Lianyungang. All the volunteers provided informed consent.
Cell Culture
HCAECs were obtained from Shanghai Chinese Academy of Sciences, Shanghai. Cells were cultured in DMEM medium containing 10% FBS and 1% penicillin/streptomycin in an atmosphere humidified with 5% CO2 at 37°C.
Cells were treated with 10 μmol/L of simvastatin (SIM), 5 μmol/L of fFerrostatin-1 (Fer-1), 20 μg/mL of (iron-saturated holo-transferrin) HTF, or 4 μM of erastin.
Cell Transfection
PDSS2 overexpression plasmids (OE), its negative control OE (NC), si-Nrf2, and its NC was provided by GenePharma, Shanghai. Cells were transfected with PDSS2 OE, NC OE, si-Nrf2, or si-NC using Lipofectamine 2000 (Invitrogen) for 48 hours.
qRT-PCR
Total RNA were collected from cells by Total RNA Kit I (Omega Bio-Tek, Norcross, GA). cDNA was synthesized from RNA using PrimeScript RT reagent Kit (Takara, Japan). PCR was performed with the SYBR Green qRT-PCR master mix (Takara) on Stratagene Mx3005P qRT-PCR system under following thermocycling conditions: 95°C for 10 minutes and then 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. GAPDH was used as loading control. The mRNA levels were calculated with 2−ΔΔCt method. Each individual experiment was conducted thrice. The sequences of the primers used in this study were listed as follows: PDSS2, F: 5′-ACCTGACCTGCCGTCTAGAA-3′ and R: 5′-TCCACCACCCTGTTGCTGTA-3′, Nrf2, F: 5′-GGCGTTAGAAAGCATCCTTCC-3′ and R: 5′-GCAGAGGGCACACTCAAAGT-3′, and GAPDH, F: 5′-AATGGACAACTGGTCGTGGAC-3′ and R: 5′-CCCTCCAGGGGATCTGTTTG-3′.
Western Blot
Total protein was harvested from cells. Protein concentrations were determined by a BCA kit. An equal amount of protein was isolated with a 12% SDS-PAGE. The isolated protein was transferred onto PVDF membranes. After sealed with non-fat milk, the membranes were incubated with primary antibodies, such as anti-PDSS2 (ab251797, 1: 1000, Abcam, Cambridge, MA), anti-Nrf2 (ab137550, 1: 1000, Abcam), anti-PKCα (ab32376, 1: 1000, Abcam), anti-p53 (ab32389, 1: 1000, Abcam), anti-FSP1 (ferroptosis-suppressor-protein 1, ab197896, 1: 1000, Abcam), and anti-GAPDH (ab9485, 1:2500, Abcam) at room temperature for 2 hours. Then the membranes were incubated with secondary goat antirabbit antibody (ab6721, 1: 2000, Abcam). Finally, the bands were visualized with an enhanced chemiluminescent kit (Pierce) and analyzed with quantified LAS3000 imaging system (Fujifilm Corporation, Japan).
CCK-8 Assay
Cell viability was determined with a CCK-8 kit. In brief, HCAECs were plated into a 24-well plate (3 × 103 cells/well). After 0, 12, 24, and 48 hours transfection, each well was added with a 10 μL CCK-8 regent and cultured for 3 hours at 37°C. The optical density was determined by a microplate reader at the wavelength of 450 nm.
Colony Formation Assay
After 48 hours transfection, HCAECs were collected, digested with trypsin–collagenase, and resuspended. Afterward, cells were planted into a 24-well plate (2 × 103 cells/well) and incubated for 14 days in 5% CO2 at 37°C. Then each well was supplemented with 2% crystal violet and cultured for another 20 minutes. Subsequently, colonies were captured with a microscope and counted.
Immunofluorescence Assay
After seeded on coverslips, cells were fixed in 4% formaldehyde and permeabilized with 0.25% Triton X-100. Then cells were incubated with 1% BSA. Afterward, cells were incubated with anti-CD31 antibody (ab76533, 1: 500, Abcam) at room temperature for 2 hours and then with goat antirabbit secondary CyTM3-goat antirabbit IgG antibody. Afterward, cells were counterstained with DAPI for 15 minutes. Finally, cells were visualized with a confocal microscope (Nikon, Japan).
Chromatin Immunoprecipitation (ChIP) Analysis
ChIP assay was conducted with an enzymatic ChIP kit. In brief, cells were fixed with 1% formaldehyde. Chromatin was prepared and digested with nuclease. Then cells were incubated with primary antibodies against PDSS2, Nrf2, and IgG. Afterward, cells were incubated at 65°C and shaken to reverse cross-links. Proteinase K was added, and the samples were incubated at 65°C for 2 hours. The results were normalized to input DNA.
Dual-Luciferase Reporter Assay
Cells were seeded in a 96-well plate. Nrf2 sequence and PDSS2-3′UTR sequence were introduced into pmirGLO vectors. Then cells were transfected with Nrf2 OE or Nrf2 KO and PDSS2-3′UTR-WT or PDSS2-3′UTR-MUT for 48 hours. The luciferase activity was determined with a luciferase kit. The luciferase activities were normalized to the Renilla luciferase activity. Each experiment was performed triplicate.
Ferrous Iron Assay
HCAECs were seeded in 24-well plate (2 × 103 cells/well). Intracellular ferrous iron levels were determined using an iron colorimetric assay kit (Abcam, United Kingdom) according to the manufacturer's protocols.
Expression and Purification of rPDSS2
Recombinant human PDSS2 (rPDSS2) was expressed using the p-SUMO-PDSS2 plasmid in E. coli and purified as previously described.
Animal Models
Eight-week-old male mice PDSS2 wild type [WT (n = 6)], PDSS2−/−, Nrf2−/−, Nrf2+/+, on C57BL/6 background (18–22 g) were provided by Nanjing Medical University (Nanjing, China). PDSS2−/− mice (n = 12) were crossed with PDSS2−/− to obtain PDSS2−/−, Nrf2−/− (n = 6) and PDSS2−/−, Nrf2+/+ (n = 6) mice. Mice were fed an AIN76A Western diet for 8 weeks to accelerate the development of AS. Mice from each group were euthanized after 8 weeks. The experimental scheme was approved by the Animal Care of the First People's Hospital of Lianyungang.
Red-Oil Staining
Aortas were stained with a freshly prepared Oil Red O working solution, differentiated by using 70% ethanol, mounted en face, and visualized by using a bright-field microscope (IX70; Olympus, Tokyo, Japan).
Statistical Analysis
Each experiment was repeated 3 times. All data were analyzed with SPSS 19.0 and represented as means ±SD. Clinicopathological data were analyzed by using Pearson χ2 tests or Fisher's exact test. Two-tailed unpaired Student's t tests were used to analyze differences between the 2 groups and 1-way analysis of variance assay followed by Duncan's post-hoc test for multigroup analysis. P < 0.05 was considered statistically significant.
RESULTS
Clinical Features
The results of clinical analysis showed that the levels of plasma glutathione (GSH) and peripheral blood mononuclear cell. GSH and oxidized low-density lipoprotein [ox-low-density lipoprotein (LDL)] were significantly increased in patients with CAD in comparison with healthy control, whereas the difference in age, gender, body mass index, systolic blood pressure, diastolic blood pressure, LDL, high-density lipoprotein, and triglycerides is of no significance (Table 1).
TABLE 1.
Clinical Features
| Items | Healthy | CAD | P |
| Age (yr) | 63 ± 12 | 68 ± 11 | NS |
| Male/female | 16/14 | 23/19 | NS |
| BMI (kg/m2) | 24.6 ± 2.3 | 24.2 ± 2.3 | NS |
| SBP (mm Hg) | 132 ± 7 | 125 ± 7 | NS |
| DBP (mm Hg) | 81 ± 6 | 78 ± 5 | NS |
| LDL (mmol/L) | 2.55 ± 0.67 | 2.45 ± 0.53 | NS |
| HDL (mmol/L) | 1.14 ± 0.18 | 1.08 ± 0.13 | NS |
| Triglycerides (mg/dL) | 123 ± 5 | 121 ± 5 | NS |
| Plasma GSH (µmol/L) | 3.89 ± 1.08 | 1.56 ± 0.68 | 0.0341* |
| PBMC GSH (ng/mg cell protein) | 2.74 ± 0.21 | 1.45 ± 0.36 | 0.0058† |
| Ox-LDL (µg/mL) | 13.87 ± 1.32 | 24.47 ± 4.53 | 0.0175* |
P < 0.05 was considered statistically significant.
† represents P < 0.01.
BMI, body mass index; DBP, diastolic blood pressure; HDL, high-density lipoprotein; PBMC, peripheral blood mononuclear cell; SBP, systolic blood pressure.
PDSS2 is Decreased in AS
To investigate the roles of PDSS2 in AS, we examined the expression of PDSS2 in AS. As showed in Figure 1A, the serum level of PDSS2 was significantly decreased in patients with AS compared with the healthy control. Moreover, a receiver operating characteristic curve was generated to evaluate the ability of PDSS2 to distinguish patients with AS from healthy subjects. The results showed that the AUC for PDSS2 was 0.9119 (Fig. 1B), suggesting that the expression of PDSS2 in synovial tissues can serve as a diagnostic biomarker for AS with high specificity and sensitivity. Furthermore, the mRNA level was significantly decreased in cells treated with ox-LDL (Fig. 1C).
FIGURE 1.

PDSS2 is decreased in AS. A, The serum level of PDSS2 determined by ELISA. B, The specificity of PDSS2 analyzed with the receiver operating characteristic curve. C, The mRNA level of PDSS2 detected by qRT-PCR. n = 3. **P < 0.01.
Ox-LDL Promotes the Ferroptosis of HCAECs
Mitochondrial damage is the main manifestation of ferroptosis. As showed in Figure 2A, ox-LDL exposure induced the mitochondrial damage of HCAECs. To further investigate the roles of oxidative stress in the development of ferroptosis, cells were treated with ferroptosis inhibitors (Fer-1 and SIM) and catalyzer (HTF and erastin). The iron and total ROS content was significantly increased by ox-LDL and erastin, which was more potent in cells treated with ox-LDL + HTF; however, Fer-1 alleviated the increase of iron and total ROS induced by ox-LDL (Figs. 2B, D). Moreover, the level of GSH was decreased by ox-LDL and ferroptosis catalyzer but increased by Fer-1 (Fig. 2C). In addition, ox-LDL-induced decrease of PDSS2 and FSP1 was facilitated by HTF and reversed by Fer-1 (Fig. 2E).
FIGURE 2.
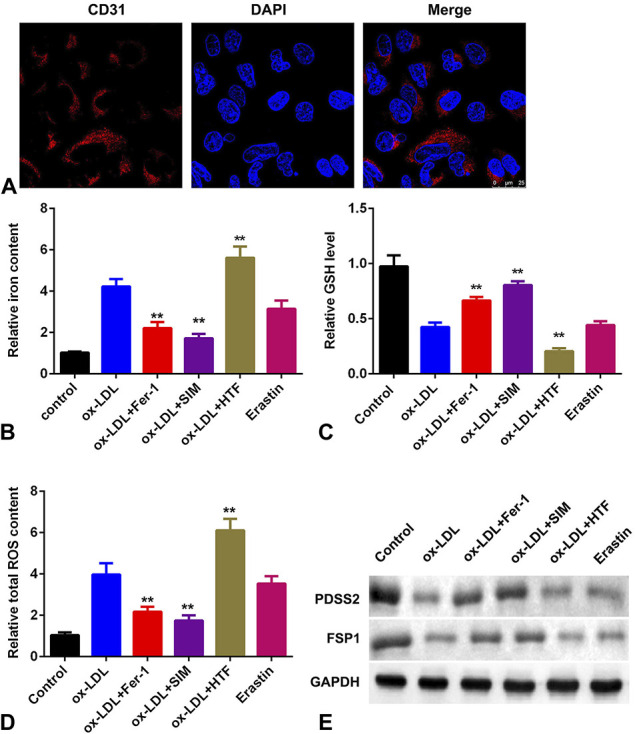
Ox-LDL promotes the ferroptosis of HCAECs. A, Mitochondrial damage detected by immunofluorescence assay. B, The level of iron content in HCAECs. C, The levels of GSH. D, The total content of ROS. E, The protein levels of PDSS2 and FSP1. n = 3. **P < 0.01.
Overexpression of PDSS2 Suppresses the Ferroptosis of HCAECs
To further investigate the potential roles of PDSS2 in AS, we examined the effects of PDSS2 on the ferroptosis of HCAECs. As showed in Figure 3A, the expression level of PDSS2 was significantly increased in cells treated with PDSS2 overexpression plasmids. Overexpression of PDSS2 decreased the content of iron and total ROS and increased the level of GSH (Fig. 3). However, knockdown of PDSS2 promoted the decrease of GSH and the increase of iron and ROS level induced by ox-LDL (Fig. 3).
FIGURE 3.

PDSS2 suppresses ox-LDL-induced ferroptosis of HCAECs. A and B, The mRNA level of PDSS2 determined by qRT-PCR. C and D, The iron content in HCAECs. E and F, The level of GSH in HCAECs. G and H, The total ROS in HCAECs. n = 3. **P < 0.01.
Overexpression of PDSS2 Promotes the Proliferation of HCAECs
To further investigate the roles of PDSS2 in AS, we examined the effects of PDSS2 on the proliferation of HCAECs. As showed in Figure 4A, overexpression of PDSS2 alleviated the increase of cell death of HCAECs induced by ox-LDL. In colony formation assay, ox-LDL decreased the proliferation of HCAECs, which was antagonized by overexpression of PDSS2 (Fig. 4). However, knockdown of PDSS2 further suppressed the proliferation of HCAECs (Fig. 4).
FIGURE 4.
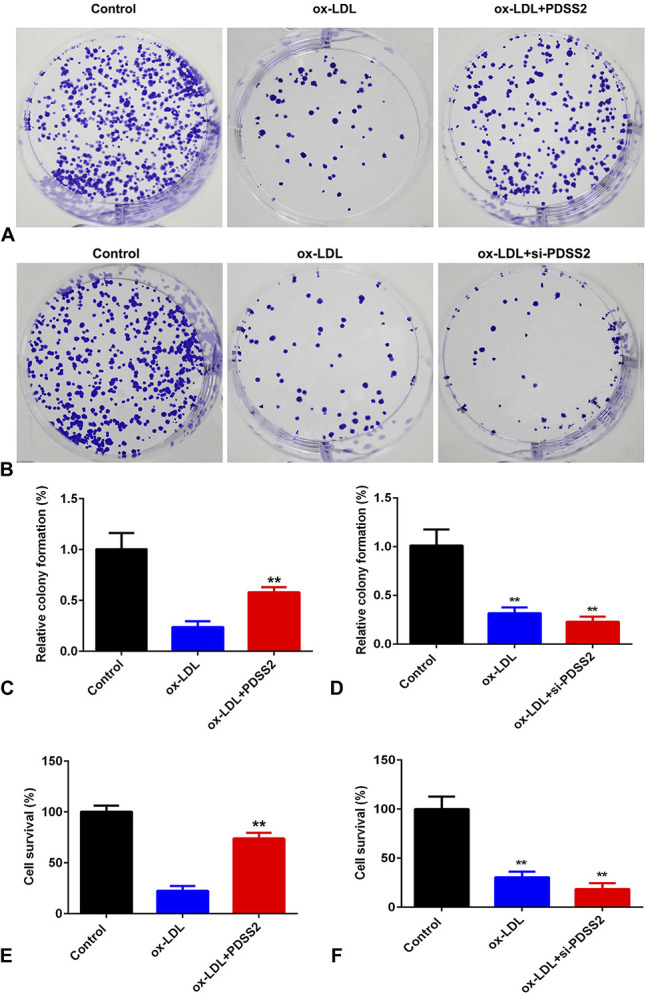
PDSS2 suppresses the death of HCAECs. A and B, Cell proliferation detected by colony formation assay. C and D, Quantification of (A) and (B), respectively. E and F, Cell survival rate determined by CCK-8 assay. n = 3. **P < 0.01.
PDSS2 Transcriptionally Activates Nrf2
To further investigate the roles of PDSS2 in AS, we explored the underlying molecular mechanisms. As showed in Figure 5A, Nrf2 was decreased in patients with AS. Furthermore, the mRNA level of Nrf2 was significantly decreased in cells treated with ox-LDL (Fig. 5B). The protein level of Nrf2 was increased by PDSS2 OE, whereas decreased by si-PDSS2 (Fig. 5C). DNA binding motif was obtained from online database JASPAR (Fig. 5D). The binding sites of Nrf2 on the promoter of PDSS2 were showed in Figure 5E. In luciferase assay, luciferase activity was decreased in cells cotransfected with PDSS2 OE, and Nrf2 3′UTR WT, whereas increased by si-PDSS2 and Nrf2 3′UTR WT (Figs. 5F, G). Moreover, Nrf2 conferred a strong affinity in the promoter region of PDSS2 (Fig. 5H).
FIGURE 5.

Nrf2 transcriptionally activates PDSS2. A, The serum levels of Nrf2 detected by ELISA. B, The level of Nrf2 detected by qRT-PCR level. C, The protein level of Nrf2 detected by Western blot. D, DNA binding motif of Nrf2 predicted by JASPAR. E, The binding sites between Nrf2 and PDSS2. F and G, The luciferase activity of HCAECs. H, The DNA affinity detected by ChIP assay. n = 3. **P < 0.01, ***P < 0.001.
PDSS2 Suppresses Ox-LDL-Induced Ferroptosis by Activating Nrf2 Pathways
Rescue assay was applied to verify the roles of PDSS2 in the development of AS. As showed in Figure 6A, the expression of Nrf2 was significantly deceased in cells transfected with si-Nrf2. Downregulation of Nrf2 increased the level of iron content. Moreover, depletion of Nrf2 increased the release of ROS and iron content and decreased the level of GSH (Figs. 6B–D).
FIGURE 6.
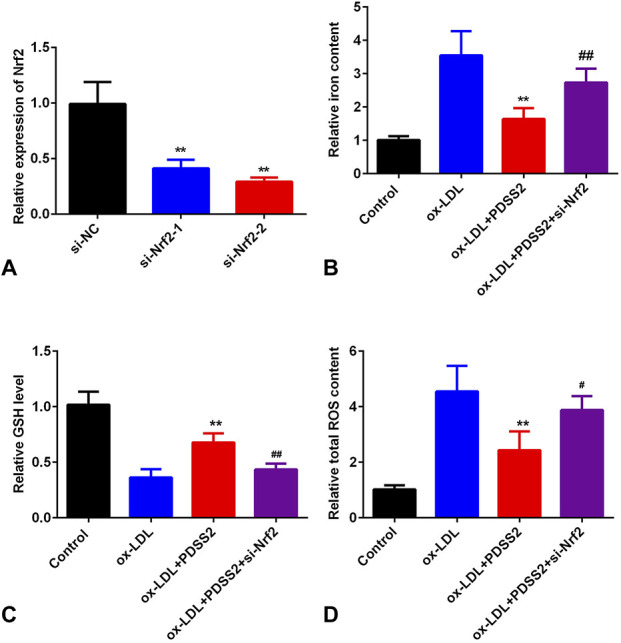
Knockdown of Nrf2 promotes ox-LDL-induced ferroptosis. A, The level of Nrf2 detected by qRT-PCR. B, The iron level in HCAECs. C, The levels of lipid peroxidation. n = 3. **P < 0.01, #P < 0.05, ##P < 0.01.
Knockdown of Nrf2 Antagonizes the Effects of PDSS2 on the Proliferation of HCAECs
We further determined the cellular function of HCAECs in AS. As showed in Figure 7A, overexpression of PDSS2 decreased cell death of HCAECs, which was reversed by Nrf2 knockdown. This was in consistence with the results from colony formation assay. Knockdown of Nrf2 alleviated Nrf2-induced increase in the proliferation of HCAECs (Fig. 7B).
FIGURE 7.

Knockdown of Nrf2 antagonizes the effects of PDSS2 on the proliferation of HCAECs. A, Cell survival determined by CCK-8. B, Cell proliferation detected by colony formation assay. n = 3. **P < 0.01, ##P < 0.01.
PDSS2 Suppresses the Atherosclerotic Lesions in Vivo
To further investigate the roles of PDSS2 in AS, we performed in vivo assay. As showed in Figure 8, the atherosclerotic plaque area was significantly increased in PDSS2−/−Nrf2−/−mice compared with PDSS2 WT mice, whereas this was alleviated in PDSS2−/−Nrf2+/+ mice. These results suggested that PDSS2/Nrf2 axis may protect against AS.
FIGURE 8.
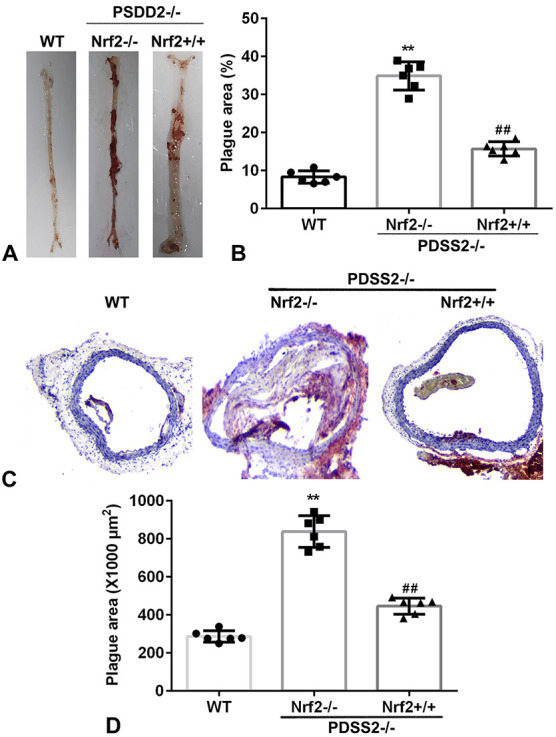
PDSS2 suppresses the atherosclerotic lesions in vivo. A, The atherosclerotic lesions determined by Oil Red O-staining assay. B, Quantification of (A). C, The atherosclerotic lesions determined by Oil Red O-staining assay. D, Quantification of (C). n = 6. **P < 0.01, ##P < 0.01.
DISCUSSION
PDSS2 has been reported to be a master regulator in AS, and its deficiency predicts poor heart function, which suggests that PDSS2 can be a possible candidate gene for AS.11,12 However, the potential roles of PDSS2 in AS is still alluring. Previous studies evidence that the deficiency of PDSS2 induces ROS, oxidative stress, and cell death.15 Thence we hypothesized that PDSS2 may participate in the progression of AS by regulating oxidative stress and cell death. In this study, PDSS2 was downregulated in patients with AS and in HCAECs exposed to ox-LDL. However, overexpression of PDSS2 activated antioxidant Nrf2/Keap1/HO-1 pathways. The activation of PDSS2/Nrf2 pathways promoted the proliferative ability of HCAECs and inhibited ferroptosis of HCAECs. To the best of our knowledge, this is the first study to unveil the potential roles of PDSS2 in AS.
Ferroptosis is identified more recently to be associated with cardiomyocyte cell death.16,17 Ferroptosis induces the release of soluble and lipid ROS through iron-dependent enzymatic reactions, which is characterized by lipid peroxidation and the decrease of GPX4 and GSH.18 Erastin, a ferroptosis catalyzer, promotes ferroptosis by blocking the antioxidant defense by decreasing the level of GSH, facilitating the release of lipid ROS. However, ferroptosis is a coin type of cell death.19 However, it is double-faced in heart disorders. The increase of ferroptosis may reduce tumor growth and contributes to myocardial ischemia/reperfusion injury.18,20 In this study, oxidative stress suppressed the activity of GSH, decreased the expression of FSP1, and increased the level of lipid peroxidation, suggesting oxidative stress promoted the ferroptosis of HCAECs. The induction of ferroptosis decreased the proliferative ability of HCAECs and promoted cell death, which is the main cause of AS.21 These results suggested that ferroptosis may be a crucial factor for AS. Nevertheless, the molecular mechanisms are still unclear.
PDSS2 functions as a tumor suppressor and has been clinically used for stratifying patients with hepatocellular carcinoma.22 In heart diseases, the deficiency of PDSS2 is associated with congenital heart disease and heart failure.11,12 Previous works regarding the role of PDSS2 in heart disorders focus on its expression profiling and functional analysis. However, the regulatory roles of PDSS2 have not been elucidated. In this study, PDSS2 was decreased in patients with AS, which suggested that PDSS2 deficiency may be associated with the progression of AS. However, overexpression of PDSS2 suppressed the release of ROS and the ferroptosis of HCAECs, which are the key factors in the initiation and progression of AS.3,16,17,21 The inhibitory effects of PDSS2 on ferroptosis-induced cell death conferred its cardiac-protective properties in AS. Moreover, overexpression of PDSS2 promoted the proliferative ability of HCAECs. Thence PDSS2 may protect against AS by suppressing ferroptosis-induced cell death and increasing the number of HCAECs. Nonetheless, the underlying molecular mechanisms are still unclear.
Nrf2 is a master regulator of the antioxidant response and collectively participates in metabolic mechanisms, including proteostasis, iron/heme metabolism, and carbohydrate and lipid metabolism.13 Nrf2 exerts multifaceted function in promoting cell survival, whereas its deficiency may contribute to oxidant resistance and cell death, including ferroptosis.23 Nrf2, as a transcription factor, is a key regulator of antiferroptotic pathways.24 For instance, Nrf2 modulates iron/heme metabolism by HMOX1 and ABCB616,25; regulates intermediate metabolism by peroxisome proliferator–activated receptor γ, aldo-ketoreductases, and aldehyde dehydrogenase 1 family member A126,27; and is involved in glutathione synthesis and metabolism by glutamate–cysteine ligase and GPX4.28,29 PDSS2 is the first key enzyme for the synthesis of coenzyme Q10, which promotes the activation of Nrf2/antioxidant response element–mediated antioxidant genes.30 Thence we hypothesized that PDSS2 activates Nrf2 pathways to inhibit oxidative stress–induced ferroptosis of HCAECs. In this study, Nrf2 was activated by PDSS2. However, depletion of Nrf2 alleviated the regulatory roles of PDSS2 in ferroptosis in AS, increased iron content, lipid peroxidation and cell death, and suppressed the synthesis of glutathione. These results suggested that PDSS2/Nrf2 axis may function as a cardiac-protective role in AS.
In conclusion, PDSS2 was decreased in patients with AS and oxidative stress–induced degeneration of HCAECs. Overexpression of PDSS2 inhibited ferroptosis and promoted the cell proliferation of HCAECs by activating Nrf2 pathways (Fig. 9). This may provide a promising therapeutic strategy for AS.
FIGURE 9.
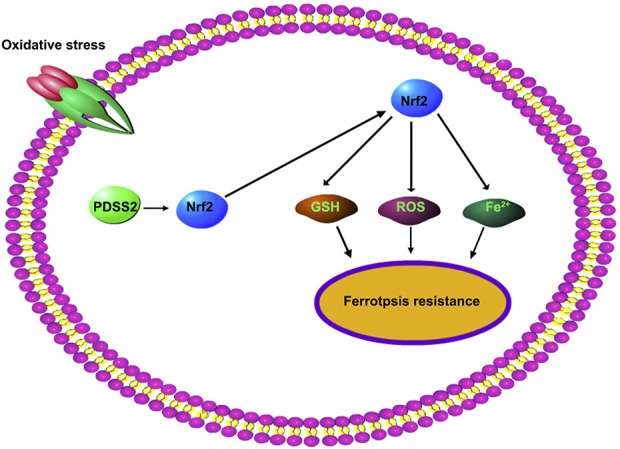
Activation of PDSS2/Nrf2 signaling confers resistance to ferroptosis in HCAECs.
Footnotes
The authors report no conflicts of interest.
Contributor Information
Kai Yang, Email: dr_Kai_Yang@outlook.com.
Hejian Song, Email: Hejian_Song@outlook.com.
Delu Yin, Email: Delu_yin@outlook.com.
REFERENCES
- 1.Lu D, Thum T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat Rev Cardiol. 2019;16:661–674. [DOI] [PubMed] [Google Scholar]
- 2.Gill SK. Cardiovascular risk factors and disease in women. Med Clin North Am. 2015;99:535–552. [DOI] [PubMed] [Google Scholar]
- 3.Kattoor AJ, Pothineni NVK, Palagiri D, et al. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017;19:42. [DOI] [PubMed] [Google Scholar]
- 4.Gimbrone MA, Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su LJ, Zhang JH, Gomez H, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koleini N, Nickel BE, Edel AL, et al. Oxidized phospholipids in Doxorubicin-induced cardiotoxicity. Chem Biol Interact. 2019;303:35–39. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Lin S, Li L, et al. PDSS2 deficiency induces hepatocarcinogenesis by decreasing mitochondrial respiration and reprogramming glucose metabolism. Cancer Res. 2018;78:4471–4481. [DOI] [PubMed] [Google Scholar]
- 8.Kanda M, Nomoto S, Oya H, et al. Decreased expression of prenyl diphosphate synthase subunit 2 correlates with reduced survival of patients with gastric cancer. J Exp Clin Cancer Res. 2014;33:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fung JM, Smith R, Brown MA, et al. Identification and characterization of a novel melanoma tumor suppressor gene on human chromosome 6q21. Clin Cancer Res. 2009;15:797–803. [DOI] [PubMed] [Google Scholar]
- 10.Hu L, Chen Q, Wang Y, et al. Sp1 mediates the constitutive expression and repression of the PDSS2 gene in lung cancer cells. Genes (Basel). 2019;10:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroeder AM, Allahyari M, Vogler G, et al. Model system identification of novel congenital heart disease gene candidates: focus on RPL13. Hum Mol Genet. 2019;28:3954–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bain CR, Ziemann M, Kaspi A, et al. DNA methylation patterns from peripheral blood separate coronary artery disease patients with and without heart failure. ESC Heart Fail. 2020;7:2468–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen QM, Maltagliati AJ. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol Genomics. 2018;50:77–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapple SJ, Cheng X, Mann GE. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013;1:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinzii CM, Garone C, Emmanuele V, et al. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013;27:612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Re DP, Amgalan D, Linkermann A, et al. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishizawa H, Matsumoto M, Shindo T, et al. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J Biol Chem. 2020;295:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Li W, Leng Y, et al. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39:210–225. [DOI] [PubMed] [Google Scholar]
- 21.Imai H, Matsuoka M, Kumagai T, et al. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–170. [DOI] [PubMed] [Google Scholar]
- 22.Kanda M, Sugimoto H, Nomoto S, et al. Clinical utility of PDSS2 expression to stratify patients at risk for recurrence of hepatocellular carcinoma. Int J Oncol. 2014;45:2005–2012. [DOI] [PubMed] [Google Scholar]
- 23.Bai YT, Chang R, Wang H, et al. ENPP2 protects cardiomyocytes from erastin-induced ferroptosis. Biochem Biophys Res Commun. 2018;499:44–51. [DOI] [PubMed] [Google Scholar]
- 24.Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaisar MA, Sivandzade F, Bhalerao A, et al. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: in vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci Lett. 2018;682:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bozaykut P, Karademir B, Yazgan B, et al. Effects of vitamin E on peroxisome proliferator-activated receptor γ and nuclear factor-erythroid 2-related factor 2 in hypercholesterolemia-induced atherosclerosis. Free Radic Biol Med. 2014;70:174–181. [DOI] [PubMed] [Google Scholar]
- 27.Foster MW, Yang Z, Gooden DM, et al. Proteomic characterization of the cellular response to nitrosative stress mediated by s-nitrosoglutathione reductase inhibition. J Proteome Res. 2012;11:2480–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian C, Gao L, Zhang A, et al. Therapeutic effects of Nrf2 activation by bardoxolone methyl in chronic heart failure. J Pharmacol Exp Ther. 2019;371:642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katunga LA, Gudimella P, Efird JT, et al. Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy. Mol Metab. 2015;4:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L. Protective effects of trimetazidine and coenzyme Q10 on cisplatin-induced cardiotoxicity by alleviating oxidative stress and mitochondrial dysfunction. Anatol J Cardiol. 2019;22:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]