

ABSTRACT
Swine influenza virus (SIV) can cause respiratory illness in swine. Swine contribute to influenza virus reassortment, as avian, human, and/or swine influenza viruses can infect swine and reassort, and new viruses can emerge. Thus, it is important to determine the host antiviral responses that affect SIV replication. In this study, we examined the innate antiviral cytokine response to SIV by swine respiratory epithelial cells, focusing on the expression of interferon (IFN) and interferon-stimulated genes (ISGs). Both primary and transformed swine nasal and tracheal respiratory epithelial cells were examined following infection with field isolates. The results show that IFN and ISG expression is maximal at 12 h postinfection (hpi) and is dependent on cell type and virus genotype.
IMPORTANCE Swine are considered intermediate hosts that have facilitated influenza virus reassortment events that have given rise pandemics or genetically related viruses have become established in swine. In this study, we examine the innate antiviral response to swine influenza virus in primary and immortalized swine nasal and tracheal epithelial cells, and show virus strain- and host cell type-dependent differential expression of key interferons and interferon-stimulated genes.
KEYWORDS: interferon-stimulated gene, interferons, swine influenza virus, swine nasal cells, swine tracheal cells
INTRODUCTION
Influenza is an orthomyxovirus that encodes 11 different proteins. These include the envelope proteins (hemagglutinin [HA] and neuraminidase [NA]) and viral RNA polymerases, matrix proteins, and nonstructural proteins (1, 2). Influenza viruses are classified into A, B, C, and D types (3). Swine influenza virus (SIV) is an acute viral infection of the respiratory tract caused by influenza type A viruses (4, 5). SIV may infect poultry and humans, but interspecies transmission is infrequent. Several subtypes of influenza A virus (IAV) circulate in swine, i.e., H1N1, H1N2, H3N1, and H3N2 (6, 7). Human-origin H1N1 in swine has resulted in the establishment of two new lineages of H1N1 and H1N2 after reassortment with triple-reassortant internal gene (TRIG) viruses containing gene segments from swine-, human-, and avian-origin influenza viruses (8–11). The TRIG strains are termed δ-lineages (12). The 2009 H1N1 pandemic virus (H1N1pdm) was transmitted to swine, leading to the establishment of a new H3 lineage; swine now have a varied pool of viruses, with 14 phylogenetic clades of HA cocirculating in swine in the United States (13).
Respiratory epithelial cells are the primary target of the influenza virus and have a critical role in the innate antiviral response to infection. IAV infection initiates by binding of hemagglutinin (HA) with sialic acid residues expressed on the cell surface. Human and swine IAVs primarily bind to α2,6 sialic acid expressed on airway epithelial cells (14, 15). The viruses are then endocytosed, involving a process that activates pattern recognition receptors (PRR), e.g., Toll-like receptors (Toll-like receptor 3 [TLR3] and TLR7) and the cytoplasmic retinoic acid-induced gene 1-like receptor (RIG-1) (16–18), and induces an antiviral response (16, 18). PRR interaction initiates signal transduction events and ultimately the release of antiviral type I IFNs (alpha interferon [IFN-α] and IFN-β), and type III IFN (IFN-λ), as well as that of cytokines and chemokines (19–21). IFNs activate the JAK-STAT signaling pathway to induce transcription of ISGs that combat viral infections (22, 23).
In this study, we examined SIV replication in primary and immortalized swine nasal and tracheal epithelial cells and determined the levels of IFN and ISG expression at early time points postinfection. We investigated four different SIV strains, i.e., A/swine/North Carolina/154072/2015 (H1N1), A/swine/North Carolina/156551/2015 (H1N2), A/swine/North Carolina/157674/2015 (H3N2), and A/Swine/MN/2009 (H1N1), isolated from swine nasal wash specimens. The glycan-binding profiles (24, 25) of the SIV were determined and the viruses sequenced. IFN and ISG expression was determined following SIV infection of primary swine nasal epithelial cells (SNECs), primary tracheal epithelial cells (STECs), immortalized swine nasal epithelial cells (SiNECs), or immortalized swine tracheal epithelial cells (SiTECs). Specifically, the expression of two key IFNs (IFN-β and IFN-γ) and several ISGs (OAS1, IRF7, GBP1, and DDX58) were determined between the respiratory cell types at 1 h postinfection (hpi), 12 hpi, and 24 hpi. Treatment of the respiratory epithelial cells with inactivated SIV did not induce substantial IFN and ISG expression, suggesting that the antiviral cytokine expression involves virus replication. The results show that IFN and ISG expression is cell type and virus specific, and oligonucleotide adenylate synthase 1 (OAS1) expression is expressed to high levels by inactivated SIV.
RESULTS
SIVs replicate in swine respiratory epithelial cell types.
We determined early antiviral responses to SIV infection in respiratory epithelial cells. Previously, SIV infection of newborn swine tracheal cells was examined for induction of JAK-STAT and MAPK signaling pathways (26). Since primary and immortalized swine nasal and tracheal cell lines were developed and shown to support the growth of human and swine nasal isolates (27), we compared how these cell lines responded to infection with the following SIV field isolates, H1N1-NC, H1N2-NC, H3N2-NC, or H1N1-MN, at a multiplicity of infection (MOI) of 0.1 for 1 hpi, 12 hpi, and 24 hpi. Following infection, the medium was removed, replaced with fresh medium, and incubated for the time points indicated. The cells were infected with H1N1-NC, H1N2-NC, or H3N2-NC because these are field isolates and host response to infection in primary swine respiratory epithelium is unknown. H1N1-MN is also a clinical isolate and was examined as a control virus (21). Importantly, analysis of intervirus segment-specific identities (Table 1) shows that the H1N1-MN virus diverged from the other clinical isolates in the HA, NP, NA, M1, and NS1/NEP segments, with sequence identities of ∼25%.
TABLE 1.
Virus segment identities
| Gene | Strain | Segment identity (%)a |
|||
|---|---|---|---|---|---|
| H1N1-NC | H1N2-NC | H1N1-MN | H3N2-NC | ||
| PB2 | H1N1-NC | 100 | |||
| H1N2-NC | 97 | 100 | 97 | ||
| H1N1-MN | 96 | 97 | 100 | 97 | |
| H3N2-NC | 98 | 100 | |||
| PB1 | H1N1-NC | 100 | |||
| H1N2-NC | 46 | 100 | 98 | ||
| H3N2-NC | 46 | 100 | |||
| PA | H1N1-NC | 100 | |||
| H1N2-NC | 93 | 100 | 93 | ||
| H3N2-NC | 99 | 100 | |||
| HA | H1N1-NC | 100 | |||
| H1N2-NC | 78 | 100 | 52 | ||
| H1N1-MN | 93 | 76 | 100 | 51 | |
| H3N2-NC | 56 | 100 | |||
| NP | H1N1-NC | 100 | |||
| H1N2-NC | 94 | 100 | 93 | ||
| H1N1-MN | 25 | 25 | 100 | 25 | |
| H3N2-NC | 97 | 100 | |||
| NA | H1N1-NC | 100 | |||
| H1N2-NC | 94 | 100 | 93 | ||
| H1N1-MN | 25 | 25 | 100 | 25 | |
| H3N2-NC | 97 | 100 | |||
| M1 | H1N1-NC | 100 | |||
| H1N2-NC | 98 | 100 | 98 | ||
| H1N1-MN | 24 | 24 | 100 | 24 | |
| H3N2-NC | 99 | 100 | |||
| NS1/NS2 | H1N1-NC | 100 | |||
| H1N2-NC | 98 | 100 | 97 | ||
| H1N1-MN | 26 | 27 | 100 | 26 | |
| H3N2-NC | 98 | 100 | |||
Percent identities between each segment among the SIV used in the study were calculated using SIAS (74).
Use of M gene expression as a surrogate for viral replication demonstrated higher M gene levels in nasal epithelial cells relative to those in tracheal cells (primary or immortalized). The different levels of replication likely reflect cell type tropism (28, 29) and/or are affected by the antiviral response to infection, i.e., expression of IFNs and ISGs (30–32).
Virus titers in the cell-free supernatants were determined at 24 hpi by plaque enumeration on MDCK cells. The data showed that H1N1-NC replicated the best in all cell types, with no statistically significant differences in titers between the cell types (Fig. 1b). For H1N2-NC infected cells, the viral titers were comparable, with significant differences observed between SiNECs and STECs (Fig. 1d). H1N1-MN virus replication was 2 to 3 log higher in all cell types compared to that of the other isolates. Among cell types, H1N1-MN virus titers were considerably higher in SiNECs and STECs than in SNECs (Fig. 1f). The H3N2-NC virus titers were significantly different between SiNECs and SNECs and were higher in SiTECs (Fig. 1h). Beta propiolactone-inactivated SIV did not produce plaques on any cell types, as expected.
FIG 1.

Swine primary and immortalized cells permit SIV replication. Relative influenza M gene expression at 12 h (open) and 24 h (filled) bars are shown for H1N1-NC (a), H1N2-NC (c), H1N1-MN (e), and H3N2-NC (g). Viral titers (log10) at 24 hpi for H1N1-NC (b), H1N2-NC (d), H1N1-MN (f), and H3N2-NC (h) are shown. Data represent mean ± standard error of the mean (SEM) of three replicates for M gene or plaque numbers at 24 hpi on MDCK cells. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
Comparison of SIV titers between cell types showed that H1N1 SIV (H1N1-NC and H1N1-MN) replicated best in all respiratory epithelial cell types (Fig. 2), while the H3N2-NC virus showed modest growth in SiTECs. H1N1-NC replicated to higher titers in SiNECs compared to those of H1N2-NC or H3N2-NC, but to lower titers compared to those of H1N1-MN. In SNECs, the H1N1-NC titer was higher than that of H3N2-NC but was not statistically different compared to those of H1N2-NC or H1N1-MN. In STECs, H1N1-MN replicated to higher titers than those of the other three isolates. For SiTECs, H1N1-NC titers were significantly higher than those of H1N2-NC (Fig. 2).
FIG 2.
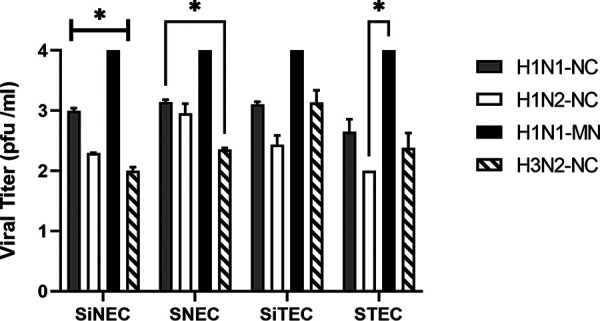
Replication of SIV isolates in swine cells. Viral titers at 24 hpi for H1N1-NC, H1N2-NC, H1N1-MN, and H3N2-NC on MDCK cells. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
Swine respiratory epithelial cells infected with SIV express type I and III IFNs.
Viral replication is affected by cellular receptors that enable viral entry. It is understood that SIV primarily binds to α2,6 sialic acid receptors, which are expressed on airway epithelial cells (14, 15). Bound viruses undergo endocytosis, a process that activates PRRs, e.g., TLR3, TLR7, and RIG-1 (16–18), inducing an antiviral response. The expressed viral genomes and gene products are recognized by surface and intracellular PRRs, which induce secretion of IFNs and ISGs that establish an antiviral state in the infected cell, as well as in adjoining tissue via paracrine signaling (33). We hypothesized that the differential replication kinetics of the SIV isolates was linked to differential induction of IFNs between SNECs, SiNECs, STECs, and SiTECs. To determine how these SIV field isolates may modulate the antiviral response, we examined the expression of type I and III IFNs in the respiratory epithelial cell types at 1 hpi, 12 hpi, and 24 hpi following infection with H1N1-NC, H1N2-NC, H1N1-MN, and H3N2-NC viruses. These cell types were mock treated, poly(I·C) treated, or treated with beta-propiolactone (BPL)-inactivated SIV (validated as inactivated by plaque assay on MDCK cells; data not shown) (see Table S1 in the supplemental material).
IFN-λ is the primary antiviral IFN against RNA virus infections (20, 34–36). IFN-λ induces expression of indoleamine oxidase1 (IDO1) in respiratory epithelial cells after influenza virus infection (37). We measured IFN-λ expression during SIV infection in the swine respiratory epithelial cells types. For H1N1-NC, infection induced IFN-λ expression at 1 hpi in nasal epithelial cell types (SNECs and SiNECs), but did not induce IFN-λ expression in tracheal cell types (STECs and SiTECs) (Fig. 3a). This may explain why tracheal cells may be more permissive than nasal epithelial cells to H1N1-NC replication. In contrast, low levels of IFN-λ were expressed at 1 hpi in SiNECs following H1N2-NC infection, but not in the other respiratory epithelial cell types (Fig. 3b). However, following H1N2- NC infection, all respiratory epithelial cell types expressed IFN-λ at 12 hpi and 24 hpi (Fig. 3b). The differences in IFN-λ expression are SIV strain specific, as the magnitude and IFN-λ expression patterns for H1N1-MN (Fig. 3c) and H3N2-NC (Fig. 3d) are different.
FIG 3.

Expression of IFN-λ. Panels show fold change in IFN-λ expression (log10) between SNECs, SiNECs, STECs, and SiTECs infected with H1N1-NC (a), H1N2-NC (b), H1N1-MN (c), and H3N2-NC (d) relative to mock-infected cells and 18S rRNA as a housekeeping control. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
IFN-β.
Type I IFNs constitute an important arm of the antiviral response against influenza viruses (30, 31, 38). IFN-β expression in H1N1-NC-infected SiNECs, SNECs, and STECs increased at 12 hpi and continued at 24 hpi. SiTECs did not express substantial IFN-β at 1 hpi (Fig. 4a), and expression was lower at 12 hpi and 24 hpi compared to that of the other respiratory epithelial cell types. In contrast, SNECs were the only cell type to express IFN-β at all time points postinfection. A similar IFN-β expression profile was seen for H1N2-NC-infected respiratory epithelial cells (Fig. 4b), except low-level IFN-β expression occurred in STECs at 1 hpi. H1N1-MN infection of SNECs and SiTECs did not induce IFN-β expression (Fig. 4c) at 1 hpi, but all other respiratory epithelial cell types expressed IFN-β at 12 hpi and 24 hpi. Following H3N2-NC infection, only STECs expressed IFN-β at 1 hpi (Fig. 4d), but all respiratory epithelial cell types expressed IFN-β at 12 hpi and 24 hpi.
FIG 4.

Expression of IFN-β. Panels show fold change in IFN-β expression (log10) between SNECs, SiNECs, STECs, and SiTECs infected with H1N1-NC (a), H1N2-NC (b), H1N1-MN (c), and H3N2-NC (d) relative to mock-infected cells and 18S rRNA as a housekeeping control. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
RIG-I (or DDX58).
Retinoic acid-inducible gene I (RIG-I), or DExD/H-Box helicase 58 (DDX58), is an innate immune receptor that senses cytoplasmic viral nucleic acids and activates a downstream signaling cascade that leads to the production of type I IFNs (16, 18). We examined RIG-I expression in SIV-infected respiratory epithelial cells at 1 hpi, 12 hpi, and 24 hpi. Early (1 hpi) RIG-I expression was only evident in nasally derived SiNECs and SNECs (Fig. 5), but all respiratory epithelial cell types expressed RIG-1 at 12 hpi and 24 hpi (Fig. 5a). This may be due to the anatomical location of nasal versus tracheal cells and the potential exposure to respiratory virus infection. Similar levels of RIG-1 expression by nasal or tracheal cells at 12 hpi and 24 hpi were expressed following H1N2-NC infection (Fig. 5b). H1N1-MN infection induced RIG-I expression in SiNECs at 1 hpi and 24 hpi, but not at 12 hpi (Fig. 5c). A similar pattern of RIG-I expression was observed between H1N1-NC- (Fig. 5a) and H3N2-NC (Fig. 5d)-infected respiratory epithelial cell types, suggesting that the pattern of RIG-I expression may not be SIV strain dependent.
FIG 5.

Expression of RIG-I. Panels show fold change in RIG-I expression (log10) between SNECs, SiNECs, STECs, and SiTECs infected with H1N1-NC (a), H1N2-NC (b), H1N1-MN (c), and H3N2-NC (d) relative to mock-infected cells and 18S rRNA as a housekeeping control. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
IRF7.
Interferon regulatory factor 7 (IRF7) is a key transcriptional regulator of type I IFNs and has a critical role in the innate immune response against DNA and RNA viruses (36–38). IRF7 deficiency has been reported to cause impaired type I IFN induction in multiple cell types (39), and thus we determined IRF7 expression in respiratory epithelial cell types infected with SIVs at 1 hpi, 12 hpi, and 24 hpi (Fig. 6). Similar patterns and kinetics of IRF7 expression were expressed for H1N1-NC- (Fig. 6a) and H1N2-NC (Fig. 6b)-infected respiratory epithelial cells, suggesting a lack of SIV strain differences mediating expression. In contrast, both H1N1-MN- (Fig. 6c) and H3N2-NC-infected (Fig. 6d) respiratory epithelial cells had distinct IRF7 expression profiles and kinetics. Of note, H1N1-MN-infected cells for the most part caused minimal IRF7 expression, particularly the trachea-derived STECs and SiTECs. A similar trend was evident for H3N2-NC-infected STECs and SiTECs compared to those infected with H1N1-NC and H1N2-NC. The differences between respiratory epithelial cell types may be associated with the expression levels of IFN-α and/or IFN-β, which regulate IRF7 and whose levels are unknown for the respiratory epithelial cell types examined here.
FIG 6.

Expression of IRF7. Panels show fold change in IRF7 expression (log10) between SNECs, SiNECs, STECs, and SiTECs infected with H1N1-NC (a), H1N2-NC (b), H1N1-MN (c), and H3N2-NC (d) relative to mock-infected cells and 18S rRNA as a housekeeping control. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
GBP1.
Guanylate binding protein 1 (GBP1) is an ISG that can inhibit influenza virus replication in cell lines (40). GBP1 is an IFN-induced GTPase that has been shown to have antiviral activities against vesicular stomatitis virus (VSV) (40), dengue virus (41), classical swine fever virus (42), influenza virus (43), and porcine reproductive and respiratory syndrome virus (44). GBP1 expression did not occur early (1 hpi) in trachea-derived STECs and SiTECs, but was expressed at 12 hpi and 24 hpi (Fig. 7). H1N1-NC infection induced GBP1 expression at 12 hpi and 24 hpi in all respiratory epithelial cell types, but H1N1-NC infection induced GBP1 at 1 hpi in SNECs (Fig. 7a). H1N2-NC infection induced GBP1 expression to similar levels in all respiratory epithelial cell types (Fig. 7b). H1N1-MN infection induced GBP1 expression in SiNECs at 1 hpi and 24 hpi, with low expression at 12 hpi (Fig. 7c). GBP1 expression was also induced in SNECs and STECs by 12 hpi, but no GBP1 expression was detectable at 24 hpi. SiTECs expressed GBP1 at 12 hpi and 24 hpi (Fig. 7c). H3N2-NC infection did not substantially induce GPB1 expression at 1 hpi, but robust GPB1 expression was evident in all respiratory epithelial cell types (Fig. 7d).
FIG 7.

Expression of GBP1. Panels show fold change in GBP1 expression (log10) between SNECs, SiNECs, STECs, and SiTECs infected with H1N1-NC (a), H1N2-NC (b), H1N1-MN (c), and H3N2-NC (d) relative to mock-infected cells and 18S rRNA as a housekeeping control. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
OAS1.
OAS1 (2′-5′-oligoadenylate synthetase 1) is induced by IFNs and encodes a protein that synthesizes 2′-5′-oligoadenylates. This protein activates latent RNase L, which results in viral RNA degradation and the inhibition of viral replication (45, 46). Multiple transcript variants of OAS arising from alternative splicing exhibit different enzymatic activities (47, 48). OAS1 activation is highly dependent on both RNA sequence and the context of activating RNA motifs (49). OAS1 expression has been shown to have antiviral activities against West Nile virus (50, 51), dengue virus (52), Japanese encephalitis virus (53), influenza virus (54), and several other RNA viruses. OAS1 expression by the respiratory epithelial cell types examined was comparatively high at 12 hpi and 24 hpi but was most highly expressed in cells of tracheal origin, i.e., STECs and SiTECs (Fig. 8). OAS1 expression by SNECs and STECs was detected at 1 hpi, but the levels were low. SIV infection of any respiratory epithelial cell type induced similar patterns and tempos of OAS1 expression following H1N1-NC (Fig. 8a), H1N2-NC (Fig. 8b), H1N1-MN (Fig. 8c), or H3N2-NC (Fig. 8d) infection.
FIG 8.

Expression of OAS1. Panels show fold change in OAS1 expression (log10) between SNECs, SiNECs, STECs, and SiTECs infected with H1N1-NC (a), H1N2-NC (b), H1N1-MN (c), and H3N2-NC (d) relative to mock-infected cells and 18S rRNA as a housekeeping control. Data represent mean ± SEM of three replicates. *, P < 0.05. Statistical methods used to determine significance are described in detail in Materials and Methods.
SIV genome differences, virus replication, and IFN antagonism.
To aid our understanding of the potential molecular mechanisms affecting virus replication and the induction of IFNs and ISGs, we examined the genomes of the SIV isolates. We analyzed the differences in all genes at the nucleotide level (Table 1), except for segments 2 and 3 of H1N1-MN, which have not been sequenced. We hypothesized that differences in IFN antagonism may inform as to why there is differential SIV replication kinetics. We focused on important genes with known roles in modulating innate immune responses, i.e., PA (55), PA-X (56, 57), NS1 (58–60), and PB1-F2 (61, 62). The polymerase subunits, PB2 and PA, were highly conserved across the three strains at the nucleotide level; however, the H3N2 PA sequence had a premature stop codon at nucleotide (nt) 664 and was predicted to lack the C-terminal 53 amino acids present in the H1N1-NC and H1N2-NC viruses. PB1 was highly divergent between H1N1-NC, H1N2-NC, and H3N2-NC viruses at the nucleotide level. The HAs from H1N1-NC, H1N2-NC, and H1N1-MN had >78% nucleotide identity, and HA from H3N2-NC was divergent, as expected. The NP, NA, MP, and NS1/NS2 gene segments were conserved for H1N1-NC, H1N2-NC, and H3N2-NC SIV, but diverged considerably for H1N1-MN.
Amino-terminal deletions of PA (PA-N155 and PA-N182) have been documented previously (63). A +1 frameshift in the PA-protein that leads to PA-X expression is a major regulator of the host response (55) and virulence of triple-reassortant H1N2 influenza infection (56). PA-X protein decreases replication and pathogenicity of SIVs (57). In contrast, an R195K mutation in PA-X has been shown to increase virulence and transmission of IAV (58), while a C-terminal 20-amino-acid truncation arising from a frameshift has been shown to alter SIV replication kinetics (58, 59). PA-X expression is highly conserved (60) and arises by alternative translation initiation at a highly conserved UCCUUUCGUC motif in PA. Sequence analysis of the putative PA-X proteins for H1N1-NC, H1N2-NC, and H3N2-NC shows identity in the PA-X C terminus (see Fig. S1 in the supplemental material). Previous studies have shown that mutations in NS1 are linked to reduced inhibition of IFN signaling (61, 62, 64). For example, dephosphorylation of Y73 and S83 residues in NS1 reduces the ability of NS1 to antagonize IFN-β via disengagement of the RIG-I signaling pathway (62). Similarly, serine 42 to proline (S42P) NS1 point mutants are unable to antagonize IFN-α and IFN-β via deactivation of IRF3 (62). The NS1 sequences between the SIVs examined in this study show that the amino acids are identical (see Fig. S2 in the supplemental material). It may be that the differential SIV replication observed (Fig. 2) is linked to differences in defective interfering (DI) particles. However, C453 and D529 in PA, which have been shown to inhibit DI particles (65), are identical for the SIVs examined (see Fig. S3 in the supplemental material), and R638, which is linked with increased production of DI particles, is also conserved for all SIV strains (Fig. S3). This refutes the hypothesis that differential replication may be due to differences in the SIV; rather, it is more likely due to differences in host cell permissiveness. Furthermore, we recently investigated the glycan-binding profiles of the SIV field isolates used in this study and showed that these SIVs bound primarily to α2,6 sialic acid linkages (66). The respiratory epithelial cells used in this study have been shown to express primarily α2,6 sialic acid linkages (27), and thus neither SIV genomes nor the ability of these viruses to binding to the respiratory epithelial cells types likely is linked to differences in virus replication, suggesting that the antiviral response by the cell types is most likely the cause. PB1-F2 has been shown to modulate the early innate response without altering pathogenicity during H1N1 infection (68). PB1-F2 in H3N2 viruses show strain-specific inhibition (69). PB1-F2 immune modulation is mediated by binding to mitochondrial antiviral signaling protein (MAVS) and inhibiting MAVS signaling (70). In this study, H1 and H3 PB1-F2 sequences differ appreciably (see Fig. S4a in the supplemental material). PB1-F2 for both H1N1-NC and H1N2-NC were considerably different from H1 PB1-F2 sequences (Fig. S4b). Comparative alignment of H3 PB1-F2 showed that H3N2-NC PB1-F2 lacked an N-terminal 10-amino-acid stretch (Fig. S4c). Thus, it is unlikely that changes in PB-F2 account for the differences observed in SIV replication.
DISCUSSION
Swine are a good model to study influenza replication because of their ability to support the reassortment and growth of avian, swine, and human influenza viruses. To understand the host response to IAV infection, we infected primary and immortalized swine nasal and tracheal cells with SIV field isolates and determined the innate antiviral response by swine respiratory epithelial cells. Our data show that SIV field isolates differentially infect swine respiratory epithelial cells. For example, H1N1-NC and H1N1-MN viruses infected all swine respiratory epithelial cell types well, while H1N2 and H3N2 replicated less well in these cell types. We examined BPL-inactivated SIV to determine if they signaled for similar antiviral responses in the swine respiratory epithelial cell types. BPL-inactivated SIV was unable to infect these cell types, as expected, but did induce a low level of antiviral responses; however, SIV infection induced the highest level of antiviral expression, so we focused on those responses.
Respiratory epithelial cell types expressed abundant IFN-λ and IFN-β following SIV infection, which usually peaked at 12 hpi. SIV infection also induced IFN and ISG expression that was largely cell type and virus specific. RIG-I expression was substantially altered in STECs and SiTECs following H1N1-NC infection, and in SiNECs and SNECs following H1N1-MN and H3N2-NC infection. Differences in IRF7 expression were significant only in STEC cells following H1N2-NC infection. Of the ISGs examined (GBP1 and OAS1), OAS1 expression had the highest induction following any SIV infection, and the OAS1 response was predominantly in the STECs and SiTECs. OAS1 expression by the respiratory epithelial cell types was maximal at 12 hpi and then declined by 24 hpi in all respiratory epithelial cell types, indicating that OAS1 may be temporally regulated. Analysis of the SIV gene segments among the SIV used in the study identified differences at the nucleotide level between H1N1-NC, H2N2-NC, H3N2-NC, and H1N1-MN SIV. However, the key genes with known antiviral activity were conserved and thus did not explain the differential SIV replication and induction of the IFN and ISG response between the viruses. Despite conservation of the PA residues linked to DI particle formation among the SIVs tested here, it is possible that the DI particles in the SIV inoculum were different. Treatment of each of the respiratory epithelial cell types with BPL-inactivated virus did induce low levels of type III IFN and ISG expression, suggesting that IFN and ISG induction was partly replication independent (data not shown). In this study, we show that SIV field isolates can replicate in undifferentiated primary immortalized nasal and tracheal cells and induce differential IFN and ISG responses that are relevant to influenza virus replication and possible reassortment.
MATERIALS AND METHODS
Cell lines and SIV infection.
SNECs and STECs were isolated from swine and are nasal epithelial cells or tracheal primary epithelial cells, respectively. SiNECs and SiTECs are immortalized swine nasal epithelial cells or swine tracheal epithelial cells from primary cells (27). These cell lines have been shown to support the replication of diverse human, swine, and avian influenza viruses, and were propagated as described (27). Briefly, the swine cell lines were grown in T25 flasks (Corning) coated with 50 μg/ml rat collagen (Sigma, St. Louis, MO) prepared in 0.02 N acetic acid (Sigma) used for coating the flasks. For SIV infection, 96-well flat-bottomed plates (Costar) were plated with 50 μg/ml rat collagen as above, coated overnight at room temperature (RT), and the medium decanted before 2 × 104 cells/well were added. The plated cells were incubated overnight at 37°C and 10% CO2, after which the cells were infected for 1 h with SIV at an MOI of 0.01 in serum-free minimal essential medium (MEM; HyClone, UT). The inoculum was decanted and infection proceeded for indicated time points in fresh MEM with 1:1,000 diluted tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin. Mock treatment consisted of cells treated with infection medium alone. Positive-control cells were treated with 50 μg/ml poly(I·C) (Sigma), a TLR3 agonist, for the indicated time points. A/swine/North Carolina/154072/2015 (H1N1-NC), A/swine/North Carolina/156551/2015 (H1N2-NC), and A/swine/North Carolina/157674/2015 (H3N2-NC) were collected during passive SIV surveillance in commercial swine farms (66), while A/Swine/MN/02749/2009 (H1N1-MN) was provided by Marie Culhane at the University of Minnesota. All isolates were passaged <3 times in MDCK cells. The viral isolates grew to similar but not identical titers in the swine and MDCK cells. Specifically, the H1N1-MN grew to higher titers than those of the other three viruses.
RNA isolation.
Total RNA was isolated from mock-treated or SIV-infected cells using the RNAdvance Cell v2 kit (Beckman Coulter, CA) at the time points indicated using solid-phase reversible immobilization (SPRI) paramagnetic beads to isolated total RNA from cultured cells. Briefly, cell pellets in 96-well plates were lysed with included lysis buffer, incubated with proteinase K for 30 min at RT, and lysates transferred to new plates. Paramagnetic beads in bead-binding solution were added to the wells and mixed to bind the total RNA. The plates were incubated on 96-well magnetic plates for 5 min at RT to separate RNA bound to beads, and supernatant was removed. The beads were washed off magnetic plates with wash buffer and applied to the magnetic plate, then washed with 70% ethanol. The ethanol was removed, and the pellets were incubated with RNase free DNase I solution for 15 min at RT. DNase activity was stopped using wash buffer followed by 70% ethanol washes per the manufacturer’s protocol, then by air-drying the washed beads. Total RNA was eluted in RNase-free molecular grade water (Sigma) and quantified using a NanoDrop 1000 spectrophotometer (Thermo Fisher, MA, USA).
qRT-PCR.
Total RNA was reverse transcribed using the LunaScript RT Supermix kit (New England Biolabs, MA, USA). Briefly, RNA samples were mixed with 5× LunaScript RT Supermix, which contained random hexamers, oligo(dT), deoxynucleoside triphosphates (dNTPs), RNase inhibitor, and Luna reverse transcriptase. Reverse transcription-quantitative PCR (qRT-PCR) runs contained undiluted cDNA, Luna universal probe qPCR master mix (New England Biolabs), and forward and reverse primers and probes. Reactions were amplified by denaturation at 95°C for 60 sec, and 40 cycles of denaturation at 95°C for 15 sec, followed by annealing at 60°C for 30 sec on a Stratagene Mx3005P instrument (Agilent, CA, USA). Swine-specific TaqMan gene expression assays were used for IFN-λ (Ss03820546_u1), IFN-β (Ss03378485_u1), RIG-I (Ss04322983_m1), IRF7 (Ss03385312_u1), GBP1 (Ss04245969_m1), and OAS1 (Ss03394660_m1).
Sanger sequencing.
SIVs used in this study were sequenced using Sanger sequencing (67). Briefly, viral RNA was reverse transcribed using primer Uni-12 (5′-AGCAAAAGCAGG-3′) and then amplified using primers MBTUni-12 (5′-ACGCGTGATCAGCRAAAGCAGG-3′) and MBTUni13 (5′-ACGCGTGATCAGTAGAAACAAGG-3′) for segments 4 to 8 and primers PB2-1 (5′-AGCRAAAGCAGGTCAATTATATTCA-3′) and PB2-2341R (5′-AGTAGAAACAAGGTCGTTTTTAAACTA-3′) for PB2, PB1-1 (5′-AGCRAAAGCAGGCAAACCATTTGAATG-3′) and PB1-2341-R (5′-AGTAGAAACAAGGCATTTTTTCATGAA-3′) for PB1, and PA-1 (5′-AGCRAAAGCAGGTACTGATYCGAAATG-3′) and PA-2233R (5′-AGTAGAAACAAGGTACTTTTTTGGACA-3′) for PA. Sequence data are provided as in File S1 in the supplemental material. For H1N1-MN SIV, segments 2 and 3 could not be sequenced.
Statistical analysis.
All data are from triplicate measurements and three independent samples. Fold changes in gene expression were calculated using the threshold cycle (ΔΔCT) method. All qPCR assay designs were as per MIQE 2 guidelines (71, 72). Prevalidated primer-probes for all genes were obtained from Applied Biosystems (ABI, CA, USA) or Integrated DNA Technologies (IDT, IA, USA). All statistical analysis was performed using one-way or two-way analysis of variance (ANOVA) in GraphPad Prism v9.0.1 with post hoc tests. Significance was determined by one-way or two-way ANOVA with the Geisser-Greenhouse correction (73), using a mixed-effects model. Multiple comparisons were corrected with Tukey’s post hoc test.
ACKNOWLEDGMENTS
We acknowledge funding in part from NIAID CEIRS contract numbers HHSN2662007000006C and HHSN272204000004C to R.A.T. and funding from the Georgia Research Alliance to R.A.T. Funding for S.S.C. was provided by NIAID CEIRS contract HHSN272201400006C and by ALSAC.
Footnotes
Supplemental material is available online only.
Contributor Information
Ralph A. Tripp, Email: ratripp@uga.edu.
Colin R. Parrish, Cornell University
REFERENCES
- 1.Petrova VN, Russell CA. 2018. The evolution of seasonal influenza viruses. Nat Rev Microbiol 16:47–60. doi: 10.1038/nrmicro.2017.118. [DOI] [PubMed] [Google Scholar]
- 2.Webster RG, Govorkova EA. 2014. Continuing challenges in influenza. Ann N Y Acad Sci 1323:115–139. doi: 10.1111/nyas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed). 2013. Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Rajao DS, Anderson TK, Gauger PC, Vincent AL. 2014. Pathogenesis and vaccination of influenza A virus in swine. Curr Top Microbiol Immunol 385:307–326. doi: 10.1007/82_2014_391. [DOI] [PubMed] [Google Scholar]
- 5.Rajao DS, Vincent AL. 2015. Swine as a model for influenza A virus infection and immunity. ILAR J 56:44–52. doi: 10.1093/ilar/ilv002. [DOI] [PubMed] [Google Scholar]
- 6.Brockwell-Staats C, Webster RG, Webby RJ. 2009. Diversity of influenza viruses in swine and the emergence of a novel human pandemic influenza A (H1N1). Influenza Other Respir Viruses 3:207–213. doi: 10.1111/j.1750-2659.2009.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webby RJ, Rossow K, Erickson G, Sims Y, Webster R. 2004. Multiple lineages of antigenically and genetically diverse influenza A virus co-circulate in the United States swine population. Virus Res 103:67–73. doi: 10.1016/j.virusres.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Krueger WS, Gray GC. 2013. Swine influenza virus infections in man. Curr Top Microbiol Immunol 370:201–225. doi: 10.1007/82_2012_268. [DOI] [PubMed] [Google Scholar]
- 9.Nelson MI, Wentworth DE, Culhane MR, Vincent AL, Viboud C, LaPointe MP, Lin X, Holmes EC, Detmer SE. 2014. Introductions and evolution of human-origin seasonal influenza a viruses in multinational swine populations. J Virol 88:10110–10119. doi: 10.1128/JVI.01080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen CW. 2002. The emergence of novel swine influenza viruses in North America. Virus Res 85:199–210. doi: 10.1016/s0168-1702(02)00027-8. [DOI] [PubMed] [Google Scholar]
- 11.Schultz-Cherry S, Olsen CW, Easterday BC. 2013. History of swine influenza, p 21–27. In Richt JA, Webby RJ (ed), Swine influenza. Springer Berlin Heidelberg, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 12.Gao S, Anderson TK, Walia RR, Dorman KS, Janas-Martindale A, Vincent AL. 2017. The genomic evolution of H1 influenza A viruses from swine detected in the United States between 2009 and 2016. J Gen Virol 98:2001–2010. doi: 10.1099/jgv.0.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajao DS, Anderson TK, Kitikoon P, Stratton J, Lewis NS, Vincent AL. 2018. Antigenic and genetic evolution of contemporary swine H1 influenza viruses in the United States. Virology 518:45–54. doi: 10.1016/j.virol.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Graaf M, Fouchier RA. 2014. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J 33:823–841. doi: 10.1002/embj.201387442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumlin U, Olofsson S, Dimock K, Arnberg N. 2008. Sialic acid tissue distribution and influenza virus tropism. Influenza Other Respir Viruses 2:147–154. doi: 10.1111/j.1750-2659.2008.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu G, Zhou Y. 2019. Cytoplasm and beyond: dynamic innate immune sensing of influenza A virus by RIG-I. J Virol 93:e02299-18. doi: 10.1128/JVI.02299-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 18.Weber F. 2015. The catcher in the RIG-I. Cytokine 76:38–41. doi: 10.1016/j.cyto.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Czyżewska-Dors E, Pomorska-Mól M, Dors A, Pluta A, Podgórska K, Kwit K, Stasiak E, Łukomska A. 2019. Proinflammatory cytokine changes in bronchoalveolar lavage fluid cells isolated from pigs infected solely with porcine reproductive and respiratory syndrome virus or co-infected with swine influenza virus. J Vet Res 63:489–495. doi: 10.2478/jvetres-2019-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K, Andreakos E, Ramos I, Smith G, Ruf-Zamojski F, Martínez-Romero C, Fribourg M, Carbajal EA, Hartmann BM, Nair VD, Marjanovic N, Monteagudo PL, DeJesus VA, Mutetwa T, Zamojski M, Tan GS, Jayaprakash C, Zaslavsky E, Albrecht RA, Sealfon SC, García-Sastre A, Fernandez-Sesma A, Coch C, Stümpel JP, Lilien-Waldau V, Wohlleber D, Kümmerer BM, Bekeredjian-Ding I, Kochs G, Garbi N, Herberhold S, Schuberth-Wagner C, Ludwig J, Barchet W, Schlee M, Hoerauf A, Bootz F, Staeheli P, Hartmann G, Hartmann E, Lozhkov AA, et al. 2017. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 46:875–890.e6. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 21.Hauser MJ, Dlugolenski D, Culhane MR, Wentworth DE, Tompkins SM, Tripp RA. 2013. Antiviral responses by swine primary bronchoepithelial cells are limited compared to human bronchoepithelial cells following influenza virus infection. PLoS One 8:e70251. doi: 10.1371/journal.pone.0070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du Y, Yang F, Wang Q, Xu N, Xie Y, Chen S, Qin T, Peng D. 2020. Influenza A virus antagonizes type I and type II interferon responses via SOCS1-dependent ubiquitination and degradation of JAK1. Virol J 17:74. doi: 10.1186/s12985-020-01348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uetani K, Hiroi M, Meguro T, Ogawa H, Kamisako T, Ohmori Y, Erzurum SC. 2008. Influenza A virus abrogates IFN-gamma response in respiratory epithelial cells by disruption of the Jak/Stat pathway. Eur J Immunol 38:1559–1573. doi: 10.1002/eji.200737045. [DOI] [PubMed] [Google Scholar]
- 24.Byrd-Leotis L, Liu R, Bradley KC, Lasanajak Y, Cummings SF, Song X, Heimburg-Molinaro J, Galloway SE, Culhane MR, Smith DF, Steinhauer DA, Cummings RD. 2014. Shotgun glycomics of pig lung identifies natural endogenous receptors for influenza viruses. Proc Natl Acad Sci U S A 111:E2241–50. doi: 10.1073/pnas.1323162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McQuillan AM, Byrd-Leotis L, Heimburg-Molinaro J, Cummings RD. 2019. Natural and synthetic sialylated glycan microarrays and their applications. Front Mol Biosci 6:88. doi: 10.3389/fmolb.2019.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delgado-Ortega M, Melo S, Punyadarsaniya D, Ramé C, Olivier M, Soubieux D, Marc D, Simon G, Herrler G, Berri M, Dupont J, Meurens F. 2014. Innate immune response to a H3N2 subtype swine influenza virus in newborn porcine trachea cells, alveolar macrophages, and precision-cut lung slices. Vet Res 45:42. doi: 10.1186/1297-9716-45-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meliopoulos V, Cherry S, Wohlgemuth N, Honce R, Barnard K, Gauger P, Davis T, Shult P, Parrish C, Schultz-Cherry S. 2020. Primary swine respiratory epithelial cell lines for the efficient isolation and propagation of influenza A viruses. J Virol 94:e01091-20. doi: 10.1128/JVI.01091-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fay EJ, Aron SL, Macchietto MG, Markman MW, Esser-Nobis K, Gale M, Jr, Shen S, Langlois RA. 2020. Cell type- and replication stage-specific influenza virus responses in vivo. PLoS Pathog 16:e1008760. doi: 10.1371/journal.ppat.1008760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Sun H, Cunningham FL, Li L, Hanson-Dorr K, Hopken MW, Cooley J, Long LP, Baroch JA, Li T, Schmit BS, Lin X, Olivier AK, Jarman RG, DeLiberto TJ, Wan XF. 2018. Tissue tropisms opt for transmissible reassortants during avian and swine influenza A virus co-infection in swine. PLoS Pathog 14:e1007417. doi: 10.1371/journal.ppat.1007417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, Yoshikai Y. 2013. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res 99:230–237. doi: 10.1016/j.antiviral.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 31.García-Sastre A. 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goraya MU, Zaighum F, Sajjad N, Anjum FR, Sakhawat I, Rahman SU. 2020. Web of interferon stimulated antiviral factors to control the influenza A viruses replication. Microb Pathog 139:103919. doi: 10.1016/j.micpath.2019.103919. [DOI] [PubMed] [Google Scholar]
- 33.Lozhkov AA, Klotchenko SA, Ramsay ES, Moshkoff HD, Moshkoff DA, Vasin AV, Salvato MS. 2020. The key roles of interferon lambda in human molecular defense against respiratory viral infections. Pathogens 9:989. doi: 10.3390/pathogens9120989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog 4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davidson S, McCabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, Hartmann R, Wack A. 2016. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol Med 8:1099–1112. doi: 10.15252/emmm.201606413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramos I, Smith G, Ruf-Zamojski F, Martinez-Romero C, Fribourg M, Carbajal EA, Hartmann BM, Nair VD, Marjanovic N, Monteagudo PL, DeJesus VA, Mutetwa T, Zamojski M, Tan GS, Jayaprakash C, Zaslavsky E, Albrecht RA, Sealfon SC, Garcia-Sastre A, Fernandez-Sesma A. 2019. Innate immune response to influenza virus at single-cell resolution in human epithelial cells revealed paracrine induction of interferon lambda 1. J Virol 93:e00559-19. doi: 10.1128/JVI.00559-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox JM, Crabtree JM, Sage LK, Tompkins SM, Tripp RA. 2015. Interferon lambda upregulates IDO1 expression in respiratory epithelial cells after influenza virus infection. J Interferon Cytokine Res 35:554–562. doi: 10.1089/jir.2014.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolff T, Ludwig S. 2009. Influenza viruses control the vertebrate type I interferon system: factors, mechanisms, and consequences. J Interferon Cytokine Res 29:549–557. doi: 10.1089/jir.2009.0066. [DOI] [PubMed] [Google Scholar]
- 39.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 40.Anderson SL, Carton JM, Lou J, Xing L, Rubin BY. 1999. Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology 256:8–14. doi: 10.1006/viro.1999.9614. [DOI] [PubMed] [Google Scholar]
- 41.Pan W, Zuo X, Feng T, Shi X, Dai J. 2012. Guanylate-binding protein 1 participates in cellular antiviral response to dengue virus. Virol J 9:292. doi: 10.1186/1743-422X-9-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li LF, Yu J, Li Y, Wang J, Li S, Zhang L, Xia SL, Yang Q, Wang X, Yu S, Luo Y, Sun Y, Zhu Y, Munir M, Qiu HJ. 2016. Guanylate-binding protein 1, an interferon-induced GTPase, exerts an antiviral activity against classical swine fever virus depending on its GTPase activity. J Virol 90:4412–4426. doi: 10.1128/JVI.02718-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu Z, Shi Z, Yan W, Wei J, Shao D, Deng X, Wang S, Li B, Tong G, Ma Z. 2013. Nonstructural protein 1 of influenza A virus interacts with human guanylate-binding protein 1 to antagonize antiviral activity. PLoS One 8:e55920. doi: 10.1371/journal.pone.0055920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niu P, Shabir N, Khatun A, Seo BJ, Gu S, Lee SM, Lim SK, Kim KS, Kim WI. 2016. Effect of polymorphisms in the GBP1, Mx1 and CD163 genes on host responses to PRRSV infection in pigs. Vet Microbiol 182:187–195. doi: 10.1016/j.vetmic.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 45.Choi UY, Kang JS, Hwang YS, Kim YJ. 2015. Oligoadenylate synthase-like (OASL) proteins: dual functions and associations with diseases. Exp Mol Med 47:e144. doi: 10.1038/emm.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mashimo T, Simon-Chazottes D, Guénet JL. 2008. Innate resistance to flavivirus infections and the functions of 2′-5′ oligoadenylate synthetases. Curr Top Microbiol Immunol 321:85–100. doi: 10.1007/978-3-540-75203-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di H, Elbahesh H, Brinton MA. 2020. Characteristics of human OAS1 isoform proteins. Viruses 12:152. doi: 10.3390/v12020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC. 2015. Overlapping patterns of rapid evolution in the nucleic acid sensors cGAS and OAS1 suggest a common mechanism of pathogen antagonism and escape. PLoS Genet 11:e1005203. doi: 10.1371/journal.pgen.1005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz SL, Park EN, Vachon VK, Danzy S, Lowen AC, Conn GL. 2020. Human OAS1 activation is highly dependent on both RNA sequence and context of activating RNA motifs. Nucleic Acids Res 48:7520–7531. doi: 10.1093/nar/gkaa513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deo S, Patel TR, Dzananovic E, Booy EP, Zeid K, McEleney K, Harding SE, McKenna SA. 2014. Activation of 2′ 5′-oligoadenylate synthetase by stem loops at the 5′-end of the West Nile virus genome. PLoS One 9:e92545. doi: 10.1371/journal.pone.0092545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rios JJ, Fleming JG, Bryant UK, Carter CN, Huber JC, Long MT, Spencer TE, Adelson DL. 2010. OAS1 polymorphisms are associated with susceptibility to West Nile encephalitis in horses. PLoS One 5:e10537. doi: 10.1371/journal.pone.0010537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin RJ, Yu HP, Chang BL, Tang WC, Liao CL, Lin YL. 2009. Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against dengue virus infection. J Immunol 183:8035–8043. doi: 10.4049/jimmunol.0902728. [DOI] [PubMed] [Google Scholar]
- 53.Zheng S, Zhu D, Lian X, Liu W, Cao R, Chen P. 2016. Porcine 2′, 5′-oligoadenylate synthetases inhibit Japanese encephalitis virus replication in vitro. J Med Virol 88:760–768. doi: 10.1002/jmv.24397. [DOI] [PubMed] [Google Scholar]
- 54.Kosmider B, Messier EM, Janssen WJ, Nahreini P, Wang J, Hartshorn KL, Mason RJ. 2012. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir Res 13:43. doi: 10.1186/1465-9921-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu G, Zhang X, Liu Q, Bing G, Hu Z, Sun H, Xiong X, Jiang M, He Q, Wang Y, Pu J, Guo X, Yang H, Liu J, Sun Y. 2017. PA-X protein contributes to virulence of triple-reassortant H1N2 influenza virus by suppressing early immune responses in swine. Virology 508:45–53. doi: 10.1016/j.virol.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Gong XQ, Sun YF, Ruan BY, Liu XM, Wang Q, Yang HM, Wang SY, Zhang P, Wang XH, Shan TL, Tong W, Zhou YJ, Li GX, Zheng H, Tong GZ, Yu H. 2017. PA-X protein decreases replication and pathogenicity of swine influenza virus in cultured cells and mouse models. Vet Microbiol 205:66–70. doi: 10.1016/j.vetmic.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Sun Y, Hu Z, Zhang X, Chen M, Wang Z, Xu G, Bi Y, Tong Q, Wang M, Sun H, Pu J, Iqbal M, Liu J. 2020. An R195K mutation in the PA-X protein increases the virulence and transmission of influenza A virus in mammalian hosts. J Virol 94:e01817-19. doi: 10.1128/JVI.01817-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu G, Zhang X, Sun Y, Liu Q, Sun H, Xiong X, Jiang M, He Q, Wang Y, Pu J, Guo X, Yang H, Liu J. 2016. Truncation of C-terminal 20 amino acids in PA-X contributes to adaptation of swine influenza virus in pigs. Sci Rep 6:21845. doi: 10.1038/srep21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi M, Jagger BW, Wise HM, Digard P, Holmes EC, Taubenberger JK. 2012. Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A virus. J Virol 86:12411–12413. doi: 10.1128/JVI.01677-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng J, Tao J, Li B, Shi Y, Liu H. 2019. The tyrosine 73 and serine 83 dephosphorylation of H1N1 swine influenza virus NS1 protein attenuates virus replication and induces high levels of beta interferon. Virol J 16:152. doi: 10.1186/s12985-019-1255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng J, Zhang C, Tao J, Li B, Shi Y, Liu H. 2018. Effects of the S42 residue of the H1N1 swine influenza virus NS1 protein on interferon responses and virus replication. Virol J 15:57. doi: 10.1186/s12985-018-0971-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muramoto Y, Noda T, Kawakami E, Akkina R, Kawaoka Y. 2013. Identification of novel influenza A virus proteins translated from PA mRNA. J Virol 87:2455–2462. doi: 10.1128/JVI.02656-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Solórzano A, Webby RJ, Lager KM, Janke BH, García-Sastre A, Richt JA. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol 79:7535–7543. doi: 10.1128/JVI.79.12.7535-7543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vasilijevic J, Zamarreno N, Oliveros JC, Rodriguez-Frandsen A, Gomez G, Rodriguez G, Perez-Ruiz M, Rey S, Barba I, Pozo F, Casas I, Nieto A, Falcon A. 2017. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog 13:e1006650. doi: 10.1371/journal.ppat.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bakre AA, Jones LP, Kyriakis CS, Hanson JM, Bobbitt DE, Bennett HK, Todd KV, Orr-Burks N, Murray J, Zhang M, Steinhauer DA, Byrd-Leotis L, Cummings RD, Fent J, Coffey T, Tripp RA. 2020. Molecular epidemiology and glycomics of swine influenza viruses circulating in commercial swine farms in the southeastern and midwest United States. Vet Microbiol 251:108914. doi: 10.1016/j.vetmic.2020.108914. [DOI] [PubMed] [Google Scholar]
- 67.Pareek CS, Smoczynski R, Tretyn A. 2011. Sequencing technologies and genome sequencing. J Appl Genet 52:413–435. doi: 10.1007/s13353-011-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meunier I, von Messling V. 2012. PB1-F2 modulates early host responses but does not affect the pathogenesis of H1N1 seasonal influenza virus. J Virol 86:4271–4278. doi: 10.1128/JVI.07243-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pena L, Vincent AL, Loving CL, Henningson JN, Lager KM, Li W, Perez DR. 2012. Strain-dependent effects of PB1-F2 of triple-reassortant H3N2 influenza viruses in swine. J Gen Virol 93:2204–2214. doi: 10.1099/vir.0.045005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiao Y, Evseev D, Stevens CA, Moghrabi A, Miranzo-Navarro D, Fleming-Canepa X, Tetrault DG, Magor KE. 2020. Influenza PB1-F2 inhibits avian MAVS signaling. Viruses 12:409. doi: 10.3390/v12040409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 72.Bustin SA, Wittwer CT. 2017. MIQE: a step toward more robust and reproducible quantitative PCR. Clin Chem 63:1537–1538. doi: 10.1373/clinchem.2016.268953. [DOI] [PubMed] [Google Scholar]
- 73.Geisser S, Greenhouse SW. 1958. An extension of Box’s results on the use of the F distribution in multivariate analysis. Ann Math Statist 29:885–891. doi: 10.1214/aoms/1177706545. [DOI] [Google Scholar]
- 74.Reche PA. 2020. Sequence identity and similarity. http://imed.med.ucm.es/Tools/sias.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4. Download JVI.00692-21-s0001.pdf, PDF file, 443 KB (442.5KB, pdf)
Table S2. Download JVI.00692-21-s0002.xlsx, XLSX file, 43 KB (42.6KB, xlsx)