

ABSTRACT
During retrovirus infection, a histone-free DNA copy of the viral RNA genome is synthesized and rapidly loaded with nucleosomes de novo upon nuclear entry. The potential role of viral accessory proteins in histone loading onto retroviral DNAs has not been extensively investigated. The p12 protein of Moloney murine leukemia virus (MMLV) is a virion protein that is critical for tethering the incoming viral DNA to host chromatin in the early stages of infection. Infection by virions containing a mutant p12 (PM14) defective in chromatin tethering results in the formation of viral DNAs that do not accumulate in the nucleus. In this report, we show that viral DNAs of these mutants are not loaded with histones. Moreover, the DNA genomes delivered by mutant p12 show prolonged association with viral structural proteins nucleocapsid (NC) and capsid (CA). The histone-poor viral DNA genomes do not become associated with the host RNA polymerase II machinery. These findings provide insights into fundamental aspects of retroviral biology, indicating that tethering to host chromatin by p12 and retention in the nucleus are required to allow loading of histones onto the viral DNA.
IMPORTANCE Incoming retroviral DNAs are rapidly loaded with nucleosomal histones upon entry into the nucleus and before integration into the host genome. The entry of murine leukemia virus DNA into the nucleus occurs only upon dissolution of the nuclear membrane in mitosis, and retention in the nucleus requires the action of a viral protein, p12, which tethers the DNA to host chromatin. Data presented here show that the tethering activity of p12 is required for the loading of histones onto the viral DNA. p12 mutants lacking tethering activity fail to acquire histones, retain capsid and nucleocapsid proteins, and are poorly transcribed. The work defines a new requirement for a viral protein to allow chromatinization of viral DNA.
KEYWORDS: chromatin immunoprecipitation, histone loading, nuclear accumulation, retroviral DNA
INTRODUCTION
The life cycle of retroviruses can be divided into two distinct phases: an early phase leading to the formation of the proviral DNA and a late phase resulting in progeny virion production. The early phase of the retrovirus life cycle begins with virus binding to cell surface receptors, followed by entry into the cytoplasm. In the context of a virion-like core particle, the single-stranded viral RNA genome is copied by the viral reverse transcriptase (RT) to generate a linear double-stranded DNA version of the genome containing so-called long terminal repeats (LTRs) at the two ends (1). This double-stranded linear viral DNA, contained within a large complex termed the preintegration complex (PIC), is then integrated into the host genomic DNA to establish the integrated provirus. Typically, a small portion of the viral DNA fails to integrate and instead gives rise to the formation of circular DNA forms containing either one or two copies of the long terminal repeats (LTRs). The majority of the 1-LTR circles form as a result of homologous recombination between the two LTRs of the linear viral DNA (2, 3). The 2-LTR circles are generated by joining the termini of the viral DNA by the host nonhomologous end joining (NHEJ) DNA repair pathways (2, 4). Because circularization mainly occurs in the nucleus, 2-LTR circles serve as a useful marker for successful viral DNA nuclear import, as their unique sequence of the LTR-LTR junction allows easy detection by PCR (5). Over time, these unintegrated DNA forms, lacking origins of DNA replication, gradually diminish with each cell division.
The timing and intracellular location of reverse transcription, the route of entry into the nucleus, and the choice of integration site all vary profoundly depending on the viral genera. Recent work has shown that HIV-1 cores enter the nucleus through enlarged nuclear pores and that reverse transcription is completed within the nucleus (6, 7). In the case of the murine leukemia viruses, the PIC is unable to enter the nucleus of nondividing cells and requires nuclear envelope breakdown during cell division to gain access to host chromosomes (8). For these viruses, recovery of the PIC in the nucleus has been shown to require association with host chromatin and retention with the host chromosomes during the reformation of the nuclear membrane at the end of mitosis (9–12).
The late phase of the retroviral life cycle begins with expression of viral genes. Following integration, the 5′ LTR, with the help of host transcription factors, functions as the viral promoter to direct transcription of viral genes by RNA polymerase II. Viral RNAs, including spliced and unspliced RNAs, then undergo nuclear export into the cytoplasm. The unspliced RNA can be used as mRNA for Gag and Gag-polymerase (Gag-Pol) translation or as viral genomic RNA to be packaged into new virions, as determined by the precise position of the 5′ end of the transcripts (13). Following assembly of viral proteins and RNA into new immature viral particles, budding occurs whereby the immature viral particle acquires the host plasma membrane. The final step involves release and maturation, when the Gag and Gag-Pol polyproteins of the virus are cleaved by the retroviral protease to generate mature particles capable of infecting new cells.
During viral particle maturation, the Gag polyprotein is cleaved by the viral protease into three main structural proteins, designated matrix (MA), capsid (CA), and nucleocapsid (NC). In many retroviral genera, additional proteins that are essential for infection are situated between MA and CA in the Gag polyprotein. One such protein is the p12 protein of the Moloney murine leukemia virus (MMLV). The MMLV p12 protein is 84 amino acids in length and plays a complex role during both early and late stages of infection (9, 11, 14–18). The N terminus of p12 contains a PPPY late domain (l-domain) motif that interacts with the HECT ubiquitin ligases to promote viral budding (14, 19). Using alanine scanning substitutions in p12, it was shown that both the N terminus and C terminus of p12 are essential for early stages of infection (14). Some mutations (PM6 and PM8) in the N-terminal region of p12 caused defects in DNA synthesis, suggesting a role for p12 early in viral entry, either before or during reverse transcription (14). Other mutations (PM5 and PM7) in the N-terminal domain allowed for normal levels of reverse transcription but blocked formation of any detectable viral circular DNAs, suggesting a defect in nuclear entry or retention. Mutations in the C-terminal domain of p12 appear to only disrupt the process of nuclear entry or nuclear accumulation, as mutant viruses with alterations in this region (PM13, PM14, and PM15) also made normal levels of linear viral DNA but no detectable circular DNAs (14). Consistent with these observations, p12 was found to be a component of the MMLV PIC (17). PICs containing wild-type (WT) p12 initially localized to the cytoplasm, but after trafficking into the nucleus, p12 accumulated on mitotic chromosomes (9, 17). The interaction between p12 and chromatin appears to be transient or labile, as chromatin decondensation resulted in the release of p12 from host chromatin (9). Several p12 C-terminal mutants (PM13, PM14, and PM15) were found to be defective in chromatin association (9, 18), a phenotype that was partially rescued by the addition of heterologous chromatin binding sequences (CBS) to p12 (10). p12 is also the main phosphoprotein in MMLV, with serine-61 of the C-terminal domain (CTD) being the major phosphorylation site in vivo (15, 16, 20, 21). Viruses harboring either phosphomimetic or phosphodeficient mutations at serine-61 showed severe defects in replication due to lack of chromatin tethering (15, 16). Prolonged passaging of a replication-defective phosphodeficient p12 mutant, SS(61,65)AA, resulted in revertants carrying additional compensatory mutations such as isoleucine substitution at M63 (M63I) that now showed levels of infectivity similar to those of wild-type virus (15). This rescued phenotype was later correlated with restoration of chromatin tethering (16). The data suggest that phosphorylated p12 mediates chromatin tethering of MMLV PIC at early stages of infection, a step that is necessary to ensure successful and persistent nuclear retention of viral DNAs prior to integration.
The state of viral DNA after reverse transcription and before integration has only recently been examined in detail. In the case of MMLV, unintegrated linear and circular viral DNAs undergo rapid association with histones after nuclear entry and before integration (22). Histone loading onto viral DNAs coincided with the loss of NC and CA from the viral DNA, raising the possibility that these two events are coordinated (22). The unintegrated MMLV DNAs are subject to rapid transcriptional silencing by host machinery including a DNA-binding protein (NP220), members of the HUSH complex (MPP8, PPHLN1, and TASOR), H3K9 methyltransferase SETDB1, and histone deacetylases HDAC1 and 4 (23). Even though HIV-1 enters the nucleus by completely different mechanisms, the unintegrated DNAs of HIV-1 are loaded with histones with kinetics similar to those seen with MMLV (24) and are similarly silenced, though apparently HIV-1 silencing utilizes a distinct set of host factors (23–25).
The relationship between the tethering of MMLV DNA to host chromatin and histone loading on DNA is not yet clear: whether MMLV p12 is required for histone loading onto viral DNAs has not been examined. In this study, we compared the kinetics of histone loading onto viral DNAs between wild-type (WT) and a p12 mutant (PM14) defective in chromatin tethering. We show that histone loading onto viral DNAs is severely reduced in the p12-PM14 mutant. Instead, mutant DNA genomes exhibit prolonged association with viral structural proteins NC and CA. The histone-poor viral DNA genomes are expressed very poorly and lack significant association with RNA polymerase II. Our findings highlight the importance of viral accessory proteins in facilitating chromatinization of viral DNAs.
RESULTS
MMLV p12 mutant defective in chromatin tethering is defective in nuclear accumulation of viral DNAs.
To investigate the requirement for p12 during histone loading onto retroviral DNAs, we utilized an MMLV reporter genome in which the viral genes were replaced with the green fluorescent protein (GFP) gene (MMLV-GFP) (Fig. 1A). Two different viral helper genomes were used to encapsidate the MMLV-GFP reporter viruses, encoding either wild-type (WT) p12 or the PM14 mutant p12 (p12-PM14) defective for tethering to host chromatin (9, 14, 17, 18). Of the set of p12 mutants originally described (14), we chose p12-PM14 because this is by far the most studied mutant and the one with the strongest and sharpest phenotype of defective nuclear entry and transduction (9–11, 16–18, 26). Of note, p12-PM14 virions contain normal levels of CA, MA, and p12 compared to the levels in WT virions (11, 14, 16, 18), indicating that the mutation does not affect the processing of the Gag precursor polyprotein. Virions were prepared by transfection of 293T cells with helper DNAs, MMLV-GFP reporter DNAs, and a construct expressing the vesicular stomatitis virus glycoprotein (VSV-G) envelope. HeLa cells were infected with comparable amounts of the two virus preparations at multiplicity of infection (MOI) of the wild-type version of approximately 0.6. Total DNA was harvested at 12 h, 24 h, or 48 h postinfection, and levels of viral DNA replication intermediates were monitored by quantitative real-time PCR (qPCR) using primers specific for GFP (to detect total viral DNA) or for the 2-LTR circles (a marker of nuclear entry). As a negative control, HeLa cells transduced with heat-inactivated (HI) virus yielded no PCR signal, indicating minimal contaminating plasmid DNA carryover from the virus preparation. In both WT and p12-PM14 virus, total viral DNA, which includes linear, circular, and integrated proviral DNA, peaked at 12 to 24 h postinfection and then decreased slightly by 48 h (Fig. 1B). The levels of total viral DNA formed by the two viruses were very similar, confirming comparable multiplicities of infection and efficiencies of entry and reverse transcription. In the WT virus, 2-LTR circles appeared at 12 h postinfection, peaked at 24 h postinfection, and decreased to low levels at 48 h, indicative of their short half-life (Fig. 1C). Levels of 2-LTR circles generated by the p12-PM14 mutant virus were very low compared to those generated by WT at all time points tested (Fig. 1C), consistent with previous reports (14, 27). Though the levels of 2-LTR circles were very low, they remained readily detectable above background (HI virus). These results suggest that the DNAs delivered by the mutant virus do not efficiently accumulate in the nucleus.
FIG 1.

MMLV p12 mutant defective in chromatin tethering has normal DNA synthesis but defective nuclear entry. (A) Schematic of MMLV single-round GFP reporter virus DNA and 2-LTR circular DNA generated during infection. Locations of PCR primers targeting GFP and 2-LTR circles are shown. (B) HeLa cells were infected with VSV-G pseudotyped wild-type (WT) or p12-PM14 mutant MMLV-GFP viruses. Total DNA were isolated from infected cells at different time points, followed by real-time quantitative PCR using primers targeting GFP (total viral DNA). Levels of total viral DNA were first normalized using the 2−ΔΔCT method to the value obtained for the GAPDH gene; the obtained values were then normalized to the values obtained from HeLa cells infected with WT virus at 12 h (set to 1). To control for potential plasmid DNA carryover in the viral supernatant, heat-inactivated (HI) virus was used in parallel. Results shown are means ± standard errors (SEs) from three independent experiments. (C) Similar experiment as that in panel B using primers targeting 2-LTR circles. Student’s t test was used for statistical analysis. *, P < 0.05 compared to the same time point in WT virus. (D) Nuclear or cytoplasmic extract prepared from HeLa cells was subjected to Western blotting with anti-histone H3 (nuclear marker) or anti-GAPDH (cytoplasmic marker). (E) HeLa cells infected with VSV-G pseudotyped WT or p12-PM14 mutant MMLV-GFP viruses were subjected to nuclear/cytoplasmic fractionation at 12 and 24 h postinfection. Total DNA were then isolated from cellular fractions followed by real-time qPCR using primers targeting GFP (total viral DNA) or GAPDH. The absolute copy number per μg of input DNA is calculated based on standard curves generated using plasmid DNA. The results shown are means ± standard deviations (SDs) from two independent experiments performed in duplicate.
To further confirm the indication that the p12-PM14 mutant virus is defective in nuclear entry or retention, MMLV-GFP infected cells were subjected to cytoplasmic/nuclear fractionation (Fig. 1D) followed by qPCR at 12 and 24 h postinfection to detect the levels of total viral DNA in these two compartments (Fig. 1E). Western blotting of nuclear and cytoplasmic fractions with anti-histone H3 and anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibodies confirmed the effectiveness of the fractionation (Fig. 1D). In the case of the WT virus, high levels of total viral DNA accumulated in the nuclear fraction at 24 h postinfection, whereas for the p12-PM14 mutant virus, total viral DNA remained predominately cytoplasmic (Fig. 1E). These results further confirmed the defect in nuclear entry/retention for the p12-PM14 mutant and are fully consistent with earlier work documenting the localization of linear and circular DNAs in wild-type and p12 mutant (27). As expected, GAPDH DNA was highly enriched in the nuclear fraction at all time points (Fig. 1E).
Histone loading onto retroviral DNAs is abolished in MMLV p12-PM14 mutant virus.
We next monitored the association of nucleosomal histones with retroviral DNAs. Infected HeLa cells were formaldehyde cross-linked at 12 h, 24 h, and 48 h postinfection, and chromatin immunoprecipitation (ChIP) assays were performed with anti-histone H3 or nonspecific IgG, followed by qPCR using primers targeting GFP or 2-LTR circles. Results are reported as levels of DNA recovered by immunoprecipitation (IP) as a percentage of the DNA input to the IP reactions. Primers targeting GAPDH were used to score fully chromatinized cellular DNA to be compared with the virus. In WT virus, the total viral DNA rapidly associated with histone H3 at 12 h, and the portion of the viral DNA loaded with histone reached levels comparable to that of the fully chromatinized GAPDH gene by just 24 h (Fig. 2). Analysis of 2-LTR circles also revealed rapid histone H3 loading, with the portion of the DNA loaded with histones also comparable to that of the GAPDH gene by 12 h (Fig. 2). In the case of p12-PM14 mutant virus, however, we observed an approximately 50-fold decrease in the portion of the total viral DNA loaded with histones (scoring GFP) compared to that of WT virus across all the time points tested, down to near background (Fig. 2). These results indicate that the viral DNAs of the p12-PM14 mutant are not available to the host machinery normally active at the loading of histones onto the incoming DNA. Furthermore, the portion of 2-LTR circles loaded with histones was approximately 10-fold lower in the p12-PM14 mutant than in WT (Fig. 2). The results suggest that the 2-LTR circles, which are generated by the p12-PM14 mutant at very low abundance, are present in a state that is poorly accessible by the histone loading machinery. As a negative control, ChIP reactions using nonspecific IgG showed no significant enrichment of any of the three DNAs (GFP, 2-LTR circles, and GAPDH), and we can reliably score the signals we see with our specific antibodies over this very low background (Fig. 2). Together, these findings suggest that histone loading onto retroviral DNAs is severely compromised in the MMLV p12 mutant defective in chromatin tethering.
FIG 2.
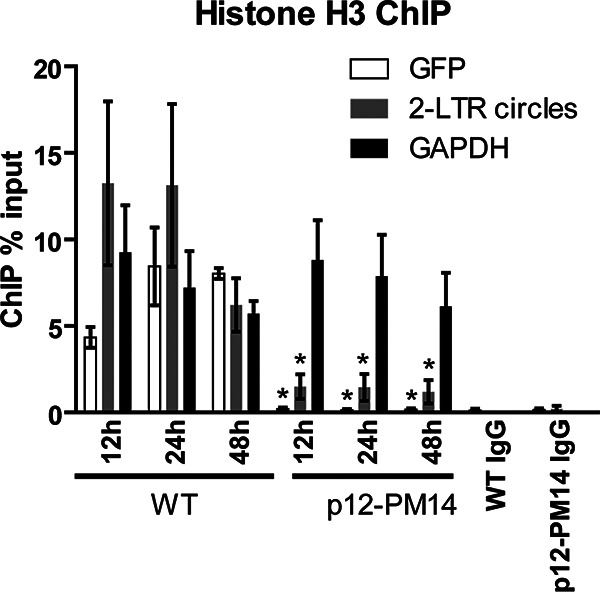
MMLV p12 mutant is defective in histone loading. Histone H3 chromatin immunoprecipitation (ChIP) analysis of chromatin harvested at indicated times following WT or p12-PM14 mutant MMLV-GFP infection of HeLa cells. ChIP data are presented as percentage of input DNA (means ± SEs from three independent experiments). Student’s t test was used for statistical analysis. *, P < 0.05 compared to the same time point in WT virus.
Retroviral DNAs derived from p12-PM14 mutant show prolonged association with NC and CA.
The NC proteins of retroviruses are small, highly basic nucleic acid-binding proteins that directly contact the RNA genome in the mature virion, as well as the retroviral DNAs in the preintegration complex (PIC) (28, 29). NC is involved in many steps of viral replication, including virion assembly, genomic RNA packaging, reverse transcription, and integration (30–32). The CA protein constitutes the major structural protein of the virion core and plays a role in early events such as nuclear entry and DNA integration (33). To test the hypothesis that histone loading may be associated with rapid disassembly of PIC components NC and CA from viral DNAs, we carried out ChIP assays outlined above using antibodies targeting MMLV NC and CA. For WT virus, both proteins were bound to viral DNA at early times after infection. The highest levels of association between NC and total viral DNA were observed at 12 h postinfection, followed by a rapid decrease to very low levels by 48 h (Fig. 3A). A similar result was obtained for CA (Fig. 3B). Notably, no association of 2-LTR circles with NC or CA was detected at any time (Fig. 3A and B), suggesting that the bulk of both viral proteins was removed before circularization. The specificity of the antibodies used for ChIP was confirmed by the lack of nonspecific binding to the DNA of the host GAPDH gene. Overall, the timing of NC and CA dissociation from the total viral DNAs coincided with the accumulation of core histones onto the DNA (Fig. 2). These results are consistent with the notion that core histone binding to viral DNA may promote the dissociation of NC and CA from the viral DNA by direct competition for DNA binding.
FIG 3.
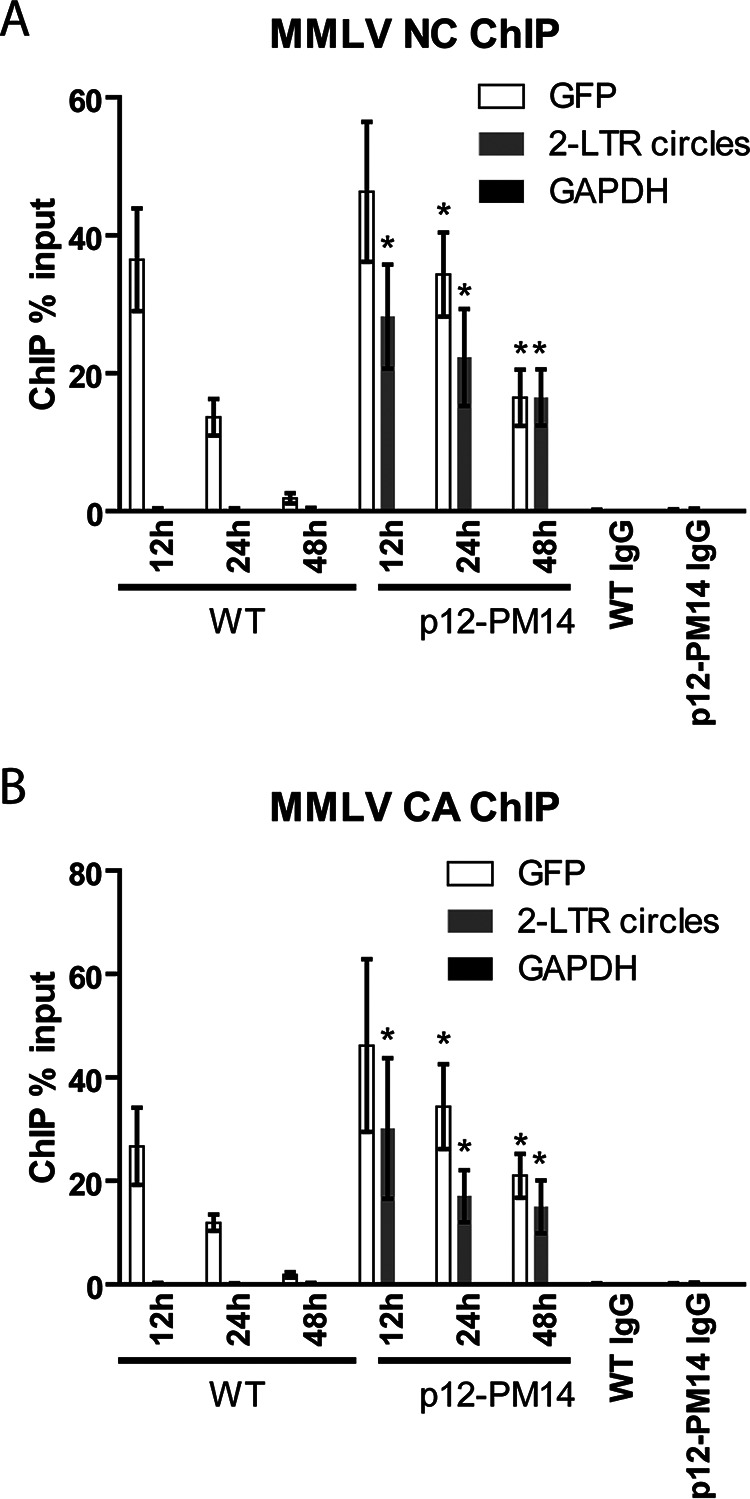
Viral capsid (CA) and nucleocapsid (NC) show delayed dissociation from total viral DNA derived from MMLV p12-PM14 mutant virus. (A) NC ChIP analysis of chromatin harvested at indicated times following WT or p12-PM14 mutant MMLV-GFP infection of HeLa cells. ChIP data are presented as percentage of input DNA (means ± SEs from three independent experiments). Student’s t test was used for statistical analysis. *, P < 0.05 compared to the same time point in WT virus. (B) Similar experiment as that in panel A using CA antibodies. Student’s t test was used for statistical analysis. *, P < 0.05 compared to the same time point in WT virus.
In the p12-PM14 mutant, similar to WT virus, the association between total viral DNA with NC or CA was highest at 12 h postinfection. However, unlike the WT virus, high levels of NC or CA remained bound to total viral DNA in the mutant even at 48 h postinfection (Fig. 3A and B). Furthermore, in sharp contrast to the WT virus, we detected robust association of NC and CA with the rare 2-LTR circles at all time points tested (Fig. 3A and B). The retention of NC and CA with the 2-LTR circles of the mutant PICs correlated with the poor chromatinization of these circular DNAs (Fig. 2). The results suggest that the state of the few circular DNAs formed in the case of the p12-PM14 virus could be very different from the state of those formed in the WT virus. Here, the circles may not be fully uncoated. The small number of circles formed by the mutant DNAs must require at least some exposure of the DNA termini to the host repair machinery to allow for end joining, but this exposure must occur in the context of a PIC with retention of CA and NC and lack of histone loading.
Histone-poor retroviral DNAs are poor templates for transcription.
Previous work has shown that the PM14 mutant virus was very poorly expressed after infection (9, 10, 14, 18). To test for expression in our experiments, HeLa cells were infected with VSV-G-pseudotyped MMLV-WT GFP or p12-PM14 mutant GFP, and GFP expression was monitored by flow cytometry analysis 48 h postinfection (Fig. 4A). Compared to that in WT virus, p12-PM14 mutant virus showed a 60-fold reduction in the percentage of GFP positive cells, close to background levels, and a 3-fold decrease in mean GFP fluorescence intensity (Fig. 4A), as previously reported (9, 10, 14, 18). We also performed ChIP assays to measure the portion of the DNA with RNA polymerase II (RNA Pol II) localized on the viral DNAs. For WT virus, the MMLV-LTR showed robust association with Pol II to a level comparable to that of the constitutively expressed GAPDH gene (Fig. 4B). Notably, we also detected a significant portion of the 2-LTR circles bound with Pol II (Fig. 4B). This is surprising given that these circular DNAs are not well expressed and suggests that Pol II is bound but is not actively transcribing on these DNA templates. Compared to that of WT, p12-PM14 virus showed a dramatic decrease in the recovery of viral reporter DNAs precipitated with antibodies to Pol II (Fig. 4B), consistent with very low levels of GFP expression in this mutant (Fig. 4A). Together, these findings indicate that the lack of histone loading onto retroviral DNAs is associated with very low levels of accessibility to Pol II and very low levels of gene expression.
FIG 4.
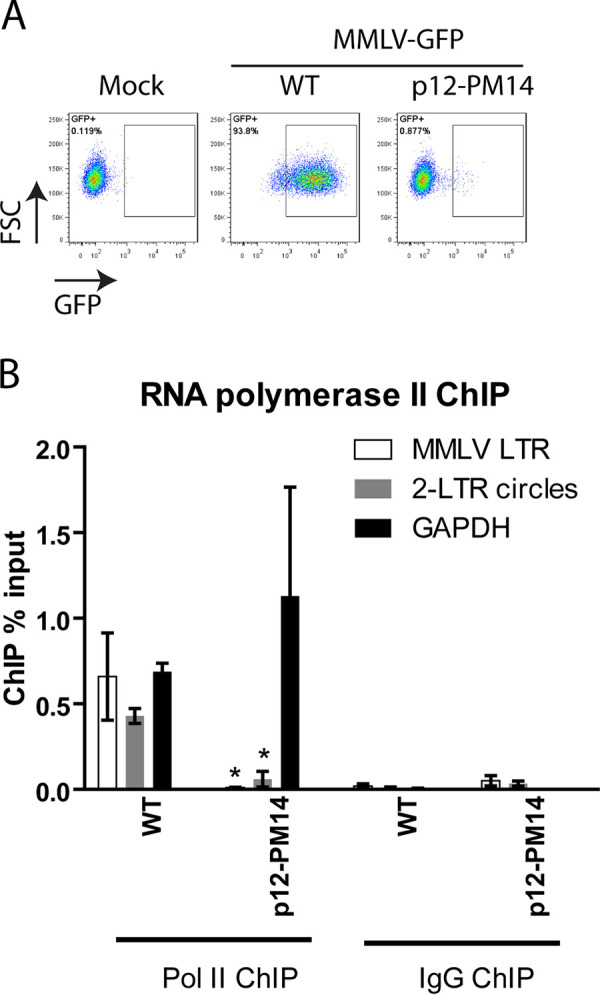
MMLV p12-PM14 mutant DNAs are poorly expressed and show decreased association with RNA polymerase II. (A) Flow cytometry analysis of HeLa cells infected with VSV-G pseudotyped WT or p12-m14 mutant MMLV-GFP viruses at 1 d postinfection. FSC, forward scatter. A representative experiment is shown. (B) RNA polymerase II ChIP analysis of chromatin harvested at 2 days following WT or p12-PM14 mutant MMLV-GFP infection of HeLa cells. ChIP data are presented as percentage of input DNA (means ± SEs from three independent experiments). Student’s t test was used for statistical analysis. *, P < 0.05 compared to WT virus.
DISCUSSION
In this study, we investigated the requirement for MMLV p12 in the chromatinization of MMLV DNA genomes by comparing the kinetics of histone loading onto MMLV-WT or the p12-PM14 mutant virus (14) previously shown to be defective in chromatin tethering and nuclear retention of PICs (9, 10, 18). In the case of WT virus, viral DNAs, including linear DNA and 2-LTR circle DNAs, rapidly associated with core histones as early as 12 h postinfection (Fig. 2). Histone loading is associated with a rapid drop in the levels of viral structural proteins NC and CA from total viral DNA at 12 h postinfection, which roughly corresponds to the time of nuclear entry (Fig. 3A and B). Moreover, we did not detect any association of WT circular DNAs with NC and CA (Fig. 3A and B), suggesting that the bulk of these viral proteins was removed before circularization. The timing of the loss of NC and CA coincided with the time of histone loading, suggesting that the two events may be coordinated. One possible model is that core histones displace NC and CA from the DNA. Retroviral NC shows high affinity for single-stranded RNA and DNA but can also bind to double-stranded DNA with lower affinity (34, 35). In contrast, CA does not bind DNA (36). CA association with the viral DNA is likely indirect, either through retaining the DNA in a cage-like structure or through bridging by another protein such as p12 or NC. In contrast to that from WT virus, total viral DNA derived from p12-PM14 mutant virus failed to associate with core histones at all time points after infection (Fig. 2) and instead showed prolonged association of viral structural proteins NC and CA, even at 48 h postinfection (Fig. 3A and B). The mutant DNA did not accumulate in the nucleus, and very few circular DNAs were formed. Although we observed much lower levels of 2-LTR circles derived from the p12-PM14 mutant virus than of those derived from WT (Fig. 1C), these residual circular DNAs were nevertheless readily detectable over background. Unlike circular DNAs derived from WT virus, which showed rapid association with core histones at 12 h postinfection (Fig. 2), the p12-PM14 mutant circular DNAs were defective in histone loading at all time points tested (Fig. 2). The mutant circles also showed prolonged association with NC and CA (Fig. 3A and B). These results were surprising and suggest that the rare 2-LTR circles generated by the p12-PM14 mutant virus may be different from circular DNAs generated from WT virus. It is possible that these circular DNAs may reside in a partially uncoated PIC that is accessible by the host NHEJ pathway mediating end joining but not accessible to the histone loading machinery.
It is unclear how uncoating, chromatin tethering, and histone loading are causally related to each other. These events are all occurring over overlapping time frames, and all seem to require a wild-type p12. It is conceivable that p12 may play a role in the gradual uncoating of PICs in the nucleus and that this is a prerequisite for the histone loading machinery to gain full access to the viral DNAs. Alternatively, p12 may promote the correct localization of viral DNAs to host chromosomes, and this localization is required for histone loading; that is, p12-dependent association of the PIC with host nucleosomes may be needed to retain the PIC during nuclear membrane reformation, and histone loading may occur only in this context.
Our detection of the association of wild-type total viral DNA with Pol II by ChIP (Fig. 4B) was to be expected, since much of the DNA is integrated and highly expressed. It was interesting that the circles were also associated with Pol II, since these unintegrated DNAs are very poorly expressed. This result suggests that the wild-type circles are accessible to histones and Pol II but that Pol II is not able to elongate, perhaps due to the state of the viral chromatin. RNA Pol II ChIP assays showed a dramatic decrease in the recovery of all viral DNAs, both total and circles, bound to Pol II in the p12-PM14 mutant (Fig. 4B). These results suggest that the DNAs delivered by the mutant p12 do not have normal access to Pol II or to histones. A portion of p12-PM14 PICs were previously detected in close proximity to chromosomes in mitotic cells, though they failed to attach to mitotic chromosomes (9, 17). These PM14 mutant PICs may not have been abundant enough or may not have achieved the complete uncoating required for full access to the histone loading machinery and to RNA polymerase II.
The p12-PM14 mutation includes substitution of serine-61, the major phosphorylation site of p12 in vivo (15, 16, 20, 21). Phosphomimetic or phosphodeficient mutations altering serine-61 result in impaired replication due to lack of chromatin tethering (15, 16), much like the PM14 mutation. Prolonged passage of a phosphodeficient p12 mutant, SS(61,65)AA, resulted in revertants carrying second-site suppressor mutations, including a particular M63I substitution, that showed increased infectivity without restoring p12 phosphorylation (15). The M63I mutation reactivated chromatin tethering of mutant p12 (16), indicating that phosphorylation per se is not required for chromatin association. We do not know if histone loading onto viral DNAs strictly requires p12 phosphorylation at serine-61 or other residues. It is also not known if second-site suppressor mutations such as M63I could rescue histone loading and replication defects of the PM14 mutant. The M63I substitution did partially rescue p12-PM15, a nearby mutation in p12 (26), and addition of heterologous chromatin binding sequences (CBS) from other viruses to p12 led to partial rescue of the PM14 phenotype (10, 18). We would predict that these modifications to the mutant p12 would restore histone loading onto the viral DNAs, but this remains to be tested.
Taken together, our findings reveal new aspects of the functions of p12 early in infection. Viral DNAs formed with mutant p12 do not become loaded with histones, retain CA and NC, and are poorly expressed. Only few circles are formed, and those circles also do not acquire histones and only inefficiently shed the associated CA and NC. The results suggest a critical requirement for functional MMLV p12 in proper uncoating and chromatinization of retroviral DNAs.
MATERIALS AND METHODS
Cell lines.
Cell lines for these experiments included human embryonic kidney (HEK)-293 and HeLa cells. All cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, 1,000 units/ml penicillin, and 100 mg/ml streptomycin.
Plasmids.
Plasmid pCMV-intron expresses wild-type Gag and Pol from NB-tropic murine leukemia virus (MLV) (37). pCMV-intron PM14 that expresses p12 mutant Gag from NB-tropic MLV was created by subcloning p12-PM14 mutant fragment from PNCS (14) into pCMV-intron via SexAI restriction enzyme digests. pMD.G expresses the vesicular stomatitis virus (VSV) envelope glycoprotein. pNCA-GFP is a replication-defective single-round MLV vector described previously (38).
Retroviral transduction assay and flow cytometry analysis.
MLV-GFP reporter viruses were produced by 293T cell transfection with 8 μg of pNCA-GFP, 4 μg of pCMV-intron or pCMV-intron p12-PM14, and 4 μg of pMD.G DNAs using Lipofectamine 3000 reagent (Thermo Scientific) according to the manufacturer’s instructions. All reporter viruses were harvested 48 h later, filtered (0.45 μm), DNase treated for 1 h (RQ1 DNase, Promega), and used directly for transduction assays as described previously (39). Twenty-four hours postinfection, cells were trypsinized, diluted using flow cytometry buffer (phosphate-buffered saline [PBS] with 1% bovine serum albumin [BSA]), and subjected to flow cytometry using automated cell analyzer (LSRII, BD Bioscience).
Chromatin immunoprecipitation.
Cells were cross-linked with 1% formaldehyde, followed by quenching with 0.125 M glycine for 5 min, lysed, and sonicated to produce an average DNA fragment size of 200 to 800 bp. Immunoprecipitations were performed by incubating 10 μg of sonicated chromatin with 1 μg of respective antibodies overnight, and protein A/G Dynabeads were added for an additional 4 h. Captured antibody-antigen complexes were washed extensively, and DNA was eluted from the beads. qPCRs were performed with specific primer pairs (see Table 1).
TABLE 1.
ChIP qPCR primers used in this study
| Primer | Sequence (5′–3′) |
|---|---|
| Total viral DNA (GFP)-F | AAGCTGACCCTGAAGTTCATCTGC |
| Total viral DNA (GFP)-R | CTTGTAGTTGCCGTCGTCCTTGAA |
| MMLV-LTR-F | AGGGTCTCCTCTGAGTGATTGACT |
| MMLV-LTR-R | TCGGACAGACACAGATAAGTTGCT |
| MMLV 2-LTR circles-F | AGGGTCTCCTCTGAGTGATT |
| MMLV 2-LTR circles-R | ATGGTGTGTGGAGGAGTATAAAG |
| GAPDH-F | ACCTTTAGCCTTGCCCTTT |
| GAPDH-R | ACATCACCCCCATCACTCAT |
Quantitative real-time PCR analysis of viral replication intermediates.
At various time points following infection, cells were washed with PBS, and total DNA was isolated using Qiagen DNeasy kit according to the manufacturer’s instructions. For analysis of viral replication intermediates, 75 ng of total DNA was mixed with SYBR green PCR master mix (Roche) containing 15 pmol of indicated primers (see Table 1). PCRs were performed in 96-well plates using 7900 fast real-time PCR system (Applied Biosystems) with the following reaction conditions: 10 min at 95°C, followed by 40 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C. For all reactions, normalized expression of viral replication intermediates was obtained by using the 2−ΔΔCT method (40) to the value obtained for the GAPDH gene.
Cell fractionation.
Cell fractionation was carried out exactly as previously described (41), and DNA from nuclear and cytoplasmic fractions were then isolated using Qiagen DNeasy kit according to the manufacturer’s instructions and subjected to viral replication intermediate analysis as above.
Western blotting.
Cell lysates prepared from nuclear and cytoplasmic fractions were subjected to SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). Membranes were blocked for 1 h at room temperature with buffer for fluorescent Western blotting (Rockland), followed by incubations with either anti-histone H3 (ab1791, Abcam, 1:5,000) or anti-GAPDH (clone 6C5, EMD Millipore, 1:10,000) overnight at 4°C. Next day, donkey anti-rabbit or mouse secondary antibodies conjugated to horseradish peroxidase (HRP) (1:10,000, Jackson ImmunoResearch Labs) were added for 1 h and all blots were subsequently developed using the enhanced chemiluminescence (ECL) method.
ACKNOWLEDGMENTS
This study was supported by NCI grant R01 CA 30488 from the National Cancer Institute. S.P.G. is an Investigator of the Howard Hughes Medical Institute.
Contributor Information
Stephen P. Goff, Email: spg1@cumc.columbia.edu.
Frank Kirchhoff, Ulm University Medical Center.
REFERENCES
- 1.Telesnitsky A, Goff SP. 1997. Reverse transcriptase and the generation of retroviral DNA. In Coffin JM, Hughes SH, Varmus HE (ed), Retroviruses. Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 2.Kilzer JM, Stracker T, Beitzel B, Meek K, Weitzman M, Bushman FD. 2003. Roles of host cell factors in circularization of retroviral DNA. Virology 314:460–467. 10.1016/s0042-6822(03)00455-0. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimura FK, Weinberg RA. 1979. Restriction endonuclease cleavage of linear and closed circular murine leukemia viral DNAs: discovery of a smaller circular form. Cell 16:323–332. 10.1016/0092-8674(79)90009-6. [DOI] [PubMed] [Google Scholar]
- 4.Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD. 2001. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J 20:3272–3281. 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 6.Dharan A, Bachmann N, Talley S, Zwikelmaier V, Campbell EM. 2020. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat Microbiol 5:1088–1095. 10.1038/s41564-020-0735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zila V, Margiotta E, Turonova B, Muller TG, Zimmerli CE, Mattei S, Allegretti M, Borner K, Rada J, Muller B, Lusic M, Krausslich HG, Beck M. 2021. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 184:1032–1046.e18. 10.1016/j.cell.2021.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roe T, Reynolds TC, Yu G, Brown PO. 1993. Integration of murine leukemia virus DNA depends on mitosis. EMBO J 12:2099–2108. 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elis E, Ehrlich M, Prizan-Ravid A, Laham-Karam N, Bacharach E. 2012. p12 tethers the murine leukemia virus pre-integration complex to mitotic chromosomes. PLoS Pathog 8:e1003103. 10.1371/journal.ppat.1003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider WM, Brzezinski JD, Aiyer S, Malani N, Gyuricza M, Bushman FD, Roth MJ. 2013. Viral DNA tethering domains complement replication-defective mutations in the p12 protein of MuLV Gag. Proc Natl Acad Sci U S A 110:9487–9492. 10.1073/pnas.1221736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wanaguru M, Barry DJ, Benton DJ, O'Reilly NJ, Bishop KN. 2018. Murine leukemia virus p12 tethers the capsid-containing pre-integration complex to chromatin by binding directly to host nucleosomes in mitosis. PLoS Pathog 14:e1007117. 10.1371/journal.ppat.1007117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wanaguru M, Bishop KN. 2018. Gammaretroviruses tether to mitotic chromatin by directly binding nucleosomal histone proteins. Microb Cell 5:385–388. 10.15698/mic2018.08.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kharytonchyk S, Monti S, Smaldino PJ, Van V, Bolden NC, Brown JD, Russo E, Swanson C, Shuey A, Telesnitsky A, Summers MF. 2016. Transcriptional start site heterogeneity modulates the structure and function of the HIV-1 genome. Proc Natl Acad Sci U S A 113:13378–13383. 10.1073/pnas.1616627113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan B, Li X, Goff SP. 1999. Mutations altering the Moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J 18:4700–4710. 10.1093/emboj/18.17.4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yueh A, Goff SP. 2003. Phosphorylated serine residues and an arginine-rich domain of the moloney murine leukemia virus p12 protein are required for early events of viral infection. J Virol 77:1820–1829. 10.1128/jvi.77.3.1820-1829.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brzezinski JD, Felkner R, Modi A, Liu M, Roth MJ. 2016. Phosphorylation requirement of murine leukemia virus p12. J Virol 90:11208–11219. 10.1128/JVI.01178-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prizan-Ravid A, Elis E, Laham-Karam N, Selig S, Ehrlich M, Bacharach E. 2010. The Gag cleavage product, p12, is a functional constituent of the murine leukemia virus pre-integration complex. PLoS Pathog 6:e1001183. 10.1371/journal.ppat.1001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wight DJ, Boucherit VC, Nader M, Allen DJ, Taylor IA, Bishop KN. 2012. The gammaretroviral p12 protein has multiple domains that function during the early stages of replication. Retrovirology 9:83. 10.1186/1742-4690-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. 2005. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J Cell Biol 168:89–101. 10.1083/jcb.200408155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pal BK, McAllister RM, Gardner MB, Roy-Burman P. 1975. Comparative studies on the structural phosphoproteins of mammalian type C viruses. J Virol 16:123–131. 10.1128/JVI.16.1.123-131.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pal BK, Roy-Burman P. 1975. Phosphoproteins: structural components of oncornaviruses. J Virol 15:540–549. 10.1128/JVI.15.3.540-549.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang GZ, Wang Y, Goff SP. 2016. Histones are rapidly loaded onto unintegrated retroviral DNAs soon after nuclear entry. Cell Host Microbe 20:798–809. 10.1016/j.chom.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Wang GZ, Cingoz O, Goff SP. 2018. NP220 mediates silencing of unintegrated retroviral DNA. Nature 564:278–282. 10.1038/s41586-018-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geis FK, Goff SP. 2019. Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced. Proc Natl Acad Sci U S A 116:23735–23742. 10.1073/pnas.1912638116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machida S, Depierre D, Chen HC, Thenin-Houssier S, Petitjean G, Doyen CM, Takaku M, Cuvier O, Benkirane M. 2020. Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome. Proc Natl Acad Sci U S A 117:6822–6830. 10.1073/pnas.1913754117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brzezinski JD, Modi A, Liu M, Roth MJ. 2016. Repression of the chromatin-tethering domain of murine leukemia virus p12. J Virol 90:11197–11207. 10.1128/JVI.01084-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan B, Fassati A, Yueh A, Goff SP. 2002. Characterization of Moloney murine leukemia virus p12 mutants blocked during early events of infection. J Virol 76:10801–10810. 10.1128/jvi.76.21.10801-10810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruceanu M, Urbaneja MA, Hixson CV, Johnson DG, Datta SA, Fivash MJ, Stephen AG, Fisher RJ, Gorelick RJ, Casas-Finet JR, Rein A, Rouzina I, Williams MC. 2006. Nucleic acid binding and chaperone properties of HIV-1 Gag and nucleocapsid proteins. Nucleic Acids Res 34:593–605. 10.1093/nar/gkj458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapadat-Tapolsky M, De Rocquigny H, Van Gent D, Roques B, Plasterk R, Darlix JL. 1993. Interactions between HIV-1 nucleocapsid protein and viral DNA may have important functions in the viral life cycle. Nucleic Acids Res 21:831–839. 10.1093/nar/21.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darlix JL, de Rocquigny H, Mauffret O, Mely Y. 2014. Retrospective on the all-in-one retroviral nucleocapsid protein. Virus Res 193:2–15. 10.1016/j.virusres.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carteau S, Batson SC, Poljak L, Mouscadet JF, de Rocquigny H, Darlix JL, Roques BP, Kas E, Auclair C. 1997. Human immunodeficiency virus type 1 nucleocapsid protein specifically stimulates Mg2+-dependent DNA integration in vitro. J Virol 71:6225–6229. 10.1128/JVI.71.8.6225-6229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poljak L, Batson SM, Ficheux D, Roques BP, Darlix JL, Kas E. 2003. Analysis of NCp7-dependent activation of HIV-1 cDNA integration and its conservation among retroviral nucleocapsid proteins. J Mol Biol 329:411–421. 10.1016/s0022-2836(03)00472-8. [DOI] [PubMed] [Google Scholar]
- 33.Campbell EM, Hope TJ. 2015. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 13:471–483. 10.1038/nrmicro3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Priel E, Aflalo E, Seri I, Henderson LE, Arthur LO, Aboud M, Segal S, Blair DG. 1995. DNA binding properties of the zinc-bound and zinc-free HIV nucleocapsid protein: supercoiled DNA unwinding and DNA-protein cleavable complex formation. FEBS Lett 362:59–64. 10.1016/0014-5793(95)00208-Q. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Yeh YS, Barbara PF. 2009. HIV-1 nucleocapsid protein bends double-stranded nucleic acids. J Am Chem Soc 131:15534–15543. 10.1021/ja9070046. [DOI] [PubMed] [Google Scholar]
- 36.Gross I, Hohenberg H, Krausslich HG. 1997. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur J Biochem 249:592–600. 10.1111/j.1432-1033.1997.t01-1-00592.x. [DOI] [PubMed] [Google Scholar]
- 37.Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res 23:628–633. 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ooi SK, Wolf D, Hartung O, Agarwal S, Daley GQ, Goff SP, Bestor TH. 2010. Dynamic instability of genomic methylation patterns in pluripotent stem cells. Epigenetics Chromatin 3:17. 10.1186/1756-8935-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GZ, Goff SP. 2015. Postentry restriction of Mason-Pfizer monkey virus in mouse cells. J Virol 89:2813–2819. 10.1128/JVI.03051-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Wang GZ, Goff SP. 2020. Yin Yang 1 is a potent activator of human T lymphotropic virus type 1 LTR-driven gene expression via RNA binding. Proc Natl Acad Sci U S A 117:18701–18710. 10.1073/pnas.2005726117. [DOI] [PMC free article] [PubMed] [Google Scholar]