

ABSTRACT
HIV persists, despite immune responses and antiretroviral therapy, in viral reservoirs that seed rebound viremia if therapy is interrupted. Previously, we showed that the BCL-2 protein contributes to HIV persistence by conferring a survival advantage to reservoir-harboring cells. Here, we demonstrate that many of the BCL-2 family members are overexpressed in HIV-infected CD4+ T cells, indicating increased tension between proapoptotic and prosurvival family members—and suggesting that inhibition of prosurvival members may disproportionately affect the survival of HIV-infected cells. Based on these results, we chose to study BCL-XL due to its consistent overexpression and the availability of selective antagonists. Infection of primary CD4+ T cells with HIV resulted in increased BCL-XL protein expression, and treatment with two selective BCL-XL antagonists, A-1155463 and A-1551852, led to selective death of productively infected CD4+ T cells. In a primary cell model of latency, both BCL-XL antagonists drove reductions in HIV DNA and in infectious cell frequencies both alone and in combination with the latency reversing agent bryostatin-1, with little off-target cytotoxicity. However, these antagonists, with or without bryostatin-1 or in combination with the highly potent latency reversing agent combination phorbol myristate acetate (PMA) + ionomycin, failed to reduce total HIV DNA and infectious reservoirs in ex vivo CD4+ T cells from antiretroviral therapy (ART)-suppressed donors. Our results add to growing evidence that bona fide reservoir-harboring cells are resistant to multiple “kick and kill” modalities—relative to latency models. We also interpret our results as encouraging further exploration of BCL-XL antagonists for cure, where combination approaches, including with immune effectors, may unlock the ability to eliminate ex vivo reservoirs.
IMPORTANCE Although antiretroviral therapy (ART) has transformed HIV infection into a manageable chronic condition, there is no safe or scalable cure. HIV persists in “reservoirs” of infected cells that reinitiate disease progression if ART is interrupted. Whereas most efforts to eliminate this reservoir have focused on exposing these cells to immune-mediated clearance by reversing viral latency, recent work shows that these cells also resist being killed. Here, we identify a “prosurvival” factor, BCL-XL, that is overexpressed in HIV-infected cells, and demonstrate selective toxicity to these cells by BCL-XL antagonists. These antagonists also reduced reservoirs in a primary-cell latency model but were insufficient to reduce “natural” reservoirs in ex vivo CD4+ T cells—adding to growing evidence that the latter are resilient in a way that is not reflected in models. We nonetheless suggest that the selective toxicity of BCL-XL antagonists to HIV-infected cells supports their prioritization for testing in combinations aimed at reducing ex vivo reservoirs.
KEYWORDS: BCL-XL antagonist, HIV, HIV persistence, HIV reservoir, kick and kill
INTRODUCTION
Although antiretroviral therapy (ART) durably suppresses HIV replication, it cannot eradicate persisting infected cells with integrated HIV proviruses. These cells compose a “viral reservoir” which seeds viral rebound when ART is interrupted, with the most well-characterized reservoir being formed in CD4+ T cells (1). An important mechanism of persistence is the maintenance of viral latency, particularly in resting memory CD4+ T cells, which prevents death from viral cytopathic effects as well as recognition and elimination by immune effectors, such as cytotoxic T-lymphocytes (CTLs) (2–4). More recent work has uncovered additional mechanisms by which reservoir-harboring cells may survive both viral- or immune-mediated cytopathicity, with the prosurvival factor BCL-2 implicated in both of these scenarios (5). With respect to viral cytopathicity, one mechanism of death is through cleavage of the host protein procaspase 8 by HIV protease to generate Casp8p41, which drives apoptosis (6). BCL-2 is able to antagonize this pathway by binding to Casp8p41, thereby preventing cell death in BCL-2high cells (7). With respect to immune-mediated cytopathicity, our group recently made a series of observations which led us to conclude that BCL-2 overexpression is one mechanism by which HIV reservoir-harboring cells resist elimination by CTL: (i) BCL-2high CD4+ T cells preferentially survive CTL killing in vitro, (ii) the inducible HIV reservoir in ex vivo CD4+ T cells is disproportionately harbored in BCL-2high CD4+ T cells, and (iii) the BCL-2 antagonist ABT-199 sensitizes ex vivo HIV reservoirs to reductions by combinations of HIV-specific T cells and latency reversing agents (LRAs) (8). While much study to date has focused on BCL-2, it is likely that this is only one of the mechanisms that govern the intrinsic susceptibility of an HIV reservoir-harboring cell to death. This idea draws support from a recent study which demonstrated a role for BIRC5, also known as survivin, in activating cellular survival programs to promote HIV persistence—BIRC5 was found to be overexpressed in HIV reservoir-harboring cells that had undergone clonal expansion (9). Inhibition of this protein resulted in a selective decrease of HIV-infected cells, suggesting that BIRC5 supports long-term survival of HIV-infected cells (9). Building upon these results, the fundamental premise of the current study is that additional prosurvival mechanisms remain to be discovered, with our current focus being on those which protect cells from viral cytopathic effects. We focus here on BCL-2 family members, as a logical extension of the current state of knowledge.
BCL-2 is the prototypical member of a family of proteins which define the pro- or antiapoptotic states of a cell through their complex interplay (reviewed in reference 10). The prodeath members (e.g., BAX) are activated in response to a range of deleterious events and act to form channels which allow cytochrome c to exit the mitochondria, which activates caspases and induces cell death (11, 12). The prosurvival proteins (e.g., BCL-2) inhibit those proapoptotic partners, leading to the balance of the original rheostat model (13–15). The expression of a number of BCL-2 family members has been reported to be modulated by HIV infection and replication, though their potential roles in HIV persistence remain largely unstudied (16). The expression of the prosurvival member BCL-2 has been reported to be reduced in CD4+ T cells by gp120 cross-linking, which induces cell apoptosis (17). HIV Env can also elicit p53-dependent Puma, followed by activating Bax and Bak to induce apoptosis (18). The HIV Tat protein has been implicated in the upregulation of a number of proapoptotic members, including the proteins Bim, Puma, and Noxa, through either activation of the FOXO3a transcriptional activator or through downregulation of BCL-2 expression, resulting in apoptosis via mitochondrial membrane permeabilization (19–21). HIV Vpr can bind to adenine nucleotide translocase (ANT), a protein that forms the inner membrane channel of the mitochondrial permeability transition pore (MPTP), then converts it into a proapoptotic pore, which leads to cell death (22, 23). Also acting to promote apoptosis, the prosurvival BCL-2 homologs BCL-XL and Bfl1/A1 have been reported to be suppressed by HIV Vpu (24). On the prosurvival side of the interaction, the proapoptotic homolog BAD has been shown to be inactivated by HIV Nef, through phosphorylation (25). Low-level HIV Vpr expression in Jurkat cells can upregulate BCL-2 and downmodulate BAX, facilitating HIV persistence (26). HIV Tat and gp120 can induce TREM-1 expression in macrophages, and TREM-1 leads to inactivation of caspase 3 and increased BCL-2 expression, thus inhibiting apoptosis (27). On the systemic level, in PBMCs from people living with HIV, BCL‐2, BCL‐XL and MCL‐1 were significantly upregulated during successful ART (28). Puma expression was enhanced in untreated HIV-infected donors, and these elevated Puma levels decreased upon ART initiation (18). Overall, these results paint a nuanced picture of apoptotic signaling in the setting of HIV infection, though despite these individual reports of BCL-2 family member modulation—often in the context of cell lines—the overall impact of BCL-2 family proteins on infected cell fate is not completely understood.
Here, we sought to define the landscape of BCL-2 family expression during HIV infection, with the aim of identifying a targetable factor that may be important to HIV persistence. Transcriptional profiling of productively HIV-infected versus HIV Gag– cells from in vitro primary CD4+ T cell infection cultures revealed that multiple BCL-2 family members were significantly differentially expressed, including both prosurvival and proapoptotic actors. We selected BCL-XL (BCL2L1), a prosurvival factor (29), for further study based on its consistent upregulation in productively HIV-infected cells along with the availability of selective inhibitors under drug development for cancer. Overexpression of BCL-XL in productively HIV-infected cells was confirmed at the protein level by flow cytometry, and we observed that two selective BCL-XL inhibitors enhanced the death of productively HIV-infected cells in vitro. Both inhibitors also consistently drove the selective death of HIV-infected cells from a primary cell model of latency, either with or without the LRA bryostatin—with very little bystander toxicity. However, when we applied the same strategies to “natural” HIV reservoirs in ex vivo CD4+ T cells from antiretroviral (ARV)-treated donors, we observed that none of these treatments were sufficient to reduce reservoir sizes. Our study thus identifies BCL-XL overexpression as a survival mechanism that can be targeted in productive infection to promote death of the HIV-infected cells, and also possibly in a latency-reactivation setting, though it remains to be determined if combining BCL-XL antagonism with immune effectors or other modes of infected cell killing can achieve a reduction in ex vivo reservoirs. The discordant results between a primary-cell latency model versus ex vivo HIV reservoirs (in closely matched experimental conditions) parallels our previous observations with CTL-based “kick and kill” combinations (8) and points to a fundamental resistance of ex vivo reservoirs to multiple modes of attack. Our study identifies BCL-XL antagonists as a novel and facile “kill” component (without the bystander toxicity of BCL-2 antagonists) that can be leveraged to study the survivability of ex vivo reservoirs. The identification of additional barriers to the elimination of these reservoirs may yield combination treatment strategies which enable the selective toxicity of BCL-XL antagonists to HIV-infected cells observed in productive infection and latency models to be brought to bear against reservoir-harboring cells.
RESULTS
Transcriptional profiling of BCL-2 family members reveals increased tension in HIV-infected versus uninfected cells.
To test whether expression of BCL-2 family proteins is altered during HIV infection, we infected CD4+ T cells from a long-term ART-suppressed donor (OM5267) with the HIV JR-CSF and assessed the expression of BCL-2 family transcripts by RNA sequencing (RNAseq). In an initial experiment, we used total CD4+ T cells for infection and sorted them into productively HIV-infected (Gag+) and HIV Gag– populations using flow cytometry (Fig. 1A). Of all the genes that we compared, we observed a cluster of BCL-2 family members (30) that were significantly upregulated (22 genes) in productively HIV-infected cells, which included the proapoptotic members BAK1 (encodes the protein BAK), BMF (encodes the protein BCL-2 modifying factor), PMAIP1 (encodes the protein NOXA), and BBC3 (encodes the protein PUMA), as well as the prosurvival members BCL2L1 (encodes BCL-XL) and BCL2A1 (encodes BCL-2-related protein A1) (Fig. 1B). Only two BCL-2 family genes were significantly underexpressed in productively HIV-infected cells, the prosurvival member MCL1 and the proapoptotic member BAX. Thus, productively HIV-infected primary CD4+ T cells were predominately characterized by overexpression of the BCL-2 family, representing both prosurvival and proapoptotic members.
FIG 1.

Transcriptional profiles reveal differences in expression of prosurvival and proapoptotic BCL-2 family genes between productively HIV-infected cells and HIV Gag– cells (not productively infected). (A) Schematic of HIV infection (in vitro) and flow cytometry sorting based on intracellular HIV-p24 (Gag) expression; Gag+ represents productively HIV-infected cells, Gag– represents uninfected and nonproductively infected cells. (B and C) Flow gating strategies for cell sorting and corresponding expression patterns of BCL-2 family genes in Gag+ versus Gag– cells. Samples were derived from total CD4+ T cells (B) or from naive CD4+ T cells differentiated into central memory T cells (C) that were then infected with HIV in vitro. The heatmaps visualize the scaled expression values based on RNA-seq of two technical replicates of Gag+ and Gag– cells (separate infection and sorting). Asterisks (*) highlight the genes that were found to be significantly differentially expressed between Gag+ cells versus Gag– cells (padj [P values that have been adjusted for multiple testing] < 0.01).
The above-described experiment utilized total CD4+ T cells and thus contained cells of diverse phenotypes and functional profiles. We next extended these results to an analogous experiment using a more homogeneous pool of cells with a central memory (TCM) phenotype, given that these cells are a major cellular reservoir of HIV (31). TCM was generated by the in vitro priming of naive CD4+ T cells, using the methodology of the well-characterized cultured TCM model of HIV latency (32, 33). With this more homogeneous cell population, we again observed a general pattern of overexpression of BCL-2 family genes in productively HIV-infected versus HIV Gag– cells, including the proapoptotic members PMAIP1, BMF, and BAK1 and the prosurvival members BCL2, BCL2A1, and BCL2L1 (Fig. 1C). As with total CD4+ T cells, we observed significant underexpression of the prosurvival gene MCL1 in productively HIV-infected cells as well as underexpression of the proapoptotic member BAX. Together, these data indicate an increased tension between proapoptotic and prosurvival BCL-2 family members in productively HIV-infected cells compared to Gag– cells, suggesting to us that the inhibition of prosurvival members may disproportionately lead to the death of productively HIV-infected cells by tipping the balance in favor of the proapoptotic members that are overexpressed in these cells. While the data propose several prosurvival members that could be targeted to test this hypothesis, we opted here to focus on BCL-XL, in part due to the availability of selective antagonists.
Selective BCL-XL antagonists disproportionately eliminate productively HIV-infected cells.
In order to evaluate if BCL2L1 overexpression translates to increased BCL-XL protein expression in productively HIV-infected versus HIV Gag– cells, protein levels were measured using flow cytometry. Activated CD4+ T cells, isolated from an HIV-negative donor, were infected with 3 different viruses—one clinical isolate from a quantitative viral outgrowth assay (QVOA), one CCR5-tropic strain (JR-CSF), and one CXCR4-tropic strain (NL4-3). One week later, infected cells were treated with a BCL-XL antagonist, either A-1155463 or A-1331852, or dimethyl sulfoxide (DMSO; negative control), under the suppression of T-20 (an HIV fusion inhibitor) in order to prevent new rounds of infection during the treatments. Then, 48 h later, cells were stained with a viability dye and anti-BCL-XL antibody. Applying the flow cytometry gating strategy shown in Fig. 2A, we observed that the BCL-XL mean fluorescence intensity MFI) in productively HIV-infected cells (HIV Gag+) was 1.86- to 2.25-fold higher than that in HIV Gag– cells (Fig. 2B), suggesting that the virus-host interaction during productive HIV infection upregulates BCL-XL expression.
FIG 2.

BCL-XL antagonists eliminate HIV-infected cells in productive infection. (A) Flow cytometry gating strategy for HIV infection with 3 different viruses—clinical isolate (QVOA virus), CCR5-tropic (JR-CSF), and CXCR4-tropic (NL4-3). Two BCL-XL antagonists were used for treatments (A-1155463 and A-1331852). (B) BCL-XL expression levels (MFI) in productively HIV-infected cells (HIV Gag+) and HIV Gag– cells. (C) BCL-XL antagonist treatments eliminate productively HIV-infected cells. Killing efficiency was calculated by the following formula: (% p24+ in DMSO condition minus % p24+ in BCL-XL antagonist condition)/(% p24+ in DMSO condition). Statistical significance was determined by two-way ANOVA using Dunnett’s multiple-comparison test. *, padj < 0.05; **, padj < 0.01; ***, padj < 0.001; ****, padj < 0.0001.
To test if HIV-infected cells could be preferentially eliminated by the two selective BCL-XL antagonists, A-1155463 and A-1331852, we measured the killing efficiency by counting the Gag+ cell percentages in each inhibitor treatment condition and then comparing this to DMSO treatment (negative control). Cell death was observed disproportionally in productively HIV-infected cells, where we observed a 12.12% average decrease in the percentage of HIV-infected cells relative to DMSO control for A-1155463 (11.60%, 16.26%, and 8.49% for OM5334 QVOA virus, JR-CSF, and NL4-3, respectively) and a 16.27% average decrease for A-1331852 (17.40%, 20.00%, and 11.42% for OM5334 QVOA virus, JR-CSF, and NL4-3, respectively) (Fig. 2C). To confirm that BCL-XL antagonists induce cell death in productively HIV-infected cells, rather than cause a reduction in HIV Gag expression, we performed a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay to assess the percentages of apoptotic cells. Treatment with either of the two selective BCL-XL antagonists resulted in a >2-fold increase in the percentage of apoptotic productively HIV-infected cells relative to the control (81.2% apoptotic for A-1155463, 82.8% apoptotic for A-1331852, and 37.9% apoptotic for the control [DMSO]); the BCL-2 inhibitor (ABT-199) showed an intermediate effect (43.2% apoptotic) (Fig. 3). These results confirm that the selective elimination of productively HIV-infected cells observed upon BCL-XL inhibitor treatment is due to cell death. Lastly, to further confirm preferential death of productively HIV-infected cells upon perturbation of BCL-XL, and to rule out a role for potential off-target effects of the BCL-XL inhibitors, we used a CRISPR-Cas9 platform with specific guide RNAs to knock out expression of BCL-XL in latency model cells and compared the effects of these knockouts on productively infected cells (from day 17 of the model) to those of treatment with the two inhibitors; we also tested CRISPR-Cas9-mediated knockout of BCL-2 and BCL-2 inhibitor (ABT-199) treatment in parallel. Efficient knockouts of both BCL-XL and BCL-2 expression were confirmed by flow cytometry (Fig. 4A and B). Similar to treatment with either of the two BCL-XL antagonists, knockout of BCL-XL expression resulted in decreased frequencies of productively infected (HIV Gag+) cells relative to a control CRISPR condition; comparable results were observed following knockout of BCL-2, though treatment with the BCL-2 inhibitor showed an intermediate effect (Fig. 4C). Additionally, measurement of p24 supernatant levels 72 h after CRISPR-Cas9-mediated knockout of BCL-XL (or BCL-2) showed reduced p24 concentrations relative to the control CRISPR condition (Fig. 4D). Altogether, these results point to BCL-XL inhibitors inducing preferential death of productively HIV-infected cells through their on-target effects on BCL-XL function.
FIG 3.
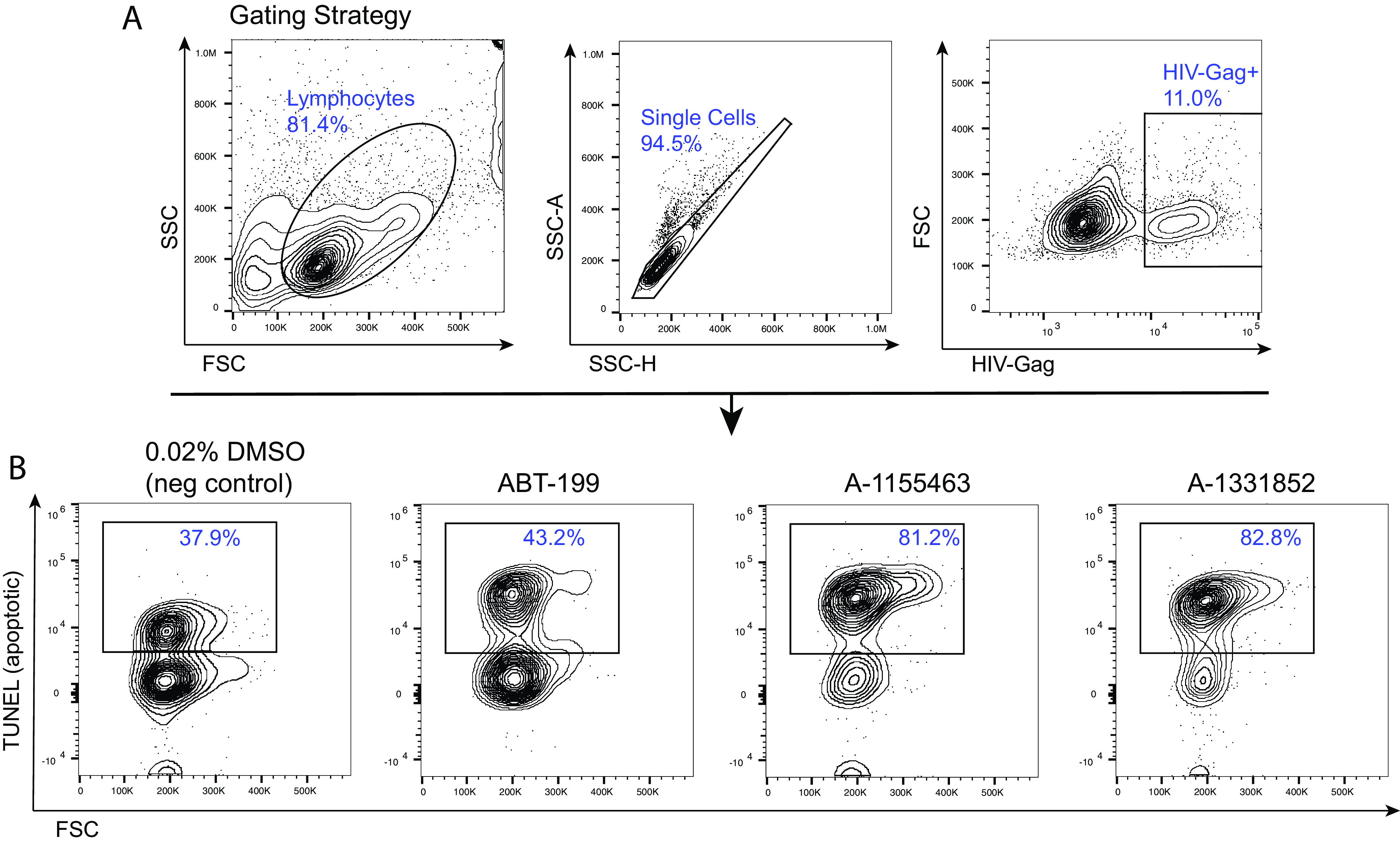
BCL-XL antagonists induce apoptosis in HIV-infected cells in productive infection. (A) Flow cytometry gating strategy for the TUNEL assay to quantify apoptotic productively HIV-infected cells (HIV Gag+) from the cultured TCM model at day 13 (B) Following 32-h treatments with one of two separate BCL-XL antagonists (A-1155463 and A-1331852), greater proportions of cells were apoptotic (TUNEL+) compared to the negative control. The BCL-2 antagonist (ABT-199) showed an intermediate level of apoptosis.
FIG 4.

CRISPR-Cas9-mediated knockout of BCL-XL and BCL-2 results in preferential death of productively HIV-infected cells. (A and B) Flow cytometry results showing reductions in BCL-2 expression or BCL-XL expression following CRISPR-Cas9-mediated knockout in latency model cells at day 17, plotted against HIV Gag expression. (C) Summary data of flow cytometry results showing reductions in productively HIV-infected cell (HIV Gag+) percentages in latency model cells following either CRISPR-mediated knockout of BCL-2 or BCL-XL, or treatment with selective BCL-2 (ABT-199) or BCL-XL (A-1155463 or A-1331852) inhibitors (32 h postknockout [post-KO] or treatment). Results are representative of three different experiments using cells from different donors. (D) ELISA results showing cell culture supernatant p24 concentrations. Cells were washed at 32 h after CRISPR-Cas9-mediated knockout of BCL-2 or BCL-XL and plated in fresh medium, and then supernatants were collected at 72 h for ELISA.
Selective BCL-XL antagonists drive reductions in total HIV DNA and infectious reservoirs in a primary cell latency model.
We next used the two selective BCL-XL antagonists to test the hypothesis that BCL-XL antagonism could sensitize HIV reservoir-harboring cells to elimination. This mirrors previous work in which we reported that the BCL-2 antagonist ABT-199 sensitized HIV reservoir-harboring cells to elimination in a primary cell latency model when used alone or in combination with an LRA (supplementary data of reference 8). The primary cell model of HIV latency (see Materials and Methods for cell generation details) typically gives rise to frequencies of latently infected cells between ∼2% and 5%, greatly exceeding the ∼0.0001% that is typical of ex vivo CD4+ T cells from ARV-treated individuals which harbor intact-inducible virus. The QVOA is the gold standard method for quantifying the inducible reservoir, and it is well suited for rare ex vivo populations (34); however, it cannot be directly applied to the much higher frequencies of infected cells in the primary cell latency model. To set the stage for direct comparisons between results from the primary cell latency model and those of ex vivo reservoirs, we therefore spiked these model cells into autologous CD4+ T cells in order to bring the frequency of latently infected cells into closer alignment with ex vivo reservoirs (aiming for ∼1,000 to 10,000 copies of HIV DNA per million CD4+ T cells). The effects of the selective BCL-XL antagonists were then assessed using an HIV eradication (HIVE) assay (Fig. 5A). HIVE assays consist of a 4-day culture with one of the selective BCL-XL antagonists in the presence of ARVs, with or without prior activation of target cells with the latency reversing agent bryostatin-1 (a protein kinase C agonist), which is washed out prior to the culture period. Surviving cells are then counted and subjected to droplet digital PCR (ddPCR) to measure total frequencies of infected cells and QVOA to measure intact-inducible reservoirs. This distinction is important, as in ex vivo CD4+ T cells from ARV-treated individuals, the large majority of HIV DNA represents defective proviruses with no potential for viral replication (34). In contrast to the BCL-2 antagonist ABT-199, which was associated with substantial levels of cell death, the selective BCL-XL antagonists A-1155463 and A-1331852 showed little or no cytotoxicity at 100 nM (Fig. 6). We were nonetheless careful to account for cell death in our QVOAs by counting only viable CD4+ T cells by flow cytometry following a 24-h drug wash-out period and using only viable cell numbers to calculate infectious units per million CD4+ T cells (IUPM).
FIG 5.

BCL-XL antagonists drive reductions in total HIV DNA and infectious reservoirs in a primary cell model of latency in “spiked” HIVE assays. (A) Schematic of a HIVE assay spiked with a primary cell latency model. (B and C) Representative spiked HIVE assay with cells from an HIV-negative donor, demonstrating reductions in HIV DNA levels as measured by ddPCR (HIV gag) (B) and reductions in infectious reservoirs (IUPM) as measured by QVOA (C) upon treatment with a BCL-2 inhibitor (ABT-199) or a BCL-XL inhibitor (A-1155463 or A-1331852), with or without Bryostatin-1, versus No Tx or Bryo only. (D and E) Representative spiked HIVE assay with cells from an HIV-positive donor, also demonstrating reductions in HIV DNA (D) and IUPM (E) upon treatment with the BCL-2 and BCL-XL inhibitors, with or without Bryostatin-1, versus No Tx or Bryo only. ddPCR results show the mean ± SD of 8 technical replicates, and QVOA results show the IUPM ± 95% confidence interval. Statistical significance was determined by one-way ANOVA for ddPCR and pairwise chi-square tests for QVOA (corrected for multiple comparisons). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (F to I) Summary data for spiked HIVE assays performed on 4 HIV-negative and 4 HIV-positive donors, with both BCL-XL inhibitors driving significant reductions in HIV DNA (F and H) and IUPM (G and I), with or without Bryostatin-1, versus No Tx or Bryo only. Black lines with dots represent the primary cell model of latency generated with ART-suppressed HIV-positive donors; gray lines with open circles represent the latency model generated with HIV-negative donors. DMSO was added to no Tx conditions at a matched concentration with +Tx conditions. Statistical significance was determined by the Wilcoxon matched-pair signed-rank test.
FIG 6.

Cell toxicity of BCL-XL antagonists on a primary cell latency model and participant-derived ex vivo cells. The percentage of cell numbers in each BCL-XL antagonist treatment compared to no Tx (black) or a combination of bryostatin-1+ BCL-XL antagonist treatment compared to bryostatin-1 (blue). Shown is the cell count percentage mean ± SD from separate HIVEs. Statistical significance was determined by one-way ANOVA. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
In an initial experiment, using model cells generated from an HIV-negative donor, we observed that combination treatments with bryostatin-1 and BCL-XL antagonists (A-1155463 or A-1331852) significantly reduced HIV DNA relative to bryostatin-1 alone (3.17-fold and 3.57-fold, respectively) as measured by gag primer/probe sets (the BCL-2 antagonist ABT-199 was used as a positive control in this experiment). Unexpectedly, latency reversal was not strictly required for reductions in HIV DNA, and we observed the following reductions without bryostatin: a 2.50-fold average reduction for A-1155463 and a 2.79-fold average reduction for A-1331852 (Fig. 5B). In this initial experiment, treatment with A-1331852 was observed to drive a significant reduction in IUPM when used alone (4.52-fold, P < 0.0001; Fig. 5C). A-1155463 alone also showed a 1.65-fold reduction in IUPM, but this was not statistically significant (Fig. 5C). Significant reductions in IUPM were also observed in treatments with each of the BCL-XL antagonists + bryostatin-1 (P < 0.01), where A-1155463 + bryostatin-1 showed a 3.59-fold decrease, and A-1331852 + bryostatin-1 showed a 3.90-fold decrease relative to bryostatin-1 alone, although these reductions were smaller in magnitude than that observed with ABT-199 + bryostatin-1, which showed a 21.18-fold decrease relative to bryostain-1 alone (Fig. 5C). Thus, these data suggest that both the BCL-XL antagonists were able to reduce the frequency of latently HIV-infected cells generated from an HIV-negative donor, either alone or in combination with bryostatin-1.
To account for potential differences in CD4+ T-cell function from HIV-negative versus people living with HIV (PLWH), we also performed HIVE assays with latency model cells generated from an ART-suppressed HIV-positive donor, spiked into autologous resting CD4+ T cells (35). As with cells from HIV-negative donors, significant reductions were observed in total HIV DNA following treatment with either of the BCL-XL antagonists or ABT-199 relative to no treatment (P < 0.001; Fig. 5D). The effects of the BCL-XL antagonists alone were more modest than those for ABT-199, with a reduction in HIV DNA of 3.01-fold, 1.32-fold, and 1.20-fold for ABT-199, A-1155463, and A-1331852, respectively (Fig. 5D). Combinations of either the BCL-XL antagonists or ABT-199 with bryostatin-1 resulted in significant and consistent reductions in HIV DNA relative to bryostatin-1 only, with reductions of 7.72-fold, 1.97-fold, and 3.12-fold, respectively (P < 0.001; Fig. 5D). We did not observe significant decreases in IUPM for BCL-XL antagonists treated alone or in combination with bryostatin-1 (Fig. 5E). However, we did observe a 21.18-fold significant reduction in IUPM in the ABT-199 + bryostatin-1 condition (P < 0.0001; Fig. 5E). Thus, results using cells from this HIV-positive donor mirrored those from the HIV-negative donor, with both showing that the two BCL-XL antagonists were effective in reducing infected cell frequencies in this spiked primary cell latency model.
To assess the generalizability of our results with BCL-XL antagonists (A-1155463 and A-1331852), we tested these agents against latency model cells generated from an additional 4 HIV-negative and 4 HIV-positive donors (Table 1). Treatment with A-1155463 alone drove statistically significant reductions in HIV DNA (1.88-fold average reduction, P = 0.01) and IUPM (2.29-fold average reduction, P = 0.05) (Fig. 5F and G), as well as when combined with bryostatin-1 (2.24-fold average reduction in HIV DNA, P = 0.02; and a 1.78-fold average reduction in IUPM, P = 0.02) (Fig. 5F to G). Treatment with A-1331852 alone drove significant reductions in HIV DNA (2.15-fold average reduction, P = 0.05) and IUPM (2.00-fold average reduction, P = 0.03) and similar decreases when combined with bryostatin-1 (2.80-fold average reduction in HIV DNA, P = 0.02; and a 1.99-fold average reduction in IUPM, P = 0.02) (Fig. 5H and I). Across these different experiments, the two BCL-XL antagonists showed little in the way of cell toxicity, in contrast to the appreciable toxicity observed with the BCL-2 antagonist ABT-199 (Fig. 5 and Fig. 6). Together, these results demonstrate that both of the BCL-XL antagonists tested were sufficient to drive statistically significant reductions in a primary cell model of HIV latency, both with and without latency reversal by bryostatin-1.
TABLE 1.
ART-suppressed participant clinical dataf
| Participant IDe | Sexa | Ageb (yrs) | Ethnicity | ART regimen | Duration of undetectable viral load (mo) | Viral load (copies/ml) | Estimated time between infectionc and ART initiation (mo) |
|---|---|---|---|---|---|---|---|
| OM5011d | M | 46 | White | 3TC/ABC/DTG | 133 | <50 | 38 |
| OM5267 | M | 28 | White | 3TC/ABC/Ral | 91 | <50 | 4 |
| OM5148d | M | 48 | White | 3TC/ABC/NVP | 149 | <50 | 57 |
| OM5365d | M | 58 | White | 3TC/ABC/DRV/RTV/ETR/MVC/ | 114 | <50 | 18 |
| WWH-B008d | M | 64 | Black | ETR/TAF/FTC/DRV/RTV | ∼47 | <50 | ∼60 |
| WWH-B011 | M | 55 | White | TAF/FTC/RPV | ∼76 | <50 | ∼264 |
M, male.
Age indicates participants’ age at time of leukapheresis.
Infection time refers to the HIV-positive detection date.
Participants used for primary cell of latency model generation and HIV-positive donor spiked HIVE assays.
All 6 participants shown in the table were used for ex vivo HIVE assays.
Clinical data of HIV-negative participants, which were used for latency model generation and HIV-negative donor spiked HIVE assays, are not available.
Selective BCL-XL antagonists fail to reduce either total HIV DNA or infectious reservoirs from ex vivo ART-suppressed HIV-infected participants’ CD4+ T cells.
We next tested whether the reductions in infected cell frequencies observed with the BCL-XL antagonist treatments in our primary cell latency model would also be recapitulated in HIVE assays targeting “natural” HIV reservoirs in ex vivo CD4+ T cells from ART-suppressed donors. A representative example of a HIVE assay performed with ex vivo samples is shown in Fig. 7A and B. In contrast to results from the primary cell model of latency, we observed a general lack of reductions in either HIV DNA or IUPM (Fig. 7B) for either of the BCL-XL antagonists, used alone or in combination with bryostatin-1. The only significant differences that we observed were increases in IUPM following treatment with bryostatin-1 (P < 0.001; Fig. 7B), which we have also reported previously (8). Thus, this initial experiment with ex vivo CD4+ T cells from a single donor showed a general lack of reduction in HIV-infected cell frequencies following treatment with BCL-XL antagonists.
FIG 7.

BCL-XL antagonists failed to drive reductions in ex vivo, latently infected CD4+ T cells in HIVE assays. HIVE assays were performed using ex vivo CD4+ T cells from ART-suppressed individual WWH-B008. (A) ddPCR results using a 5′ end primer/probe set (HIV gag) for a representative HIVE assay. Shown are means ± SD of 8 replicates. (B) QVOA results from the same representative HIVE assay (WWH-B008), showing IUPM ± 95% confidence interval. Statistical significance determined by one-way ANOVA for ddPCR and a pairwise Chi-square test for QVOA (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (C) Levels of HIV DNA (HIV gag) and (D) IUPM, comparing A-1155463 (100 nM) versus no Tx, and bryostatin-1+A-463 versus bryostatin-1 (n = 6). (F) Levels of HIV DNA (HIV gag) and IUPM, comparing treatment with A-1331852 (100 nM) versus no Tx, and bryostatin-1+A-852 versus bryostatin-1 (n = 6). DMSO was added to no Tx conditions at a matched concentration with +Tx conditions. Statistical significance was determined by Wilcoxon matched-pair signed rank test. HIVE assays were performed using ex vivo CD4+ T cells from ART-suppressed individual WWH-B011. ddPCR results using a (G) 5′ end primer/probe set (HIV gag), (H) 3′ end primer/probe set (HIV env), or (I) calculating double positives for a representative HIVE assay. Shown are means ± SD of 8 replicates. (J) QVOA results from the same representative HIVE assay (WWH-B011), showing IUPM ± 95% confidence interval. Statistical significance was determined by one-way ANOVA for ddPCR and a pairwise Chi-square test for QVOA (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
We extended these results by performing HIVE assays with ex vivo resting CD4+ T cells from 6 long-term ART-suppressed HIV-positive donors (Table 1). Across this study population, we observed a lack of significant differences in either HIV DNA or IUPM when comparing untreated conditions to treatment with any of the BCL-XL antagonists tested either alone or in combination with bryostatin-1 (Fig. 7C to F), while the increase in IUPM observed with bryostatin-1 treatment in Fig. 7B was found to be consistent across this population (Fig. 7D and F). Additionally, we also tested the same concept in combination with a more potent LRA combination, phorbol myristate acetate (PMA) and ionomycin, and consistently observed the same lack of effects for the BCL-XL antagonists. Again, we were not able to observe a significant difference in either HIV DNA, which was measured by gag primers/probe (Fig. 7G) or env primers/probe (Fig. 7H), or “intact” provirus copies by calculating the double positives (Fig. 7I), or IUPM (Fig. 7J) when comparing untreated conditions to treatment with any of the BCL-XL antagonists tested either alone or in combination with PMA + ionomycin. Thus, in contrast to the primary cell latency model, neither of the BCL-XL antagonists (A-1155463 and A-1331852) was sufficient to drive reductions in ex vivo viral reservoirs—including when combined with potent LRAs.
DISCUSSION
The intrinsic apoptotic pathway is one of the main contributors to CD4+ T-cell depletion during productive HIV infection (36–38). BCL-2 and some of its family member proteins, including BCL-XL, can protect productively HIV-infected cells from apoptosis and can contribute to the maintenance of the HIV reservoir under ART. Furthermore, some LRAs—including PKC agonists—have the unintended effect of enhancing the antiapoptotic activity of BCL-2 (and perhaps other antiapoptotic proteins), which may need to be overcome to facilitate the clearance of infected cells (39). Previously, we investigated the role of BCL-2 in helping to maintain HIV reservoirs under ART by conferring a survival advantage (8). In the current study, we first tested the expression of proapoptotic and prosurvival BCL-2 family proteins during productive HIV infection by using a transcriptome sequencing (RNA-seq approach). We consistently observed the overexpression of the proapoptotic proteins Bak (BAK1 coding protein), Bmf (BMF coding protein), and Noxa (PMAIP1 coding protein) and the prosurvival proteins BCL-XL (BCL2L1 coding protein) and A1 (BCL2A1 coding protein) in productively HIV-infected CD4+ T cells, compared to HIV Gag- cells, for both total CD4+ and naive-derived central memory CD4+ T cells. We selected BCL-XL for further investigation in the context of both productive and latent HIV infection based on these initial data and the availability of drugs in clinical development to target this molecule. BCL-XL inhibits cell apoptosis by competing with the proapoptotic protein BAX/Bak (40–42). In brief, as previously demonstrated by Kale et al. (43), BID is first activated by caspase-8-mediated cleavage into cleaved BID (cBID), which comprises two fragments, BID-P7 and BID-P15; then, rapid high-affinity binding to cell membranes dissociates the P7 fragment into solution and favors insertion of the P15 fragment (tBID [truncated BID]) into the membrane. Next, membrane-bound tBID recruits inactive BAX from the cytosol and then activates BAX, which inserts into the lipid bilayer, oligomerizing and permeabilizing the mitochondrial outer membrane, thereby releasing intermembrane space proteins, including cytochrome c and SMAC, to induce cell apoptosis. When BCL-XL is overexpressed, active tBID and BAX can recruit BCL-XL to the membrane, resulting in inhibition of both pro- and antiapoptotic proteins by mutual sequestration. BCL-XL prevents tBID from activating BAX and prevents BAX from oligomerizing, resulting in the inhibition of mitochondrial outer membrane permeablization (MOMP) (43). The affinity of BCL-XL is higher for tBID than for active BAX, and BAX-mediated membrane permeabilization is completely inhibited by BCL-XL in sufficient quantities (44). On the side of BAK, it was shown that BCL-XL has a higher-association affinity for BAK than for BAX (41), and BCL-XL neutralizes BAK-mediated cell death; BAK is held in check only by Mcl-1 and BCL-XL and can only induce apoptosis if freed from both (45). In our RNA-seq results, both BCL-XL and BAK were significantly overexpressed in productively HIV-infected cells; thus, as BCL-XL can prevent BAX-mediated apoptosis, this may lead to long-term survival of some of the HIV-infected cells, indicating a potential mechanism of HIV persistence.
The role of BCL-XL during HIV infection has not been well studied, but lessons can be learned from the setting of cancer, especially the study of solid tumors, where BCL-XL overexpression is correlated with drug resistance or cancer persistence. As BCL-XL is essential for platelet survival (46–49), development and utilization of BCL-XL antagonists need to be evaluated for safety and off-target toxicity. In high-risk B-cell acute lymphoblastic leukemia (B-ALL), loss of functional transcription factor IKZF1 (IKAROS) and increased expression of casein kinase II (CK2) results in increased expression of BCL-XL, which is associated with resistance to doxorubicin treatment (50). In the differentiated thyroid carcinoma (DTC) diagnosis, BCL-XL expression was found to be high and was confirmed as an independent prognostic factor for persistent disease (51). BCL-XL is also a key survival factor in activation of stromal fibroblasts (ASFs) as well as in senescent cholangiocytes. Treatment with the BCL-XL-specific inhibitor A-1331852 reduces liver fibrosis, possibly by a dual effect on activated fibroblasts and senescent cholangiocytes (52). Under the condition of endoplasmic reticulum (ER) stress, stimulator of interferon genes (STING) induction was enhanced in epidermal growth factor receptor (EGFR) drug-tolerant persister cells, and this also links to a BCL-XL-dependent mitochondrial protection and cell survival in the absence of UFMylation. Cell death following ER stress induction in EGFR tyrosine kinase inhibitor persister cells could be triggered by using the BCL-XL inhibitor A-1331852 (53). In vivo, selective inhibition of BCL-XL (by WEHI-539 or A-1155463), but not BCL-2, resulted in a decrease in tumor growth rate in an orthotopic swarm rat chondrosarcoma (SRC) model (54). A-1155463 was tested in vivo and showed a reversible thrombocytopenia in mice and inhibited H146 small cell lung cancer xenograft tumor growth following multiple doses (55). A-1155463 and A-1331852 showed the potential to enhance the efficacy of docetaxel in solid tumors and avoid the exacerbation of neutropenia (56). In addition to this, the BCL-XL antagonists were also tested as senolytic agents and induced apoptosis in senescent cells (57). Given the overexpression of BCL-XL in HIV-infected cells in our study, this evidence suggests that BCL-XL may also contribute to HIV-infected cell persistence, which warrants further investigation.
Given the potential of BCL-XL to contribute to HIV persistence, we were interested in testing whether BCL-XL antagonists are able to eradicate productively HIV-infected cells, as well as long-lived HIV reservoir-harboring cells. In in vitro productive infection, both of the selective BCL-XL antagonists tested showed some efficacy in infected cell killing, which was preferential for productively HIV-infected cells (where BCL-XL expression was upregulated). Thus, we moved to a primary cell model of HIV latency (33). Selective BCL-XL antagonists were used alone (to sensitize to cell apoptosis without cell activation) or in combination with a latency reversing agent to reactivate virus and promote viral cytopathic effects (kick and kill). Since this primary cell model of HIV latency was mainly used to test the concept of the antagonists against the target cells only—whether they can sensitize the HIV reservoir-harboring cells to apoptosis—no immune-effector cells were added. We observed significant reductions in HIV-infected cell frequencies, both total HIV DNA level and replication-competent virus level (IUPM), providing initial support for the concept of utilizing BCL-XL antagonists for HIV cure strategies.
This prompted us to test this strategy against HIV reservoirs in ex vivo CD4+ T cells, representing long-lived cells potentially selected by immune and other pressures. Similar to our recent work with BCL-2 antagonism ABT-199 (8), the reductions in infected cells observed in the latency model system did not extend to measurable reductions in the ex vivo reservoir, whether BCL-XL antagonists were used alone or in combination with bryostatin-1. While the reasons underlying this difference are currently unclear, they fit a pattern in our recent results, which have indicated that success in various kick and kill combinations that are effective against primary cell models of latency has not translated into success against reservoirs in ex vivo CD4+ T cells from ART-treated donors, including CTL + LRAs (58), BCL-2 antagonists + LRAs (8), and now BCL-XL antagonists ± LRAs. While additional research in this area is needed, the results fit a model that we have recently proposed which postulates that long-lived reservoir-harboring cells in people living with HIV have been selected not only with respect to proviral latency, but also with respect to the intrinsic survivability of the reservoir harboring itself (reviewed in reference 59). Either or both of these factors may have contributed to our results, where we acknowledge that limitations in latency reversal in ex vivo reservoirs may have also limited the impact of BCL-XL antagonists, even in the case of PMA + ionomycin stimulation. Our results call for further study into these mechanisms. We suggest that BCL-XL antagonists will be a useful tool in this regard given their ease of use, low toxicity to bystander cells, and clear dichotomy between the model and ex vivo system.
Finally, we would propose the overall interpretation of our results as a partial success of BCL-XL antagonists, encouraging of their further testing and development. The ability to drive reductions in ex vivo HIV reservoirs appears to be a high bar to clear, and we interpret activity against the latency model as motivation for future studies testing BCL-XL antagonists in combination with immune effectors such as CTL (shown to be partially effective in combination with BCL-2 antagonists), novel LRA combinations, and other emerging agents which show promise in terms of overcoming remaining barriers to the elimination of reservoir-harboring cells.
MATERIALS AND METHODS
Agents—latency-reversing agents, chemical agents, and antibodies.
LRAs and BCL-XL antagonists were used at the following concentrations: bryostatin-1 dissolved in DMSO and used at 10 nM (Sigma-Aldrich); PMA and ionomycin (both Sigma-Aldrich) dissolved in DMSO and used at 50 ng/ml and 1 μM, respectively; anti-CD3 (OKT3; BioLegend) and anti-CD28 (CD28.2; BioLegend) antibodies used at 1 μg/ml each; ABT-199 (MedChemExpress; catalog no. HY-15531), A-1155463 (MedChemExpress; catalog no. HY-19725), and A-1331852 (MedChemExpress; catalog no. HY-19741) were dissolved in DMSO and used at 100 nM each. Antibodies for cell staining included fixable viability dye (aqua; Thermo Fisher), anti-human CD3 (clone SK7; BioLegend; 1:200), anti-human CD4 (clone RPA-T4; BD Biosciences; 1:200), anti-human CD8 (clone RPA-T8; BioLegend; 1:200), anti-human CD45RA (clone HI100; BioLegend; 1:200), anti-human CCR7 (clone G043H7; BioLegend; 1:200), anti-human CD69 (clone FN50; BioLegend; 1:200), anti-human HLA-DR (clone L243; BioLegend; 1:200), anti-human BCL-XL (clone 7B2.5; Invitrogen; 1:50), and p24 antibodies (anti-HIV core antigen, clone KC57; Beckman Coulter; 1:200).
HIV infection and flow cytometry staining for BCL-XL expression.
Total CD4+ T cells were enriched from HIV-negative donor peripheral blood mononuclear cells (PBMCs) by magnetic negative selection following the manufacturer’s instructions (Stemcell Technologies). These cells were then activated with 1 μg/ml each of anti-CD3 and anti-CD28 antibodies in RPMI-10 medium supplemented with 50 IU/ml of interleukin-2 (IL-2) (R10-50). Activated CD4+ T cells were infected with an HIV strain (JR-CSF, NL4-3, or supernatant collected from a QVOA p24-positive well from donor OM5334 [named OM5334 QVOA virus]) by spinoculation at 2,000 × g for 2 h. Cells were then incubated in R10-50 at 37°C for 7 days, with half medium changes every other day. Cells were collected and treated with DMSO (1:1,000 as control), ABT-199, A-1155463, or A-1331852 in R10-50 for 48 h. Cells were then stained with antibodies against human CD3, CD4, CD8, BCL-XL, and p24, with a fixable live/dead dye staining, and then analyzed by flow cytometry (Attune NxT). Data were analyzed using FlowJo software.
RNA-seq sample acquisition.
Cultured TCM CD4+ T cells were generated as previously described (32; also see below). Total CD4+ T cells or TCM CD4+ cells were activated and infected as described above. When the infection rate went over 10%, cells were collected and stained with antibodies against human CD3, CD4, CD8, and p24, with a fixable live/dead dye staining, and then sorted by flow cytometry (SONY9000) directly into 15-ml collection tubes for HIV+ (p24+) and HIV– (p24–) cells (Fig. 1A). Total RNA was immediately extracted using the miRNeasy FFPE kit (Qiagen), and RNA quality and concentration were determined using an Agilent Bioanalyzer 2100. Library preparation was performed using the methods of TruSeq RNA sample preparation (nonstranded and poly-A selection), and sequencing was run on a HiSeq 4000 instrument (Illumina) with a single read clustering and 100 cycles of sequencing.
RNA-seq data analysis.
The raw sequencing reads in BCL format were processed through bcl2fastq 2.19 (Illumina) for FASTQ conversion and demultiplexing. Reads were aligned with default parameters to the human reference genome (GRCh38.p12) with STAR (2.6.0c) (60). Gene abundance was calculated with featureCounts using composite gene models from Gencode release 28. Differentially expressed genes were determined with DESeq2 using Wald tests (q < 0.01) (61). Expression heatmaps of the genes encoding proteins of the BCL-2 family were generated using variance-stabilized data, with the values being centered and scaled across each gene. The gene counts and code for the analysis and visualization of the bulk RNA-seq data can be found at https://github.com/abcwcm/RenHIV (62).
Generation of cultured TCM CD4+ primary T cells.
Latency model cells were generated as previously described (32). In brief, 5 × 106 naive CD4+ T cells from HIV– or HIV+ donors were isolated by magnetic negative selection (Stemcell Technologies) and then cultured at 106 cells/ml in 96-well plates using R-10 medium (RPMI 1640 medium supplemented with 10% fetal bovine serum [FBS], 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin) supplemented with 12.5 μl/ml of Dynabeads human T-activator CD3/CD28 (Invitrogen), 2 μg/ml anti-human IL-12 (PeproTech), 1 μg/ml anti-human IL-4 (PeproTech), and 10 ng/ml transforming growth factor β1 (TGF-β1). After 3 days, Dynabeads were removed by magnetic selection, and cells were washed, followed by culture in R-10 medium supplemented with 30 IU/ml of IL-2 (R-10-30). The medium was changed on days 4 and 5 with fresh R-10-30 medium.
Infection of cultured TCM cells to generate latency model.
On day 7 of the above-described cultured TCM generation protocol, 1/5th of the cells were infected with HIVNL4-3 at a multiplicity of infection (MOI) of 0.6 by spinoculating at 2,000 × g for 2 h at 37°C. Cells were then resuspended in R-10-30 with the other 3/5th of the cells and placed back in the incubator, while the remaining 1/5th were set aside as an uninfected control. On day 10, cells were recounted, washed, and resuspended in R-10-30 at 1 M cells/ml and plated in 96-well round-bottom plates to crowd infection. On day 13, cells were washed and resuspended at 1 M cells/ml and then transferred to culture flasks in R-10-30 supplemented with 1 μM raltegravir and 0.5 μM nelfinavir at 1 M cells/ml. On day 17, CD4 positive cells were isolated by magnetic positive selection following the manufacturer’s instructions (Life Technologies) and then used in the various assays. A small portion of the cells were reactivated with CD3/CD28 Dynabeads and stained for intracellular Gag to determine infection percentages and ensure quality controls.
Cultured TCM primary cell latency model spiked HIV eradication (HIVE) assays.
HIVE assays were set up as previously described (58). Briefly, >15 M CD4+ T cells were pulsed with or without bryostatin-1 for 2 h and then washed and cocultured with or without either BCL-XL antagonists (A-1155463 or A-1331852) in XVIVO-15 medium (Lonza) supplemented with 1 μM tenofovir disoproxil fumarate, 1 μM nevirapine, 1 μM emtricitabine, 10 μM T-20, 10U/ml human DNase I (ProSpec), and 0.1 nM IL-7 (HIVE medium). Primary latency model cells were spiked into newly isolated, autologous resting CD4+ T cells to achieve a frequency of ∼1,000 to 10,000 copies of HIV DNA per million CD4+ T cells. Following a 4-days coculture, CD4+ T cells were isolated and rested for 24 h in R10-50 medium at 37°C to allow for an ARV washout period. Aliquots of pre- and post- CD4 enrichment samples were collected and stained for viability and memory phenotype/activation status with antibodies against CD3, CD4, CD8, CD45RA, CCR7, CD69, and a viability dye (Invitrogen Technologies) and then analyzed by flow cytometry. Following the overnight culture, a small aliquot of cells was mixed with CountBright absolute counting beads and viability dye to obtain a count of total, live CD4+ T cells by flow cytometry. This viable cell count was used to determine cell numbers for ddPCR and QVOA plating strategies (Fig. 5A).
Ex vivo HIV reservoir eradication (HIVE) assays.
HIVE assays with ex vivo resting CD4+ T cells from long-term ART-suppressed donors were set up as described above, without the cultured TCM primary cell latency model spiked in.
Digital droplet PCR.
The ddPCR measuring total HIV DNA (HIVEs) was performed as previously described (63), with slight modifications. For each PCR, 5 units of restriction enzyme BsaJI (New England Biolabs [NEB]) was directly mixed with 300 ng of DNA, ddPCR supermix (no dUTP) for probes (Bio-Rad), and final concentrations of 900 nM primers and 250 nM probe. Primers/probes are listed in Table 2; droplets were prepared using the QX200 droplet generator (Bio-Rad) following the manufacturer’s instructions. Sealed plates were cycled using the following program: 95°C for 10 min; 40 cycles of 94°C for 30 s, 60°C for 1 min; and 98°C for 10 min. Reactions were analyzed using the QX200 droplet reader, and the number of template molecules per μl of starting material was estimated using the Quantalife ddPCR software. Eight technical replicates were run per sample, and we consistently applied a predetermined exclusion criterion to outliers that deviated from mean values by >2× the standard deviation.
TABLE 2.
Primers and probes used for ddPCR
| Primer/probe namea | Sequence |
|---|---|
| HIV-gag; Fwd primer | TCTCGACGCAGGACTCG |
| HIV-gag; Rev primer | TACTGACGCTCTCGCACC |
| HIV-gag; probe | FAM-CTCTCTCCTTCTAGCCTC-MGBNFQ |
| RPP30; Fwd primer | GATTTGGACCTGCGAGCG |
| RPP30; Rev primer | GCGGCTGTCTCCACAAGT |
| RPP30; probe | VIC-CTGAACTGAAGGCTCT-MGBNFQ |
| HIV multiplex primer/probe | sequence |
| HIV-gag; Fwd primer | TCTCGACGCAGGACTCG |
| HIV-gag; Rev primer | TACTGACGCTCTCGCACC |
| HIV-gag; probe | FAM-CTCTCTCCTTCTAGCCTC-IABkFQ |
| HIV-env (RRE); Fwd primer | AGTGGTGCAGAGAGAAAAAAGAGC |
| HIV-env (RRE); Rev primer | GTCTGGCCTGTACCGTCAGC |
| HIV-env (RRE); probe | HEX- CCTTGGGTTCTTGGGA-IABkFQ |
| HIV-env (RRE); hypermutant probe | IABkFQ-CCTTAGGTTCTTAGGAGC-IABkFQ |
| House-keeping multiplex Primer/Probe | |
| RPP30-shearing; Fwd primer | CCATTTGCTGCTCCTTGGG |
| RPP30-shearing; Rev primer | CATGCAAAGGAGGAAGCCG |
| RPP30-shearing; probe | FAM-GGAAAGGAGCAAGGTTC-IABkFQ |
| RPP30; Fwd primer | GATTTGGACCTGCGAGCG |
| RPP30; Rev primer | GCGGCTGTCTCCACAAGT |
| RPP30; probe | HEX-CTGAACTGAAGGCTCT-IABkFQ |
Fwd, forward; Rev, reverse.
For HIVE in Fig. 7G to I, a modified intact proviral DNA assay (IPDA) (64) was applied. For each PCR, the same ddPCR supermix and final concentrations of primers and probes as described above was used, but with 5 units of restriction enzyme Xho I (NEB) mixed with 750 ng DNA. Primers and probes were used in 2 separate PCR systems: (i) housekeeping multiplex with RPP30 and RPP30-shearing and (ii) HIV multiplex with gag and HIV-env (RRE). Primers/probes were listed in Table 2. The PCR program is as follows: 95°C for 10 min; 45 cycles of 94°C for 30 s, 53°C for 1 min; and 98°C for 10 min. DNA input of housekeeping multiplex is 100-fold diluted from the input of HIV-multiplex. Total gag, env, or intact provirus copies were calculated by multiplying the dilution factors, and intact provirus copies were corrected with the shearing percentage calculated from the housekeeping multiplex. Eight technical replicates were run per sample and applied with a predetermined exclusion criterion to outliers that deviated from mean values by >2× the standard deviation.
Quantitative viral outgrowth assays (QVOAs).
QVOAs were performed using a previously described protocol (65), with slight modifications depending on the application. Live cells counted by flow cytometry were distributed into either 3- or 2-fold serial dilutions with 12 replicates per dilution. This was determined based on the numbers of viable cells recovered at the end of each HIVE assay and the baseline IUPM values of the donor. Cells were then stimulated with 2 μg/ml of phytohemagglutinin (PHA; Thermo Fisher Scientific) + 1 M PBMCs (HIV-negative donor, irradiated at 5,000 rads). The next day, 1 M CCR5+MOLT-4 cells were added along with a half medium change. Cultures were then incubated for 14 days, with half medium changes with R10-50 every 3 to 4 days. We performed p24 enzyme-linked immunosorbent assay (ELISA) on supernatant 15 days after the PHA stimulation. For each condition, values for cells/well, number of positive wells, and total wells tested were entered into a limiting dilution analyzer (http://bioinf.wehi.edu.au/software/elda/) (66) to calculate the maximal likelihood IUPM and a corresponding 95% confidence interval.
TUNEL assay.
The APO-BrdUTUNEL assay kit, with Alexa Fluor488 anti-BrdU (Invitrogen/Thermo Fisher; catalog no. 23210) was used to detect apoptotic HIV Gag+ cells from the latency model described above. The protocol was followed as suggested by the manufacturer, with the following amendments: (i) in step 1.3, Cytofix/Cytoperm (BD; catalog no. 554714) was used in place of 4% PFA. After the Cytofix/Cytoperm was removed, cells were washed once with phosphate-buffered saline (PBS) and then stained for expression of HIV Gag by KC57 RD-1 (Beckman Coulter; catalog no. 6604667) at a 1/100 dilution in PBS; cells were then washed again with PBS. (ii) In step 1.7, the 30-minute ethanol treatment was preformed in lieu of longer ethanol treatments. (iii) In step 2.5, the DNA labeling solution was applied for 1 h at 37°C in lieu of longer room temperature incubations. (iv) In step 2.10, cell cycle information was not needed, and therefore the propidium iodide solution was not used; in its place, we used PBS as suggested by the manufacturer’s technician. All samples were read on the Attune NxT flow cytometer within 1 h of the final staining.
CRISPR knockout of BCL-XL and BCL-2.
To knock out BCL-XL or BCL-2 expression, we duplexed Alt-R CRISPR Cas9 tracrRNA with two guide CRISPR RNAs (crRNAs) used against the BCL2L1 gene at position 31721915 sequence ACGAGTTTGAACTGCGGTAC PAM CGG and position 31722063 sequence GAGACCCCCAGTGCCATCAA PAM TGG, or one guide cRNA used against the BCL2 gene at position 63318177 sequence TGTGTGTGGAGAGCGTCAAC PAM CGG (Integrated DNA technologies [IDT]). A computationally validated Alt-R CRISPR-Cas9 negative crRNA was used as a CRISPR control. Duplexed guides were incubated with Alt-R S.p. Cas9 nuclease V3 for 20 min at room temperature, mixed with HIV JR-CSF-infected CD4+ T cells from day 17 of the latency model in P3 primary cell nucleofector solution (Lonza), and pulsed using the Lonza 4D nucleofector—pulse code EO 115 for human stimulated T cells. Cells were incubated in R10-50 medium for 3 days, and then knockout of BCL-2 and BCL-XL expression was confirmed by flow cytometry.
Quantification and statistical analysis.
Statistical analyses were performed using Prism 8 (GraphPad), and the statistical analysis methods used are reported in the figure legends. All ddPCR data were analyzed by ordinary one-way analysis of variance (ANOVA), with Tukey’s multiple-comparison test if the overall ANOVA F-test was significant; statistics for the summary data sets for HIV DNA were performed using the mean of 8 replicates per condition. QVOAs were run at the end of each HIVE assay, and the IUPM was calculated as described above; pairwise comparisons were made with a Chi-square test following the method of Hu and Smyth (66) using ELDA software (http://bioinf.wehi.edu.au/software/elda/). All comparisons between HIVE conditions used a paired nonparametric test (2-tailed) Wilcoxon matched-pair signed rank test. A P value of less than 0.05 was considered significant.
Study approval.
People living with HIV were recruited from either the Maple Leaf Medical Clinic in Toronto, Canada, through a protocol approved by the University of Toronto Institutional Review Board (IRB), or Whitman-Walker Health in Washington, DC (Table 1). Additional use of deidentified samples was reviewed and approved by the George Washington University (Washington, DC) and Weill Cornell Medicine (New York) Institutional Review Boards. All subjects were adults and gave written informed consent prior to their participation. Leukapheresis samples were used immediately, if possible, or cryopreserved in liquid nitrogen; cells were not left in culture prior to the initiation of experiments.
ACKNOWLEDGMENTS
This work was supported by the NIH-funded R01 awards AI31798 and AI147845 and the R56 award AI52764. It was also supported in part by the Martin Delaney BELIEVE Collaboratory (NIH grant 1UM1AI26617) and the NIH-funded Center for AIDS Research grants (P30 AI117970), which are both supported by the following NIH cofunding and participating institutes and centers: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, FIC, and OAR.
The following reagents were obtained from the NIH AIDS Research and Reference Reagent Program: IL-2, pNL4-3, and CCR5+ MOLT-4 cells. Reagents for HIV p24 ELISAs were obtained from the NCI’s AIDS and Cancer Virus Program.
R.B.J. and Y.R. conceptualized the study. R.B.J., Y.R., S.H.H., and A.B. developed the methodology. R.B.J., Y.R., S.H.H., A.B.M., W.D.C.A., T.K., L.L., T.M.M., D.C., R.T., A.R.W., and T.R. conducted the investigation. P.Z., F.D., and D.B. provided bioinformatic analysis for the RNA-seq data. C.K. and E.B. recruited study participants and provided clinical samples. R.B.J. and Y.R. wrote the original draft of the manuscript, with edits provided by all coauthors. R.B.J. acquired funding. R.B.J. and A.B. supervised the study.
Y.R., S.H.H., and R.B.J. declare that they are inventors on a patent that claims the use of BCL-2 and BCL-XL antagonists as therapies for HIV. R.B.J. declares that he has received payments for his role on the scientific advisory board of AbbVie, Inc.
Contributor Information
R. Brad Jones, Email: rbjones@med.cornell.edu.
Guido Silvestri, Emory University.
REFERENCES
- 1.Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC. 1999. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A 96:15109–15114. 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 3.Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1:1284–1290. 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Blanco MA, Cullen BR. 1991. Molecular basis of latency in pathogenic human viruses. Science 254:815–820. 10.1126/science.1658933. [DOI] [PubMed] [Google Scholar]
- 5.Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA. 2007. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med 204:1665–1675. 10.1084/jem.20070618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sainski AM, Natesampillai S, Cummins NW, Bren GD, Taylor J, Saenz DT, Poeschla EM, Badley AD. 2011. The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak. J Virol 85:7965–7975. 10.1128/JVI.02515-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Natesampillai S, Cummins NW, Nie Z, Sampath R, Baker JV, Henry K, Pinzone M, O'Doherty U, Polley EC, Bren GD, Katzmann DJ, Badley AD. 2018. HIV protease-generated Casp8p41, when bound and inactivated by Bcl2, is degraded by the proteasome. J Virol 92:e00037-18. 10.1128/JVI.00037-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Y, Huang SH, Patel S, Alberto WDC, Magat D, Ahimovic D, Macedo AB, Durga R, Chan D, Zale E, Mota TM, Truong R, Rohwetter T, McCann CD, Kovacs CM, Benko E, Wimpelberg A, Cannon C, Hardy WD, Bosque A, Bollard CM, Jones RB. 2020. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J Clin Invest 130:2542–2559. 10.1172/JCI132374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo HH, Ahmad R, Lee GQ, Gao C, Chen HR, Ouyang Z, Szucs MJ, Kim D, Tsibris A, Chun TW, Battivelli E, Verdin E, Rosenberg ES, Carr SA, Yu XG, Lichterfeld M. 2018. Anti-apoptotic protein BIRC5 maintains survival of HIV-1-infected CD4(+) T cells. Immunity 48:1183–1194.e5. 10.1016/j.immuni.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardwick JM, Soane L. 2013. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol 5:a008722. 10.1101/cshperspect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. 1998. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A 95:4997–5002. 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, Newmeyer DD. 1999. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol 147:809–822. 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossé T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. 1998. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature 391:496–499. 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 14.Kim CN, Wang X, Huang Y, Ibrado AM, Liu L, Fang G, Bhalla K. 1997. Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res 57:3115–3120. [PubMed] [Google Scholar]
- 15.Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. 1993. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol 4:327–332. [PubMed] [Google Scholar]
- 16.Chandrasekar AP, Cummins NW, Badley AD. 2019. The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence. Clin Microbiol Rev 33:e00107-19. 10.1128/CMR.00107-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto F, Oyaizu N, Kalyanaraman VS, Pahwa S. 1997. Modulation of Bcl-2 protein by CD4 cross-linking: a possible mechanism for lymphocyte apoptosis in human immunodeficiency virus infection and for rescue of apoptosis by interleukin-2. Blood 90:745–753. 10.1182/blood.V90.2.745. [DOI] [PubMed] [Google Scholar]
- 18.Perfettini JL, Roumier T, Castedo M, Larochette N, Boya P, Raynal B, Lazar V, Ciccosanti F, Nardacci R, Penninger J, Piacentini M, Kroemer G. 2004. NF-kappaB and p53 are the dominant apoptosis-inducing transcription factors elicited by the HIV-1 envelope. J Exp Med 199:629–640. 10.1084/jem.20031216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen D, Wang M, Zhou S, Zhou Q. 2002. HIV-1 Tat targets microtubules to induce apoptosis, a process promoted by the pro-apoptotic Bcl-2 relative Bim. EMBO J 21:6801–6810. 10.1093/emboj/cdf683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dabrowska A, Kim N, Aldovini A. 2008. Tat-induced FOXO3a is a key mediator of apoptosis in HIV-1-infected human CD4+ T lymphocytes. J Immunol 181:8460–8477. 10.4049/jimmunol.181.12.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sastry KJ, Marin MC, Nehete PN, McConnell K, el-Naggar AK, McDonnell TJ. 1996. Expression of human immunodeficiency virus type I tat results in down-regulation of bcl-2 and induction of apoptosis in hematopoietic cells. Oncogene 13:487–493. [PubMed] [Google Scholar]
- 22.Ferri KF, Jacotot E, Blanco J, Este JA, Kroemer G. 2000. Mitochondrial control of cell death induced by HIV-1-encoded proteins. Ann N Y Acad Sci 926:149–164. 10.1111/j.1749-6632.2000.tb05609.x. [DOI] [PubMed] [Google Scholar]
- 23.Halestrap AP, Brenner C. 2003. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 10:1507–1525. 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 24.Akari H, Bour S, Kao S, Adachi A, Strebel K. 2001. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J Exp Med 194:1299–1311. 10.1084/jem.194.9.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolf D, Witte V, Laffert B, Blume K, Stromer E, Trapp S, d’Aloja P, Schurmann A, Baur AS. 2001. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat Med 7:1217–1224. 10.1038/nm1101-1217. [DOI] [PubMed] [Google Scholar]
- 26.Conti L, Rainaldi G, Matarrese P, Varano B, Rivabene R, Columba S, Sato A, Belardelli F, Malorni W, Gessani S. 1998. The HIV-1 vpr protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: possible implications for the pathogenesis of AIDS. J Exp Med 187:403–413. 10.1084/jem.187.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan Z, Fan X, Staitieh B, Bedi C, Spearman P, Guidot DM, Sadikot RT. 2017. HIV-related proteins prolong macrophage survival through induction of Triggering receptor expressed on myeloid cells-1. Sci Rep 7:42028. 10.1038/srep42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balestrieri E, Grelli S, Matteucci C, Minutolo A, d’Ettorre G, Di Sora F, Montella F, Vullo V, Vella S, Favalli C, Macchi B, Mastino A. 2007. Apoptosis-associated gene expression in HIV-infected patients in response to successful antiretroviral therapy. J Med Virol 79:111–117. 10.1002/jmv.20768. [DOI] [PubMed] [Google Scholar]
- 29.Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. 1999. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem 274:2225–2233. 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 30.Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59. 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 31.Ledford H. 2009. Ahead of the pack. Nature 459:286–287. 10.1038/nj7244-286a. [DOI] [PubMed] [Google Scholar]
- 32.Bosque A, Planelles V. 2009. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113:58–65. 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martins LJ, Bonczkowski P, Spivak AM, De Spiegelaere W, Novis CL, DePaula-Silva AB, Malatinkova E, Trypsteen W, Bosque A, Vanderkerckhove L, Planelles V. 2016. Modeling HIV-1 latency in primary T cells using a replication-competent virus. AIDS Res Hum Retroviruses 32:187–193. 10.1089/aid.2015.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. 2013. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155:540–551. 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macedo AB, Resop RS, Martins LJ, Szaniawski MA, Sorensen ES, Spivak AM, Nixon DF, Jones RB, Planelles V, Bosque A. 2018. Influence of biological sex, age, and HIV status in an in vitro primary cell model of HIV latency using a CXCR4 tropic virus. AIDS Res Hum Retroviruses 34:769–777. 10.1089/AID.2018.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ameisen JC, Capron A. 1991. Cell dysfunction and depletion in AIDS: the programmed cell death hypothesis. Immunol Today 12:102–105. 10.1016/0167-5699(91)90092-8. [DOI] [PubMed] [Google Scholar]
- 37.Laurent-Crawford AG, Krust B, Muller S, Riviere Y, Rey-Cuille MA, Bechet JM, Montagnier L, Hovanessian AG. 1991. The cytopathic effect of HIV is associated with apoptosis. Virology 185:829–839. 10.1016/0042-6822(91)90554-o. [DOI] [PubMed] [Google Scholar]
- 38.Terai C, Kornbluth RS, Pauza CD, Richman DD, Carson DA. 1991. Apoptosis as a mechanism of cell death in cultured T lymphoblasts acutely infected with HIV-1. J Clin Invest 87:1710–1715. 10.1172/JCI115188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.French AJ, Natesampillai S, Krogman A, Correia C, Peterson KL, Alto A, Chandrasekar AP, Misra A, Li Y, Kaufmann SH, Badley AD, Cummins NW. 2020. Reactivating latent HIV with PKC agonists induces resistance to apoptosis and is associated with phosphorylation and activation of BCL2. PLoS Pathog 16:e1008906. 10.1371/journal.ppat.1008906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. 2008. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 6:e147. 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ku B, Liang C, Jung JU, Oh BH. 2011. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res 21:627–641. 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volkmann N, Marassi FM, Newmeyer DD, Hanein D. 2014. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ 21:206–215. 10.1038/cdd.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kale J, Osterlund EJ, Andrews DW. 2018. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25:65–80. 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135:1074–1084. 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 45.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19:1294–1305. 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. 2007. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 14:943–951. 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 47.Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR, White MJ, Kruse EA, Lane RM, Ellis S, Nurden P, Mason KD, O'Reilly LA, Roberts AW, Metcalf D, Huang DC, Kile BT. 2011. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med 208:2017–2031. 10.1084/jem.20110750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, Flygare JA, Gibbons P, Kersten WJ, Kulasegaram S, Moss RM, Parisot JP, Smith BJ, Street IP, Yang H, Huang DC, Watson KG. 2013. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol 9:390–397. 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 49.Huska JD, Lamb HM, Hardwick JM. 2019. Overview of BCL-2 family proteins and therapeutic potentials. Methods Mol Biol 1877:1–21. 10.1007/978-1-4939-8861-7_1. [DOI] [PubMed] [Google Scholar]
- 50.Song C, Ge Z, Ding Y, Tan BH, Desai D, Gowda K, Amin SG, Gowda R, Robertson G, Yue F, Huang S, Spiegelman V, Payne J, Reeves M, Gurel Z, Iyer S, Dhanyamraju PK, Xiang M, Kawasawa YI, Cury NM, Yunes JA, McGrath M, Schramm J, Su RJ, Yang Y, Zhao Z, Lyu X, Muschen M, Payne KJ, Gowda C, Dovat S. 2020. IKAROS and CK2 regulate expression of BCL-XL and chemosensitivity in high-risk B-cell acute lymphoblastic leukemia. Blood 136:1520–1534. 10.1182/blood.2019002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez-Brocca MA, Castilla C, Navarro E, Amaya MJ, Travado P, Japon MA, Saez C. 2007. Clinicopathological correlations of Bcl-xL and Bax expression in differentiated thyroid carcinoma. Clin Endocrinol 68:190–197. 10.1111/j.1365-2265.2007.03018.x. [DOI] [PubMed] [Google Scholar]
- 52.Moncsek A, Al-Suraih MS, Trussoni CE, O’Hara SP, Splinter PL, Zuber C, Patsenker E, Valli PV, Fingas CD, Weber A, Zhu Y, Tchkonia T, Kirkland JL, Gores GJ, Mullhaupt B, LaRusso NF, Mertens JC. 2018. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(-/-)) mice. Hepatology 67:247–259. 10.1002/hep.29464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terai H, Kitajima S, Potter DS, Matsui Y, Quiceno LG, Chen T, Kim TJ, Rusan M, Thai TC, Piccioni F, Donovan KA, Kwiatkowski N, Hinohara K, Wei G, Gray NS, Fischer ES, Wong KK, Shimamura T, Letai A, Hammerman PS, Barbie DA. 2018. ER stress signaling promotes the survival of cancer “persister cells” tolerant to EGFR tyrosine kinase inhibitors. Cancer Res 78:1044–1057. 10.1158/0008-5472.CAN-17-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Jong Y, Monderer D, Brandinelli E, Monchanin M, van den Akker BE, van Oosterwijk JG, Blay JY, Dutour A, Bovee J. 2018. Bcl-xl as the most promising Bcl-2 family member in targeted treatment of chondrosarcoma. Oncogenesis 7:74. 10.1038/s41389-018-0084-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, Czabotar PE, Deshayes K, Fairbrother WJ, Flygare JA, Hymowitz SG, Jin S, Judge RA, Koehler MF, Kovar PJ, Lessene G, Mitten MJ, Ndubaku CO, Nimmer P, Purkey HE, Oleksijew A, Phillips DC, Sleebs BE, Smith BJ, Smith ML, Tahir SK, Watson KG, Xiao Y, Xue J, Zhang H, Zobel K, Rosenberg SH, Tse C, Leverson JD, Elmore SW, Souers AJ. 2014. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett 5:1088–1093. 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, Jin S, Smith M, Xue J, Zhang H, Oleksijew A, Magoc TJ, Vaidya KS, Albert DH, Tarrant JM, La N, Wang L, Tao ZF, Wendt MD, Sampath D, Rosenberg SH, Tse C, Huang DC, Fairbrother WJ, Elmore SW, Souers AJ. 2015. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 7:279ra40. 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, Kirkland JL. 2017. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 9:955–963. 10.18632/aging.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang SH, Ren Y, Thomas AS, Chan D, Mueller S, Ward AR, Patel S, Bollard CM, Cruz CR, Karandish S, Truong R, Macedo AB, Bosque A, Kovacs C, Benko E, Piechocka-Trocha A, Wong H, Jeng E, Nixon DF, Ho YC, Siliciano RF, Walker BD, Jones RB. 2018. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest 128:876–889. 10.1172/JCI97555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang S-H, McCann CD, Mota TM, Wang C, Lipkin SM, Jones RB. 2019. Have cells harboring the HIV reservoir been immunoedited? Front Immunol 10:1842. 10.3389/fimmu.2019.01842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zumbo P, Dündar F. 2020. abcwcm/RenHIV: major release. Zenodo v1.0.0. https://zenodo.org/record/4289911#.YJs6G6FlC00.
- 63.Strain MC, Lada SM, Luong T, Rought SE, Gianella S, Terry VH, Spina CA, Woelk CH, Richman DD. 2013. Highly precise measurement of HIV DNA by droplet digital PCR. PLoS One 8:e55943. 10.1371/journal.pone.0055943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bruner KM, Wang Z, Simonetti FR, Bender AM, Kwon KJ, Sengupta S, Fray EJ, Beg SA, Antar AAR, Jenike KM, Bertagnolli LN, Capoferri AA, Kufera JT, Timmons A, Nobles C, Gregg J, Wada N, Ho YC, Zhang H, Margolick JB, Blankson JN, Deeks SG, Bushman FD, Siliciano JD, Laird GM, Siliciano RF. 2019. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 566:120–125. 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laird GM, Eisele EE, Rabi SA, Lai J, Chioma S, Blankson JN, Siliciano JD, Siliciano RF. 2013. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog 9:e1003398. 10.1371/journal.ppat.1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347:70–78. 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]