

Loeys-Dietz (LDS) syndromes (types 1–5) are autosomal dominant conditions with pathogenic variants in TGFβR1, TGFβR2, SMAD3, TGFβ2 and TGFβ3 that affect the cardiovascular, musculoskeletal and craniofacial systems. Aortic root dissection is a major cause of mortality in these individuals. Aortic root dilation is often present in childhood and may be recognized in utero. Our case highlights the prenatal ultrasound features of LDS.
Case History:
A 19 year-old primigravida with a diagnosis of LDS type 4 due to a TGFβ2 (c.988C>T) pathogenic variant presented for prenatal care at 26 weeks gestation. The patient’s clinical features included hypertelorism, high-arched palate, aortic root enlargement (3.6 cm, z- score for body surface area of 4.0) and hypermobile joints. She declined amniocentesis for prenatal diagnosis. A fetal echocardiogram at 26 weeks demonstrated moderate aortic root dilation (7.8 mm, Z-score 4.9), mild dilation of the main pulmonary artery (8.2 mm, Z-score 2.2), normal biventricular size and systolic function (Figure 1A) which led to prenatal suspicion for LDS type 4. Her anatomy ultrasound at 31weeks showed no abnormalities with estimated fetal weight (EFW) of 1517 grams (13%); however prominent eyes were noted (Figure 1B). EFW was 24th percentile at 34 weeks in a subsequent ultrasound. The mother took metoprolol during pregnancy and had echocardiograms with stable aortic root dimensions of 3.6–3.7 cm. She underwent a non-contrast magnetic resonance study from brain to pelvis at 32 weeks demonstrating aortic root dimension 3.9 cm and tortuous extracranial internal cerebral arteries without aneurysm.
Figure 1A:

Fetal echocardiogram at 26 weeks with moderate aortic root dilation (7.8 mm z score 4.9)
Figure 1B:

3D ultrasound at 31 weeks gestation of fetal face with prominent eyes
An uncomplicated cesarean delivery was performed at 37 weeks with a male infant weighing 2805 grams (37%). Postnatal examination revealed hypertelorism with prominent eyes, mild retrognathia, translucent skin, joint hypermobility and arachnodactyly (Figure 2A, B, C). Uvula and palate were normal. Genetic testing confirmed LDS type 4. Postnatal echocardiography confirmed aortic root dilation (1.22 cm Z-score 2.34), pulmonary artery dilation (1.2 cm z-score 3.8) and, additionally showed a small secundum atrial septal defect (ASD) and a moderate-sized patent ductus arteriosus (PDA). Propranolol was started in the hospital after birth and at his 14 month follow-up visit, the PDA had resolved, however the ASD and aortic root/pulmonary artery dilation were still present. Maternal postpartum course was uneventful.
Figure 2:
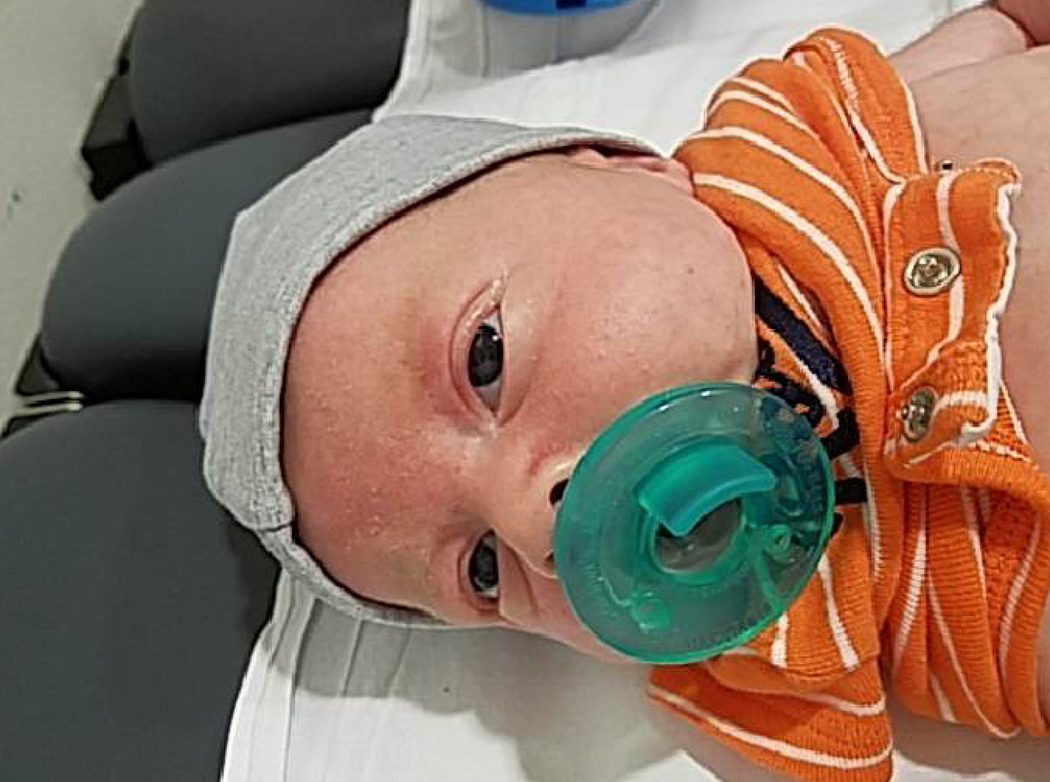


Postnatal clinical exam findings in neonate: 2A: hypertelorism, prominent eyes, mild retrognathia. Figure 2B: translucent skin. Figure 2C: arachnodactyly.
Discussion:
Recognition of fetal aortic root dilation has dual importance in the neonate and mother. For the neonate, the diagnosis of LDS was suspected in our particular case because of the maternal diagnosis; however, if fetal aortic root dilation is recognized with no known family history, diagnostic testing for LDS and neonatal Marfan syndrome (FBN1) should be considered. If diagnosis is suspected or confirmed, post-natal referral to pediatric cardiology with genetic expertise is recommended. Although some controversy exists about medication use in very young children, animal models demonstrate that medical therapy from birth is the most effective way to slow aortic root growth.6 In regards to maternal care, fetal aortic root dilation should prompt an examination for potential maternal connective tissue disorder because 25% of cases are inherited from a parent. This work-up includes maternal echocardiogram and examination by provider with expertise in genetics. A maternal diagnosis of LDS influences pregnancy management with anesthesia and delivery considerations due to potential cardiovascular complications of aortic root aneurysm and dissection, other arterial aneurysm and dissection, and possible cervical neck instability.7
This is the first case describing prenatal ultrasound features in LDS type 4. Previous cases have described prenatal features of type 1 and 2 LDS; all of these cases describe cardiovascular findings (Table S1). 3–5,8 Therefore, if LDS is known maternally, a fetal echocardiogram is recommended. Our case demonstrates the importance of in utero recognition as this can be a severe disease in the neonate and can have life-threatening complications in a pregnant mother.
Supplementary Material
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/uog.22003
References:
- 1.Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, De Backer J, Muiño-Mosquera L, Naudion S, Zordan C, Morisaki T, Morisaki H, Von Kodolitsch Y, Dupuis-Girod S, Morris SA, Jeremy R, Odent S, Adès LC, Bakshi M, Holman K, LeMaire S, Milleron O, Langeois M, Spentchian M, Aubart M, Boileau C, Pyeritz R, Milewicz DM, Montalcino Aortic Consortium. International registry of patients carrying TGFBR1 or TGFBR2 mutations: results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet. 2016; 9: 548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akutsu K, Morisaki H, Okajima T, Yoshimuta T, Tsutsumi Y, Takeshita S, Nonogi H, Ogino H, Higashi M, Morisaki T. Genetic analysis of young adult patients with aortic disease not fulfilling the diagnostic criteria for Marfan syndrome. Circ J. 2010. May;74(5):990–7. [DOI] [PubMed] [Google Scholar]
- 3.Gindes L, Berkenstadt M, Reznik-Wolf H, Pras E, Achiron R. Prenatal diagnosis of Loeys-Dietz syndrome. Ultraschall Med. 2014; 35(5): 391–394. [PubMed] [Google Scholar]
- 4.Kawazu Y, Inamura N, Kayatani F, Okamoto N, Morisaki H. Prenatal complex congenital heart disease with Loeys-Dietz syndrome. Cardiol Young. 2012. February;22(1):116–9. [DOI] [PubMed] [Google Scholar]
- 5.Viassolo V, Lituania M, Marasini M, Dietz H, Benelli F, Forzano F, Faravelli F. Fetal aortic root dilation: a prenatal feature of the Loeys-Dietz syndrome. Prenat Diagn. 2006. November;26(11):1081–3. [DOI] [PubMed] [Google Scholar]
- 6.Habashi JP, Judge DP, Holm T, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006; 312(5770): 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russo ML, Sukhavasi N, Mathur V, Morris SA. Obstetric Management of Loeys-Dietz Syndrome. Obstet Gynecol. 2018. June;131(6):1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valenzuela I, Fernández-Alvarez P, Munell F, Sanchez-Montanez A, Giralt G, Vendrell T, Tizzano EF. Arthrogryposis as neonatal presentation of Loeys-Dietz syndrome due to a novel TGFBR2 mutation. Eur J Med Genet. 2017. June;60(6):303–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.