

It has been more than 100 years since Beaston and Boyd independently reported that bilateral oophorectomy was an effective treatment or adjuvant for breast cancer, observations that together initiated the use of endocrine therapy for this disease.1,2 Remarkably, these advances were made before the discovery of 17β-estradiol (E2) or the establishment of its role as a mitogen in breast cancer cells. Prophetically, Boyd surmised that some substance produced by the ovaries was having a positive impact on tumor growth and survival, but it took nearly 75 years to advance endocrine therapy beyond surgical oophorectomy and/or radiation-induced ovarian ablation. Notable in this regard was the work of Elwood Jensen, William Hoekstra, Jack Gorski, and David Toft, who, in the 1960s, defined the biochemical entity that we now know to be the estrogen receptor (ER), and the subsequent studies by Bert O'Malley, which demonstrated that ER was, in fact, a ligand-regulated transcription factor.3-6 For many years prior to these discoveries, and absent any understanding of the molecular mechanism of ER action, there had been considerable interest and success in developing estrogen-like compounds for use as emergency contraceptives.7 In the early 1970s, Craig Jordan was one of the first to capitalize on the availability of these drugs, which displayed different pharmacologic attributes (ie, mimicked or opposed the action of estrogens in reproductive tissues).8,9 He demonstrated that one such antiestrogen, tamoxifen, which earlier had shown efficacy in the treatment of metastatic breast cancer, actually functioned as a direct competitive inhibitor of ER, and its activity in models of carcinogen-induced mammary tumors suggested that it was likely to have activity in the adjuvant setting.10-12 This body of work also led to the establishment of a simple model of ER pharmacology that posited that upon binding an agonist, ER underwent a biochemical transformation that enabled it to regulate target gene transcription and that antagonists functioned simply by competitively inhibiting agonist binding to the receptor.9 Within the confines of this model, it was considered that, notwithstanding improvements in affinity and pharmaceutical properties of drugs, additional discovery in this area would likely lead to only incremental advances in efficacy.
With the emergence and clinical success of aromatase inhibitors in the early 2000s, there was little interest in the pharmaceutical industry in the continued development of ER modulators for breast cancer.13,14 However, there continued to be considerable research defining the mechanism of action of ER and in identifying the molecular determinants of ER pharmacology. This was driven in large part by two pharmacologic curiosities: (1) tamoxifen and another drug raloxifene, while functioning as antagonists in breast cancer cells, exhibited different degrees of estrogenic activity in different cells, and (2) in both patients and preclinical models of breast cancer, there was evidence that resistance to tamoxifen occurred when something happened in cells that enabled them to switch from recognizing tamoxifen as an antagonist to an agonist.15-18 These observations framed the important question as to how the same drug, acting through the same receptor, could have different activities in different cells. Leveraging insights from our work and from other investigators, we developed a contemporary model to explain the molecular pharmacology of ER, which holds that (1) the overall conformation of ER is influenced by the nature of the ligand to which it is bound, (2) differences in ER conformation allow the differential presentation of protein-protein interaction surfaces on the receptor, and (3) the relative and absolute expression of functionally distinct receptor-interacting proteins (coregulators) dictate how differently conformed ER-ligand complexes are recognized in cells (Fig 1).19-21 Thus, with respect to drug discovery, the primary exploitable feature of ER is the ability to use small-molecule ligands to manipulate its conformation, and this engenders different coregulator interactions.21-23 Reflecting their ability to induce different alterations in ER structure and manifest tissue-selective agonist and antagonist activities, tamoxifen and raloxifene were reclassified as selective estrogen receptor modulators (SERMs).24 These insights informed the discovery of new SERMs with unique clinical profiles such as lasofoxifene, bazedoxifene, pipendoxifene, ospemifene, and more recently H3B-6545, some of which are being evaluated as breast cancer therapeutics.25-29
FIG 1.
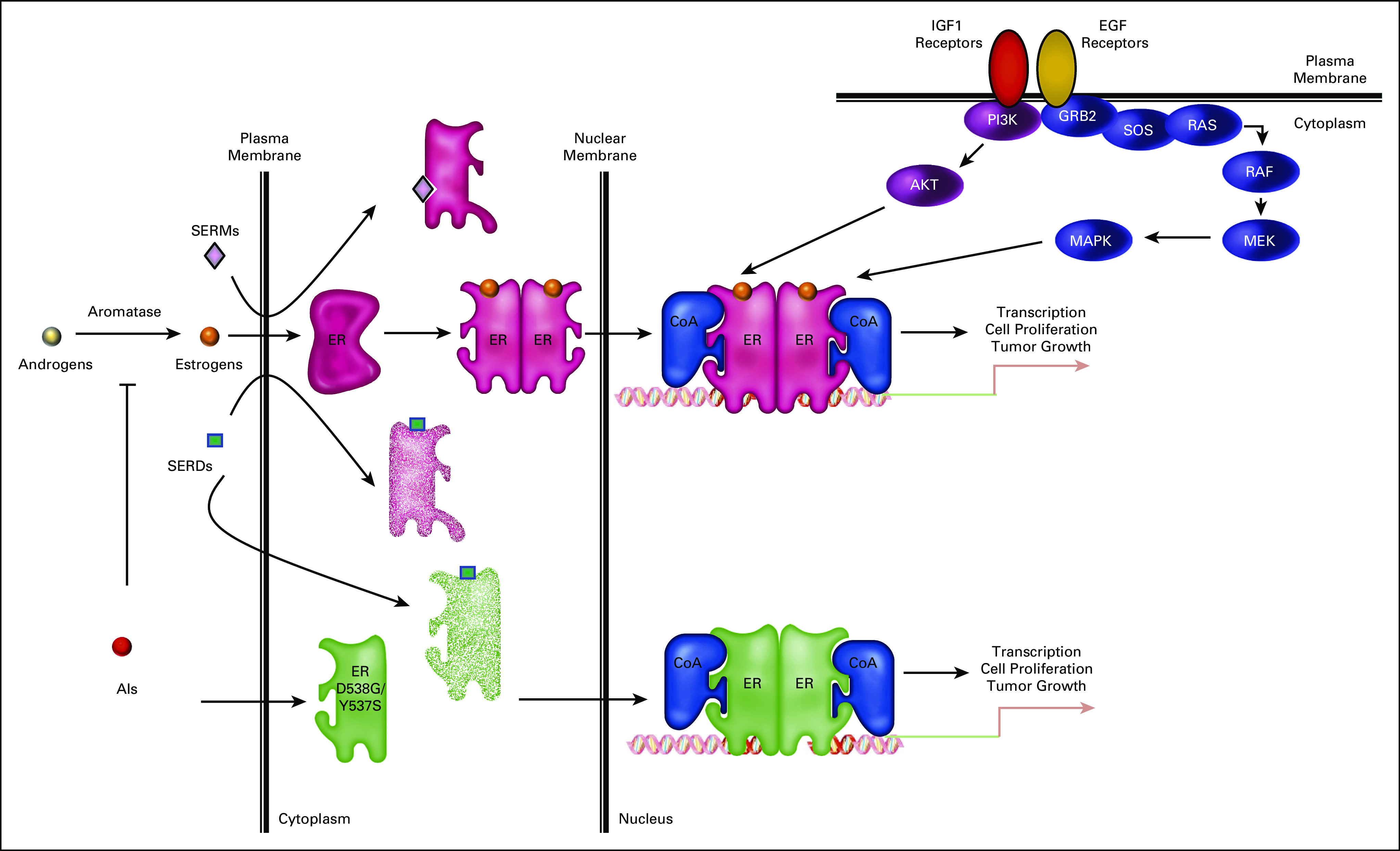
Endocrine therapy landscape for ER-positive breast cancer. SERMs such as tamoxifen inhibit ER-positive breast tumor growth by competitively blocking estrogen binding to the receptor, whereas AIs function by blocking the production of estrogens from androgens. These treatments are not curative as growth factor signaling pathways can mediate resistance by activating the receptor-CoA complex in the absence of hormone or when the receptor is occupied by an SERM. Treatment escape following AI therapy, and to a lesser extent SERM therapy, is also often accompanied by the expression of constitutively active ER mutants, most commonly ER-D538G and ER-Y537S. SERDs, such as fulvestrant and elacestrant (RAD1901), can circumvent some of the therapeutic liabilities of SERMs and AIs by degrading both wild-type and mutant receptors. AIs, aromatase inhibitors; AKT, protein kinase B; CoA, coactivator; EGF, epidermal growth factor; ER, estrogen receptor; GRB2, growth factor receptor-bound protein 2; IGF1, insulin-like growth factor 1; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; PI3K, phosphoinositide 3-kinase; RAF, Raf oncogene; RAS, Ras oncogene; SERD, selective estrogen receptor downregulator or degrader; SERM, selective estrogen receptor modulator; SOS, son of sevenless homolog 1.
Whereas SERMs have found utility in the treatment and prevention of osteoporosis, dyspareunia, and other symptoms associated with estrogen deprivation (menopause), there emerged a disappointingly long list of next-generation SERMs that failed to show efficacy as second-line endocrine therapies in breast cancer.30 Several related discoveries provided a potential explanation for these drug failures. The first was the observation that certain ER coregulators (eg, SRC1 and SRC3) were significantly overexpressed in advanced disease, which decreased the inhibitory activity of even the most antagonistic SERMs.31 Further, it was demonstrated that breast cancer cells likely use the same mechanism, overexpression of coregulators, to circumvent the estrogen deprivation afforded by aromatase inhibitors.32,33 Even more problematic from a therapeutic point of view was the observation that coregulators and ER itself were posttranslationally modified upon the activation of several intracellular signaling pathways (eg, mitogen-activated protein kinase and phosphoinositide 3-kinase) and that this enabled ER to direct target gene transcription absent a canonical small-molecule ligand.34,35 These insights reduced enthusiasm for further development of SERMs as therapeutics in advanced disease.
The observation that coregulator biology is frequently dysregulated in breast cancer and that ER can activate transcription absent a ligand suggested that removal of ER, rather than solely inhibiting its classical activities, may have particular utility in breast cancer. Indeed, this idea was supported by the observation in preclinical models that fulvestrant, a first-in-class selective estrogen receptor downregulator or degrader (SERD), was effective in animal models of endocrine therapy–resistant breast cancer.36 Fulvestrant is approved for use as a second-line endocrine therapy, and despite the fact that it is an injectable with considerable pharmaceutical liabilities, its effectiveness in metastatic disease has validated the general SERD approach.37-40 The early clinical experience with fulvestrant drove the search for effective oral SERDs, which have increased efficacy in the setting of metastatic disease and which would also be suitable for use in the adjuvant setting in patients at high risk for recurrence. To our knowledge, our group identified the first oral SERD etacstil (DPC974), whose development, despite evidence of clinical efficacy, was discontinued for business reasons.41 However, the clear understanding of the mechanism of action of this drug led others to pursue this therapeutic modality with the result that there are currently at least 13 oral SERDs (and two new classes of SERM) in clinical development for breast cancer (Fig 2). As described in the accompanying article, RAD1901 (elacestrant), a drug our group repurposed for breast cancer, having identified it to be an oral SERD, is the furthest along in development.42-44 The positive clinical activity of RAD1901 in breast cancer reported by Bardia et al42 bodes well for the success of the oral SERDs as a class and is instructive with respect to biomarkers that may predict positive response in patients. Preliminary efficacy data should be available for several other drugs of this class later this year.
FIG 2.

ER modulators that are currently approved for use in the treatment or prevention of ER-positive breast cancers or that are currently in clinical development. Those drugs (SERDs and SERMs) that are currently approved for clinical use in breast cancer are highlighted in green. Investigational drugs that are currently in development for breast cancer are as follows: AZD9833 (NCT04588298; SERENA-3), RG6171 (giredestrant) (NCT04576455), SAR439859 (amcenestrant) (NCT03284957; AMEERA-1), ZN-c5 (NCT03560531), G1T48 (rintodestrant) (NCT03455270), LY3844356 (NCT04188548; EMBER), D-0502 (NCT03471663), SHR9549 (NCT03596658), OP-1250 (NCT04505826), LSZ102 (NCT02734615), ARV-471 (NCT04072952), bazedoxifene (NCT02448771), RAD1901 (elacestrant) (NCT03778931; EMERALD), lasofoxifene (NCT03781063; ELAINE-1), and H3B-6545 (NCT04288089). The relative SERD or SERM activity reflects the authors' summary of the available literature and may change as more data become available from comparative studies. ER, estrogen receptor; SERD, selective estrogen receptor downregulator or degrader; SERM, selective estrogen receptor modulator.
In addition to wild-type ER (wtER), there has emerged considerable interest in understanding how SERDs work in breast (and gynecologic) cancers harboring ESR1 mutations, recently found to occur in approximately 40% of the metastatic lesions in patients who have progressed on aromatase inhibitors.45-47 These mutations, which occur within the ligand-binding domain of ER, alter the pharmacology of the receptor, facilitate constitutive coregulator binding, and permit ligand-independent transcriptional activity.46 They may also endow upon the receptor neomorphic pathologic activities.48 Whereas it is believed that these mutants are directly involved in the regulation of processes that affect disease pathology, it remains to be determined how, given their low allelic frequency and the fact that they likely are coexpressed with wtER in most cells, they affect cancer cell biology. Indeed, we have recently demonstrated that the pharmacology of the most commonly occurring mutants is normalized in cells when wtER is present.49 Intriguing to us is the possibility that the presence of ESR1 mutations in tumor cells may be a predictive biomarker that reads on the acute estrogen dependency of a tumor and thus may serve as a positive predictor of response to SERDs or even some SERMs. It is notable in this regard that although the sample size was small, it was reported in the accompanying study that the response to the SERD, elacestrant (RAD1901), was greater in patients in which an ESR1 mutation was detected (ORR 33% in patients harboring ESR1 mutations v 19% in all comers).42 It may be possible to identify those patients who will most likely respond to second-line endocrine therapies by virtue of being able to detect ESR1 mutations in biopsied metastatic tumors or in circulating tumor DNA.
It is likely that most of the SERDs in development will demonstrate efficacy in cancers harboring either wild-type or mutant ESRs, and thus there is a need to consider how they can be distinguished in a clinically meaningful manner. Key differentiators will likely be tumor exposure, target engagement (ER turnover), and tolerability, issues that to date have significantly limited progress in this area. Some drugs appear to be associated with significant GI issues, and bradycardia is a potential liability of at least two SERDs in development.50 These issues could limit the use of some SERDs in the adjuvant setting and possibly also in advanced disease, where they would likely be used in combination with other drugs that have their own inherent liabilities (eg, CDK4/6, PI3K, and mTOR inhibitors).51-53 Another important distinguishing feature of SERDs will be their differential ability to cross the blood-brain barrier, where they would be anticipated to inhibit the growth or progression of metastatic lesions. However, the significant functional differences in the murine and human blood-brain barriers make it difficult to predict human brain exposure of the various drugs at the current time. Further, some SERDs, by mechanisms that remain elusive, also protect against bone loss, and in the adjuvant setting, this is not only likely to be beneficial to bone health but may also decrease or prevent secondary metastasis.50,54 Clearly, the positive or negative activity of these drugs in other ER-target tissues needs to be considered in evaluating their likely benefit in patients.
Finally, looking to the future, all the existing SERDs and SERMs were developed with the understanding that the most important target in breast tumors is ER expressed within the cancer cells. Thus, drugs were generally optimized for activity in cellular models of luminal breast cancer, and then activities were confirmed in xenograft tumor models as a surrogate for activity in metastatic disease. However, this established and traditional discovery path does not reflect the fact that in addition to cancer cells, ER is expressed in most cells within the tumor microenvironment and that the impact of inhibiting this receptor in these cells remains unknown. There is clearly a need to define how estrogens, SERMs, and SERDs affect tumor immunity through actions in T cells, macrophages, and other cells in the tumor stroma. Somewhat forgotten are the results of early studies using endocrine therapy that demonstrated that tamoxifen has considerable efficacy in patients whose tumors were biochemically ER-negative.10 As compelling were data suggesting that although overall response to tamoxifen and the high-affinity ER agonist diethylstilbestrol were equivalent in patients with breast cancer, those patients taking the estrogen had a better overall survival.55,56 Also important to consider is the most updated data from the Women's Health Initiative, which reported a reduced incidence of breast cancer in postmenopausal women receiving supplemental estrogen therapy.57,58 The ongoing studies with SERDs (and SERMs) in breast cancer and the correlative studies associated with these trials may help to address some of these complex issues. However, it is our strongly held opinion that in the process of developing the next generation of endocrine therapies, we should take a step back and define the activities of ER in different cellular components of the tumor microenvironment and how they are influenced by different ER modulators. With this information in hand, we should be in a position to identify new drugs that retain their antagonist and/or SERD activity on ER within the tumor cells, but which also exert favorable activities in tumor-associated cells that contribute to the biology of tumors.
Donald P. McDonnell
Employment: Duke University
Stock and Other Ownership Interests: Zentalis, G1 Therapeutics, Viba Therapeutics, Rappta Therapeutics, X-RAD Therapeutics
Honoraria: Novartis
Consulting or Advisory Role: Zentalis, G1 therapeutics, Bristol-Myers Squibb, Rappta Therapeutics
Research Funding: Bristol-Myers Squibb, Novartis, Zentalis
Patents, Royalties, Other Intellectual Property: Inventor on two patents (assigned to Duke) licensed to Radius Health covering the use of Rad1901 for Breast cancer. I am an inventor on two patents (assigned to Duke) that covers the use of lasofoxifene for ESR1-mutant breast cancers. Licensed to Sermonix
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Suzanne E. Wardell
Consulting or Advisory Role: Zentalis
Research Funding: Zentalis, Bristol-Myers Squibb
Patents, Royalties, Other Intellectual Property: I am listed as an inventor on a patent for the use of RAD1901 in metastatic breast cancer
Ching-Yi Chang
Research Funding: Novartis Institutes for BioMedical Research, Bristol-Myers Squibb
Patents, Royalties, Other Intellectual Property: Sermonix. Patent application for the use of lasofoxifene as treatment for breast cancer
John D. Norris
Consulting or Advisory Role: G1 Pharmaceuticals, Celgene
Research Funding: G1 Therapeutics, Celgene
No other potential conflicts of interest were reported.
See accompanying article on page 1360
SUPPORT
Some of the work described in this manuscript was supported by a DOD Innovator grant, BC170954.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: Donald P. McDonnell
Data analysis and interpretation: Donald P. McDonnell
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Next-Generation Endocrine Therapies for Breast Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Donald P. McDonnell
Employment: Duke University
Stock and Other Ownership Interests: Zentalis, G1 Therapeutics, Viba Therapeutics, Rappta Therapeutics, X-RAD Therapeutics
Honoraria: Novartis
Consulting or Advisory Role: Zentalis, G1 therapeutics, Bristol-Myers Squibb, Rappta Therapeutics
Research Funding: Bristol-Myers Squibb, Novartis, Zentalis
Patents, Royalties, Other Intellectual Property: Inventor on two patents (assigned to Duke) licensed to Radius Health covering the use of Rad1901 for Breast cancer. I am an inventor on two patents (assigned to Duke) that covers the use of lasofoxifene for ESR1-mutant breast cancers. Licensed to Sermonix
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Suzanne E. Wardell
Consulting or Advisory Role: Zentalis
Research Funding: Zentalis, Bristol-Myers Squibb
Patents, Royalties, Other Intellectual Property: I am listed as an inventor on a patent for the use of RAD1901 in metastatic breast cancer
Ching-Yi Chang
Research Funding: Novartis Institutes for BioMedical Research, Bristol-Myers Squibb
Patents, Royalties, Other Intellectual Property: Sermonix. Patent application for the use of lasofoxifene as treatment for breast cancer
John D. Norris
Consulting or Advisory Role: G1 Pharmaceuticals, Celgene
Research Funding: G1 Therapeutics, Celgene
No other potential conflicts of interest were reported.
REFERENCES
- 1.Beatson G.On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treatment with illustrative cases Lancet 2162–1651896 [PMC free article] [PubMed] [Google Scholar]
- 2.Boyd S.On oophorectomy in the treatment of cancer Br Med J 2890–8961987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glascock RF, Hoekstra WG.Selective accumulations of tritium-labelled hexoestrol by the reproductive organs of immature female goats and sheep Biochem J 72673–6821959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen EV, Jacobson HI.Basic guides to the mechanism of estrogen action Rec Prog Horm Res 18387–4141962 [Google Scholar]
- 5.Toft D, Gorski J.A receptor molecule for estrogens: Isolation from the rat uterus and preliminary characterization Proc Natl Acad Sci U S A 551574–15811966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shyamala G, Gorski J.Estrogen receptors in the rat uterus: Studies on the interaction of cytosol and nuclear binding sites J Biol Chem 2441097–11031968 [PubMed] [Google Scholar]
- 7.Harper MJK, Walpole AL.A new derivative of triphenylethylene: Effect on implantation and mode of action in rats J Reprod Fert 13101–1191967 [DOI] [PubMed] [Google Scholar]
- 8.Jordan VC, Collins MM, Rowsby L, et al. A mono-hydroxylated metabolite of tamoxifen with potent antioestrogen activity J Endocrinol 75305–3161977 [DOI] [PubMed] [Google Scholar]
- 9.Jordan VC, Koerner S.Tamoxifen (ICI 46,474) and the human carcinoma 8S oestrogen receptor Eur J Cancer 11205–2061975 [DOI] [PubMed] [Google Scholar]
- 10.Furr BJ, Jordan VC.The pharmacology and clinical uses of tamoxifen Pharmacol Ther 25127–2051984 [DOI] [PubMed] [Google Scholar]
- 11.Colletta AA, Wakefield LM, Howell FV, et al. The growth inhibition of human breast cancer cells by a novel synthetic progestin involves the induction of transforming growth factor beta J Clin Invest 87277–2831991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cole MP, Jones CT, Todd ID.A new anti-oestrogenic agent in late breast cancer: An early clinical appraisal of ICI46474 Br J Cancer 25270–2751971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson ER, Clyne C, Rubin G, et al. Aromatase—A brief overview Annu Rev Physiol 6493–1272002 [DOI] [PubMed] [Google Scholar]
- 14.Simpson ER, Davis SR.Aromatase and the regulation of estrogen biosynthesis—Some new perspectives Endocrinology 1424589–45942001 [DOI] [PubMed] [Google Scholar]
- 15.Plotkin D, Lechner JJ, Jung WE, et al. Tamoxifen flare in advanced breast cancer JAMA 2402644–26461978 [PubMed] [Google Scholar]
- 16.Gottardis MM, Jordan VC.Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration Cancer Res 485183–51871988 [PubMed] [Google Scholar]
- 17.Canney PA, Griffiths T, Latief LN, et al. Clinical significance of tamoxifen withdrawal response. Lancet. 1987;1:36. doi: 10.1016/s0140-6736(87)90717-3. [DOI] [PubMed] [Google Scholar]
- 18.Grese TA, Sluka JP, Bryant HU, et al. Molecular determinants of tissue selectivity in estrogen receptor modulators Proc Natl Acad Sci U S A 9414105–141101997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonnell DP, Wardell SE.The molecular mechanisms underlying the pharmacological actions of ER modulators: Implications for new drug discovery in breast cancer Curr Opin Pharmacol 10620–6282010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDonnell DP, Wardell SE, Norris JD.Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer J Med Chem 584883–48872015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norris JD, Paige LA, Christensen DJ, et al. Peptide antagonists of the human estrogen receptor Science 285744–7461999 [DOI] [PubMed] [Google Scholar]
- 22.McDonnell DP, Clemm DL, Hermann T, et al. Analysis of estrogen receptor function in vitro reveals three distinct classes of antiestrogens Mol Endocrinol 9659–6691995 [DOI] [PubMed] [Google Scholar]
- 23.Wijayaratne AL, Nagel SC, Paige LA, et al. Comparative analyses of the mechanistic differences among antiestrogens Endocrinology 1405828–58401999 [DOI] [PubMed] [Google Scholar]
- 24.Termine JD, Wong M.Post-menopausal women and osteoporosis: Available choices for maintenance of skeletal health Maturitas 30241–2451998 [DOI] [PubMed] [Google Scholar]
- 25.Ke HZ, Qi H, Crawford DT, et al. Lasofoxifene (CP-336,156), a selective estrogen receptor modulator, prevents bone loss induced by aging and orchidectomy in the adult rat Endocrinology 1411338–13442000 [DOI] [PubMed] [Google Scholar]
- 26.LaCroix AZ, Powles T, Kent Osborne C, et al. Breast cancer incidence in the randomized PEARL trial of lasofoxifene in postmenopausal osteoporotic women J Natl Cancer Inst 1021706–17152010 [DOI] [PubMed] [Google Scholar]
- 27.Cotreau MM, Stonis L, Dykstra KH, et al. Multiple-dose, safety, pharmacokinetics, and pharmacodynamics of a new selective estrogen receptor modulator, ERA-923, in healthly postmenopausal women J Clin Pharmacol 42157–1652002 [DOI] [PubMed] [Google Scholar]
- 28.Komm BS, Kharode YP, Bodine PVN, et al. Bazedoxifene acetate: A selective estrogen receptor modulator with improved selectivity Endocrinology 1463999–40082005 [DOI] [PubMed] [Google Scholar]
- 29.Puyang X, Furman C, Zheng GZ, et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERalpha(WT) and ERalpha(MUT) breast cancer Cancer Discov 81176–11932018 [DOI] [PubMed] [Google Scholar]
- 30.Johnston SR.Endocrinology and hormone therapy in breast cancer: Selective oestrogen receptor modulators and downregulators for breast cancer—Have they lost their way? Breast Cancer Res 7119–1302005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osborne CK, Bardou V, Hopp TA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer J Natl Cancer Inst 95353–3612003 [DOI] [PubMed] [Google Scholar]
- 32.Masamura S, Santner SJ, Heitjan DF, et al. Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells J Clin Endocrinol Metab 802918–29251995 [DOI] [PubMed] [Google Scholar]
- 33.Santen R, Jeng MH, Wang JP, et al. Adaptive hypersensitivity to estradiol: Potential mechanism for secondary hormonal responses in breast cancer patients J Steroid Biochem Mol Biol 79115–1252001 [DOI] [PubMed] [Google Scholar]
- 34.Power RF, Mani SK, Codina J, et al. Dopaminergic and ligand-independent activation of steroid hormone receptors Science 2541636–16391991 [DOI] [PubMed] [Google Scholar]
- 35.Wu RC, Smith CL, O'Malley BW.Transcriptional regulation by steroid receptor coactivator phosphorylation Endocr Rev 26393–3992005 [DOI] [PubMed] [Google Scholar]
- 36.Lee ES, Schafer JM, Yao K, et al. Cross-resistance of triphenylethylene-type antiestrogens but not ICI 182,780 in tamoxifen-stimulated breast tumors grown in athymic mice Clin Cancer Res 64893–48992000 [PubMed] [Google Scholar]
- 37.DeFriend DJ, Howell A, Nicholson RI, et al. Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer Cancer Res 54408–4141994 [PubMed] [Google Scholar]
- 38.Robertson JF, Llombart-Cussac A, Rolski J, et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: Results from the FIRST study J Clin Oncol 274530–45352009 [DOI] [PubMed] [Google Scholar]
- 39.Robertson JF, Semiglazov V, Nemsadze G.Effects of fulvestrant 250 mg in premenopausal women with oestrogen receptor-positive primary breast cancer Eur J Cancer 4364–702007 [DOI] [PubMed] [Google Scholar]
- 40.Robertson JF, Steger GG, Neven P, et al. Activity of fulvestrant in HER2-overexpressing advanced breast cancer Ann Oncol 211246–12532010 [DOI] [PubMed] [Google Scholar]
- 41.Connor CE, Norris JD, Broadwater G, et al. Circumventing tamoxifen resistance in breast cancers using antiestrogens that induce unique conformational changes in the estrogen receptor Cancer Res 612917–29222001 [PubMed] [Google Scholar]
- 42.Bardia A, Kaklamani V, Wilks S, et al. Phase I study of elacestrant (RAD1901), a novel selective estrogen receptor degrader, in ER-positive, HER2-negative advanced breast cancer J Clin Oncol39:1360-1370, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wardell SE, Nelson ER, Chao CA, et al. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader Endocr Relat Cancer 22713–7242015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel HK, Tao N, Lee KM, et al. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. 2019;21:146. doi: 10.1186/s13058-019-1230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeselsohn R, De Angelis C, Brown M, et al. The evolving role of the estrogen receptor mutations in endocrine therapy-resistant breast cancer. Curr Oncol Rep. 2017;19:35. doi: 10.1007/s11912-017-0591-8. [DOI] [PubMed] [Google Scholar]
- 46.Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer Nat Genet 451439–14452013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaillard SL, Andreano KJ, Gay LM, et al. Constitutively active ESR1 mutations in gynecologic malignancies and clinical response to estrogen-receptor directed therapies Gynecol Oncol 154199–2062019 [DOI] [PubMed] [Google Scholar]
- 48.Jeselsohn R, Bergholz JS, Pun M, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations Cancer Cell 33173–186.e52018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andreano KJ, Baker JG, Park S, et al. The dysregulated pharmacology of clinically relevant ESR1 mutants is normalized by ligand-activated WT receptor Mol Cancer Ther 191395–14052020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel HK, Bihani T.Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment Pharmacol Ther 1861–242017 [DOI] [PubMed] [Google Scholar]
- 51.Clark AS, Karasic TB, DeMichele A, et al. Palbociclib (PD0332991)-a selective and potent cyclin-dependent kinase inhibitor: A review of pharmacodynamics and clinical development JAMA Oncol 2253–2602016 [DOI] [PubMed] [Google Scholar]
- 52.Andre F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer N Engl J Med 3801929–19402019 [DOI] [PubMed] [Google Scholar]
- 53.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer N Engl J Med 366520–5292012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wright LE, Guise TA.The microenvironment matters: Estrogen deficiency fuels cancer bone metastases Clin Cancer Res 202817–28192014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304:1621. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- 56.Peethambaram PP, Ingle JN, Suman VJ, et al. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer: An updated analysis Breast Cancer Res Treat 54117–1221999 [DOI] [PubMed] [Google Scholar]
- 57.Chlebowski RT, Aragaki AK, Anderson GL.Menopausal hormone therapy influence on breast cancer outcomes in the Women's Health Initiative J Natl Compr Canc Netw 13917–9242015 [DOI] [PubMed] [Google Scholar]
- 58.Manson JE, Aragaki AK, Rossouw JE, et al. Menopausal hormone therapy and long-term all-cause and Cause Specific mortality: The Women's Health Initiative randomized trials JAMA 318927–9382017 [DOI] [PMC free article] [PubMed] [Google Scholar]