

INTRODUCTION
In 2002, we published a review in Journal of Clinical Oncology examining the potential of KIT inhibition to treat advanced cancer.1 At that time, imatinib was the first and only US Food and Drug Administration–approved kinase inhibitor and there were many unanswered questions (Table 1). Now, nearly 20 years later, there are five FDA-approved KIT-targeted kinase inhibitors, including imatinib. We have learned a great deal more about KIT and how mutations affect its function. We have also elucidated how specific secondary KIT mutations confer drug resistance in patients. This review will explore the journey of therapeutic KIT targeting that began with imatinib 20 years ago. We will discuss the basic research and clinical findings that informed the development of additional KIT inhibitors and how they were successfully integrated into patient care to combat resistance. We will make comparisons between the two malignancies that have been most significantly affected by KIT-targeted therapies, GI stromal tumor (GIST) and mast cell malignancies, to highlight the importance of genetic profiling in informing treatment success. Furthermore, we will discuss the lessons learned through the development of the five FDA-approved KIT targeted therapies (Table 2) and predictions for the future of the field.
TABLE 1.
Outstanding Questions 2000 Versus 2020

TABLE 2.
Summary of FDA-Approved KIT-Targeted Therapies

CONTEXT
Key Objective
Imatinib, the first kinase inhibitor for cancer treatment, was developed over 20 years ago. In that time, imatinib as a KIT-targeted therapy revolutionized the treatment of patients with KIT-driven malignancies, primarily GI stromal tumor and systemic mastocytosis, and led to the development of additional KIT inhibitors that have significantly improved patient outcomes. We explore the history of KIT-targeted therapies beginning with imatinib.
Knowledge Generated
Numerous lessons have been learned from the initial preclinical and clinical studies with imatinib and other KIT inhibitors. The clinical use of imatinib has also provided the basis to understand the molecular properties of KIT and its interactions with drugs, allowing for rational design of more successful KIT inhibitors.
Relevance
The development of imatinib, as well as later-line KIT-targeted kinase inhibitors, has transformed the way we treat GI stromal tumors and mast cell malignancies. Further understanding of KIT biology and resistance mechanisms will further inform and refine our treatment of KIT-driven diseases.
HISTORICAL DEVELOPMENTS
Precision medicine, or targeted therapy, as a paradigm for cancer treatment is a relatively recent development. It began with the discovery that recurrent activating mutations in oncogenes can drive cancer development and sustain cell proliferation and survival. Chemical compounds were identified that have the ability to block the activity of mutant enzymes and thus reduce tumor viability, revolutionizing cancer treatment. The first, and most significant, of these compounds was imatinib (originally known as CGP57418B or STI-571), an inhibitor of BCR-ABL1, the oncogenic driver in chronic myeloid leukemia (CML).2 After extensive testing in preclinical models, imatinib demonstrated both safety and efficacy in CML, moving quickly from phase I through III, as it surpassed the standard of care (interferon plus cytarabine).3-5 Based on these studies, in 2001, imatinib set a new record for fastest FDA approval (Fig 1).
FIG 1.

Significant milestones in KIT-targeted treatment (1990-2020). Events shown chronologically from top to bottom. Events relevant to mast cell disease shown on the left and those for GIST on the right. AL, activation loop; FDA, US Food and Drug Administration; GIST, GI stromal tumor; ISM, indolent systematic mastocytosis; SM, systemic mastocytosis.
As the first successful targeted cancer therapy, imatinib opened the door for a new way of thinking about the treatment of cancers driven by distinct oncogenic mutations. At that time, evidence was building that KIT serves as an oncogene in several cancers and was therefore a logical therapeutic target. KIT, a member of the type III receptor tyrosine kinase (RTK) family, was originally discovered through the viral oncogene v-kit, encoded by the Harvey-Zuckerman-4 strain of the feline sarcoma virus.6-8 Activating human KIT mutations were first identified in systemic mastocytosis (SM) cell lines and in humans with SM9,10 (Fig 1). A few years later, Hirota et al reported activating KIT mutations in GI mesenchymal tumors that came to be known as GIST. Unlike BCR-ABL1, which is a fusion gene product, mutations in KIT were point mutations or small indels distributed throughout the kinase domain, and the location of mutations differed by tumor type.
From these early studies, it was apparent that KIT mutations are important in both SM and GIST, but there are important differences. It is now appreciated that > 80% of SM cases in adults have KIT mutations with the majority occurring in the activation loop (AL, KIT exon 17), the dominant being D816V (Fig 2). Rarely adult SM patients have mutations affecting the KIT extracellular (encoded by KIT exons 8 and 9), transmembrane (KIT exon 10), or juxtamembrane (JM, KIT exon 11) domains. Interestingly, these mutations are much more frequent in pediatric SM cases, of which 75% are KIT mutant.11,12 In contrast, more than 70% of mutations in GIST involve the JM domain.13-15 Mutations of the KIT extracellular domain and kinase domain (KIT exons 13 or 17) are found in a minority of GIST.14,16,17 Notably, the D816V mutation typical of adult mast cell neoplasms has not been observed as a primary mutation in GIST, but other AL (KIT exon 17) mutations do rarely occur (Fig 2).14,15,18
FIG 2.

Frequency of activating primary KIT mutations observed in KIT-mutant GIST and mast cell malignancies. Relative frequency of KIT mutations seen in KIT-mutant cases only. KIT mutations are seen in > 80% of adult mast cell malignancies, 75% of pediatric mast cell malignancies, and 75%-80% of GIST; EC, TM, JM, BP, and AL. Dotted shading indicates mutations in tumors that typically respond to imatinib treatment. AL, activation loop; BP, binding pocket; EC, extracellular; GIST, GI stromal tumor; JM, juxtamembrane; TM, transmembrane.
The clinical success of imatinib in CML and the growing interest in targeting KIT led to efforts to identify a KIT inhibitor suitable for clinical testing. Originally, KIT was not an identified target of imatinib, but based on its activity against the homologous PDGFRA and PDGFRB RTKs, the ability of imatinib to inhibit ligand-activated KIT was investigated. This was the first example of drug repurposing to inhibit a target other than that for which it was originally designed. Imatinib was found to inhibit not only wild-type KIT with similar potency to the BCR-ABL1 and PDGFRA/B but also potently the KIT JM–mutant kinase activity, like those most commonly described in GIST.19,20 No activity, however, was seen against KIT with compound JM + D816V mutations for reasons that were unknown at the time.21,22 Further preclinical testing demonstrated that imatinib decreased proliferation and survival in the first KIT-mutant GIST cell line, indicating the oncogenic dependence of GIST cells on the kinase activity of mutated KIT.23
As the previous century ended, the stage was set for testing imatinib in GIST and mast cell neoplasms. The available preclinical data predicted that imatinib might be effective for treating GIST, which typically expresses imatinib-sensitive KIT JM mutations, but was unlikely to be effective against the majority of mast cell neoplasms that express the imatinib-resistant KIT D816V. In retrospect, these preclinical results not only were excellent predictors of the primary response outcomes in human studies of imatinib but also predicted how compound mutations in different parts of the kinase domain differentially affect kinase conformation and thus drug binding. As discussed below, the limitations of imatinib drove serial efforts to produce more potent KIT inhibitors for patients with GIST and SM.
IMATINIB, THE FIRST CLINICALLY EFFECTIVE KIT INHIBITOR
GIST Clinical Studies
The first clinical success with imatinib in GIST was seen in the treatment of a single patient with heavily chemotherapy pretreated, KIT JM–mutant GIST.24 This small proof-of-concept trial was initiated amid concerns over drug absorption in patients with GIST, many of whom had undergone resection of the gut (stomach, small bowel, or colon), as well as drug metabolism, since these patients often had liver metastases and/or prior hepatic surgery.25-27 Despite these concerns, the first patient's tumor showed a complete metabolic response and a 52% decrease in size after one month. Imatinib was well-tolerated, and the side effect profile was consistent with those reported in CML.24 Based on these results, two randomized phase I-II studies were opened in the United States and Europe in 2000.28-30 In both studies, the objective response rate (ORR) was approximately 67% and no new imatinib safety signals were noted.27,31 Two large randomized phase III studies confirmed the efficacy of imatinib in GIST, with an ORR of approximately 50%, a median progression-free survival (PFS) of around 2 years, and a median overall survival (OS) of 4-5 years.32-35 More recently, long-term follow-up of these phase III studies has estimated 10-year PFS and OS rates of 8% and 20%, respectively.36,37 Imatinib was granted FDA approval for treatment of unresectable, recurrent, and/or metastatic GIST in February 2002, just nine months after its initial approval for CML38 (Fig 1). Additional accelerated and subsequently regular approvals for the adjuvant treatment of patients following complete gross resection of GIST were granted in 2008 and 2012, respectively.39
Mastocytosis
Although the majority of KIT-mutant GIST have imatinib-sensitive mutations, the converse is true in SM. The D816V mutation is resistant, and only a small number of other KIT mutations are imatinib sensitive (Fig 2).20,22,40-43 These preclinical observations were confirmed in the initial clinical studies in mastocytosis, where the majority of patients who had meaningful responses to imatinib were those with co-existent eosinophilia. Further analysis of these exceptional responders, as well as patients with hypereosinophilic syndrome without a diagnosis of SM, identified a chromosomal translocation producing the imatinib-sensitive FIP1L1-PDGFRA fusion kinase as the underlying molecular basis for response.44,45 Based on these observations, in 2008, the WHO reclassified these cases as myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, FGFR1, or PCM-JAK2.
In contrast, SM patients with the typical D816V had minimal or no response to imatinib, whereas the rare patients with imatinib-sensitive mutations involving KIT exons 8, 9, 10, or 17 were observed to have very good responses to imatinib.46-50 In addition, some responses were noted in SM patients with no molecular analysis of KIT or PDGFRA mutations. In October 2006, the FDA approved imatinib for treatment of patients with aggressive SM whose disease had no detectable KIT D816V mutation or had an unknown KIT mutational status (Fig 1).51 However, in retrospect, it is likely that many of these cases had imatinib-sensitive mutations that were not identified because of an incomplete spectrum of mutational analysis or insensitive technologies for detecting mutations in a background of normal cells.12,52,53
Lessons Learned From the Initial Experience in Treating KIT-Mutant Malignancies With Imatinib
Despite initial concerns that a potent KIT inhibitor might cause unacceptable short-term or long-term toxicities, especially in terms of myelosuppression, the data from the initial imatinib studies in CML and GIST revealed an acceptable safety profile. Many patients have now been treated for more than a decade or two without any known long-term side effects. This important finding from the early imatinib studies generated interest in developing additional KIT kinase inhibitors.36,54,55
The results from the early imatinib studies in treating GIST and SM demonstrated a strong relationship between the underlying oncogenic mutation and drug response. In GIST, the best outcomes were seen with patients with KIT JM–mutant tumors.56,57 Randomized phase III studies demonstrated that patients with KIT exon 9-mutant GIST respond better to high-dose imatinib (800 mg daily), whereas patients with KIT JM–mutant GIST had similar outcomes with standard (400 mg daily) or high doses.32,58 Additionally, patients with GIST lacking KIT mutations rarely responded to imatinib, but analysis of the few responsive cases revealed imatinib-sensitive PDGFRA mutations as the basis for response.57,59,60 However, the majority of PDGFRA-mutant GIST have an imatinib-resistant mutation, PDGFRA D842V, which is homologous to the KIT D816V mutation seen in SM.59,60 Based on these observations, a molecular classification of GIST was proposed and has been used to guide therapy and also to focus discovery efforts on subsets of GIST lacking any definable oncogenic mutations16,26 After two decades of research, the pathogenic cause of more than 99% cases of GIST can be identified and used to guide therapy.61
As noted above, imatinib was successfully repurposed from the BCR-ABL1 kinase inhibitor development program to target KIT. This paradigm has been extended in other diseases, where a kinase inhibitor used to target one particular kinase can be clinically expanded to homologous oncogenic kinases in the same or different diseases.62,63 Adoption of imatinib as a KIT inhibitor helped lay the foundation for what has become a molecularly focused, histology agnostic approach to drug development.64
When imatinib was identified using high-throughput chemical compound screens, there were no crystal structures for ABL1 or KIT. When Schindler et al65 reported the crystal structure of the ABL1 catalytic domain complexed with an imatinib analog, it was revealed that imatinib bound to the inactive conformation of ABL1 Subsequently, a similar mode of imatinib binding to the inactive KIT structure was reported (Fig 1).66 Thus, imatinib is classified as a type II kinase inhibitor (binds to the inactive structure).67 These results suggested that mutations of the AL that stabilize the active conformation of KIT would result in imatinib resistance, explaining the differential activity of imatinib against the typical GIST-associated KIT JM mutations (inactive conformation favored) versus the typical SM KIT D816V mutation (active conformation strongly favored).68,69 Based on these and other considerations, structural biology-guided drug design became standard practice in drug development programs.67,70-72
Consistent with the observation that imatinib binds to the inactive KIT conformation, secondary mutations involving the KIT AL were discovered to be a common cause of acquired imatinib resistance in GIST. The other major class of secondary resistance mutations involves the KIT ATP (and imatinib) binding pocket (Fig 3).73-78 Largely parallel to conclusions from the analysis of secondary ABL1 mutations in imatinib-resistant CML, these observations supported that KIT-mutant GIST remained strongly dependent upon KIT signaling.79-82 This conclusion led to the hypothesis that imatinib-resistant GIST might be effectively treated using alternative KIT tyrosine kinase inhibitors (TKIs) that could overcome AL or ATP binding pocket mutations.
FIG 3.
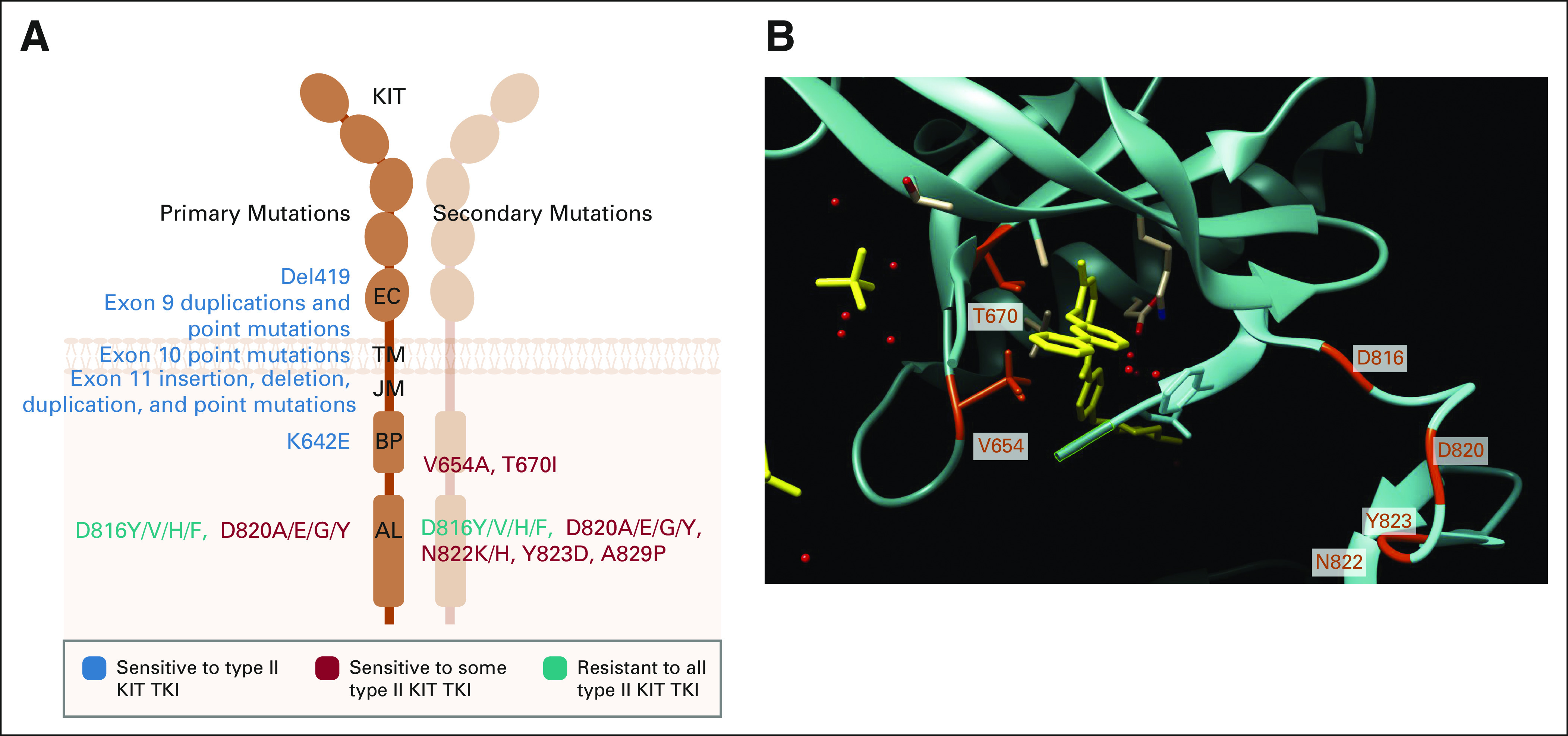
KIT primary mutations are largely sensitive to type II KIT TKIs, whereas KIT secondary mutations confer resistance. (A) Schematic of locations of primary and secondary KIT mutations observed in mast cell malignancies and GIST with their corresponding sensitivity to type II KIT TKIs. Created using BioRender. (B) Three-dimensional model of KIT (blue-green) in an inactive conformation bound to imatinib (yellow). Common secondary resistance mutation sites are highlighted in red-orange (T670 and V654 within the binding pocket) and in cyan (D816, D820, N822, and Y823 within the AL). Molecular graphics and analyses performed using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.132 AL, activation loop; BP, binding pocket; EC, extracellular; GIST, GI stromal tumor; JM, juxtamembrane; TKI, tyrosine kinase inhibitor; TM, transmembrane.
Development of Additional Type II Inhibitors for Imatinib-Resistant GIST
By the time resistance to imatinib was fully appreciated, many other kinase inhibitors had been created. The discovery that drug-resistant, KIT-mutant GIST remained KIT-dependent led to the development of salvage treatments for imatinib-resistant GIST. The first of these to be tested clinically was sunitinib, originally known as SU11248 and developed as an inhibitor of FLT3, which is an RTK closely related to KIT.77,83-85 Promising activity in a phase I-II study led to a randomized, double-blind, placebo-controlled, multicenter, international trial,77,86,87 which led to the 2007 FDA approval of sunitinib for treatment of patients with GIST with disease progression or intolerance to imatinib.88 A number of other repurposed KIT inhibitors were tested for treatment of imatinib-resistant GIST, including dasatinib, sorafenib, and nilotinib.89-93 However, neither dasatinib nor sorafenib advanced to phase III studies (because of insufficient activity and competing kinase development programs, respectively). A phase III study to test the superiority of nilotinib versus imatinib for the front-line treatment of GIST was terminated early because of futility.94 In contrast, another multitargeted kinase inhibitor with activity against KIT, regorafenib, was successfully tested in both a phase II and a placebo-controlled, double-blind, multicenter, international phase III study.95,96 Despite the lack of a survival benefit in this or in the sunitinib study, real-world data from a large patient-reported registry strongly suggest that the availability of additional lines of therapy after front-line imatinib has improved OS for patients with advanced GIST during the 2000-2020 time period.97
The results of these studies showed the limitations of serial treatment with type II inhibitors, as the ORR decreased from around 50% with front-line imatinib to only 4.5% with third-line regorafenib. There was a corresponding decrease in median PFS, dropping from approximately 20-22 months with imatinib to 4.8 months with regorafenib.26,34,35,37,87,96 These results are explained by in vitro studies showing that sunitinib is active against all secondary KIT ATP-binding pocket mutations (V654A and T670I), but has minimal activity against AL mutations (D816V, D820A, etc).77,98 In contrast, regorafenib has limited activity against secondary KIT mutations located in the ATP binding pocket, but clinically useful activity against (some but not all) AL mutations.98 Given the widespread heterogeneity of clones with different secondary mutations between lesions and within a given lesion,99 it is likely that we are nearing the limits of what conventional type II inhibitors can deliver in the setting of advanced, drug-resistant GIST (Fig 3).
Attempts to Develop Type I KIT Inhibitors to Treat GIST
Based on the above observations, several type I inhibitors that target the active conformation of KIT have been tested for treatment of advanced GIST. Ponatinib, a TKI approved for treatment of CML and acute lymphoblastic leukemia, showed promising activity in vitro against secondary KIT AL mutations, but failed to demonstrate sufficient activity in a phase II study of drug-resistant GIST.100 The results from this study have not been fully reported, but based on in vitro data, it is likely that ponatinib lacked sufficient potency against the common KIT V654A (ATP binding pocket) secondary mutation to produce meaningful disease control.101
More recently, a rationally designed type I KIT inhibitor, avapritinib (formerly BLU-285), has been clinically developed. This compound was designed by optimizing activity against the prototypical KIT AL mutation, D816V, an approach that differed from testing repurposed compounds against wild-type or KIT JM–mutant kinases. The end result of this screening approach yielded a compound with marked potency against all KIT AL mutations, including KIT D816V.102-104 Avapritinib showed promising activity against drug-resistant KIT-mutant GIST in a phase I study, but overall clinical activity was limited by a lack of potency against KIT ATP binding pocket mutations.105 Recently, the top-line data of a phase III randomized study of avapritinib versus regorafenib for patients with GIST who experience treatment failure during prior imatinib and sunitinib therapy were reported (VOYAGER, ClinicalTrials.gov identifier: NCT03465722). Unfortunately, avapritinib did not confer treatment benefit in terms of median PFS compared with regorafenib. However, it should be noted that avapritinib has strong clinical activity against the PDGFRA D842V mutation, homologous to KIT D816V, which is found as a primary mutation in 7%-10% of all primary GIST.104,106 In January 2020, avapritinib was FDA-approved for treatment of GIST with a PDGFRA exon 18 mutation (Fig 1).107
Development of Type I KIT Inhibitors for Mastocytosis
The primary KIT AL mutation, D816V, in patients with SM presented an urgent clinical challenge that required the development of type I KIT inhibitors from the start. In vitro studies of the type I inhibitor dasatinib demonstrated activity against D816V-mutant KIT.22,41 However, when this agent was tested in a phase II study of patients with indolent or advanced SM,108 the ORR was 33% (11 of 33), and the only two complete responses were in patients whose disease lacked the KIT D816V mutation. The partial responses observed in the other nine patients were due to improved symptoms only, with no objective evidence of decreased neoplastic mast cell numbers using laboratory or pathology testing methods.
Subsequently, several new type I inhibitor therapies for treatment of KIT D816V+ advanced SM have shown substantial clinical activity. Midostaurin (formerly PKC412), a staurosporine derivative, emerged from a chemical screen of protein kinase C inhibitors and was later found to also inhibit VEGFRs, PDGFRs, KIT, and FLT3. Activity of midostaurin against KIT D816V was demonstrated in several preclinical models including cell lines and malignant mast cells isolated from patients with SM.109-112 Much like during the development of imatinib to treat GIST, these preclinical observations led to the testing of midostaurin in a single patient with mast cell leukemia as part of a compassionate use protocol (Fig 1). This patient had evidence of a partial clinical and molecular response with an 80% decrease in the level of detectable KIT D816V in the peripheral blood.113 Based in part on the results from this single patient treatment protocol, midostaurin was evaluated in two phase II studies, an open-label, international, multisite study and a study that reported patient outcomes with a 10-year median follow-up time.114,115 Overall, midostaurin treatment was found to be well-tolerated and highly effective. The initial response rate of patients with SM to midostaurin was 60%-69%.114,115 The median OS was 28.7 months, and the median PFS was 14.1 months.115 Moreover, with a 10-year follow-up, little toxicity was observed.114 Based on these results, the FDA approved midostaurin for treatment of advanced SM in 2017 (Fig 1).116
The novel type I inhibitor, avapritinib, also demonstrated promising in vitro and clinical activity against D816V-mutant mast cells. In the latest update from a phase I study (EXPLORER, ClinicalTrials.gov identifier: NCT02561988) of avapritinib in advanced SM, the ORR was 75%, with 70% reporting complete or partial responses.117 Notably, avapritinib induced responses in patients regardless of prior midostaurin therapy, and these responses were rapid and long-lasting. In the phase I EXPLORER study, 25% of patients achieved a complete molecular response of the KIT D816V mutation using digital droplet PCR (sensitivity approximately 0.17%), a new response benchmark in the disease.117,118 Avapritinib is undergoing further testing in a multicenter phase II study (PATHFINDER, ClinicalTrials.gov identifier: NCT03580655). Based on its activity in advanced SM and favorable side effect profile, avapritinib is also being examined in patients with indolent or smoldering SM whose symptoms are inadequately controlled by standard therapy (PIONEER, ClinicalTrials.gov identifier: NCT03731260).119 This study includes a randomized, double-blind, placebo-controlled component. The clinical evidence to date suggests that avapritinib has a high potential to be approved as an additional therapy for SM, potentially for both advanced and indolent SM.
To date, clinical mechanisms of resistance to midostaurin or avapritinib in SM are not well-understood. In vitro studies of these agents have suggested that the previously described KIT V654A or T670I secondary mutations may result in midostaurin or avapritinib resistance.103,120 Increased variant allele frequency of non-KIT mutations such as K/NRAS, RUNX1, IDH2, or NPM1 has also been associated with clinical resistance to midostaurin.121 In addition to the problems with emergence of drug-resistant mastocytosis clones, there still remains the challenge of how best to treat patients who have SM with an associated hematological neoplasm.122
Ripretinib, the Most Recently Approved KIT Inhibitor
Ripretinib (formerly DCC-2618) emerged from a program to develop novel inhibitors that bind to the switch control region of kinases, rather than the ATP-binding pocket.123-126 This discovery program used the known KIT structure to develop compounds that bind to the kinase switch pocket, therefore preventing the AL access to this region and thereby locking the kinase into the inactive state. In addition, ripretinib binds to the KIT AL to further secure it in the inactive state. Unlike all of the previously discussed inhibitors, ripretinib is not a competitive ATP inhibitor and thereby retains potency, even in the presence of physiological levels of ATP.127 In preclinical studies, ripretinib had excellent potency against all tested KIT AL mutations and was also active, although less so, against KIT ATP–binding pocket mutations.127
Ripretinib was initially tested in a phase I study (ClinicalTrials.gov identifier: NCT02571036) that included patients with both GIST and advanced SM in which the recommended to phase II dose of 150 mg once daily was determined.128 This novel inhibitor had a favorable safety and tolerability profile and was active in patients with GIST whose tumors were refractory to multiple previous TKIs (data about efficacy in SM are not yet reported). Ripretinib was further evaluated in a double-blind, randomized, placebo-controlled study (INVICTUS, ClinicalTrials.gov identifier: NCT03353753) of adult patients with GIST who had progression or intolerance during prior therapies, which included, at a minimum, imatinib, sunitinib, and regorafenib. Ripretinib was associated with 85% reduction in the risk of death or progression when compared with placebo and was associated with an acceptable safety profile.129 Based on these results, the FDA approved ripretinib in May 2020 for the treatment of adult patients with advanced GIST who had received prior treatment with three or more kinase inhibitors, including imatinib.130 Currently, the activity of ripretinib to treat patients earlier in their disease course is being tested in a global, randomized, open-label, phase III study comparing the safety and efficacy of ripretinib versus sunitinib in patients with advanced GIST following imatinib (INTRIGUE, ClinicalTrials.gov identifier: NCT03673501). The primary end point is PFS, and key secondary objectives include ORR and OS. Despite the impact of the COVID-19 pandemic, it is anticipated that accrual to this study will be completed in 2021.131
In conclusion, it has been two decades since the first KIT inhibitor was approved for treatment of a KIT-mutant disease. The very first kinase inhibitor approved as a cancer therapy, imatinib, has provided immense insights into how to manage the treatment of KIT-mutant neoplasms. At the time of its approval, we had little knowledge of either primary or secondary resistance mechanisms. We now understand the importance of molecular profiling of tumors to predict drug response as specific mutations, but not necessarily KIT overexpression or autocrine signaling, can confer sensitivity or resistance to KIT inhibitors.
In GIST, imatinib is still the first-line therapy for KIT-mutant patients, the majority of which present with imatinib-sensitive mutations (encoded in KIT exons 8, 9, 11, and 13). Secondary resistance because of intra-allelic KIT mutations emerging during imatinib treatment required the application of new drugs for second- and third-line treatment to combat imatinib resistance. Contrarily, the driving KIT mutation observed in the majority of SM, D816V, confers primary resistance to type II inhibitors like imatinib. For this reason, imatinib is not part of the treatment regimen for this form of SM, but other KIT inhibitors have been developed for this disease, such as midostaurin and avapritinib.
Early on, drug repurposing was the main approach to drug discovery. This approach led to the approval of three additional KIT inhibitors (sunitinib, regorafenib, and midostaurin) to treat GIST and SM. However, greater understanding of primary and secondary KIT mutations inspired more sophisticated approaches to rationally design KIT inhibitors with greater potency. Ripretinib and avapritinib are two such inhibitors that emerged from these KIT-focused drug development programs. There are now five FDA-approved inhibitors to treat KIT-mutant disease, with another, avapritinib, likely to be approved in the near future for KIT D816V+ SM (Table 2).
The overarching lesson learned from imatinib, beginning with BCR-ABL1 and translated to KIT, is that a detailed understanding of both the target and its mechanisms of drug escape is necessary to further advance the field. The introduction of each subsequent KIT inhibitor, first preclinically and then in clinical trials, provided further insight into how drug development should proceed. Preclinical studies have shown that the type I TKIs, avapritinib and midostaurin, will be thwarted by secondary KIT mutations just like imatinib, and this has been seen in early clinical data from patients treated with avapritinib, as discussed above. It is not yet clear if this will be the case with ripretinib, but it is likely, given that the PFS with this agent is only slightly more than 6 months. Thus, two decades on, the lessons from imatinib, the first kinase inhibitor, continue to be carried forward in the ongoing battle against kinase inhibitor resistance, leaving new outstanding questions for basic and clinical researchers to answer (Table 1).
Christopher L. Corless
Employment: Omics Data Automation
Leadership: Omics Data Automation
Stock and Other Ownership Interests: Guardant Health, Omics Data Automation
Honoraria: Roche
Consulting or Advisory Role: Roche, Thermo Fisher Scientific Biomarkers, Cepheid, Amgen
Research Funding: Aileron Therapeutics, Arvinas
Travel, Accommodations, Expenses: Roche, Thermo Fisher Scientific, Cepheid
Open Payments Link: https://openpaymentsdata.cms.gov/physician/1233373
Michael C. Heinrich
Stock and Other Ownership Interests: MolecularMD
Honoraria: Novartis
Consulting or Advisory Role: MolecularMD, Novartis, Blueprint Medicines, Deciphera
Patents, Royalties, Other Intellectual Property: Patent on treatment of GIST-licensed to Novartis
Expert Testimony: Novartis
No other potential conflicts of interest were reported.
AUTHOR CONTRIBUTIONS
Conception and design: Michael C. Heinrich
Financial support: Michael C. Heinrich
Administrative support: Christopher L. Corless, Michael C. Heinrich
Collection and assembly of data: Lillian R. Klug, Michael C. Heinrich
Data analysis and interpretation: Christopher L. Corless
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Inhibition of KIT Tyrosine Kinase Activity: Two Decades After the First Approval
The following represents disclosure information provided by the authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Christopher L. Corless
Employment: Omics Data Automation
Leadership: Omics Data Automation
Stock and Other Ownership Interests: Guardant Health, Omics Data Automation
Honoraria: Roche
Consulting or Advisory Role: Roche, Thermo Fisher Scientific Biomarkers, Cepheid, Amgen
Research Funding: Aileron Therapeutics, Arvinas
Travel, Accommodations, Expenses: Roche, Thermo Fisher Scientific, Cepheid
Open Payments Link: https://openpaymentsdata.cms.gov/physician/1233373
Michael C. Heinrich
Stock and Other Ownership Interests: MolecularMD
Honoraria: Novartis
Consulting or Advisory Role: MolecularMD, Novartis, Blueprint Medicines, Deciphera
Patents, Royalties, Other Intellectual Property: Patent on treatment of GIST-licensed to Novartis
Expert Testimony: Novartis
No other potential conflicts of interest were reported.
REFERENCES
- 1.Heinrich MC, Blanke CD, Druker BJ, et al. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies J Clin Oncol 201692–17032002 [DOI] [PubMed] [Google Scholar]
- 2.Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative Cancer Res 56100–1041996 [PubMed] [Google Scholar]
- 3.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia N Engl J Med 3441031–10372001 [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome N Engl J Med 3441038–10422001 [DOI] [PubMed] [Google Scholar]
- 5.O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia N Engl J Med 348994–10042003 [DOI] [PubMed] [Google Scholar]
- 6.Besmer P, Murphy JE, George PC, et al. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family Nature 320415–4211986 [DOI] [PubMed] [Google Scholar]
- 7.Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand EMBO J 63341–33511987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson DM, Lyman SD, Baird A, et al. Molecular cloning of mast cell growth factor, a hematopoietin that is active in both membrane bound and soluble forms Cell 63235–2431990 [DOI] [PubMed] [Google Scholar]
- 9.Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product J Clin Invest 921736–17441993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder Proc Natl Acad Sci USA 9210560–105641995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kristensen T, Vestergaard H, Moller MB.Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay J Mol Diagn 13180–1882011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arock M, Sotlar K, Akin C, et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis Leukemia 291223–12322015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors Science 279577–5801998 [DOI] [PubMed] [Google Scholar]
- 14.Corless CL, Barnett CM, Heinrich MC.Gastrointestinal stromal tumours: Origin and molecular oncology Nat Rev Cancer 11865–8782011 [DOI] [PubMed] [Google Scholar]
- 15.Barnett CM, Corless CL, Heinrich MC.Gastrointestinal stromal tumors: Molecular markers and genetic subtypes Hematol Oncol Clin North Am 27871–8882013 [DOI] [PubMed] [Google Scholar]
- 16.Corless CL, Fletcher JA, Heinrich MC.Biology of gastrointestinal stromal tumors J Clin Oncol 223813–38252004 [DOI] [PubMed] [Google Scholar]
- 17.Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors Am J Pathol 156791–7952000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Longley BJ, Jr, Metcalfe DD, Tharp M, et al. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis Proc Natl Acad Sci USA 961609–16141999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors J Pharmacol Exp Ther 295139–1452000 [PubMed] [Google Scholar]
- 20.Heinrich MC, Griffith DJ, Druker BJ, et al. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor Blood 96925–9322000 [PubMed] [Google Scholar]
- 21.Heinrich MC, Wait CL, Yee KWH, et al. STI571 inhibits the kinase activity of wild type and juxtamembrane c-kit mutants but not the exon 17 D816V mutation associated with mastocytosis. Blood. 2000;96:173b. [Google Scholar]
- 22.Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies Cancer Res 66473–4812006 [DOI] [PubMed] [Google Scholar]
- 23.Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: Biological and clinical implications Oncogene 205054–50582001 [DOI] [PubMed] [Google Scholar]
- 24.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor N Engl J Med 3441052–10562001 [DOI] [PubMed] [Google Scholar]
- 25.Dematteo RP, Heinrich MC, El-Rifai WM, et al. Clinical management of gastrointestinal stromal tumors: Before and after STI-571 Hum Pathol 33466–4772002 [DOI] [PubMed] [Google Scholar]
- 26.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors N Engl J Med 347472–4802002 [DOI] [PubMed] [Google Scholar]
- 27.Van Oosterom AT, Judson I, Verweij J, et al. STI571, an active drug in metastatic gastrointestinal stromal tumors (GIST) an EORTC phase I study. Proc Am Soc Clin Oncol. 2001;20:1a. [Google Scholar]
- 28.Demetri G, von Mehren M, Blanke CD, et al. Antitumor effects of an oral selective tyrosine kinase inhibitor, imatinib mesylate, in patients with advanced gastrointestinal stromal tumors N Engl J Med 347472–4802002 [DOI] [PubMed] [Google Scholar]
- 29.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT J Clin Oncol 26620–6252008 [DOI] [PubMed] [Google Scholar]
- 30.Van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: A phase I study Lancet 3581421–14232001 [DOI] [PubMed] [Google Scholar]
- 31.Blanke CD, von Mehren M, Joensuu H, et al. Evaluation of the safety and efficacy of an oral molecularly-targeted therapy, STI571, in patients with unresectable or metastatic gastrointestinal stromal tumors (GISTs) expressing c-kit (CD117) Proc Am Soc Clin Oncol. 2001;20:1a. [Google Scholar]
- 32.Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 study by Cancer and Leukemia Group B and Southwest Oncology Group J Clin Oncol 265360–53672008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033 J Clin Oncol 26626–6322008 [DOI] [PubMed] [Google Scholar]
- 34.Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial Lancet 3641127–11342004 [DOI] [PubMed] [Google Scholar]
- 35.Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1,640 patients J Clin Oncol 281247–12532010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casali PG, Zalcberg J, Le CA, et al. Ten-year progression-free and overall survival in patients with unresectable or metastatic GI stromal tumors: Long-term analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup Phase III Randomized Trial on imatinib at two dose levels J Clin Oncol 351713–17202017 [DOI] [PubMed] [Google Scholar]
- 37.Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: Analysis of phase 3 SWOG Intergroup Trial S0033 JAMA Oncol 3944–9522017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dagher R, Cohen M, Williams G, et al. Approval summary: Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors Clin Cancer Res 83034–30382002 [PubMed] [Google Scholar]
- 39.FDA grants imatinib (Gleevec) full approval for adjuvant treatment of GIST. Oncology. 2012;26:264, 309. [PubMed] [Google Scholar]
- 40.Frost MJ, Ferrao PT, Hughes TP, et al. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant Mol Cancer Ther 11115–11242002 [PubMed] [Google Scholar]
- 41.Shah NP, Lee FY, Luo R, et al. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis Blood 108286–2912006 [DOI] [PubMed] [Google Scholar]
- 42.Akin C, Longley J, Brockow K, et al. Effects of the tyrosine kinase inhibitor STI571 on mutated kit and neoplastic cells. Blood. 2001;96:747a. [Google Scholar]
- 43.Zermati Y, Desepulveda P, Feger F, et al. Effects of the tyrosine kinase inhibitor STI571 on the kinase activity of wild type and various mutated c-kit receptors found in mast cell neoplasms Blood 96310a–311a2001 [DOI] [PubMed] [Google Scholar]
- 44.Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome N Engl J Med 3481201–12142003 [DOI] [PubMed] [Google Scholar]
- 45.Pardanani A, Tefferi A.Imatinib therapy for hypereosinophilic syndrome and eosinophilia-associated myeloproliferative disorders Leuk Res 2847–522004suppl 1 [DOI] [PubMed] [Google Scholar]
- 46.Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis Leuk Res 331481–14842009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akin C, Fumo G, Yavuz AS, et al. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib Blood 1033222–32252004 [DOI] [PubMed] [Google Scholar]
- 48.Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—Iin vitro and in vivo responsiveness to imatinib therapy Leuk Res 30373–3782006 [DOI] [PubMed] [Google Scholar]
- 49.Pardanani A, Elliott M, Reeder T, et al. Imatinib for systemic mast-cell disease Lancet 362535–5362003 [DOI] [PubMed] [Google Scholar]
- 50.Heinrich MC, Joensuu H, Demetri GD, et al. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases Clin Cancer Res 142717–27252008 [DOI] [PubMed] [Google Scholar]
- 51.Ustun C, DeRemer DL, Akin C.Tyrosine kinase inhibitors in the treatment of systemic mastocytosis Leuk Res 351143–11522011 [DOI] [PubMed] [Google Scholar]
- 52.Corless CL, Harrell P, Lacouture M, et al. Allele-specific polymerase chain reaction for the imatinib-resistant KIT D816V and D816F mutations in mastocytosis and acute myelogenous leukemia J Mol Diagn 8604–6122006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erben P, Schwaab J, Metzgeroth G, et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis Ann Hematol 9381–882014 [DOI] [PubMed] [Google Scholar]
- 54.Druker BJ, Talpaz M, Resta D, et al. Clinical efficacy and safety of an Abl specific tyrosine kinase inhibitor as targeted therapy for chronic myelogenous leukemia. Blood. 1999;94:368a. [Google Scholar]
- 55.Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia N Engl J Med 376917–9272017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group Eur J Cancer 40689–6952004 [DOI] [PubMed] [Google Scholar]
- 57.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor J Clin Oncol 214342–43492003 [DOI] [PubMed] [Google Scholar]
- 58.Debiec-Rychter M, Sciot R, Le CA, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours Eur J Cancer 421093–11032006 [DOI] [PubMed] [Google Scholar]
- 59.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors Science 299708–7102003 [DOI] [PubMed] [Google Scholar]
- 60.Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors Gastroenterology 125660–6672003 [DOI] [PubMed] [Google Scholar]
- 61.Bannon AE, Klug LR, Corless CL, et al. Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors Expert Rev Mol Diagn 17445–4572017 [DOI] [PubMed] [Google Scholar]
- 62.Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors Cancer Discov 5850–8592015 [DOI] [PubMed] [Google Scholar]
- 63.Rodig SJ, Shapiro GI.Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases Curr Opin Investig Drugs 111477–14902010 [PubMed] [Google Scholar]
- 64.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children N Engl J Med 378731–7392018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase Science 2891938–19422000 [DOI] [PubMed] [Google Scholar]
- 66.Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase J Biol Chem 27931655–316632004 [DOI] [PubMed] [Google Scholar]
- 67.Wodicka LM, Ciceri P, Davis MI, et al. Activation state-dependent binding of small molecule kinase inhibitors: Structural insights from biochemistry Chem Biol 171241–12492010 [DOI] [PubMed] [Google Scholar]
- 68.Vendome J, Letard S, Martin F, et al. Molecular modeling of wild-type and D816V c-Kit inhibition based on ATP-competitive binding of ellipticine derivatives to tyrosine kinases J Med Chem 486194–62012005 [DOI] [PubMed] [Google Scholar]
- 69.Klug LR, Kent JD, Heinrich MC.Structural and clinical consequences of activation loop mutations in class III receptor tyrosine kinases Pharmacol Ther 191123–1342018 [DOI] [PubMed] [Google Scholar]
- 70.Cowan-Jacob SW, Möbitz H, Fabbro D.Structural biology contributions to tyrosine kinase drug discovery Curr Opin Cell Biol 21280–2872009 [DOI] [PubMed] [Google Scholar]
- 71.Bailey FP, Andreev VI, Eyers PA.The resistance tetrad: Amino acid hotspots for kinome-wide exploitation of drug-resistant protein kinase alleles Methods Enzymol 548117–1462014 [DOI] [PubMed] [Google Scholar]
- 72.Kim C, Kim E.Rational drug design approach of receptor tyrosine kinase type III inhibitors Curr Med Chem 267623–76402019 [DOI] [PubMed] [Google Scholar]
- 73.Chen LL, Trent JC, Wu EF, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors Cancer Res 645913–59192004 [DOI] [PubMed] [Google Scholar]
- 74.Tamborini E, Bonadiman L, Greco A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient Gastroenterology 127294–2992004 [DOI] [PubMed] [Google Scholar]
- 75.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation Clin Cancer Res 114182–41902005 [DOI] [PubMed] [Google Scholar]
- 76.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors J Clin Oncol 244764–47742006 [DOI] [PubMed] [Google Scholar]
- 77.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor J Clin Oncol 265352–53592008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate Clin Cancer Res 121743–17492006 [DOI] [PubMed] [Google Scholar]
- 79.Al-Ali HK, Heinrich MC, Lange T, et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib Hematol J 555–602004 [DOI] [PubMed] [Google Scholar]
- 80.Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance Blood 993472–34752002 [DOI] [PubMed] [Google Scholar]
- 81.Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis Blood 102276–2832003 [DOI] [PubMed] [Google Scholar]
- 82.Shah N, Nicoll J, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 83.Abrams TJ, Lee LB, Murray LJ, et al. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer Mol Cancer Ther 2471–4782003 [PubMed] [Google Scholar]
- 84.Ikezoe T, Yang Y, Nishioka C, et al. Effect of SU11248 on gastrointestinal stromal tumor-T1 cells: Enhancement of growth inhibition via inhibition of 3-kinase/Akt/mammalian target of rapamycin signaling Cancer Sci 97945–9512006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prenen H, Cools J, Mentens N, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate Clin Cancer Res 122622–26272006 [DOI] [PubMed] [Google Scholar]
- 86.Demetri GD, Heinrich MC, Fletcher JA, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure Clin Cancer Res 155902–59092009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial Lancet 3681329–13382006 [DOI] [PubMed] [Google Scholar]
- 88.Rock EP, Goodman V, Jiang JX, et al. Food and Drug Administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma Oncologist 12107–1132007 [DOI] [PubMed] [Google Scholar]
- 89.Montemurro M, Schoffski P, Reichardt P, et al. Nilotinib in the treatment of advanced gastrointestinal stromal tumours resistant to both imatinib and sunitinib Eur J Cancer 452293–22972009 [DOI] [PubMed] [Google Scholar]
- 90.Cauchi C, Somaiah N, Engstrom PF, et al. Evaluation of nilotinib in advanced GIST previously treated with imatinib and sunitinib Cancer Chemother Pharmacol 69977–9822012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: A phase II study of Korean Gastrointestinal Stromal Tumors Study Group Invest New Drugs 302377–23832012 [DOI] [PubMed] [Google Scholar]
- 92.Montemurro M, Gelderblom H, Bitz U, et al. Sorafenib as third- or fourth-line treatment of advanced gastrointestinal stromal tumour and pretreatment including both imatinib and sunitinib, and nilotinib: A retrospective analysis Eur J Cancer 491027–10312013 [DOI] [PubMed] [Google Scholar]
- 93.Montemurro M, Cioffi A, Domont J, et al. Long-term outcome of dasatinib first-line treatment in gastrointestinal stromal tumor: A multicenter, 2-stage phase 2 trial (Swiss Group for Clinical Cancer Research 56/07) Cancer 1241449–14542018 [DOI] [PubMed] [Google Scholar]
- 94.Blay JY, Shen L, Kang YK, et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): A randomised phase 3 trial Lancet Oncol 16550–5602015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: A multicenter phase II trial J Clin Oncol 302401–24072012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial Lancet 381295–3022013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Call JW, Wang Y, Montoya D, et al. Survival in advanced GIST has improved over time and correlates with increased access to post-imatinib tyrosine kinase inhibitors: Results from Life Raft Group Registry. Clin Sarcoma Res. 2019;9:4. doi: 10.1186/s13569-019-0114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Serrano C, Marino-Enriquez A, Tao DL, et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours Br J Cancer 120612–6202019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST J Pathol 21664–742008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heinrich MC, vonMehren M, Demetri GD, et al. A phase 2 study of ponatinib in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after failure of tyrosine kinase inhibitor (TKI) therapy: Initial report. J Clin Oncol. 2014;32 suppl; abstr 10506. [Google Scholar]
- 101.Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients Clin Cancer Res 205745–57552014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weisberg E, Meng C, Case AE, et al. Comparison of effects of midostaurin, crenolanib, quizartinib, gilteritinib, sorafenib and BLU-285 on oncogenic mutants of KIT, CBL and FLT3 in haematological malignancies Br J Haematol 187488–5012019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Apsel Winger B, Cortopassi WA, Garrido Ruiz D, et al. ATP-competitive inhibitors midostaurin and avapritinib have distinct resistance profiles in exon 17-mutant KIT Cancer Res 794283–42922019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Evans EK, Gardino AK, Kim JL, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9:eaao1690. doi: 10.1126/scitranslmed.aao1690. [DOI] [PubMed] [Google Scholar]
- 105.Heinrich MC, Jones R, von Mehren M, et al. Clinical activity of avapritinib in ≥ fourth-line (4L+) and PDGFRA Exon 18 gastrointestinal stromal tumors (GIST) J Clin Oncol. 2019;37 suppl; abstr 11022. [Google Scholar]
- 106.Heinrich MC, Jones RL, von Mehren M, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial Lancet Oncol 21935–9462020 [DOI] [PubMed] [Google Scholar]
- 107.Dhillon S.Avapritinib: First approval Drugs 80433–4392020 [DOI] [PubMed] [Google Scholar]
- 108.Verstovsek S, Tefferi A, Cortes J, et al. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis Clin Cancer Res 143906–39152008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gotlib J, Berubé C, Growney JD, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood1062865–28702005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants Gastroenterology 128270–2792005 [DOI] [PubMed] [Google Scholar]
- 111.Gleixner KV, Mayerhofer M, Aichberger KJ, et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA), and evaluation of cooperative drug effects Blood 107752–7592005 [DOI] [PubMed] [Google Scholar]
- 112.Growney JD, Clark JJ, Adelsperger J, et al. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412 Blood 106721–7242005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gotlib J, Berube C, Growney JD, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation Blood 1062865–28702005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.DeAngelo DJ, George TI, Linder A, et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial Leukemia 32470–4782017 [DOI] [PubMed] [Google Scholar]
- 115.Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis N Engl J Med 3742530–25412016 [DOI] [PubMed] [Google Scholar]
- 116.Kasamon YL, Ko CW, Subramaniam S, et al. FDA approval summary: Midostaurin for the treatment of advanced systemic mastocytosis Oncologist 231511–15192018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gotlib J, Radia DH, George TI, et al. Pure pathologic response is associated with improved overall survival in patients with advanced systemic mastocytosis receiving avapritinib in the phase I EXPLORER study Blood 13637–382020 [Google Scholar]
- 118.Gotlib J, Radia DH, George TI, et al. Ep1079 avapritinib induces responses in patients (pts) with advanced systemic mastocytosis (Advsm), regardless of prior midostaurin therapy. HemaSphere. 2020;4:2. [Google Scholar]
- 119.Akin C, Elberink H, Gotlib J, et al. PIONEER: A randomized, double-blind, placebo-controlled, phase 2 study of avapritinib in patients with indolent or smoldering systemic mastocytosis (SM) with symptoms inadequately controlled by standard therapy. J Allergy Clin Immunol. 2020;145:AB336. [Google Scholar]
- 120.Grunewald S, Klug LR, Muhlenberg T, et al. Resistance to avapritinib in PDGFRA-driven GIST is caused by secondary mutations in the PDGFRA kinase domain Cancer Discov 11108–1252021 [DOI] [PubMed] [Google Scholar]
- 121.Jawhar M, Schwaab J, Schnittger S, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis Leukemia 30136–1432016 [DOI] [PubMed] [Google Scholar]
- 122.Pardanani A.Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management Am J Hematol 94363–3772019 [DOI] [PubMed] [Google Scholar]
- 123.Chan WW, Wise SC, Kaufman MD, et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036 Cancer Cell 19556–5682011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eide CA, Adrian LT, Tyner JW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile Cancer Res 713189–31952011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bai Y, Bandara G, Ching CE, et al. Targeting the KIT activating switch control pocket: A novel mechanism to inhibit neoplastic mast cell proliferation and mast cell activation Leukemia 27278–2852012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Heinrich MC, Wise S, Hood M, et al. In vitro activity of novel KIT/PDGFRA switch pocket kinase inhibitors against mutations associated with drug-resistant GI stromal tumors. J Clin Oncol 28, 2010 (suppl 15; abstr 10007)
- 127.Smith BD, Kaufman MD, Lu WP, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants Cancer Cell 35738–751.e92019 [DOI] [PubMed] [Google Scholar]
- 128.Janku F, Abdul Razak AR, Chi P, et al. Switch control inhibition of KIT and PDGFRA in patients with advanced gastrointestinal stromal tumor: A phase I study of ripretinib J Clin Oncol 383294–33032020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial Lancet Oncol 21923–9342020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dhillon S.Ripretinib: First approval Drugs 801133–11382020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nemunaitis J, Bauer S, Blay JY, et al. Intrigue: Phase III study of ripretinib versus sunitinib in advanced gastrointestinal stromal tumor after imatinib Future Oncol 164251–42642020 [DOI] [PubMed] [Google Scholar]
- 132.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis J Comput Chem 251605–16122004 [DOI] [PubMed] [Google Scholar]
- 133.Kindler T, Breitenbuecher F, Marx A, et al. Efficacy and safety of imatinib in adult patients with c-kit-positive acute myeloid leukemia Blood 1033644–36542004 [DOI] [PubMed] [Google Scholar]
- 134.Heidel F, Cortes J, Rucker FG, et al. Results of a multicenter phase II trial for older patients with c-Kit-positive acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS) using low-dose Ara-C and Imatinib Cancer 109907–9142007 [DOI] [PubMed] [Google Scholar]
- 135.Cortes J, Giles F, O'Brien S, et al. Results of imatinib mesylate therapy in patients with refractory or recurrent acute myeloid leukemia, high-risk myelodysplastic syndrome, and myeloproliferative disorders Cancer 972760–27662003 [DOI] [PubMed] [Google Scholar]
- 136.Chevallier P, Hunault-Berger M, Larosa F, et al. A phase II trial of high-dose imatinib mesylate for relapsed or refractory c-kit positive and BCR-ABL negative acute myeloid leukaemia: The AFR-15 trial Leuk Res 331124–11262009 [DOI] [PubMed] [Google Scholar]
- 137.Einhorn LH, Brames MJ, Heinrich MC, et al. Phase II study of imatinib mesylate in chemotherapy refractory germ cell tumors expressing KIT Am J Clin Oncol 2912–132006 [DOI] [PubMed] [Google Scholar]
- 138.Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma Br J Cancer 921398–14052005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wyman K, Atkins MB, Prieto V, et al. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy Cancer 1062005–20112006 [DOI] [PubMed] [Google Scholar]
- 140.Kim KB, Eton O, Davis DW, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma Br J Cancer 99734–7402008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Handolias D, Hamilton AL, Salemi R, et al. Clinical responses observed with imatinib or sorafenib in melanoma patients expressing mutations in KIT Br J Cancer 1021219–12232010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification J Clin Oncol 292904–29092011 [DOI] [PubMed] [Google Scholar]
- 143.Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin J Clin Oncol 313182–31902013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Coit DG, Thompson JA, Albertini MR, et al. Cutaneous melanoma, version 2.2019, NCCN clinical practice guidelines in oncology J Natl Compr Canc Netw 17367–4022019 [DOI] [PubMed] [Google Scholar]
- 145.Goodman VL, Rock EP, Dagher R, et al. Approval summary: Sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma Clin Cancer Res 131367–13732007 [DOI] [PubMed] [Google Scholar]
- 146.FDA approves stivarga for GIST. Cancer Discov. 2013;3:OF1. [Google Scholar]
- 147.Avapritinib approved for GIST subgroup. Cancer Discov. 2020;10:334. doi: 10.1158/2159-8290.CD-NB2020-002. [DOI] [PubMed] [Google Scholar]