

Abstract
PURPOSE
CD19 chimeric antigen receptor (CD19-CAR) T cells induce high response rates in children and young adults (CAYAs) with B-cell acute lymphoblastic leukemia (B-ALL), but relapse rates are high. The role for allogeneic hematopoietic stem-cell transplant (alloHSCT) following CD19-CAR T-cell therapy to improve long-term outcomes in CAYAs has not been examined.
METHODS
We conducted a phase I trial of autologous CD19.28ζ-CAR T cells in CAYAs with relapsed or refractory B-ALL. Response and long-term clinical outcomes were assessed in relation to disease and treatment variables.
RESULTS
Fifty CAYAs with B-ALL were treated (median age, 13.5 years; range, 4.3-30.4). Thirty-one (62.0%) patients achieved a complete remission (CR), 28 (90.3%) of whom were minimal residual disease−negative by flow cytometry. Utilization of fludarabine/cyclophosphamide–based lymphodepletion was associated with improved CR rates (29/42, 69%) compared with non–fludarabine/cyclophosphamide–based lymphodepletion (2/8, 25%; P = .041). With median follow-up of 4.8 years, median overall survival was 10.5 months (95% CI, 6.3 to 29.2 months). Twenty-one of 28 (75.0%) patients achieving a minimal residual disease−negative CR proceeded to alloHSCT. For those proceeding to alloHSCT, median overall survival was 70.2 months (95% CI, 10.4 months to not estimable). The cumulative incidence of relapse after alloHSCT was 9.5% (95% CI, 1.5 to 26.8) at 24 months; 5-year EFS following alloHSCT was 61.9% (95% CI, 38.1 to 78.8).
CONCLUSION
We provide the longest follow-up in CAYAs with B-ALL after CD19-CAR T-cell therapy reported to date and demonstrate that sequential therapy with CD19.28ζ-CAR T cells followed by alloHSCT can mediate durable disease control in a sizable fraction of CAYAs with relapsed or refractory B-ALL (ClinicalTrials.gov identifier: NCT01593696).
INTRODUCTION
CD19 chimeric antigen receptor (CAR) T cells (CD19-CAR) have demonstrated impressive early response rates in B-cell acute lymphoblastic leukemia (B-ALL),1-6 but limited long-term follow-up data are available. In a global phase II trial of tisagenlecleucel in children and young adults (CAYAs) with relapsed or refractory B-ALL, a complete remission (CR) rate of 82% was achieved by day 28 postinfusion. However, with limited median follow-up of 13.1 months, only 59% of these patients remained in remission, the majority relapsing with CD19-negative disease.2,7 Allogeneic hematopoietic stem-cell transplantation (alloHSCT) improves long-term disease-free survival in patients with relapsed or refractory B-ALL rendered into remission with chemotherapy,8,9 but the role for consolidative alloHSCT in patients following CD19-CAR remains unclear1,3,10 and clinical practice varies.
CONTEXT
Key Objective
What is the role of consolidative hematopoietic stem-cell transplant (HSCT) following CD19-targeted CAR T cells for long-term remission in children and young adults (CAYAs) with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL)?
Knowledge Generated
Our article reports long-term outcomes of 50 CAYAs treated with CD19-CAR T cells for B-ALL. We demonstrate that CAYAs receiving post-CAR HSCT were associated with long-term disease-free survival and a low risk of post-HSCT relapse. Additionally, we provide data supporting the superiority of fludarabine/cyclophosphamide for lymphodepletion over alternative regimens. We also show the feasibility and efficacy of CD19-CAR T cells in the treatment of active CNS disease.
Relevance
We demonstrate durable relapse-free survival in those who underwent post-CAR HSCT, providing evidence that HSCT is important for maintaining remissions in CAYAs receiving CD19-CAR T cells for B-ALL. Our data additionally support treatment of active CNS disease, expanding the therapeutic index for CAR T cells.
Following our initial report of a phase I dose-escalation trial of autologous CD19.28ζ-CAR T cells in 20 CAYAs with relapsed or refractory B-ALL,1 we treated 30 additional patients in an expansion phase, which included treatment of active CNS disease and incorporated intensified lymphodepletion strategies to test the hypothesis that alternative chemotherapy regimens could more effectively reduce disease burden, thereby reducing cytokine release syndrome (CRS) severity, and potentially improve response to CD19-CAR. With a median follow-up of 4.8 years, we report long-term outcomes of 50 CAYAs with B-ALL who received autologous CD19.28ζ-CAR T cells. Additionally, a sizable fraction of patients received alloHSCT following CD19-CAR, providing an opportunity to evaluate the role of alloHSCT in CAYAs following CD19-CAR.
METHODS
Patients and Objectives
This was a single-center, phase I dose-escalation study of CD19.28ζ-CAR T cells for CAYAs (age 3-30 years) conducted in the Pediatric Oncology Branch of the National Cancer Institute (NCI). The Protocol (online only) was approved by the NCI Institutional Review Board and registered at ClinicalTrials.gov identifier: NCT01593696. This report incorporates data from all patients with B-ALL who received CD19.28ζ-CAR T cells on-study from July 2012 through March 2016 with a data cutoff of October 1, 2019. See the Data Supplement, online only for additional details.
The primary objective of the phase I trial was to define the maximum tolerated dose of CD19-CAR T cells, to describe the toxicity of the regimen, and to determine the feasibility of generating CD19-CAR T cells in this population, which has been previously reported.1 The trial was subsequently amended to add additional objectives, which specifically included determining the safety of administering cells in two groups of patients with CD19+ disease, stratified by high- and low-burden disease, and to evaluate the impact of alternative lymphodepleting regimens on toxicity and outcomes. Secondary objectives sought to define response rate; to measure expansion and persistence of CD19-CAR T cells in the peripheral blood, bone marrow, and CSF; to describe the toxicity of administration of CD19-CAR T cells in those with CNS disease; and to identify biological correlates of clinical outcomes and toxicity including cytokine values. The impact of alloHSCT on survival was retrospectively analyzed on a separate NCI Institutional Review Board–approved protocol evaluating CAR T-cell–related outcomes (ClinicalTrials.gov identifier: NCT03827343).
Study Design
CAR T-cell manufacturing was done as previously described (Data Supplement).1 Two dose levels were utilized: 1 × 106 transduced CAR T-cells/kg once on day 0 (DL1), and 3 × 106 transduced CAR T-cells/kg once on day 0 (DL2). For the initial 21 patients, pre-CAR T-cell lymphodepletion consisted of standard low-dose fludarabine (25 mg/m2 × 3 days) and cyclophosphamide (900 mg/m2 × 1 day) administered on days −4 to −2 (LD-Flu/Cy). From patient 22 onward, with a goal of reducing disease burden prior to proceeding with CAR T-cell infusion (day 0), those with high-burden disease (defined by ≥ 25% bone marrow blasts, circulating peripheral blasts, or lymphomatous disease) received one of the three alternative regimens used: (1) HD-Flu/Cy, high-dose fludarabine (30 mg/m2 once a day × 4 days) and cyclophosphamide (1,200 mg/m2 once a day × 2 days) from days −5 to −2; (2) FLAG, fludarabine (25 mg/m2 × 5 days) and cytarabine (2,000 mg/m2/day × 5 days) (days −6 to −2) with filgrastim (5 μg/kg/dose daily) starting on day −7; and (3) Ifos/Etop, ifosfamide (1,800 mg/m2 once a day × 5 days; with MESNA) and etoposide (100 mg/m2 once a day × 5 days) (days −6 to −2).
Toxicity and Efficacy Evaluations
Adverse events were captured using Common Terminology Criteria for Adverse Events (version 4.0) through 30 days of post-CAR infusion or resolution. CRS was prospectively graded using the Lee scale following its development (with initial patients retrospectively graded),11 and American Society of Transplantation and Cellular Therapy CRS consensus grading12 was retrospectively assigned for CRS and neurotoxicity.
Baseline bone marrow for pre-CAR disease assessment was performed prior to lymphodepletion. Initial response evaluations were performed at day 28 ± 4 days post-CAR T-cell infusion with routine surveillance thereafter. Minimal residual disease (MRD) negativity was defined as < 0.01% detectable leukemic blasts among mononuclear cells by flow cytometry (FC).13,14 Patients with sufficient samples available had retrospective next-generation sequencing (NGS) analysis using the recently US Food and Drug Administration–approved clonoSEQ assay.15
Correlative Studies
FC was used to quantitate CD19-CAR T cells in blood, marrow, and CSF using the anti-idiotype mAb 136.20.1 as described,16 and circulating CAR T-cell numbers were calculated on the basis of absolute lymphocyte counts. Cytokine profiling and T-cell subsets are described in the Data Supplement.
Statistical Analysis
Descriptive statistics were computed to summarize patient and disease characteristics. Kaplan-Meier survival curves17 were used to show event-free survival (EFS) and overall survival (OS) in B-ALL. In addition, we evaluated the EFS and OS for patients who proceeded to alloHSCT, starting at the day of alloHSCT (HSCT day 0). The cumulative incidence of relapse after alloHSCT was calculated on the basis of a competing risk of death from any cause before a relapse could be identified18 (Data Supplement).
RESULTS
Patients
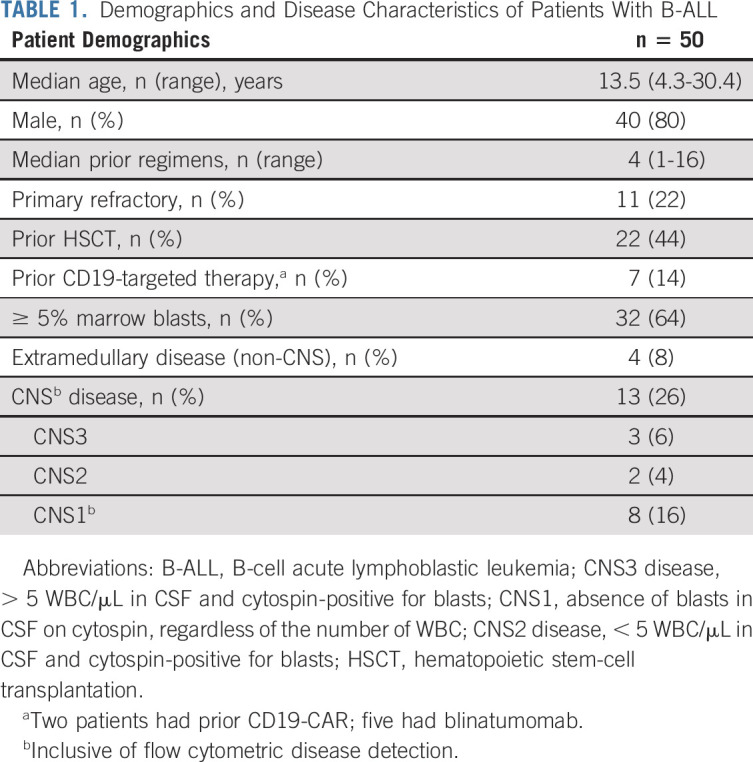
Among 53 enrolled patients, 51 had B-ALL and two had diffuse large B-cell lymphoma (Data Supplement; Table 1). One patient with B-ALL developed invasive fungal disease that precluded infusion. All subsequent analyses are based on the infused population with B-ALL (n = 50). The median age at infusion was 13.5 years (range, 4.3-30.4 years). Thirty-three patients (66.0%) had ≥ M2 marrow (≥ 5% marrow blasts), and 22 (44%) had undergone at least one prior alloHSCT. Two patients had received a prior CD19-CAR T-cell therapy and five had prior blinatumomab. Thirteen patients had CNS involvement at infusion (CNS1 with flow cytometric disease only [n = 8], CNS2 disease [n = 2], and CNS3 [n = 3]), which included a patient with extensive leptomeningeal involvement. Five patients were treated at DL2, and 45 patients were treated at DL1, including two patients who did not meet the target dose.
TABLE 1.
Demographics and Disease Characteristics of Patients With B-ALL
Toxicity
CRS occurred in 35 (70.0%) patients; nine patients (18.0%) had CRS grade 3-4 (Table 2). Seven patients received tocilizumab and four also received steroids. The median time to CRS onset was 5 days (range, 1-12 days). Neurotoxicity occurred in 10 (20.0%) patients, with four patients experiencing severe neurotoxicity (seizure, n = 2; grade 3 dysphasia, n = 2). All had resolution of symptoms to baseline, including patients with CNS3 disease. One patient had cardiac arrest during CRS with full recovery and has been previously reported.1,11 Disease burden was associated with CRS severity. Specifically, among responders receiving Flu/Cy (standard or low-dose, n = 29), one of 15 (6.67%) patients with an M1 marrow had grade 3-4 CRS versus eight of 14 (57.1%) patients with a ≥ M2 marrow had grade 3-4 CRS (P = .005). Among the nine patients with grade 3-4 CRS, all but one patient had ≥ M2 marrow. Neurotoxicity was also associated with high-grade CRS and high-disease burden. Specifically, seven of 10 patients with neurotoxicity had grade 3-4 CRS and eight of 10 had ≥ M2 marrow.
TABLE 2.
Toxicity and Response (N = 50)
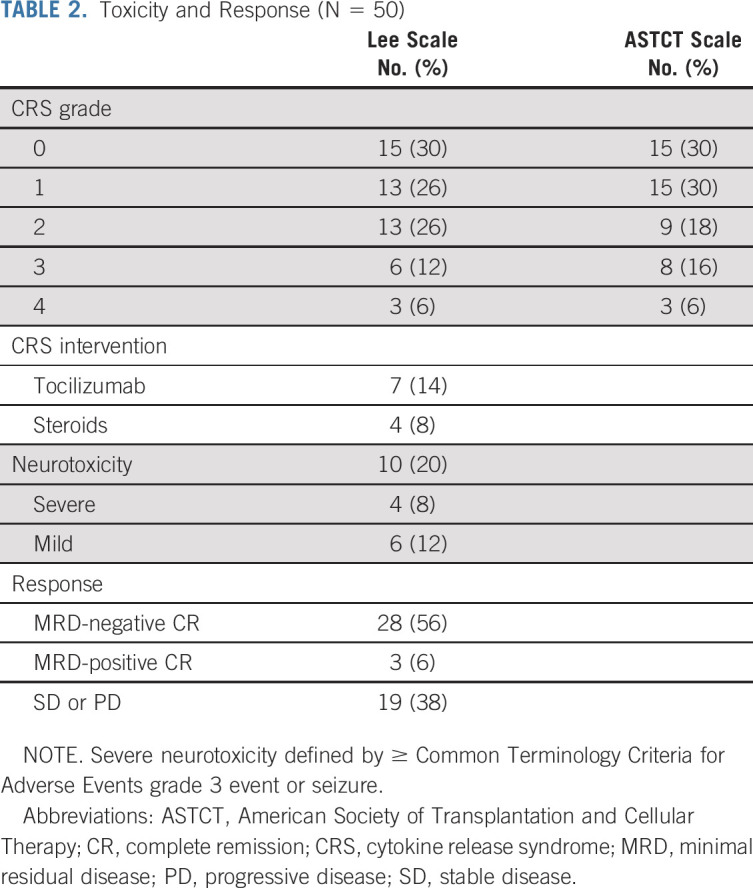
Response
Thirty-one of 50 treated patients with B-ALL (62.0%) achieved a CR, among whom 28 patients (90.3%) were MRD-negative by FC (56.0% MRD-negative CR rate overall) (Fig 1A). Among 12 patients with B-ALL who were MRD-negative by FC and had concurrent NGS MRD testing, five were NGS-positive at the day-28 assessment, with MRD levels ranging from 1 × 101 to 1 × 103 cells/million. CR rates were higher in patients who were primary refractory (P = .0035), had fewer prior lines of therapy (P = .033), had an M1 marrow (MRD-positive, < 5% blasts) (P = .0007), or received Flu/Cy lymphodepletion (P = .041) (Fig 1B). Specifically, patients with low-burden disease (M1 marrow) had higher rates of CR (16/17, 94.1%) than those with high-burden disease (≥M2 marrow; 15/33, 45.5%) (P = .0007). Duration in remission is shown in Figure 1C and was affected by alloHSCT as further described.
FIG 1.
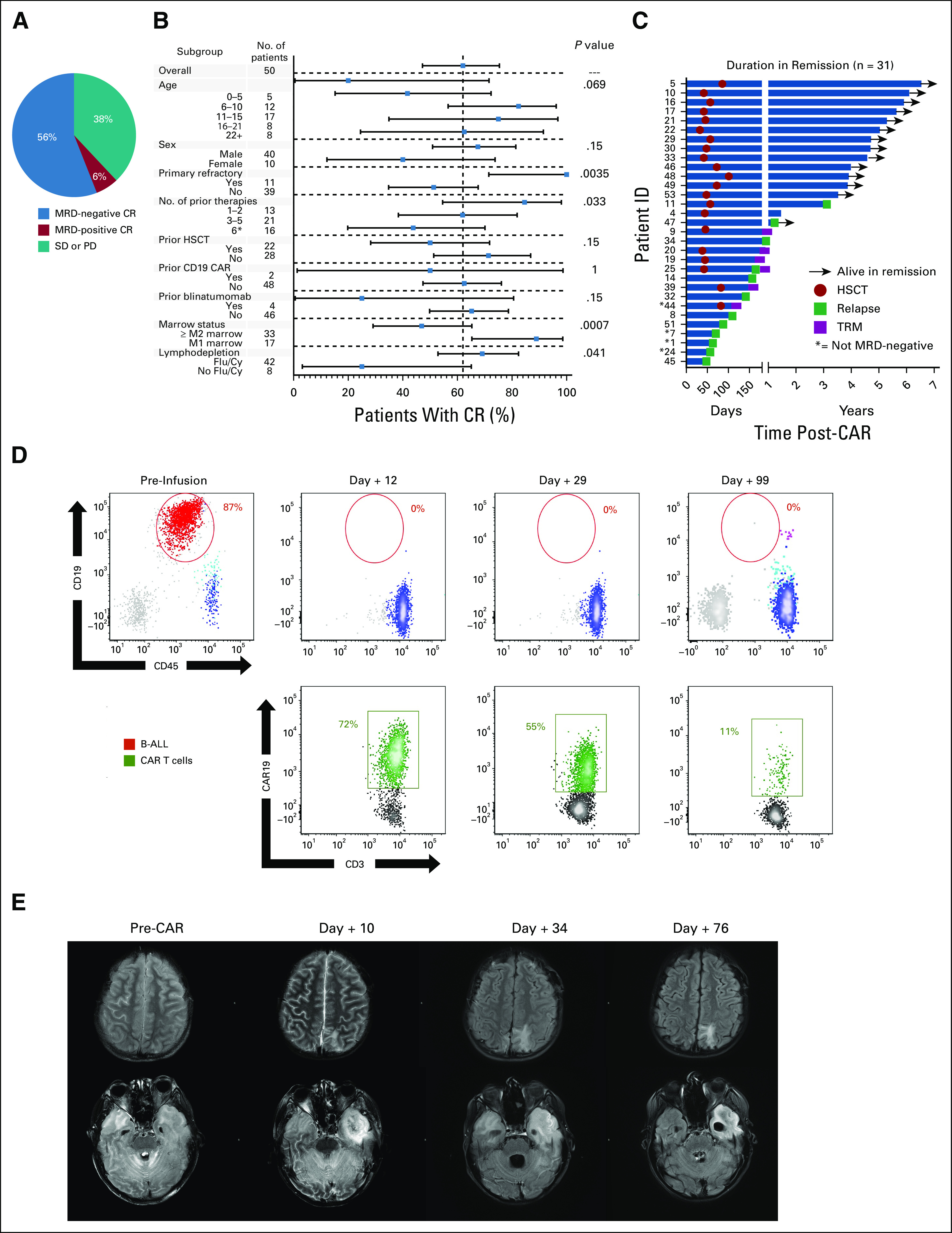
Response to CD19.28ζ CAR T-cell therapy. (A) Rate of those with MRD-positive or MRD-negative CR and those who had SD or PD among those with acute lymphoblastic leukemia. (B) Fraction of CR (MRD-positive and MRD-negative) according to patient demographics, disease characteristics, and treatment course. Squares represent the observed CR rate with the lines representing the 95% CIs for the difference of proportions from the reference category. The forest plots are based on the fraction and a Clopper-Pearson exact two-tailed 95% CI. (C) Duration in continuous remission among those who achieved a CR, indicating those who remain alive in remission, when HSCT was performed, the time of relapse, and those who experienced TRM. (D) Flow cytometric analysis of the CSF in patient 45 showing progressive CAR T-cell expansion (green) along with leukemia regression (red) in a patient with extensive CNS disease. (E) Brain magnetic resonance imaging findings from before, during, and after CAR T-cell infusion corresponding with patient 45, whose concurrent flow cytometry samples are shown in (D). Top panel (postcontrast axial fluid-attenuated inversion recovery) shows the evolution of intraparenchymal edema related to the leptomeningeal involvement to encephalomalacia. Bottom panel shows the evolution of widespread patchy white matter injury or edema involving the cerebrum and cerebellum with extensive involvement of the left temporal lobe (pretreatment). At day +10, neurologic symptoms included encephalopathy and right gaze preference. Necrosis had developed in the portion of the left temporal lobe that was most edematous. Diffusion-weighted images (not shown) demonstrated restricted diffusion corresponding to the area that is dark on fluid-attenuated inversion recovery. By day +76, the necrosis in the left temporal lobe had evolved to encephalomalacia (not ex vacuo expansion of the left temporal horn). B-ALL, B-cell acute lymphoblastic leukemia; CR, complete remission; HSCT, hematopoietic stem-cell transplant; MRD, minimal residual disease; PD, progressive disease; SD, stable disease; TRM, transplant-related mortality.
CNS B-ALL involvement was effectively treated in all who had CRS and a concurrent marrow response, with one patient having residual low-level CNS flow cytometric disease (CNS1). There was resolution of leptomeningeal enhancement when present and evidence of CAR T-cell trafficking to the CNS in the majority of patients (Figs 1D and 1E). Notably, three of 13 patients with CNS involvement also had evidence of non-CNS extramedullary (EM) disease, including one patient who had an M1 marrow with extensive EM involvement, including renal masses. Two of the three were rendered into an MRD-negative CR with resolution of both CNS and non-CNS EM disease at 1 month.
CAR T-Cell Expansion and Cytokine Profiling
CR was associated with higher CAR T-cell expansion and grade 3-4 CRS (Figs 2A-2C; Data Supplement). Twenty-nine patients had evidence of CAR T-cell trafficking to the CNS with 0.3 to 81% of T cells in the CSF as CAR T-cell–positive. We observed no difference in the response rate associated with expression of the T-cell exhaustion markers PD1, TIM3, or LAG3 on infused CAR T cells (Fig 2D). Extended immunophenotyping of the CAR T-cell product revealed that CD4+ and CD8+ CAR T-cell populations were balanced between central and effector immunophenotypes (Figs 2E and 2F). Peak levels for the majority of evaluated cytokines were differentially elevated between those with low- and high-grade CRS (Figs 2G-2I, Data Supplement).
FIG 2.
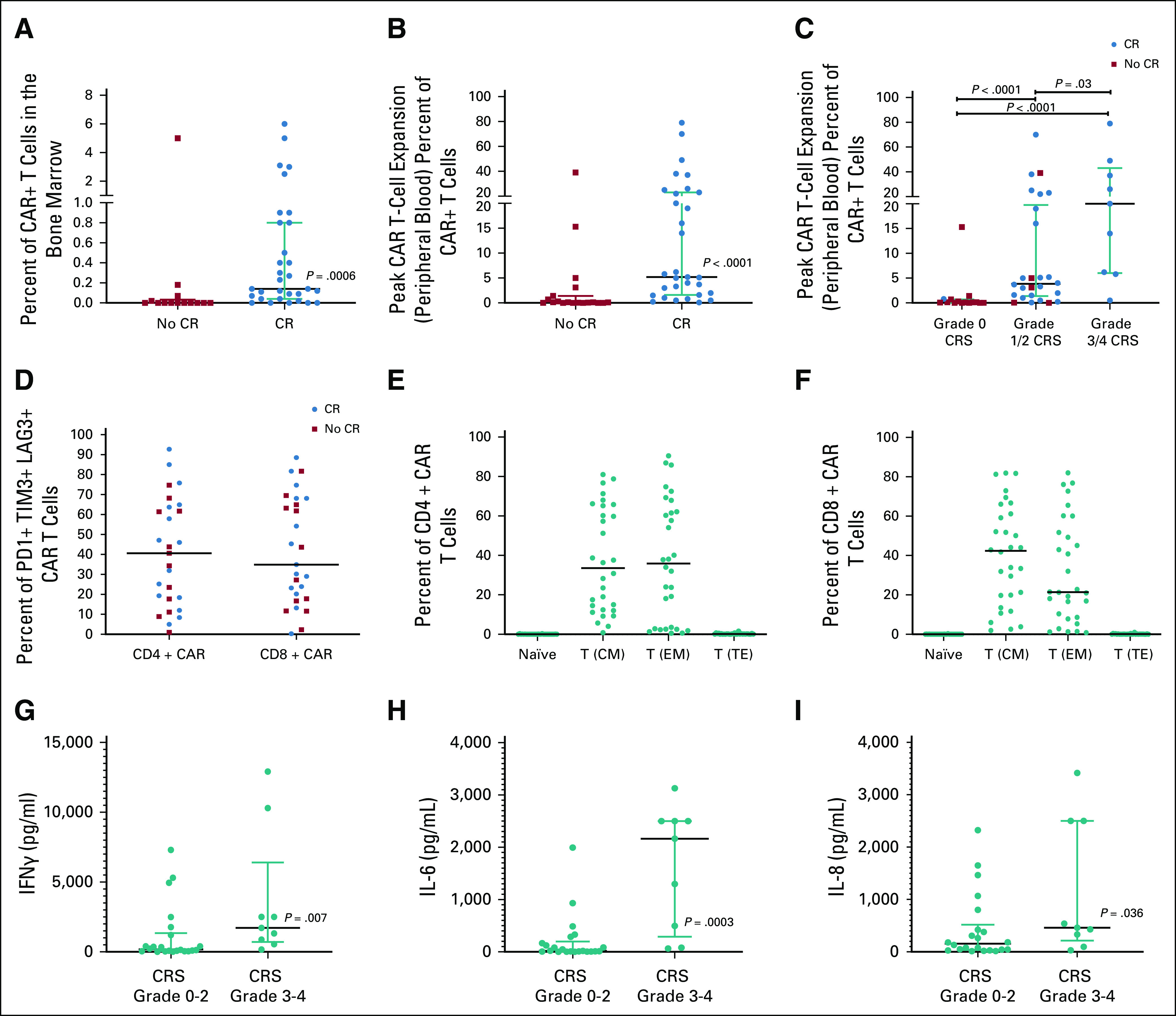
(A) Median and interquartile range of CAR T-cell expansion (as a percentage of T cells that were CAR T-cell–positive) in the bone marrow at day 28 (± 4 days) post-CAR T-cell infusion, stratified by those who achieved a CR and those who did not (lower limit of CAR T-cell detection by flow cytometry is 0.05% of CD3+ T cells). (B) Median and interquartile range of peak CAR T-cell expansion (as a percentage of T cells that were CAR T-cell–positive) in the peripheral blood within the first 28 days (± 4 days) post-CAR T-cell infusion, stratified by those who achieved a CR and those who did not. (C) Median and interquartile range of peak CAR T-cell expansion (as a percentage of T cells that were CAR T-cell–positive) in the peripheral blood within the first 28 days (± 4 days) post-CAR T-cell infusion, stratified by those who had grade 0, grade 1/2, and grade 3/4 CRS. (D) Percentage of CAR T cells positive for PD1, TIM3, or LAG3 as stratified by CD4 and CD8 populations with red indicating those who achieved a CR and blue indicating those who did not. (E) Proportion of the CD4+ CAR T-cell product that are naïve T cells; T (CM), T (EM), and T (TE). (F) Proportion of the CD8+ CAR T-cell product that are naïve T cells; T (CM), T (EM), and T (TE). (G) Peak IFNγ values stratified by CRS grade 0-2 versus CRS grade 3-4 in those who achieved a CR. (H) Peak IL-6 values stratified by CRS grade 0-2 versus CRS grade 3-4 in those who achieved a CR. (I) Peak IL-8 values stratified by CRS grade 0-2 versus CRS grade 3-4 in those who achieved a CR. CR, complete remission; CRS, cytokine release syndrome; EM, extramedullary; IFN, interferon; IL, interleukin; PD, progressive disease; T (CM), central memory T cells; T (EM), effector memory or effector T cells; T (TE), terminal effector T cells.
Lymphodepleting Regimen
Among 50 patients with B-ALL, 35 patients received LD-Flu/Cy and 15 patients were treated with alternative regimens, including HD-Flu/Cy (n = 7), FLAG (n = 6), or Ifos/Etop (n = 2). The CR rate was higher among those who received any Flu/Cy regimen (P = .041) with CR in four of seven (57.1%) patients receiving HD-Flu/Cy and in 25 of 35 (71.4%) receiving LD-Flu/Cy. Cumulatively, 29 of 42 (69.0%) patients receiving any Flu/Cy regimen achieved a CR. Responses using alternative lymphodepleting regimens were suboptimal with zero of two (0.0%) patients receiving Ifos/Etop and two of six (33.3%) patients receiving FLAG chemotherapy achieving CR.
Long-Term Outcomes
With a median follow-up of 4.8 years (range, 3.5-7.2 years), the median OS for the entire cohort was 10.5 months (95% CI, 6.3 to 29.2 months). Median EFS was 3.1 months (95% CI, 0.9 to 7.7 months), with 3-month EFS of 52.0% (95% CI, 37.4 to 64.7) and 6-month EFS of 38% (95% CI, 24.8 to 51.1) (Fig 3A).
FIG 3.
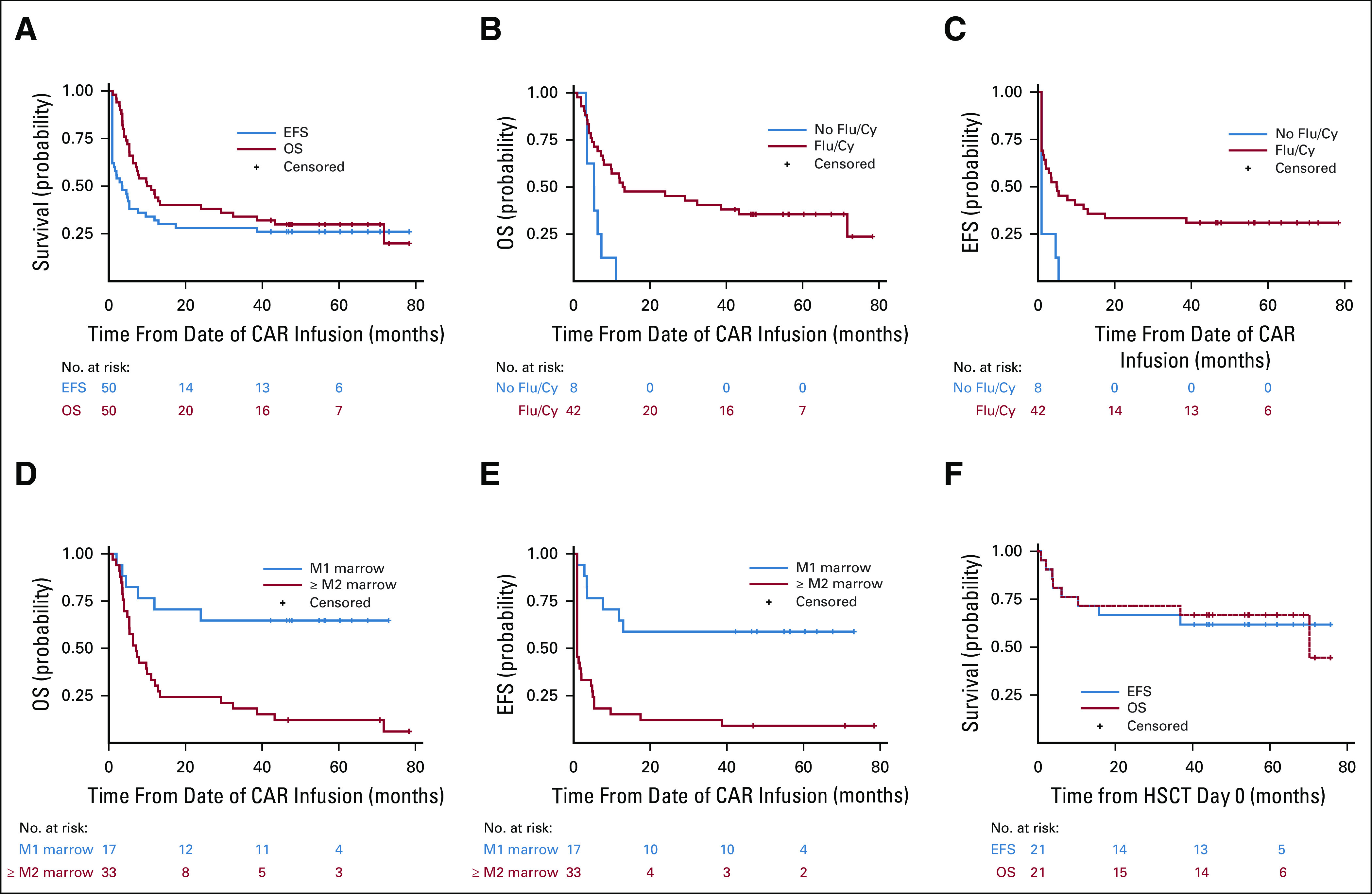
Continuous remission, OS, and EFS. (A) OS and EFS among patients with B-cell acute lymphoblastic leukemia, starting from CAR infusion at day 0. (B) OS, (C) EFS among patients who received Flu/Cy–based pre-CAR T-cell lymphodepletion versus those who did not. (D) OS, (E) EFS among patients who had an M2 marrow (≥ 5% marrow disease) or higher compared with those who had an M1 marrow (< 5%) disease. (F) OS and EFS among patients who proceeded to a consolidative allogeneic HSCT, from HSCT day 0. CAR, chimeric antigen receptor; EFS, event-free survival; Flu/Cy, fludarabine/cyclophosphamide; HSCT, hematopoietic stem-cell transplant; OS, overall survival.
Receipt of any Flu/Cy regimen was associated with an improved EFS and OS (Figs 3C and 3D). Median EFS was not reached in patients treated with an M1 marrow, whereas median EFS was 0.9 months (95% CI, 0.9 to 2.0 months) in patients treated with ≥ M2 marrow (P ≤ .0001) (Figs 3E and 3F). Importantly, only eight of 15 patients achieving CR proceeded to HSCT among this high-burden disease cohort.
Role of Consolidative HSCT
Twenty-one of 28 (75.0%) patients achieving MRD-negative CR proceeded to a consolidative alloHSCT. The median time to alloHSCT was 54 days from CAR infusion (range, 42-97 days). In four patients, this represented a second alloHSCT. All 17 patients for whom this was a first HSCT underwent myeloablative conditioning (Data Supplement). The median OS from HSCT day 0 was 70.2 months (95% CI, 10.4 months to not estimable), and median EFS was not reached (Fig 3F). The 5-year EFS following alloHSCT was 61.9% (95% CI, 38.1 to 78.8). Eight patients died between 0.8 and 71 months following alloHSCT, causes of which included transplant-related complications and/or graft-versus-host disease or infection (n = 6), complications of second malignancy 3 years post-HSCT (n = 1)19, and relapsed disease (n = 1). A total of two patients relapsed after alloHSCT, and the cumulative incidence of relapse after alloHSCT, with death as a competing risk, was 4.8% (95% CI, 0.3 to 20.3) at 12 months and 9.5% (95% CI, 1.5 to 26.8) at 24 months.
All seven who achieved an MRD-negative CR and did not proceed to a consolidative HSCT experienced relapse at a median of 152 days post-CAR infusion (range, 94-394 days), highlighting the importance of consolidative alloHSCT following this construct. Six patients did not proceed to HSCT because of concern for second alloHSCT–associated risks. In one patient with underlying trisomy 21, a first HSCT was deferred because of concern for toxicity. No patient achieved a CR following a second CAR T-cell infusion (Data Supplement).
CD19 Antigen Expression and Prior CD19 Targeting
With a limited sample size (n = 42), CD19 antigen site density at enrollment was not significantly different between responders and nonresponders (median CD19 site density 7,603 v 8,175, respectively [P = .82]). Among those who had received prior CD19-targeted therapy, one of two who had received prior CD19-CAR T cells and four of five who had received prior blinatumomab were nonresponders. CD19 immunophenotype on leukemia blasts at relapse for patients achieving MRD-negative CR and not proceeding to HSCT was CD19+ (n = 3), CD19−/dim (n = 3), and CD19 unknown (n = 1) blasts.
DISCUSSION
Nearly all published reports detailing activity of CD19-CAR in B-ALL have focused on CR rates at day 28, which occur in approximately 60%-100% of patients.1-4,20-25 Long-term follow-up data from these studies are largely lacking. However, even with limited follow-up, high relapse rates have been observed and a clear plateau in survival has not been demonstrated. Additional confounding data regarding long-term outcomes is the fact that a significant fraction of patients rendered into remission with CD19-CAR receive additional treatment, including alloHSCT. One previous series examined outcomes in adults with a median age of 44 years (range, 23-74) and concluded that post-CAR alloHSCT did not improve survival.3 However, it remains unknown whether alloHSCT after CAR T-cell therapy affects outcome in the CAYA population.
With a median follow-up of 4.8 years, this report provides the longest duration of follow-up of any series following treatment with CD19-CAR for B-ALL. In CAYA patients with relapsed or refractory B-ALL treated with autologous T cells expressing a CD19.28ζ CAR, we demonstrate that patients who achieved a CR and proceeded to a consolidative alloHSCT had a relapse rate of < 10% at 24 months post-CAR, a 5-year EFS of 61.9%, and a median OS of 70.2 months from HSCT. All patients who did not undergo post-CAR T-cell alloHSCT experienced relapse. The contrast to outcomes reported by Park et al3 using a similar CAR T-cell construct, where HSCT did not lead to improved outcomes, and may relate to the higher morbidity and mortality associated with alloHSCT for B-ALL in adults, along with variability in conditioning regimen and graft choice.26 Importantly, the decision to undergo alloHSCT and the choice of preparative regimen were not dictated by our protocol, and therefore, this study is not capable of providing information regarding the optimal regimen for use in this setting. Nonetheless, the results provide strong evidence that a treatment regimen that incorporates autologous CD19.28ζ CAR T cells to achieve remission followed by a consolidative alloHSCT is associated with a remarkably low relapse rate in a very high-risk patient population. The high relapse rates for those not proceeding to alloHSCT may relate, in part, to the shorter persistence of the CD28 costimulatory domain construct,1,3,27 supporting a role for utilization of this construct as a bridge to HSCT. However, given that patients who experience long-term persistence of CD19.BBζ-CARs may relapse with CD19-negative B-ALL, consolidative alloHSCT could improve outcomes following alterative CAR T-cell constructs regardless of persistence. Prospective trials that directly incorporate consolidative alloHSCT following CAR T cells are warranted for further evaluation in improving outcomes for these high-risk patients.
This series also provides additional data to support the use of CD19-CAR for the treatment of CNS leukemia. We demonstrate that CNS2 and CNS3 disease can be safely and effectively treated, potentially expanding the therapeutic indications for CAR T cells and raising the prospect that CD19-CAR could provide effective therapy without toxicity from standard radiation and intensified chemotherapy-based approaches. These results support further study of CAR T cells in patients with B-ALL and active CNS involvement.
Additionally, we provide data confirming the importance of Flu/Cy–based lymphodepletion in improving responses to CD19-CAR (Figs 3C and 3D), as previously reported,25,28 and confirm previous data demonstrating that patients with low-burden or primary refractory disease experience superior long-term outcomes following CD19-CAR therapy compared with heavily pretreated patients with high-burden disease (Figs 3E and 3F).3
Given the role of CD19-CAR T cells as a bridge to HSCT, it warrants consideration of alternative approaches for remission induction. Blinatumomab, for instance, is US Food and Drug Administration–approved for children and adults with relapsed or refractory B-ALL. However, despite its more ready availability, which is not dependent on manufacturing time or success thereof, the efficacy of blinatumomab in children is lower than in adults receiving blinatumomab29 and also lower than remission rates following CD19-CAR T cells, using any construct, particularly for those with high-burden disease.30,31 Additionally, there is no role for blinatumomab in active CNS disease, and there are limited data for its use in EM disease. Therefore, selection of CAR T cells over blinatumomab may be advantageous in patients with higher-burden disease and EM disease or as a salvage for blinatumomab nonresponders. Although patients with high-burden disease in our study had lower response rates, this may be related to the utilization of non–Flu/Cy–based lymphodepletion, which would not be endorsed in future studies. Of note, our results raise the concern that prior CD19 targeting may affect response to CD19-CAR T cells. Notably, the global registration study for tisagenlecleucel excluded patients who had received prior CD19-targeted therapies,2 and recent studies suggest that blinatumomab prior to CD19-CAR T cells may negatively affect outcomes.32
In summary, we demonstrate that CD19.28ζ CAR T cells followed by a consolidative alloHSCT can provide long-term durable disease control in CAYAs with relapsed or refractory B-ALL. Following alloHSCT, we observed a significant long-term EFS with an apparent plateau and a low relapse rate, providing support for this sequential approach for long-term cure.
ACKNOWLEDGMENT
We gratefully acknowledge the study participants and their families, referring medical care teams, the faculty and staff of the NIH Clinical Center who provided their expertise in the management of the study participants, including Joanne Derdak, CRNPs and the data managers, Showri Kakumanu and Ekaterina Nikitina, and patient care coordinators involved with this work. The experience of the first 21 patients who were treated on this trial was previously published in Lee et al. Lancet 2014. PMID: 25319501.
Nirali N. Shah
Research Funding: Lentigen
Daniel W. Lee
Employment: Karyopharm Therapeutics
Consulting or Advisory Role: Harpoon Therapeutics, Amgen, Celgene
Research Funding: Kite/Gilead
Patents, Royalties, Other Intellectual Property: CAR T cell for pediatric and adult high-grade gliomas and other tumors
Travel, Accommodations, Expenses: Kite/Gilead
Pamela L. Wolters
Stock and Other Ownership Interests: Bristol-Myers Squibb
Crystal L. Mackall
Stock and Other Ownership Interests: Lyell Immunopharma, Alimera Sciences, Apricity Health
Consulting or Advisory Role: Bryology, Vor Biopharma, Apricity Health, TPG, Alimera Sciences, PACT Pharma, Nektar, Lyell Immunopharma, NeoImmuneTech
Patents, Royalties, Other Intellectual Property: I am an inventor on numerous patents related to chimeric antigen receptor therapeutics and received royalties from NIH for the CD22-CAR patent licensed to Juno therapeutics.
Travel, Accommodations, Expenses: NeoImmuneTech, Roche, Nektar
Other Relationship: Lyell Immunopharma
No other potential conflicts of interest were reported.
DISCLAIMER
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
SUPPORT
Supported by NCI Intramural Research Program (ZIA BC 011498)—C.L.M.; Ludwig Institute for Cancer Research (SU2C-AACR-DT1113, 5P30CA124435L)—C.L.M.; Parker Institute for Cancer Immunotherapy—C.L.M.; NCI Intramural Program (ZIA BC 011823) N.N.S; St Baldrick's Foundation Scholar Award—D.W.L.; NCI Intramural Program (ZIA BC 011498). Supported in part by the Intramural Research Program, National Cancer Institute and NIH Clinical Center, National Institutes of Health. Supported by a St Baldrick's/Stand Up 2 Cancer Pediatric Dream Team Translational Cancer Research Grant. Stand Up 2 Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. C.L.M is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program.
CLINICAL TRIAL INFORMATION
N.N.S. and D.W.L. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Nirali N. Shah, Daniel W. Lee, Staci Martin, Pamela L. Wolters, David F. Stroncek, Crystal L. Mackall
Provision of study materials or patients: Nirali N. Shah, Daniel W. Lee, Crystal L. Mackall
Collection and assembly of data: Nirali N. Shah, Daniel W. Lee, Bonnie Yates, Constance M. Yuan, Staci Martin, Pamela L. Wolters, Cindy P. Delbrook, Maryalice Stetler-Stevenson, Terry J. Fry, David F. Stroncek, Crystal L. Mackall
Data analysis and interpretation: Nirali N. Shah, Daniel W. Lee, Constance M. Yuan, Haneen Shalabi, Staci Martin, Pamela L. Wolters, Seth M. Steinberg, Eva H. Baker, Maryalice Stetler-Stevenson, Crystal L. Mackall
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Long-Term Follow-Up of CD19 CAR T-Cell Therapy in Children and Young Adults With B-ALL
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Nirali N. Shah
Research Funding: Lentigen
Daniel W. Lee
Employment: Karyopharm Therapeutics
Consulting or Advisory Role: Harpoon Therapeutics, Amgen, Celgene
Research Funding: Kite/Gilead
Patents, Royalties, Other Intellectual Property: CAR T cell for pediatric and adult high-grade gliomas and other tumors
Travel, Accommodations, Expenses: Kite/Gilead
Pamela L. Wolters
Stock and Other Ownership Interests: Bristol-Myers Squibb
Crystal L. Mackall
Stock and Other Ownership Interests: Lyell Immunopharma, Alimera Sciences, Apricity Health
Consulting or Advisory Role: Bryology, Vor Biopharma, Apricity Health, TPG, Alimera Sciences, PACT Pharma, Nektar, Lyell Immunopharma, NeoImmuneTech
Patents, Royalties, Other Intellectual Property: I am an inventor on numerous patents related to chimeric antigen receptor therapeutics and received royalties from NIH for the CD22-CAR patent licensed to Juno therapeutics.
Travel, Accommodations, Expenses: NeoImmuneTech, Roche, Nektar
Other Relationship: Lyell Immunopharma
No other potential conflicts of interest were reported.
REFERENCES
- 1.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial Lancet 385517–5282015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia N Engl J Med 378439–4482018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia N Engl J Med 378449–4592018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults Blood 1293322–33312017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curran KJ, Margossian SP, Kernan NA, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL Blood 1342361–23682019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner RA, Ceppi F, Rivers J, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy Blood 1342149–21582019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hay KA, Gauthier J, Hirayama AV, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy Blood 1331652–16632019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pulsipher MA, Peters C, Pui CH.High-risk pediatric acute lymphoblastic leukemia: To transplant or not to transplant? Biol Blood Marrow Transpl 17S137–S14820111 suppl [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khandelwal P, Millard HR, Thiel E, et al. Hematopoietic stem cell transplantation activity in pediatric cancer between 2008 and 2014 in the United States: A Center for International Blood and Marrow Transplant Research report Biol Blood Marrow Transpl 231342–13492017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summers C, Annesley C, Bleakley M, et al. Long term follow-up after SCRI-CAR19v1 reveals late recurrences as well as a survival advantage to consolidation with HCT after CAR T cell induced remission. Blood. 2018;132(1 suppl):967. [Google Scholar]
- 11.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome Blood 124188–1952014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells Biol Blood Marrow Transpl 25625–6382019 [DOI] [PubMed] [Google Scholar]
- 13.Cherian S, Stetler-Stevenson M. Flow cytometric monitoring for residual disease in B lymphoblastic leukemia post T cell engaging targeted therapies. Curr Protoc Cytom. 2018;86:e44. doi: 10.1002/cpcy.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao W, Salem D, McCoy CS, et al. Early recovery of circulating immature B cells in B-lymphoblastic leukemia patients after CD19 targeted CAR T cell therapy: A pitfall for minimal residual disease detection Cytometry B Clin Cytom 94434–4432018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ClonoSEQ cleared for residual cancer testing. Cancer Discov. 2018;8:OF6. doi: 10.1158/2159-8290.CD-NB2018-136. [DOI] [PubMed] [Google Scholar]
- 16.Jena B, Maiti S, Huls H, et al. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan EL, Meier P.Nonparametric estimation from incomplete observation J Am Stat Assoc 53457–4811958 [Google Scholar]
- 18.Gooley TA, Leisenring W, Crowley J, et al. Estimation of failure probabilities in the presence of competing risks: New representations of old estimators Stat Med 18695–7061999 [DOI] [PubMed] [Google Scholar]
- 19.Good ML, Malekzadeh P, Kriley IR, et al. Intrahepatic cholangiocarcinoma as a rare secondary malignancy after allogeneic hematopoietic stem cell transplantation for childhood acute lymphoblastic leukemia: A case report. Pediatr Transpl. 2020;24:e13653. doi: 10.1111/petr.13653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia Blood 1302317–23252017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias Blood 1184817–48282011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia N Engl J Med 3681509–15182013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacoby E, Bielorai B, Avigdor A, et al. Locally produced CD19 CAR T cells leading to clinical remissions in medullary and extramedullary relapsed acute lymphoblastic leukemia Am J Hematol 931485–14922018 [DOI] [PubMed] [Google Scholar]
- 25.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients J Clin Invest 1262123–21382016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saadeh SS, Litzow MR.Hematopoietic stem cell transplant in adults with acute lymphoblastic leukemia: The present state Expert Rev Hematol 11195–2072018 [DOI] [PubMed] [Google Scholar]
- 27.Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors Nat Med 21581–5902015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirayama AV, Gauthier J, Hay KA, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells Blood 1331876–18872019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia N Engl J Med 376836–8472017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia J Clin Oncol 344381–43892016 [DOI] [PubMed] [Google Scholar]
- 31.Brown PA, Ji L, Xu X, et al. A randomized phase 3 trial of blinatumomab vs. chemotherapy as post-reinduction therapy in high and intermediate risk (HR/IR) first relapse of B-acute lymphoblastic leukemia (B-ALL) in children and adolescents/young adults (AYAs) demonstrates superior efficacy and tolerability of blinatumomab: A report from Children's Oncology Group Study AALL1331 Blood 134LBA-12019suppl 2 [Google Scholar]
- 32.Pillai V, Muralidharan K, Meng W, et al. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy Blood Adv 33539–35492019 [DOI] [PMC free article] [PubMed] [Google Scholar]