

SUMMARY
The presence of DNA in the cytoplasm is normally a sign of microbial infections and is quickly detected by cyclic GMP-AMP synthase (cGAS) to elicit anti-infection immune responses. However, chronic activation of cGAS by self-DNA leads to severe autoimmune diseases for which no effective treatment is available yet. Here we report that acetylation inhibits cGAS activation and that the enforced acetylation of cGAS by aspirin robustly suppresses self-DNA-induced autoimmunity. We find that cGAS acetylation on either Lys384, Lys394, or Lys414 contributes to keeping cGAS inactive. cGAS is deacetylated in response to DNA challenges. Importantly, we show that aspirin can directly acetylate cGAS and efficiently inhibit cGAS-mediated immune responses. Finally, we demonstrate that aspirin can effectively suppress self-DNA-induced autoimmunity in Aicardi-Goutières syndrome (AGS) patient cells and in an AGS mouse model. Thus, our study reveals that acetylation contributes to cGAS activity regulation and provides a potential therapy for treating DNA-mediated autoimmune diseases.
INTRODUCTION
Cytosolic DNA is a potent danger signal that triggers robust innate immune responses because the presence of DNA in the cytoplasm is normally a sign of microbial infection or tissue damage (Barbalat et al., 2011). Recognition of cytosolic DNA is an important mechanism for host defense. Cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS) has been identified as a key sensor that mediates immune responses upon cytoplasmic DNA challenge. Upon DNA binding, the activated cGAS catalyzes the synthesis of cyclic GMP-AMP (cGAMP) from ATP and guanosine triphosphate (GTP) (Gao et al., 2013; Sun et al., 2013; Wu et al., 2013b). cGAMP then acts as a second messenger to bind and activate the endoplasmic reticulum protein STING (also known as MITA, MPYS, and ERIS) (Ishikawa and Barber, 2008; Jin et al., 2008; Sun et al., 2009; Zhong et al., 2008), which, in turn, mediates the activation of the downstream effectors TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3) to produce type I interferon (IFN) (Bowie, 2012). Type I IFN plays critical roles in antiviral responses by inducing the expression of a number of interferon-stimulated genes (ISGs) (Gao et al., 2015).
In addition to microbial infection, cellular damage or reverse transcription of endogenous retroviruses can also generate self-DNA in the cytoplasm (Ahn and Barber, 2014; Kassiotis and Stoye, 2016; O’Neill, 2013). Metazoans have evolved DNases that clear self-DNA to prevent inappropriate activation of cGAS-mediated immune responses. For example, DNA 3′ repair exonuclease 1 (TREX1) degrades cytosolic DNA, and loss-of-function mutations of TREX1 have been identified in human patients with autoimmune disorders such as Aicardi-Goutières syndrome (AGS) and lupus (Ahn and Barber, 2014; Aicardi and Goutières, 1984; Crow and Manel, 2015; Lisnevskaia et al., 2014). AGS patients harboring TREX1 mutations are known to accumulate cytoplasmic self-DNAs that chronically stimulate cGAS-mediated type I IFN production (Crow et al., 2006; Gao et al., 2015; Gray et al., 2015; Stetson et al., 2008). The excessively produced IFN drives systemic inflammation and other autoimmune responses (Elkon and Wiedeman, 2012). Deletion of Trex1 in mice results in severe cGAS-dependent autoimmunity because deletion of Cgas rescues the lethality and autoimmune conditions in Trex−/− mice (Gao et al., 2015; Gray et al., 2015). These studies suggest that cGAS inhibition could be used for treating self-DNA-mediated autoimmune diseases. A limited understanding of how cGAS activation is regulated, however, has prevented the development of effective treatments.
Post-translational modifications (PTMs) such as acetylation and phosphorylation are known to be critical for regulation of protein functions (Cohen, 2000; Verdin and Ott, 2015). Protein acetylation plays important roles in various biological processes, including immune responses (Shakespear et al., 2011). In the current study, we found that acetylation at one of three lysine residues, Lys384, Lys394, or Lys414, contributes to keeping cGAS inactive. cGAS is deacetylated in response to DNA challenge. Importantly, we show that aspirin, a drug that is well-tolerated in humans, can robustly enforce the acetylation of cGAS and led to cGAS inhibition. Furthermore, we demonstrate that aspirin ameliorates DNA-mediated autoimmune responses in mice and in AGS patient cells.
RESULTS
Acetylation-Mimicking Mutation of cGAS Blocks cGAS Activation
Because acetylation is a key PTM that plays important roles in many biological processes (Verdin and Ott, 2015), and acetylation of cGAS in quiescent cells has been implied in proteomic studies (Choudhary et al., 2009; Zhao et al., 2010), we investigated whether acetylation is involved in the regulation of cGAS activity. To do so, we first created a FLAG-cGAS stable THP-1 (human monocytic cells) cell line and examined cGAS acetylation in quiescent cells by performing liquid chromatography-mass spectrometry (LC-MS) with the immunoprecipitated FLAG-cGAS. We identified six lysine (K) acetylation sites of cGAS: K7, K50, K384, K392, K394, and K414 (Figure S1A). Interestingly, three of these six sites are conserved across species (Figure S1B). To study the function of acetylation of these sites, we mutated each site by replacing a lysine with a glutamine (Q), which mimics the acetylation state on lysine (Zhao et al., 2010), and tested the effect of these mutations on cGAS activity in HEK293T cells that stably express STING (Sun et al., 2013). cGAS-STING pathway activation can be reflected by phosphorylation of the downstream transcription factor IRF3 (Sun et al., 2013). We found that K-to-Q mutation of each of the three conserved sites K384, K394 or K414 (cGASK384Q, cGASK394Q, or cGASK414Q)—led to a dramatic reduction in cGAS activity, whereas the mutations of the non-conserved sites—cGASK7Q, cGASK50Q, or cGASK392Q—had no effect (Figure 1A). By replacing a lysine with an arginine (R), we further found that K-to-R mutation of K384 or K394 did not affect cGAS activation, whereas substitution of K414 with R also inhibited cGAS activity (Figure 1B). These data suggest that, although K414 appeared to be a critical residue of cGAS, acetylation on these conserved lysine sites could possibly inhibit cGAS activation. Next we used CRISPR/Cas9 to delete endogenous cGAS in THP-1 cells. As expected, these cells failed to induce IRF3 phosphorylation and IFN production upon stimulation by introducing herring testis DNA (HT-DNA) into the cells. This deficiency could be rescued by wild-type cGAS but not acetylation-mimicking cGAS (Figures 1C and 1D). Using simultaneous K-to-Q mutation of cGAS on K384, K394, and K414 (cGAS3KQ), we obtained similar results (Figures 1E and 1F). These data further suggest that acetylation may inhibit cGAS activation.
Figure 1. Acetylation Inhibits cGAS-Mediated Type I IFN Production.

(A and B) Plasmids encoding FLAG-tagged wild-type (WT) human cGAS and its acetylation-mimetic (K-to-Q) mutants (A) or K-to-R mutants (B) were transfected into HEK293T cells that stably expressed STING (HEK293T-STING). Shown is immunoblot analysis of phosphorylation of IRF3 in the cells.
(C–H) cGAS knockout THP-1 cells were infected with a lentivirus carrying WT or acetylation-mimetic cGAS to make cell lines expressing different cGAS mutants. The cells were differentiated with phorbol-12-myristate-13-acetate (PMA) for 72 h, followed by transfection of HT-DNA (2 μg/mL) for 6 h (C and D) or 3 h (G) or for the indicated time (E, F, and H). IRF3 phosphorylation was detected by immunoblotting (C and E). Total RNAs were extracted, and the IFNB1 mRNA levels were analyzed by qPCR (D and F). The production of cGAMP was detected by LC-MS/MRM. Data are mean ± SEM from triplicates (technical replicates); unpaired t test (D and F–H).
Anti-α-tubulin (A–C and E) blots indicate loading of lanes. Data represent at least three independent experiments.
See also Figure S1.
To study the activity of cGAS i n the above cells, we used LC-MS/multiple reaction monitoring (LC-MS/MRM) to measure the production of cGAMP (Figure S1C). In contrast to wild-type cGAS, the acetylation-mimicking cGAS mutants cGASK384Q, cGASK394Q, cGASK414Q, and cGAS3KQ failed to synthesize cGAMP upon stimulation by the introduced HT-DNA (Figures 1G and 1H).
Acetylation Suppresses cGAS Activity
To unambiguously determine the effect of acetylation on cGAS at these residues, we utilized a system to genetically incorporate acetyl-lysine into a recombinant cGAS protein (Neumann et al., 2008) at each of the conserved sites to create cGASLys384Ac, cGASLys394Ac, and cGASLys414Ac (Figure 2A). We also created cGASLys392Ac as a control because the K392Q mutation did not inhibit cGAS activation (Figure 1A). Additionally, we generated site-specific anti-acetylation antibodies using the peptides FSHIEK(384)AcEILNNHGKSK(394)AcT and RKDCLKLMK(414)Ac YLLEQ to detect the K384 and/or K394 and K414 acetylated human cGAS, respectively (Figure 2B). We performed an in vitro cGAMP synthesis assay by incubating the acetylated cGAS proteins cGASLys384Ac, cGASLys394Ac, or cGASLys414Ac with the substrates ATP and GTP in the presence of HT-DNA. Consistent with the K-to-Q mutation results, cGASLys384Ac, cGASLys394Ac, or cGASLys414Ac failed to synthesize cGAMP, whereas the unmodified cGAS (cGASNon-Ac) and cGASLys392Ac effectively produced cGAMP (Figures 2C, S2A, and S2B). Thus, these data suggest that acetylation may inhibit cGAS activity.
Figure 2. Acetylation Suppresses cGAS Activity.

(A) A schematic describing the generation of site-specific acetylated recombinant cGAS protein with an acetyl-lysine tRNA, which incorporates the acetyl-lysine on the amber codon (uracil, adenine, guanine [UAG]).
(B) Immunoblot analysis of the acetylated recombinant cGAS proteins with site-specific cGAS acetylation antibodies and the pan-acetyl-lysine antibody. The anti-His blot indicates loading of lanes.
(C) Ion exchange chromatography analysis of cGAMP production from an in vitro cGAMP synthesis assay.
(D) Real-time association and dissociation of non-acetylated or acetylated recombinant cGAS proteins with biotin-EGFP (EGFP-coding DNA sequence) were recorded by ForteBio Octet red 96. Processed kinetics data show binding affinity constants (equilibrium dissociation constant, KD).
(E) FLAG-tagged WT cGAS was expressed in HEK293T cells. The cell lysates were then incubated with recombinant His-tagged cGAS proteins, cGASNon-Ac, cGASLys384Ac, cGASLys394Ac, or cGASLys414Ac. The cGAS-cGAS interaction was examined by pulling down His-tagged cGAS protein and immunoblotting with anti-FLAG antibody.
(F) cGAS knockout THP-1 cells were infected with a lentivirus carrying WT or acetylation-mimetic cGAS to make cell lines expressing different cGAS mutants. Cells were differentiated with PMA for 72 h, followed by cycloheximide (CHX) (100 μg/mL) treatment for the indicated time. cGAS was immunoblotted. The anti-GAPDH blot indicates loading of lanes.
(G) Immunofluorescence analysis of cGAS (red) in PMA-differentiated FLAG-cGAS or FLAG-cGAS3KQ expressing THP-1 cells. Hoechst (blue) stained the nuclei.
Data represent at least three independent experiments.
See also Figure S2.
Previous structural studies suggested that these sites (K384, K394, and K414) are involved in the DNA binding of cGAS (Gao et al., 2013; Kranzusch et al., 2013; Li et al., 2013; Figure S2C). We then tested the above acetylated cGAS proteins for their DNA-binding ability using biolayer interferometry and found that single-site acetylation of cGAS slightly reduced cGAS-DNA binding affinity (Figures 2D and S2D). These data are consistent with a previous report showing that the mutations on some of these sites had only a limited effect on DNA binding of cGAS (Li et al., 2013). Next we detected whether cGAS mutations affect the dimerization of cGAS because cGAS is known to bind to DNA as a dimer (Zhang et al., 2014). To do so, FLAG-tagged wild-type cGAS was expressed in HEK293T cells. The cell lysates were then incubated with recombinant His-tagged cGAS proteins: cGASNon-Ac, cGASLys384Ac, cGASLys394Ac, or cGASLys414Ac. The cGAS-cGAS interaction were examined by pulling down His-tagged cGAS protein and immunoblotting with anti-FLAG antibody. Our data showed that cGAS dimerization was not likely to be affected by cGAS acetylation (Figure 2E). Using the cGAS K-to-Q mutant-rescued cell lines, we found that these acetylation-mimicking mutants had similar protein stability and localization compared with that of wild-type cGAS (Figures 2F and 2G). It is possible that acetylation interferes with cGAS enzymatic activity.
Deacetylation of cGAS upon DNA Treatment Is Involved in cGAS Activation
To explore how cGAS acetylation is regulated in response to DNA challenge, we treated FLAG-cGAS stable THP-1 cells with HT-DNA and checked the acetylation of cGAS with site-specific antibodies. We found that acetylation of cGAS on K384 and/or K394 and K414 was reduced after HT-DNA treatment (Figure 3A), suggesting that DNA treatment leads to deacetylation of cGAS. This was confirmed by examining the acetylation of the endogenous cGAS protein in response to HT-DNA treatment and infection with herpes simplex virus (HSV)-1, a DNA virus (Figures 3B and S3A). Using MS to analyze cGAS acetylation before and after DNA virus or HT-DNA treatment, our data further supported the above observations (Figures S3B and S3C; Table S2).
Figure 3. Deacetylation of cGAS upon DNA Treatment Is Involved in cGAS Activation.

(A) PMA-differentiated THP-1 cells that stably express FLAG-cGAS were left untreated or treated with HT-DNA (2 μg/mL) for 3 h. FLAG-cGAS was immunoprecipitated, and the acetylation of cGAS was analyzed with site-specific cGAS acetylation antibodies as indicated.
(B) PMA-differentiated THP-1 cells were infected with HSV-1 (MOI = 10:1) for 6 h. cGAS was immunoprecipitated, and the acetylation of cGAS was analyzed with site-specific cGAS acetylation antibodies as indicated.
(C) PMA-differentiated THP-1 cells were treated with HT-DNA (2 μg/mL) for 3 h. The interaction between HDAC3 and cGAS was examined by immunoprecipitation (IP) and immunoblotting.
(D) Immunofluorescence analysis of cGAS (green) and HDAC3 (red) in human primary macrophages that were left untreated or stimulated with HT-DNA (1 μg/mL) for 2 h. Hoechst (blue) stained the nuclei.
(E) THP-1 cells were transfected with control siRNAs or HDAC3 siRNAs for 24 h, followed by PMA treatment for another 48 h. Cells were then treated with HT-DNA (2 μg/mL) for 3 h. cGAS was immunoprecipitated, and acetylation of cGAS was analyzed with site-specific cGAS acetylation antibodies.
※ indicates non-specific bands (B and E). WCL, whole cell lysate (A–C and E). Anti-α-tubulin blots indicate loading of lanes (C and E). Data represent three independent experiments.
It is known that deacetylation is mediated by deacetylases, sirtuins (SIRTs), and histone deacetylases (HDACs) (Shakespear et al., 2011). Interestingly, when we examined the cGAS-immunoprecipitated complex by LC-MS analysis, we found that HDAC3 protein was co-precipitated with cGAS (Table S1). This interaction of cGAS and HDAC3 was confirmed by western blotting (Figure S3D). As reported previously, the nuclear deacetylase HDAC3 can also localize in the cytoplasm (Yuan et al., 2005), and we found that the interaction of HDAC3 and cGAS is stabilized by DNA treatment (Figures 3C and 3D). Importantly, knockdown of HDAC3 prevented deacetylation of cGAS in response to HT-DNA stimuli and attenuated DNA-induced IRF3 phosphorylation (Figure 3E). Consistently, knockdown of HDAC3 also reduced HT-DNA-induced cGAMP production in cells (Figure S3E). These data suggest that acetylation contributes to cGAS activity inhibition and that deacetylation of cGAS upon DNA treatment is involved in cGAS activation.
Enforced Acetylation of cGAS by Aspirin
We next investigated whether enforced acetylation of cGAS could inhibit its activity. Aspirin, a non-steroidal anti-inflammatory drug (NSAID), is known to be able to acetylate proteins such as cyclooxygenase (COX, also known as prostaglandin-endoperoxide synthase) (Roth and Majerus, 1975; Vane and Botting, 2003). For COX acetylation, aspirin acetylates the hydroxyl group of serine and forms an ester bond (O-link). It is known that amido bonds are more stable than ester bonds; we therefore hypothesized that aspirin may acetylate the amino group of lysine to form an amido bond (N-link). We then tested whether aspirin could acetylate cGAS at the above lysine sites and inhibit its activation. Apparently, aspirin is indeed able to acetylate cGAS because incubation of aspirin with the recombinant cGAS protein led to a remarkable modification of cGAS (Figure 4A). Using our site-specific cGAS acetylation antibodies, we confirmed that aspirin can acetylate cGAS recombinant protein at K384 and/or K394 and K414 (Figure 4B). By testing the cytotoxicity of aspirin, we treated THP-1, human peripheral blood mononuclear cells (PBMCs), and mouse bone marrow cells with aspirin and found that the concentration of 4 mM aspirin did not significantly affect cell viability (Figures S4A–S4C). We thus used aspirin at 4 mM to treat cells. Importantly, treating THP-1 cells with aspirin enforced acetylation of the endogenous cGAS (Figures 4C and 4D). We further found that aspirin treatment enforced and maintained the high level of cGAS acetylation, even in the presence of DNA treatment (Figure S4D). Moreover, aspirin treatment also acetylated mouse cGAS (m-cGAS) at the corresponding sites (Figure S4E; Table S2). Thus, aspirin can be used to induce inhibitory acetylation on cGAS.
Figure 4. Aspirin Directly Acetylates Cgas.
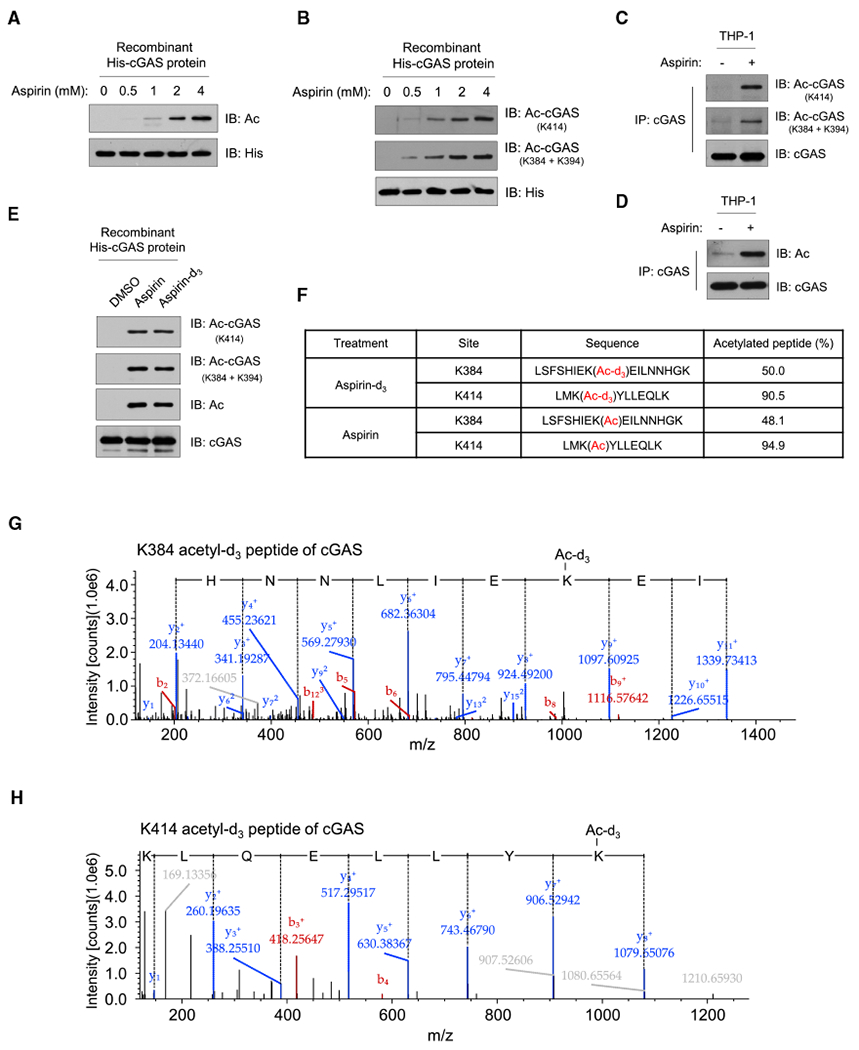
(A and B) Incubation of recombinant cGAS protein with aspirin. Shown is immunoblot analysis of cGAS acetylation with a pan-acetyl-lysine antibody (A) or site-specific cGAS acetylation antibodies (B).
(C and D) PMA-differentiated THP-1 cells were treated with DMSO (–) or aspirin (4 mM) for 24 h, and cGAS was immunoprecipitated. cGAS acetylation was analyzed by immunoblotting with site-specific cGAS acetylation antibodies (C) or pan-acetyl-lysine antibody (D).
(E) Incubation of recombinant cGAS protein with aspirin (4 mM) or isotopically labeled aspirin-d3 (4 mM). Shown is immunoblot analysis of cGAS acetylation with a pan-acetyl-lysine antibody or site-specific cGAS acetylation antibodies.
(F–H) PMA-differentiated FLAG-cGAS-expressing THP-1 cells were treated with aspirin-d3 (4 mM) or aspirin (4 mM) for 24 h, and FLAG-cGAS was immunoprecipitated. The percentage of cGAS acetylation of the indicated sites (F) and the representative K384 (G) and K414 (H) acetylated peptides of cGAS with acetyl-d3 were analyzed by Thermo Scientific Q Exactive HF Hybrid Quadrupole-Orbitrap mass spectrometer. The y ion peaks are shown in blue, and the b ion peaks are shown in red. The partial amino acid sequence (from left to right, C-terminal to N-terminal) deduced from y ions is shown on the spectrum. The mass difference between y9 and y8 ions (G) or y8 and y7 ions (H) is the mass of the acetyl-d3-modified lysine residue.
Anti-His blots (A and B) indicate loading of lanes. Data represent at least three independent experiments.
To further demonstrate that aspirin can directly acetylate cGAS, we used an isotopically labeled aspirin (aspirin-d3) according to a previous report (Tatham et al., 2017). By incubating recombinant cGAS protein with aspirin-d3, we found that, similar to the regular aspirin, this isotopically labeled aspirin can directly acetylate cGAS protein (Figure 4E). We next confirmed that aspirin-d3 treatment led to a high level of cGAS acetylation with an acetyl-d3 group in cells by MS (Figures 4F–4H; Table S2). These data collectively suggest that aspirin treatment can directly induce cGAS acetylation and that aspirin could be used to inhibit cGAS activity.
Aspirin Inhibits cGAS-Mediated IFN Production
We then tested the effect of aspirin on DNA-induced activation of cGAS and found that aspirin had a significant inhibitory effect on IRF3 phosphorylation induced by the DNA virus HSV-1 but not the RNA virus vesicular stomatitis virus (VSV) (Figures 5A and 5B). We obtained a similar result using aspirin-d3 (Figure 5C). Thus, aspirin inhibits DNA-mediated activation of the cGAS-STING pathway.
Figure 5. Aspirin Inhibits cGAS-Mediated IFN Production.
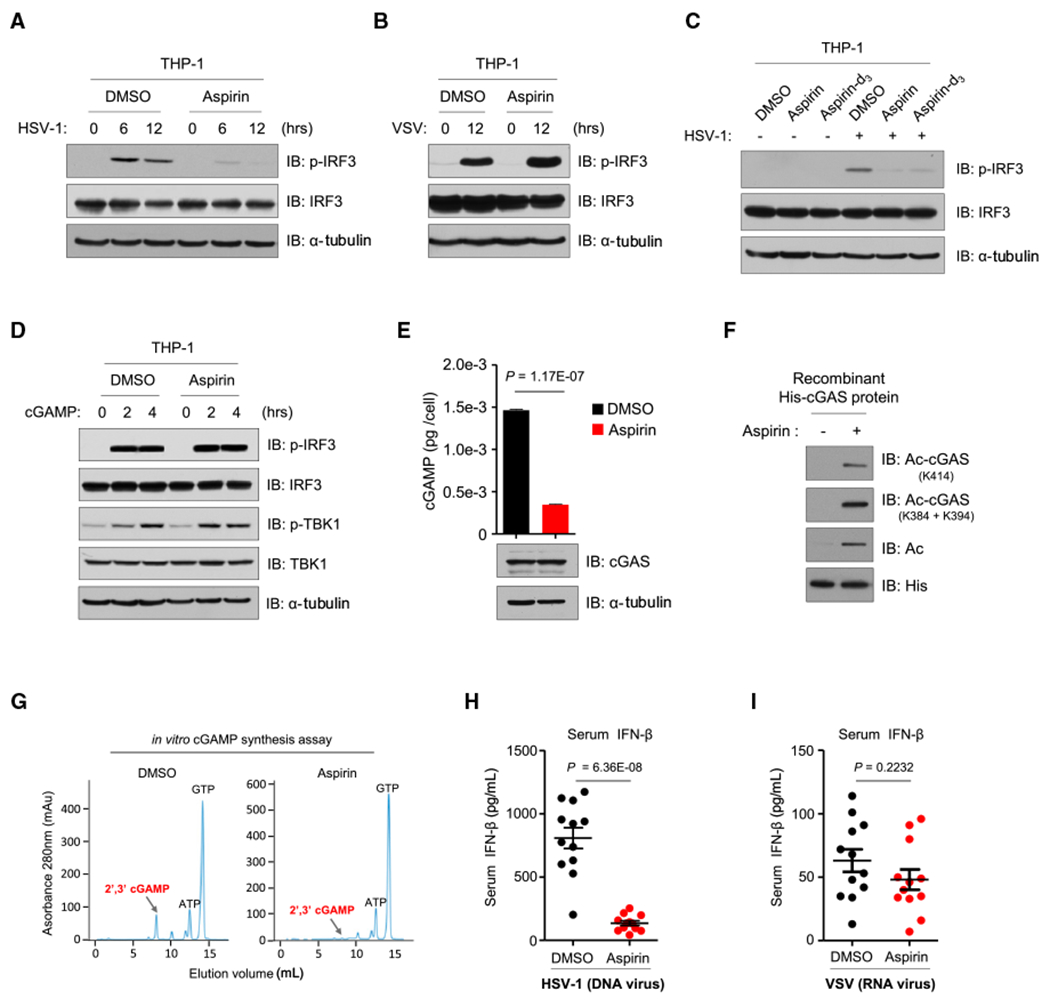
(A and B) PMA-differentiated THP-1 cells were infected with HSV-1 (MOI = 10:1) (A) or VSV (MOI = 10:1) (B) after DMSO or aspirin (4 mM) pretreatment, and the phosphorylation of IRF3 was analyzed by immunoblotting.
(C) PMA-differentiated THP-1 cells were infected with HSV-1 (MOI = 10:1) after DMSO, aspirin (4 mM), or aspirin-d3 (4 mM) pretreatment, and the phosphorylation of IRF3 was analyzed by immunoblotting.
(D) PMA-differentiated THP-1 cells were pre-treated with DMSO or aspirin (4 mM) for 24 h, followed by treatment with cGAMP (1 μg/mL) for the indicated hours. Shown is immunoblot analysis of the phosphorylation of IRF3 and TBK1.
(E) PMA-differentiated THP-1 cells were treated with DMSO or aspirin for 24 h, followed by a 2-h treatment of HT-DNA (1 μg/mL). cGAMP production was quantified by LC-MS/MRM; data are mean ± SEM from triplicates (technical replicates), unpaired t test. Shown is immunoblot analysis of cGAS in the corresponding protein samples (bottom).
(F and G) Immunoblot analysis (F) and in vitro cGAMP synthesis assay (G) of recombinant cGAS proteins in the presence of DMSO or aspirin (4 mM). Site-specific cGAS acetylation antibodies and the pan-acetyl-lysine antibody were used to analyze cGAS acetylation. The anti-His blot indicates loading of lanes (F).
(H and I) Wild-type C57BL/6J mice were given a daily intraperitoneal (i.p.) injection of DMSO (n = 12) or aspirin (50 mg/kg, n = 12) for 2 days, followed by i.p. injection of either HSV-1 (H) or VSV (I) for 6 h. Serum from mice was obtained for ELISA analysis of IFN-β concentration. Data are mean ± SEM, unpaired t test.
Anti-α-tubulin blots indicate loading of lanes (A–E). Combined data of two independent experiments are shown (H and I); other data represent three independent experiments.
See also Figure S5.
To confirm that the inhibitory effect of aspirin on the cGAS-STING pathway is through inhibiting cGAS, we first tested whether aspirin has any effect on cGAMP-mediated activation of the pathway. We found that aspirin did not inhibit cGAMP-activated phosphorylation of IRF3 and TBK1 (Figure 5D) or cGAMP-induced IFN production (Figure S5A). We then used another STING activator, cyclic diguanylate (c-di-GMP) (Burdette et al., 2011), to challenge the aspirin-treated cells and found that aspirin did not inhibit IFN production activated by c-di-GMP (Figure S5B). These data demonstrate that the inhibitory effect of aspirin is most likely through blocking cGAS activity. Consistently, aspirin suppressed cGAMP synthesis in HT-DNA-stimulated cells without significantly affecting the protein level of cGAS (Figures 5E and S5C). By incubating aspirin with recombinant cGAS protein, we found that aspirin acetylated the protein (Figure 5F) and blocked the production of cGAMP (Figure 5G). Taken together, these results suggest that aspirin acetylates cGAS and inhibits its activity.
Next, we studied whether aspirin can specifically inhibit cGAS-mediated IFN production in vivo by injecting mice with aspirin prior to HSV-1 or VSV infection. We measured the serum levels of IFN-β and found that aspirin significantly inhibited HSV-1-but not VSV-induced IFN-β production in mice (Figures 5H and 5I). Thus, aspirin blocks cGAS-mediated type I IFN production both in vitro and in vivo.
Aspirin Suppresses DNA-Mediated Autoimmunity in Mice
To determine whether aspirin could be used to treat self-DNA-induced autoimmunity, we used Trex1−/− mice, which exhibit similar autoimmune disorders as human patients (Morita et al., 2004). We first isolated bone marrow cells from both wild-type and Trex1−/− mice. As expected, Trex1−/− bone marrow cells showed highly elevated levels of ISGs compared with wild-type cells. Treating Trex1−/− bone marrow cells with aspirin strongly inhibited the expression of ISGs (Figure S6A). We further calculated the half-maximal inhibitory concentration (IC50) of aspirin for suppressing ISG expression by performing a dose-response curve experiment (Figure 6A). According to a previous study (di Palma et al., 2006), we gave mice a daily aspirin injection (50 mg/kg) for 10 days and monitored the body weight of mice during the process. We observed barely any weight loss in the treated mice (Figure S6B). We then injected Trex1−/− mice intraperitoneally (i.p.) with aspirin to test whether aspirin has a therapeutic effect to alleviate the disease phenotype in these mice. Because it has been reported that aspirin also targets other inflammation-related pathways, such as COX-1 and COX-2 (Vane and Botting, 2003) and nuclear factor κB (NF-κB) (Kopp and Ghosh, 1994; Yin et al., 1998), we also used diclofenac sodium, a potent inhibitor of COX-1 and COX-2 (Grabocka and Bar-Sagi, 2016), and salicylic acid, a metabolite of aspirin (acetylsalicylic acid) (Hutt et al., 1986) known to inhibit the NF-κB pathway (Kopp and Ghosh, 1994; Yin et al., 1998), as control compounds (Figures S6C and S6D). Because salicylic acid does not have an acetyl group, it cannot acetylate cGAS protein (Figure S6E). Consistently, salicylic acid did not obviously inhibit DNA virus-induced IRF3 phosphorylation (Figure S6F). Because NF-κB is involved in the induction of IFN and ISGs (Chen et al., 2016b; Pfeffer, 2011), it is possible that salicylic acid could partially inhibit DNA-induced IFN production and ISG expression by inhibiting NF-κB. Indeed, salicylic acid had an inhibitory effect on the expression of ISGs in Trex1−/− bone marrow cells (Figure S6G). However, compared with aspirin, this inhibitory effect was much weaker, especially in lower concentrations (Figures 6A and S6G). In line with these observations, after daily injection of these drugs for 1 week, we detected ISG expression levels in mouse hearts, a widely used indicator for disease phenotype (Gao et al., 2015; Gray et al., 2015), and found that, although aspirin significantly reduced ISG expression in the hearts of Trex1−/− mice, salicylic acid had a weak effect, and diclofenac sodium did not have any effect (Figure 6B). By detecting m-cGAS acetylation in spleens, which express much higher levels of m-cGAS than the hearts of these mice, we found that aspirin injection led to obviously increased m-cGAS acetylation in mouse tissue (Figure 6C). As expected, neither salicylic acid nor diclofenac sodium acetylated cGAS in mouse tissue (Figure 6C). We also detected m-cGAS acetylation in spleens from both wild-type and Trex−/− mice. Consistent with our finding that DNA treatment led to the deacetylation of cGAS, our results showed that, in the spleens of Trex1−/− mice, m-cGAS acetylation levels were reduced (Figure S6H). Further, daily injection with aspirin, but not salicylic acid or diclofenac sodium, significantly prolonged the survival of Trex1−/− mice (Figure 6D). Thus, these data suggest that the inhibitory effect of aspirin is unlikely to be through inhibition of the COX-1/2 pathway and that acetylation is important for aspirin-mediated inhibition on self-DNA-induced autoimmunity.
Figure 6. Aspirin Suppresses DNA-Mediated Autoimmunity in Mice.

(A) Bone marrow cells from Trex1−/− mice were treated with aspirin at the indicated concentrations for 2 days. The mRNA levels of ISGs, as indicated, were analyzed by qPCR (relative to that of WT bone marrow cells). The IC50 was calculated by Statistical Package for the Social Sciences (SPSS). Data are mean ± SEM from triplicates (technical replicates).
(B) Trex1−/− mice (n = 6) were given daily administration (i.p.) of aspirin (50 mg/kg), salicylic acid (50 mg/kg), diclofenac sodium (1 mg/kg), or DMSO for 7 days, and then the mRNA levels of ISGs in the mouse hearts were analyzed by qPCR. Data are mean ± SEM, unpaired t test.
(C) 3-week old Trex1−/− mice were given daily treatment of aspirin (50 mg/kg), salicylic acid (50 mg/kg), diclofenac sodium (1 mg/kg), or DMSO for 1 week by i.p. injection. m-cGAS in the spleen was immunoprecipitated, and the acetylation of m-cGAS was analyzed with pan-acetyl-lysine antibody; anti-α-tubulin blots indicate loading of lanes. * indicates non-specific bands.
(D) 3-week old Trex1−/− mice were administered (i.p.) aspirin (50 mg/kg), salicylic acid (50 mg/kg), diclofenac sodium (1 mg/kg), or DMSO daily. Survival curves of mice are shown. Statistical analysis was performed with a log rank (Mantel-Cox) test.
(E) Trex1−/− mice (n = 6) were given daily administration (i.p.) of MS-275 (20 mg/kg) or DMSO for 10 days, mRNA levels of ISGs in mouse hearts were analyzed by qPCR. Data are mean ± SEM, unpaired t test.
Data represent at least two independent experiments (A–C and E); combined data of at least two independent experiments are shown (D).
See also Figure S6.
Because we showed that HDAC3 mediated the deacetylation of cGAS in response to DNA, we next tested whether inhibiting HDAC3 could be effective in ameliorating the autoimmune phenotype in Trex1−/− mice. To do so, we first treated Trex1 knockdown L929 cells with two HDAC inhibitors, RGFP966 and MS-275. RGFP966 is a selective inhibitor of HDAC3 (Malvaez et al., 2013), and MS-275 can target HDAC1 and HDAC3 (Tatamiya et al., 2004). By measuring the expression levels of ISGs in the treated cells, we found that, although RGFP966 had a mild inhibitory effect on ISG expression, MS-275 inhibited ISG efficiently (Figure S6I). These data are consistent with our original cell experiments showing that knockdown of HDAC3 only partially blocked cGAS activation (Figures 3E and S3E), which indicated that other deacetylases may also contribute to cGAS deacetylation. Given the strong inhibitory effect of MS-275 and that MS-275 is a drug approved by the Food and Drug Administration (FDA), we next used MS-275 to treat Trex1−/− mice. Our results showed that MS-275 can inhibit ISG levels in the hearts of Trex1−/− mice (Figure 6E). Thus, targeting HDAC could also have an inhibitory effect on self-DNA-induced autoimmune responses in Trex1−/− mice.
Aspirin Suppresses DNA-Mediated Autoimmunity in Human Patient Cells
To further study the effectiveness of aspirin in treating AGS, we recruited an AGS patient and his healthy elder brother from an AGS family (Guo et al., 2014) and isolated PBMCs from the brothers. We first confirmed the frameshift mutation (c.459_460insA) of TREX1 in the AGS sibling, whose PBMCs had very little TREX1 expression (Figures 7A and 7B) and highly elevated levels of ISGs (Figure 7C). Treatment of the AGS patient’s PBMCs with aspirin significantly reduced the levels of ISGs (Figure 7C). MS-275 treatment also reduced the elevated ISG levels in patient cells (Figure S7A). Thus, our data further support the idea that aspirin could be used to treat self-DNA-induced autoimmune responses.
Figure 7. Aspirin Suppresses DNA-Mediated Autoimmunity in Human Patient Cells.

(A) Schematic diagram showing the frameshift mutation of TREX1 in the AGS patient (AGS, bottom) in comparison with his healthy brother (Healthy, top). The frameshift mutation (c. 459-460insA, red A in the diagram) results in a premature stop codon (red *) at the position of the 156th amino acid. The dark gray color in the schematic indicates different regions according to Uniprot: Q9NSU2-1. The first region (position 75–76) is described as a substrate-binding region, the second region (position 109–118) is a proline-rich region, and the last region (position 291–369) is necessary for endoplasmic reticulum localization.
(B) Immunoblot analysis of TREX1 and cGAS in PBMCs from the healthy brother and the AGS patient; the anti-α-tubulin blot indicates loading of lanes.
(C) PBMCs from the healthy brother and the AGS patient were treated with DMSO or aspirin (4 mM) for 2 days. The mRNA levels of ISGs were detected by qPCR. Data are mean ± SEM from triplicates (technical replicates), unpaired t test.
Data represent at least two independent experiments (B and C).
See also Figure S7.
Aspirin Suppresses cGAS-Mediated Type I Interferonopathies
To determine whether aspirin-mediated cGAS inhibition presents a therapeutic strategy for other cGAS-related primary interferonopathies, we used small interfering RNAs (siRNAs) to knock down the expression of Dnase2a or Rnaseh2b in L929 cells. It has been reported previously that the loss-of-function mutations of Dnase2a or Rnaseh2b lead to type I interferonopathies (Crow and Manel, 2015; Gao et al., 2015; Mackenzie et al., 2016); deficiency of Dnase2a or Rnaseh2b resulted in significantly elevated ISG expression, and, importantly, elevated ISG expression was efficiently inhibited by aspirin (Figures S7B–S7E). STING-associated vasculopathy with onset in infancy (SAVI) is a recently characterized form of AGS-like disease caused by auto-activating mutations in STING (Liu et al., 2014; Melki et al., 2017). Because this disease affects a step downstream of cGAS activation, we tested whether aspirin has any effect in an SAVI model. To do so, we created a gain-of-function-mutated STING-driven cell model. We found that aspirin treatment had no effect on STING-mediated IRF3 phosphorylation, whereas BX-795, a known TBK1 inhibitor (Clark et al., 2009), strongly inhibited IRF3 phosphorylation (Figure S7F). These data indicate that aspirin only affects cGAS- but not STING-mediated immune responses.
DISCUSSION
Detection of cytosolic DNA by cGAS is critical for the immune system to sense and fight against infection. However, deregulated chronic activation of cGAS by self-DNA is the primary cause for several devastating autoimmune diseases, including lupus and AGS (Crow and Manel, 2015; Gao et al., 2015; Gray et al., 2015; Lisnevskaia et al., 2014; Mackenzie et al., 2016), but so far, there is no effective therapy for treating these diseases (Crow et al., 2015). In the current study, we report a novel regulation of cGAS activity. Our findings also indicate that aspirin could be used to inhibit cGAS activation and treat self-DNA-induced autoimmune diseases.
Aspirin is widely used to treat inflammation by inhibiting COX. In our study, using another specific COX inhibitor, diclofenac sodium, we found that diclofenac sodium had no effect on cGAS activation. This result suggests that aspirin-mediated inhibition of cGAS activation is not related to its effect on COX. It is known that cGAS activates downstream IRF3 and NF-κB signaling via STING (Sun et al., 2013) and that both IRF3 and NF-κB are involved in the production of IFN (Chen et al., 2016b). It has also been reported that NF-κB can regulate the expression of some ISGs (Pfeffer, 2011). As expected, salicylic acid had a partial inhibitory effect on ISG expression, possibly through inhibition of the NF-κB pathway. In contrast to salicylic acid, however, aspirin may inhibit the activation of both IRF3 and NF-κB by targeting cGAS. Therefore, aspirin, but not salicylic acid, was effective in promoting the survival of Trex1−/− mice. Moreover, using isotopically labeled aspirin, we demonstrated direct acetylation of cGAS by aspirin. Our findings collectively suggest that aspirin functions as a tangible cGAS inhibitor.
Aspirin has been shown to acetylate many protein targets in addition to cGAS. It is possible that aspirin also targets other proteins in addition to cGAS in vivo. However, we showed that aspirin strongly inhibited cGAS-mediated TBK1-IRF3 signaling but did not affect STING activator cGAMP- or c-di-GMP-induced downstream TBK1-IRF3 signaling. We also showed that aspirin did not inhibit SAVI-associated STING mutant-mediated IRF3 activation. In addition, aspirin only had a marginal effect on RNA virus-induced IFN production in mice. These data indicate that the inhibitory effect of aspirin on the DNA-cGAS-STING-TBK1-IRF3 axis is mainly achieved through inhibition of cGAS.
With an acetyl-lysine-incorporating protein synthesis system, we generated recombinant acetylated cGAS proteins and showed that the acetylation on these residues strongly inhibits the activity of cGAS. It has been reported that these acetylation sites are involved in the DNA binding of cGAS (Civril et al., 2013; Gao et al., 2013; Kranzusch et al., 2013; Zhang et al., 2014; Zhou et al., 2018). Using recombinant acetylated cGAS proteins, we found that the acetylation on each of the individual sites slightly reduced the DNA binding activity of cGAS. These data are consistent with a previous study showing that the mutations on some of these residues have only a limited effect on DNA binding of cGAS (Li et al., 2013). Accumulating evidence suggests that several regions of cGAS protein are important for cGAS-DNA binding (Civril et al., 2013; Gao et al., 2013; Kranzusch et al., 2013; Li et al., 2013; Zhang et al., 2014; Zhou et al., 2018). Therefore, when K384, K394, or K414 is acetylated, other regions of cGAS, such as the N-terminal region (Du and Chen, 2018; Lee et al., 2017; Tao et al., 2017), could still contribute to DNA binding. Binding of DNA to cGAS led to deacetylation of cGAS, promoting cGAS activation.
Our study suggests that K414 is a critical residue for cGAS function. Interestingly, a recent study reported that, in resting cells, cGAS undergoes K48-linked ubiquitination at K414, leading to p62-dependent selective autophagic degradation of cGAS and that, upon DNA virus infection, this inhibitory ubiquitination is removed to promote cGAS activation (Chen et al., 2016a). It is likely that, on K414, acetylation may compete with ubiquitination to maintain a proper level of cGAS protein in resting cells. K27-linked ubiquitination on K384 of cGAS has been shown to potentiate cGAS activity in response to DNA virus infection (Wang et al., 2017). In response to DNA treatment, HDAC-mediated removal of K384 acetylation may allow the K27-linked ubiquitination on K384 to further promote cGAS activation. Together, K384 and K414 of cGAS are critical residues for cGAS regulation by multiple PTMs. Future studies are necessary to further investigate the complex interplay of these PTMs.
The deletion of Cgas eliminated the lethality and autoimmune phenotypes in Trex1−/− mice, emphasizing the importance of cGAS inhibition in treating these diseases (Gao et al., 2015; Gray et al., 2015). cGAS has been shown to be regulated by phosphorylation, glutamylation, and sumoylation (Hu et al., 2016; Seo et al., 2015; Xia et al., 2016), but further efforts are needed to develop chemicals that can alter these modifications on cGAS to inhibit its activity. Screening of cGAS inhibitors based on the cGAS crystal structure has been reported recently, but none of these inhibitors have been tested in mouse AGS models or human AGS patients (An et al., 2015). In this study, we revealed that acetylation inhibits cGAS activity. This regulation of cGAS seems to be evolutionally conserved because the acetylation sites we identified are conserved across species. More importantly, we demonstrated that a commonly used NSAID, aspirin, could inhibit cGAS activation through acetylation and be used to treat AGS and potentially other DNA-mediated autoimmune diseases. Particularly, the effective dosage of aspirin needed for inhibiting cGAS falls well below the upper limit for its use in humans. Therefore, our findings provide a feasible therapy for treating AGS and other autoimmune diseases.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Tao Li (tli@ncba.ac.cn).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Trex1+/− C57BL/6 mice were from D. Barnes and T. Lindahl (Cancer Research UK) (Morita et al., 2004; Yang et al., 2007). All animal experiments were performed in accordance with the National Institutes of Health guide for the care and use of laboratory animals and with the approval of the Institutional Animal Care and Use Committee of the National Center of Biomedical Analysis. Stock solutions of aspirin or the control compounds in DMSO was further diluted in PBS containing 5% (vol/vol) Cremophor EL (C5135, Sigma-Aldrich) before intraperitoneal (i.p.) injection. 3-week old Trex1−/− mice were given aspirin, salicylic acid, diclofenac sodium, MS-275 or DMSO treatment, respectively. After daily administration for 7 or 10 days, mice were sacrificed and hearts of mice were collected for quantitative polymerase chain reaction (qPCR) analysis of the expression levels of ISGs. Spleens were collected for immunoblot analysis of cGAS acetylation. To evaluate the effect of aspirin on the lethality of Trex1−/− mice, 3-week old Trex1−/− mice were given daily administration of aspirin, salicylic acid, diclofenac sodium or DMSO for 7 weeks. All the analyses were performed blindly. Trex1−/− mice were randomly allocated into experimental groups for further treatment. Cell samples were allocated based on the genotype of interest.
Cells
HEK293T and HeLa cells were cultured in DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS), 2 mM glutamine, penicillin (100 U/mL) and streptomycin (100 μg/mL). Murine bone marrow cells, THP-1 and L929 cells were cultured in RPMI-1640 medium supplemented with 10% (vol/vol) FBS. 2 mM glutamine, penicillin (100 U/mL) and streptomycin (100 μg/mL).
HEK293T, L929 and PMA (100 ng/mL)-differentiated THP-1 cells were transfected using VigoFect (Vigorous Biotechnology) or Lipofectamine 2000 (Invitrogen) following the manufacture’s instructions. For treatment of aspirin or salicylic acid, stock solution was diluted to the final concentrations in Opti-MEM (GIBCO), the pH value of which was adjusted to be under 7 (Charifson and Walters, 2014). The cells were first washed with PBS for three times and then cultured with the aspirin- or salicylic acid-containing Opti-MEM for 1-2 days before further treatment. The cytotoxicity was analyzed by CellTiter 96® AQueous One Solution Cell Proliferation Assay (G3580, Promega) according to the manufacture’s instruction.
For cGAMP or c-di-GMP stimulation, cells were incubated for 30 min at 37°C with cGAMP or c-di-GMP in permeabilization buffer (50 mM HEPES pH 7, 100 mM KCl, 3 mM MgCl2, 0.1 mM DTT, 85 mM sucrose, 0.2% BSA, 1 mM ATP and 0.1 mM GTP) with 1 μg/mL digitonin (Sigma, D141). The permeabilization buffer was replaced with RPMI-1640 medium and cells were cultured for indicated time before further analysis.
To make FLAG-cGAS-stable cells, lentiviral stocks were generated in HEK293T. THP-1 cells were transduced with a pCDH-Puro lentivirus carrying FLAG-cGAS expression sequence in the presence of 5 mg/mL polybrene (Sigma-Aldrich). After 24-hour culturing, transduced cells were selected with puromycin (2 μg/mL, Amresco). Endogenous cGAS was deleted in THP1 cells using the lentiCRISPR system. Single-guide RNA (sgRNA) sequences of cGAS were designed by the online tool from Dr. Feng Zhang’s lab (http://zlab.bio/guide-design-resources). To rescue the expression of cGAS, cGAS−/− THP1 cells were infected with pCDH-CopGFP lentivirus carrying sgRNA-resistant wild-type or the mutant cGAS expression sequences. Rescued cells (GFP+) were sorted by flow cytometry (FACS Aria II, BD Biosciences) 3 days after infection. To establish L929-FLAG-m-cGAS stable cells, lentiviral stocks were generated in HEK293T. L929 cells were infected with pCDH-CopGFP lentivirus. Cells that stably expressing FLAG-m-cGAS (GFP+) were sorted by flow cytometry (FACS Aria II, BD Biosciences) 3 days after infection.
THP-1, L929, HeLa and HEK293T cells were obtained from ATCC. All the cell lines have been tested to be mycoplasma-free by PCR.
Viruses
HSV-1 was propagated and titered by plague assay on Vero cells. 6-week old female C57BL/6 mice were given daily administration of aspirin (50 mg/kg) or DMSO for two days before infected with HSV-1 (107 pfu per mouse) or VSV (107 pfu per mouse) by i.p. injection. Serum was collected 6 h after viral infection, and was measured for cytokine production by ELISA. All the analyses were performed blindly. Mice were randomly allocated into experimental groups for virus infection.
METHODS DETAILS
Quantitative Polymerase Chain Reaction (qPCR)
Cells were collected and the total RNA was extracted with TRIzol (Thermo Fisher Scientific). The diluted RNA was reverse-transcripted (PrimeScript RT Master Mix, RR036A, TAKARA) and analyzed with qPCR (FastStart Universal SYBR Green Master (Rox), 04913914001, Roche) for the expression of genes on StepOnePlus Real-Time PCR System (Applied Biosystems). The primers were synthesized from Invitrogen and listed in Table S3, GAPDH and Hprt were used for normalization.
ELISA
Mice serum was collected and measured for IFN-β concentration using a mouse IFN-β ELISA kit (439408, Biolegend) according to the manufacturer’s protocol. PMA-differentiate THP-1 cells were treated with 20 μM diclofenac sodium or DMSO for 24 h and collected for measuring PGE2 concentration using Human PGE2 ELISA kit (EH4233, FineTest).
Isolation of PBMCs from AGS patient
After written informed consent was obtained, we collected the peripheral blood of the patient and his healthy brother with the help of the department of blood transfusion, affiliated hospital of Academy of Military Medical Sciences (AMMS). Human blood samples were obtained following the National Institutes of Health Guide for the use of human samples and with the approval of the Scientific Investigation Board of affiliated hospital of AMMS. Human primary PBMCs were isolated with HISTOPAQUE-1077 (Sigma-Aldrich) according to the manufacturer’s protocols. Cells were maintained in RPMI-1640 medium supplemented with 10% (vol/vol) FBS, 2 mM glutamine, penicillin (100 U/mL) and streptomycin (100 μg/mL).
Mass Spectrometry
To identify cGAS acetylation in FLAG-cGAS-expressing THP-1 cells and quantification of HSV-1-induced cGAS acetylation change in these cells, desalting and MS analysis of digested peptides was carried out as previously described (Wang et al., 2014). In brief, desalted peptides were analyzed using a TripleTOF 5600+ mass spectrometer (AB Sciex) coupled to an ekspert nanoLC 425 (Eksigent). TripleTOF 5600+ raw data were processed using MS Data Converter (AB Sciex) to generate .mgf files. All Mascot searches were performed against a downloaded SwissProt database (released in Jan 2015, 547,357 proteins, 194,874, 700 residues) with the taxonomy of Homo sapiens. The percentage of acetylation was calculated by peptides counts. The quantification of aspirin-, aspirin-d3- or HT-DNA-induced cGAS acetylation change in FLAG-cGAS-expressing THP-1 cells and aspirin-induced cGAS acetylation in L929-FLAG-m-cGAS cells were performed on the Thermo Scientific Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer. The MS data were analyzed with Proteome Discover 2.1. The percentage of acetylation was calculated by peptides peak area.
Immunoblotting and Immunoprecipitation
The total cell lysates were prepared in M2 buffer (20 mM Tris-HCl pH 7.5, 0.5% Nonidet P-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol) supplemented with complete protease inhibitor cocktail (Roche, 04693132001). Cell lysates were separated by SDS-PAGE and analyzed by immunoblotting. Proteins were visualized by enhanced chemiluminescence according to the manufacturer’s instructions (ThermoFisher Scientific).
To detect cGAS acetylation, cells were lysed in M2 buffer supplemented with complete protease inhibitor cocktail, 1 μM trichostatin A (TSA) and 10 mM nicotinamide (NAM), followed by sonication and centrifugation at 16,000 × g for 10 min at 4°C. The supernatants were immunoprecipitated with cGAS antibody or FLAG antibody for 12 h at 4°C. The immunoprecipitants were washed six times with M2 buffer and boiled in 1 × SDS-loading buffer for immunoblot analysis.
To detect interaction between cGAS and HDACs, cells were lysed in M2 buffer supplemented with complete protease inhibitor cocktail, followed by centrifugation at 16,000 × g for 10 min at 4°C. The supernatants were immunoprecipitated with anti-HDAC3 or anti-FLAG antibody for 12 h at 4°C. The immunoprecipitants were washed six times with M2 buffer and boiled in 1 × SDS-loading buffer for immunoblot analysis.
To detect cGAS acetylation in vivo, 3-week old Trex1−/− mice were given daily treatment of aspirin (50 mg/kg), salicylic acid (50 mg/kg), diclofenac sodium (1 mg/kg) or DMSO for 1 week by i.p. injection, respectively. Spleens of mice were homogenized with handheld homogenizer (PT 1200 E, Kinematica) and lysed in RIPA buffer (50 mM Tris-HCl pH 7.5,1 % Nonidet P-40, 150 mM NaCl, 5 mM EDTA, 0.5% Deoxycholate Sodium) supplemented with complete protease inhibitor cocktail, 1 μM TSA and 10 mM NAM, followed by sonication and centrifugation at 16,000 × g for 30 min at 4°C. The supernatants were immunoprecipitated with mouse specific cGAS antibody for 12 h and then incubated with Protein G Sepharose (17-0618-01, GE Healthcare) for 3 h at 4°C. The immunoprecipitants were washed six times with RIPA buffer and boiled in 1 × SDS-loading buffer for immunoblot analysis.
Purification of site-specific acetylated cGAS recombinant proteins
The site-specific acetylated cGAS recombinant proteins were generated according to a previous report (Neumann et al., 2008). In brief, Escherichia coli strain, BL21 (DE3), was transformed with plasmids pAcKRS-3 and pCDF PylT-1 carrying the ORF for cGAS with an amber codon at the desired site. The cells were first grown overnight in LB medium supplemented with 50 μg/mL kanamycin and 50 μg/mL spectinomycin (LB-KS) at 37°C. 5 mL overnight cultured bacteria were then inoculated into 100 mL LB-KS for further culturing. When the OD600 reached 0.4~0.6, 20 mM nicotinamide (NAM) and 10 mM acetyl-lysine were added and 30 min later, the protein expression was induced at 16-18°C overnight by adding 0.5 mM IPTG. Cells were harvested after induction, and were washed with ice-cold PBS containing 20 mM NAM, the proteins were purified with HisTrap FF (GE Healthcare, 17-5319-01) according to the manufacturer’s protocol.
In vitro cGAMP synthesis assay
According to a previously published protocol (Li et al., 2013), unmodified cGAS protein (cGASNon-Ac) and lysine-acetylated cGAS protein (cGASLys384Ac, cGASLys394Ac, cGASLys414Ac and cGASLys392Ac) were incubated with HT-DNA (20 μg/mL) in reaction buffer (20 mM HEPES, pH 7.5, 5 mM MgCl2, 2 mM ATP, 2 mM GTP) at 37°C for 2 h. The samples were diluted by 5-fold and centrifuged at 16,000 × g for 10 min, and the supernatant was filtered with a 10 kD ultrafiltration filter (Millipore), and analyzed by ion exchange chromatography with a MonoQ column (GE Healthcare).
cGAMP extraction
cGAMP extraction from cells was carried out according to previous report (Wu et al., 2013a). After HT-DNA stimulation, the cells were washed with PBS and lysed with 700 μL of cold extraction solvent (40:40:20 (v/v/v) methanol-acetonitrile-water). The cell lysates were stored at −20°C for 30 min and then centrifuged at 16,000 × g for 5 min. The supernatants were evaporated at room temperature and the pellets were resuspended in 100 μL of ammonium acetate buffer (10 mM ammonium acetate, 0.05% acetate in water). After centrifugation at 16,000 × g for 10 min, the supernatants were quantified by liquid chromatography-MS/multiple reaction monitoring (LC-MS/MRM).
Quantification of cGAMP using LC-MS/MRM
The LC–MS/MS system consisted of an ekspert ultraLC 110-XL system (AB Sciex) and a Qtrap6500 triple quadropole mass spectrometer (AB Sciex). The Qtrap6500 triple quadropole mass spectrometer was operated in positive ionization mode for MRM analysis of cGAMP. The source conditions were set as follows: ionspray voltage was 5.5 kV, ion source temperature was 550°C, curtain gas was 20 (arbitrary units), collision gas was Medium (CAD) and the dwell time for cGAMP was 100 ms. The optimized ion transitions were: cGAMP m/z 675 → 524; m/z 675 → 506; m/z 675 → 136. The parameters, entrance potential (EP), declustering potential (DP), collision energy (CE) and collision exit potential (CXP), were set as 7 V, 90 V, 29 V, and 18 V, respectively.
RNA Interference
For siRNA-mediated knockdown in THP1 cells, siRNAs were electroporated into undifferentiated cells at a final concentration of 100 nM using the Amaxa Transfection Device following the manufacturer’s instructions. 24 h after electroporation, cells were differentiated with PMA for 48 h followed by transfection of HT-DNA. For L929 cells, siRNAs were transfected with Lipofectamine RNAiMAX (Invitrogen) at a final concentration of 100 nM. After 24-hour siRNA transfection, cells were treated with aspirin for 48 h. Drug-containing medium was changed every 24 h. Mouse Dnase2a (siG151201033118), mouse Rnaseh2b (siG170516091413), human HDAC3 (siB1032283530) and control siRNAs were purchased from RiboBio. Mouse Trex1 (MSS238570, 5′-ACCGACAGACUCACAUACUGCUGAA-3′) siRNA was from Invitrogen.
IC50 Determination
Bone marrow cells from Trex1−/− mice were collected and treated with Red Blood Cells Lysis Buffer (155 mM NH4Cl, 12 mM KHCO3 and 100 μM EDTA). Cells were washed and resuspended in medium containing different concentration of aspirin or salicylic acid. Drug-containing medium was changed every 24 h. 48 h later, cells were collected and the mRNA levels of ISGs were analyzed by qPCR. Data were analyzed by GraphPad Prism and IC50 values were calculated with SPSS (Version 21).
DNA Binding Assay
The real-time binding assay was performed by biolayer interferometry using ForteBio Octet red 96. Briefly, Streptavidin (SA) Biosensor from ForteBio were used to capture 20 nM biotin-ISD (biotinylated interferon stimulatory DNA) onto the surface of the SA biosensor. After reaching base line, sensors were subjected to the association step containing 15.625, 31.25, 62.5, 125, 250 or 500 nM His-tagged non-acetylated or acetylated recombinant cGAS proteins for 60 s and then dissociated for 60 s. All reagents were diluted in ForteBio kinetics buffer (50 mM Tris-HCl pH7.5, 150mM NaCl, 0.02% Tween 20, 0.1 mg/mL BSA). Biotin-labeled EGFP-coding sequence (Biotin-EGFP, primer sequences for EGFP cloning are listed in Table S3) was also used as a longer DNA to perform this assay.
Immunofluorescence
To detect the colocalization of HDAC3 and cGAS, human primary macrophages were seeded on coverslips in 24-well plates. After transfection with HT-DNA, the cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked in 3% BSA for 1 h. Cells were then incubated with primary antibodies (such as rabbit anti-cGAS (Cell Signaling Technology) and mouse anti-HDAC3 (Santa Cruz)) overnight at 4°C,. Alexa Fluor 488- and 546-conjugated secondary antibodies and Hoechst (Life technologies) were incubated for 1 h before the images were acquired using a ZEISS LSM 880 (Zeiss) confocal microscope.
To detect the localization of acetylated cGAS, PMA-differentiated FLAG-cGAS or FLAG-cGAS3KQ-expressing THP1 cells were seeded on coverslips in 24-well plates. After fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked in 3% BSA for 1 h, cells were then incubated with anti-FLAG antibodies overnight at 4°C. Alexa Fluor 546-conjugated secondary antibody and Hoechst (Life technologies) were incubated for 1 h before the images were acquired using a ZEISS LSM 880 (Zeiss) confocal microscope.
cGAS dimerization
HEK293T cells were transfected with FLAG-tagged WT cGAS by VigoFect (Vigorous Biotechnology). After 24 h, the cell lysates were prepared in M2 buffer supplemented with complete protease inhibitor cocktail. Cell lysates were then incubated with recombinant His-tagged cGAS proteins, cGASNon-Ac, cGASLys384Ac, cGASLys394Ac, or cGASLys414Ac for 12 h. Then the cell lysates were incubated with Ni-NTA agarose beads (QIAGEN) for 4 h to pull down His-tagged cGAS proteins. The cGAS-cGAS interaction were analyzed by immunoblotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to estimate sample size. A standard two-tailed unpaired Student’s t test was used for statistical analysis of two groups. Statistical analysis of survival curves was performed with a log-rank (Mantel–Cox) test. Statistical analyzed data are expressed as mean ± standard error of the mean (SEM). A p value < 0.05 is considered as statistically significant. We performed the statistical analyses using GraphPad Prism.
DATA AND SOFTWARE AVAILABILITY
All the data are available from the authors upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-phospho-TBK1/NAK (Ser172) | Cell Signaling Technology | Cat# 5483P; RRID: AB_10693472 |
| Rabbit monoclonal anti-TBK1 | Cell Signaling Technology | Cat# 3504S; RRID: AB_2255663 |
| Rabbit monoclonal anti-cGAS (mouse specific) | Cell Signaling Technology | Cat# 31659 |
| Rabbit monoclonal anti-cGAS | Cell Signaling Technology | Cat# 15102; RRID: AB_2732795 |
| Rabbit polyclonal anti-Acetylated-Lysine | Cell Signaling Technology | Cat# 9441; RRID: AB_331805 |
| Mouse monoclonal anti-Acetylated-Lysine | Cell Signaling Technology | Cat# 9681S; RRID: AB_331799 |
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Rabbit monoclonal anti-HDAC3 | Abcam | Cat# ab32369; RRID: AB_732780 |
| Rabbit monoclonal anti-IRF3 (phospho S386) | Abcam | Cat# ab76493; RRID: AB_1523836 |
| Rabbit monoclonal anti-IRF3 | Abcam | Cat# ab68481; RRID: AB_11155653 |
| Mouse monoclonal anti-TREX-1 | Santa Cruz Biotechnology | Cat# sc-271870; RRID: AB_10708266 |
| Mouse monoclonal anti-β-Actin | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_626632 |
| Rabbit polyclonal anti-HA-probe | Santa Cruz Biotechnology | Cat# sc-805; RRID: AB_631618 |
| Mouse monoclonal anti-HDAC3 | Santa Cruz Biotechnology | Cat# sc-376957; RRID: AB_2715509 |
| Rabbit polyclonal anti-His-tag | MEDICAL & BIOLOGICAL LABORATORIES | Cat# PM032; RRID: AB_10209426 |
| Rabbit polyclonal anti-cGAS | This paper | N/A |
| Rabbit polyclonal anti-Ac-cGAS (K384+K394) | This paper | N/A |
| Rabbit polyclonal anti-Ac-cGAS (K414) | This paper | N/A |
| Bacterial and Virus Strains | ||
| HSV-1 | Prof. Jiahuai Han (Xiamen U. China) | N/A |
| VSV | Prof. Hui Zhong (Molecular Genetics Department Beijing Institute of Biotechnology, China) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phorbol-12-myristate-13-acetate (PMA) | Merck | Cat# 524400; CAS No.16561-29-8 |
| ATP Solution (100 mM) | ThermoFisher Scientific | Cat# R0441 |
| GTP Solution (100 mM) | ThermoFisher Scientific | Cat# R0461 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596018 |
| Nε-Acetyl-L-lysine | Sigma-Aldrich | Cat# A4021; CAS No.692-04-6 |
| HT-DNA | Sigma-Aldrich | Cat# D6898 |
| Poly(I:C) | Sigma-Aldrich | Cat# P1530 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636; CAS No.98-92-0 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | Cat# H9268; CAS No. 28728-55-4 |
| Histopaque®-1077 | Sigma-Aldrich | Cat# 10771 |
| Puromycin hydrochloride | Amresco | Cat# J593-25MG; CAS No.58-58-2 |
| 2′3′-cGAMP | Invivogen | Cat# tlrl-cga23 |
| c-di-GMP | Invivogen | Cat# tlrl-cdg |
| Recombinant Human TNF-alpha Protein | R&D System | Cat# 210-TA |
| Trichostatin A (TSA) | Cell Signaling Technology | Cat# 9950S; CAS No.58880-19-6 |
| Cycloheximide (CHX) | Cell Signaling Technology | Cat# 2112S; CAS No. 66-81-9 |
| Aspirin | Ouhe Technology | Cat# 50-78-2; CAS No.50-78-2 |
| Aspirin-d3 | Santa Cruz Biotechnology | Cat# sc-480464; CAS No.921943-73-9 |
| Salicylic acid | Alfa Aesar | Cat# 30782; CAS No. 69-72-7 |
| Diclofenac sodium | Aladdin | Cat# D129332; CAS No. 15307-79-6 |
| MS-275 | MedChemExpress | Cat# HY-12163; CAS No. 209783-80-2 |
| RGFP966 | Selleck | Cat# S7229; CAS No. 1396841-57-8 |
| BX-795 | Selleck | Cat# S1274; CAS No. 702675-74-9 |
| Ni-NTA Agarose beads | QIAGEN | Mat. No. 1018244 |
| Critical Commercial Assays | ||
| IFN-β ELISA kit | Biolegend | Cat# 439408 |
| Human PGE2 ELISA Kit | FineTest | Cat# EH4233 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | Cat# CRL-11268 |
| Human: THP-1 | ATCC | Cat# TIB-202 |
| Human: Human primary PBMCs | This paper | N/A |
| Human: HeLa | ATCC | Cat# CCL-2 |
| Mouse: L929 | ATCC | Cat# CCL-1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Trex1+/− C57BL/6J | Morita et al., 2004 | N/A |
| Oligonucleotides | ||
| Human cGAS-targeted sgRNA: CACCGAAGTGCGACTCCGCGTTCAG, AAACCTGAACGCGGAGTCGCACTTC | This paper | N/A |
| Biotinylated interferon stimulatory DNA: sense: 5′-Biotin-TACAGATCTACTAGTGATCTATGACTGATCTGTACATGATCTACA-3′, anti-sense: 5′-TGTAGATCATGTACAGATCAGTCATAGATCACTAGTAGATCTGTA-3′ | Thermo Fisher Scientific | N/A |
| siRNA: mouse Trex1 | Invitrogen | Cat#: MSS238570 |
| siRNA: human HDAC3 | RiboBio | Cat#: siB1032283530 |
| siRNA: mouse Dnase2a | RiboBio | Cat#: siG151201033118 |
| siRNA: mouse Rnaseh2b | RiboBio | Cat#: siG170516091413 |
| qPCR pimers, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| pAcKRS-3 | Prof. Jason W. Chin (Cambridge, UK) | N/A |
| pCDF PylT-1 | Prof. Jason W. Chin (Cambridge, UK) | N/A |
| Human cGAS cDNA construct | Prof. Jea U. Jung (USC, USA) | N/A |
| Mouse Cgas cDNA construct | Prof.Russell E. Vance (UC Berkeley, USA) | N/A |
| STING cDNA construct | Prof. Hongbing Shu (Wuhan U, China) | N/A |
| lentiCRISPR v2 | Addgene | Plasmid #52961 |
| Software and Algorithms | ||
| GraphPad Prism 5.0 | GraphPad Software | https://www.graphpad.com/ |
| MS Data Converter | AB Sciex | |
| Proteome Discover 2.1 | ThermoFisher Scientific | |
| SPSS | N/A | https://www.ibm.com/analytics/spss-statistics-software |
ACKNOWLEDGMENTS
We sincerely thank the patient and his brother for participating in this research. We thank Cancer Research Technology Limited, UK for providing the Trex1+/− mice, Prof. Jason W. Chin (Cambridge, UK) for providing the pAcKRS-3 and pCDF PylT-1 plasmids, Prof. Jea U. Jung for providing cGAS cDNA constructs, Prof. Hongbing Shu (Wuhan University, China) for providing the STING cDNA construct, Prof. Jiahuai Han (Xiamen University, China) for providing the HSV-1 virus, and Prof. Hui Zhong for providing the VSV virus. We thank Prof. Yixian Zheng (Carnegie Institution for Science) for helpful discussions and critical reading of the manuscript. We also thank Bing-Zhen Cao and Hong-Wei Guo for help with contacting the AGS patients. This work was supported by grants from The National Key Research and Development Program of China (2017YFC1601100 and 2017YFA0505602) and National Natural Science Foundation of China (81771708, 81872153, 31570837, and 81472273). X.-M.Z.’s ORCID number is 0000-0003-0207-4532.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and three tables and can be found with this article online at https://doi.org/10.1016/j.cell.2019.01.016.
REFERENCES
- Ahn J, and Barber GN (2014). Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol 31, 121–126. [DOI] [PubMed] [Google Scholar]
- Aicardi J, and Goutières F (1984). A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann. Neurol 15, 49–54. [DOI] [PubMed] [Google Scholar]
- An J, Woodward JJ, Sasaki T, Minie M, and Elkon KB (2015). Cutting edge: Antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol 194, 4089–4093. [DOI] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, and Barton GM (2011). Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol 29, 185–214. [DOI] [PubMed] [Google Scholar]
- Bowie A (2012). The STING in the tail for cytosolic DNA-dependent activation of IRF3. Sci. Signal 5, pe9. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charifson PS, and Walters WP (2014). Acidic and basic drugs in medicinal chemistry: a perspective. J. Med. Chem 57, 9701–9717. [DOI] [PubMed] [Google Scholar]
- Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang RF, and Cui J (2016a). TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 64, 105–119. [DOI] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016b). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, and Hopfner KP (2013). Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Plater L, Peggie M, and Cohen P (2009). Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J. Biol. Chem 284, 14136–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P (2000). The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci 25, 596–601. [DOI] [PubMed] [Google Scholar]
- Crow YJ, and Manel N (2015). Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. (2006). Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet 38, 917–920. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 167A, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Palma A, Matarese G, Leone V, Di Matola T, Acquaviva F, Acquaviva AM, and Ricchi P (2006). Aspirin reduces the outcome of anticancer therapy in Meth A-bearing mice through activation of AKT-glycogen synthase kinase signaling. Mol. Cancer Ther 5, 1318–1324. [DOI] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon KB, and Wiedeman A (2012). Type I IFN system in the development and manifestations of SLE. Curr. Opin. Rheumatol 24, 499–505. [DOI] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013). Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, and Chen ZJ (2015). Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 112, E5699–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabocka E, and Bar-Sagi D (2016). Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 167, 1803–1813.e1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, and Stetson DB (2015). Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutières Syndrome. J. Immunol 195, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Li C, Wang M, Mao N, Wang S, Chang G, and Cao B (2014). Clinicopathological and genetic analysis of Aicardi-Goutieres syndrome. Chin. J. Neurol 47, 96–100. [Google Scholar]
- Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, Yin L, and Shu HB (2016). Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 45, 555–569. [DOI] [PubMed] [Google Scholar]
- Hutt AJ, Caldwell J, and Smith RL (1986). The metabolism of aspirin in man: a population study. Xenobiotica 16, 239–249. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, and Cambier JC (2008). MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell. Biol 28, 5014–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassiotis G, and Stoye JP (2016). Immune responses to endogenous retro-elements: taking the bad with the good. Nat. Rev. Immunol 16, 207–219. [DOI] [PubMed] [Google Scholar]
- Kopp E, and Ghosh S (1994). Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265, 956–959. [DOI] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS, Berger JM, and Doudna JA (2013). Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Park EB, Lee J, Choi BS, and Kang SJ (2017). The N terminus of cGAS de-oligomerizes the cGAS:DNA complex and lifts the DNA size restriction of core-cGAS activity. FEBS Lett 591, 954–961. [DOI] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, and Li P (2013). Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisnevskaia L, Murphy G, and Isenberg D (2014). Systemic lupus erythematosus. Lancet 384, 1878–1888. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014). Activated STING in avascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaitė Ž, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. (2016). Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 35, 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, and Wood MA (2013). HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 110, 2647–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki I, Rose Y, Uggenti C, Van Eyck L, Frémond ML, Kitabayashi N, Rice GI, Jenkinson EM, Boulai A, Jeremiah N, et al. (2017). Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol 140, 543–552.e5. [DOI] [PubMed] [Google Scholar]
- Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, and Barnes DE (2004). Gene-targeted mice lacking the Trex1 (DNase III) 3′–>5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol 24, 6719–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Peak-Chew SY, and Chin JW (2008). Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol 4, 232–234. [DOI] [PubMed] [Google Scholar]
- O’Neill LA (2013). Immunology. Sensing the dark side of DNA. Science 339, 763–764. [DOI] [PubMed] [Google Scholar]
- Pfeffer LM (2011). The role of nuclear factor kappaB in the interferon response. J. Interferon Cytokine Res 31, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth GJ, and Majerus PW (1975). The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J. Clin. Invest 56, 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, and Jung JU (2015). Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 13, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakespear MR, Halili MA, Irvine KM, Fairlie DP, and Sweet MJ (2011). Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 32, 335–343. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, and Jiang Z (2009). ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 106, 8653–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Zhang XW, Jin J, Du XX, Lian T, Yang J, Zhou X, Jiang Z, and Su XD (2017). Nonspecific DNA Binding of cGAS N Terminus Promotes cGAS Activation. J. Immunol 198, 3627–3636. [DOI] [PubMed] [Google Scholar]
- Tatamiya T, Saito A, Sugawara T, and Nakanishi O (2004). Isozyme-selective activity of the HDAC inhibitor MS-275. Cancer Res 64, 567. [Google Scholar]
- Tatham MH, Cole C, Scullion P, Wilkie R, Westwood NJ, Stark LA, and Hay RT (2017). A Proteomic Approach to Analyze the Aspirin-mediated Lysine Acetylome. Mol. Cell. Proteomics 16, 310–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, and Botting RM (2003). The mechanism of action of aspirin. Thromb. Res 110, 255–258. [DOI] [PubMed] [Google Scholar]
- Verdin E, and Ott M (2015). 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol 16, 258–264. [DOI] [PubMed] [Google Scholar]
- Wang H, Gau B, Slade WO, Juergens M, Li P, and Hicks LM (2014). The global phosphoproteome of Chlamydomonas reinhardtii reveals complex organellar phosphorylation in the flagella and thylakoid membrane. Mol. Cell. Proteomics 13, 2337–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Huang L, Hong Z, Lv Z, Mao Z, Tang Y, Kong X, Li S, Cui Y, Liu H, et al. (2017). The E3 ubiquitin ligase RNF 185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 13, e1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Cheng YY, Yin H, Song XN, Li WW, Zhou XX, Zhao LP, Tian LJ, Han JC, and Yu HQ (2013a). Oxygen promotes biofilm formation of Shewanella putrefaciens CN32 through a diguanylate cyclase and an adhesin. Sci. Rep 3, 1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013b). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, and Fan Z (2016). Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol 17, 369–378. [DOI] [PubMed] [Google Scholar]
- Yang YG, Lindahl T, and Barnes DE (2007). Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886. [DOI] [PubMed] [Google Scholar]
- Yin MJ, Yamamoto Y, and Gaynor RB (1998). The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396, 77–80. [DOI] [PubMed] [Google Scholar]
- Yuan ZL, Guan YJ, Chatterjee D, and Chin YE (2005). Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, and Chen ZJ (2014). The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep 6, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. (2010). Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, and Shu HB (2008). The adaptor protein MITA links virussensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550. [DOI] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, and Kranzusch PJ (2018). Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data are available from the authors upon request.