

Abstract
Resistance to chemotherapy remains a critical barrier to effective cancer treatment. Although cisplatin is one of the most commonly used chemotherapeutic agents in the treatment of non-small cell lung cancer (NSCLC), mechanisms of resistance to this drug are not fully understood. Here, we report a novel cisplatin-resistance mechanism involving SET Domain Containing 2 (SETD2), a histone H3 lysine 36 (H3K36) trimethyltransferase, and cAMP-responsive element-binding protein 1 (CREB1). A549 cells selected in vivo to give brain metastases exhibited cisplatin resistance and decreased expression of phosphorylated CREB1. Next-generation sequencing (NGS) analysis identified a missense mutation in SETD2 (p.T1171K), and we demonstrated that SETD2-mediated trimethylation of H3K36 (H3K36me3) and CREB1 phosphorylation are critical for cellular sensitivity to cisplatin. Moreover, we showed that suppression of SETD2 or CREB1 and ectopic expression of mutant SETD2 conferred cisplatin resistance through inhibition of H3K36me3 and ERK activation in NSCLC cells. Our results provide evidence that SETD2 and CREB1 contribute to cisplatin cytotoxicity via regulation of the ERK signaling pathway, and their inactivation may lead to cisplatin resistance.
Introduction
Cisplatin remains one of the most commonly used chemotherapeutic agents [1], and it improves survival in many cancer types [2, 3]. Although platinum-based combination chemotherapy is standard treatment for non-small cell lung cancer (NSCLC), response rates are usually low (less than 40%) due to presence of intrinsic resistance [4, 5]. Acquired resistance to chemotherapy is responsible for even lower response rates in the second-line setting, where objective responses are observed only in about 10–20% of patients. The mechanisms by which cells develop resistance to platinum compounds are not fully understood. Understanding mechanisms of drug resistance may provide clues to novel therapeutic strategies aimed at increasing efficacy of chemotherapy against advanced lung cancer.
In this study, we found that highly metastatic NSCLC cells exhibited cisplatin resistance, accompanied by decreased level of phosphorylated CREB1 (pCREB1) and acquired missense mutation in SETD2 (p.T1171K). SETD2/HYPB (Huntingtin-interacting protein B), a histone methyltransferase specialized in trimethylation of lysine 36 in histone H3, is the human ortholog of yeast Set2 [6, 7]. SETD2 functions as a transcriptional regulator [8] and is often mutated in clear cell renal cell carcinoma (ccRCC) and acute leukemia [9, 10]. Recent studies suggest that SETD2 is a tumor suppressor gene in various human cancers [11–13] and also identified SETD2 mutations in 9% of lung adenocarcinomas [14]. Cyclic AMP-response element-binding protein 1 (CREB1) is a phosphorylation-dependent transcription factor that belongs to the leucine zipper family of DNA-binding protein. Several protein kinases including PKA, CaMKIV, p70S6K, and RSK2 phosphorylate CREB1 on serine 133 [15]. Phosphorylation of CREB1 is required for CREB1-mediated transcription [16], and CREB1 expression is critical for cell cycle and survival [17]. Here, we reported that SETD2-mediated trimethylation of histone 3 (H3K36me3) and CREB1 phosphorylation are critical for cellular sensitivity to cisplatin. We demonstrated that suppression of SETD2 or CREB1 and ectopic expression of mutant SETD2 (mtSETD2) conferred cisplatin resistance through inhibition of H3K36me3 and ERK in NSCLC cells.
Results
NSCLC cells metastatic to the brain acquire resistance to cisplatin
In a previous study, metastatic derivatives of A549 cells obtained from brain metastases were generated (R1−R4 cells; metastatic potential from low to high) through intracardiac injections in mice [18]. Since the metastatic process occurs through an increase in subclonal mutations [19], which may cause a decrease in sensitivity to treatments, we compared survival of the A549 metastatic derivatives upon treatment with several anticancer drugs. We found that highly metastatic R3 and R4 cells were much less sensitive to cisplatin than A549 parental (R0) and the less metastatic R1 and R2 cells (Fig. 1a); in contrast, no significant difference in cellular viability was observed when treated with etoposide, doxorubicin, or taxol (Supplementary Figure S1A–C). PARP cleavage was significantly inhibited in R4 cells upon cisplatin exposure, whereas low concentrations of cisplatin were able to induce PARP cleavage in R0 cells. Cisplatin also induced caspase-3 cleavage and the expression of γH2AX, a DNA damage marker, in R0 cells but not in R4 cells (Fig. 1b and Supplementary Figure S1D). The TUNEL assay demonstrated that R0 cells are more sensitive to cisplatin-induced apoptosis than R4 cells, at different concentrations of cisplatin (Fig. 1c, d and Supplementary Figure S1E–F).
Fig. 1.
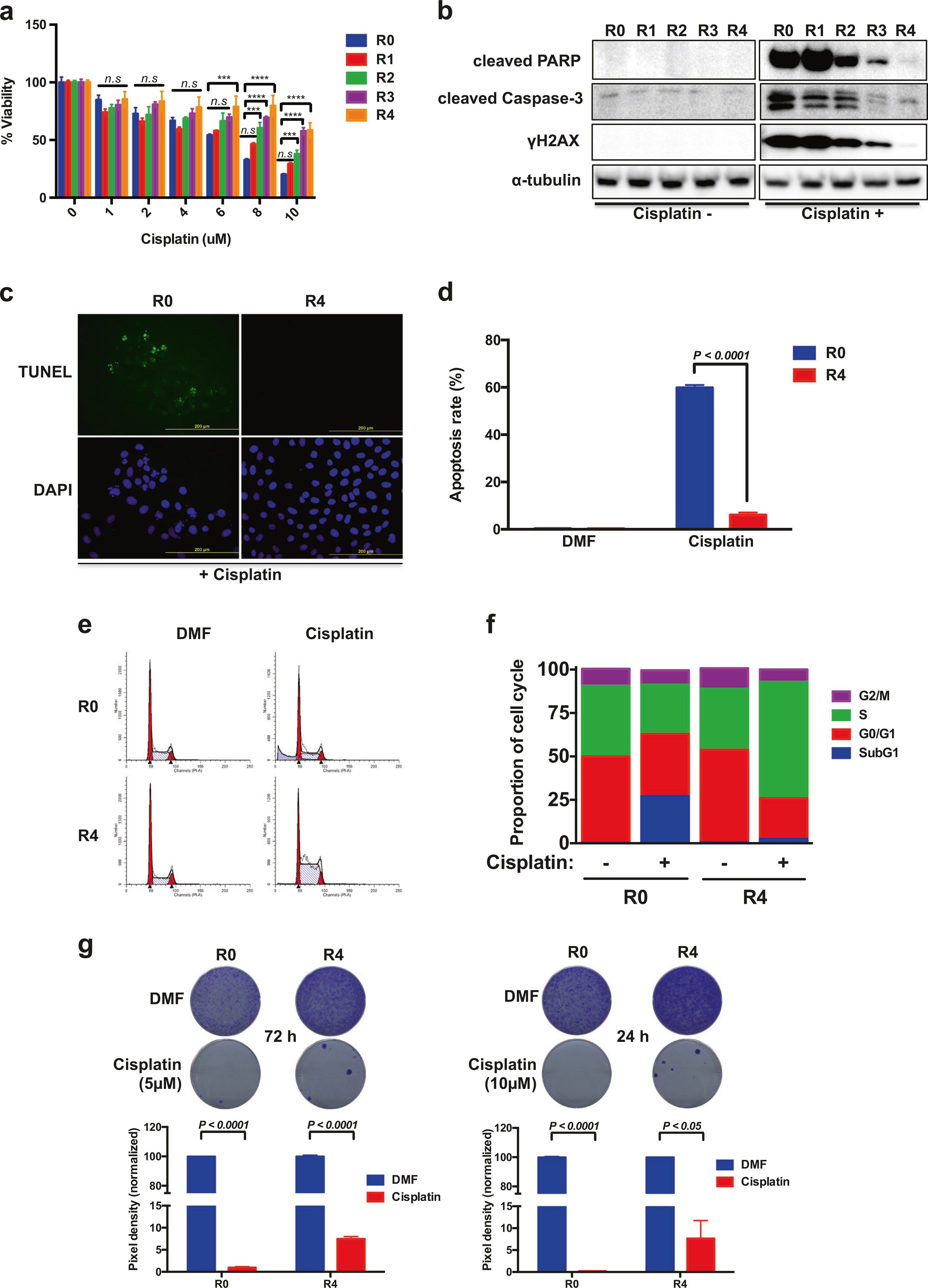
A549 cells metastatic to the brain develop cisplatin resistance. a Cell viability reveals that highly metastatic A549 cells are more resistant. Data are means ± s.d. from three independent experiments (n = 4). Statistical significances compared with control are denoted as ***P < 0.001; ****P < 0.0001 by two-way ANOVA with Dunnett’s multiple comparisons test. b Levels of cleaved PARP, cleaved Caspase-3, and γH2AX in metastatic series of A549 (R0 to R4). Cells were exposed to 10 μM cisplatin and harvested after 24 h incubation. c Representative images of TUNEL staining of A549-R0 and -R4 cells after cisplatin treatment (20 μM) for 24 h. d Representative histogram showing the percentage of apoptotic cells (TUNEL-positive cells). e A549-R0 and -R4 cells were exposed to cisplatin (15 μM) for 24 h and cell cycle distribution was determined by flow cytometry analysis. f The percentage in each phase of the cell cycle is shown. Data are means ± s.d. from n = 3. g Cell survival of R0 and R4 cells with cisplatin treatment was evaluated by clonogenic assay. Pixel density quantification of clonogenic assay shown as histogram. d, g Data are means ± s.d. from three independent experiments, and statistical significance was determined by two-way ANOVA with Sidak’s multiple comparisons test
We further assessed the impact of cisplatin on the cell cycle: there was an increase in the sub-G1 population in R0 cells with cisplatin treatment, and although R4 cells were arrested in S-phase, cisplatin did not increase the sub-G1 fraction in these cells (Fig. 1e, f). Moreover, cisplatin-resistant clones were detected by clonogenic survival assays in R4 cells (Fig. 1g). These observations indicate that A549 cells exposed to serial intracardiac injections develop cisplatin resistance during metastatic seeding to the brain.
ERK activation is required for cisplatin-induced apoptosis in metastatic NSCLC
Since A549 cells carry a KRAS mutation, resulting in the activation of the MAPK/ERK pathway, we assessed whether the activation of MAPK pathway was relevant to cisplatin-sensitivity. The phosphorylation levels of ERK and p38, but not JNK, were significantly higher in R0 cells compared to R4 cells upon cisplatin treatment (Fig. 2a). We next examined whether inhibition of ERK or p38 activity affects cisplatin-induced apoptosis. Inhibition of ERK activation by U0126 significantly attenuated cisplatin-induced PARP cleavage and γH2AX expression in R0 cells, whereas SB203580-mediated p38 suppression was less effective (Fig. 2b). The simultaneous inhibition of ERK and p38 resulted in similar effect as with U0126 treatment alone (Supplementary Figure S2A). Because lung cancer has higher PARP expression than normal lung tissue [20] and PARP1 expression is required for lung adenocarcinoma metastases to bones and brain [21], we determined whether PARP contributes to cisplatin resistance in R4 cells. Olaparib, a PARP inhibitor, did not increase cisplatin sensitivity or PARP cleavage in R4 cells (Supplementary Figure S2B). We then investigated the significance of ERK activation in cisplatin-induced apoptosis by cell cycle analysis, TUNEL, and clonogenic survival assays. U0126 significantly reduced cisplatin-induced sub-G1 arrest (Fig. 2c, d) and apoptosis (Fig. 2e, f). As shown in Fig. 2g and Supplementary Figure S2C, ERK inhibition was associated with a decreased sensitivity to cisplatin while the inhibition of p38 slightly but reproducibly promoted clonogenicity only at low concentrations in R0 cells. Moreover, even though U0126 reduced the proliferation rate (Supplementary Figure S2D), ERK inhibition increased the IC50 of cisplatin (from 4.8 to 9.9 μM at 48 h, and from 3.9 to 8.2 μM at 72 h) in R0 cells (Supplementary Figure S2E).
Fig. 2.
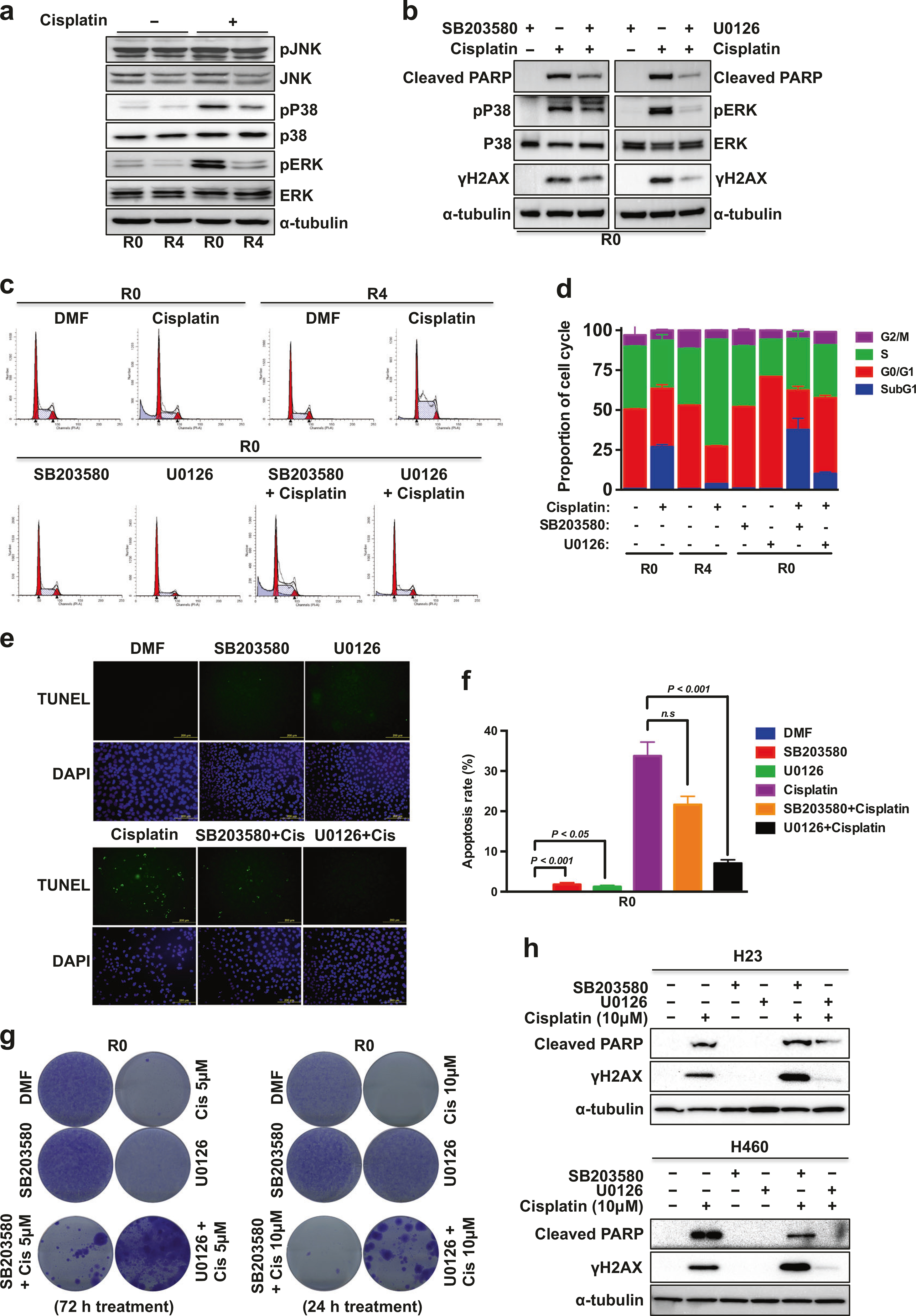
Inhibition of ERK activation attenuates cisplatin-induced apoptosis. a, b WB of cells grown in 10 μM cisplatin with/without inhibitor (10 μM) for 24 h. c Cell cycle distribution analysis. d Representative cell cycle histogram of (c) demonstrating the percentage of the cells in each phase. Data are means ± s.d. from n = 3. e Representative images of TUNEL staining of R0 cells after 15 μM cisplatin treatment for 24 h with/without inhibitor (10 μM). f Representative histogram showing the percentage of apoptotic cells (TUNEL-positive cells). Data are means ± s.d. from three independent experiments, and statistical significance was determined by one-way ANOVA with Dunnett’s multiple comparisons test. g Cell survival of A549-R0 cells under cisplatin treatment with/without inhibitor (10 μM) was assessed by clonogenic assays. h WB of H23 or H460 cells were grown in cisplatin with/without inhibitor (10 μM) for 24 h
Furthermore, cisplatin-induced PARP cleavage and γH2AX expression were also robustly reduced by ERK inhibition in two other KRAS-mutated NSCLC cell lines, H23 and H460 (Fig. 2h). CCK-8 assays showed that ERK inhibition significantly increased the IC50 of cisplatin (from 10 to 20 μM in H23, and from 5.4 to 21 μM in H460) (Supplementary Figure S3A–B), and more surviving colonies were detected in U0126-cotreated H23 and H460 cells (Supplementary Figure S3C–D). These results indicate that ERK activation is critical for cisplatin sensitivity in KRAS mutant NSCLC cells.
SETD2 is mutated in metastatic NSCLC cells
To further elucidate the molecular mechanisms of cisplatin resistance caused by metastatic progression, we performed targeted exome sequencing using a custom NGS panel of 206 genes that are frequently mutated in cancer or related with lung cancer (Supplementary Table S1). The R0 and R4 cells yielded 3.4 million and 3.5 million reads with a mean read depth of 128 and 145, respectively. After variant filtering, we detected three somatic mutations that were found only in R4 cells and not in R0 cells (Table 1, Supplementary Table S2, and Fig. 3a, b). The SETD2 mutation acquired in R4 cells was flagged as deleterious and probably damaging, according to SIFT and PolyPhen prediction analyses. While several mechanisms of resistance to EGFR tyrosine kinase inhibitor have been reported [22], there is no study of acquired cisplatin resistance in relation to EGFR mutations. FANCC is one of the 13 FANC (Fanconi anemia complementation group) family members and associated with DNA repair [23]. FANCC mutation can cause aberrant splicing, which result in inactivating FANCC protein [24] and primary embryonic fibroblast cells from FANCC knockout mice were hypersensitive to cisplatin [25]. Therefore, FANCC c.1534+8 G > T mutation is unlikely related to cisplatin resistance in lung cancer.
Table 1.
Genetic variants found in R4 cells
| Gene | Variant | Variant location | Consequence/type | Total read depth | Variant freq | SIFT/PolyPhen | COSMIC ID/dbSNP |
|---|---|---|---|---|---|---|---|
| SETD2 | chr3:47162614G > T p.T1171K c.3512C > A |
exon 3 | Missense substitution | 266 | 22% | deleterious(0) probably_damaging (0.98) |
— |
| EGFR | chr7:55211142A > G p.K129E c.385A > G |
exon 3 | Missense substitution | 314 | 16% | tolerated(1) benign(0.005) |
COSM 5041204 |
| FANCC | chr9:97864140C > A c.1534 + 8G > T |
exon 15 | Splice region | 126 | 10% | — | — |
Fig. 3.
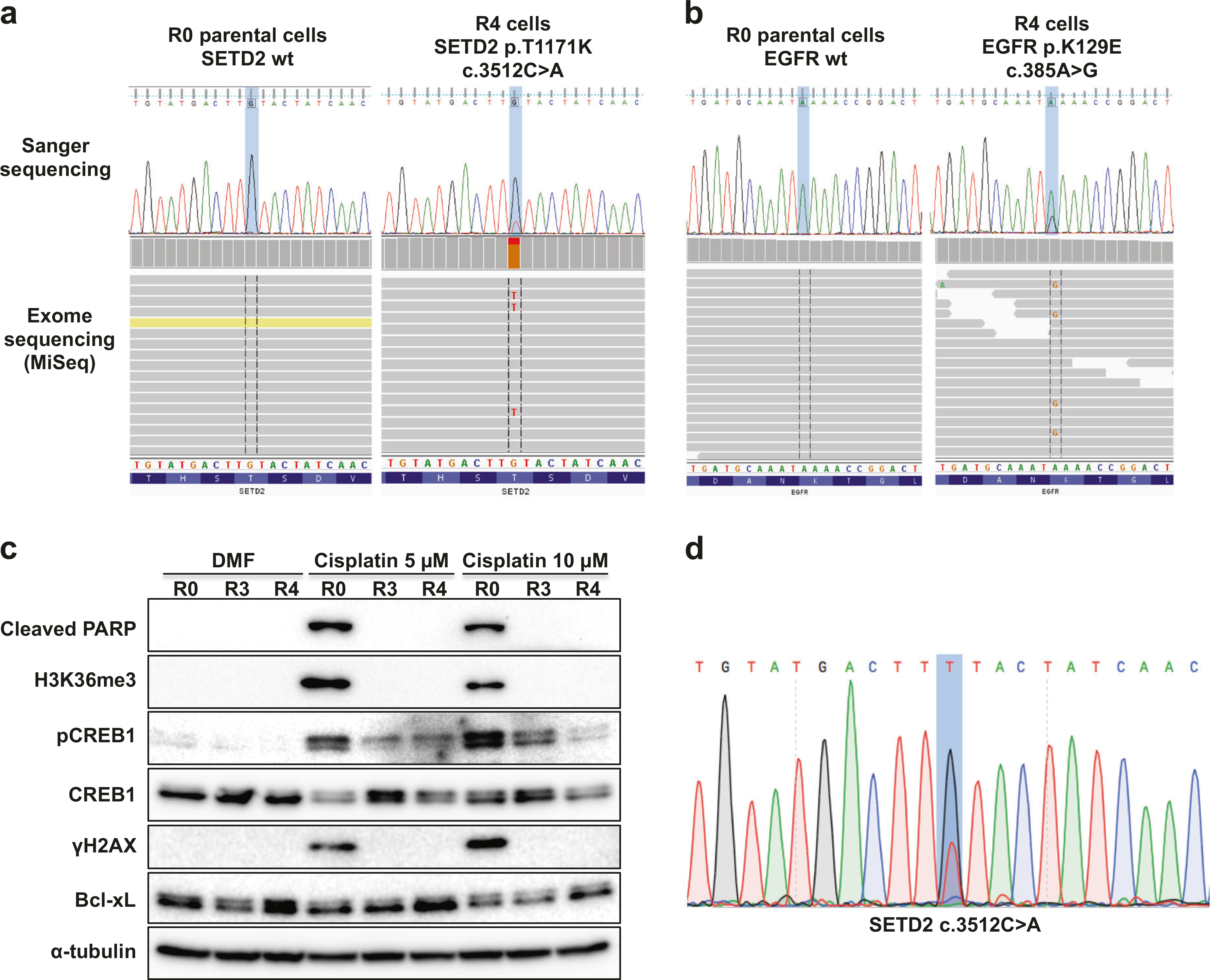
Cisplatin-resistant brain metastases of A549 cells acquire SETD2 mutation and display impaired CREB activation. a Identification of SETD2 mutation by MiSeq sequencing in A549-R4 cells, using IVG. b Identification of EGFR mutation by MiSeq sequencing in A549-R4 cells. c WB analysis of the indicated proteins in the A549-R0, -R3, and -R4 cells treated with DMF (control) or cisplatin for 24 h. d Sanger sequencing confirmation of SETD2 mutation in the A549-R4 surviving clones in clonogenic assay after exposure to cisplatin (10 μM)
Cisplatin-induced apoptosis is associated with the presence of wild-type SETD2 and CREB1 activity in NSCLC cells
SETD2 is mutated frequently in diverse human cancers, and the most frequently detected mutations confer loss of function [26]; SETD2 is therefore considered a tumor suppressor gene. KRAS or RAF are commonly mutated (25%; 5/20) in lung adenocarcinomas harboring SETD2 mutations [14], and given the role of ERK pathway in cisplatin sensitivity (Fig. 2), we therefore examined the impact of SETD2 inactivation on cisplatin cytotoxicity. In R0 cells, cisplatin induced H3K36me3, whereas H3K36me3 was markedly reduced in cisplatin-resistant R4 clones that also expressed somewhat higher levels of the antiapoptotic protein Bcl-xL (Fig. 3c and Supplementary Figure S4A). These results strongly suggest that SETD2 plays a role in cisplatin-mediated cell apoptosis. Further sequencing analyses revealed that cisplatin-resistant cells isolated from clonogenic assays (Fig. 1g) harbored the identical SETD2 mutation (Fig. 3d).
In addition, we performed reverse-phase protein microarray analyses to identify molecules associated with the enhanced metastatic potential of A549 subclones, as reported previously [27]. We found that pCREB1 expression was reduced by 55%, whereas phosphorylated-EGFR (Y1068, Y1148, and Y1173) and total levels of EGFR were not changed in R4 compared to R0. We then investigated whether CREB1 phosphorylation was associated with cisplatin sensitivity. Western blot (WB) analysis showed that the levels of pCREB1 induced by cisplatin were significantly reduced in the resistant cells, especially R4 (Fig. 3c and Supplementary Figure S4A). Collectively, these results led us to hypothesize that trimethylation of H3K36 and CREB1 phosphorylation may be required for sensitization to cisplatin, and SETD2 mutation or decreased CREB1 activity may confer cisplatin resistance in the highly metastatic NSCLC cells.
SETD2 or CREB1 knockdown confers cisplatin resistance
To verify whether the activities of SETD2 and CREB1 were important for cisplatin sensitivity, we established stable cell lines expressing the short hairpin RNAs (shRNAs) of SETD2 (shSETD2) or CREB1 (shCREB1) in the A549-parent R0 cells. Knockdown of either SETD2 or CREB1 enhanced cell viability and clonogenicity, and led to a significant decrease in the sub-G1 population when treated with cisplatin in R0 cells (Fig. 4a–d). These results indicate that both the SETD2 mutation and the decreased CREB1 activity may impede cisplatin-induced apoptosis in NSCLC cells. WB showed that the cleavage of PARP caused by cisplatin was considerably inhibited in R0 cells stably transfected with two different shRNAs of SETD2 (Supplementary Figure S4B), or CREB1, compared to controls (Fig. 4e). Interestingly, however, downregulation of SETD2 or of CREB1 expression did not alter γH2AX levels. Because SETD2 is required for murine embryonic stem cell differentiation through ERK activation [8], we examined whether SETD2 knockdown affected ERK activation upon cisplatin treatment. Knockdown of SETD2 effectively reduced cisplatin-induced ERK activation, and upregulated Bcl-xL expression. In addition, knockdown of CREB1 also decreased pERK level and increased Bcl-xL. Interestingly, CREB1 phosphorylation was not affected in the SETD2-knockdown R0 cells, even though H3K36me3 expression caused by cisplatin was drastically decreased in the CREB1-knockdown R0 cells (Fig. 4e). To further elucidate the role of CREB1 in cisplatin-induced apoptosis in NSCLC, we also stably transfected shCREB1 into H23 cells. As shown in Fig. 4f, downregulation of CREB1 suppressed not only PARP cleavage and γH2AX expression, but also pERK under cisplatin treatment in the CREB1 knockdown H23 cells. Cell viability was enhanced upon cisplatin treatment and the cisplatin-induced sub-G1 population was markedly reduced in the CREB1 knockdown H23 cells (Supplementary Figure S5).
Fig. 4.
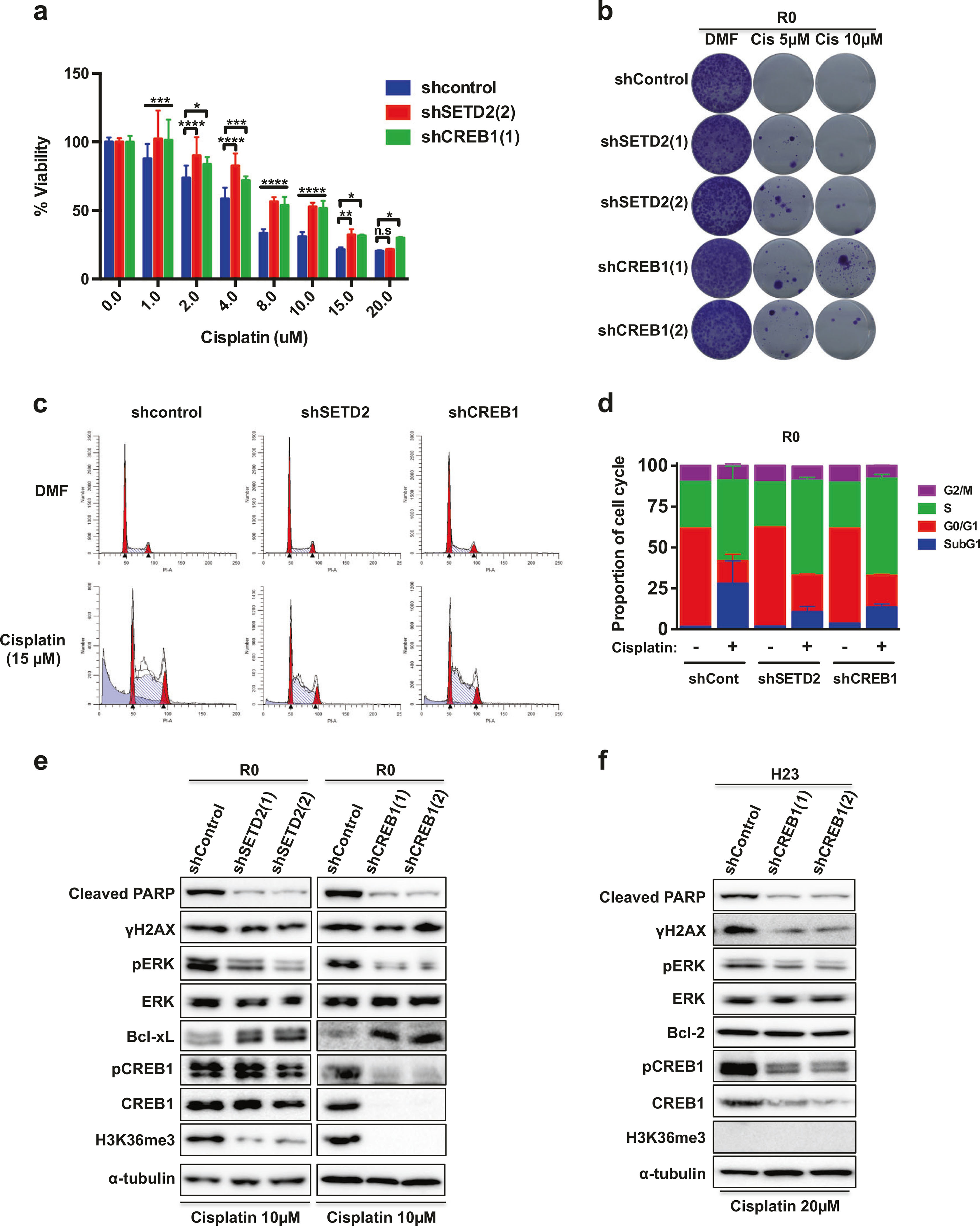
The activities of both SETD2 and CREB1 are essential for cisplatin-induced cell death. a Viability assay of SEDT2 or CREB1 knockdown A549-R0 cells. Data are means ± s.d. from three independent experiments (n = 4). Statistically significant differences compared with control are denoted as *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 by two-way ANOVA with Dunnett’s multiple comparisons test. b Clonogenic assay. SETD2 or CREB1 knockdown A549-R0 and control cells were treated with cisplatin for 24 h. c Cell cycle distribution analysis of A549-R0 cells was performed using flow cytometry. d Representative cell cycle histogram of (c) demonstrating the percentage of the cells in each phase. Data are means ± s.d. from n = 3. e SETD2 or CREB1 knockdown A549-R0 cells were exposed to cisplatin for 24 h, and WB analysis was performed. f CREB1 knockdown H23 cells were exposed to cisplatin for 24 h, and WB analysis was performed. A549 and H23 cells transfected with pLKO.1 were used as a control
Mutant SETD2 confers cisplatin resistance through inhibition of the H3K36me3-ERK pathway
To assess the role of the acquired SETD2 mutation (p. T1171K) in cisplatin resistance of NSCLC cells, we established A549, H460, H157, and H358 cell lines with stable expression of the mutant SETD2 (mtSETD2). TUNEL assay revealed that cisplatin-induced apoptosis was reduced in mtSETD2-overexpressing A549 (Fig. 5a, b) and H460 (Fig. 5c, d) cells. Flow cytometry analysis confirmed that exposure of A549 and H460 cells to cisplatin caused an increase in the sub-G1 fraction (from 1.54 to 27.6%, and from 1.43 to 17.5%, respectively) in control cells, whereas the proportion of sub-G1 fraction induced by cisplatin was decreased in mtSETD2-expressing A549, H460, H157, and H358 cells (Fig. 5e–h and Supplementary Figure S6A–B). Clonogenic assay showed that there were surviving colonies in mtSETD2-expressing A549 and H460 cells, compared to none in their control cells treated with cisplatin (Fig. 5i). Mutant SETD2 expression enhanced cell viability in cisplatin-treated H157 and H358 cells (Supplementary Figure S6C). Cisplatin-induced PARP or Caspase-3 cleavage, H3K36me3, γH2AX expression, and ERK activation were suppressed by mtSETD2 overexpression. In addition, Bcl-xL expression was upregulated in mtSETD2-expressing A549, H460, and H157 cells. The expression of pCREB1, as in SETD2 knockdown R0 cells, did not change significantly in those cell lines (Fig. 5j and Supplementary Figure S6D). These results suggest that the SETD2 mutation contributes to cisplatin resistance in NSCLC cells, and mtSETD2 likely acts in a dominant-negative fashion.
Fig. 5.
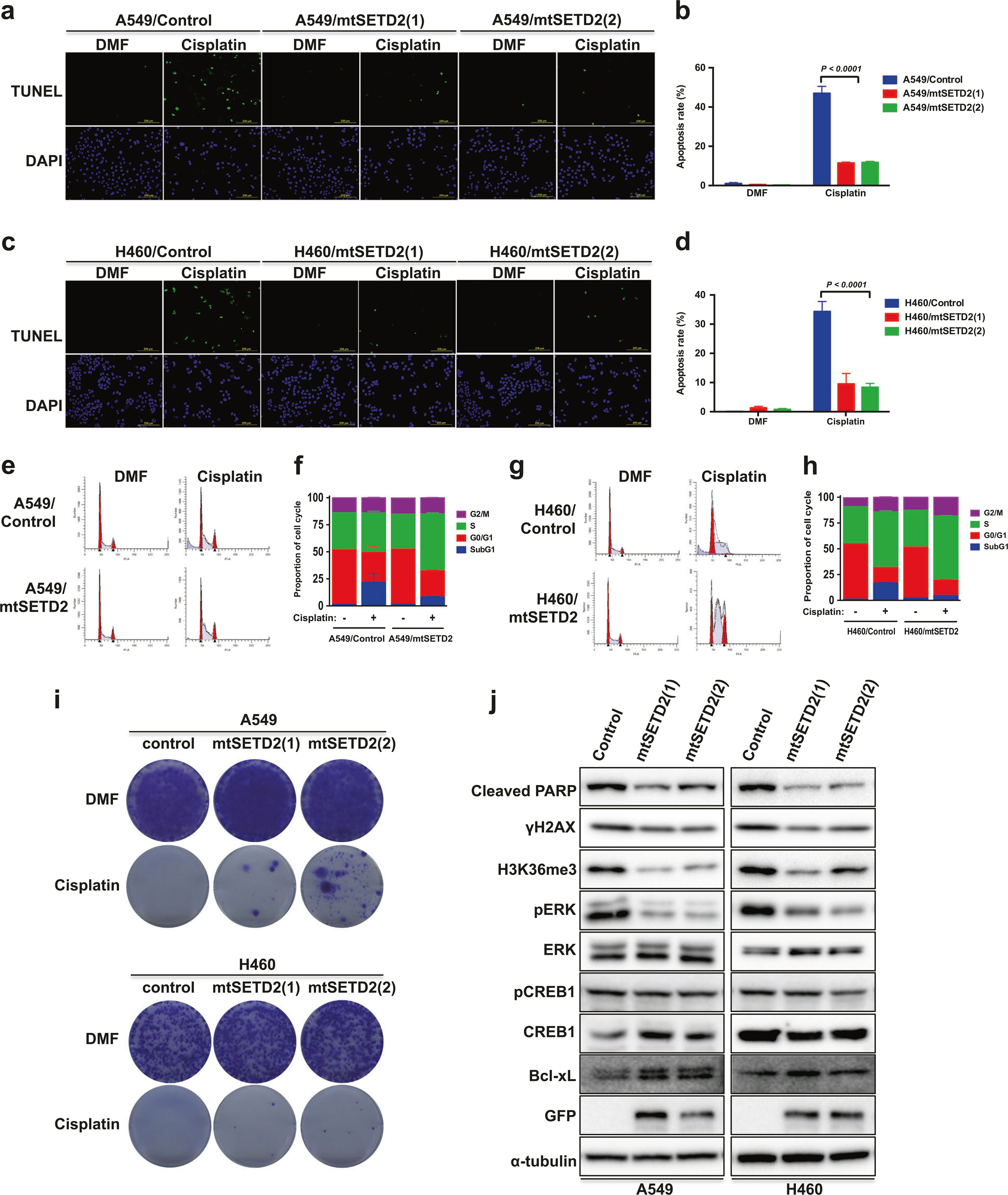
Ectopic expression of mutant SETD2 inhibits cisplatin-induced apoptosis in NSCLC cells. a Representative images of TUNEL staining of A549 or mtSETD2-expressing A549 cells after 15 μM cisplatin treatment for 24 h. A549 and H460 cells transfected with pBI-MCS-EGFP were used as control. b Representative histogram showing the percentage of apoptotic cells (TUNEL-positive cells). c Representative images of TUNEL staining of H460 or mtSETD2-expressing H460 cells after 15 μM cisplatin treatment for 24 h. d Representative histogram showing the percentage of apoptotic cells (TUNEL-positive cells). Data (b,d) are means ± s.d. from three independent experiments, and statistical significance was determined by two-way ANOVA with Sidak’s multiple comparisons test. e A549 or mtSETD2-expressing A549 cells were exposed to 15 μM cisplatin for 24 h and the cell cycle distribution was determined by flow cytometry analysis. f Representative cell cycle histogram of (e) demonstrating the percentage of the cells in each phase. Data are means ± s.d. from n = 3. g H460 or mtSETD2-expressing H460 cells were exposed to 15 μM cisplatin for 24 h and the cell cycle distribution was determined by flow cytometry analysis. h Representative cell cycle histogram of (g) demonstrating the percentage of the cells in each phase. Data are means ± s.d. from n = 3. i Cell survival of mtSETD2-expressing A549 or H460 cells under 10 μM cisplatin treatment for 24 h was assessed by clonogenic assay. j Mutant SETD2-expressing A549 or H460 cells were exposed to 10 μM cisplatin for 24 h, and WB analysis was performed
Systematic assessment of SETD2 and CREB1 levels in relation to cisplatin sensitivity and identification of SETD2 mutation in human tumors
To examine the correlation between SETD2 or CREB1 and cisplatin sensitivity, we used the Oncomine database (http://www.oncomine.org) and CellMiner (http://discover.nci.nih.gov/cellminer). Two primary filters (CREB1 and SETD2 mutation) were applied and only one dataset was identified (Garnett’s group [28]) in Oncomine. 281 differentially regulated genes out of 126,234 genes were identified with cut-off values for fold change (>2) and P value (<0.0001), from 732 human cancer cell lines. CREB1 was one of the dysregulated genes: mRNA levels of CREB1 were low in diverse human carcinomas (Supplementary Table S3), and then we further grouped gene expression analysis data by cisplatin sensitivity. We found that most of cisplatin-resistance cell lines expressed low levels of CREB1 while CREB1 expression was increased in cisplatin-sensitive cell lines (Supplementary Table S4A). Moreover, positive correlations between CREB1 (mRNA expression or copy number) and cisplatin-sensitivity were found in the NCI CellMiner databases (Supplementary Figure S7A–B).
CellMiner of the NCI-60 cell lines also showed that cisplatin-sensitivity is correlated negatively with SETD2 mutation (homozygous deleterious) and positively with wild-type SETD2 expression (Supplementary Figure S7C–D). However, we could not find any significant correlation between CREB1 expression and SETD2 or KRAS gene mutation status in Oncomine dataset (Supplementary Table S4B–C).
To investigate the relationship between SETD2 mutation and cancer metastasis, we examined the NGS data from Caris Life Science for a total 17,913 cases in 39 human solid tumors and classified cases according to SETD2 mutation status. Pathogenic SETD2 mutations were identified (211 of 17,913 cases; 1.2%) in 23 different tumor types and found in 46 of 3083 (1.5%) NSCLC cases; however, the SETD2 p.T1171K was not among this group. Further, we divided SETD2 mutation-positive cancers originating from primary site or metastatic site. Interestingly, SETD2 mutations were found with higher frequency in metastatic breast, kidney, melanoma, and thymic carcinoma, and 18 out of 46 (39.1%, P value = 0.0136 chi square) metastatic NSCLC had SETD2 mutation (Supplementary Dataset). However, the number of NSCLC cases is too small to allow any conclusion. We also examined DNA both from FFPE tumor and matching blood in 36 thymic carcinoma patients [29]. Remarkably, two patients had SETD2 mutations and SETD2 p.T1171K was observed in one patient (Supplementary Table S5).
Discussion
In this study, we utilized a brain metastasis model of the A549 lung adenocarcinoma cells to study mechanisms of cisplatin resistance in NSCLC. Our results indicate that SETD2 mutation and CREB1 inactivation mediate cisplatin resistance in highly metastatic NSCLC. Most NSCLC patients are diagnosed at a late stage, when local treatments are no longer curative. Chemotherapy has limited efficacy in advanced NSCLC, and few reports have shown that a specific genetic alteration is involved in causing cisplatin resistance during metastasis.
SETD2 mutations were first reported in ccRCC [9], and have since been discovered in several other human cancers [12, 14, 30]. Little is known about the potential contribution of SETD2 mutations to chemoresistance and metastasis in NSCLC. We found that the SETD2 mutation (p.T1171K) that occurred in brain-metastasized A549 cells resulted in the loss of methyltransferase activity, and those cells exhibited cisplatin resistance. Cisplatin-induced apoptosis was attenuated, and cell survival was enhanced in these highly metastatic cells. Consistent with our findings, somatic mutations in SETD2 are gained during relapse in acute lymphoblastic leukemia, suggesting SETD2 inactivation may lead to clonal survival and chemotherapy tolerance [10]. Isogenic leukemia cell lines with CRISPR-mediated frameshift deletions in SETD2 and conditional SETD2 knockout mice were resistant to DNA-damaging agents, but cisplatin was not tested, through impairment of DNA damage response [31]. In addition, three discrete SETD2 mutations with different regional distributions were validated and missense mutations of SETD2 were identified at metastatic sites of renal cell carcinoma patients [26]. Xiang et al. showed that miR-106b-5p targets and inactivates the tumor suppressor gene SETD2, and that upregulation of SETD2 expression resulted in a G0/G1 cell cycle arrest in ccRCC [32].
MAPK activation is a major signaling component that contributes to the fate of cells in response to diverse extracellular stimuli [33]. Although ERK1/2 can be activated by several growth factors and plays a role in prosurvival, accumulating reports also support that ERK activation contributes to cell death in different cell types, including in vivo mouse models, and in response to different stimuli [34–38]. Moreover, recent studies showed that endometrial cancer patients with low pERK1/2 expression had significantly shorter relapse-free survival and OS [39]. In our study, ERK inhibition through suppression of MEK decreased cisplatin-induced PARP cleavage, DNA damage response (γH2AX), and the percentage of the sub-G1 fraction, and enhanced cell viability and clonogenicity of A549, H23, and H460 cells. It is conceivable that ERK activation may be required for sensitivity to cisplatin treatment in NSCLC. Clinical trials with MEK inhibitors, selumetinib and trametinib, showed that ERK targeting had limited clinical activity and monotherapy did not improve the outcome of KRAS mutant NSCLC patients compared to docetaxel [40, 41]. We demonstrated that a decrease in cisplatin-induced apoptosis in SETD2 knockdown cells was associated with low levels of pERK. Zhang et al. suggested that Setd2−/− murine embryonic stem cells express aberrantly low levels of ERK activity [8], and that ectopic expression FGFR3 rescued the defective ERK pathway. However, there was no significant difference in FGFR3 protein expression in our cell lines under cisplatin treatment (Supplementary Figure S8).
A number of reports described CREB1 overexpression or activation in various human cancer [42–45]. However, the exact role and mechanisms underlying CREB1-mediated cancer progression still remain to be clarified, and little is known about the function of CREB1 on chemosensitivity. In the present study, we demonstrated that cisplatin-induced pCREB1 expression is significantly downregulated in highly metastatic A549 (R3 and R4) NSCLC cells, and that knockdown of CREB1 by shRNA confers cisplatin resistance via attenuation of ERK activation in A549 and H23 cells. Consistent with our results, CREB1 phosphorylation is required for retinoic acid (RA)-mediated apoptosis, and ectopic expression of dominant-negative CREB1 confers RA and doxorubicin resistance in neuroblastoma cells [46]. Nagao et al. showed that dexamethasone-induced CREB1 activation and it is required for glucocorticoid-induced apoptosis [47]. Because the transactivation of Dot1 mediated by CREB1 leads to hypermethylation of histone H3K79 [48], we propose that regulation of H3K36me3 by CREB1 in R0 cells (Fig. 4) might be modulated by a similar molecular machinery.
Recently we have begun realizing that heterogeneity may be at the base of resistance to systemic therapies. Since comutations on TP53, EGFR, BRAF, or KRAS were detected in lung adenocarcinoma patients along with SETD2 mutations [14], and SETD2 also known as a dual-function methyltransferase for histones and microtubules [49], multiple mechanisms seem to be at play and may also contribute to cisplatin resistance. Although the activities of SETD2 and CREB1 might be interconnected, inactivation of either one can confer cisplatin-insensitivity (Fig. 6). Further studies investigating the clinical relevance of SETD2 and the potential link between SETD2 mutations and CREB1 phosphorylation are warranted.
Fig. 6.
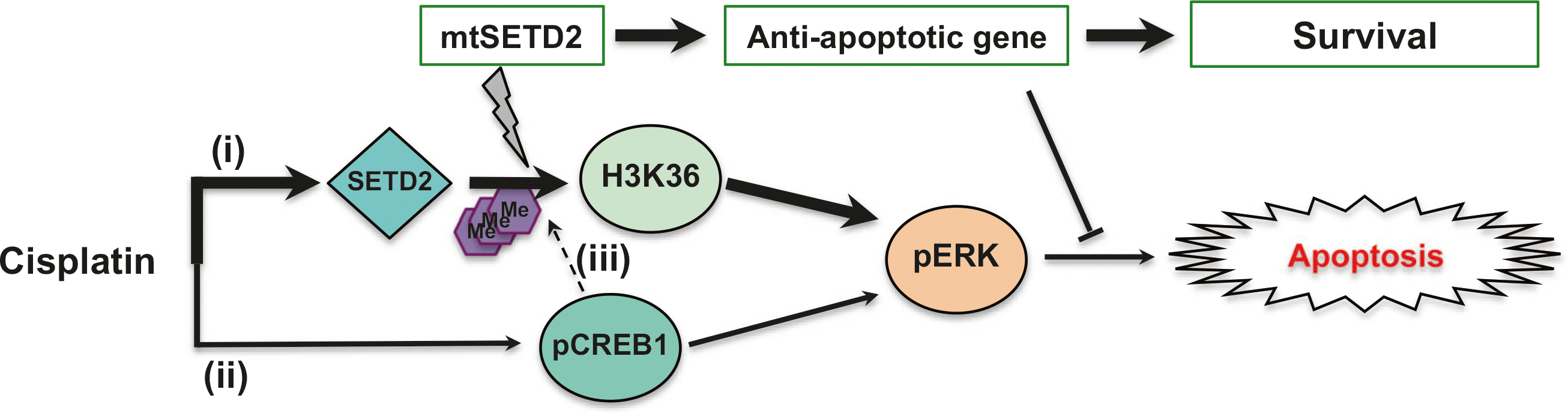
Proposed schematic representation of the role of SETD2 and CREB1 in the regulation of the cisplatin-induced apoptosis. Cisplatin induces cell death through SETD2-mediated trimethylation of H3K36 (H3K36me3) and ERK activation primarily (i) or CREB1 activation (ii). According to our results, resistance to cisplatin can occur through two different mechanisms, blockade of CREB1 activation or SETD2 mutation by epigenetic effectors inhibiting cisplatin-induced trimethylation H3K36 and ERK phosphorylation. Since CREB1 transactivates methyltransferase [48], CREB1 may play an important role in H3K36 methylation (iii). Me¸ methyl group
Materials and methods
Cell culture and reagents
All human cancer cell lines (A549, NCI-H23, -H157, -H358, and -H460) were purchased from ATCC (Manassas, VA, USA), and maintained in RPMI-1640 with 10% FBS and 2% of penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Metastatic series of A549-R0 to -R4 were established by intracardiac injection in nude mice, as described previously [18]. Cis-Diammineplatinum (II) dichloride, Etoposide, Doxorubicin hydrochloride, Paclitaxel, and Dimethylformamide (DMF) were purchased from Sigma (St. Louis, MO, USA). U0126 and SB203580 were obtained from Cell Signaling (Danvers, MA, USA). Puromycin and Olaparib were from InvivoGen (San Diego, CA, USA) and Selleckchem (Houston, TX, USA) respectively. Cisplatin was dissolved in DMF whereas other compounds were dissolved in dimethyl sulfoxide.
Western blotting
For WB analysis, cells were lysed in 1× lysis buffer (60 mM Tris (pH 7.4), 25 mM HEPES, 150 mM NaCl, 10% glycerol, 5 mM EDTA, 1% Triton X-100, 5 mM Na3VO4, 50 mM NaF, 2 pills protease inhibitor cocktail tablets (Roche)) on ice for 45 min and centrifuged at 15,000 rpm for 15 min. Cell lysates were separated by electrophoresis on 4–20% SDS-PAGE gels, transferred to PVDF membrane and incubated with the following primary antibodies: mouse anti-HA and anti-α-Tubulin (Sigma); rabbit anti-pErk1/2, -Erk1/2, -pJNK, -JNK, -pP38, -P38, -cleaved PARP, -cleaved caspase-3, -phospho-Histone H2A.X (Ser139), -Bcl-2, -CREB1 (Cell Signaling); rabbit anti-H3K36me3 and -pCREB1 (Abcam, Cambridge, MA, USA); mouse anti-Bcl-xL and rabbit anti-FGFR3 antibody (Santa Cruz Biotechnology, Dallas, TX, USA). The bound antibodies were visualized with a suitable secondary antibody conjugated with horseradish peroxidase using enhanced chemiluminescence reagent WESTSAVE up (AXXORA, Farmingdale, NY, USA) and the PXi6 imaging system (Syngene, Cambridge, UK).
Cell cytotoxicity and apoptosis
The CCK-8 kit (VitaScientific, MD, USA) was used to determine cell viability. Two to four thousand cells were plated in 96-well plates and incubated overnight. Drugs were added at the indicated concentrations and incubated for 48 or 72 h. Inhibitors were added 1 h prior to cisplatin addition. CCK-8 solution was added and followed by incubation for 1 h at 37 °C. The absorbance at 450 nm was measured by Glomax (Promega, Madison, WI, USA).
TUNEL assays were done using the In Situ Cell Death Detection Kit (Sigma) to assess cell apoptosis. Cells (8×104) were plated in 12-well plates with cover slip and incubated for 24–48 h. Inhibitors were added 1 h prior to cisplatin treatment, and cells were fixed with 4% paraformaldehyde and permeabilized by 0.1% Triton X-100. Labeled cells were mounted in VECTASHIELD Hardset Mounting Medium (Vector Laboratories, Burlingame, CA, USA) and examined using an immunofluorescence microscope (Olympus, Japan).
Clonogenic assay
Cell survival ability was determined using clonogenic assays in six-well plates. Cells (4×103) were plated and incubated overnight and then cisplatin was added. Inhibitors were added 1 h prior to cisplatin treatment, and growth medium was refreshed at the indicated time points. Three weeks to 28 days later, cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. Images of colony formation were acquired by scanner (Epson, Japan), and pixel density was quantified using Photoshop.
Cell cycle
Cells (8×105) were treated with either DMF or cisplatin with/without inhibitors (10 μM, 1 h prior to cisplatin treatment) for 24 h. Following incubation, cells were fixed with ethanol, and cell cycles were analyzed using FACSCalibur Flow Cytometer (BD Bioscience, San Jose, CA, USA). Acquired data were processed using the FlowJo software (Tree Star).
Site-directed mutagenesis
Human SETD2 expression plasmid (SETD2-GFP) was obtained from Addgene (Cambridge, MA, USA) and the SETD2 T1171K mutation was introduced using the QuickChange II XL Site-Directed Mutagenesis kit (Agilent, Savage, MD, USA) and primers with the sequences: forward (5′- GATGGGGTTGATAGTAAAAGTCATACAGA TGTG-3′), reverse (5′- CACATCTGTATGACTTTTACTATCAACCCCATC-3′). The following PCR conditions were used for mutant strand synthesis: 95 °C/1 min; 18 cycles of 95 °C/50 s, 60 °C/50 s, 68 °C/12 min; 68 °C/7 min. The insert size was confirmed with BglII and PstI digestion, and finally the corrected sequence was verified by sequencing (Genewiz, NJ, USA).
Stable cell lines and transfection
pLKO.1 from Addgene, two CREB1 shRNA (TRCN0000226468, TRCN0000226467) and SETD2 shRNA (TRCN0000003032, TRCN0000003030) were purchased from Sigma, and these plasmids were used to generate stable knockdown cell lines. A549 or H23 cells were transfected with 2 μg of each plasmid using X-tremGENE 9 DNA transfection reagent (Sigma) in six-well plates and selected by 600 μg/ml of puromycin 48 h after transfection. Cells transfected with pLKO.1 were used as a control.
A549, H460, H157, and H358 cells were transfected with 1 μg of pBI-MCS-EGFP (Addgene) or mutant SETD2 (mtSETD2)-GFP plasmid for overexpressing stable cell lines. Forty-eight hours after transfection, mtSETD2-GFP transfected cells were selected by 5–800 μg/ml of Geneticin (Gaithersburg, MD, USA). Cells transfected with pBI-MCS-EGFP were used as a control.
Next-generation sequencing
Genomic DNA was extracted from the R0 and R4 cell pellets using the DNeasy Blood and Tissue kit (Qiagen, Germantown, MD, USA) and quantified using the Quantifluor ONE dsDNA kit (Promega) and the NanoDrop spectrophotometer (Thermo Scientific). DNA sample (3 μg) were sheared to a target size of 200 bp using a Covaris M220 focused-ultrasonicator (Covaris, Woburn, MA, USA), and used to construct individual indexed libraries using NEB-Next library reagents (New England Biolabs, Ipswich, MA, USA), the SureSelect XT hybridization reagent kit (Agilent Technologies), and Personal Genome Diagnostics custom probes (PGDx). Quality and quantity of the indexed libraries were assessed using a 2100 Bioanalyzer (Agilent Technologies) and were combined in an equimolar pool. Paired end 2 × 150 bp sequencing was performed on the Illumina MiSeq according to the manufacturer’s protocol. Our custom panel targets the exons of 206 cancer-related genes and evaluates a subset of genes for copy number variation and translocations (Supplementary Table S1). Alignment to the human reference genome 19 (GRCh37, UCSC hg19 assembly), quality and adapter trimming, and variant calling were executed using the MiSeq Reporter software on the MiSeq or the PGDx server analysis pipeline. Annotations and filtering of all the variants were completed on the VariantStudio v2.2.1 software (Illumina, San Diego, CA, USA). Variants were filtered by a mapping quality score greater than 30, read depth greater than 30, and variant frequency greater than 0.10. All synonymous and noncoding (intron) variants found outside of splicing regions were also removed. Furthermore, variants found in both R0 and R4 cells were removed to discover unique somatic mutations in each cell line. Each filtered variant location was examined concurrently in the Integrative Genomics Viewer (IGV, Broad Institute) between the R0 and R4 reads for verification and visual inspection.
SETD2 mutation data in patient samples were generated from the NextSeq Illumina platform by Caris Life Science (Irving, TX, USA), a custom-designed SureSelect XT assay used to enrich 592 whole-gene targets (Agilent). All variants are detected with >99% confidence based on allele frequency and amplicon coverage with an average sequencing depth of coverage of >500 and with an analytic sensitivity of 5%. Manual microdissection was performed on most cases prior to testing to enrich tumor content. Then tumor cases were divided into wild-type and mutated groups (Pathogenic, Benign, VUS; Variant of unknown significance) for SETD2 gene status.
SETD2 gene status was evaluated in 36 samples of patients with advanced refractory or recurrent thymic carcinomas. Most patients were heavily pretreated and had extensive metastatic disease [29].
Quantitative RT-PCR (qRT-PCR) analysis
Total RNA was isolated using RNeasy Plus Mini Kit (Qiagen) and qRT-PCR was carried out using an ABI 7900HT Fast Real-time PCR system (Applied Biosystems, Foster City, CA, USA) with QuantiTect SYBR Green RT-PCR kit (Qiagen). Reactions were run in triplicate in three independent experiments. Primer sequences were 5′-GGAGGCTCAGAAACAACCAGC-3′ and 5′-TGGGATAACCTGGTGGGTAA-3′ for SETD2; 5′-CAATGACCCCTTCATTGACC-3′ and 5′-GATCTCGCTCCTGGAAGATG-3′ for GAPDH.
Statistics
All data represent the means ± s.d. of three independent experiments, and statistical significance was determined by one- or two-way ANOVA using GraphPad Prism 6 software (GraphPad Software, Inc, San Diego, CA, USA). A P value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
We acknowledge the assistance provided by the Flow Cytometry Shared Resource of Lombardi Comprehensive Cancer Center for FACS. We thank Professor Vincent Notario for critical reading of the manuscript.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41388–018-0429–3) contains supplementary material, which is available to authorized users.
References
- 1.Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222:385–6. [DOI] [PubMed] [Google Scholar]
- 2.Pignon JP, Tribodet H, Scagliotti GV, Douillard JY, Shepherd FA, Stephens RJ, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26:3552–9. [DOI] [PubMed] [Google Scholar]
- 3.Advanced Bladder Cancer Meta-analysis C. Neoadjuvant chemotherapy in invasive bladder cancer: a systematic review and meta-analysis. Lancet. 2003;361:1927–34. [DOI] [PubMed] [Google Scholar]
- 4.Sandler AB, Nemunaitis J, Denham C, von Pawel J, Cormier Y, Gatzemeier U, et al. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2000;18:122–30. [DOI] [PubMed] [Google Scholar]
- 5.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–98. [DOI] [PubMed] [Google Scholar]
- 6.Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, et al. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005;280:35261–71. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Xie S, Zhou Y, Xie Y, Liu P, Sun M, et al. H3K36 histone methyltransferase Setd2 is required for murine embryonic stem cell differentiation toward endoderm. Cell Rep. 2014;8:1989–2002. [DOI] [PubMed] [Google Scholar]
- 9.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RM, et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70:4287–91. [DOI] [PubMed] [Google Scholar]
- 12.Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46:287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al Sarakbi W, Sasi W, Jiang WG, Roberts T, Newbold RF, Mokbel K. The mRNA expression of SETD2 in human breast cancer: correlation with clinico-pathological parameters. BMC Cancer. 2009;9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–63. [DOI] [PubMed] [Google Scholar]
- 16.Carlezon WA Jr., Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–45. [DOI] [PubMed] [Google Scholar]
- 17.Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7:351–62. [DOI] [PubMed] [Google Scholar]
- 18.Moon YW, Rao G, Kim JJ, Shim HS, Park KS, SSAn. et al. LAMC2 enhances the metastatic potential of lung adenocarcinoma. Cell Death Differ. 2015;22:1341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–21. [DOI] [PubMed] [Google Scholar]
- 20.Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of poly (ADP-Ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer. 2010;1:812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi EB, Yang AY, Kim SC, Lee J, Choi JK, Choi C, et al. PARP1 enhances lung adenocarcinoma metastasis by novel mechanisms independent of DNA repair. Oncogene. 2016;35:4569–79. [DOI] [PubMed] [Google Scholar]
- 22.Maione P, Sacco PC, Sgambato A, Casaluce F, Rossi A, Gridelli C. Overcoming resistance to targeted therapies in NSCLC: current approaches and clinical application. Ther Adv Med Oncol. 2015;7:263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–20. [DOI] [PubMed] [Google Scholar]
- 24.Verlinsky Y, Rechitsky S, Schoolcraft W, Strom C, Kuliev A. Preimplantation diagnosis for Fanconi anemia combined with HLA matching. JAMA. 2001;285:3130–3. [DOI] [PubMed] [Google Scholar]
- 25.Kachnic LA, Li L, Fournier L, Willers H. Fanconi anemia pathway heterogeneity revealed by cisplatin and oxaliplatin treatments. Cancer Lett. 2010;292:73–79. [DOI] [PubMed] [Google Scholar]
- 26.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao G, Pierobon M, Kim IK, Hsu WH, Deng J, Moon YW, et al. Inhibition of AKT1 signaling promotes invasion and metastasis of non-small cell lung cancer cells with K-RAS or EGFR mutations. Sci Rep. 2017;7:7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giaccone G, Kim C, Thompson J, McGuire C, Kallakury B, Chahine JJ, et al. Pembrolizumab in patients with thymic carcinoma: a single-arm, single-centre, phase 2 study. Lancet Oncol. 2018;19:347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013;125:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mar BG, Chu SH, Kahn JD, Krivstov AV, Koche R, Castellano CA, et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood. 2017;130:2631–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiang W, He J, Huang C, Chen L, Tao D, Wu X, et al. miR-106b-5p targets tumor suppressor gene SETD2 to inactive its function in clear cell renal cell carcinoma. Oncotarget. 2015;6:4066–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. [DOI] [PubMed] [Google Scholar]
- 34.Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20:147–55. [DOI] [PubMed] [Google Scholar]
- 35.Yang R, Piperdi S, Gorlick R. Activation of the RAF/mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase pathway mediates apoptosis induced by chelerythrine in osteosarcoma. Clin Cancer Res. 2008;14:6396–404. [DOI] [PubMed] [Google Scholar]
- 36.Shenoy K, Wu Y, Pervaiz S. LY303511 enhances TRAIL sensitivity of SHEP-1 neuroblastoma cells via hydrogen peroxide-mediated mitogen-activated protein kinase activation and upregulation of death receptors. Cancer Res. 2009;69:1941–50. [DOI] [PubMed] [Google Scholar]
- 37.Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, et al. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:11569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jo SK, Cho WY, Sung SA, Kim HK, Won NH. MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis. Kidney Int. 2005;67:458–66. [DOI] [PubMed] [Google Scholar]
- 39.Mizumoto Y, Kyo S, Mori N, Sakaguchi J, Ohno S, Maida Y, et al. Activation of ERK1/2 occurs independently of KRAS or BRAF status in endometrial cancer and is associated with favorable prognosis. Cancer Sci. 2007;98:652–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5:1630–6. [DOI] [PubMed] [Google Scholar]
- 41.Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–81. [DOI] [PubMed] [Google Scholar]
- 42.Wu D, Zhau HE, Huang WC, Iqbal S, Habib FK, Sartor O, et al. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene. 2007;26:5070–7. [DOI] [PubMed] [Google Scholar]
- 43.Seo HS, Liu DD, Bekele BN, Kim MK, Pisters K, Lippman SM, et al. Cyclic AMP response element-binding protein overexpression: a feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer Res. 2008;68:6065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crans-Vargas HN, Landaw EM, Bhatia S, Sandusky G, Moore TB, Sakamoto KM. Expression of cyclic adenosine monophosphate response-element binding protein in acute leukemia. Blood. 2002;99:2617–9. [DOI] [PubMed] [Google Scholar]
- 45.Kovach SJ, Price JA, Shaw CM, Theodorakis NG, McKillop IH. Role of cyclic-AMP responsive element binding (CREB) proteins in cell proliferation in a rat model of hepatocellular carcinoma. J Cell Physiol. 2006;206:411–9. [DOI] [PubMed] [Google Scholar]
- 46.Jiang M, Zhu K, Grenet J, Lahti JM. Retinoic acid induces caspase-8 transcription via phospho-CREB and increases apoptotic responses to death stimuli in neuroblastoma cells. Biochim Biophys Acta. 2008;1783:1055–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagao K, Iwai Y, Miyashita T. RCAN1 is an important mediator of glucocorticoid-induced apoptosis in human leukemic cells. PLoS ONE. 2012;7:e49926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Z, Kong Q, Kone BC. CREB trans-activation of disruptor of telomeric silencing-1 mediates forskolin inhibition of CTGF transcription in mesangial cells. Am J Physiol Ren Physiol. 2010;298:F617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL, et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell. 2016;166:950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.