

Abstract
Skeletal muscle is the most abundant tissue in the human body and regulates a variety of functions including locomotion and whole-body metabolism. Skeletal muscle has a plethora of mitochondria, the organelles that are essential for aerobic generation of ATP which provides the chemical energy to fuel vital functions such as contraction. The number of mitochondria in skeletal muscle and their function decline with normal aging and in various neuromuscular diseases and in catabolic conditions such as cancer, starvation, denervation, and immobilization. Moreover, compromised mitochondrial function is also associated with metabolic disorders including type 2 diabetes mellitus. It is now clear that maintaining mitochondrial content and function in skeletal muscle is vital for sustained health throughout the lifespan. While a number of staining methods are available to study mitochondria, transmission electron microscopy (TEM) is still the most important method to study mitochondrial structure and health in skeletal muscle. It provides critical information about mitochondrial content, cristae density, organization, formation of autophagosomes, and any other abnormalities commonly observed in various disease conditions. In this article, we describe a detailed protocol for sample preparation and analysis of mouse skeletal muscle mitochondria by TEM.
Keywords: Transmission electron microscopy, Skeletal muscle, Mitochondria, Autophagy, Myopathy, Atrophy, Oxidative metabolism
Background
Skeletal muscle is a highly plastic tissue that undergoes morphological and metabolic adaptations in response to a number of extracellular cues. A number of perturbations including resistance or endurance exercise stimulates mitochondrial biogenesis leading to increased metabolic capacity and resistance to fatigue ( Li et al., 2008 ; Sandri, 2008). By contrast, during aging, inactivity, and in many catabolic disease states, skeletal muscle mitochondrial number and function decline, leading to increased fatigability and insulin resistance (Sandri, 2008). An accumulation of dysfunctional mitochondria may also result in progressive reactive oxygen species-induced damage, producing a further impairment of oxidative capacity in skeletal muscle ( Bonnard et al., 2008 ).
Mitochondria exist as a reticular membrane network that is located in different subcellular compartments in skeletal muscle. The subsarcolemmal (SS) mitochondria, account for 10-15% of the mitochondrial volume and lie directly beneath the sarcolemmal membrane, whereas the intermyofibrillar (IMF) mitochondria are located in close contact with the myofibril (Takahashi and Hood, 1996). Mitochondria are double membrane structures containing an intermembrane space between the outer and inner membranes as well as the inner matrix compartment, where most of the metabolic processes take place. The inner membrane is highly folded, forming so-called cristae, to accommodate its large surface area. The five complexes that make up the respiratory chain where oxidative phosphorylation takes place are embedded within the inner mitochondrial membrane. In this process, a proton gradient across the inner membrane is coupled to ATP synthesis at complex V ( Peterson et al., 2012 ). In addition to producing ATP for cross-bridge cycling between actin and myosin, mitochondria are a source of free radicals which regulate skeletal muscle physiology ( Peterson et al., 2012 ).
Transmission electron microscopy (TEM) is a powerful technique for ultrastructural studies (Watson, 1958). TEM has been very useful in studying mitochondrial structure in skeletal muscle in both physiological and pathological conditions ( Picard et al., 2013 ). For example, TEM can provide information about mitochondrial content, organization, cristae structure, and vacuolization as observed in some neuromuscular disorders such as Amyotrophic lateral sclerosis ( Picard et al., 2013 ). In many muscle wasting conditions, mitochondrial content is reduced through autophagy, also known as mitophagy. In this regard, TEM has been found to be an important approach to study autophagosome formation (Sandri, 2008). We have developed an efficient protocol that can be easily adapted in any laboratory to study the ultrastructure of mouse mitochondria in skeletal muscle by TEM ( Paul et al., 2010 ; Hindi et al., 2014 and 2018). In the following sections, we provide a step-wise protocol for sample preparation and analysis of SS and IMF mitochondria in skeletal muscle. A similar protocol can be used for studying other organelles in skeletal muscle by TEM as well.
Materials and Reagents
Glass specimen vials (Electron Microscopy Sciences, catalog number: 72630-05)
Razor blades, Double Edge Coated, Washed Version (Electron Microscopy Sciences, Personna, catalog number: 72000-WA)
Transfer pipette (Fisher Scientific, catalog number: 13-711-9BM)
Nitrile gloves
Glass strips, ultramicrotomy grade, 6.4 x 25 x 400 mm (Electron microscopy sciences, catalog number: 71012)
Glass slides (Fisher Scientific, catalog number: 12-550-15)
Syringes 1 ml (BD, catalog number: 329652)
Syringes 30 ml (BD, catalog number: 302833)
0.22 μm syringe filter (Merck, catalog number: SLGV033RS)
Conical centrifuge tube, 50 ml (VWR, catalog number: 21008-169)
Conical centrifuge tube, 15 ml (VWR, catalog number: 21008-089)
Non-sterile urine specimen container (Electron Microscopy Sciences, catalog number: 64231-10)
Filter paper, Qualitative Grade 1 Circles (GE Healthcare, Whatman, catalog number: 1001-090)
Glass stirring rod (United Scientific Supplies, catalog number: GSR012)
Wood applicators (Electron microscopy Sciences, catalog number: 72300)
Flat, silicone embedding mold (Electron Microscopy Sciences, catalog number: 70900)
Glass knife boat, 6.4 mm (Electron Microscopy Sciences, catalog number: 71007)
Glass knife box (Electron Microscopy Sciences, catalog number: 71010)
N95 respirator, with valve (VWR, catalog number: 89201-510)
Metal loop, perfect loop (Electron Microscopy Sciences, catalog number: 70944)
Grids, tabbed, copper, 200 mesh (Ted Pella, catalog number: 3HGC200)
Grid storage box, tabbed (Ted Pella, catalog number: 161)
Petri dish, glass, 100 x 20 mm (Corning, catalog number: 70165-102)
Glutaraldehyde, EM grade, 8% (Polysciences, catalog number: 00216-30)
Sodium phosphate monobasic monohydrate, NaH2PO4 H2O (Sigma-Aldrich, catalog number: S9638)
Sodium phosphate dibasic anhydrous, Na2HPO4 (Sigma-Aldrich, catalog number: S9763)
Osmium tetroxide, 10 x 1 g (Electron Microscopy Sciences, catalog number: 19110)
Ethanol, 200 Proof (Decon Labs, catalog number: 2701)
EMbed-812 kit, includes: EMbed-812, DDSA, NMA, and DMP-30 (Electron Microscopy Sciences, catalog number: 14120)
Sodium borate (MP Biomedicals, catalog number: 0219030980)
Toluidine blue O (Amresco, catalog number: 0672-25G)
Uranyl acetate dihydrate powder (depleted) (Electron Microscopy Sciences, catalog number: 22400)
NaOH pellets (Amresco, catalog number: 0583-500G)
Lead Nitrate, Pb(NO3)2 (Electron Microscopy Sciences, catalog number: 17900)
Sodium citrate, Na3(C6H5O7)·2H2O (Electron Microscopy Sciences, catalog number: 21140)
Propylene oxide, EM grade (Electron Microscopy Sciences, catalog number: 20401)
Dental wax (Electron Microscopy Sciences, catalog number: 72660)
3% glutaraldehyde (see Recipe 1)
0.1 M sodium phosphate buffer pH 7.4 (see Recipe 2)
1% osmium tetroxide (see Recipe 3)
Ethanol dilutions (see Recipe 4)
Embedding media (see Recipe 5)
1% toluidine blue stain (see Recipe 6)
4% uranyl acetate stock (aq) (see Recipe 7)
1 N-NaOH (see Recipe 8)
Reynold’s Lead Citrate (see Recipe 9)
Precautions/Hazards: As with any chemicals and reagents handled in the lab, users should be aware of how to use and manipulate them safely. Please refer to each chemical’s Material Safety Data Sheet (MSDS) for detailed information about precautions and hazards. Electron microscopy uses quite a few hazardous chemicals, such as: glutaraldehyde, osmium tetroxide, propylene oxide, uranyl acetate, lead citrate, and others. Please handle these chemicals using the proper personal protective equipment (PPE), ventilation conditions, and dispose of these chemicals in accordance with your institution’s Department of Environmental Health and Safety.
Equipment
Amber, wide-mouth glass bottle, 125 ml (VWR, catalog number: 10861-846)
Clear, media bottle, 1 L (Corning, PYREX®, catalog number: 1399-1L)
Graduated cylinder, 1,000 ml (VWR, catalog number: 65000-012)
Graduated cylinder, 25 ml (VWR, catalog number: 65000-002)
Magnetic stirring bar (VWR, catalog number: 58948-025)
General-Purpose Liquid-In-Glass Thermometer (VWR, catalog number: 89095-626)
Negative-action, curved self-closing tweezers (Electron Microscopy Sciences, catalog number: 72864-D)
Eyelash manipulator (Electron Microscopy Sciences, catalog number: 71182)
1,000 ml Glass Griffin beaker (VWR, catalog number: 10754-960)
50 ml Glass Griffin beakers (VWR, catalog number: 10754-946)
Glass funnel, 100 mm (VWR, catalog number: 10546-048)
50 ml volumetric flask (VWR, catalog number: 10123-996)
100 ml volumetric flask, amber (VWR, catalog number: 10124-022)
-20 °C Freezer (VWR, catalog number: 97014-903)
4 °C Refrigerator (VWR, catalog number: 14236-525)
Clinical rotator, variable speed tube rotator (Cole-Parmer, Stuart, catalog number: SB3)
Culture tube holder for clinical rotator, variable speed tube rotator, 12 mm offering a rolling action for tubes (Cole-Parmer, Stuart, catalog number: SB3/3)
pH meter, SymPhony B10P (VWR, catalog number: 89231-662)
pH probe, refillable, glass (VWR, catalog number: 89231-580)
Precision Balance, AV212C (Ohaus, out of production)
Precision Balance (OHAUS, catalog number: 30122632)
Hotplate stirrer (Fisher Scientific, catalog number: SP88857200P)
Vacuum oven (Electron Microscopy Sciences, catalog number: 63235-10)
Ultramicrotome (Reichert-Jung, model: Ultracut E, out of production, eBay or other second-hand markets)
New ultramicrotome (Leica Microsystems, model: Leica EM UC7)
Glass knifemaker (LKB, model: LKB Type 7801B, out of production, eBay or other second-hand markets)
New glass knifemaker, Leica EM KMR3 (Leica Microsystems,, catalog number: Leica EM KMR3)
Light microscope, Olympus CX31 (Olympus, catalog number: CX31)
Diamond knife, Diatome wet ultra 45°, 3.5 mm (Electron Microscopy Sciences, model: Diatome Ultra)
Phillips CM10 transmission electron microscope retrofitted with a new digital camera (Phillips, catalog number: CM10)
High Definition CCD Camera for TEM retrofitted to Phillips CM10 scope (Advanced Microscopy Techniques, catalog number: BioSprint)
Procedure
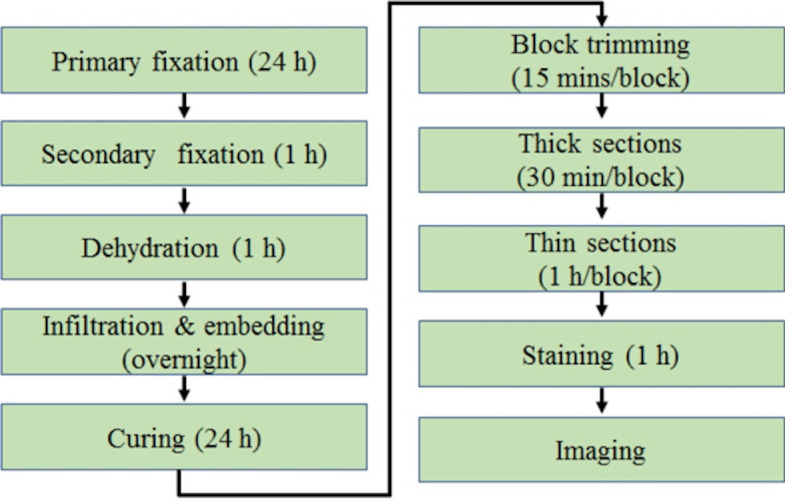
The basic steps for using TEM to analyze mitochondrial ultrastructure in skeletal muscle are presented in Figure 1.
Figure 1. The schematic view of general procedures for TEM of skeletal muscle from start to finish.
-
Primary fixation
The goal of primary fixation with glutaraldehyde is to preserve the ultrastructure of the skeletal muscle tissue in a physiologically relevant state by cross-linking proteins, primarily reacting with nucleophiles and other amines.
Following Recipe 1, prepare the 3% glutaraldehyde solution.
Remove the muscle tissue and immediately submerge it in 3% glutaraldehyde in a glass specimen vial and label accordingly (see Note 1).
Place the specimen vials on a clinical rotator for 30 min at room temperature.
After 30 min, quickly transfer the muscle to filter paper and cut it into the desired size (for mouse soleus, the muscle is cut in half for transverse and longitudinal sections, respectively) using a razor blade or scalpel with a light pulling motion to slice, do not crush tissue (see Note 2).
As quickly as possible, place each new section of tissue into its own specimen vial filled with 3% glutaraldehyde and label the vials appropriately.
Place the specimen vials on a clinical rotator and leave for 24 h at room temperature.
-
Secondary fixation
The purpose of secondary fixation with osmium tetroxide is two-fold, it cross-links lipids preserving their structure and it effectively stains the tissue by adding electron-dense material, enhancing contrast.
Following Recipe 2, prepare 0.1 M sodium phosphate buffer pH 7.4.
Remove the 3% glutaraldehyde from the specimen vials with a transfer pipette and transfer the solution to a proper waste container.
Add phosphate buffer to the specimen vials and return the samples to the clinical rotator for 5 min.
Dispose of the phosphate buffer in the proper waste container and repeat Step B3 twice more, on the third wash before disposing of the phosphate buffer, have your 1% osmium solution ready so that your sample does not dry out.
Following Recipe 3, prepare 1% osmium tetroxide (see Note 3).
Under a chemical fume hood with complete attention and care, dispense 1% osmium tetroxide into the specimen vials, cap the vials, and then return to the clinical rotator for 1 h.
The specimens should appear black.
Under a fume hood, remove the 1% osmium tetroxide solution from the specimen vials and dispose of the solution into the appropriate waste container.
Dispose of the plastic caps in an appropriate waste container as well and then use new plastic caps on your specimen vials for subsequent steps.
Add the phosphate buffer to the specimen vials and return the samples to the clinical rotator for 5 min.
Dispose of the phosphate buffer in the proper waste container and repeat Step B10 twice more.
-
Dehydration
Water must be removed from the tissue through graded alcohol dehydration to allow for the infiltration of embedding media.
Following Recipe 4, prepare all ethanol dilutions.
Transfer 10% ethanol solution to the specimen vials and return them to the clinical rotator for 5 min.
Remove 10% ethanol and repeat Step C2 with 25%, 50%, 75%, and 95% ethanol solutions.
Transfer 100% ethanol to the specimen vials and return them to the clinical rotator for 5 min.
Remove 100% ethanol and repeat Step C4 twice more.
Under a chemical fume hood, transfer 100% propylene oxide to the specimen vials and cap the lids tightly (lids may pop off if not secured).
Return the samples to the clinical rotator for 5 min.
Remove the 100% propylene oxide and dispose of it in the proper waste container.
Repeat Steps C6, C7, and C8 twice more.
-
Infiltration and embedding
Propylene oxide is equally miscible in both alcohol and embedding media, thereby clearing the alcohol and facilitating infiltration of the embedding media into the depths of the tissue.
Following Recipe 5, prepare the embedding media.
In a chemical fume hood, prepare a 2:1 (propylene oxide:embedding media) solution, the total volume you make will depend on how many samples you have (you want enough solution to completely cover your sample, even when rotating).
Transfer 2:1 (propylene oxide:embedding media) solution to the specimen vials, cap them, and place them on the clinical rotator for 1 h (see Note 4).
Remove 2:1 solution from the specimen vials and dispose of it in a plastic waste container.
In a chemical fume hood, prepare a 1:1 (propylene oxide:embedding media) solution, the same total volume you used with the 2:1 solution can be used here as well.
Transfer 1:1 (propylene oxide:embedding media) solution to the specimen vials, cap them, and return the vials to the clinical rotator. Leave the samples on the clinical rotator overnight and store the remaining embedding media in the -20 °C freezer.
The next day, dispose of the 1:1 solution in the same waste container and bring the embedding media to room temperature.
In a chemical fume hood, prepare a 1:2 (propylene oxide:embedding media) solution, using the same total volume as above.
Transfer 1:2 (propylene oxide:embedding media) solution to the specimen vials cap them, and place them on the clinical rotator for 1 h.
Remove 1:2 solution from the specimen vials and dispose of it in the designated waste container.
Transfer 100% embedding media to the specimen vials and return them to the rotator for 1 h.
Remove 100% embedding media and dispose of it in the designated waste container.
Transfer 100% embedding media to the specimen vials, leave the lids off, and place in a vacuum oven pressurized to 20 PSI for 1 h set to room temperature.
Specimens are now ready for curing and there is no need to remove the remaining 100% media from the vials, as the specimens will be transferred directly from the vials to the mold.
-
Curing
The viscous embedding media must be cured so that the block containing the tissue becomes hard enough for ultrathin sectioning.
Set a vacuum oven to 60 °C and 0 PSI, then confirm the temperature with a thermometer before proceeding.
With a pencil, write sample information on a small piece of paper that can fit into the base of the mold to be cured with the sample (will be visible through the cured plastic).
Fill the individual wells of the plastic mold halfway with 100% embedding media using a wood applicator to drizzle the media into the wells of the mold.
With fine negative-action tweezers, transfer the piece of paper with sample information into the bottom portion of the mold (the part that is farthest away from where the sample will rest).
Next, with fine negative-action tweezers, transfer the blackened, osmicated (hard) sample from the specimen vial to the mold (see Note 5).
Orient the sample accordingly to ensure that the sample can be sectioned in a cross-section or in a longitudinal section with ease (see Figure 6 for schematic on how it should look in the block).
Repeat Steps E4, E5, and E6 with the remaining samples.
Transfer enough 100% embedding media to fully fill the wells.
Ensure that the samples are correctly oriented one last time, as adding media can distort their position slightly.
Carefully, place the plastic mold into the oven to cure for at least 24 h (see Note 6).
-
Glass knife fabrication
Fabricating your own glass knives is a cost-saving technique, allowing you to preserve and extend the life of your expensive diamond knife for only cutting ultrathin sections. Pre-fabricated glass knives can be purchased if this equipment is not available at your institution.
Wearing nitrile gloves, clean your glass strip (6.4 x 25 x 400 mm) with detergent and a brush and then rinse it thoroughly with tap water (see Note 7).
Using a squirt bottle, rinse another time with distilled water.
Leave the glass strip on paper towels to dry in a relatively clean, dust-free environment.
Retract the Rear Glass Holder (Figure 2A) by rotating the Rear Glass Holder Knob (Figure 2A) and pull out the knob to lock it in place.
Ensure that the Breaking Knob (Figure 2B) on the front is turned fully counterclockwise.
Rotate the Locking Lever (Figure 2A) back to the rear position, fully raising the scorer.
Push the Scoring Shaft (Figure 2B) all the way in.
Make sure that the Score Selector (Figure 2B) above the Scoring Knob is set to.
Transfer a cleaned, dry glass strip to the glass knife maker and place it flush against the White Guide Plate (Figure 2B) and then push the strip against the first Lateral Arresting Stud (Figure 2C) (used for 25 mm wide strips).
While holding the glass strip in place, swing the Locking Lever gently forward until the Vertical Support Studs (Figure 2C) come into contact with the glass strip.
Remove your hands and then press the Locking Lever down further until there is significant resistance.
Place the Glass Fork (Figure 2A) under the left side of the glass strip to catch your glass square.
Pull the glass Scoring Shaft Knob in a swift fluid, smooth movement across the face of the glass strip until it stops to score the glass (Figure 3A).
Rotate the Breaking Knob clockwise until the glass breaks, you will be able to visually and audibly sense this.
Reset the Breaking Knob fully counterclockwise.
Rotate the Locking Lever back to the rear position, fully raising the scorer.
Push the Scoring Shaft Knob back in completely.
Clean off the glass break area with a brush.
Place the Dampening Pressure Adjustment Lever (Figure 2C) on the dot.
Adjust the Forward and Rear Glass Holder adjustment knobs to fit the glass square.
Place the square piece of glass that you just made at a 45° angle onto the Breaking Pins and leave the Glass Fork underneath the square (Figure 3B).
Disengage the Rear Glass Holder Knob from Step F4 by pressing it in and then rotating it clockwise to bring the Rear Glass Holder in contact with the corner of the glass square.
The glass square should sit with its corners between the forward and rear glass holders.
Gently pull the Locking Lever to the forward position until the Support Studs come into contact with the glass square.
Press the Locking Lever down further until there is significant resistance.
Pull the Scoring Shaft Knob in a swift fluid, smooth movement across the face of the glass square until it stops to score the glass.
Rotate the Dampening Pressure Adjustment Lever until the damping pad contacts the corner of glass in contact with the Front Glass Holder.
Rotate the Breaking Knob clockwise until the glass breaks; you will be able to visually and audibly hear this.
Reset the Breaking Knob fully counterclockwise.
Rotate the Locking Lever back to the rear position, fully raising the scorer.
Push the Scoring Shaft Knob back in completely.
Reset the Dampening Pressure Adjustment Lever back to the dot.
Rotate the Rear Glass Holder Knob counter-clockwise to retract the Rear Glass Holder away from the glass triangles and pull the knob out to lock it in place.
Use the Glass Fork to lift the left and right glass knives up and out of the knife maker and place the knives in a glass knife holder.
Clean off the glass break area with a brush.
Repeat Steps F1-F35 to make more glass knives.
To attach a plastic boat to the knife, heat some dental wax in a glass beaker on a hot plate to set to 100 °C.
Once the wax has fully melted, use a wood applicator to apply wax to the edges of the plastic boat and immediately align the plastic boat onto the back of your glass knife (see Note 8).
With the wood applicator, transfer more melted wax to the edges of the attached boat to seal completely (Figure 3D).
Test the wax seal by transferring distilled water with a transfer pipette into the plastic boat, creating a convex meniscus of water.
Leave the glass knife with the plastic boat for 5 min and then check the water level to determine if there are leaks.
If leak-free, empty water and store it in your glass knife holder for future use.
-
Trim block
Trimming the block into the trapezoidal pyramid shape enhances the thick and thin sections you are able to produce by reducing the frictional and resistive forces on your knives.
Remove the plastic mold from the oven (from Step E10) and let it cool for at least 30 min.
Once cool, place the plastic block with your tissue sample into the Specimen Holder (Figure 4A) that is set into the Specimen Holder Adapter (Figures 4A and 5A).
Clamp the Specimen Holder and Adapter (Figure 4A) to ensure that the specimen block does not move.
Press the green On-Off Main Switch (Figure 5A) on the power supply to the ultramicrotome and switch on the lights using the Multi-Position Illumination Switch (Figure 5C), both the overhead light and the light under the stage will turn on.
Bring the specimen into focus by turning the Focus Knob (Figure 5D) while looking through the Wide-Field Binocular Eyepieces.
The objective is to trim the specimen block into a trapezoidal pyramid that is as small as possible while including your specimen in the center (see Note 9) (Figure 6).
While looking through the Wide-Field Binocular Eyepieces (Figure 5B) and using a razor blade, trim the face of the block until you bring your sample into plane. You should see the lighter-colored resin and then the dark shadow of your sample in the trimmed slices that you are creating.
Next, while looking through the Wide-Field Binocular Eyepieces, use a razor blade to remove excess resin from all four sides of the block around your sample to sculpt your pyramid.
Unclamp the Specimen Holder from the Adapter, place the Specimen Holder onto the Specimen Arm (Figures 5C and 5D) of the ultramicrotome and use the screw to secure the Specimen Holder tightly into place.
Unclamp and remove the Specimen Holder Adapter, place your Knife Holder (Figures 4C and 5B) onto the ultramicrotome stage, and clamp the Knife Holder into place.
Make sure that the Clearance Angle Adjustment Knob (Figure 4C) is adjusted to the proper clearance angle, which should correspond to the angle written on the box that came with your diamond knife (6°).
Use the Knife Clamping Screw (Figure 4C) to clamp a glass knife onto the Knife Holder.
Use the East-West Stage Control Knob (Figure 5C) and the North-South Knife Feed Knob (Figure 5B) to bring the knife holder with your glass knife close to your block, without touching.
Adjust your Specimen Holder so that the leading edge of your sample (the part that is cut first and the base of your pyramid) is parallel with the knife edge.
Using the North-South Knife Feed Knob on the ultramicrotome, slowly bring the glass knife closer to the sample block. Your ultramicrotome is equipped with a light that shines up from the base, illuminating the gap between the knife and the block face. As you bring the knife closer to the block face, this illuminated area will dim more and more, signifying that the two surfaces are almost touching.
Set the Manual Fine-Feed Knife Control Knob (Figure 5C) to 1 μm.
While looking through the Wide-Field Binocular Eyepieces, rotate the Manual Fine-Feed Knife Control Knob to advance the sample to the knife edge while rotating the Handwheel (Figure 5D) for the Specimen Arm for a full cycle.
Repeat Step G17 until you begin to see sections of the sample face coming off onto the edge of the glass knife and then continue until the full face of your sample is being sectioned.
Now, the face is trimmed, smooth, and ready for thick sections.
Use the North-South Knife Feed Knob to move the Knife Holder away from your sample.
Unscrew the Knife Clamping Screw to loosen your glass knife and remove from the Knife Holder.
-
Cut thick sections
The purpose of cutting thick sections and then staining them is to confirm the orientation of the tissue sample before proceeding.
Following Recipe 6, prepare the 1% toluidine blue stain.
Turn on a hot plate to approximately 100 °C.
Place a glass knife with a plastic boat and tighten the Knife Clamping Screw (Figure 4C) to clamp the knife into place.
Use the East-West Stage Control Knob (Figure 5C) and the North-South Knife Feed Knob (Figure 5B) to bring the knife holder with your glass knife close to your block, without touching.
Adjust your Specimen Holder (Figure 5D) so that the leading edge of your sample (the part that is cut first and the base of your pyramid) is parallel with the knife edge.
Using the North-South Knife Feed Knob on the ultramicrotome, slowly bring the glass knife closer to the sample block. Your ultramicrotome is equipped with a light that shines up from the base, illuminating the gap between the knife and the block face. As you bring the knife closer to the block face, this illuminated area will dim more and more, signifying that the two surfaces are almost touching.
Fill the plastic boat with distilled water until the water is convex.
Place the Suction Syringe (Figure 5D) into the boat beneath the water line.
While looking through the Wide-Field Binocular Eyepieces (Figure 5B), use the Suction Knob (Figure 5D) to adjust the water level until you see an even silver reflection over the surface of the water (see Notes 10 and 11) (Figure 7).
Set the Manual Fine-Feed Knife Control Knob (Figure 5C) to 1 μm.
While looking through the Wide-Field Binocular Eyepieces, rotate the Manual Fine-Feed Knife Control Knob to advance the sample to the knife edge while rotating the Handwheel for the Specimen Arm (Figure 5D) for a full cycle.
Repeat Step H17 until you begin to see sections of the sample face coming off the edge of the glass knife onto the surface of the water and then continue until the full face of your sample is being sectioned (see Note 12).
Once you get a full-faced section, use a metal loop to fish out the part-sections and the full section you just cut and discard them by rotating the metal loop in a beaker of distilled water.
Set the Manual Fine-Feed Knife Control Knob to 0.5 μm.
While looking through the Wide-Field Binocular Eyepieces, rotate the Manual Fine-Feed Knife Control Knob to advance the sample to the knife edge while rotating the Handwheel for the Specimen Arm for a full cycle.
Discard the first couple of sections into the beaker of water.
Cut 5-10 more 0.5 μm sections.
Label two glass slides with the pertinent sample information and apply a drop (~2 cm diameter) of distilled water to the slide.
Use the metal loop to transfer individual sections to the drop of water on the glass slide and rotate the loop to transfer.
After you finish distributing the sections between the two glass slides, place the glass slides onto the hot plate (see Note 13).
After the water has fully evaporated, the sections are ready to be stained.
While the slides are still on the hot plate, add 1% toluidine blue stain drop-wise to completely cover the dried sections and leave for 2 min (see Note 14).
Wash the slides with running water to remove as much excess stain from the slides as possible and allow them to dry.
After allowing the slides to dry, view the sections with a light microscope (see Notes 15 and 16).
If satisfied with the orientation of the sections, then no adjustment is necessary and you can move on to cutting thin sections.
Use the North-South Knife Feed Knob to move the Knife Holder away from your sample.
Unscrew the Knife Clamping Screw to loosen your glass knife and remove it from the Knife Holder.
-
Cut thin sections
In order to form a high-resolution image, the electron beam must be able to penetrate the section without a significant loss of speed, requiring an ultrathin section.
Place a diamond knife onto the Knife Holder (Figure 5B) and tighten the Knife Clamping Screw (Figure 4C) to clamp the knife into place.
Use the East-West Stage Control Knob (Figure 5C) and the North-South Knife Feed Knob (Figure 5B) to bring the knife holder with your glass knife close to your block, without touching.
Adjust the Specimen Holder (Figure 5D) so that the leading edge of your sample (the part that is cut first and the base of your pyramid) is parallel with the knife edge.
Using the North-South Knife Feed Knob on the ultramicrotome, slowly bring the glass knife closer to the sample block. Your ultramicrotome is equipped with a light that shines up from the base, illuminating the gap between the knife and the block face. As you bring the knife closer to the block face, this illuminated area will dim more and more, signifying that the two surfaces are almost touching.
Fill the diamond knife boat with distilled water until the water is convex.
Place the Suction Syringe (Figure 5D) into the boat beneath the water line.
While looking through the Wide-Field Binocular Eyepieces (Figure 5B), use the Suction Knob (Figure 5D) to adjust the water level until you see an even silver reflection over the surface of the water (Figure 7).
Set the Manual Fine-Feed Knife Control Knob (Figure 5C) to 0.5 µm.
While looking through the Wide-Field Binocular Eyepieces, rotate the Manual Fine-Feed Knife Control Knob to advance the sample to the knife edge while rotating the Handwheel for the Specimen Arm (Figure 5D) for a full cycle.
Repeat Step I9 until you begin to see sections of the sample face coming off the edge of the glass knife onto the surface of the water and then continue until the full face of your sample is being sectioned (see Note 17).
Once you get a full-faced section, use a metal loop to fish out the part-sections and the full-faced section you just cut and discard by rotating the metal loop in a beaker of distilled water.
Next, on the power supply, set the value on the Semi-Thin Sectioning Control (Figure 5A) to 0.50 μm.
On the power supply, adjust the Cutting Speed Control (Figure 5A) to 1.0 mm/sec (this corresponds to the speed that the ultramicrotome arm uses on the down stroke).
Press down on the Motor Drive Control Lever (Figure 5D) that begins the automated advancement and rotation of the Specimen Arm.
Look through the Wide-Field Binocular Eyepieces and observe the full-faced sections coming off the knife (see Note 18).
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.40 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.30 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the binocular lenses, sections after this adjustment should begin to display colors that correspond to the approximate thickness of the section and the refractive index of the embedding material. At this point, they will most likely be yellow.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.25 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look yellow to blue.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.20 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look blue to purple.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.17 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look purple.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.14 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look purple to gold.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.12 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look gold.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.11 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look purely gold (see Note 19).
If satisfied, then continue cutting these sections until you have your desired quantity and continue to Step I47. If not satisfied, then continue to Step I40.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.10 µm.
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look gold to silver.
After 3-4 sections, look through the Wide-Field Binocular Eyepieces and once the Specimen Arm completes its down stroke sending a new section into the boat, adjust the Semi-Thin Sectioning Control to 0.09 µm (90 nm).
Use a metal loop to fish out the sections you just cut and discard them by rotating the metal loop in a beaker of distilled water.
When observed in the Wide-Field Binocular Eyepieces, sections after this adjustment should begin to look silver.
Continue cutting sections until you are satisfied, typically 15-20 homogenously colored sections are what to aim for.
Use the North-South Knife Feed Knob to move the Knife Holder away from your sample.
Use the negative-action forceps to pick up a copper grid with your dominant hand and place the eyelash manipulator tool in your non-dominant hand.
While looking through the Wide-Field Binocular Eyepieces, bring these tools into view.
Submerge the grid under the surface of the water and begin to advance it directly under your section of choice (see Note 20).
At the same time, slightly submerge your eyelash manipulator tool next to your section of choice and gently guide the section over your grid.
Hold the section in place as best as you can and slowly raise the grid beneath the section until the section rests relatively centered on top of the grid and remove from the water.
With your section-mounted grid still resting in the forceps, take a triangle of filter paper and gently lower it next to the outer rim of the grid. Water should begin transferring from the grid to the filter paper (see Note 21).
After the water is removed as best that you can, place the grid into the grid box.
Continue collecting sections from the boat of your diamond knife, typically 10 grids for each sample is plenty.
Allow grids to fully dry in your grid box overnight prior to staining.
-
Stain grids
Staining the sections with uranyl acetate and lead citrate enhances the contrast for countless cellular structures.
Turn on a hot plate/stirrer and adjust the temperature to ~200 °C.
Place a 1 L beaker filled with distilled water and a magnetic stir bar on top of the hot plate (Figure 8).
Bring the water to a boil and then turn off the hot plate while leaving the stirrer on (see Note 22).
Distribute the water into four 50 ml beakers and allow to cool to ~25 °C (see Note 23).
Following Recipe 7, prepare 1% uranyl acetate solution.
Following Recipe 8, prepare 1 N-NaOH solution.
Following Recipe 9, prepare Reynold’s lead citrate solution.
Use a 1 ml syringe, with the needle removed and a 0.22 μm syringe filter attached, to distribute a drop of uranyl acetate into a glass Petri dish, with the bottom filled with dental wax (see Note 24).
With negative-action forceps, place your grid, section-side down, onto the drop of uranyl acetate.
Set a timer for 30 min and cover the Petri dish with something that blocks light (see Note 25).
After 30 min, use negative-action forceps to pick up the grid and then dunk the grid into one of the 50 ml beakers filled with previously boiled, distilled water. Dunk the grid into and out of the water ~30 times.
Advance to the next 50 ml beaker filled with previously boiled, distilled water and dunk the grid into and out of the water ~30 times (use these same two beakers for subsequent uranyl acetate washes).
Set the negative-action forceps onto the benchtop and apply a triangle of filter paper gently to the outer rim of the copper grid to remove water from the surface of the grid.
Use a second glass Petri dish, with dental wax in the bottom, and place pellets of sodium hydroxide around the area of the dish where you will add drops of lead citrate. Close the lid immediately while preparing the lead citrate.
Use a 1 ml syringe, with the needle removed and a 0.22 µm syringe filter attached, to distribute a drop of lead citrate onto the bottom of the glass Petri dish. Cover immediately.
Transfer the grid that was resting in the negative-action forceps section-side down onto the drop of lead citrate. Close immediately and set a timer for 5 min.
After 5 min, use negative-action forceps to pick up the grid and then dunk the grid into one of the 50 ml beakers filled with previously boiled, distilled water. Dunk the grid into and out of the water ~30 times.
Advance to the next 50 ml beaker filled with boiled, distilled water and dunk the grid into and out of the water ~30 times (use these same two beakers for subsequent lead citrate washes).
Set the negative-action forceps onto the benchtop and apply a triangle of filter paper gently to the outer rim of the copper grid to remove water from the surface of the grid.
Transfer the grid back into the grid box and allow it to dry for at least a couple of hours prior to viewing in the electron microscope.
-
Capturing images with the transmission electron microscope
The goal of imaging with the transmission electron microscope is to provide high-resolution images of skeletal muscle and mitochondrial ultrastructure in an unbiased manner, taking enough images to get a true sense of the microanatomy.
Press the Panel On-Off Knob (Figure 9D) to illuminate the screen displaying the microscope settings.
Press the HT (High Tension) button (Figure 9D) where a green light above the button will illuminate and check the emission gauge to ensure that this returns closer to ~0 before proceeding.
Turn the Filament Knob (Figure 9D) clockwise ~23 steps with associated clicks as you turn the knob to reach saturation. This will take approximately 2 min and the scope will beep when this occurs. The viewing screen of the microscope should be illuminated in bright green light (see Note 26).
Remove the Specimen Holder (Figure 9B) from the microscope by rotating the holder approximately a quarter turn clockwise and then pull it out completely.
Place the Specimen Holder onto the Specimen Holder Stand (Figure 9F) and use the clip tool to raise the specimen clip at the end of the Specimen Holder (see Note 27).
Use the negative-action forceps to pick up and transfer your grid from the grid box to the circular grid holder at the bottom of the Specimen Holder, ensuring that the grid rests completely within the area and is not resting slightly outside.
Use the clip tool to lower the clip down onto your grid, holding the grid in place.
Insert the Specimen Holder halfway into the scope and stop. A red Pre-vacuum Indicator Light (Figure 9B) will turn on and you will hear the pre-vacuum click on.
Wait for the red Pre-vacuum Indicator Light to go out and then continue to insert the Specimen Holder fully into the high vacuum chamber by gently rotating the holder counter-clockwise and the vacuum will literally pull the specimen holder in (see Note 28).
The grid will now be in view on the viewing screen with a low magnification.
Move around your viewing area to find your sample area of interest by rotating the two grips (-x and -y) (Figure 9C).
Once in the desired area, increase the magnification with the Magnification Knob (Figure 9D). The screen will display your current level of magnification.
As you zoom in, you will have to adjust the viewing area with the two grips to keep it centered as much as possible.
After the magnification is increased to ~500x, there will be an audible beep prompting you to flip the Condenser Aperture Lever (Figure 9B), switching from the low magnification aperture to high magnification.
As you continue to increase the magnification, the muscle section will begin to come into view; however, the screen will get darker and darker because you have not increased the intensity of the electron beam (see Note 29).
As you zoom in, gently adjust the electron beam intensity by rotating the Intensity Knob (Figure 9C) so that you can see the muscle ultrastructure clearly.
Bring the muscle ultrastructure into focus with the Focus Knob (Figure 9D).
Turn on the computer monitor and click on the AMT software icon.
Before switching on the software and lifting the viewing screen, as a rule of thumb, adjust the intensity of the electron beam to 0.500, which is displayed on the screen next to ‘meter’ (see Note 30).
Lift the Fluorescent Screen (Figure 9C) using the lever and click on ‘Click for Live Image’, a gray-scale image of your sample will appear on the monitor (See Note 31).
The software is automatically set to ‘QualityLive’ and is in ‘Survey’ mode.
If you want to adjust the focus slightly better, click on the ‘Focus’ button that is under the ‘Survey’ button in the software, which will zoom in the image allowing you to get a finer focus.
Click the ‘Survey’ button to return to the preview screen.
If satisfied, click on ‘Final Image’.
Name your image with the relevant information and save the image in a designated folder.
Once finished taking images, close the software and lower the Fluorescent Screen using the lever.
Decrease your zoom with the magnification knob, while rotating the electron beam intensity counter-clockwise to compensate.
An audible beep ~500x will prompt you to flip the Condenser Aperture Lever from the high magnification aperture back to the low magnification one.
Continue to decrease the magnification to ~75x and then re-center the viewing area with the -x and -y axis grips.
Remove the Specimen Holder from the high vacuum chamber and remove your grid.
Place the specimen holder back into the high vacuum chamber exactly as was described above, waiting for the pre-vacuum to kick off before inserting fully.
Rotate the filament knob counter-clockwise until the viewing screen is no longer illuminated and there will be an audible beep.
Depress the high-tension button and the green light will go off.
Pull out the panel dim knob to turn off the screen.
Figure 6. The schematic view of how to trim the blocks prior to sectioning.
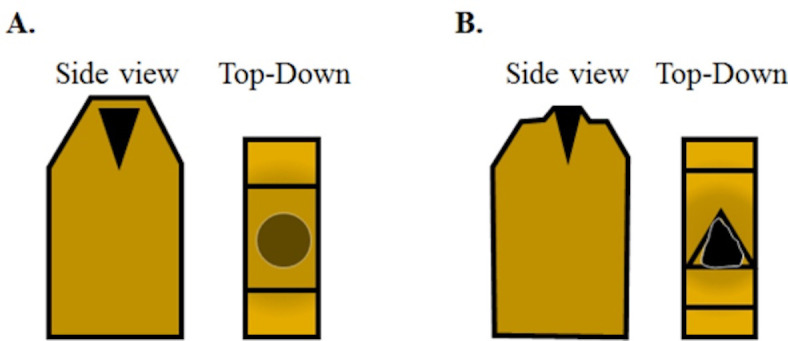
A. A fully cured block with an osmicated muscle sample aligned to be cut in cross-section should appear like this. From the top, looking down the osmicated muscle will look relatively less dark due to the layer of plastic between the tissue and the top of the block. Using a razor blade, the top of the block face needs to be trimmed down to expose the tissue sample. B. Initially, the trimmed layers will feature plastic only and eventually you will begin to see the black tissue in the trimmed layer with surrounding plastic once the tissue is exposed. Next, trim the plastic around the sample block to create a trapezoidal pyramid. The sample block is now ready to be faced using a glass knife.
Figure 2. Glass knife maker with labeled parts.

A. Angled side view with labeled parts. B. Angled frontal view with labeled parts. C. Close-up of angled side view with labeled parts.
Figure 3. Fabrication of glass knives.

A. Glass strip after being scored and broken into a glass square; B. Glass square aligned between the rear and forward glass holders with the dampening pressure adjustment lever adjusted and the glass fork underneath to catch the glass; C. Glass square, prior to scoring with the scorer above, being pressed onto the breaking pins with the locking lever pressed down fully; D. Several glass knives without boats and one with a plastic boat attached with dental wax in the glass knife holder.
Figure 4. Reichert-Jung Ultramicrotome with specimen holder in position to be trimmed and knife holder in position to cut specimens.

A. Specimen holder clamped into the adaptor and guide track for trimming of the embedded specimen. B. This is what the specimen block looks like while looking through the binocular eye pieces. C. The knife holder with a diamond knife is clamped into place and adjusted for cutting thin sections. D. This is what the diamond knife edge looks like while looking through the binocular eye pieces.
Figure 5. Reichert-Jung Ultramicrotome with labeled parts.

A. Power supply; B. Front view of the ultramicrotome; C. Angled left side view; D. Angled right side view.
Figure 7. Views through binocular lens showing proper water level in the diamond knife prior to sectioning.

The convex surface of water overfilling the diamond knife boat (A). Using the ultramicrotome syringe, the water level is lowered until there is a silvery, reflective surface as shown (B). After lowering the water and attempting to cut sections, if any water collects on the face of the block, the water may need to be lowered slightly further as shown (B), where you can see a slight shadow forming at the knife edge. However, if the water is lowered too much, it will pull away from the knife edge and will need to be refilled.
Figure 8. Setup for staining grids.

A. General equipment for staining grids. B. A close-up showing the use of sodium hydroxide pellets with two grids being stained with lead citrate, the lid is closed immediately after floating the grids on drops of lead citrate to prevent precipitation of lead citrate.
Figure 9. Phillips CM10 Transmission Electron Microscope.

A and B. A general overview of the electron microscope and its parts. C. The left control panel is mainly used to control the intensity of the electron beam and for adjusting the screens for focusing images with the objective lenses or capturing images by lifting the fluorescent screen. D. The right control panel houses most of the functions needed for adjusting magnification, focusing, and the screen for monitoring the electron microscope system as a whole. E. The vacuum screen shows a schematic of the vacuum system with the various pumps, valves, and pressure gauges used to monitor the performance of the system. F. The specimen holder on the specimen holder stand with the clip opening tool inserted and the grid clip lifted.
Data analysis
For most of our studies focused on the effect of knocking out or knocking in a signaling molecule and subjecting the muscle to various conditions that induce hypertrophy or atrophy, we perform TEM analysis on a minimum of n = 3 samples per group. We subjected TRAF6ff and TRAF6mko mice to denervation of the sciatic nerve for 10 days and then utilized TEM to study the ultrastructure of the TA muscle between these groups ( Paul et al., 2010 ). With TEM, we were able to complement our molecular biological experimental results by showing a reduction in atrophy and autophagy in the denervated TRAF6mko mice when compared to control TRAF6ff mice. Ultrastructurally, it was noted that the denervated muscle of TRAF6ff mice showed disorganization of SS and IMF mitochondria and an increase in autophagic vacuoles with the fusion of mitochondria to autophagosomes; whereas, this phenotype was largely absent in the denervated muscle of TRAF6mko mice ( Paul et al., 2010 ). In another study, we looked at the role of transgenic overexpression of the TWEAK cytokine in mice and used TEM to evaluate the ultrastructure of the soleus muscle in the naïve condition. It was found that there was a drastic decrease in the size and number of mitochondria in the TWEAK-TG mice when compared to wild-type littermates at 6 months of age ( Hindi et al., 2014 ). This was quantified by counting the SS mitochondria in three samples per group utilizing at least 10 pictures per sample magnified at 2,200x. This was repeated with the IMF mitochondria ( Hindi et al., 2014 ). We present the data as mean ± standard deviation (SD) and use a paired Student’s t-test to determine statistical differences among the different groups with a P < 0.05 being considered as statistically significant. Most recently, we used TEM to qualitatively compare the mitochondrial ultrastructure in the soleus muscle of Tak1fl/fl mice to those of muscle-specific knockout or Tak1mKO mice. We found that the Tak1mKO mice had an abundance of vacuolated mitochondria and an increased proportion of enlarged mitochondria with disturbances in the structure of their cristae when compared to the Tak1fl/fl mice ( Hindi et al., 2018 ). Our TEM observations were combined with biochemical analyses conducted between these groups to conclude that TAK1 is required for mitochondrial homeostasis in the skeletal muscle of these mice. Please see Figure 10 for examples of electron micrographs in the subsarcolemmal and intermyofibrillar regions of mouse skeletal muscle.
Figure 10. TEM micrographs of mouse skeletal muscle mitochondria.

A. Intermyofibrillar mitochondria from sagittal sections (Scale bar = 1 μm); B. Subsarcolemmal mitochondria from sagittal sections (Scale bar = 1 μm); C. Healthy subsarcolemmal mitochondria with well-defined, uniform cristae (Scale bar = 500 nm); D. Several enlarged subsarcolemmal mitochondria with non-uniform cristae (Scale bar = 500 nm).
Notes
Any mouse skeletal muscle tissue can be used for this analysis. Typically, in the skeletal muscle field, the mouse hind limb muscles are isolated for study, each muscle with its own characteristic fiber type composition. We use the mouse tibialis anterior (TA) and mouse soleus for TEM analyses due to their distinct differences in fiber type composition. The TA muscle has been characterized as a fast-glycolytic muscle, while the soleus has been characterized as a slow-oxidative muscle ( Schiaffino et al., 2011 ; Kammoun et al., 2014 ). Therefore, it is necessary to compare the same muscle between experimental groups. For a helpful video on how to isolate hind limb muscles from mice, please see the video in Hindi et al., 2017 . After isolating the muscle, it is imperative that you submerge the whole muscle into the glutaraldehyde as quickly as possible to preserve the muscle in its physiological state.
It must be emphasized that you don’t need very much of your tissue sample for TEM. The mouse soleus muscle is a relatively small muscle when compared to the mouse TA muscle. For the soleus, we typically cut the muscle in half mid-belly and use one half for our cross-section analysis and the other half for our sagittal section analysis without the need to further trim the tissue down. On the other hand, the mouse TA muscle is much larger and needs to be trimmed down to a thin strip, similar in size to the diameter of the soleus. If you need help with this visualization, use your mouse soleus muscle as a comparison. From there, divide your strip into two parts, one for cross-section analysis and one for sagittal section analysis.
Many of the chemicals in TEM tissue processing are highly toxic, especially osmium tetroxide. Osmium tetroxide is capable of fixing your corneas, so make sure that these steps are done in a properly ventilated chemical fume hood and personal protective equipment is worn.
Ensure that the black samples remain mobile in the various dilutions and that they aren’t stuck to the side of the vial. This can be helped by adjusting the rotator speed to rotate more appropriately with the increasingly viscous solutions. After adding the next media solution, briefly shake or flick samples to dislodge them from the bottom or side.
Try to avoid introducing bubbles into the block, especially around the sample itself. If there is a bubble at the bottom, then it’s not a big deal. If there is a bubble close to the sample, use a syringe needle to move and then lift it out of the mold.
If not given enough time to cure, the plastic won’t be hard enough to yield proper thin sections. This will be realized immediately when trimming the block with your razor blade. If the plastic appears to be giving too much to the pressure of the blade or there is significant resistance when trimming as if the blade is sticking (not simply because the block is hard), then this is a good indication that the block could use additional time in the oven to finish curing. Simply add the blocks back to the oven and give them some more time. Alternatively, if the oven maintains the temperature well without fluctuation, there is no problem leaving the tissue blocks in for longer periods of time. Often, I have placed the blocks into the oven to cure over the weekend and then returned to trim them on Monday without issue.
Do not touch the glass with your bare hands, the oils on your hands will ruin the quality of the glass when sectioning. In addition, if you feel that you need a supplement to go along with the instructions and figures, search for ‘LKB Type 7801B videos’ and there are a couple that may help illustrate these steps.
Attaching the plastic boats to the glass knives with wax can be a challenge at first. Practice is important. Transferring enough wax to the edges of the plastic boat and then attaching the boat to the glass as fast as possible before the wax begins to harden is the difficult part.
A trapezoidal pyramid is desired so that as the block encounters the diamond knife, there is a decrease in the surface area encountered by the blade as the blade cuts the full section. This relieves pressure/friction and ensures a smoother section with as little sectioning artifact as possible. This also preserves your knife edge for as long as possible. One video that does a pretty good job of demonstrating the trimming process that can be applied here is in Jenny, 2011.
Adjusting the water level to the point where there is a silver reflection is difficult for the first time, but is noticeable. Practice moving the water up and down as you go from convex to silver reflection and then to concave as you see the water begin to pull away from the knife edge. Please refer to the video by Soplop et al., 2009 that is helpful for both thick and thin sectioning and shows how the sections float on the surface of the water in the knife boat as they are sectioned.
If water has pulled away from the knife edge, the water level is too low and needs to be increased. Sometimes there is only a small portion of the knife where this has happened and in which case you can use a tool with an eye-lash adhered to the end to brush water up onto the edge.
When you begin trying to cut sections, you may notice that water has pulled up onto the face of your sample block most likely via surface tension. If this happens, then you need to stop, dry off the face with a piece of filter paper and then adjust your water level a little lower. Sometimes it needs to be a little lower closer to the knife edge to where you see a shadow closer to the knife edge, but not to the point where the water has pulled off the edge.
You don’t want the water on the hot plate to evaporate too quickly or else the sections will dry unevenly on the glass plate and will be bunch up in a wrinkly manner. If you notice this occurring, adjust the temperature so that they dry relatively slowly.
Similarly, you don’t want the hot plate to be hot enough to where it boils on contact, which sends toluidine blue stain bubbling everywhere. Adjust accordingly. You want a ring of yellowish gold, dried stain to develop, ~2 min. At this point, you wash.
With the light microscope, the main objective is to determine that your sections are oriented appropriately. You want to see that the section is free from obvious defects and that it is a true cross-section or longitudinal section.
If the sections look more oblique in the light microscope, then you can adjust the chuck that is holding the block in the way that you think necessary to correct the sectioning angles. At this point, depending on how drastic your changes are, you may need to re-face the block with the original glass knife in the previous ‘trimming step’.
When manual advancing the ultramicrotome arm and then rotating the arm to cut a section, ensure that this is a slow, smooth rotation. You don’t want to rotate the ultramicrotome arm to quickly or aggressively, else you may decrease the life of your diamond knife.
If the sample pyramid is small enough and the face was aligned parallel to the edge of the diamond knife, the sections will come off the knife and form a ribbon of sections that don’t float away from one another. This can be helpful when you collect the sections with a grid. In addition, you may be wondering why we are using the Semi-thin Sectioning Control rather than the Ultra-thin Sectioning Control. Our ultramicrotome model is relatively old and the Ultra-thin Sectioning Control feature relies on a thermal advance feature that we found to be inaccurate, most likely due to the age of the device. While it may take longer as you slowly decrease the section thickness from 0.5 µm to 0.100 µm (100 nm), the quality and consistency of our sections improved. So feel free to try out your Ultra-thin Sectioning Control feature, which may save some time if it is working properly.
Gold sections provide higher contrast, but not as much resolution. Silver sections provide high resolution, but with less contrast. Most of the time, gold sections are desired for skeletal muscle unless you’re trying to resolve difficult structures like the sarcoplasmic reticulum, T-tubules, etc. where silver sections may be more preferable.
The ultrathin sections are extremely fragile and care should be taken with the hair tools to gently guide and not destroy your sections. It may take some time to get the fine dexterity to comfortably mount your thin sections on grids without frustration. Practice is the best way.
When attempting to dry the grids with the triangle of filter paper, don’t place the filter paper directly onto your section. There is typically an outer ring of thicker copper where you should aim to rest the filter paper so that your section doesn’t get damaged.
Boiling the distilled water removes carbon dioxide from the water. The goal here is to prevent the precipitation of our heavy metal stains that can leave our grids with aggregated clumps of metal that interfere with the image quality. This can be quite frustrating after getting this far into the process. Take every precaution.
If you don’t allow your water to cool enough, you will notice that the contrast and staining looks relatively ‘flat’ when looking at the sections in the scope. Be patient and let it cool.
Filtering your heavy metal stains directly before use will hopefully remove any aggregates that are in the solution and lead to better staining.
Uranyl acetate is photosensitive and should be protected from light when in use and when not in use in dark colored bottles. Lead citrate is sensitive to carbon dioxide and should be stored in a bottle with a tight seal and parafilm to prevent aggregation. During the staining process, adding sodium hydroxide is a necessary step to remove as much carbon dioxide from the staining dish as possible while the reaction is taking place.
This scope uses a tungsten filament, which is why the filament knob is turned that many steps and in quick succession. If there were a LaB6 filament, this would be a much slower, more gradual process. The biggest difference between these two is the longevity and the price of the filaments. Tungsten filaments are good for ~100 h of use, whereas the LaB6 filaments yield ~1,000+ h of use and are considerably more expensive.
Be careful with the specimen holder and especially the specimen holder tool, which is very fragile and will break if too much force is applied. Be careful not to let your fingers come into contact with the lower, shiny half of the specimen holder. Oil from your hands will ruin the device if this is not done.
After the red light goes off, you want to gently rotate the specimen holder with constant pressure and rotation. If this is done too quickly and aggressively, the pressure in this chamber may increase and the high tension may kick off as a safety mechanism. Then you would need to start over. If you want to monitor the pressure of the chamber when inserting the specimen holder, press the button next to where it says vacuum on the ready screen, where you will see a schematic of the vacuum systems and pressure values on the bottom. The Ion Getter Pump (IGP) values will correspond to the pressure in the chamber that you are inserting the specimen holder. Try to keep this as low as possible (< 50).
Do not adjust the beam intensity too quickly or you may burn a hole in your sample grid. Always work on the right side (or clockwise) of crossover. If you rotate the intensity knob counter-clockwise, you will decrease the intensity and at a certain point you will be able to visualize the filament itself. This is known as crossover. If you continue to rotate the intensity knob counter-clockwise, it will get brighter and brighter. This would be the ‘left’ or ‘counter-clockwise’ side of crossover. We want to work on the right side where increasing the intensity is a result of rotating clockwise.
This CM10 Phillips Electron Microscope has been retrofitted with a new camera and uses AMT software to acquire images. Adjusting the electron beam intensity to 0.500 ensures that the camera won’t be saturated or burned by a beam that is too intense. One can adjust the intensity accordingly once the image is being looked at in the software and it will indicate when saturation or low signal occurs by the histogram in the software.
There is a fluorescent screen where your EM image is projected for manual adjustment by looking at it in the viewing screen, like looking through the objectives of a light microscope. This is an older electron microscope retrofitted with a computer and digital camera for imaging, allowing high resolution images. The camera is located at the bottom of the microscope and the beam is coming from above. The fluorescent screen needs to be flipped out of the way so that the digital camera can capture the final image.
Recipes
-
3% glutaraldehyde
In the fume hood, crack open the scored 10 ml vial of 8% glutaraldehyde and empty contents into a 50 ml conical test tube
Dilute to 3% glutaraldehyde by adding 16.6 ml of phosphate buffer
Invert several times to mix and it’s ready for immediate use
-
0.1 M sodium phosphate buffer pH 7.4
Measure 800 ml of distilled water in a graduated cylinder and add a magnetic stir bar
Turn on the stirrer
Weigh 3.1 g of sodium phosphate monobasic monohydrate (NaH2PO4·H2O) and add to the graduated cylinder
Weigh 10.9 g sodium phosphate dibasic anhydrous (Na2HPO4) and add to the graduated cylinder
Check the pH using a pH meter and ensure that it has a pH of 7.4
Volume up to 1,000 ml and store in a glass bottle
-
1% osmium tetroxide
Open the container containing the scored ampoules with 1 g of osmium tetroxide under the fume hood
Prepare 100 ml of 0.1 M phosphate buffer in a clean tight-sealing, amber glass bottle
Drop the scored ampoule into the glass bottle and using a clean glass rod, break open the ampoule into several pieces
Seal the glass bottle immediately with a cap and swirl several times
Allow the osmium crystals to dissolve completely at room temperature (can be left in the hood overnight)
Once dissolved, store your amber bottle inside its own air-tight container (a metal can with lid or any air-tight container large enough to fit the amber bottle inside) and then place in a 4 °C refrigerator until ready to use
-
Ethanol dilutions
Prepare ethanol dilutions (10%, 25%, 50%, 75%, 95%, 100%):
For 100%, pour 50 ml of 200 Proof ethanol into a 50 ml conical centrifuge tube and label accordingly
For 95%, measure 47.5 ml of 200 Proof ethanol in a graduated cylinder and then transfer to a 50 ml conical centrifuge tube. Volume up to the 50 ml graduation with distilled water and label accordingly
For 75%, measure 37.5 ml of 200 Proof ethanol in a graduated cylinder and then transfer to a 50 ml conical centrifuge tube. Volume up to the 50 ml graduation with distilled water and label accordingly
For 50%, measure 25.0 ml of 200 Proof ethanol in a graduated cylinder and then transfer to a 50 ml conical centrifuge tube. Volume up to the 50 ml graduation with distilled water and label accordingly
For 25%, measure 12.5 ml of 200 Proof ethanol in a graduated cylinder and then transfer to a 50 ml conical centrifuge tube. Volume up to the 50 ml graduation with distilled water and label accordingly
For 10%, measure 5.0 ml of 200 Proof ethanol in a graduated cylinder and then transfer to a 50 ml conical centrifuge tube. Volume up to the 50 ml graduation with distilled water and label accordingly
-
Embedding media
Turn on the hot plate to 60 °C and place the two anhydrides, DDSA and NMA, on top to reduce their viscosity
Measure 40 ml of EMbed 812 in a 50 ml conical centrifuge tube and then pour into a disposable urine specimen container
Using a new 50 ml conical centrifuge tube, measure 17 ml of warmed DDSA and then pour into the same disposable urine specimen container
Using a new 50 ml conical centrifuge tube, measure 26 ml of warmed NMA and pour into the same disposable urine specimen container
Using a graduated transfer pipette, transfer 1 ml of DMP-30 to the urine specimen container
Using the same transfer pipette, ensure that any remaining volume of EMbed 812, DDSA, and NMA is removed and transferred to the mixture in the urine specimen container
Cap the 50 ml conical centrifuge tubes and discard
Thoroughly stir the embedding mixture in the urine specimen container with a wooden applicator until homogenous
The embedding media can be stored in the -20 °C freezer until ready to use
If you’re ready, simply prepare propylene oxide solutions as needed and with respect to how many samples you are processing
-
1% toluidine blue stain
Weigh 2 g of sodium borate and dissolve in 100 ml of distilled water
Weigh 1 g of toluidine blue powder and dissolve in sodium borate solution
Filter the stain solution using a syringe filter and ready for use
-
4% uranyl acetate stock (aq)
Wear appropriate personal protective equipment including at least N-95 respirator
Boil 500 ml of distilled water in a beaker on a hot plate to remove CO2 and then turn off the hot plate
In the fume hood, weigh 4 g of uranyl acetate dihydrate (depleted) and transfer to 100 ml volumetric flask
Transfer 100 ml of near-boiling water to the amber glass bottle and place on a stirrer with magnetic stir bar to stir overnight
Filter solution with a Whatman #1 filter into a clean, amber glass bottle
Cap and label the bottle appropriately
This solution can be stored in a 4 °C refrigerator for months.
Make 1% working solution by diluting 2 ml of 4% uranyl acetate solution into 6 ml of previously boiled distilled water, CO2-free
Filter the 1% uranyl acetate solution immediately prior to use using a 0.22 μm syringe filter placing filtered drops directly into your staining dish
-
1 N-NaOH
Measure 0.40 g of sodium hydroxide
Dissolve in 8 ml of water
Volume up to 10 ml of solution
-
Reynold’s lead citrate (Reynold’s, 1963)
Wear appropriate personal protective equipment including at least an N-95 respirator
Boil 500 ml of distilled water in a beaker on a hot plate to remove CO2 and then let cool
In the fume hood, weigh 1.33 g of lead nitrate, Pb(NO3)2 and transfer to a 50 ml volumetric flask
In the fume hood, weigh 1.76 g of sodium citrate, Na3(C6H5O7)·2H2O and transfer to the same volumetric flask
Add 30 ml of the cooled CO2-free distilled water from above to the volumetric flask and cap
Shake the suspension vigorously for 1 min and then let it stand for 30 min with intermittent shaking to ensure complete conversion of lead nitrate to lead citrate. The solution will remain cloudy
Carefully add 1 N-NaOH 1 ml at a time (~8 ml total) swirl and check the pH using a pH meter. The pH should be 12.0 ± 0.1. If the measurement is above 12.1, then start over
The solution will go from cloudy to clear and should not have any turbidity
Volume up to 50 ml total and store in a tightly-sealed glass bottle
Acknowledgments
The authors would like to thank Yann S. Gallot for his thorough reading of the manuscript and the helpful comments. The authors would also like to declare no conflicts of interest or competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Bonnard C., Durand A., Peyrol S., Chanseaume E., Chauvin M. A., Morio B., Vidal H. and Rieusset J.(2008). Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest 118(2): 789-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hindi L., McMillan J. D., Afroze D., Hindi S. M. and Kumar A.(2017). Isolation, culturing, and differentiation of primary myoblasts from skeletal muscle of adult mice. Bio-protocol 7(9): e2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hindi S. M., Mishra V., Bhatnagar S., Tajrishi M. M., Ogura Y., Yan Z., Burkly L. C., Zheng T. S. and Kumar A.(2014). Regulatory circuitry of TWEAK-Fn14 system and PGC-1alpha in skeletal muscle atrophy program. FASEB J 28(3): 1398-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hindi S. M., Sato S., Xiong G., Bohnert K. R., Gibb A. A., Gallot Y. S., McMillan J. D., Hill B. G., Uchida S. and Kumar A.(2018). TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight 3(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jenny A.(2011). Preparation of adult Drosophila eyes for thin sectioning and microscopic analysis . J Vis Exp(54): 2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kammoun M., Cassar-Malek I., Meunier B. and Picard B.(2014). A simplified immunohistochemical classification of skeletal muscle fibres in mouse. Eur J Histochem 58(2): 2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H., Malhotra S. and Kumar A.(2008). Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med(Berl) 86(10): 1113-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paul P. K., Gupta S. K., Bhatnagar S., Panguluri S. K., Darnay B. G., Choi Y. and Kumar A.(2010). Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 191(7): 1395-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peterson C. M., Johannsen D. L. and Ravussin E.(2012). Skeletal muscle mitochondria and aging: a review. J Aging Res 2012: 194821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Picard M., White K. and Turnbull D. M.(2013). Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: a quantitative three-dimensional electron microscopy study. J Appl Physiol(1985) 114(2): 161-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reynolds E. S.(1963). The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. The Journal of Cell Biology 17 (1), 208-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandri M.(2008). Signaling in muscle atrophy and hypertrophy. Physiology(Bethesda) 23: 160-170. [DOI] [PubMed] [Google Scholar]
- 13. Schiaffino S. and Reggiani C.(2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91(4): 1447-531. [DOI] [PubMed] [Google Scholar]
- 14. Soplop N. H., Patel R. and Kramer S. G.(2009). Preparation of embryos for Electron Microscopy of the Drosophila embryonic heart tube . J Vis Exp(34): 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takahashi M. and Hood D. A.(1996). Protein import into subsarcolemmal and intermyofibrillar skeletal muscle mitochondria. Differential import regulation in distinct subcellular regions. J Biol Chem 271(44): 27285-27291. [DOI] [PubMed] [Google Scholar]
- 16. Watson M. L.(1958). Staining of tissue sections for electron microscopy with heavy metals. J Biophys Biochem Cytol 4(4): 475-478. [DOI] [PMC free article] [PubMed] [Google Scholar]