

Abstract
Vitamin C (ascorbic acid: AA) uptake in neurons occurs via the sodium-dependent vitamin C transporter-2 (SVCT2), which is highly expressed in the central nervous system (CNS). During chronic neuroinflammation or infection, CNS levels of lipopolysaccharide (LPS) and LPS-induced tumor necrosis factor-α (TNFα) are increased. Elevated levels of LPS and TNFα have been associated with neurodegenerative diseases together with reduced levels of AA. However, little is known about the impacts of LPS and TNFα on neuronal AA uptake. The objective of this study was to examine the effect of LPS and TNFα on SVCT2 expression and function using in vitro and in vivo approaches. Treatment of SH-SY5Y cells with either LPS or TNFα inhibited AA uptake. This reduced uptake was associated with a significant decrease in SVCT2 protein and mRNA levels. In vivo exposure to LPS or TNFα also decreased SVCT2 protein and mRNA levels in mouse brains. Both LPS and TNFα decreased SLC23A2 promoter activity. Further, the inhibitory effect of LPS on a minimal SLC23A2 promoter was attenuated when either the binding site for the transcription factor Sp1 was mutated or cells were treated with the NF-κB inhibitor, celastrol. We conclude that inflammatory signals suppress AA uptake by impairing SLC23A2 transcription through opposing regulation of Sp1 and NF-κB factors.
1. Introduction
Vitamin C (ascorbic acid: AA) is an essential micronutrient for cellular function, growth, and development, serving as a cofactor for an array of biological reactions and as a pleiotropic intracellular antioxidant [1, 2]. AA also serves as a first-line antioxidant defense to neutralize reactive oxygen species (ROS) by promoting the regeneration of endogenous antioxidants [3]. Brain tissue is susceptible to free radical damage and oxidative stress, since the brain is the most metabolically active organ in the body, and for this reason, the brain contains the highest concentration of vitamin C [3]. Accumulation of vitamin C in the brain cells occurs by a two-step mechanism, first by absorption across the choroid plexus and second by concentration into neurons and glia [4, 5]. The human sodium-dependent vitamin C transporter-2 (hSVCT2, the product of the SLC23A2 gene) controls these steps [4, 5]; knockout of murine SVCT2 results in undetectable levels of AA in the mouse brain [6].
Deficiencies of vitamin C could play a major role in brain dysfunction and neurodegeneration. Plasma vitamin C levels are found to be significantly lower in patients with neurodegenerative diseases [3, 7–10]. For instance, in Alzheimer's disease (AD), reduced vitamin C levels may accelerate amyloid-beta (Aβ) accumulation and cognitive impairment [3, 7, 8, 11]. Reciprocally, restoration of vitamin C levels and maintaining its homeostasis appear to safeguard against cognitive decline and the progression of AD neuropathology [12]. Therefore, studies aimed at understanding the underlying molecular mechanisms that control vitamin C homeostasis in the CNS may prove essential for developing strategies to counteract conditions of disease-enhanced oxidative stress through optimization of vitamin C homeostasis.
Neuroinflammation plays a pivotal role in the pathophysiology of many neurodegenerative diseases [13–16]. Chronic neuroinflammation and systemic bacterial infection lead to increased levels of proinflammatory cytokines like TNFα, IL-6, and IL-1β [17–20]. Lipopolysaccharide (LPS) is a cell wall-derived endotoxin of most gram-negative bacteria that is capable of inducing a strong neuroinflammatory response [21, 22]. Recent studies have shown that LPS and bacterial components are associated with plaques in postmortem AD brains [21, 23]. In addition, LPS is present in a septic patient's blood plasma [24–26], where it is assumed to play an important role in systemic inflammatory response syndrome, and also, some evidence indicates that sepsis is associated with lower blood vitamin C levels [27–29]. LPS is known to affect the function and expression of certain neuronal transporters [30–32]; however, the effect of LPS and subsequently induced TNFα on AA uptake and SVCT2 expression has been overlooked. Therefore, we studied the impact of LPS and TNFα exposure on neuronal AA uptake using both in vitro (SH-SY5Y cells) and in vivo (mouse) models.
2. Materials and Methods
2.1. Materials
The 14C-AA (specific activity 2.8 mCi/mmol; radiochemical purity > 98%) used in vitamin uptake analysis was acquired from PerkinElmer, Inc. (Boston, MA). LPS (E. coli 0111:B4) was purchased from Millipore Sigma (St. Louis, MO). Human TNFα was bought from Invitrogen (Carlsbad, CA), and murine TNFα was from PeproTech, Inc. (Rocky Hill, NJ). Antibodies were obtained from the following sources: anti-β-actin antibodies (ThermoFisher Scientific, Huntington Beach, CA), anti-NF-κB p65 and anti-IKKαβ antibodies (Abcam, Cambridge, MA), and anti-laminin antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). LI-COR (Lincoln, NE) IRDyes 800CW and 680LT goat anti-mouse and anti-rabbit secondary antibodies were used for western blot. Celastrol was ordered from InvivoGen, Inc. (San Diego, CA). Services provided by Integrated DNA Technologies (San Diego, CA) were used to synthesize oligonucleotide primers (Table 1). All other molecular biology-grade chemicals, reagents, and materials used in this study were from commercial sources.
Table 1.
Oligonucleotide primer combinations used to amplify coding region of the respective genes by RT-qPCR.
| Gene name | Forward and reverse primers (5′-3′) |
|---|---|
| Real-time PCR primers | |
| hSVCT2 | TCTTTGTGCTTGGATTTTCGAT; ACGTTCAACACTTGATCGATTC |
| hRFVT2 | CCCTGGTCCAGACCCTA; ACACCCATGGCCAGGA |
| hSp1 | CCATACCCCTTAACCCCG; GAATTTTCACTAATGTTTCCCACC |
| hβ-actin | CATCCTGCGTCTGGACCT; TAATGTCACGCACGATTTCC |
| mSVCT2 | AACGGCAGAGCTGTTGGA; GAAAATCGTCAGCATGGCAA |
| mNLRP3 | ATTACCCGCCCGAGAAAGG; TCGCAGCAAAGATCCACACAG |
| mTNFα | CATCTTCTCAAAATTCGAGTGACAA; TGGGAGTAGACAAGGTACAACCC |
| mβ-actin | ATCCTCTTCCTCCCTGGA; TTCATGGATGCCACAGGA |
| hnRNA primers | |
| mSVCT2 | ACTCTTGTCCATGGCTCTGG; GGGCAAAATCTTCGTTGGGT |
| mβ-actin | AGATGACCCAGGTCAGTATC; GAGCAGAAACTGCAAAGAT |
2.2. Culturing of SH-SY5Y Cells and AA Uptake Analysis
Human-derived neuroblastoma SH-SY5Y cells (ATCC, Manassas, VA) were given DMEM-F12 medium (ATCC) (with 20% fetal bovine serum (FBS) and penicillin-streptomycin added) and cultured in a temperature-controlled CO2 incubator at 37°C. In assays treating SH-SY5Y cells with LPS or TNFα, overnight serum starvation was followed by exposure either with LPS (20 μg/ml) or TNFα (20 ng/ml) or LPS plus celastrol (100 nM) in DMEM-F12 (with 0.5% FBS and no antibiotics added). SH-SY5Y cells were subjected to pretreatment with celastrol (100 nM) for 5 h before LPS treatment. After 48 h of LPS or TNFα or LPS plus celastrol treatment, 14C-AA uptake was performed in vitro [33, 34]. Briefly, SH-SY5Y cells were incubated (30 min) with 14C-AA (0.1 μCi) in Krebs-Ringer (KR) buffer at 37°C in a water bath, and then, sample lysates were prepared for radioactivity determination using a liquid scintillation counter [33, 34].
2.3. Animal Studies
For in vivo experiments, adult male C57BL/6 mice aged 8-12 weeks (Jackson Laboratory, Bar Harbor, ME) were administered either a single injection of LPS (5 mg/kg body weight; 100 μl of PBS) [33, 35] or TNFα (15 μg/mouse; 100 μl of PBS) [34, 36] or vehicle alone (100 μl of PBS) intraperitoneally (IP). After 72 h, mouse brains were removed immediately after euthanization and protein and RNA extracted. The animal protocol gained approval from the Institutional Animal Care and Use Committee (IACUC), Veteran Administration Medical Center, Long Beach, CA, and University of California, Irvine, CA.
2.4. Real-Time PCR (RT-qPCR) Analysis
Total RNA was prepared from SH-SY5Y cells and mouse brain exposed to either LPS or TNFα or LPS plus celastrol and their respective controls using the TRIzol reagent (Life Technologies). One microgram of total RNA sample was reverse-transcribed (RT) to cDNA using the i-Script cDNA synthesis kit (Bio-Rad, CA). RT-qPCR analysis was then performed using the cDNA and gene-specific primers (Table 1) in a CFX96 Real-Time iCycler (Bio-Rad) [33, 34]. Simultaneously amplified β-actin acted as a base for comparison to normalize the relative expression of different mRNAs, which were quantified using a relative relationship method (Bio-Rad) [33, 34].
2.5. Heterogeneous Nuclear RNA (hnRNA) Analysis
Table 1 shows the gene-specific hnRNA primers for Slc23a2 that were utilized in RT-qPCR analysis of total RNA prepared using LPS- or TNFα-treated mouse brain samples and parallel controls [37]. DNase I (Invitrogen) was added to the RNA samples for digestion before they were reverse-transcribed using i-Script cDNA synthesis kit reagents (Bio-Rad). β-Actin was again used to normalize RT-qPCR data, which was calculated as described above.
2.6. Transfection and Promoter Assays
SH-SY5Y cells were cultured on twelve-well plates (Corning) and cotransfected using Lipofectamine 2000 (3 μl/well; Invitrogen) with full-length, minimal, or mutant SLC23A2 promoter constructs (3 μg plasmid DNA/well) [38, 39] and Renilla luciferase-thymidine kinase (pRL-TK, 100 ng/well; Promega). Cells were left to incubate for 24 h before being treated with either LPS or TNFα or LPS plus celastrol for an additional 48 h. Then, the samples were processed following the Promega Dual-Luciferase Reporter Assay System. In short, each sample was lysed using passive lysis buffer (Promega), and a luminometer detected both the firefly and Renilla luciferase activities sequentially [38, 39].
2.7. Western Blotting
SH-SY5Y cells and mouse brain total protein were prepared by homogenization in RIPA (Radioimmunoprecipitation Assay) Buffer (Sigma) with 1X protease inhibitor cocktail (Roche, Nutley, NJ). The nuclear and cytosolic fractions from LPS- or LPS plus celastrol-treated SH-SY5Y cells were obtained using the NE-PER nuclear and cytoplasmic extraction kit (ThermoFisher Scientific). Total protein (60 μg) was separated using 4-12% NuPAGE Bis-Tris protein gels (Invitrogen) and transferred to a PVDF membrane. After protein transfer, the membrane was blocked for 10 min at room temperature in LI-COR Odyssey Blocking Buffer and then probed with previously characterized primary SVCT2 antibodies (1 : 500 dilution) [40], anti-IKKαβ antibodies (1 : 1000 dilution; Abcam), anti-NF-κβ p65 antibodies (1 : 1000 dilution; Abcam), anti-laminin antibodies (1 : 300 dilution; Santa Cruz Biotechnology), and anti-β-actin mouse monoclonal antibody (1 : 3000 dilution; ThermoFisher Scientific) used. The respective secondary antibodies (anti-rabbit IRDye-800 and anti-mouse IRDye-680, LI-COR Biosciences) were used in 1 : 30,000 dilutions [33, 34, 40]. Odyssey Infrared imaging system (LI-COR Biosciences) software was used to quantify the densitometry of specific band signal intensities normalized against β-actin.
2.8. Statistical Analyses
Carrier-mediated AA uptake analysis data from these investigations are presented as the means ± SE of at least 3 to 4 separate investigations with multiple determinations and represent a percentage relative to simultaneously performed untreated controls. RT-qPCR, western blot, and promoter assays were determined from at least 3 different batches of cells or 3 pairs of mouse samples. Student's t-test with P < 0.05 set as statistically significant was chosen to perform statistical analysis.
3. Results
3.1. Effect of LPS on hSVCT2 Function In Vitro
Recent studies have shown detectable levels of LPS in the AD brain [21, 23]. LPS is a potent inflammatory stimulator, which can affect the neuronal transport of many different substrates [30–32]. To assess the effect of LPS on AA uptake, we measured hSVCT2 mRNA levels by RT-qPCR after exposure of cells to various concentrations of LPS (10-50 μg/ml for 48 h). Data showed a concentration-dependent decrease in hSVCT2 mRNA expression relative to untreated control SH-SY5Y cells (Figure 1). To address the specificity of the LPS effect, we also determined mRNA levels of the brain-specific human riboflavin transporter-2 (hRFVT2) [41]. There was no significant change in hRFVT2 mRNA in SH-SY5Y cells treated with LPS (20 μg) compared to untreated cells (100 ± 11 and 117 ± 20 for control and LPS treatment, respectively).
Figure 1.
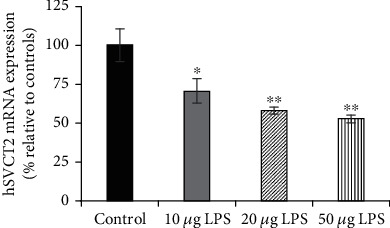
Effect of different concentrations of LPS on hSVCT2 mRNA expression in SH-SY5Y cells. SH-SY5Y cells were serum-deprived overnight and exposed to different concentrations of LPS (10, 20, and 50 μg/ml). After 48 h, total RNA was prepared to carry out RT-qPCR. Data are means ± SE of at least 6 separate determinations utilizing multiple batches of SH-SY5Y cells. ∗∗P < 0.01; ∗P < 0.05.
An LPS treatment paradigm of 20 μg/ml for 48 h also caused a significant (P < 0.001) inhibition of AA uptake (Figure 2(a)), coupled with a significant (P < 0.05) decrease in hSVCT2 protein expression (Figure 2(b)). The action of LPS was then interrogated at the level of the SLC23A2 promoter, by monitoring promoter activity of a luciferase reporter construct (pGL3-SLC23A2). This reporter construct was transiently transfected into SH-SY5Y cells, and then, cells were treated (24 h after transfection) with LPS (20 μg/ml for 48 h) before firefly luciferase activity was determined. LPS treatment caused significantly (P < 0.01) reduced SLC23A2 promoter activity when compared to untreated SH-SY5Y cells (Figure 2(c)). These data suggest that LPS decreases hSVCT2 function via transcriptional regulation.
Figure 2.
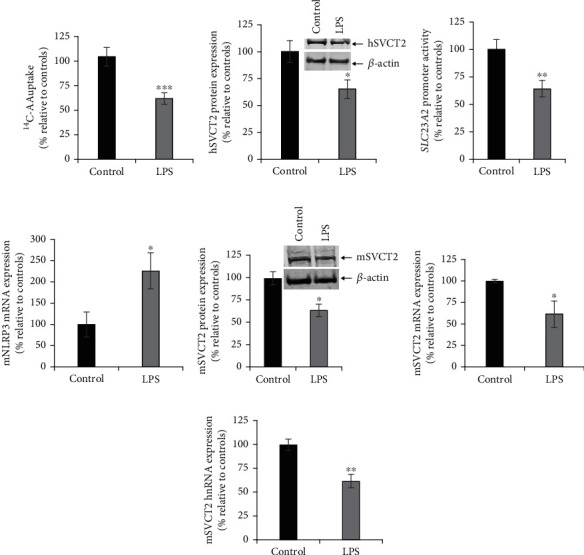
Effect of exposure of SH-SY5Y cells and mouse brain with LPS on different aspects of SVCT2 functional expression. (a) SH-SY5Y cells were serum-deprived overnight and exposed to LPS (20 μg/ml). After 48 h, AA uptake was determined. (b) SH-SY5Y cells were exposed to LPS for 48 h, and the protein was prepared to perform western blot analysis to determine the hSVCT2 protein expression levels. (c) SLC23A2 full-length promoter activity was determined in LPS-treated SH-SY5Y cells. (d, f) Total RNA isolated from LPS-administered and control mouse brain were used to determine the mSVCT2 and NLRP3 mRNA expression levels by RT-qPCR. (e) Protein samples prepared from mouse brain tissue of LPS (5 mg/kg body weight; 72 h) exposed and controls were subjected to western blotting to determine mSVCT2 protein expression levels. (g) Total RNA prepared from LPS-injected and control mouse brain were subjected to RT-qPCR to determine the mSVCT2 hnRNA expression levels. Values are means ± SE of at least 3-5 independent investigations utilizing multiple batches of SH-SY5Y cells or at least 3-5 pairs of mice. ∗∗∗P < 0.001; ∗∗P < 0.01; ∗P < 0.05.
3.2. Effect of LPS on SVCT2 Function In Vivo
Next, we examined whether similar effects occurred in vivo. LPS (5 mg/kg body weight; single dose [33, 35]) was administered intraperitoneally to wild-type (WT) mice, and responses were compared with vehicle (PBS)-injected controls. To monitor inflammation, the expression of nucleotide-binding, oligomerization domain- (NOD-) like receptor family, pyrin domain containing 3 (NLRP3) was examined. NLRP3 mRNA levels were found to be significantly (P < 0.05) increased in LPS-administered brain samples 72 h after injection (Figure 2(d)). These data demonstrate activation of an inflammatory marker following LPS administration in mouse brain samples. As expected, TNFα mRNA expression was also significantly (P < 0.0001) increased in LPS-administered mouse brain compared to controls (100 ± 19 and 351 ± 33 for control and LPS-administered mouse brains, respectively). Levels of mSVCT2 protein, mRNA, and heterogeneous nuclear RNA (hnRNA) were then determined in control and LPS-injected animals. Results showed that the expression levels of mSVCT2 protein, mRNA, and hnRNA were all markedly reduced in LPS-injected mouse brain samples versus controls (Figures 2(e)–2(g)). The latter represents the initial products of gene transcription, reflecting the rate of transcription of a given gene [37]. Collectively, these findings suggest that LPS also decreases the mSVCT2 functional expression in vivo, and this occurs via a transcriptional mechanism.
3.3. Effect of TNFα on SVCT2 Function
Elevated levels of proinflammatory cytokines such as TNFα, IL-6, and IL-1β in the brain and blood are linked to neuroinflammation and systemic bacterial infection [17–20]. TNFα is upregulated in AD brain samples and in the blood of patients infected with bacteria [18, 20]. Still, there is little evidence to describe the effect of TNFα on SVCT2 expression and function in neuronal systems. Treatment of SH-SY5Y cells with TNFα (20 ng/ml) significantly (P < 0.001) inhibited AA uptake (Figure 3(a)). This inhibition in uptake was again accompanied by marked decreases in the hSVCT2 protein (Figure 3(b)) and mRNA (Figure 3(c)) expression levels, as well as a significant reduction in SLC23A2 promoter activity (Figure 3(d)). To assess responses to TNFα in vivo, mice were injected intraperitoneally with TNFα (15 μg/mouse) [34, 36], followed by evaluation of mSVCT2 protein, mRNA, and hnRNA expression levels in mouse brain after 72 h. Results showed a significant (P < 0.05 for all) decrease in mSVCT2 protein, mRNA, and hnRNA in TNFα-administrated mouse brain samples compared to control mouse brain samples (Figures 3(e)–3(g)). Together, these results suggest that the TNFα-mediated decrease in SVCT2 functional expression also occurs via transcriptional mechanism(s).
Figure 3.
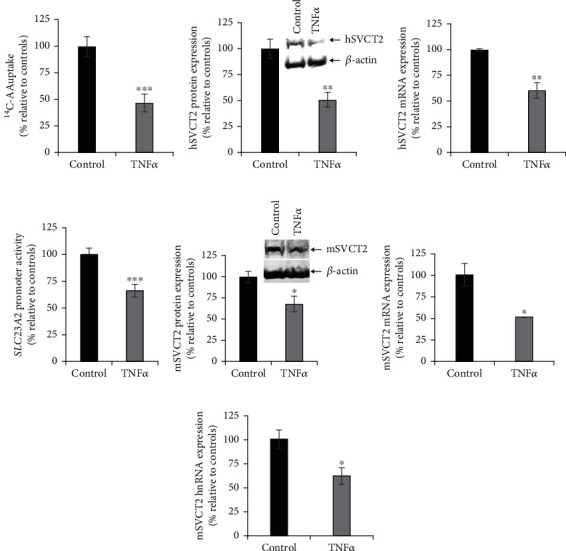
Effect of exposure of SH-SY5Y cells and mouse brain with TNFα on different aspects of SVCT2 functional expression. (a) SH-SY5Y cells were serum-deprived overnight and exposed to TNFα (20 ng/ml). After 48 h, AA uptake was performed. (b) SH-SY5Y cells were pretreated with TNFα for 48 h, and the total protein was prepared and used to perform western blotting to determine hSVCT2 protein expression levels. (c) Total RNA isolated from TNFα pretreated SH-SY5Y cells was utilized to determine the level of hSVCT2 mRNA expression by RT-qPCR. (d) SLC23A2 full-length promoter activity was determined in TNFα pretreated SH-SY5Y cells. (e) Protein samples from mouse brain tissue of TNFα (15 μg/mouse; 72 h) exposed and control mice were isolated to perform western analysis to determine mSVCT2 protein expression levels. (f) Total RNA isolated from TNFα-injected and control mouse brain was used to determine the mSVCT2 mRNA expression by RT-qPCR. (g) Total RNA samples prepared from TNFα-administered and control mouse brain were subjected to determination of the mSVCT2 hnRNA expression levels by RT-qPCR. Values are means ± SE of at least 3-5 separate investigations using different batches of SH-SY5Y cells or at least 3-4 pairs of mice. ∗∗∗P < 0.001; ∗∗P < 0.01; ∗P < 0.05.
3.4. Role of the Transcription Factor Sp1 in the Effect of LPS on Neuronal AA Uptake
As shown above, the full-length SLC23A2 promoter activity is inhibited by LPS in SH-SY5Y cells (Figure 2(c)). To investigate the molecular basis for this effect in greater depth, we tested whether the LPS inhibitory effect was also apparent on specific regions of the promoter. First, we assessed LPS action on a SLC23A2 minimal promoter reporter construct (-97 bp to +102 bp; Figure 4(a)) transiently expressed in SH-SY5Y cells. The SLC23A2 minimal (WT) promoter activity was significantly (P < 0.001) inhibited following LPS treatment compared with controls (Figure 4(b)). The minimal promoter region contains one Sp1-binding and two KLF-binding sites. It has been previously established that both transcription factors, Sp1 and KLF, are necessary to drive the basal transcriptional activity of the SLC23A2 promoter [39, 42]. Therefore, we tested the role of mutations at these sites on the inhibitory LPS effect. Mutant minimal SLC23A2 promoter constructs were transiently transfected into SH-SY5Y cells. After 24 h of transfection, cells were exposed to LPS for 48 h. Mutational ablation of either KLF-binding site (KLF1 or KLF2) had no effect on the inhibitory action of LPS (Figure 4(b)). In contrast, mutational ablation of the Sp1-binding site led to a loss of the LPS inhibitory effect on the SLC23A2 promoter activity (Figure 4(b)). Based on this result, we examined the effect on Sp1 protein and mRNA expression in SH-SY5Y cells after exposure to LPS. LPS treatment resulted in significantly (P < 0.05 for protein and P < 0.001 for mRNA) decreased human Sp1 protein and mRNA levels compared with untreated SH-SY5Y cells (Figures 4(c) and 4(d)). These data suggest that the transcription factor Sp1 mediates the LPS-induced inhibition of neuronal AA uptake.
Figure 4.
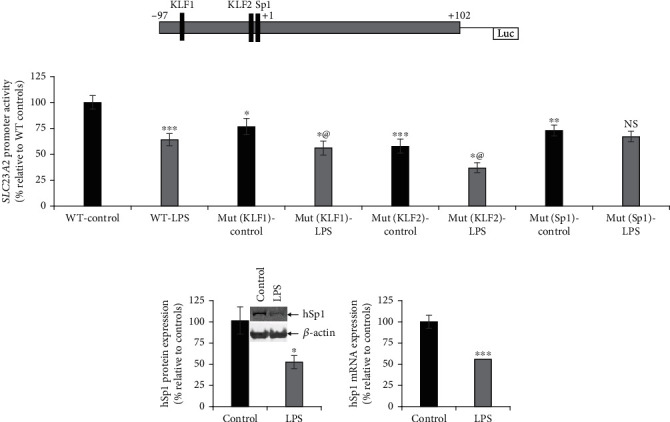
Effect of pretreating SH-SY5Y cells with LPS on the activity of SLC23A2 minimal (WT) and mutant promoter constructs. (a) Schematic depiction of SLC23A2 minimal promoter and the locations of Sp1- and KLF-binding sites. (b) SLC23A2 minimal (WT), Sp1- and KLF-binding sites mutated promoter construct activities were determined in LPS-exposed and control SH-SY5Y cells (KLF1 designates the site 5′ of the second site named KLF2). Western blot analysis was done to determine the expression levels of Sp1 protein (c), and RT-qPCR was done to determine the level of Sp1 mRNA (d) in LPS-exposed cells. @: cells exposed to LPS treatment significantly decreased compared to KLF1 or KLF2 controls. NS: not significant. Values are means ± SE of at least 4 separate experiments. ∗∗∗P < 0.001; ∗∗P < 0.01; ∗P < 0.05.
3.5. NF-κB Signaling Regulates the Inhibitory Effect of LPS
The NF-κB inflammatory signaling pathway is a part of the regulatory mechanism that mediates the action of LPS on gene expression [43–45]. LPS activates the NF-κB pathway in SH-SY5Y cells by driving nuclear translocation of NF-κB and promoting degradation of IKKαβ in the cytoplasm (Figures 5(a) and 5(b)). Both these actions were blocked by celastrol, which can act as a NF-κB inhibitor (Figures 5(a) and 5(b)). As Sp1 and NF-κB are often involved in coordinated regulation of gene expression [46–48], we examined whether NF-κB was engaged by LPS to repress hSVCT2 expression. The addition of celastrol to inhibit NF-κB action markedly reversed the effect of LPS-induced inhibition on AA uptake (Figure 5(c)). Celastrol markedly increased the hSVCT2 protein, mRNA expression levels, and SLC23A2 promoter activity (Figures 5(d)–5(f)). Collectively, these data support the concept that Sp1 and NF-κB signaling pathway coordinate to regulate SLC23A2 promoter activity in neuronal cells, where NF-κB is activated (Figures 5(a) and 5(b)) and Sp1 is inhibited (Figure 4) by elevated LPS.
Figure 5.
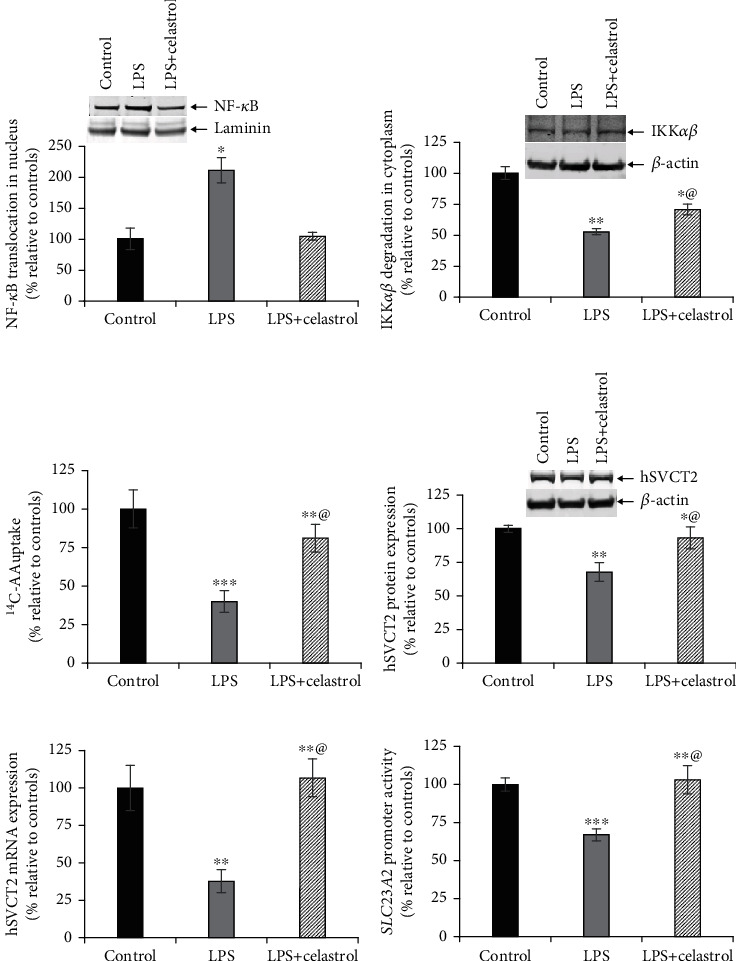
NF-κB signaling pathway plays a role in mediating the LPS-induced inhibitory effect on SVCT2 functional expression. SH-SY5Y cells were pretreated with celastrol (100 nM) for 5 h before LPS treatment; then 48 h later, the NF-κB expression in the nucleus (a), IKKαβ expression in the cytoplasm (b), carrier-mediated AA uptake (c), hSVCT2 protein expression (d), hSVCT2 mRNA expression (e), and SLC23A2 promoter activity (f) were determined. @: cells exposed to LPS plus celastrol treatment significantly recovered versus LPS alone-treated cells. Values are means ± SE of at least 3 separate investigations. ∗∗∗P < 0.001; ∗∗P < 0.01; ∗P < 0.05.
4. Discussion
The highest concentration of vitamin C is found in the brain, and its levels can be markedly lower in the plasma of patients with neurodegenerative disease [3, 9, 10]. Expression levels of SVCT2 are also markedly lower in human and mouse brain tissue with AD pathology (unpublished observations), which is a possible explanation for this deficit. Recent studies have shown that LPS and other bacterial products are associated with amyloid-beta (Aβ) plaques in AD brains [21, 23], suggesting that abnormal buildup of these bacterial components may be an additional factor to trigger chronic neuroinflammation during the disease course. Brain vitamin C dyshomeostasis induced by inflammation may therefore serve as a mechanism linking inflammation to exacerbated disease phenotypes. LPS and TNFα both affect neuronal gene function and expression [30–32, 49, 50]. Here, we investigated their roles in regulating AA uptake and SVCT2 expression in neuronal systems.
Both in vitro and in vivo assays suggest that the lower levels of SVCT2 functional expression observed upon LPS exposure in neuronal systems are mediated through transcriptional regulation of the SLC23A2 gene. Substantial evidence shows that Sp1 drives the basal activity of SLC23A2 promoter in different cellular systems [33, 39, 42] and has also been implicated in transporter regulation in inflammatory conditions [51]. In our investigation, we have used a SLC23A2 minimal promoter construct expressed in neuronal cells [38, 39] to demonstrate that Sp1 mutation attenuated the LPS-induced decrease in firefly luciferase activity (Figure 4). LPS also markedly decreased the Sp1 protein and mRNA expression, signifying that LPS degrades Sp1 and thus reduces SLC23A2 activity [52].
Sp transcription factors often interact with NF-κB signals mediated at the same DNA binding sites [46–48]. NF-κB is a pleiotropic regulator of many genes responsible for host defense, inflammatory response, and apoptosis [53–56]. The observation that celastrol reversed the inhibitory action of LPS and nuclear translocation of NF-κB implies a convergent regulation of hSVCT2 promoter activity by these dual and often dueling transcription factors. Further work will be needed to resolve the details of how these factors may exert their opposing influences on hSVCT2 promoter activity, possibly even via the very same DNA binding sites. It is worth mentioning that NF-κB is a redox-sensitive transcription factor and regulates SVCT2 mRNA expression in response to redox-state unsteadiness [57], and also, nitric oxide (NO) regulates SVCT2 expression via the NF-κB signaling pathway [58].
In summary, our findings suggest that LPS and TNFα downregulate the functional expression of SVCT2, the major vitamin C transporter in the brain. These actions may contribute to the low levels of AA observed during neuroinflammatory insults.
Acknowledgments
The present study was supported by the National Institutes of Health grant DK107474 (VSS).
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
No potential conflict of interest was reported by the authors.
Authors' Contributions
Conceptualization was performed by V.S.S., M.K., and J.S.M.; formal analysis was performed by T.T. and V.S.S.; investigation and writing (review and editing) were performed by T.T., A.A., M.K., J.S.M., and V.S.S.; writing (original draft preparation) was performed by T.T., A.A., J.S.M., and V.S.S.; and visualization was performed by T.T., A.A., and V.S.S. All authors have read and approved the manuscript.
References
- 1.Packer L., Fuchs J. Vitamin C in Health and Disease. New York, NY, USA: Marcel Dekker Inc; 1997. [Google Scholar]
- 2.Carr A. C., Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. The American Journal of Clinical Nutrition. 1999;69(6):1086–1107. doi: 10.1093/ajcn/69.6.1086. [DOI] [PubMed] [Google Scholar]
- 3.Monacelli F., Acquarone E., Giannotti C., Borghi R., Nencioni A. Vitamin C, Aging and Alzheimer's disease. Nutrients. 2017;9(7):p. 670. doi: 10.3390/nu9070670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice M. E. Ascorbate regulation and its neuroprotective role in the brain. Trends in Neurosciences. 2000;23(5):209–216. doi: 10.1016/S0166-2236(99)01543-X. [DOI] [PubMed] [Google Scholar]
- 5.Bürzle M., Suzuki Y., Ackermann D., et al. The sodium-dependent ascorbic acid transporter family SLC23. Molecular Aspects of Medicine. 2013;34(2-3):436–454. doi: 10.1016/j.mam.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Sotiriou S., Gispert S., Cheng J., et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Medicine. 2002;8(5):514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 7.Murakami K., Murata N., Ozawa Y., et al. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of Alzheimer's disease. Journal of Alzheimer's Disease. 2011;26(1):7–18. doi: 10.3233/JAD-2011-101971. [DOI] [PubMed] [Google Scholar]
- 8.Dixit S., Bernardo A., Walker J. M., et al. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chemical Neuroscience. 2015;6(4):570–581. doi: 10.1021/cn500308h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charlton K. E., Rabinowitz T. L., Geffen L. N., Dhansay M. A. Lowered plasma vitamin C, but not vitamin E, concentrations in dementia patients. The Journal of Nutrition, Health & Aging. 2004;8(2):99–107. [PubMed] [Google Scholar]
- 10.Rinaldi P., Polidori M. C., Metastasio A., et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiology of Aging. 2003;24(7):915–919. doi: 10.1016/S0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 11.Harrison F. E., Hosseini A. H., McDonald M. P., May J. M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacology, Biochemistry, and Behavior. 2009;93(4):443–450. doi: 10.1016/j.pbb.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison F. E. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer's disease. Journal of Alzheimer's Disease. 2012;29(4):711–726. doi: 10.3233/JAD-2012-111853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang F., Liu J., Shi J. S. Anti-inflammatory activities of resveratrol in the brain: role of resveratrol in microglial activation. European Journal of Pharmacology. 2010;636(1-3):1–7. doi: 10.1016/j.ejphar.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y. N., Lai C. C., Chiu C. T., Lin J. J., Wang J. Y. L-ascorbate attenuates the endotoxin-induced production of inflammatory mediators by inhibiting MAPK activation and NF-κB translocation in cortical neurons/glia Cocultures. PLoS One. 2014;9(7, article e97276) doi: 10.1371/journal.pone.0097276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim S. H., Noh M. Y., Kim H. J., et al. A therapeutic strategy for Alzheimer's disease focused on immune-inflammatory modulation. Dementia and neurocognitive disorders. 2019;18(2):33–46. doi: 10.12779/dnd.2019.18.2.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sochocka M., Koutsouraki E. S., Gasiorowski K., Leszek J. Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer's disease: a new approach to therapy. CNS & Neurological Disorders Drug Targets. 2013;12(6):870–881. doi: 10.2174/18715273113129990072. [DOI] [PubMed] [Google Scholar]
- 17.Bauer J., Strauss S., Schreiter-Gasser U., et al. Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer's disease cortices. FEBS Letters. 1991;285(1):111–114. doi: 10.1016/0014-5793(91)80737-N. [DOI] [PubMed] [Google Scholar]
- 18.Wen L., Chiu C., Huang Y., Chang C., Wang J. Rapid glia expression and release of proinflammatory cytokines in experimental _Klebsiella pneumoniae_ meningoencephalitis. Experimental Neurology. 2007;205(1):270–278. doi: 10.1016/j.expneurol.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Alasmari F., Ashby C. R., Jr., Hall F. S., Sari Y., Tiwari A. K. Modulation of the ATP-binding cassette B1 transporter by neuro-inflammatory cytokines: role in the pathogenesis of Alzheimer's disease. Frontiers in Pharmacology. 2018;9:p. 658. doi: 10.3389/fphar.2018.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grammas P., Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer's disease. Neurobiology of Aging. 2001;22(6):837–842. doi: 10.1016/S0197-4580(01)00276-7. [DOI] [PubMed] [Google Scholar]
- 21.Zhan X., Stamova B., Jin L. W., DeCarli C., Phinney B., Sharp F. R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology. 2016;87(22):2324–2332. doi: 10.1212/WNL.0000000000003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B., He Y., Ma J., et al. Mild cognitive impairment has similar alterations as Alzheimer's disease in gut microbiota. Alzheimer's & Dementia. 2019;15(10):1357–1366. doi: 10.1016/j.jalz.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y., Jaber V., Lukiw W. J. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer's disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Frontiers in Cellular and Infection Microbiology. 2017;7:p. 318. doi: 10.3389/fcimb.2017.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cepinskas G., Wilson J. X. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. Journal of Clinical Biochemistry and Nutrition. 2008;42(3):175–184. doi: 10.3164/jcbn.2008026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Opal S. M. The clinical relevance of endotoxin in human sepsis: a critical analysis. Journal of Endotoxin Research. 2002;8(6):473–476. doi: 10.1177/09680519020080060401. [DOI] [PubMed] [Google Scholar]
- 26.Venet C., Zeni F., Viallon A., et al. Endotoxaemia in patients with severe sepsis or septic shock. Intensive Care Medicine. 2000;26(5):538–544. doi: 10.1007/s001340051201. [DOI] [PubMed] [Google Scholar]
- 27.Armour J., Tyml K., Lidington D., Wilson J. X. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. Journal of Applied Physiology (1985) 2001;90(3):795–803. doi: 10.1152/jappl.2001.90.3.795. [DOI] [PubMed] [Google Scholar]
- 28.Schorah C. J., Downing C., Piripitsi A., et al. Total vitamin C, ascorbic acid, and dehydroascorbic acid concentrations in plasma of critically ill patients. The American Journal of Clinical Nutrition. 1996;63(5):760–765. doi: 10.1093/ajcn/63.5.760. [DOI] [PubMed] [Google Scholar]
- 29.Wu F., Tyml K., Wilson J. X. Ascorbate inhibits iNOS expression in endotoxin- and IFN gamma-stimulated rat skeletal muscle endothelial cells. FEBS Letters. 2002;520(1-3):122–126. doi: 10.1016/S0014-5793(02)02804-1. [DOI] [PubMed] [Google Scholar]
- 30.Ogura M., Nakamichi N., Takano K., et al. Functional expression of a glutamine transporter responsive to down-regulation by lipopolysaccharide through reduced promoter activity in cultured rat neocortical astrocytes. Journal of Neuroscience Research. 2006;83(8):1447–1460. doi: 10.1002/jnr.20855. [DOI] [PubMed] [Google Scholar]
- 31.Hsueh P. T., Wang H. H., Liu C. L., Ni W. F., Chen Y. L., Liu J. K. Expression of cerebral serotonin related to anxiety-like behaviors in C57BL/6 offspring induced by repeated subcutaneous prenatal exposure to low-dose lipopolysaccharide. PLoS One. 2017;12(6, article e0179970) doi: 10.1371/journal.pone.0179970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wittmann G., Mohácsik P., Balkhi M. Y., Gereben B., Lechan R. M. Endotoxin-induced inflammation down-regulates L-type amino acid transporter 1 (LAT1) expression at the blood-brain barrier of male rats and mice. Fluids Barriers CNS. 2015;12(1):p. 21. doi: 10.1186/s12987-015-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian V. S., Sabui S., Moradi H., Marchant J. S., Said H. M. Inhibition of intestinal ascorbic acid uptake by lipopolysaccharide is mediated via transcriptional mechanisms. Biochimica et Biophysica Acta - Biomembranes. 2018;1860(2):556–565. doi: 10.1016/j.bbamem.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian V. S., Sabui S., Subramenium G. A., Marchant J. S., Said H. M. Tumor necrosis factor alpha reduces intestinal vitamin C uptake: a role for NF-κB-mediated signaling. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2018;315(2):G241–g248. doi: 10.1152/ajpgi.00071.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Y., Liu Y., Hao W., et al. Stimulation of TLR4 attenuates Alzheimer's disease-related symptoms and pathology in tau-transgenic mice. Journal of Immunology. 2016;197(8):3281–3292. doi: 10.4049/jimmunol.1600873. [DOI] [PubMed] [Google Scholar]
- 36.Arda-Pirincci P., Oztay F., Bayrak B. B., Yanardag R., Bolkent S. Teduglutide, a glucagon-like peptide 2 analogue: a novel protective agent with anti-apoptotic and anti-oxidant properties in mice with lung injury. Peptides. 2012;38(2):238–247. doi: 10.1016/j.peptides.2012.09.030. [DOI] [PubMed] [Google Scholar]
- 37.Elferink C. J., Reiners J. J., Jr. Quantitative RT-PCR on CYP1A1 heterogeneous nuclear RNA: a surrogate for the in vitro transcription run-on assay. BioTechniques. 1996;20(3):470–477. doi: 10.2144/19962003470. [DOI] [PubMed] [Google Scholar]
- 38.Reidling J. C., Subramanian V. S., Dahhan T., Sadat M., Said H. M. Mechanisms and regulation of vitamin C uptake: studies of the hSVCT systems in human liver epithelial cells. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2008;295(6):G1217–G1227. doi: 10.1152/ajpgi.90399.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reidling J. C., Rubin S. A. Promoter analysis of the human ascorbic acid transporters SVCT1 and 2: mechanisms of adaptive regulation in liver epithelial cells. The Journal of Nutritional Biochemistry. 2011;22(4):344–350. doi: 10.1016/j.jnutbio.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heskett C. W., Teafatiller T., Hennessey C., et al. Enteropathogenic Escherichia coli infection inhibits intestinal ascorbic acid uptake via dysregulation of its transporter expression. Digestive Diseases and Sciences. 2020;66(7):2250–2260. doi: 10.1007/s10620-020-06389-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foley A. R., Menezes M. P., Pandraud A., et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137(1):44–56. doi: 10.1093/brain/awt315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiao H., May J. M. Regulation of the human ascorbate transporter SVCT2 exon 1b gene by zinc- finger transcription factors. Free Radical Biology & Medicine. 2011;50(9):1196–1209. doi: 10.1016/j.freeradbiomed.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexandrov P., Zhai Y., Li W., Lukiw W. Lipopolysaccharide-stimulated, NF-kB-, miRNA-146a- and miRNA-155-mediated molecular-genetic communication between the human gastrointestinal tract microbiome and the brain. Folia Neuropathologica. 2019;57(3):211–219. doi: 10.5114/fn.2019.88449. [DOI] [PubMed] [Google Scholar]
- 44.Yao C., Purwanti N., Karabasil M. R., et al. Potential down-regulation of salivary gland AQP5 by LPS via cross-coupling of NF-κB and p-c-Jun/c-Fos. The American Journal of Pathology. 2010;177(2):724–734. doi: 10.2353/ajpath.2010.090282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan W., Yu C., Hsuchou H., Kastin A. J. The role of cerebral vascular NFkappaB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cellular Physiology and Biochemistry. 2010;25(6):623–630. doi: 10.1159/000315081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao X., Moerman A. M., Barger S. W. Neuronal kappa B-binding factors consist of Sp1-related proteins. Functional implications for autoregulation of N-methyl-D-aspartate receptor-1 expression. Journal of Biological Chemistry. 2002;277(47):44911–44919. doi: 10.1074/jbc.M204292200. [DOI] [PubMed] [Google Scholar]
- 47.Hirano F., Tanaka H., Hirano Y., et al. Functional interference of Sp1 and NF-kappaB through the same DNA binding site. Molecular and Cellular Biology. 1998;18(3):1266–1274. doi: 10.1128/MCB.18.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu A., Hoffman P. W., Lu W., Bai G. NF-κB site interacts with Sp factors and up-regulates the NR1 promoter during neuronal differentiation. The Journal of Biological Chemistry. 2004;279(17):17449–17458. doi: 10.1074/jbc.M311267200. [DOI] [PubMed] [Google Scholar]
- 49.Stellwagen D., Beattie E. C., Seo J. Y., Malenka R. C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. The Journal of Neuroscience. 2005;25(12):3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Motagally M. A., Lukewich M. K., Chisholm S. P., Neshat S., Lomax A. E. Tumour necrosis factor alpha activates nuclear factor kappaB signalling to reduce N-type voltage-gated Ca2+ current in postganglionic sympathetic neurons. The Journal of Physiology. 2009;587(11):2623–2634. doi: 10.1113/jphysiol.2009.172312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kekuda R., Saha P., Sundaram U. Role of Sp1 and HNF1 transcription factors in SGLT1 regulation during chronic intestinal inflammation. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2008;294(6):G1354–G1361. doi: 10.1152/ajpgi.00080.2008. [DOI] [PubMed] [Google Scholar]
- 52.Ye X., Liu S. F. Lipopolysaccharide down-regulates Sp1 binding activity by promoting Sp1 protein dephosphorylation and degradation. The Journal of Biological Chemistry. 2002;277(35):31863–31870. doi: 10.1074/jbc.M205544200. [DOI] [PubMed] [Google Scholar]
- 53.Baeuerle P. A., Henkel T. Function and activation of NF-kappaB in the immune system. Annual Review of Immunology. 1994;12(1):141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 54.Li N., Karin M. Is NF-κB the sensor of oxidative stress? The FASEB Journal. 1999;13(10):1137–1143. doi: 10.1096/fasebj.13.10.1137. [DOI] [PubMed] [Google Scholar]
- 55.Pahl H. L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 56.Yamamoto Y., Gaynor R. B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. The Journal of Clinical Investigation. 2001;107(2):135–142. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Savini I., Rossi A., Catani M. V., Ceci R., Avigliano L. Redox regulation of vitamin C transporter SVCT2 in C2C12 myotubes. Biochemical and Biophysical Research Communications. 2007;361(2):385–390. doi: 10.1016/j.bbrc.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Portugal C. C., da Encarnação T. G., Socodato R., et al. Nitric oxide modulates sodium vitamin C transporter 2 (SVCT-2) protein expression via protein kinase G (PKG) and nuclear factor-κB (NF-κB) The Journal of Biological Chemistry. 2012;287(6):3860–3872. doi: 10.1074/jbc.M111.260166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.