

Abstract
Adiponectin is an adipokine that exerts insulin-sensitizing and anti-inflammatory roles in insulin target tissues including liver. While the insulin-sensitizing function of adiponectin has been extensively investigated, the precise mechanism by which adiponectin alleviates diet-induced hepatic inflammation remains elusive. Here, we report that hepatocyte-specific knockout (KO) of the adaptor protein APPL2 enhanced adiponectin sensitivity and prevented mice from developing high-fat diet–induced inflammation, insulin resistance, and glucose intolerance, although it caused fatty liver. The improved anti-inflammatory and insulin-sensitizing effects in the APPL2 hepatocyte–specific KO mice were largely reversed by knocking out adiponectin. Mechanistically, hepatocyte APPL2 deficiency enhances adiponectin signaling in the liver, which blocks TNF-α–stimulated MCP-1 expression via inhibiting the mTORC1 signaling pathway, leading to reduced macrophage infiltration and thus reduced inflammation in the liver. With results taken together, our study uncovers a mechanism underlying the anti-inflammatory role of adiponectin in the liver and reveals the hepatic APPL2–mTORC1–MCP-1 axis as a potential target for treating overnutrition-induced inflammation in the liver.
Introduction
Obesity is a major medical problem worldwide. It is associated with insulin resistance, type 2 diabetes, and nonalcoholic fatty liver disease (NAFLD). It is currently believed that overnutrition-induced chronic inflammation is one of the key mechanisms underlying obesity-induced metabolic diseases (1,2).
Adiponectin is an adipose tissue–derived hormone that possesses antidiabetes and anti-inflammatory functions (3). Circulating levels of adiponectin are inversely associated with obesity and type 2 diabetes. Administration of adiponectin into diabetic mouse models restores insulin sensitivity by improving glucose metabolism (4). Adiponectin has also been shown to exert an anti-inflammatory role by suppressing inflammatory responses in macrophages, adipocytes, and endothelial cells (5). A number of studies reveal an inverse correlation of circulating adiponectin levels with liver inflammation (6) and NAFLD (7); however, the molecular mechanism underlying the anti-inflammatory action of adiponectin, especially in hepatocytes, remains largely unknown.
Adiponectin exerts its beneficial effects via binding to its plasma membrane receptors, including AdipoR1, AdipoR2, and T-cadherin (3,8). While AdipoR1 is ubiquitously expressed in most tissues, AdipoR2 is primarily expressed in the liver (9) and T-cadherin is absent in hepatocytes under physiological conditions (10). APPL1 and APPL2 are two scaffold proteins that bind directly to AdipoR1/R2. The binding of APPL1 with AdipoR1 and AdipoR2 positively mediates adiponectin signaling (11) and facilitates the cross talk between adiponectin and insulin signal pathways (12). APPL2, an APPL1 isoform displaying 54% identity in protein sequences (13), interacts with multiple receptors and signaling molecules in cells, including AdipoR1 and AdipoR2 (13), Rab5 (14), NuRD/MeCP1 (14), GIPC1 (15), and FSHR (16). In contrast to APPL1, APPL2 negatively regulates adiponectin signaling and function by blocking the binding of APPL1 to the adiponectin receptors (13). Muscle-specific ablation of APPL2 increases insulin sensitivity and promotes insulin-induced glucose uptake (17). Human studies have linked single nucleotide polymorphisms on the Appl2 gene with high cardiovascular risk (18), obesity (19), and NAFLD occurrence (20). However, the physiological role of APPL2 in liver is currently unknown.
In the current study, we report the function of APPL2 in diet-induced inflammation and insulin resistance in the liver. Our results demonstrate a critical role of adiponectin signaling in preventing inflammation and macrophage infiltration in the liver and identifies TNF-α–induced MCP-1 as a key target of adiponectin’s action in hepatocytes.
Research Design and Methods
Animal Experiments
APPL2HepKO mice and their Loxp controls were fed a regular diet or a high-fat diet (HFD). For HFD feeding, 8-week-old male knockout (KO) mice and their control littermates were fed a 60% HFD (60% fat; Research Diets, New Brunswick, NJ) for 12 weeks. For examination of insulin signaling in vivo, the KO and Loxp control mice fed HFD for 12 weeks were fasted overnight and then injected with 1 unit/kg human insulin (Humulin R; Eli Lilly, Indianapolis, IN) or an equal volume of saline. Five minutes postinjection, liver and epididymal fat tissues were collected and stored at −80°C. For collection of skeletal muscle tissues, the mice were injected for 10 min. Hyperinsulinemic-euglycemic clamp studies were performed on animals fed with HFD for 8 weeks as previously described (21).
Statistical Analysis
The Western blot images were quantified with the National Institutes of Health ImageJ software. All results are presented as mean ± SD, and P values were calculated with Student t test, one-way ANOVA, or two-way ANOVA with Tukey multiple comparison test with use of the GraphPad Prism 7 software (*P < 0.05, **P < 0.01, and ***P < 0.001).
Data and Resource Availability
The data sets generated during the current study are available from the corresponding author on reasonable request.
Results
Generation and Characterization of Hepatocyte-Specific APPL2 KO Mice
To investigate the physiological role of APPL2 in liver, we generated hepatocyte-specific APPL2 KO (APPL2HepKO) mice by crossing APPL2 Loxp mice (Supplementary Fig. 1A and B) with albumin promoter–controlled cre-expressing mice. APPL2 deficiency in the liver was confirmed by Western blot analysis (Supplementary Fig. 1C). The KO mice were viable and fertile, born at the expected Mendelian ratios, and showed no significant differences in their body weight (Supplementary Fig. 1D), food intake (Supplementary Fig. 1E), and body composition (Supplementary Fig. 1F) compared with their control littermates under normal chow feeding.
To determine effects of APPL2 on glucose and lipid metabolism, we performed glucose and insulin tolerance tests. APPL2HepKO mice showed a trend of improvement in glucose tolerance (Supplementary Fig. 1G) and significant enhancement of insulin sensitivity (Supplementary Fig. 1H) compared with their Loxp wild-type littermates, suggesting that hepatic APPL2 plays an inhibitory role in systematic insulin action. Consistently, there was a trend of reduced fasting insulin levels in the KO mice in comparison with their controls (Supplementary Fig. 1I). No changes in fasting glucose levels (Supplementary Fig. 1J) and pyruvate-stimulated glucose production (Supplementary Fig. 1K) were observed between the KO and control mice. A trend of reduced TAG levels, but not free fatty acid (FFA) levels, was observed in the sera of the KO mice (Supplementary Fig. 1L), suggesting that APPL2 regulates TAG metabolism in the liver.
To investigate the role of APPL2 in regulating energy homeostasis, we measured O2 consumption and found a significant increase in O2 consumption in APPL2HepKO mice compared with their controls (Supplementary Fig. 1N). Since there were no changes in food intake (Supplementary Fig. 1E), body weight (Supplementary Fig. 1D), and resting metabolic rate (Supplementary Fig. 1P), this enhancement was likely due to elevated locomotor activity (Supplementary Fig. 1M). The KO mice also showed a significant increase in respiratory quotient (Supplementary Fig. 1O), suggesting carbohydrate as a favorable energy source for APPL2-deficient mice.
APPL2HepKO Mice Are Resistant to HFD-Induced Insulin Resistance
For investigation of the effect of APPL2 on diet-induced insulin resistance, APPL2HepKO mice and the Loxp controls were fed with an HFD for 14 weeks. The KO mice displayed no significant difference in body weight (Supplementary Fig. 2A), food intake (Supplementary Fig. 2B), body composition (Supplementary Fig. 2C), fasting glucose levels (Fig. 1B), or glucose tolerance (Fig. 1C) compared with their Loxp control littermates. However, APPL2 deficiency led to reduced fasting insulin levels (Fig. 1A) and greatly improved insulin tolerance (Fig. 1D), suggesting an inhibitory role of APPL2 in regulation of insulin sensitivity. The effects of hepatic APPL2 on insulin sensitivity appear cell autonomous, as no significant difference in serum levels of adiponectin and leptin was observed between the KO and control mice (Supplementary Fig. 2D and E).
Figure 1.
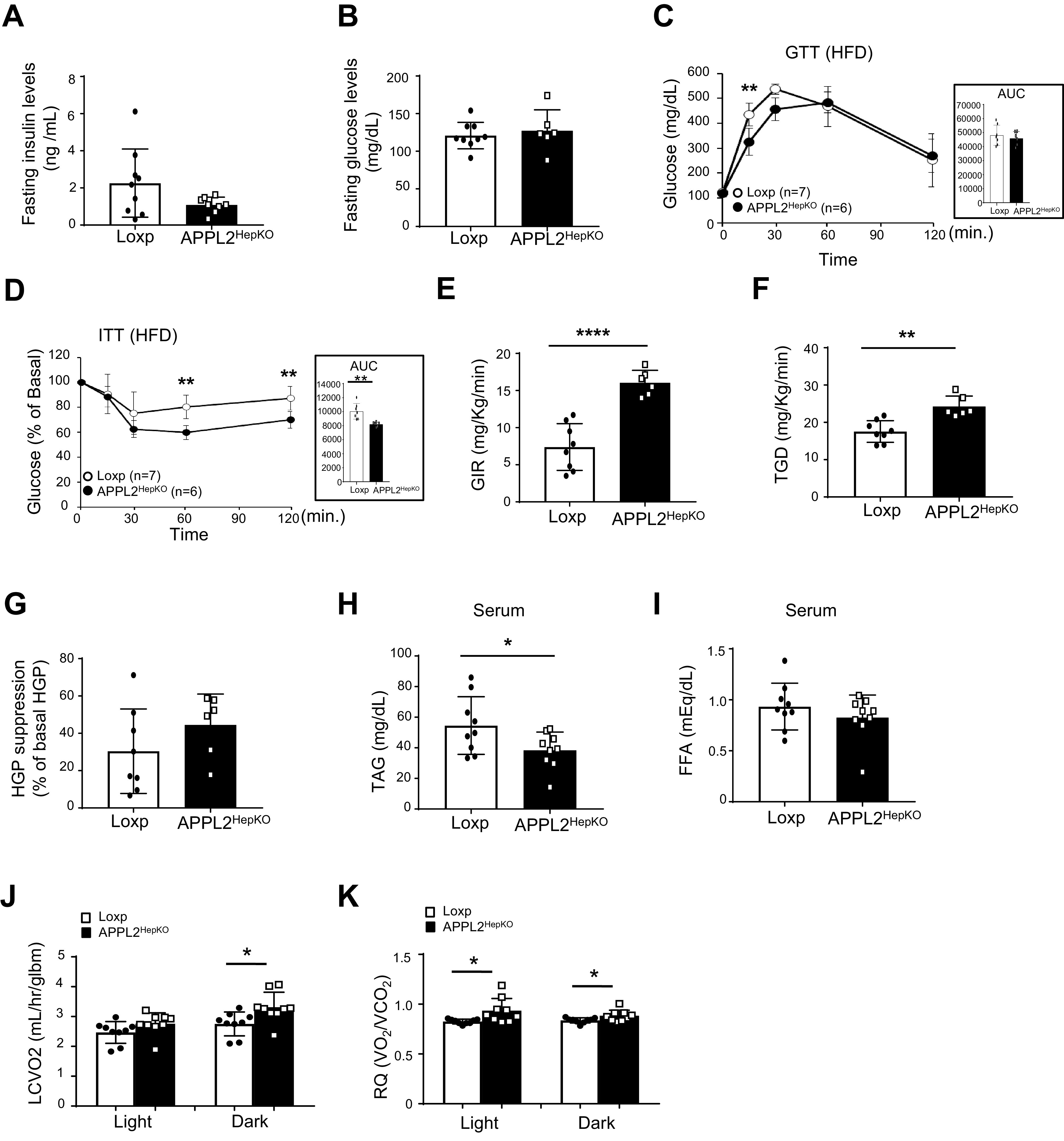
Deletion of hepatic Appl2 gene protects the mice from HFD-induced insulin resistance. A: Fasting blood insulin levels of APPL2HepKO and Loxp mice under 14 weeks HFD. B: Fasting blood glucose levels. C: Glucose tolerance test (GTT) on male mice. D: Insulin tolerance test (ITT) on the male mice. E–G: Hyperinsulinemic-euglycemic clamp study. Glucose infusion rates (GIR) (E), total glucose disposal (TGD) (F), and suppression of HGP (G) during hyperinsulinemic-euglycemic clamp. Serum levels of TAG (H) and FFA (I). O2 consumption (J) and respiratory quotient (K) with calorimetric analysis. All results are presented as mean ± SD, and P values were calculated with use of two-way ANOVA or Student t test (*P < 0.05, **P < 0.01, and ****P < 0.0001). AUC, area under the curve; glbm, grams lean body mass; LCVO2, adjusted oxygen consumption now accounting for lean mass; RQ, respiratory quotient.
By hyperinsulinemic-euglycemic clamp study, we found that glucose infusion rates and total glucose disposal rates were significantly enhanced in HFD-fed APPL2HepKO mice in comparison with the controls (Fig. 1E and 1F). Insulin-induced suppression of hepatic glucose production (HGP) was increased in the KO mice (Fig. 1G). Consistent with improved systemic insulin sensitivity, APPL2 deficiency resulted in reduction of serum TAG and FFA levels (Fig. 1H and I). These findings indicate that deletion of the Appl2 gene in hepatocytes enhances insulin sensitivity in insulin target tissues such as muscle, fat, and the liver.
While the resting metabolic rate and core body temperature of APPL2HepKO mice were comparable with those of the Loxp mice (Supplementary Fig. 2G and H), APPL2HepKO mice showed a significant increase in respiratory quotient (Fig. 1K), a trend of increased activity (Supplementary Fig. 2F), and enhanced UCP-1 expression in brown adipose tissue (Supplementary Fig. 2P). O2 consumption levels of the KO mice were elevated during the dark cycle (Fig. 1J). These data suggest that enhanced activity- and diet-induced thermogenesis caused the increase in energy expenditure in APPL2HepKO mice.
Hepatic APPL2 Deficiency Reduces HGP and Promotes Lipid Accumulation
Consistent with enhanced insulin sensitivity in the liver (Fig. 1G), HFD-fed APPL2HepKO mice showed a significant decrease in HGP in the pyruvate tolerance test (Fig. 2A). Blood glucagon levels and hepatic glucagon signaling were comparable between the KO and Loxp mice (Supplementary Fig. 2J and K). These data suggest that APPL2 deficiency causes a reduction in gluconeogenesis and this effect is independent of systemic glucagon levels and hepatic glucagon signaling. In line with this, the KO mice had significantly lower mRNA levels of glucose-6 phasphatase (G6PC) compared with the controls (Fig. 2B) but had no changes in gene expression related to glycolysis (Supplementary Fig. 2I). These findings reveal hepatic APPL2 to be a regulator of HGP in the liver.
Figure 2.
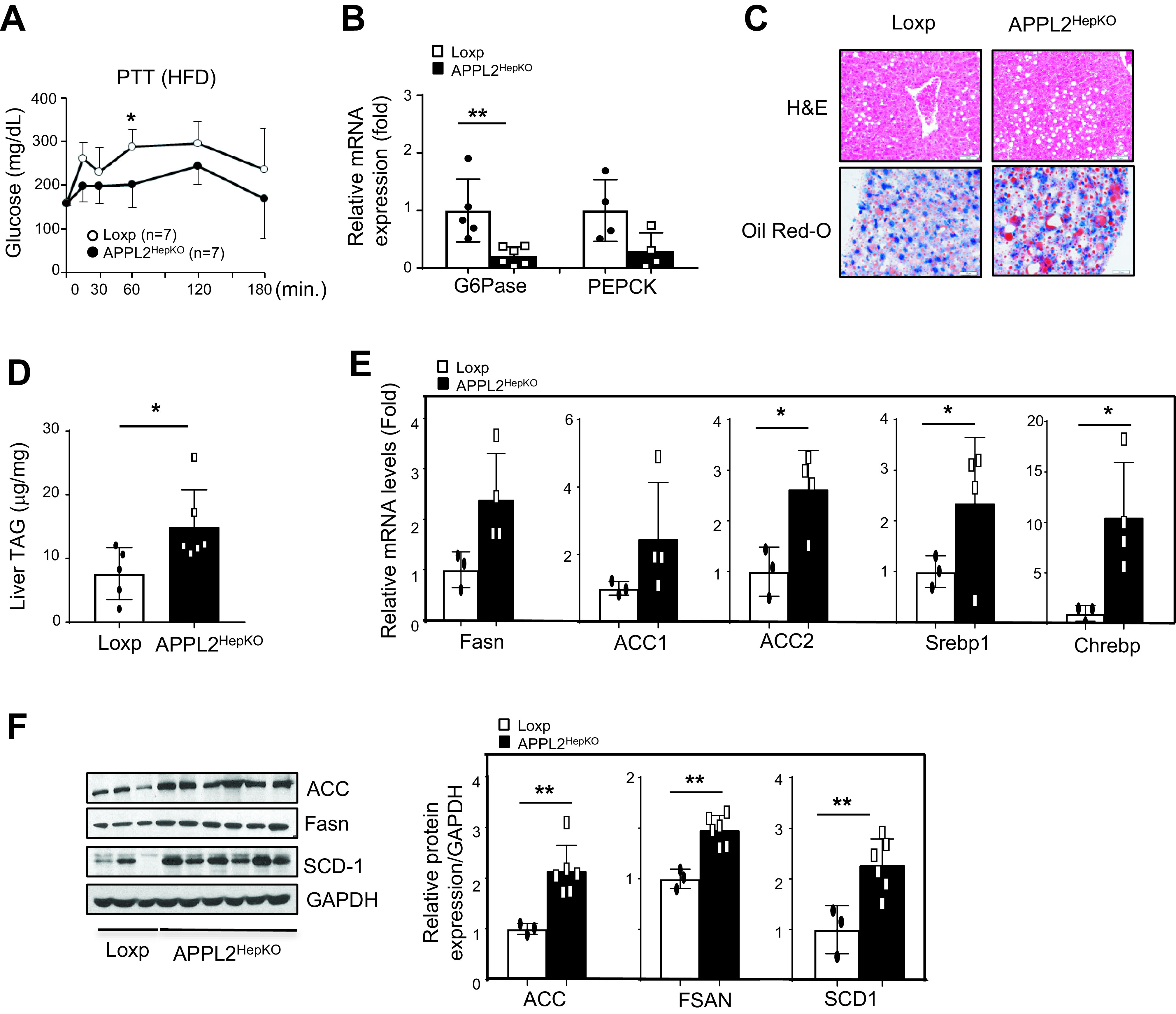
APPL2HepKO mice have reduced gluconeogenesis and enhanced lipogenesis under HFD feeding. A: Pyruvate tolerance test (PTT) of the KO and Loxp mice under 14 weeks HFD. B: mRNA expression of gluconeogenic genes in the mouse liver tissues. C: Hematoxylin-eosin (H&E) staining of the mouse liver tissues. Lipid accumulation in liver tissues was detected by Oil Red O staining. The scale bar indicates 50 μm. D: TAG contents in the mouse liver tissues. E: mRNA levels of lipogenic genes in liver tissues. F: Protein levels of lipogenic genes in the mouse liver tissues. Bar graphs represent the relative protein expression over the levels of GAPDH. All results are presented as mean ± SD, and P values were calculated with use of two-way ANOVA or Student t test (*P < 0.05 and **P < 0.01). hr, hour
We next investigated the effects of hepatic APPL2 deficiency on lipid metabolism. While hepatic KO of the Appl2 gene led to a reduction of serum TAG and FFA (Fig. 1H and I), elevated lipid accumulation (Fig. 2C) and TAG contents (Fig. 2D) were detected in the liver of the KO mice in comparison with their controls under HFD feeding. Interestingly, hepatic levels of diglyceride, which is implicated in promoting hepatic lipotoxicity (22) and insulin resistance (23), were comparable between two genotypes of mice (Supplementary Fig. 2O). Consistent with the increased lipid accumulation, genes involved in lipogenesis such as carbohydrate-responsive element–binding protein (CHREBP), sterol regulatory element–binding transcriptional factor 1 (SREBP1), and acetyl-CoA carboxylase 2 (ACC2) were significantly increased in the liver of the KO mice (Fig. 2E). The levels of proteins involved in lipogenesis such as ACC, FASN, and stearoyl-CoA desaturase (SCD-1) were also increased in APPL2-deficient liver (Fig. 2F). Of note, no significant difference was observed in the expression of other lipid metabolic genes between the KO and control mice, including lipid uptake (CD36 and lipoprotein lipase [LPL]) and lipid export (ABCA1, ABCA6, APOB, and APOE) as well as fatty acid oxidation and ketone body generation (carnitine palmitoyltransferase 1 [CPT1], acyl-CoA dehydrogenase medium chain [ACADM], medium-chain acyl-CoA dehydrogenase [MCAD], and hydroxymethylglutaryl-CoA synthase [HMGS2]) (Supplementary Fig. 2L–N). Together, these data indicate that deletion of the Appl2 gene in hepatocytes protected the mice from diet-induced hyperlipidemia despite these mice displaying enhanced lipogenesis and more lipid accumulation in the liver.
Hepatic APPL2 Deficiency Promotes Insulin Sensitivity and Adiponectin Signaling in Liver
To investigate the mechanisms underlying hepatic APPL2-regulated glucose and lipid metabolism, we investigated the effects of APPL2 deficiency on insulin sensitivity in the liver. Insulin-stimulated Akt phosphorylation at Thr308 and Ser473 was significantly enhanced in the liver, epididymal fat, and skeletal muscle of the KO mice (Fig. 3A and Supplementary Fig. 3A and B), concurrently with enhanced phosphorylation of GSK3β, a downstream target of Akt (Fig. 3A). These data indicate that APPL2 negatively regulates insulin signaling in mouse liver. Additionally, we investigated the role of APPL2 on adiponectin signaling by treating primary hepatocytes isolated from the KO mice or their controls with 50 μmol/L AdipoRon, an adiponectin mimetic (24). AdipoRon had a greater stimulatory effect on AMPK phosphorylation in APPL2 KO primary hepatocytes in comparison with the control cells (Fig. 3B). In contrast, overexpression of APPL2 blocked adiponectin-stimulated AMPK phosphorylation in primary hepatocytes (Fig. 3C).
Figure 3.
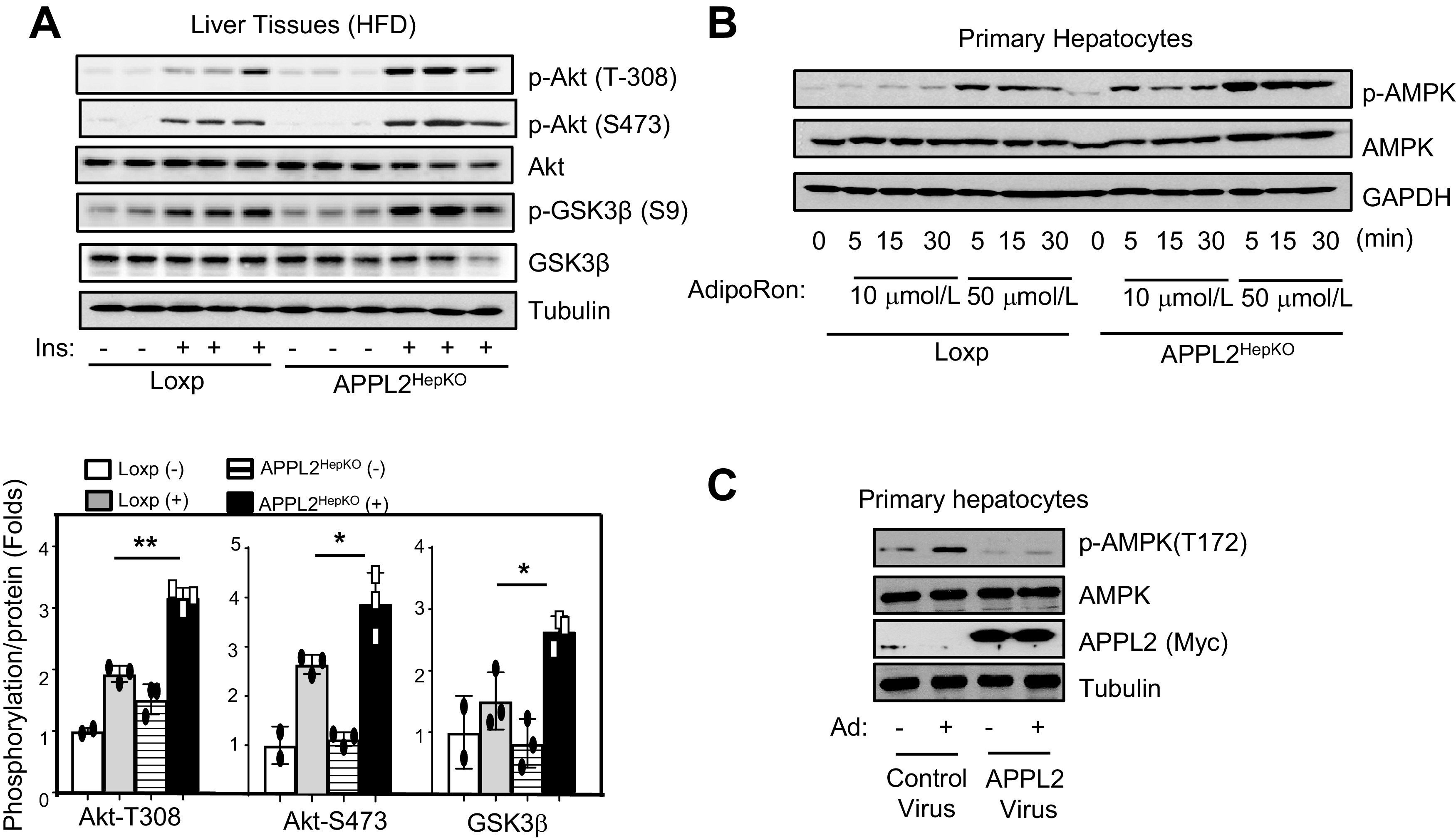
Hepatic APPL2 deficiency promotes insulin and adiponectin signaling. A: Insulin signaling in liver tissue of APPL2HepKO or Loxp mice. Bar graphs represent the ratios of insulin-stimulated phosphorylation (p-) to the levels of total proteins. B: Adiponectin signaling in the primary hepatocytes. Isolated primary hepatocytes were treated with 10 μmol/L or 50 μmol/L AdipoRon at indicated times. C: Adiponectin (Ad) signaling in primary hepatocytes overexpressing APPL2 protein. APPL2 was overexpressed in wild-type primary hepatocytes by infection of the cells with adenovirus encoding GFP or GFP plus APPL2 for 24 h. After 2 h serum starvation, full-length adiponectin (5 μg/mL) was used to treat the cells for 15 min. All results are presented as mean ± SD, and P values were calculated with use of Student t test (*P < 0.05 and **P < 0.01).
To investigate the regulation of APPL2 expression under overnutrient conditions, we detected APPL2 levels in the livers of mice fed HFD or chow diet. While hepatic APPL2 expression was similar between HFD- and chow-fed mice (Supplementary Fig. 6A), higher levels of APPL2 were detected on the plasma membrane of liver tissues from HFD-fed mice in comparison with those under chow diets (Supplementary Fig. 6B), suggesting that accumulation of APPL2 on the plasma membrane under HFD conditions might sequester and thereby prevent APPL1 from interacting with adiponectin receptors. Consistent with this, systemic injection of adiponectin in mice induced significantly higher levels of APPL1 binding to the adiponectin receptors in livers of the KO mice in comparison with the controls (Supplementary Fig. 6C). Together, these results reveal an inhibitory role of APPL2 in regulating adiponectin signaling and insulin sensitivity in hepatocytes.
APPL2HepKO Mice Are Resistant to HFD-Induced Inflammation in Liver Tissues
Because APPL2 deficiency promoted fatty liver (Fig. 2C), we examined whether the enhanced lipid accumulation in the liver is associated with increased inflammation. Surprisingly, levels of F4/80 staining, an indicator of macrophages, were significantly less in the liver of the KO mice in comparison with the controls (Fig. 4A), indicating that hepatic APPL2 deficiency prevents diet-induced macrophage infiltration in mouse liver. While there was no significant difference in the mRNA levels of several M2 macrophage markers between APPL2HepKO and control mice (Supplementary Fig. 4A), expression levels of M1 macrophage markers, such as F4/80, were significantly reduced in the liver tissues of the KO mice in comparison with their controls (Fig. 4B). Additionally, we found that this anti-inflammatory effect of APPL2 deficiency in liver seems to have limited impact on systemic inflammation, as the levels of inflammatory factors in sera were comparable between the Loxp and the KO mice (Supplementary Fig. 4B). Collectively, these data demonstrate that APPL2 deficiency in hepatocytes protects mice against diet-induced liver inflammation in spite of severe fatty liver.
Figure 4.
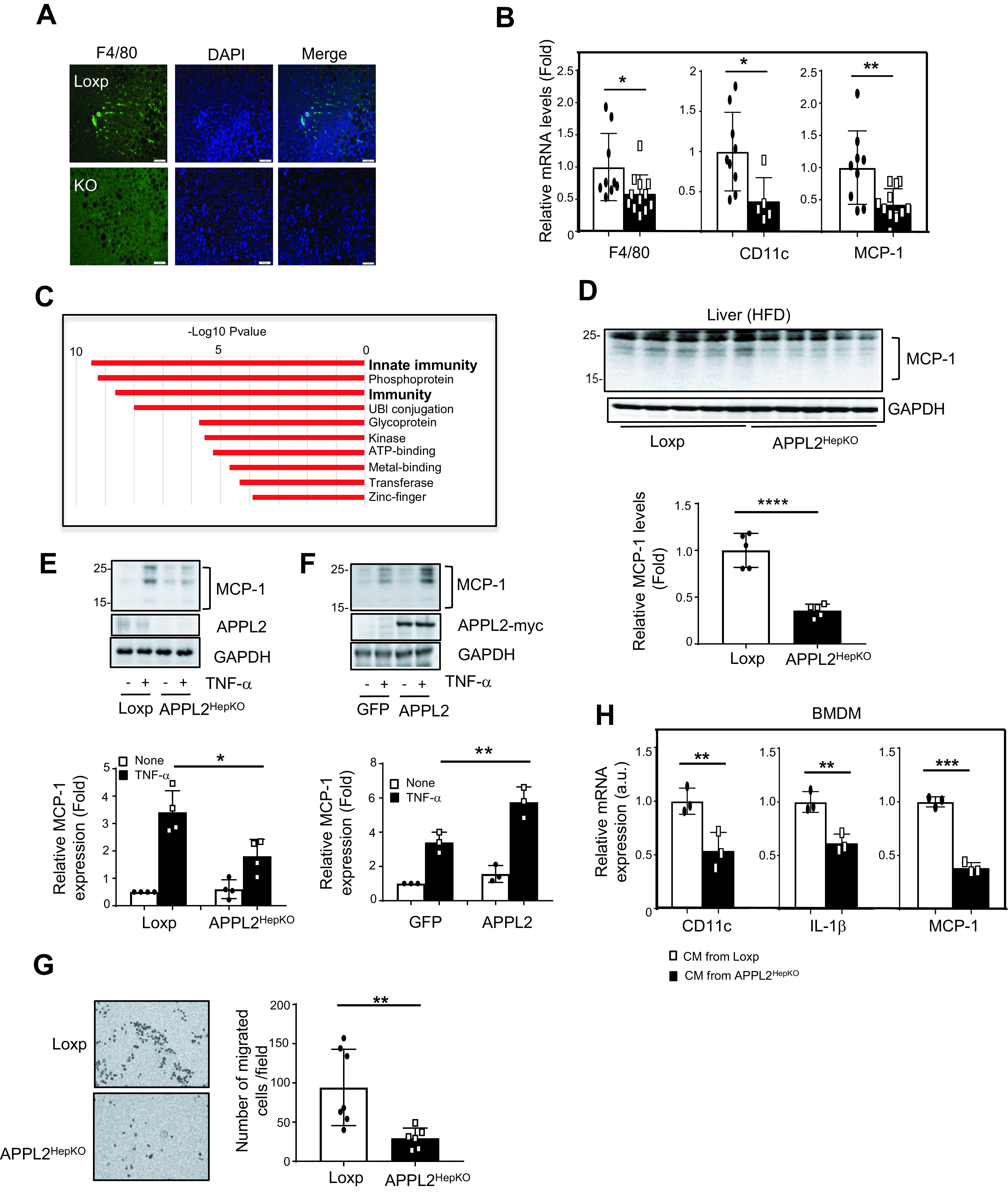
Hepatic APPL2 deficiency alleviates diet-induced inflammation in liver. A: Immunofluorescence of macrophage marker, F4/80 (green), and nucleus staining with DAPI (blue) in the liver of APPL2HepKO and Loxp mice under HFD. The scale bar indicates 100 μm. B: mRNA levels of M1 macrophage markers in the mouse livers. C: Gene functional analysis with significantly changed gene set from RNA-sequencing analysis. D: MCP-1 protein expression in liver tissues. E: TNF-α–stimulated MCP-1 expression in the mouse primary hepatocytes. TNF-α (10 ng/mL) was used to treat primary mouse hepatocytes for 24 h. F: TNF-α–stimulated MCP-1 expression in primary hepatocytes expressing GFP or GFP plus APPL2. G: Migration assay of BMDMs to the mouse primary hepatocytes. Picture of migrated macrophages on the Transwell membrane (left) and graph for the number of migrated cells per field. H: Proinflammatory gene expressions in the BMDMs incubated with conditioned media (CM) collected from TNF-α–treated hepatocytes. All results are presented as mean ± SD, and P values were calculated with Student t test (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). a.u., arbitrary units.
To define the mechanism by which APPL2 deficiency prevents diet-induced inflammation, we performed RNA-sequencing analysis to profile gene expression in the livers of HFD-fed KO and control mice. This study led to identification of 438 genes whose expression levels were significantly different (P < 0.05) between the KO and the control mice. Gene functional analysis revealed that genes involved in innate immunity, phosphoproteins, and immunity were the top three systems with significant changes in gene expression (Fig. 4C). Quantitative RT-PCR experiments showed that many of the genes involved in innate immunity were reduced in the liver of the KO mice in comparison with the controls (Supplementary Fig. 4C). Given that hepatic MCP-1, a macrophage-attracting factor, is known to stimulate innate immunity and inflammation in the liver (25,26), we examined MCP-1 levels in the liver tissues and found that both mRNA (Fig. 4B) and protein (Fig. 4D) levels of MCP-1 were significantly reduced in the KO mice in comparison with the controls. The reduction of MCP-1 expression is specific to the liver, since the levels of this cytokine were comparable in epididymal white adipose tissue, subcutaneous white adipose tissue, and skeletal muscle tissues between the KO and control mice (Supplementary Fig. 4D–F)
TNF-α is one of the major stimulators of the innate immune response in hepatocytes (27). High levels of TNF-α are reported in obese humans (28,29) and mice with diet-induced obesity (30) and are associated with chronic liver inflammation. While TNF-α–stimulated MCP-1 secretion has been reported in vascular endothelial cells (31) and adipocytes (32), it is unclear whether TNF-α can stimulate MCP-1 expression in hepatocytes. By treating mouse primary hepatocytes with TNF-α, we found that TNF-α stimulation greatly enhanced MCP-1 expression (Fig. 4E). This stimulatory effect was significantly reduced in APPL2-deficient hepatocytes (Fig. 4E). Conversely, overexpression of APPL2 in mouse primary hepatocytes remarkably promoted TNF-α–stimulated MCP-1 expression (Fig. 4F). These data demonstrate that TNF-α is a stimulator of MCP-1 expression in mouse hepatocytes and that APPL2 promotes the effect of TNF-α on MCP-1 expression.
Consistent with the role of MCP-1 as a potent factor attracting monocytes/macrophages (33,34), less macrophage infiltration was observed in liver tissues of HFD-fed KO mice in comparison with the controls (Fig. 4A). To determine the impact of APPL2-promoted MCP-1 expression in liver, we treated primary hepatocytes isolated from the KO and control mice with TNF-α to stimulate inflammatory cytokine expression. The cells were then incubated with bone marrow–derived macrophages (BMDMs), and the migration rates of BMDMs were detected. BMDMs cocultured with APPL2-deficient primary hepatocytes showed a significantly reduced migration rate compared with those cocultured with control hepatocytes (Fig. 4G). To further determine whether the APPL2-induced hepatic cytokines play a role in liver inflammation, we incubated BMDMs with conditioned medium collected from TNF-α–treated hepatocytes isolated from control or KO mice. As shown in Fig. 4H, the mRNA levels of CD11c, IL-1β, and MCP-1 were significantly lower in the BMDMs treated with conditioned media from APPL2-deficient hepatocytes than in those from controls. Taken together, these findings indicate that hepatic APPL2 is a key molecule promoting MCP-1 production in hepatocytes, leading to enhanced macrophage infiltration, proinflammatory gene expression, and consequent liver inflammation.
APPL2 Blocks the Anti-inflammatory Effect of Adiponectin in Hepatocytes
We next investigated the role of adiponectin in TNF-α−stimulated MCP-1 expression in hepatocytes. Treating mouse primary hepatocytes with AdipoRon, a synthetic small-molecule agonist for adiponectin receptors (24), completely blocked TNF-α−stimulated MCP-1 expression (Fig. 5A). While abrogation of APPL2 expression in hepatocytes ameliorated TNF-α–stimulated MCP-1 expression, AdipoRon treatment further diminished the role of TNF-α in MCP-1 expression (Fig. 5A). Conversely, overexpression of APPL2 in hepatocytes reduced the inhibitory effect of adiponectin on TNF-α–induced MCP-1 expression (Fig. 5B). These results suggest that TNF-α–stimulated MCP-1 expression in hepatocytes is a target of adiponectin action and that APPL2 promotes HFD-induced liver inflammation by blocking adiponectin signaling in hepatocytes.
Figure 5.
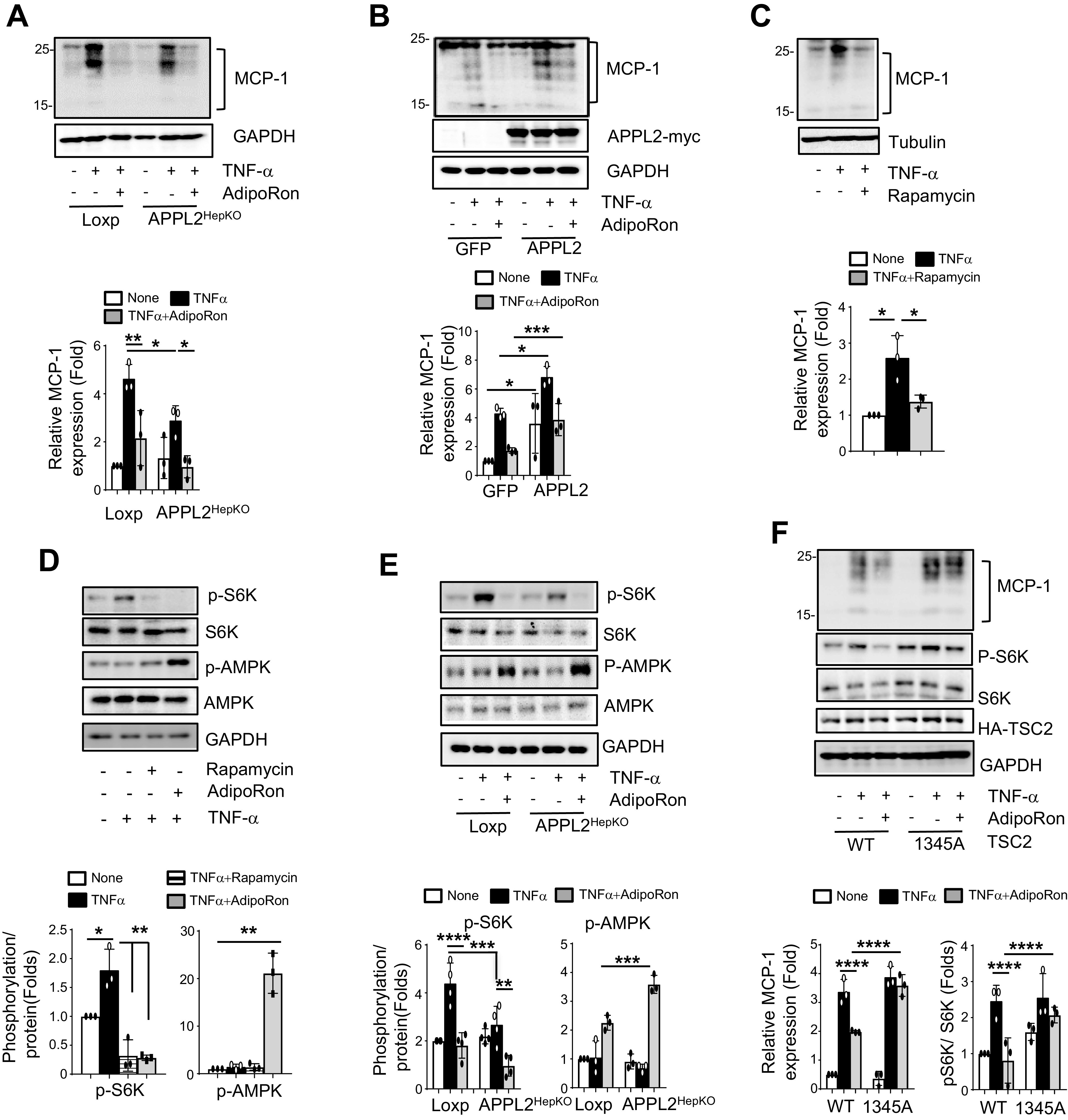
Adiponectin suppresses TNF-α–induced MCP-1 expression and liver inflammation. A: TNF-α–induced MCP-1 expression in primary mouse hepatocytes pretreated with 50 μmol/L AdipoRon or vehicle (DMSO) for 1 h followed by 10 ng/mL TNF-α treatment for 24 h. B: Overexpression of APPL2 diminishes the inhibitory effect of adiponectin on MCP-1 expression. Primary hepatocytes overexpressing GFP control or GFP plus APPL2 were pretreated with AdipoRon (50 μmol/L) for 1 h, followed by 20 ng/mL TNF-α treatment for 6 h. C: Rapamycin treatment is sufficient to block TNF-α–induced MCP-1 expression in hepatocytes. Primary hepatocytes were pretreated with rapamycin (20 nmol/L) for 1 h, followed by TNF-α stimulation for 24 h. D: AdipoRon or rapamycin inhibits TNF-α–induced mTORC1 activation. Wild-type primary hepatocytes were pretreated with 50 μmol/L AdipoRon, 20 nmol/L rapamycin, or DMSO (vehicle) for 1 h, followed by treatment without or with 20 ng/mL TNF-α for 1 h. E: Depletion of hepatic APPL2 reduces TNF-α–stimulated mTORC1 activation in hepatocytes. APPL2-deficient hepatocytes were pretreated with 50 μmol/L AdipoRon or DMSO for 1 h, followed by 20 ng/mL TNF-α for 1 h. F: Overexpression of a dominant-negative mutant of TSC2 blocks the inhibitory effect of adiponectin on TNF-α–stimulated MCP-1 expression. Plasmids containing wild-type or dominant-negative mutant (S1345A) of TSC2 were transfected into TSC2 KO cell lines. Twenty-four hours posttransfection, cells were pretreated with 50 μmol/L AdipoRon or DMSO vehicle for 1 h, followed by 100 ng/mL TNF-α treatment. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, p-, phosphorylated; WT, wild type.
To examine the signaling pathway(s) mediating TNF-α–induced MCP-1 expression, we treated primary hepatocytes with various inhibitors, including IKKβ inhibitor for NF-κB, rapamycin for mTORC1, LY294002 for phosphatidylinositol 3-kinase, SB203580 for p38 MAPK, and PD98059 for MAPKK. While treatment with inhibitors for IKKβ, p38 MAPK, phosphatidylinositol 3-kinase, and MAPKK had marginal effects on TNF-α–stimulated MCP-1 expression (Supplementary Fig. 5), inhibition of mTORC1 signaling by rapamycin abrogated TNF-α–induced MCP-1 expression (Fig. 5C), which is comparable to the effect of AdipoRon treatment (Fig. 5A). Consistently, TNF-α treatment stimulated phosphorylation of S6K1, a downstream signal molecule of mTORC1, and rapamycin treatment was sufficient to block the effect of TNF-α–induced S6K1 activation to the same extent as for AdipoRon (Fig. 5D). Collectively, these data indicate that mTORC1-S6K1 signaling mediates TNF-α–stimulated MCP-1 expression in hepatocytes.
We next investigate the mechanism by which adiponectin blocks TNF-α–stimulated mTORC1-S6K1 signaling in hepatocytes. It has been reported that mTORC1 signaling can be inhibited by AMPK via phosphorylation and activation of TSC2, a negative regulator of mTORC1, in HEK293 (human embryonic kidney) cells (35). Since the expression levels of APPL2 are negatively associated with adiponectin-stimulated AMPK phosphorylation in primary hepatocytes (Figs. 3B and C and 5E), we asked whether AMPK-induced phosphorylation of TSC2 at Ser1345 is the major converging point by which adiponectin and mTORC1 pathways regulate TNF-α–induced MCP-1 expression in hepatocytes. As showed in Fig. 5F, AdipoRon treatment failed to abrogate TNF-α−stimulated MCP-1 expression in cells overexpressing a dominant-negative mutant (S1345A) of TSC2 in comparison with cells overexpressing wild-type TSC2. These data suggest that adiponectin suppresses TNF-α–stimulated mTORC1-S6K1 signaling by promoting AMPK-regulated TSC2 activity.
Deletion of ADIPOQ Gene in APPL2HepKO Mice Largely Diminishes the Anti–Insulin Resistance and Anti-Inflammation Phenotypes
To investigate the contribution of adiponectin to the antiinsulin resistance and anti-inflammation phenotypes observed in APPL2HepKO mice, we deleted ADIPOQ gene in APPL2-deficient mice. Under HFD conditions, hepatic APPL2 and adiponectin double KO (APPL2HepKO/AdKO) mice showed body weight gain and fasting glucose levels similar to those of APPL2HepKO mice (data not shown). While there was no difference in the glucose tolerance between the double KO mice and APPL2HepKO mice (Fig. 6A), the double KO mice showed impaired insulin tolerance compared with APPL2-deficient mice (Fig. 6B). Strikingly, deletion of the adiponectin gene in APPL2HepKO mice reversed the HFD-induced proinflammatory gene expression (MCP-1 and TNF-α) and M1 macrophage infiltration markers (F4/80 and CD11c) in comparison with APPL2-deficient mice (Fig. 6C). Consistent with the stimulatory role of adiponectin in lipogenesis (36), APPL2 deficiency–induced lipid accumulation was reduced in the liver of the double KO mice (Supplementary Fig. 7). These data demonstrate that the anti-inflammatory and anti–insulin resistance phenotypes observed in the APPL2-deficient mice are largely due to enhanced adiponectin action in hepatocytes.
Figure 6.
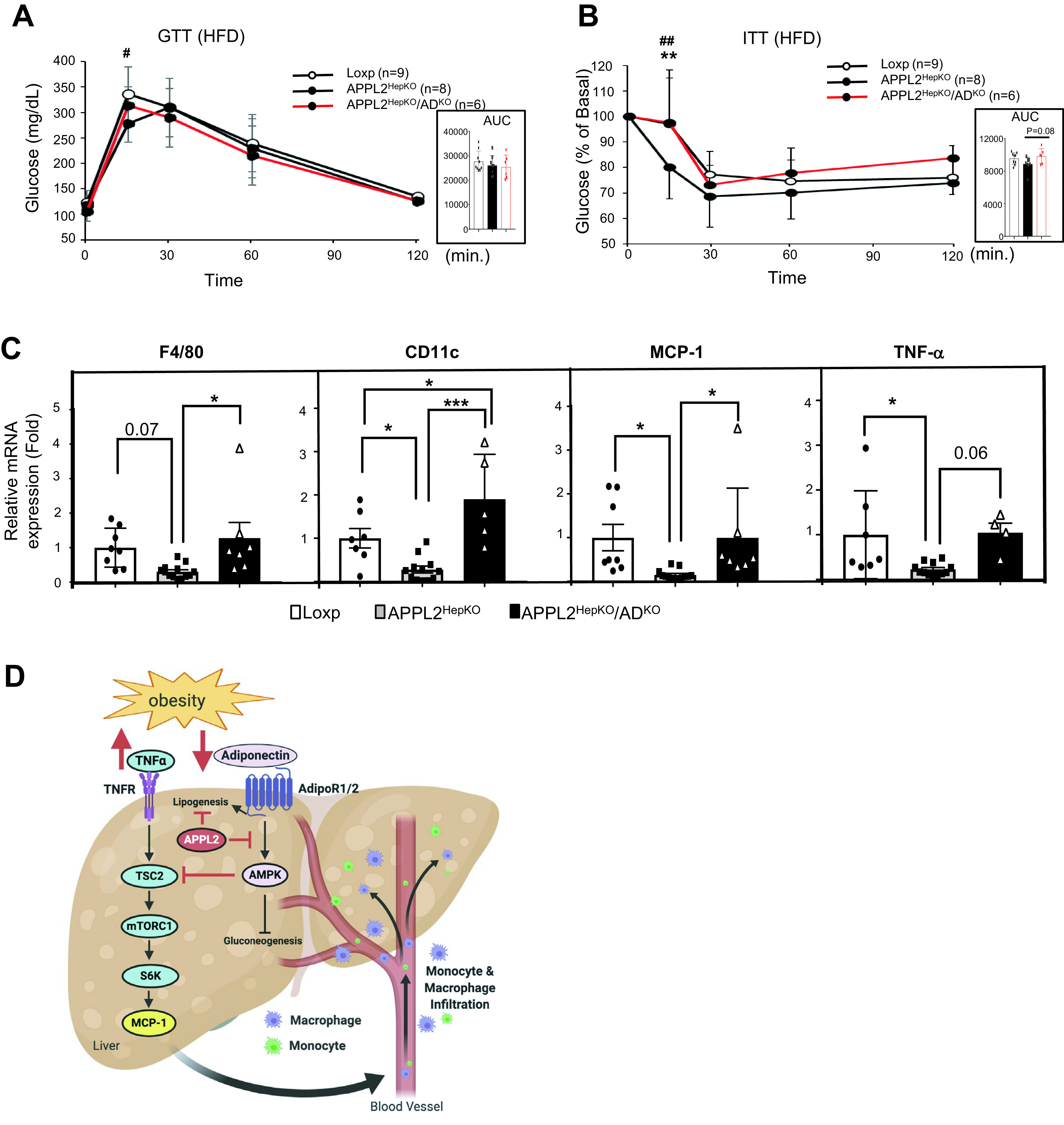
Deletion of the adiponectin gene in APPL2HepKO mice reverses the anti–insulin resistance and anti-inflammation phenotypes. A: Glucose tolerance test (GTT) with APPL2HepKO and APPL2HepKO/AdKO mice under HFD (*P < 0.01, Loxp vs. APPL2HepKO). B: Insulin tolerance test (ITT) (**P < 0.001, Loxp vs. APPL2HepKO; ##P < 0.001, APPL2HepKO vs. APPL2HepKO/AdKO). C: mRNA expression of M1 macrophage–related genes in the mouse livers. D: Schematic diagram to demonstrate the mechanism by which hepatic adiponectin signaling prevents diet-induced hepatic inflammation and insulin resistance in liver tissue. Figure created with BioRender (biorender.com). All results are presented as mean ± SD, and P values were calculated with use of two-way ANOVA, for A and B, or one-way ANOVA, for C (*P < 0.05 and ***P < 0.001). AUC, area under the curve.
Discussion
The liver is a major target organ of adiponectin action (3,7,9,37). While the insulin-sensitizing role of adiponectin has been extensively investigated in the liver (4,12,37,38), the mechanisms underlying the anti-inflammatory action of adiponectin are still largely unexplored. In the current study, we show that hepatocyte-specific disruption of APPL2 enhanced adiponectin signaling and protected mice from diet-induced insulin resistance. Despite more lipid accumulation in the liver, APPL2HepKO mice are resistant to diet-induced macrophage infiltration and liver inflammation. Mechanistically, obesity-induced APPL2 membrane translocation blocks adiponectin–AdipoR1/2–APPL1–AMPK signaling in hepatocytes. This inhibition mitigates the inhibitory effect of adiponectin on TNF-α–triggered MCP-1 expression in liver via the mTORC1-S6K1 pathway and induces diet-induced liver inflammation and insulin resistance (Fig. 6D).
The liver is a major organ for developing overnutrition-induced chronic inflammation (26). As heterogeneous tissue, various cell types including hepatocytes participate in the progression of chronic inflammation in liver (39). In this study, we reveal that deletion of the Appl2 gene in hepatocytes is sufficient to protect mice from diet-induced macrophage infiltration and suppress innate immunity–related gene expression in the liver. We also demonstrate a paracrine role of hepatic APPL2 in regulation of the liver microenvironment by showing hepatic APPL2 deficiency to be sufficient to reduce microphage migration and APPL2-controlled hepatocytic secretome contributing to the inflammatory status of monocytes/macrophages. Collectively, our studies reveal that hepatic APPL2 is a key molecule regulating the gene expression in innate immunity and controlling macrophage infiltration and liver inflammation.
MCP-1, also known as CCL2, is a potent monocytes/macrophages-attracting factor for recruiting immune cells to peripheral tissues in response to inflammation (25,40). While elevated MCP-1 in the liver has been shown to contribute to the development of hepatic steatosis and insulin resistance in HFD-fed mice (26,33,40,41) as well as liver injury in humans (42), the mechanism underlying diet-induced hepatic MCP-1 expression is unknown. Among the suppressed innate immunity–related genes in the liver of APPL2HepKO mice, MCP-1 is a downstream target of APPL2 in hepatocytes. This is evidenced by the fact that APPL2HepKO mice showed reduced MCP-1 expression in both liver tissues and primary hepatocytes and that overexpression of APPL2 in primary hepatocytes is sufficient to promote TNF-α–stimulated MCP-1 expression. Since enhanced serum levels of TNF-α are observed in obese humans (29,43,44) and rodents (45–47), our finding uncovers a link between TNF-α and hepatic MCP-1 expression and demonstrates a role of APPL2 in promoting this regulation.
Hepatic TAG accumulation is associated with nonalcoholic steatohepatitis development (48). However, it remains unclear whether TAG accumulation is sufficient to promote liver injury and inflammation. Cases of hepatic TAG accumulation in human subjects are not always associated with liver injury, suggesting that factors other than TAGs are required for triggering of hepatic inflammation (49). Consistently, FFAs, diglycerides, and endotoxins have been reviewed as lipotoxic molecules, as they can induce liver injury in the presence of TAG accumulation (50). In this study, we showed that deletion of Appl2 gene in hepatocytes was sufficient to induce more fat accumulation, less inflammation, and improved insulin sensitivity in comparison with the control mice under HFD conditions. Interestingly, the levels of diglyceride in liver tissues of APPL2HepKO mice were comparable with those of control mice. These results suggested that TAG accumulation, without elevation of lipotoxic intermediates, is not sufficient for inducing inflammation. In support of this view, APPL2HepKO mice were less responsive to TNF-α–stimulated MCP-1 expression and had reduced macrophage infiltration in the liver. The protective effects of APPL2 deficiency on diet-induced inflammation are largely dependent on hepatic adiponectin signaling, since adiponectin treatment blocked TNF-α–induced MCP-1 expression and overexpression of APPL2 was sufficient to impair the inhibitory role of adiponectin in hepatic inflammation. Deletion of the adiponectin gene in APPL2HepKO mice reversed the diet-induced anti-inflammatory and severe fatty liver phenotypes in APPL2HepKO mice. Together, our study indicates that TAG accumulation in liver is uncoupled with inflammation as long as hepatic adiponectin signaling is intact.
It has been well-known that adiponectin possesses anti-inflammatory and antifibrogenic properties in the liver (7,51). While several signaling pathways have been suggested to mediate the anti-inflammatory effect of adiponectin in liver, such as PPARα (52) and IL-10 (53), the molecular targets and the underlying mechanism of adiponectin’s anti-inflammatory action in liver remain largely unclear. By using APPL2HepKO mice as a model system, we demonstrate that enhanced adiponectin signaling by deletion of hepatic Appl2 gene is the major mechanism by which APPL2HepKO mice are protected from diet-induced macrophage infiltration and liver inflammation. First, adiponectin-stimulated AMPK phosphorylation was enhanced and blocked in APPL2-deficient and APPL2-overexpressed hepatocytes, respectively. Additionally, the anti-inflammatory phenotype of APPL2HepKO mice is largely dependent on adiponectin expression, since the deletion of ADIPOQ gene in the KO mice reverses the expression of F4/80, CD11c, MCP-1, and TNF-α in the liver. Furthermore, treating primary hepatocytes with adiponectin was sufficient to suppress TNF-α–stimulated MCP-1 expression, and overexpression of APPL2 reduced the inhibitory effect of adiponectin in primary hepatocytes. These results demonstrate that MCP-1 is a direct target of adiponectin action in hepatocytes and that APPL2 promotes MCP-1 expression and inflammation in liver by blocking adiponectin signaling.
Several signal pathways have been proposed to mediate TNF-α action, including NF-κB (32,54), p38 MAPK (55), AKT/PKB (31), and AP-1 (54) pathways in adipocytes and vascular endothelial cells. Our study suggests that these pathways play a limited role in mediating TNF-α–stimulated MCP-1 expression in primary hepatocytes. Instead, we found that TNF-α–induced and adiponectin-suppressed MCP-1 expression is largely dependent on mTORC1-S6K1 signaling. This is demonstrated by the fact that treating primary hepatocytes with rapamycin, an inhibitor of mTORC1 (56), was sufficient to block TNF-α–stimulated MCP-1 expression. In line with this, levels of TNF-α–stimulated S6K1 phosphorylation were reduced in primary hepatocytes isolated from APPL2HepKO mice. While mTORC1 signaling is required for TNF-α–stimulated MCP-1 expression in HeLa cells (57), our study provides the first evidence that the mTORC1-S6K1 pathway in hepatocytes mediates TNF-α signaling to promote MCP-1 expression.
Given that adiponectin-stimulated AMPK phosphorylation, which has been reported to inhibit mTORC1-S6K signaling in C2C12 myotubes (58), was enhanced in APPL2-deficient hepatocytes, we investigate the role of adiponectin-induced APMK activity in mediating the inhibitory role of adiponectin in TNF-α–stimulated mTORC1–MCP-1 signaling. By overexpression of TSC2 (S1345A), a dominant-negative mutant that blocks AMPK-induced TSC2 phosphorylation and promotes mTORC1 signaling (35), we found that the inhibitory effect of adiponectin on TNF-α–stimulated MCP-1 expression was diminished. These data suggest that TSC2, a regulatory molecule upstream of mTORC1, is at the crossroads of adiponectin-AMPK signaling and TNFα–mTORC1–MCP-1 signaling in hepatocytes. Consistent with this view, overexpression of APPL2, which is sufficient to inhibit adiponectin-stimulated AMPK phosphorylation in hepatocytes, promotes TNFα-induced MCP-1 expression. In conclusion, our data pinpoint the mechanism by which hepatic adiponectin signaling blocks the stimulatory effect of TNF-α on MCP-1 expression by targeting on TSC2, a site upstream of mTORC1, in hepatocytes.
The role of adiponectin in regulating hepatic lipid metabolism remains controversial. Adiponectin-deficient mice were resistant to HFD-induced fatty liver (59), although the KO mice exhibited HFD-induced insulin resistance (60–62). Consistently, adiponectin transgenic mice exhibited improved insulin sensitivity but an increase in adiposity (38). Overexpression of adiponectin in ob/ob mice caused a significant expansion of the fat mass but improved systemic insulin sensitivity (63). These studies suggest a stimulatory role of adiponectin in both insulin sensitivity and lipogenesis. On the other hand, several studies reported an inhibitory role of adiponectin in hepatic lipid accumulation. Adiponectin-activated AMPK inhibits acetyl-CoA carboxylase (ACC), a rate-limiting enzyme in de novo lipogenesis (64). Overexpression of AdipoR2 in liver enhanced fatty acid oxidation and reduced TAG levels via PPAR-α pathways (52). By using APPL2HepKO mice as a model system, we demonstrate that enhanced adiponectin signaling in hepatocytes promotes insulin sensitivity, leading to an increase in hepatic de novo lipogenesis and lipid accumulation. Consistent with this view, we observed that the lipid accumulation in the liver of APPL2HepKO mice was alleviated in the liver tissue from APPL2HepKO/AdKO mice. It is interesting to note that APPL2HepKO mice were resistant to diet-induced hyperlipidemia, despite the fact that they have more severe fatty liver compared with the controls. This could be due to enhanced insulin sensitivity in other peripheral tissues such as skeletal muscle tissues. With results taken together, we demonstrate that hepatic adiponectin signaling controls systemic lipid metabolism by promoting lipid biosynthesis in the liver and normalizing lipid levels in circulation.
Adiponectin can improve liver glucose metabolism by lowering HGP and suppressing gluconeogenesis gene expression (4,37,65). Consistent with the inhibitory role of APPL2 in adiponectin signaling and glucose uptake in skeletal muscles (13,17), we show that deletion of Appl2 gene in hepatocytes alleviated HFD-induced HGP and improved insulin sensitivity. The mechanism underlying the beneficial effects of APPL2 deficiency on insulin sensitivity could be due to increased adiponectin signaling observed in the primary hepatocytes of APPL2HepKO mice. In agreement with this, the anti–insulin resistance phenotype of APPL2HepKO mice is largely dependent on adiponectin expression, since deletion of ADIPOQ gene in APPL2-deficient mice reversed the improved insulin sensitivity. It is worth noting that human genetic studies reveal associations of single nucleotide polymorphisms in Appl2 gene with higher cardiovascular disease risk (18), overweight and obesity (19), NAFLD (20), and type 2 diabetes (66). Our study suggests that targeting APPL2, an inhibitory protein in adiponectin signaling (13), is an attractive approach to enhancing insulin sensitivity and improving glucose metabolism.
In summary, we demonstrate an in vivo role of hepatic APPL2 in promoting diet-induced inflammation and insulin resistance. By identifying innate immune factors, such as MCP-1, as targets of adiponectin action in hepatocytes, we reveal a mechanism by which adiponectin and adiponectin signaling prevent diet-induced chronic inflammation in the liver.
Article Information
Acknowledgments. The authors are thankful for technical support by University of Texas Health Science Center at San Antonio (UTHSCSA) Biobanking and Genome Analysis Core.
Funding. This work was supported in part by American Diabetes Association Basic Science Award (grant 7-13-BS-043 to L.Q.D.), National Institutes of Health R01 grants (DK102965 to L.Q.D. and DK114479 to F.L.), DOM Clinical and Innovative Therapeutic Award Pilot Program (grant G1100-22100 to J.R.), a Biomedical Research grant from the Semp Russ Foundation of the San Antonio Area Foundation (grant 161926 to J.R.), and T32 Biology of Aging and Age-Related Diseases Training Grant (T32-AG021890-17 to J.T.H.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. L.Q.D. supervised the project, contributed to discussion, and reviewed and edited the manuscript. F.L. contributed to discussion and reviewed and edited the manuscript. J.R. performed experiments, conducted data analysis, and wrote the manuscript. J.T.H. performed experiments and edited the manuscript. Z.L., F.D., H.X., X.X., Y.Z., C.C., X.G., and J.L.Z. performed experiments. S.L., R.J.L., M.A.A.-G., A.K., and R.A.D. contributed to discussion. L.Q.D. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 78th Scientific Sessions of the American Diabetes Association, Orlando, FL, 22–26 June 2018.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.14195288.
References
- 1. Duan Y, Zeng L, Zheng C, et al. Inflammatory links between high fat diets and diseases. Front Immunol 2018;9:2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu Y, Wu T, Wu J, et al. Chronic inflammation exacerbates glucose metabolism disorders in C57BL/6J mice fed with high-fat diet. J Endocrinol 2013;219:195–204 [DOI] [PubMed] [Google Scholar]
- 3. Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol 2016;8:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 2001;7:947–953 [DOI] [PubMed] [Google Scholar]
- 5. Fantuzzi G. Adiponectin in inflammatory and immune-mediated diseases. Cytokine 2013;64:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Masaki T, Chiba S, Tatsukawa H, et al. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology 2004;40:177–184 [DOI] [PubMed] [Google Scholar]
- 7. Buechler C, Wanninger J, Neumeier M. Adiponectin, a key adipokine in obesity related liver diseases. World J Gastroenterol 2011;17:2801–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol 2016;8:93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003;423:762–769 [DOI] [PubMed] [Google Scholar]
- 10. Adachi Y, Takeuchi T, Sonobe H, Ohtsuki Y. An adiponectin receptor, T-cadherin, was selectively expressed in intratumoral capillary endothelial cells in hepatocellular carcinoma: possible cross talk between T-cadherin and FGF-2 pathways. Virchows Arch 2006;448:311–318 [DOI] [PubMed] [Google Scholar]
- 11. Mao X, Kikani CK, Riojas RA, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 2006;8:516–523 [DOI] [PubMed] [Google Scholar]
- 12. Ryu J, Galan AK, Xin X, et al. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep 2014;7:1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang C, Xin X, Xiang R, et al. Yin-Yang regulation of adiponectin signaling by APPL isoforms in muscle cells. J Biol Chem 2009;284:31608–31615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miaczynska M, Christoforidis S, Giner A, et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 2004;116:445–456 [DOI] [PubMed] [Google Scholar]
- 15. Varsano T, Dong MQ, Niesman I, et al. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol 2006;26:8942–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nechamen CA, Thomas RM, Dias JA. APPL1, APPL2, Akt2 and FOXO1a interact with FSHR in a potential signaling complex. Mol Cell Endocrinol 2007;260-262:93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng KK, Zhu W, Chen B, et al. The adaptor protein APPL2 inhibits insulin-stimulated glucose uptake by interacting with TBC1D1 in skeletal muscle. Diabetes 2014;63:3748–3758 [DOI] [PubMed] [Google Scholar]
- 18. Ma XW, Ding S, Ma XD, Gu N, Guo XH. Genetic variability in adapter proteins with APPL1/2 is associated with the risk of coronary artery disease in type 2 diabetes mellitus in Chinese Han population. Chin Med J (Engl) 2011;124:3618–3621 [PubMed] [Google Scholar]
- 19. Jiang S, Fang Q, Yu W, et al. Genetic variations in APPL2 are associated with overweight and obesity in a Chinese population with normal glucose tolerance. BMC Med Genet 2012;13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barbieri M, Esposito A, Angellotti E, Rizzo MR, Marfella R, Paolisso G. Association of genetic variation in adaptor protein APPL1/APPL2 loci with non-alcoholic fatty liver disease. PLoS One 2013;8:e71391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H, Bai J, Dong F, et al. Hepatic DsbA-L protects mice from diet-induced hepatosteatosis and insulin resistance. FASEB J 2017;31:2314–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol 2008;19:295–300 [DOI] [PubMed] [Google Scholar]
- 23. Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med 2010;16:400–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okada-Iwabu M, Yamauchi T, Iwabu M, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013;503:493–499 [DOI] [PubMed] [Google Scholar]
- 25. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 2011;54:2185–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kitade H, Chen G, Ni Y, Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients 2017;9:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 2016;13:301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem 2008;114:183–194 [DOI] [PubMed] [Google Scholar]
- 29. Alzamil H. Elevated Serum TNF-α is related to obesity in type 2 diabetes mellitus and is associated with glycemic control and insulin resistance. J Obes 2020;2020:5076858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hofmann C, Lorenz K, Braithwaite SS, et al. Altered gene expression for tumor necrosis factor-alpha and its receptors during drug and dietary modulation of insulin resistance. Endocrinology 1994;134:264–270 [DOI] [PubMed] [Google Scholar]
- 31. Murao K, Ohyama T, Imachi H, et al. TNF-alpha stimulation of MCP-1 expression is mediated by the Akt/PKB signal transduction pathway in vascular endothelial cells. Biochem Biophys Res Commun 2000;276:791–796 [DOI] [PubMed] [Google Scholar]
- 32. Zhu J, Yong W, Wu X, et al. Anti-inflammatory effect of resveratrol on TNF-alpha-induced MCP-1 expression in adipocytes. Biochem Biophys Res Commun 2008;369:471–477 [DOI] [PubMed] [Google Scholar]
- 33. Obstfeld AE, Sugaru E, Thearle M, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010;59:916–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009;29:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577–590 [DOI] [PubMed] [Google Scholar]
- 36. Liu J, Guo M, Zhang D, et al. Adiponectin is critical in determining susceptibility to depressive behaviors and has antidepressant-like activity. Proc Natl Acad Sci U S A 2012;109:12248–12253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Combs TP, Marliss EB. Adiponectin signaling in the liver. Rev Endocr Metab Disord 2014;15:137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Combs TP, Pajvani UB, Berg AH, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 2004;145:367–383 [DOI] [PubMed] [Google Scholar]
- 39. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun 2020;4:478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006;116:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 2006;116:115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Czaja MJ, Geerts A, Xu J, Schmiedeberg P, Ju Y. Monocyte chemoattractant protein 1 (MCP-1) expression occurs in toxic rat liver injury and human liver disease. J Leukoc Biol 1994;55:120–126 [DOI] [PubMed] [Google Scholar]
- 43. Zahorska-Markiewicz B, Janowska J, Olszanecka-Glinianowicz M, Zurakowski A. Serum concentrations of TNF-alpha and soluble TNF-alpha receptors in obesity. Int J Obes Relat Metab Disord 2000;24:1392–1395 [DOI] [PubMed] [Google Scholar]
- 44. Goyal R, Faizy AF, Siddiqui SS, Singhai M. Evaluation of TNF-α and IL-6 levels in obese and non-obese diabetics: pre- and postinsulin effects. N Am J Med Sci 2012;4:180–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91 [DOI] [PubMed] [Google Scholar]
- 46. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997;389:610–614 [DOI] [PubMed] [Google Scholar]
- 47. Ventre J, Doebber T, Wu M, et al. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes 1997;46:1526–1531 [DOI] [PubMed] [Google Scholar]
- 48. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 1998;114:842–845 [DOI] [PubMed] [Google Scholar]
- 50. Semova I, Biddinger SB. Triglycerides in nonalcoholic fatty liver disease: guilty until proven i. Trends Pharmacol Sci 2021;42:183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park PH, Sanz-Garcia C, Nagy LE. Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr Pathobiol Rep 2015;3:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 2007;13:332–339 [DOI] [PubMed] [Google Scholar]
- 53. Wolf AM, Wolf D, Avila MA, et al. Up-regulation of the anti-inflammatory adipokine adiponectin in acute liver failure in mice. J Hepatol 2006;44:537–543 [DOI] [PubMed] [Google Scholar]
- 54. Chen YM, Chiang WC, Lin SL, Wu KD, Tsai TJ, Hsieh BS. Dual regulation of tumor necrosis factor-alpha-induced CCL2/monocyte chemoattractant protein-1 expression in vascular smooth muscle cells by nuclear factor-kappaB and activator protein-1: modulation by type III phosphodiesterase inhibition. J Pharmacol Exp Ther 2004;309:978–986 [DOI] [PubMed] [Google Scholar]
- 55. Chen XL, Grey JY, Thomas S, et al. Sphingosine kinase-1 mediates TNF-alpha-induced MCP-1 gene expression in endothelial cells: upregulation by oscillatory flow. Am J Physiol Heart Circ Physiol 2004;287:H1452–H1458 [DOI] [PubMed] [Google Scholar]
- 56. Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991;253:905–909 [DOI] [PubMed] [Google Scholar]
- 57. Nakatsumi H, Matsumoto M, Nakayama KI. Noncanonical pathway for regulation of CCL2 expression by an mTORC1-FOXK1 axis promotes recruitment of tumor-associated macrophages. Cell Rep 2017;21:2471–2486 [DOI] [PubMed] [Google Scholar]
- 58. Wang C, Mao X, Wang L, et al. Adiponectin sensitizes insulin signaling by reducing p70 S6 kinase-mediated serine phosphorylation of IRS-1. J Biol Chem 2007;282:7991–7996 [DOI] [PubMed] [Google Scholar]
- 59. Liu Q, Yuan B, Lo KA, Patterson HC, Sun Y, Lodish HF. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc Natl Acad Sci U S A 2012;109:14568–14573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maeda N, Shimomura I, Kishida K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 2002;8:731–737 [DOI] [PubMed] [Google Scholar]
- 61. Kubota N, Terauchi Y, Yamauchi T, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem 2002;277:25863–25866 [DOI] [PubMed] [Google Scholar]
- 62. Nawrocki AR, Rajala MW, Tomas E, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem 2006;281:2654–2660 [DOI] [PubMed] [Google Scholar]
- 63. Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 2007;117:2621–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 2007;100:328–341 [DOI] [PubMed] [Google Scholar]
- 65. Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002;8:1288–1295 [DOI] [PubMed] [Google Scholar]
- 66. Klaus VS, Schriever SC, Peter A, et al. Correlation guided Network Integration (CoNI) reveals novel genetic regulators of hepatic metabolism. 31 January 2020 [preprint]. BioRxiv: 2020;1:29.924944 [Google Scholar]