

Abstract
Humans are living ecosystems composed of human cells and microbes. The microbiome is the collection of microbes (microbiota) and their genes. Recent breakthroughs in the high-throughput sequencing technologies have made it possible for us to understand the composition of the human microbiome. Launched by the National Institutes of Health in USA, the human microbiome project indicated that our bodies harbor a wide array of microbes, specific to each body site with interpersonal and intrapersonal variabilities. Numerous studies have indicated that several factors influence the development of the microbiome including genetics, diet, use of antibiotics, and lifestyle, among others. The microbiome and its mediators are in a continuous cross talk with the host immune system; hence, any imbalance on one side is reflected on the other. Dysbiosis (microbiota imbalance) was shown in many diseases and pathological conditions such as inflammatory bowel disease, celiac disease, multiple sclerosis, rheumatoid arthritis, asthma, diabetes, and cancer. The microbial composition mirrors inflammation variations in certain disease conditions, within various stages of the same disease; hence, it has the potential to be used as a biomarker.
Keywords: Microbiome, Microbiota, Metagenomics, Dysbiosis, Microbe–immune cell interactions
1. Introduction to the Human Microbiome
The term “microbiome” was introduced by the Nobel Laureate Joshua Lederberg and McCray in 2001, and it is used interchangeably with the term “microbiota” [1]. The two terms, however, have distinct meanings: Microbiota is defined as the entirety of microbes (bacteria, archaea, fungi, and viruses) that are found in a particular environment at a particular time [2], whereas the term microbiome refers to the collection of genomes encoded by the microbiota [2].
Microorganisms inhabit numerous human body sites including epithelial barriers and body fluids such as the skin, mouth, saliva, nose, vagina, vaginal secretion, seminal fluid, mammary glands, and the largest number residing in the gut due to the bioavailability of nutrients [3]. It is estimated that collectively the human gut microbiota are at least as numerous as the total cells in the human body by ten to one [4, 5], consisting of about 100 trillion microbes that represent around 5000 distinct species and make up to 2 kg of the human body weight [3]. The human microbiome is often referred to, as “our second genome” due to its size and sheer vastness [5]. In particular, the gut microbiota is now considered an “organ” of its own and is nicknamed “our second brain” due to its role in various immunological, neuronal, and endocrine responses [6].
Studies of the diversity in the human microbiome initially started in 1680, when Antoine van Leeuwenhoek compared his own oral and fecal samples using self-made microscopes [7]. He observed that the two habitats had distinct microbes and this observation led him to compare samples from healthy and diseased individuals where he also noted striking differences in their microbial communities [7, 8]. However, owing to the limitation of the in vitro microbial cultivation, the complex and dynamic nature of our microbiota was not fully recognized up until recently. This was facilitated by the advent of high-throughput sequencing technologies and the undertaking of large-scale metagenomics endeavors such as the Human Microbiome Project (HMP) that was initiated by the National Institutes of Health in the USA [9] and the Metagenomes Human Intestinal Tract (MetaHIT) project developed by the European Commission [10]. These projects have expanded our understanding of the Human microbiome. The MetaHIT study reported gut metagenomes from stool samples of predominantly healthy cohort of 124 European adults [11]. Whereas, in an effort to define the healthy human microbiome, the HMP researchers recruited 242 volunteers (129 males, 113 females) and sampled tissues from 15 body sites in men and 18 body sites in women. By incorporating several complementary techniques and analyses including 16S ribosomal RNA (rRNA) gene sequencing, whole-genome shotgun sequencing (WGS) and aligning the assembled sequences to the reference microbial genomes [12, 13] they were able to define the microbiome composition of each body site they sampled from [9]. They showed that, various body sites show a tenfold difference in the estimated microbial richness, with the gut being the richest ecosystem [9]. They also confirmed that a high interindividual variation of the microbiome exists and concluded that a healthy microbiome can be achieved by maintaining a balance between all the factors that contribute to the microbiome makeup (will be discussed later).
The most extensive research has been conducted with the gut ecosystem, in order to assess the impact of the gut microbes on human health and predisposition to various diseases [11, 14]. The gut microbiomes described as part of the MetaHIT cohorts indicated that a large proportion of the microbial genes are shared between individuals of this cohort and that more than 99% of these genes are bacterial, representing 1000–1150 bacterial species [11]. A healthy gut microbiota is mainly composed of strict anaerobes dominated by two main phyla: Bacteroidetes and Firmicutes [13].
In a healthy individual, many other body sites are occupied by microbial communities although not as well studied as the gut [13]. One of the largest human-associated microbial habitats is the skin surface, and is colonized primarily by Corynebacterium, Propionibacterium, and Staphylococcus [15]. The composition of the microbial communities shows more similarity “within habitats” as compared to “between habitats.” For example, samples collected from the oral communities show similar composition between different individuals, whereas compared with other communities within the same person, the oral ecosystem looks vastly different [13]. The oral and salivary microbiomes mirror the gut microbiota in terms of complexity and diversity [13].
An ecologically stable vaginal microbiome is dominated by species of Lactobacillus (L. crispatus, L. iners, L. jensenii, or L. gasseri) or by other microbes including Gardnerella vaginalis and Atopobium spp., among others and is classified in community state types [16]. This microbial diversity is affected by race, ethnicity, pregnancy, hormonal changes, sexual activities, and hygiene practices, among others [16–18]. The vaginal microbiome plays an important role in the prevention of genital and urinary tract infections and in prevention sexually transmitted diseases, and multiple studies have shown that an abnormal vaginal microbial composition is associated with an increased risk of HIV infection [19].
There are body sites particularly difficult to characterize, due to the low microbial load in healthy individuals, for example, the lung microbiota has a low microbial density, especially in the absence of an infection or pulmonary disease, as under normal conditions, the human lungs harbor approximately 2.2 × 103 bacterial genomes per cm2 [20]. It is also worth-noting that the identification of these microbial genomes remains tedious due to the technical challenges associated with the sampling from such sites. Similarly, habitats such as mammary glands, breast milk [21], placenta [22], and amniotic fluid [23] are of great interest due to their potential role in establishment of the early infant microbiome, but most studies have relied on traditional culture dependent approaches and/or single-cell techniques, such as FISH or microfluidics to study their microbiomes. Development of better sampling techniques facilitating the use of larger-scale high-throughput metagenomics studies are thus required to establish the exact composition and functionality of these challenging microbial habitats [23].
2. Universal Properties of the Human Microbiome
Based on the HMP and other research studies the “universal” properties of a healthy human microbiome have been identified. Dr. Lita Proctor, the coordinator of the HMP, pointed out few of these properties [13]. First, unlike the human genome, the human microbiome is not inherited but acquired upon birth. Newborns are described as “microbe magnets,” where the microbiota of the babies born vaginally resembles closely the mother’s vaginal microbiota, whereas those born via cesarean section (C-section) derive their microbiota from the mother’s skin [13] or the surrounding environment [24]. A second universal property is that, the adult microbial community composition is dictated by specific body sites, each body habitat such as gut, skin, oral cavity and urogenital tract, has a distinct microbial composition, regardless of the host gender, age, weight, or any other factor. HMP 16S rRNA data revealed clustering of specific microbial taxa within each of the body sites, and this clustering was driven by various factors including temperature, humidity, and pH, among others [25]. The final universally applicable property is that the gut microbiome changes over the lifetime, advances in metagenomics techniques have facilitated our understanding of the microbiome composition and its variation from infancy to old age. The ELDERMET project reported that the microbiomes in elderly people (aged 65 and over) are quite distinct from the microbiomes of middle-aged adults [26].
3. Inception of the Microbial Colonization
One of the most essential processes in the human life is the initial colonization of the infant’s gut by microbes, as this sets the stage for the establishment of a stable microbiome later in life [27]. Microbial contact during prenatal life and the inoculum transferred during birth and breastfeeding imprint the infant’s microbiota and the immune system [24, 28]. The gut microbiota of neonates is low in diversity and is dominated by two major phyla such as Proteobacteria and Actinobacteria, while in the weeks following birth, the microbiota becomes more diverse and dominated by Firmicutes and Bacteroidetes [11, 28–30]. Between one to two years of life, the microbial succession begins to vary, coinciding with the transition to a more diverse type of diet [29]. By the end of the second year of age, the taxonomic composition of the gut microbiome stabilizes and converges toward a characteristic adult microbiome [28]. Therefore, the first 2 years of life represents a vital period for any diet-based alteration to potentially impact the human health and immunity in later life.
4. Major Factors Influencing the Development of the Microbiome
Several pre and postnatal factors can significantly alter the composition of the gut microbiota including genetics; the mode of delivery at birth; the method of infant feeding; the use of medications, especially antibiotics; and diet, among others.
4.1. Birth Mode
Numerous studies have shown that the mode of delivery (vaginally or by C-section) has a strong influence on early gut colonization [24, 28]. Vaginally delivered infants are exposed to their mother’s vaginal and fecal microbiota and have an abundance of Lactobacillus and Bifidobacterium species [24]. Whereas, the very first microbial inoculum received by the infant born via a C-section is distinct; and these newborns become quickly colonized by the bacteria acquired from the skin and the surrounding hospital environment [24]. It is also suggested that the microbial composition in those infants may be disturbed for the first few months in life [31, 32]. Analysis of the meconium in the first few days after birth revealed that the microbial communities of the infant’s gut mirror either those of the mother’s vagina (Lactobacillus, Prevotella, or Sneathia) in the case of vaginal delivery, or the mother’s skin microbiota (Staphylococcus, Corynebacterium, and Propionibacterium) in the case of a C-section [24]. Vaginally delivered infants also share the higher proportion of their gut microbial 16S gene sequences with their own mothers as compared to other mothers [32].
4.2. Feeding Mode
The mode of feeding is another factor that influences the development of the microbiota in the infant’s gut. Not only is mother’s milk the major source of bacteria colonizing the infant’s gut, but it is also considered a major determinant and contributor to the child health status [33]. Analysis of breast milk samples revealed the presence of Streptococcus and Staphylococcus genus, and those are considered among the early colonizers of the infant’s gut [34]. Additional phyla such as Bifidobacterium and Lactobacillus were detected in the breast milk samples and also in the gut of breastfed infants in higher counts as compared to the formula-fed infants [35–37] indicating a major role of breast milk in seeding the gut microbiota early in life. The gut microbial diversity increases with the infant’s age and during the gradual shift from milk to solid food where a higher abundance of Bacteroides and Clostridium species in the toddler’s gut starts to be observed.
4.3. Antibiotics
The use of broad-spectrum antibiotics in infants and young children has negative effects on the composition of the gut microbiota, which results in significant drops in taxonomic richness, diversity and evenness of the gut microbial communities [38, 39]. Prenatal/or maternal antibiotic exposure is shown to increase the abundance of many pathogenic communities (pathobionts) such as
Staphylococcus, Streptococcus, Serratia, and Parabacteroides [40].
Early antibiotic exposure in neonates (such as Ampicillin and Gentamicin administered within the first 48 h of birth) results in significantly lower levels of Bifidobacterium and Lactobacillus when compared to untreated controls [41].
4.4. Preterm Birth
Compared to full term born infants, the preterm born infant’s microbiota has a reduced microbial diversity and a decreased abundance of bacteria such as Bifidobacterium and Bacteroides species [42]. In addition, potentially pathogenic bacteria such as Enterococcus species together with Proteobacteria species increase proportionally as a result of the reduction of the beneficial microbes [41]. Preterm birth is also associated with inflammatory intestinal disorders such as Neonatal Necrotizing Enterocolitis which has been linked to microbial dysbiosis and an increase in Proteobacteria is considered a predictive marker for the disease [43].
4.5. Host Genetics
The contribution of the host genetic makeup in shaping the early microbiota remains a subject of many speculations, as defining the host genotype–microbiota interaction in humans remains complicated. However specific bacteria found in the gut microbiota may be influenced in part, by the genetic makeup of the host, impacting as a result our metabolism and ultimately the health status.
Family studies with varying degrees of relatedness along with the twin cohorts’ studies provided a great input on the role of genetics in the microbiome composition. The microbial diversity is similar within family members as compared to unrelated individuals and monozygotic twins share certain the expression of certain heritable taxa compared to the dizygotic twins [44]. However, the overall variance in the microbiome composition is also affected for a few percent by host genetics and mostly determined by environmental factors [45]. A recent study in southern China, showed that the major influence on microbiome composition was due to the geographical localization in different districts and that these differences could simply be accounted by environmental or life style differences among districts [46]. Detailed genome-wide association studies are required to provide a greater insight into the role of the genetic makeup in the microbiome composition and in the mechanism underlying the microbiota–host genetic interactions.
4.6. Environmental Factors
Differences in lifestyle due to the geographical location, religion and culture are among the factors impacting the development of an early gut microbiota [47]. Infants from the Northern part of Europe have higher abundance of Bifidobacterium, Clostridium and Atopobium species in their gut microbiome, while infants living in southern Europe have higher percentages of Eubacteria, Lactobacillus and Bacteroides [36]. Similar differences in the gut microbial composition have been reported between German and Finnish infants [36], and between Swedish and Estonian infants [48]. Family dynamics may also act as a relevant environmental factor, where having siblings usually results in a higher percentage of Bifidobacterium as compared to single infants [49]. Exposure to household pets was shown to play a role in shaping the gut microbiome of infants [50]. An emerging spectrum of environmental factors including maternal or infant’s exposures to toxins/pollutants and xenobiotic substances are important to be considered and warrant more studies in order to determine their role in the development of the early gut microbiota.
5. Tools Used to Study the Microbiome
Recent advancements in the development of sequencing technologies and bioinformatics tools, fostered the growth of new omics technologies used to study the microbiome composition and its interaction with the host. Omics technology helps to explore the microbiome at various levels including gene expression, gene abundance, protein expression and microbial metabolites [51]. This knowledge is an essential tool used to study the role of the microbiome in many diseases and pathological conditions using various models (described later).
5.1. 16S rRNA Gene Sequencing
This is a gene-specific technique, where the bacterial taxonomic profile of a sample is characterized by sequencing specific hypervariable regions of the 16S rRNA gene [52]. Amplicons are generated using universal primers used to anneal with the conserved regions of the 16S rRNA gene sequences [43]. Those hypervariable region sequences are highly polymorphic, therefore allowing the classification of the bacterial taxa from phyla to species levels [52]. Illumina MiSeq and ThermoFisher Ion Torrent Personal genomic instruments are the widely used sequencers with a capacity to generate 300–400 bp reads [53]. Sequence reads can be analyzed using the traditional way of operation taxonomic units-based clustering approach using QIIME pipeline or calling amplicon sequence variants using DADA2 pipeline [54, 55]. The other available tools used to detect the taxonomic profiles using 16S data are LEA-seq, 16S based oligo typing, Long-read 16S, UNOISE2, and Deblur [56–59]. GreenGenes and SILVA are the most commonly used databases to profile the taxonomic differences among various samples [60, 61]. Eukaryotic organisms (such as fungi and protists) also play an important role in the microbiome. They can be analyzed in a similar manner to 16S studies by either looking at the hypervariable regions of the 18S rRNA or the internal transcribed spacer (ITS) regions between rRNA genes [62–74].
5.2. Metagenomics: Which Microbes Are There?
This technology is based on the shotgun sequencing of the whole genomic DNA and is used to study all the genetic materials in the microbiota [51]. Metagenomic study provides information about the taxonomic profile, functional composition and gene abundance of the microbiota [75]. It can also provide information up to species/strain level compared to 16S rRNA gene sequencing [75]. One challenge of metagenomic shotgun sequencing is that of retrieving enough data pertaining to the microbiota from low biomass samples. These low biomass samples are host tissues which present a low concentration of microbes relative to host somatic cells. Because shotgun sequencing is nontargeted, and the size of a mammalian genome is three orders of magnitude larger than that of a bacterial genome, the sequencing effort for a low biomass sample may end up being dominated by the host DNA.
Although metagenomics can explore the functional abundance (relative abundance of the microbial genes), it lacks information about the microbial activity (gene expression in live microbes) [75]. Therefore, recent studies are using metatranscriptomics (RNA-based) along with metagenomics (DNA-based) to explore the functionality of the active/viable microbes and their role in the host physiology [51]. Metagenomic sequencing reads can be taxonomically classified using, basically, two approaches, namely, the alignment of reads to reference bacterial genomes, or a comparison of snippets, or “words,” of genome of a fixed small size (known as k-mers) to the ones found in reference bacterial genomes. The HUMAnN pipeline [76] infers presence and relative abundance (quantity) of species by alignment to precomputed clade-specific genes, which are genes exclusive to a species or other higher-level taxonomic levels. Metabolic reconstruction using hits to reference are performed using ASGARD [77]. Classification of reads coming from species which are not in a reference database can be done by k-mer spectrum analysis using Kraken [78] and by assembling the reads into contiguous sequences, called contigs, followed by naive gene prediction and classification using a functional database like InterPro [79].
5.3. Meta transcriptomics: What Are Those Microbes Doing?
The advent development in the field of RNA-sequencing has provided more exciting opportunities in the field of transcriptomics, especially microbial transcriptomics. Metatranscriptomics reflect the functional activity of microbial communities by assessing the community-wide gene expression [80]. Sample processing for metatranscriptomics is laborious compared to the metagenomics library preparation. After total RNA extraction, bacterial rRNA is removed using specific magnetic probes of rRNAs. The prokaryotic RNA content is only 1–5% of the total RNA content and is enriched with mRNA that is converted into cDNA before proceeding with the RNA sequencing protocol [81]. Sequence reads are mapped to reference genomes, and the expressed genes are identified based on the sequence reads covering these regions. Raw Sequence data is filtered using Trimmomatic and mapped using BOWTIE and GEM [82]. The differentially expressed genes are identified using CuffDiff [83]. HUMAnN and MG-RAST are the most extensively used pipelines for metatranscriptomics analysis [82, 84–86].
This approach can give a panorama for assessing the microbial activity of the human microbiome, the antisense RNAs and small noncoding RNAs, and facilitates a deeper understanding of the host–microbiome interactions in abnormal and diseased states [81].
5.4. Metabolomics
Defined as the analysis of the metabolomic profiles using untargeted and targeted liquid chromatography–mass spectrometry (LC-MS) methods or nuclear magnetic resonance spectroscopy (NMR); metabolomics is used to explain the mechanism of microbial bioactivity [53]. Of all the previously described omics approaches, the metabolomics approach acts as a bridging gap between the microbiome and host physiology. Metabolomic profiles can be analyzed with biofluids such as plasma, serum, urine, tissue extracts, and fecal supernatant [87, 88]. The output of the metabolomics data allows correlations between microbiota, their metabolites and metabolic routes using multivariate analysis. It can also be used as a diagnostic and prognostic marker for the diagnosis and treatment of various diseases.
6. Microbiota Derived-Metabolites and Small Molecules
After completion of the HMP [9], Cimermancic et al. used an advanced computational approach with a moving aim from “who’s there?” to “what are they doing?” and identified more than 14,000 biosynthetic gene clusters in the human microbiome [89]. Of which, 3118 genes were present in one or more of the 752 sequenced samples [9]. While microbes are well known to produce diverse natural molecules mainly enzymes and antimicrobial agents, the recent genome mining have discovered numerous novel unexpected biosynthetic gene clusters and distinct natural molecules, which contribute to the microbial social and competitive interaction with other organisms [89–92].
The microbiota produces a range of molecules from various chemical classes including low molecular weight metabolites, to high molecular weight ribosomally synthesized posttranslationally modified peptides and proteins (RiPP), short-chain fatty acids (SCFAs) and diverse lipopolysaccharide (LPS) [92]. SCFAs contain acetate (C2), propionate (C3), and butyrate (C4) and are mainly produced by Butyrivibrio, Clostridium, Faecalibacterium, and Bifidobacterium species [93]. Vitamins like vitamin B and vitamin K are produced by various gut microbes including Propionibacterium freudenreichii, Salmonella enterica, Listeria innocua, and Enterobacter species, among others [94–96].
Those metabolites possess a wide range of activities; for example, microcin a narrow-spectrum antibacterial RiPP produced by Escherichia coli inhibits the growth of other closely related microbes; while the LPS and glycolipids released by the gut microbiota target various host immune cells; and the SCFAs produced by Bacteroidetes and Firmicutes stimulate a range of host processes such as epithelial cell proliferation, energy metabolism, host immune system (discussed later). We have summarized some of the natural molecules or metabolites produced by different microbiota species that mediate microbe-microbe interaction and host–microbe interactions in Table 1.
Table 1.
Selected microbial associated metabolites from the human microbiota
| Metabolite | Related bacteria | Body site | Potential functions |
|---|---|---|---|
| Antimicrobial | |||
| Ruminococcin A, microcin, clostridiolysin S, zwittermicin, cytolysin, epidermin | Ruminococcus gnavus, Escherichia coli, Clostridium sporogenes, Bacillus cereus∗, Enterococcus faecalis, Staphylococcus epidermidis, Streptococcus pyogenes | Gut | Inhibit the growth of pathogens, promote commensal colonization in gut |
| Lactocillin | Lactobacillus gasseri | Vagina | Antibacterial peptide, inhibit the growth other pathogens |
| Epidermin | Staphylococcus epidermidis | Skin | Antimicrobial peptide |
| SA-FF22 | Streptococcus pyogenes | Skin/oral | Antimicrobial peptide |
| Streptolysin | |||
| Tilivalline | Klebsiella oxytoca | Gut | Peptide, cytotoxic |
| Cereulide | Bacillus cereus | Gut | Cytotoxic |
| Staphyloxanthin | Staphylococcus aureus | Skin | Virulence factor |
| Mycolactone | Mycobacterium ulcerans | Skin | Polyketide, cytotoxic |
| Immunomodulatory | |||
| α-Galactosylceramide | Bacteroides fragilis | Gut | Essential for myelin synthesis, Stimulate host immune systems |
| Short-chain fatty acids: propionic acid, acetate, butyrate, indolepropionic acid |
Bacteroides spp., Clostridium spp., Eubacterium spp., Butyrivibrio, Faecalibacterium spp., Clostridium sporogenes | Gut | Modulate host immune system, Energy to epithelial cells |
| Vitamins: Vitamin K, vitamin B12, biotin, folate, riboflavin, etc. |
Bifidobacterium spp., Propionibacterium freudenreichii, Salmonella enterica, Listeria innocua, Enterobacter spp. | Gut | Sources of vitamins, stimulate immune system, Exert epigenetic effects |
| Lipids: LPS, peptidoglycan, acylglycerols, triglycerides, Polysaccharide A |
Bifidobacterium spp. Roseburia, Lactobacillus, Clostridium spp., Bacillus fragilis | Gut | Induce inflammation, Enhance immune system, influence gut permeability |
| Polyamines: Putrescine, cadaverine, spermine |
Campylobacter jejuni, Clostridium saccharolyticum | Gut | Modulate immune system, anti-inflammatory and antitumoral effects |
| Bile acids: Cholate, muricholate, etc. |
Lactobacillus spp., Bifidobacterium spp., Clostridium spp., Bacteroides | Gut | Influence intestinal permeability |
| Muramyl dipeptides and tripeptides | Fusobacterium nucleatum | Oral | Modulate immune system |
| Siderophore | |||
| Corynebactin | Corynebacterium spp. | Skin | Peptide, Siderophore |
| Staphyloferrin B | Staphylococcus aureus | Skin | Peptide, Siderophore |
| Neurotransmitter | |||
| D-lactate Ammonia Acetyl-choline, Gamma-amino Butyrate, acetylcholine, Norepinephrine, serotonin, Dopamine |
Lactobacillus spp., Enterobacter spp., Bacillus spp. |
Gut | Influence blood-brain barrier, Neurotoxic |
| Lactobacillus spp., Bifidobacterium spp. Escherichia spp., Streptococcus spp., Bacillus spp. | Gut | Influence human hormones, Neurotransmitter, Influence human behaviors |
|
| Unknown functions | |||
| Skatole, p-cresol, phenyllactic acid, listeriolysin S, clostridiolysin S, δ-aminovaleric acid, β-aminoisobutyric acid | Clostridium spp., Bifidobacterium spp., Listeria monocytogenes, C. sporogenes, Clostridium spp. | Gut | Unknown |
| Mutanobactin, coproporphyrin III, corynomycolic acid | Streptococcus mutans, Propionibacterium acnes, Corynebacterium spp. | Mouth/skin | Unknown |
7. Microbiome–Host Interactions: A Bidirectional Relationship
The recent developments of the high-throughput technologies and availability of the germ-free mice or humanized mice models have shed light into the interaction of the microbiota and/or its mediators with the immune system, and contributed to a better understanding of the molecular mechanisms governing these interactions [97–100]. These models have provided important insights into how the microbiota influence the host immune signaling in mammalian cells [101–103]. Most studies focused on the interaction between the gut microbiota and the immune system, since the gut is the most studied ecosystem. These studies showed that the microbiota–immune cell interaction begins at birth, where the microbiota educates the immune system, and in turn the immune system shapes the composition of the gut microbiota [104]. This interaction can be local or systemic.
7.1. Local Interactions
A great example of the local interaction of the microbiome with the immune system is represented in the gut (Fig. 1). The intestinal mucosa is protected by an array of physical, biochemical barriers and immunological elements. The physical barrier is made of the mucus layer consisting of a glycosylated gel composed of mucin molecules that are secreted by Goblet cells, the intestinal epithelial cells (IECs) connected by tight junctional complexes for protection against pathogen translocation. The microbiota is considered a part of the physical intestinal mucosal barrier as well [105]. The biochemical barriers are made of bile and gastric acid, defensins, lectins, and lysozymes secreted by Paneth cells [106]. The immunological elements include the antimicrobial peptides which are mainly produced by Paneth cells and act against a wide array of invading pathogens [107]. In addition, the secretory IgA is produced by the plasma cells in the lamina propria and is transported to the lumen, where it blocks invading pathogens by preventing their attachment to the epithelial cell receptors [108]. All immune cells residing in the lamina propria such as macrophages, neutrophils, dendritic cells (DCs), IECs, T and B lymphocytes, T-regulatory cells (TRegs) and plasma cells orchestrate an immune protection against invading pathogens and an immune tolerance for the microbiota (Fig. 1) [108–111].
Fig. 1.
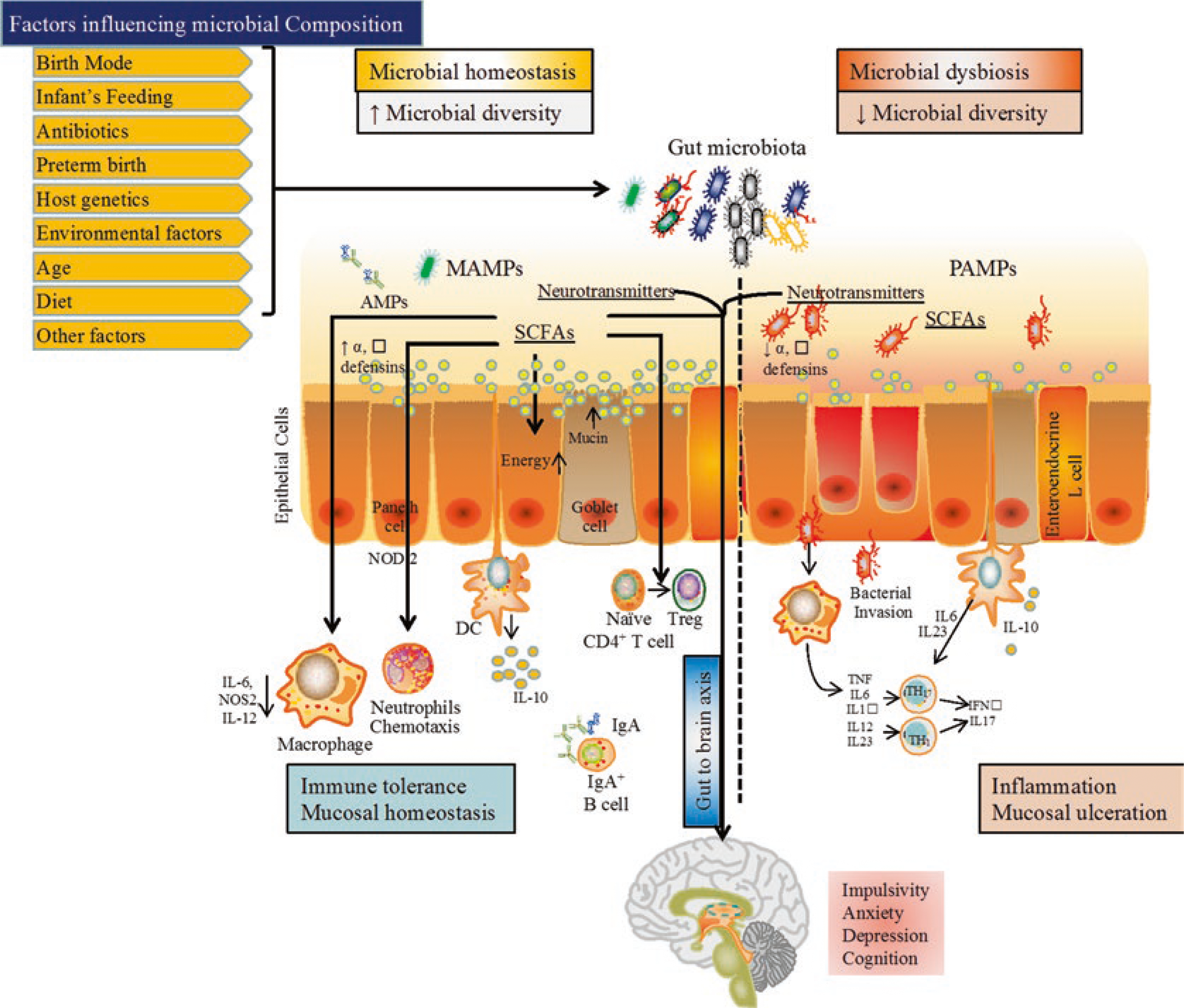
Integrative view of the gut microbiome. MAMPs microbial associated molecular patterns, PAMPs pathogens associated molecular patterns, SCFAs short chain fatty acids, DC dendritic cells, Il interleukin, Treg regulatory T-cells, Th T-helper
Pathogenic microbes and the microbiota interact with the immune cells through similar conserved ligands that signal through various host pattern recognition receptors (PRRs) such as the Toll-like receptors family (TLR) [112–114]. Those receptors recognize various microbe-associated molecular patterns (MAMPs) including lipopolysaccharides, peptidoglycans, flagellin, and formylated peptides [115, 116]. Innate immune cells have TLRs that recognize MAMPs leading to the activation of Nuclear factor-Kappa B (NFκB) which triggers the cytokine production [117]. Moreover, the upregulation of these cytokines against the PAMPs [112] leads to T-cells activation, a function that is facilitated by the gut microbiota that acts as an intermediate connector between the innate and the adaptive immunity (Fig. 1) [112, 113].
7.2. Systemic Interactions
Beneficial microbes such as Lactobacillus and Bifidobacterium promotes the upregulation of potential transmembrane proteins to protect the tight junctions of IECs and reduces gut permeability [118, 119]. Impairment in the barrier leads to an increase in the gut permeability and promotes translocation of bacteria through the intestinal barriers (Fig. 1) [120]. Continuous exposure of bacteria and bacterial LPS to the mucosal immune cells leads to the secretion of cytokines as a primary local immune response which in turn activates the vagus nerve, the spinal afferent neurons [121], and as a result the central nervous system (CNS) [122, 123]. Gut microbiota indirectly affects the CNS by releasing their metabolites in the form of neurotransmitters, for example, Lactobacillus reuteri and Bifidobacterium infantis produces histamine and serotonin, respectively [124]. Serotonin synthesis is mainly dependent on the availability of tryptophan, an essential amino acid acquired through diet [125]. In patients with depression, a reduction in the levels of tryptophan was observed [126]. Increased levels of tryptophan have been observed as a result of the oral administration of B. infantis in rats [127], but whether similar effect will be observed in patients with depression is yet to be described.
Dietary tryptophan, an essential amino acid is metabolized by the gut microbiota in order to produce natural ligands including indole-3-aldehyde and indole-3-propionic acid [128]. Through these metabolites, the gut microbiota is able to regulate the activity of the astrocytes (microglial cells of brain) by activating the astrocyte aryl hydrocarbon receptors (AHR) [129].
Moreover, the dietary tryptophan and nicotinamide are involved in regulating the mammalian target of rapamycin pathway (mTOR), which is involved in cell proliferation, survival, protein synthesis and transcription among other functions. In an experimental mouse model with Angiotensin Converting Enzyme 2 (ACE-2) mutation, reduced activity of the mTOR pathway is observed in epithelial cells which results in increase severity of colitis [130]. However, when the mice diet is supplemented with tryptophan, the colitis symptoms are reduced [130].
8. Mediators of the Microbiome–Host Interactions
The continuous cross talk between the microbiota and our immune system is mediated by a wide array of signaling mechanisms that involve many classes of microbial products [131] including microbiota-derived nucleic acids, metabolites, and small molecules:
8.1. Microbiota-Derived Nucleic Acids and the Immune System
The microbial DNA is structurally diverse from the human DNA and shows a remarkable sequence diversity between microbial species [132]. Microbial DNA utilizes its unique properties such as the higher frequency of CpG or CG dinucleotides and the presence of unmethylated cytosine as compared to being methylated as in the mammalian DNA [133–135]. The unmethylated CpG motifs in the microbial DNA are sensed by TLR9, which is expressed in IECs, plasmacytoid dendritic cells (PDCs) and B cells in humans [136]. IECs express TLR9 on both the apical and basolateral sides, and distinct responses are triggered depending on which side the stimulus is coming from (Lee et al. 2006). Stimulation of TLR9 on the apical side of the IECs is known to inhibit their inflammatory response and instead it induces tolerance toward other MAMPs [137]. However, the basolateral stimulation, leads to the induction of cell signaling pathways including the mitogen activated protein kinases (MAPKs), NFκB and the production of proinflammatory cytokines, interferons, and chemokines [138, 139]. In general, the apical stimulation of TLR9 is known to improve the barrier functions of the gut mucosa [140].
8.2. Microbiome Associated Metabolites-Host Interactions
Inside the GI tract, the gut microbiota facilitates digestion, fermentation of foods and produces a number of gastrointestinal metabolites, while the IECs orchestrate the intestinal defense mechanisms and mediate the interaction between the microbial mediators and our immune system [141]. A key example of those microbial metabolites includes the SCFAs, vitamins, neurotransmitters, microbial cell components such as the cell envelope or capsular components which can also act as microbial mediators to stimulate the immune functions as discussed in the previous section [128, 142–144]. Taken together >50% of the fecal and urinary metabolites are either derived from or modified by the gut microbiome [145] aiming to modulate the host cellular functions [146–148].
Microbial metabolites stimulate the host immune response through several ways [92, 149, 150], (a) by providing energy to the intestinal cells, (b) by manipulating the immune cells transcriptional programming, (c) by driving immune modulation through unknown mechanism.
Mounting evidences indicate that the SCFAs serve as a key energy source for the IECs and can influence their metabolism and integrity [149]. It was shown that, in germ-free mice, in absence of butyrate, the IECs suffer an altered energy metabolism which leads to autophagy activation due to the nutrient starvation in those cells [149, 151]. However, the monoinoculation of the germ-free mice with butyrate producing bacteria, was shown to rescue those cells from autophagy confirming the important role of butyrate in producing energy in the IECs [149, 151].
In tandem of being a direct energy source, the microbial metabolites may also tune the transcriptional programming of immune cells through the epigenetic landscape in order to control the immune function and the gut microbial composition [149]. One example is the butyrate concentration in the gut which can influence the transcription of mucin-related genes in the goblet cells (Fig. 1) and in turn modulate the transcription of many proinflammatory mediators such as IL-6, NOS2, IL-12 [152, 153]. The gut microbial composition can also be influenced by other microbial metabolites through the activation of the Nlrp6 inflammasome signaling pathway resulting in the secretion of proinflammatory cytokine, IL-18 and synthesis of some antimicrobial peptides [154, 155]. These mediators prevent the growth of the invading pathogens and maintain the gut tolerance for the commensal microbes.
Part of the protective effect of the microbial metabolites is mediated by their capacity to modulate directly or indirectly the function and the regulatory network of the host immune cells [150]. For example, the recognition of microbial derived butyrate and nicotinic acid (NA) by the G protein-coupled receptor GPR109A, expressed on the epithelial cells, macrophages, and dendritic cells, is critical for development of TReg cells [156]. Through the GPR109A receptor, both butyrate and NA can trigger IL-18 and IL-10 secretion, which leads to the development of TReg from naïve CD4+ cells and fine-tune the functionality of the epithelial cells to provide protection against pathogens [156, 157]. On other hand, altered gut microbiota results in a weakened immune cell response against infection, deregulated metabolism and compromised immune homeostasis [158]. The interaction of microbial mediators with the immune cells are just beginning to be revealed, with different microbial metabolites being sensed through different receptors in order to modulate cell programming, metabolic pathways, epigenetic modification [155]; however, the mode of action remains to be defined.
9. Models Developed to Study the Microbiome–Host Interactions
As discussed previously, the host and the microbiome are in a mutualistic relationship [159]. To study this interaction, many models were developed and used, most of them focus on modeling the gut environment as it is the most studied body site [160–162]. Some of these models are described below:
9.1. In Vitro Models
The human gut environment plays a vital role in the metabolism of nutrients directly and indirectly through the gut microbiota [163]. In vitro models of multicellular cell lines can be cocultured with lymphocytes and monocytes to mimic the gut ecosystem [161]. Developing those cell-culture models (described below), in combination with a microbial consortium can help fill the gap in understanding the gut–microbial interactions.
9.1.1. Caco-2 Cell Line
It was established in 1970 from gastrointestinal tumors, widely used as an intestinal epithelial barrier model in microbiological, pharmacological and nutritional fields [164]. Caco-2 is mainly used as a model to study the interaction of commensal or pathogenic bacteria and their metabolites with the cells [164]. This model was used as a three-stage culture system, to assess adherence of the gut microbiota to the epithelial cells and measure cytokine expression [165]. This study revealed that Atopobium species as the predominantly adherent organism, and Bifidobacterium longum showed a significant modulation of the expression levels of IL-4, IL-10, TGF-β2, IL-1α, and tumor necrosis factor alpha (TNF-α) [165]. Caco-2 cells were also used to show a positive modulation of the gut microbiota after using prebiotic oligosaccharides that result in proinflammatory cytokines reduction [166].
9.1.2. HT-29 Cell Line
HT-29 cell line was isolated in 1964 from a human colon adenocarcinoma by Fogh et al., [167]. These enterocytes-like cells have the capability to produce cytokines such as interleukins and TNF-α, which makes it an important model to study the host–microbiome interactions [168]. A coculture model of Caco-2/HT29 cells was used to assess the immune functions and measure the improvement in the gut barrier using prebiotic fibers [169]. Caco-2 and HT-29 cells were also used to study the role of a bacterial metabolite, butyrate, in activating the activator protein-1 (AP-1) which is a potential target for cancer therapy [170]. In another study, HT-29 cells were used as a colon cancer model to study the metastatic therapeutic effect of nisin, a bacteriocin derived from Lactobacillus lactis [171].
9.1.3. IPEC-1 and IPEC-J2
IPEC cell lines (IPEC-1 and IPEC-J2) are porcine IECs isolated from the jejunum of a unsuckled neonatal piglet [172]. IPEC-1 cells were isolated from the ileal and jejunal tissue while the IPEC-J2 cells were isolated from the jejunum [172]. It is a unique and nontumorigenic cell line that can mimic the human physiology. It can be used as a supreme to study epithelial transport, interactions with the enteric bacteria, and to assess the effects of probiotic microorganisms [173]. IPEC-J2 was used to identify the role of butyrate in the epithelial cell integrity against LPS-induced damage [174]. This study confirmed that butyrate restored the LPS induced impairment in gut barrier via activation of Akt-mediated protein synthesis pathway by promoting claudin expression [174]. In another study, IPEC-J2 was used as a model to study the protective effect of Lactobacilli against enterotoxigenic E. coli (ETEC) which destruct the gut barrier. L. reuteri showed a substantial protection against ETEC by conserving tight junction structures of IPEC-J2 [175].
9.1.4. IEC-6 and IEC-18
IEC-6 cells were isolated from the rat small intestine and are considered a good model to study bacterial adhesion [176]. Adhesion of multiple Lactobacillus strains was tested using Caco-2 and IEC-6 cells and results showed that L. plantarum shows a higher ability of adherence [162]. In another study, L. reuteri showed a protective effect against LPS-induced inflammation [177]. Butyrate induced a higher expression of the Heat shock protein 25 in those cells reflecting its protective effect [178]. Moreover, the LPS derived from enteropathogenic E. coli and four types of Bifidobacteria species are shown to induce autophagy in those cells [179], which renders this model very useful to study the probiotic effect of the gut microbiota in various diseases.
9.2. Ex Vivo Models
Ex vivo means out of live beings, which can represent as a model between in vitro and in vivo [180] using human or animal cells, tissue, or organs to evaluate the host–microbiota interactions.
9.2.1. InTESTine™ System by TNO
TIMs (TNO intestinal Model) are model systems used to mimic the gut, TIM-1 simulates the stomach, Tiny-TIM simulates the small intestines and TIM-2 model simulates the physiological conditions of the colon [181]. TNO recently developed InTESTine™, a new in vitro intestinal model using healthy porcine intestinal tissue. It contains a mucus layer which enables to utilize in experiments involving interaction of single microbe or a microbial consortium. Till date, there is no publications using this system, but still it provides a novel way to study the host–microbiome interactions.
9.2.2. Ussing Chamber
An Ussing chamber is an apparatus to measure epithelial membrane properties. It is widely used to quantify transport and barrier functions in a variety of living tissues. The main advantage of this chamber is the ability to study different segments of the intestines which makes it a powerful ex vivo model [168]. It contains electrodes to measure the voltage and short-circuit current in order to determine both cell permeability and ions transport. This system is mainly used to study host–microbiota interaction via microbial toxins and microbiota [168]. The effect of Enterococcus faecalis on a colonic mucus mounted on Ussing chamber was studied to understand the role of bacterial invasion; the study revealed that an adhesion product of E. faecalis promotes its translocation in the colon mucus [182]. The probiotic role of Lactobacillus plantarum has also been studied using this model [183].
9.2.3. Stem Cell-Based Organoids
Intestinal organoids are cultured from adult intestinal stem cells or induced pluripotent stem cells, which represent the new sophisticated gut model [184, 185]. Directed differentiation methods allow the cell-specific responses, assessing the intestinal barrier functions and the host–microbe interactions [186]. Human enteroids have been widely used to study host–pathogen interactions including enterohemorrhagic E. coli, enterotoxigenic E. coli, enteroaggregative E. coli, and Cholera toxin [187–189] as well as host interactions with L. rhamnosus GG, L. acidophilus, and Bacteroides thetaiothaomicron [190–192].
The apical to basolateral polarity conserved even after exposure to the microbes and their components is considered the main advantage of the ex vivo models; however, the short-term survival is the main disadvantage of these models.
9.3. In Vivo Models
Animal model-based research provide opportunities to manipulate, engineer, and study the host–microbiome interplay with a certain level of experimental control which is not feasible in human recruited experiments [160]. Below are few models used for microbiome studies:
9.3.1. Fruit Fly
Fruit fly, Drosophila melanogaster has been widely used as a model system to study the host-innate immunity and microbial pathogenesis [117, 193]. Around 5–30 different bacterial species are found in the fruit fly and the predominant microbial members of the Drosophila gut are aerotolerant Acetobacteraceae, Lactobacillales, and Enterobacteriaceae families [194]. Due to their similarity to the mammalian gut and our ability to culture these bacteria in the laboratory, the availability of various mutants, its affordable price renders Drosophila is considered a very useful model to study host–microbe mechanisms associated with innate immunity modulation, probiotic role and diet [195–197].
9.3.2. Zebrafish
Zebrafish is a small freshwater fish mainly used as a vertebrate model that is simple to study host–microbe interactions. It has a diverse microbiota with gamma proteobacteria and fusobacteria species being the most abundant members of the zebrafish’s gut [198]. A study between conventionally raised zebrafish and germ-free zebrafish indicated several new insights into the host–microbe interactions related mechanisms [199].
9.3.3. Mice
Laboratory mouse has been widely used in understanding the role of gut microbiota in host physiology, such as angiogenesis, metabolism of nutrients, cognitive health, hepatic and immune functions [92, 200–203]. Almost 99% of mouse genetics are shared with human host and the gut microbiome members from phylum to family levels [204]. These characteristics made lab mouse as prime choice of animal model to evaluate host–microbe interactions.
The most commonly used inbred mice strain is C57BL/6.129, C3H and BALB/c are the other inbred strains used in microbiome studies. The other concept of mice model is to use mouse with less complex predefined microbial consortia. The germ-free mice colonized with a defined microbial cocktail (isolated from conventional mice) to produce mice with standardized gut microbiota are called gnotobiotic mice models, which can serve as an ideal tool in host–microbe interactions [205]. This defined cocktail consists of eight aerobic bacteria, were E. coli, var. mutabilis, Streptococcus faecalis, Lactobacillus acidophilus, Lactobacillus salivarius, group N Streptococcus, Bacteroides distasonis, a Clostridium sp., and a fusiform bacterium; anaerobes were excluded due to difficulties in cultivation [206].
10. Microbial Dysbiosis and Immune Dysregulation
A healthy individual maintains a unique balance between the microbiome, immune system for protection from invading bacteria [207]. However, in some cases pathogenic bacteria increases in numbers replacing the beneficial bacteria (microbial dysbiosis) leading to a significant impact on our health [208].
The cross talk between the host microbiota and our immune system begins at birth [24, 28]. In order to achieve homeostasis, the intestinal microbiota and our mucosal immunity interact constantly as summarized in Fig. 1.
Various chronic immune-mediated and metabolic diseases such as inflammatory bowel disease, celiac disease, multiple sclerosis, rheumatoid arthritis, asthma, diabetes, and certain types of cancers are associated with dysbiosis in the gut [207–209]. The availability of high-throughput techniques has significantly advanced our capacity to sequence the microbiome and demonstrated variable degrees of dysbiosis in the above diseases [207–209]. For example, subjects with colorectal cancer (CRC) suffer from microbial dysbiosis when compared to normal controls [210, 211]. CRC adenoma is characterized by a high proportion of pathogenic bacteria, such as Pseudomonas, Helicobacter, and Acinetobacter and relatively lower abundance of beneficial bacteria, such as butyrate-producing bacteria [212]. As the disease advances from adenoma to carcinoma, a significant increase in Bacteroides massiliensis, Bacteroides ovatus, Bacteroides vulgatus, and E. coli was observed [213]. The promotion of inflammation due to bacterial dysbiosis partially explains the induction of tumorigenesis [214, 215].
11. Microbiome and Cancer
The microbiome composition modulates metabolism, inflammation, immunity and other functions of the organism. It also affects cancer initiation, progression and response to therapy [216]. Genetically transformed cells require a favorable microenvironment to form tumors. The microbiome modulates the tumor microenvironment and tumor progression both locally on the epithelial barriers in which it resides and systemically.
Based on epidemiological studies, a role of the microbiome and dysbiosis (i.e., a change in microbiome composition associated to disease) has been proposed for gastrointestinal and lung cancer. The only bacterial species recognized by the International Agency for Research on Cancer (IARC) as a group 1 human carcinogen is Helicobacter pylori that is associated to stomach cancer [217]. Other bacterial species were observed within the neoplastic lesions or positively correlated with cancer risk in epidemiologic studies. The association of bacteria with cancer may not be an evidence of their direct role in carcinogenesis but rather be a biomarker for coregulated or keystone bacterial species modulating the microbiome composition. Also, the presence of changes in the microbiome composition in the cancer patients may be induced by the presence of the tumor rather than being a cause of it. None of the bacteria associated with human cancer but H. pylori has been identified with certainty as a human carcinogen by showing that their elimination from the host prevent disease [218]. However, mouse and human studies suggest that several bacterial species (e.g., Fusobacterium nucleatum, enteropathogenic Escherichia coli, enterotoxigenic Bacteroides fragilis, Enterococcus faecalis, Streptococcus gallolyticus, Helicobacter hepaticus, and Salmonella enterica) may play a role in carcinogenesis [214, 215, 218]. The microbiome modulates locally inflammation and immunity at all epithelial barrier surfaces, where commensals and host cells are in proximity [219]. In addition, various bacterial products including toxins and metabolites, are genotoxic or target proliferation and repair of epithelial cells [216].
11.1. Colorectal Cancer
Various species of Fusobacterium (F. nucleatum), orally associated anaerobic gram-negative bacteria that form biofilms in the mouth, can colonize the colon and are enriched in human colonic adenomas and adenocarcinomas [214, 215]. Various mechanisms have been proposed for the procarcinogenic effect of F. nucleatum: (1) recruitment of tumor-promoting myeloid cells; (2) modulation of autophagy resulting in chemoresistance; (3) binding of Fap2 protein to the TIGIT inhibitory receptor resulting in inhibition of NK and T cells activity; and (4) association of FadA adhesin to E-cadherin, thus activating β-catenin/Wnt signaling in epithelial cells [214, 215, 220, 221]. Bacterial biofilms, in some patients containing Fusobacteria, are often present in carcinomas of the ascending colon and produce the polyamine metabolite N1,N12-diacetylspermine that favor epithelial proliferation, diminish E-cadherin expression and activate STAT3 and IL-6 [222, 223]. Fusobacteria and their associated microbiome have also been observed in liver metastases of colon carcinoma patients and remain associated to the tumors derived from these metastases when they are passaged several times as xenographs in immune deficient mice [224]. Elimination of cancer-associated Fusobacteria by antibiotics treatment reduces tumor xenograph growth [224].
E. coli is also over-represented in the gut microbiome of some patients with colitis and colon cancer [225]. The colibactin toxin from E. coli strains expressing the pks pathogenicity island impedes SUMOylation of p53 and favors cellular senescence associated to the secretion of tumor promoting proinflammatory and growth factors [225]. Enterotoxigenic Bacteroides fragilis (ETBF), is a low abundance commensal of the human intestine that when overexpressed becomes a pathobiont causing diarrhea and associated with inflammatory bowel disease and cancer [218]. ETBT toxin degrades E-cadherin and activates β-catenin enhancing proliferation of epithelial cells. It also permeabilizes the mucosa and induces STAT3 phosphorylation in both epithelial cells and inflammatory cells. In animals it induces by expansion of both Tregs and Th17 cells and favors colon carcinogenesis [218]. Whether the microbiome affects cancer on other epithelial surfaces including the skin, the oropharyngeal cavity, the lungs, and the urogenital tract remains to be determined. In addition to bacteria and viruses, the presence of fungi producing carcinogenic acetaldehyde has been associated the oral and esophageal cancer observed in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), a syndrome due to mutations in the AIRE gene [226]. Other members of our microbiome such as protists and helminths are also able to orchestrate gut immunity and could play a role in gastrointestinal cancer [227].
11.2. Tumors in Tissues Not Colonized by the Microbiome
The microbiome may also affect the development of tumors in sterile tissues. For example, certain bacteria modulate estrogen metabolism via β-glucuronidases and β-glucuronides and regulate endometrial and breast cancer through a noninflammatory pathway [228]. Microbial β-glucuronidases are also involved in the metabolism of xenobiotics and can produce genotoxic and procarcinogenic metabolites of heterocyclic aromatic amines from burned meat or environmental pollutants [229]. A systemic effect of the microbiome on carcinogenesis has been shown in mouse models. For example, Helicobacter hepaticus colonic infection results in an increased incidence of prostate and mammary carcinoma in APCmin/+/Rag2−/− mice and also promotes chemical and viral liver carcinogenesis [230, 231]. The variable incidence of tumors in genetic models of carcinogenesis observed has been attributed to diversity of microbiome composition in different animal facilities (e.g., thymic lymphoma in mice genetically deficient for the ataxia telangiectasia mutated (Atm) gene [232]. Recently, the presence of a microbiome within solid tumors that originate or reside in sterile tissues, such as pancreatic cancer, lung cancer, and colorectal carcinoma distant metastases has been demonstrated [224, 233–236]. The presence of proteobacteria and other bacterial phyla such as Bacteroidetes and Firmicutes was reported for human and murine pancreatic cancer where they can affect the response to chemotherapy and the local immunological responses within the tumors by suppressing innate and adaptive immune response and preventing effective response to immune checkpoint inhibitor therapy [233–235]. Variovorax and Acidovorax species were found to be increased in lung carcinoma lesions and in particular that Acidovorax is preferentially associated with p53 mutated squamous cell carcinoma of the lung [236].
11.3. Bacteria as Cancer Drugs
Bacteria can also be used as cancer drugs. The New York surgeon William Coley in the late XIX century, based on clinical observations that acute infections are in some case associated with tumor regression, successfully treated patients with soft tissue sarcoma patients with “Coley’s toxins,” a preparation of Streptococcus pyogenes and gram-negative Bacillus prodigiosus (Serratia marcescens) [237]. Other bacteria have then been used for cancer treatment (i.e., Corynebacterium parvum and the streptococcal preparation OK-432); bacille Calmette–Guérin, an attenuated Mycobacterium bovis strain is still used for local therapy of superficial bladder carcinoma [238]. Salmonella, Escherichia, and Clostridium species accumulate in tumors when delivered systemically and are being developed as anticancer drugs [239]. The hypoxic and necrotic areas present in large tumors are poorly accessible to drugs and also resistant to radiation induced DNA damage [239]. When spores of obligate anaerobic bacteria such as Clostridium spp. are administered, they germinate and deliver antitumor activity only when they reach the hypoxic tumor tissues [240, 241]. Conversely, well oxygenated smaller tumors and metastases are more susceptible to the antitumor effect of facultative anaerobes (i.e., Salmonella and Escherichia genera) [242]. Ideal anticancer bacterial preparations need to be selectively toxic for tumor cells, able to reach and proliferate in tumor area not well accessed by traditional therapies, maintained under control with a sufficient latency by the immune response, and genetically modifiable to engineer their tropism [239]. Bacteria-derived vectors can also be used to deliver cytokines, antigens, toxins, antibodies and antitumor genes [239].
12. Microbiome Impact on Cancer Drug Metabolism and Efficacy of Treatment
The microbiome regulate the metabolism and pharmacokinetics of certain enteral or parenteral anticancer drugs [243]. Intestinal host and bacterial enzymes regulate absorption and bioavailability of certain oral drugs [244]. Gut microbial enzymes induce drug bio-transformation prevalently by catalyzing reduction and hydrolysis [245]. The drug metabolism can also be indirectly affected by the ability of drugs and other xenobiotics to modify the composition and gene expression of the gut microbiome [246]. Although the gut microbiome is known to be involved in the metabolism of more than 40 drugs, this effect has been described only for a few anticancer drugs: (1) nitroreduction of the radiation sensitizer misonidazole; (2) hydrolysis of the antimetabolite methotrexate; and (3) deconjugation of the liver detoxified form of the topoisomerase I inhibitor irinotecan (CPT-11) [247].
The gut microbiome modulates in the intestine and liver the expression of genes encoding molecules involved in xenobiotic metabolism and sensing [248, 249]. Germ-free are characterized by enhanced xenobiotic metabolism than conventional mice [248, 250]. Intestinal microbiome diversity may explain the heterogeneous response of patients to drugs and their toxicity [251].
12.1. Microbiome and Chemotherapy
The pharmacokinetics, efficacy, and toxicity of chemotherapy drugs can be modulated by the microbiota [252]. In mice, tumor associated γ-proteobacteria produce cytidine deaminase and degrade the drug gemcitabine [233]. The antibiotic ciprofloxacin reestablished sensitivity to gemcitabine in mice with a colon carcinoma tumor experimentally colonized with E. coli [233]. Proteobacteria have been described to be present in many human pancreatic ductal adenocarcinoma [233].
The chemical structure and activity of chemotherapy drugs can also be modified by direct interaction with bacteria. Thirty different drugs were incubated in vitro with either nonpathogenic gram-negative Escherichia coli or gram-positive Listeria welshimeri: the activity of ten of them was decreased whereas the activity of six other was improved [253].
Oxaliplatin and cisplatin have severely reduced antitumor activity when used in germ-free mice or antibiotics-treated mice [254]. The gut microbiome is endowing the antitumor activity of these platinum compounds by licensing myeloid cells in the tumor to produce, reactive oxygen species (ROS) needed for drug-induced DNA damage [254]. Genetic deficiency for the Cybb gene encoding the gp91phox chain of NADPH oxidase two or treatment with either the ROS scavenger N-acetyl cysteine or antibodies depleting myeloid cells make the mice resistant to the antitumor effect of oxaliplatin [254]. Cisplatin antitumor activity is restored by the administration of Lactobacillus acidophilus to antibiotic-treated mice [255]. Acetovanillone, an inhibitor of NADPH oxidases, protects mice from nephrotoxicity induced by cisplatin, suggesting that similar mechanisms may be involved both in the therapeutic and toxic effect of cisplatin [256].
The alkylating chemotherapy drug cyclophosphamide (CTX) increases intestinal mucosa permeability in mice allowing gram-positive bacteria (e.g., L. johnsonii, L. murinus, and Enterococcus hirae) to translocate to the mesenteric lymph nodes [257]. The translocated bacteria potentiate the antitumor immune response evoked by the immunogenic tumor cells killed by CTX [257]. Thus, the antitumor effect of CTX is dramatically reduced in germ-free mice and in antibiotics-treated mice [257].
12.2. Microbiome and Immunotherapy
Cancer immunotherapy has represented a major improvement in cancer treatment in the last few years. Many patients, however, fail to respond and not all type of tumors respond to immunotherapy. Thus, there is an urgent medical need to increase the proportion of cancer patients responding to immunotherapy. Recent data in animals and in clinical trial claim that the composition of gut microbiome maybe an important variable determining immunotherapy efficacy [254, 258, 259] and raise the intriguing prospect that targeting the microbiome in the patients may enhance the response to immunotherapy.
Already over 10 years ago it was shown that the gut microbiome was required for effective adoptive T cell therapy of cancer in mice that were preconditioned with total body irradiation (TBI) [260]. Translocation to the mesenteric lymph nodes of bacteria and activated dendritic cells following mucosal damage induced by TBI was required for expansion and antitumor toxicity of the transferred CD8+ T cells [260]. More recently, it was shown that the gut microbiome was necessary for antitumor effect of intratumorally injected TLR9 agonist CpG oligonucleotide (CpG-ODN) [254]. CpG-ODN treatment induces production of proinflammatory cytokines including TNF and IL-12 followed by TNF-mediated hemorrhagic necrosis [261]. In germ-free or antibiotic treated mice, the production of proinflammatory cytokines by tumor associated myeloid cells in response to CpG-ODN is impaired, preventing the TNF-mediated necrosis and the subsequent antitumor immune response [254]. In lymphoid organ phagocytes from germ-free mice, genes encoding proinflammatory factors have been shown to have an epigenetically closed chromatin conformation and to be unresponsive to microbial stimulation [262]. Thus, colonization of mice with the commensal microbiome primes myeloid cells for responses to inflammatory stimuli by altering their chromatin conformation. This priming is reminiscent of the mechanisms involved in the phenomenon of trained resistance in innate effector cells [262, 263]. The presence of the gram-negative Alistipes and the gram-positive Ruminococcus genera in the fecal microbiota positively correlated with the amount of TNF produced by tumor associated myeloid cells in response to CpG-ODN. Conversely, the Lactobacillus genus including L. murinus, L. intestinalis, and L. fermentum species known to be anti-inflammatory when used as probiotics negatively correlated with TNF production [254]. Administration of Alistipes shahii and L. fermentum by oral gavage to mice enhanced and decreased TNF production, respectively, by tumor-associated myeloid cells [254]. Thus, antibiotics by depleting the gut microbiome reverse the priming of myeloid cells for response to CpG-ODN while administration of bacterial strains that were positively and negatively correlated with TNF production increased or decreased TNF production, respectively.
Recently, the composition of the gut microbiome was demonstrated to modulate the efficacy of immune checkpoint inhibitors (ICI) therapies (anti-CTLA-4 and anti-PD1/PD-L1) both in animals and clinical trials [258, 259, 264–266].
12.3. Immune Checkpoint Blockers: Anti-CTLA4
It was shown that the absence of gut microbiome in antibiotic-treated or in germ-free mice abolishes the antitumor activity of anti-CTLA-4 [258]. The presence in the microbiome of certain bacterial species, mostly of the Bacteroides genus, was reported in mice to correlate with anti-CTLA-4 therapy efficacy [258]. Gavage of B. thetaiotaomicron or B. fragilis to microbiome-depleted mice activated dendritic cells and induced a Th1 response resulting in efficient antitumor activity of anti-CTLA-4 therapy [258]. Interestingly, treatment with both B. fragilis and Burkholderia cepacia decreased anti-CTLA-4-induced subclinical colitis while enhancing the anticancer response [258]. Thus, by targeting the microbiome it may be possible to dissociate the favorable effect on anticancer therapy from therapy-induced toxicity. Treatment of mice with antibiotics that spare Bacteroidales and Burkholderiales (e.g., vancomycin) improve the efficacy of anti-CTLA-4 therapy [258, 267]. A recent study reports the first series of ICI-associated colitis successfully treated with fecal microbiota transplantation.
The fecal microbiome of the melanoma patients responding to anti-CTLA-4 showed abundance of certain Bacteroides species [258]. When germ-free mice were colonized with this favorable microbiome showed enrichment for B. thetaiotaomicron or B. fragilis they start responding to anti-CTLA-4 [258]. In another trial, the presence of the Bacteroidetes phylum was identified as a prognostic factor for reduced intestinal toxicity [268]. A different study also identified in melanoma patients a microbiome pattern that was associated with both anti-CTLA-4 tumor response and colitis [269]. In this study, the response correlated with Faecalibacterium and other firmicutes while Bacteroidales correlated with poor antitumor response and less toxicity [269].
12.4. Immune Checkpoint Blockers: Anti-PD1/PD-L1
The anticancer activity of anti-PD1/PD-L1 is not fully dependent on the presence of the gut microbiome, but it can be enhanced by the presence of certain bacterial species [259, 265, 266]. Mice from different vendors sustain the growth of B16 melanoma at different rates and anti-PD-L1 therapy is more effective in the mice with slow tumor growth that are characterized by a higher number of tumor-infiltrating CD8+ T cells suggesting a stronger antitumor immunity before treatment [259]. Cohousing of the different mice transmit the ability to slow the growth of the tumor and the responsiveness to anti-PD-L1 suggesting a role of the microbiome [259]. Indeed, responsiveness correlated with the Bifidobacterium genus (B. breve, B. longum, and B. adolescentis) [259]. The therapeutic activity of anti-PD-L1 in poorly responsive mice could be enhanced by a commercial probiotic preparation of various Bifidobacterium species (including B. breve and B. longum) [259]. These results suggest that Bifidobacterium spp. in the fecal microbiome of mice sustain the development of antitumor immunity that, when enhanced by anti-PD-L1, prevents cancer progression.
The role of the microbiome in anti-PD1 therapy has been recently evaluated in clinical trials treating patients with melanoma and, lung and renal carcinoma [264–266, 270]. While the oral microbiome composition did not distinguish responder and nonresponder patients, the analysis of the fecal microbiota by shotgun metagenomic sequencing showed that certain enterotypes and microbial species in the fecal microbiome were associated with a favorable response [264]. In one of these studies the fecal microbiome of the responder patients had a higher alpha-diversity, possibly indicating a healthier and varied microbiome that better sustained the establishment of cancer immunity [265]. Surprisingly, however, the sets of fecal bacteria identified in these trials as associated with favorable response to anti-PD1 were completely not overlapping. Increased representation of Akkermansia muciniphila and a trend for a higher representation of Enterococcus hirae characterized lung and renal cancer patients that responded to anti-PD1 [266]. In melanoma patients, Faecalibacterium species were reported in one study to be associated with anti-PD1 response [264] while another study identified Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium [265]. Combined analysis of the data of the three studies with a common bioinformatic platform failed to identify any bacterial species associated with response in all three studies and did not resolve these discrepancy [271]. However, strong support for a role of patients’ microbiome in anti-PD1 response was provided by the finding in all these studies that after colonization of antibiotic-treated or germ-free mice with patients’ fecal microbiomes response to anti-PD1/PD-L1 was observed in those mice that have received the microbiome from responder patients but not in those that have received the microbiome from nonresponder patients [264–266]. Furthermore, the poor response to anti-PD1 of mice that have been associated with the microbiome from nonresponder patients could be rescued by colonization with some of the bacterial species shown to be associated with favorable response (i.e., A. muciniphila alone or combined with E. hirae) [266]. Notably, mice and patients with what has been identified in the different studies as a favorable microbiome had a higher cytotoxic CD8+ T cell infiltration in the tumor microenvironment at treatment baseline suggesting the presence of a preexisting antitumoral immunity that would allow a response to anti-PD1/PD-L1 therapy [264–266]. A further evidence for a role of the microbiome composition in the patients’ response to anti-PD1 was provided by the observation in a few clinical trials in different geographical localization that the microbiome alteration induced by antibiotic treatment of the patients within a month before initiation of the treatment resulted in a lower likelihood of response to the anti-PD1 therapy [266, 272].
Noteworthy, although these studies pinpointed the importance of the microbiota in response to therapy, they identified different bacterial players. These discordant results may be in part explained by the following: the heterogeneity of microbiota composition due to many factors including genetics, lifestyle, and previous therapies; the small patient cohorts studied from geographically distant populations; the different cancer types studied; and variability in criteria for therapy response. Also, most likely the observed regulation by the microbiota of anti-PD1 therapy is not mediated by a single bacterial species but rather is dependent on multiple changes in the microbiota ecology and metabolism that together stimulate cancer immunity. The species or group of species that have been highlighted in these studies could be considered just biomarkers of the ecological changes and, because of the small-sized and heterogeneous cohorts, different bacterial species may have reached significance.
13. Microbiome as a Biomarker, a Target for Cancer and Cancer Therapy Efficacy
Although it has been known for many years that the microbiome and/or specific bacterial species affect tumor formation and progression, the ability of the microbiome composition to modulate cancer therapy effectiveness and toxicity has been a recent finding based on both preclinical and clinical evidence.
The presence of certain bacteria can be used as biomarkers for cancer diagnosis and they could be targeted for cancer progression. The presence of H. pylori in the stomach is considered a risk factor for development of gastric cancer and its eradication is considered efficacious in cancer prevention although it may lead to dysbiosis, metabolic alteration and possibly susceptibility to other cancers of the upper gastrointestinal tract [273–275]. Presence and amount of Fusobacterium nucleatum has been proposed as a possible diagnostic and prognostic marker in colorectal cancer [214, 215, 276]. Recently it has been suggested that microbiome dysbiosis of the tongue coat could be used as a diagnostic marker for liver and pancreatic carcinoma [277, 278].
The recent evidence of the role of the microbiome in modulating cancer therapy has raised the expectation that targeting the microbiome may represent a novel modality to improve therapy efficacy particularly for the new immunotherapy protocols. Ideally, we should aim to modify the microbiome from a procarcinogenic and immunosuppressive composition to one that would enhance antitumor immunity while preventing therapy- and cancer-associated toxicity and comorbidities. However, formidable roadblocks remain. Most data supporting the role of the microbiome in modulating cancer therapy are from experimental mouse models and relatively few and contradictory data exist in humans. Mouse physiology and immunity are not perfect models for human findings and laboratory mice live in a very different environment than patients living in open communities. Recently, it has been suggested that mice colonized with microbiome from wild or pet mice might better replicate the patient response than conventional laboratory mice [279, 280]. Transfer of human microbiome in mice has been used extensively to characterize the mechanisms by which responder patients’ microbiome enhance the response to immunotherapy [258, 264–266]. However, the human microbiome in mice does not perfectly mimic the donor microbiome, it is relatively unstable, and the physiological and immune response of the mice to the human microbiome may not be identical to that of the patients.
The results of the clinical trials in patients treated with anti-PD1 have identified bacterial species that might be associated with response to the therapy (although stringent analysis excluding false discovery rate may limit the significance of these associations [271]) but unfortunately different species have been identified in the various trials [264–266]. In part these contradictory results may be due to the large variability of the composition of the human microbiome among individuals and geographical differences in microbiome composition. For example, it has been shown that while the microbiome composition can be used to predict susceptibility to metabolic disease within Chinese populations living in same district but not across districts [46]. Thus, a major challenge remains to identify reliable microbiome-related biomarkers for prediction of response and stratification of patients across clinical centers. It should also be considered that the microbiome may modulate cancer therapy response not because of a single bacterial species but due to a particular ecology and metabolism of the gut microbiome to which a large number of bacterial species and functions may participate and together affect anticancer immunity. Thus, the bacterial species that have been found in different clinical centers to correlate with response may just be the tip of the iceberg of complex ecological changes that reached statistical significance due to small-sized and heterogeneous cohorts analyzed.
Because of our inability to identify with accuracy the composition of a microbiome that would sustain cancer, it remains difficult to plan therapeutic approaches to target the microbiome in order to improve immunotherapy response. Fecal transplant trials have been planned or initiated to treat patients undergoing immunotherapy. A case series has been reported of immune checkpoint inhibitors-associated colitis successfully treated with fecal transplant from an unrelated healthy donor [267]. However, in the case of fecal transplant for enhancing response to therapy, the inability to identify a favorable microbiome composition has led to clinical protocols based on the transfer of microbiome from cancer patients that have successfully responded to anti-PD1 therapy (ClinicalTrials.gov Identifiers: NCT03353402, NCT03341143 and NCT03772899). If we would be able to identify a favorable microbiome composition for immunotherapy response, the use of healthy balanced fecal microbiome from healthy donors rather than possibly dysbiotic fecal microbiome from sick and treated patients would seem to be preferable. Also, whereas in fecal transplant for opportunistic infections (e.g., Clostridium difficile), most of the preexisting recipient microbiome is depleted by antibiotics treatment [281], the data showing that antibiotics treatment before immune checkpoint therapy may decrease the response rate [266, 272] would suggest to avoid this treatment in fecal transplant aimed to enhance therapy response.
Rather than a crude effort using total fecal transplant with the risk of transferring bacteria that could act as pathogen or pathobionts, it would be ideal to characterize mechanisms of microbiome enhancement of cancer therapy shared by different bacterial species and to identify ecologically balanced consortia of commensal bacteria that favor a clinical response and could be utilized in any clinical center. One approach is to develop standardized microbial ecosystem therapeutics (MET) composed of a pool of bacterial isolates from healthy donors’ fecal microbiome, selected for having minimal antibiotic resistance and lacking any known virulence determinants; this pool of isolates is then formulated into an ecosystem and tested in vitro using chemostat technology [282]. A clinical trial is planned to use one of these preparations orally (MET-4) in solid tumor patients receiving anti-PD1/PD-L1 therapies (ClinicalTrials.gov Identifier: NCT03686202). In a more targeted approach, a consortium of human commensals from healthy human feces was assembled that in mice induce interferon-γ secreting CD8+ T cells and enhance host resistance against Listeria monocytogenes infection and the therapeutic efficiency of immune checkpoint inhibitors in syngeneic tumor models [283]. On the basis of these findings, a clinical trial has been announced using VE800, a lyophilized preparation of eight bacterial strains with immunostimulating properties together with anti-PD1 therapy in patients with advanced or metastatic cancer (https://www.vedantabio.com/news-media/press-releases/detail/2492). In a more restrictive approach, clinical trials (ClinicalTrials.gov Identifiers: NCT03775850 and NCT03595683) have been initiated in patients with different type of advanced cancer and treated with immune checkpoint inhibitors using EDP1503, an orally delivered monoclonal microbial product (a Bifidobacterium strain) that correspond to one of the bacterial species identified in clinical studies to correlate with favorable response to anti-PD1 [265].
14. Future Directions and Conclusions
The microbiome research has come a long way in a short time. However, despite the remarkable work carried out in the field, there are still huge gaps in our understanding of how the microbiota shapes our host immunity and how the immune system regulates our microbiota. These questions will require innovation of more tools and new approaches. Regardless of the pending questions, a number of ongoing clinical trials are using microbiota with the aim of improving the outcomes of various chronic diseases (https://clinicaltrials.gov).
As discussed in the preceding sections, the microbiota profoundly affects our host immune system and thus has a great potential to be used as a new biomarker. The microbial composition can mirror inflammation variations in certain disease conditions, within various stages of the same disease; hence, it has the potential to be used as a biomarker.
Using microbes as early diagnostic biomarkers will facilitate disease intervention at the onset of the disease and will help monitor the progress of any therapeutic strategy. The understanding of the microbiome’s influence on the immune responses is vital as we move forward in this era of precision medicine. It is also important to understand in more depth, the complex factors that affect the gut microbiome and how they interact, in order to develop modern strategies to manipulate the microbiome aiming to delay disease onset, disease progression, or improve therapeutic responses.
Moving forward, more efforts should be made to integrate human and microbial multiomics data (such as genetics, transcriptomics, epigenetics, proteomics, and metabolomics) using current bioinformatics and technological approaches. Special attention should be given to standardize our clinical assessment used to classify patients as well as to use universal collection and storage of biological specimens. Well-planned longitudinal studies and integrated analyses of multiple dimensions of data in diverse populations are vital to complete the association and impact on treatment outcomes.
References
- 1.Lederberg J, McCray AT (2001) Ome sweet omics: a genealogical treasury of words. Scientist 15:8 [Google Scholar]
- 2.Marchesi JR, Ravel J (2015) The vocabulary of microbiome research: a proposal. Microbiome 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flint HJ (2012) The impact of nutrition on the human microbiome. Nutr Rev 70(Suppl 1):S10–S13 [DOI] [PubMed] [Google Scholar]
- 4.Sender R, Fuchs S, Milo R (2016) Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164:337–340 [DOI] [PubMed] [Google Scholar]
- 5.Grice EA, Segre JA (2012) The human microbiome: our second genome. Annu Rev Genomics Hum Genet 13:151–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochoa-Reparaz J, Kasper LH (2016) The second brain: is the gut microbiota a link between obesity and central nervous system disorders? Curr Obes Rep 5:51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Leeuwenhoek A (1684) An abstract of a letter from Antonie van Leeuwenhoek, Sep. 12, 1683. About animals in the scrurf of the teeth. Philos Trans R Soc Lond:568–574 [Google Scholar]
- 8.Dobell C (1920) The discovery of the intestinal protozoa of man. Proc R Soc Med 13:1–15 [PMC free article] [PubMed] [Google Scholar]
- 9.Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, JE ME, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M (2009) The NIH human microbiome project. Genome Res 19:2317–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrlich SD, The MetaHIT Consortium (2011) MetaHIT: the European Union project on metagenomics of the human intestinal tract. In: Nelson KE (ed) Metagenomics of the human body. Springer, New York, pp 307–316 [Google Scholar]
- 11.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Human Microbiome Jumpstart Reference Strains C, Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, Feldgarden M, Gevers D, Haas BJ, Madupu R, Ward DV, Birren BW, Gibbs RA, Methe B, Petrosino JF, Strausberg RL, Sutton GG, White OR, Wilson RK, Durkin S, Giglio MG, Gujja S, Howarth C, Kodira CD, Kyrpides N, Mehta T, Muzny DM, Pearson M, Pepin K, Pati A, Qin X, Yandava C, Zeng Q, Zhang L, Berlin AM, Chen L, Hepburn TA, Johnson J, McCorrison J, Miller J, Minx P, Nusbaum C, Russ C, Sykes SM, Tomlinson CM et al. (2010) A catalog of reference genomes from the human microbiome. Science 328:994–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al Khodor S, Reichert B, Shatat IF (2017) The microbiome and blood pressure: can microbes regulate our blood pressure? Front Pediatr 5:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Program NCS, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA (2009) Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, Brotman RM, Davis CC, Ault K, Peralta L, Forney LJ (2011) Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A 108(Suppl 1):4680–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, Sun CL, Goltsman DS, Wong RJ, Shaw G, Stevenson DK, Holmes SP, Relman DA (2015) Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci U S A 112:11060–11065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fettweis JM, Brooks JP, Serrano MG, Sheth NU, Girerd PH, Edwards DJ, Strauss JF, The Vaginal Microbiome C, Jefferson KK, Buck GA (2014) Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology 160:2272–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cone RA (2014) Vaginal microbiota and sexually transmitted infections that may influence transmission of cell-associated HIV. J Infect Dis 210(Suppl 3):S616–S621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO (2010) Disordered microbial communities in asthmatic airways. PLoS One 5:e8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toscano M, De Grandi R, Grossi E, Drago L (2017) Role of the human breast milk-associated microbiota on the newborns’ immune system: a mini review. Front Microbiol 8:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J (2014) The placenta harbors a unique microbiome. Sci Transl Med 6:237ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Buhimschi CS, Temoin S, Bhandari V, Han YW, Buhimschi IA (2013) Comparative microbial analysis of paired amniotic fluid and cord blood from pregnancies complicated by preterm birth and early-onset neonatal sepsis. PLoS One 8:e56131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107:11971–11975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huse SM, Ye Y, Zhou Y, Fodor AA (2012) A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7:e34242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O’Connor M, Harnedy N, O’Connor K, Henry C, O’Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP, O’Toole PW (2011) Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A 108(Suppl 1):4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO (2007) Development of the human infant intestinal microbiota. PLoS Biol 5:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI (2012) Human gut microbiome viewed across age and geography. Nature 486:222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backhed F (2011) Programming of host metabolism by the gut microbiota. Ann Nutr Metab 58(Suppl 2):44–52 [DOI] [PubMed] [Google Scholar]
- 31.Salminen S, Gibson GR, McCartney AL, Isolauri E (2004) Influence of mode of delivery on gut microbiota composition in seven year old children. Gut 53:1388–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, Bjorksten B, Engstrand L, Andersson AF (2014) Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 63:559–566 [DOI] [PubMed] [Google Scholar]
- 33.Fernandez L, Langa S, Martin V, Maldonado A, Jimenez E, Martin R, Rodriguez JM (2013) The human milk microbiota: origin and potential roles in health and disease. Pharmacol Res 69:1–10 [DOI] [PubMed] [Google Scholar]
- 34.Collado MC, Delgado S, Maldonado A, Rodriguez JM (2009) Assessment of the bacterial diversity of breast milk of healthy women by quantitative real-time PCR. Lett Appl Microbiol 48:523–528 [DOI] [PubMed] [Google Scholar]
- 35.Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, Welling GW (2000) Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr 30:61–67 [DOI] [PubMed] [Google Scholar]
- 36.Fallani M, Young D, Scott J, Norin E, Amarri S, Adam R, Aguilera M, Khanna S, Gil A, Edwards CA, Dore J, Other Members of the IT (2010) Intestinal microbiota of 6-week-old infants across Europe: geographic influence beyond delivery mode, breast-feeding, and antibiotics. J Pediatr Gastroenterol Nutr 51:77–84 [DOI] [PubMed] [Google Scholar]
- 37.Rinne M, Kalliomaki M, Arvilommi H, Salminen S, Isolauri E (2005) Effect of probiotics and breastfeeding on the bifidobacterium and lactobacillus/enterococcus microbiota and humoral immune responses. J Pediatr 147:186–191 [DOI] [PubMed] [Google Scholar]
- 38.Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6:e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dethlefsen L, Relman DA (2011) Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108(Suppl 1):4554–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chernikova DA, Koestler DC, Hoen AG, Housman ML, Hibberd PL, Moore JH, Morrison HG, Sogin ML, Zain-Ul-Abideen M, Madan JC (2016) Fetal exposures and perinatal influences on the stool microbiota of premature infants. J Matern Fetal Neonatal Med 29:99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fouhy F, Guinane CM, Hussey S, Wall R, Ryan CA, Dempsey EM, Murphy B, Ross RP, Fitzgerald GF, Stanton C, Cotter PD (2012) High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob Agents Chemother 56:5811–5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrett E, Kerr C, Murphy K, O’Sullivan O, Ryan CA, Dempsey EM, Murphy BP, O’Toole PW, Cotter PD, Fitzgerald GF, Ross RP, Stanton C (2013) The individual-specific and diverse nature of the preterm infant microbiota. Arch Dis Child Fetal Neonatal Ed 98:F334–F340 [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Hoenig JD, Malin KJ, Qamar S, Petrof EO, Sun J, Antonopoulos DA, Chang EB, Claud EC (2009) 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J 3:944–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE (2014) Human genetics shape the gut microbiome. Cell 159:789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, Shilo S, Lador D, Vila AV, Zmora N, Pevsner-Fischer M, Israeli D, Kosower N, Malka G, Wolf BC, Avnit-Sagi T, Lotan-Pompan M, Weinberger A, Halpern Z, Carmi S, Fu J, Wijmenga C, Zhernakova A, Elinav E, Segal E (2018) Environment dominates over host genetics in shaping human gut microbiota. Nature 555:210–215 [DOI] [PubMed] [Google Scholar]
- 46.He Y, Wu W, Zheng HM, Li P, McDonald D, Sheng HF, Chen MX, Chen ZH, Ji GY, Zheng ZD, Mujagond P, Chen XJ, Rong ZH, Chen P, Lyu LY, Wang X, Wu CB, Yu N, Xu YJ, Yin J, Raes J, Knight R, Ma WJ, Zhou HW (2018) Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med 24:1532–1535 [DOI] [PubMed] [Google Scholar]
- 47.Gupta VK, Paul S, Dutta C (2017) Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol 8:1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sepp E, Julge K, Vasar M, Naaber P, Bjorksten B, Mikelsaar M (1997) Intestinal microflora of Estonian and Swedish infants. Acta Paediatr 86:956–961 [DOI] [PubMed] [Google Scholar]
- 49.Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA, Stobberingh EE (2006) Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118:511–521 [DOI] [PubMed] [Google Scholar]
- 50.Tun HM, Konya T, Takaro TK, Brook JR, Chari R, Field CJ, Guttman DS, Becker AB, Mandhane PJ, Turvey SE, Subbarao P, Sears MR, Scott JA, Kozyrskyj AL, Investigators CS (2017) Exposure to household furry pets influences the gut microbiota of infant at 3–4 months following various birth scenarios. Microbiome 5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guan NMLL (2016) Gut microbiome and omics: a new definition to ruminant production and health. Anim Front 6:8–12 [Google Scholar]
- 52.Van de Peer Y, Chapelle S, De Wachter R (1996) A quantitative map of nucleotide substitution rates in bacterial rRNA. Nucleic Acids Res 24:3381–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mullish BH, Osborne LS, Marchesi JR, McDonald JA (2018) The implementation of omics technologies in cancer microbiome research. Ecancermedicalscience 12:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tikhonov M, Leach RW, Wingreen NS (2015) Interpreting 16S metagenomic data without clustering to achieve sub-OTU resolution. ISME J 9:68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, Rosenbaum M, Gordon JI (2013) The long-term stability of the human gut microbiota. Science 341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eren AM, Borisy GG, Huse SM, Mark Welch JL (2014) Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci U S A 111:E2875–E2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franzen O, Hu J, Bao X, Itzkowitz SH, Peter I, Bashir A (2015) Improved OTU-picking using long-read 16S rRNA gene amplicon sequencing and generic hierarchical clustering. Microbiome 3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, Knight R (2017) Deblur rapidly resolves single-nucleotide community sequence pat terns. mSystems 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciardo DE, Schar G, Bottger EC, Altwegg M, Bosshard PP (2006) Internal transcribed spacer sequencing versus biochemical profiling for identification of medically important yeasts. J Clin Microbiol 44:77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pryce TM, Palladino S, Price DM, Gardam DJ, Campbell PB, Christiansen KJ, Murray RJ (2006) Rapid identification of fungal pathogens in BacT/ALERT, BACTEC, and BBL MGIT media using polymerase chain reaction and DNA sequencing of the internal transcribed spacer regions. Diagn Microbiol Infect Dis 54:289–297 [DOI] [PubMed] [Google Scholar]
- 64.Rittenour WR, Ciaccio CE, Barnes CS, Kashon ML, Lemons AR, Beezhold DH, Green BJ (2014) Internal transcribed spacer rRNA gene sequencing analysis of fungal diversity in Kansas City indoor environments. Environ Sci Process Impacts 16:33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shittu OB, Adelaja OM, Obuotor TM, Sam-Wobo SO, Adenaike AS (2016) PCR-Internal Transcribed Spacer (ITS) genes sequencing and phylogenetic analysis of clinical and environmental Aspergillus species associated with HIV-TB co infected patients in a hospital in Abeokuta, southwestern Nigeria. Afr Health Sci 16:141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Banos S, Lentendu G, Kopf A, Wubet T, Glockner FO, Reich M (2018) A comprehensive fungi-specific 18S rRNA gene sequence primer toolkit suited for diverse research issues and sequencing platforms. BMC Microbiol 18:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper MK, Phalen DN, Donahoe SL, Rose K, Slapeta J (2016) The utility of diversity profiling using Illumina 18S rRNA gene amplicon deep sequencing to detect and discriminate toxoplasma gondii among the cyst-forming coccidia. Vet Parasitol 216:38–45 [DOI] [PubMed] [Google Scholar]
- 68.De Filippis F, La Storia A, Blaiotta G (2017) Monitoring the mycobiota during Greco di Tufo and Aglianico wine fermentation by 18S rRNA gene sequencing. Food Microbiol 63:117–122 [DOI] [PubMed] [Google Scholar]
- 69.Dorigo U, Berard A, Humbert JF (2002) Comparison of eukaryotic phytobenthic community composition in a polluted river by partial 18S rRNA gene cloning and sequencing. Microb Ecol 44:372–380 [DOI] [PubMed] [Google Scholar]
- 70.Hancock JM, Vogler AP (1998) Modelling the secondary structures of slippage-prone hypervariable RNA regions: the example of the tiger beetle 18S rRNA variable region V4. Nucleic Acids Res 26:1689–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hwang UW, Ree HI, Kim W (2000) Evolution of hypervariable regions, V4 and V7, of insect 18S rRNA and their phylogenetic implications. Zool Sci 17:111–121 [DOI] [PubMed] [Google Scholar]
- 72.Rosser TG, Griffin MJ, Quiniou SM, Khoo LH, Pote LM (2014) 18S rRNA gene sequencing identifies a novel species of Henneguya parasitizing the gills of the channel catfish (Ictaluridae). Parasitol Res 113:4651–4658 [DOI] [PubMed] [Google Scholar]
- 73.Saki J, Foroutan-Rad M, Asadpouri R (2016) Molecular characterization of cryptosporidium spp. in wild rodents of Southwestern Iran using 18s rRNA gene nested-PCR-RFLP and sequencing techniques. J Trop Med 2016:6834206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanabe AS, Nagai S, Hida K, Yasuike M, Fujiwara A, Nakamura Y, Takano Y, Katakura S (2016) Comparative study of the validity of three regions of the 18S-rRNA gene for massively parallel sequencing-based monitoring of the planktonic eukaryote community. Mol Ecol Resour 16:402–414 [DOI] [PubMed] [Google Scholar]
- 75.Clooney AG, Fouhy F, Sleator RD, A OD, Stanton C, Cotter PD, Claesson MJ (2016) Comparing apples and oranges?: next generation sequencing and its impact on Microbiome analysis. PLoS One 11:e0148028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methe B, Schloss PD, Gevers D, Mitreva M, Huttenhower C (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alves JM, Buck GA (2007) Automated system for gene annotation and metabolic pathway reconstruction using general sequence databases. Chem Biodivers 4: 2593–2602 [DOI] [PubMed] [Google Scholar]
- 78.Wood DE, Salzberg SL (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, Gough J, Haft DR, Huang H, Letunic I, Lopez R, Luciani A, Madeira F, Marchler-Bauer A, Mi H, Natale DA, Necci M, Nuka G, Orengo C, Pandurangan AP, Paysan-Lafosse T, Pesseat S, Potter SC, Qureshi MA, Rawlings ND, Redaschi N, Richardson LJ, Rivoire C, Salazar GA, Sangrador-Vegas A, Sigrist CJA, Sillitoe I, Sutton GG, Thanki N, Thomas PD, Tosatto SCE, Yong SY, Finn RD (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47:D351–D360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bikel S, Valdez-Lara A, Cornejo-Granados F, Rico K, Canizales-Quinteros S, Soberon X, Del Pozo-Yauner L, Ochoa-Leyva A (2015) Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput Struct Biotechnol J 13:390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bashiardes S, Zilberman-Schapira G, Elinav E (2016) Use of Metatranscriptomics in Microbiome research. Bioinform Biol Insights 10:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marco-Sola S, Sammeth M, Guigo R, Ribeca P (2012) The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods 9:1185–1188 [DOI] [PubMed] [Google Scholar]
- 83.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 7:562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Glass EM, Wilkening J, Wilke A, Antonopoulos D, Meyer F (2010) Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb Protoc 2010:pdb prot5368. [DOI] [PubMed] [Google Scholar]
- 85.Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, Lindon JC, Nicholson JK (2007) Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc 2:2692–2703 [DOI] [PubMed] [Google Scholar]
- 88.Gratton J, Phetcharaburanin J, Mullish BH, Williams HR, Thursz M, Nicholson JK, Holmes E, Marchesi JR, Li JV (2016) Optimized sample handling strategy for metabolic profiling of human feces. Anal Chem 88:4661–4668 [DOI] [PubMed] [Google Scholar]
- 89.Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG, Fischbach MA (2014) Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158:412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T (2013) antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–W212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA (2014) A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158:1402–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Donia MS, Fischbach MA (2015) HUMAN MICROBIOTA. Small molecules from the human microbiota. Science 349:1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ (2002) The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 217:133–139 [DOI] [PubMed] [Google Scholar]
- 94.Degnan PH, Taga ME, Goodman AL (2014) Vitamin B12 as a modulator of gut microbial ecology. Cell Metab 20:769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beulens JW, Booth SL, van den Heuvel EG, Stoecklin E, Baka A, Vermeer C (2013) The role of menaquinones (vitamin K(2)) in human health. Br J Nutr 110:1357–1368 [DOI] [PubMed] [Google Scholar]
- 96.Biesalski HK (2016) Nutrition meets the microbiome: micronutrients and the microbiota. Ann N Y Acad Sci 1372:53–64 [DOI] [PubMed] [Google Scholar]
- 97.Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Backhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, Backhed F, Isolauri E, Salminen S, Ley RE (2012) Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150:470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ley RE, Peterson DA, Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837–848 [DOI] [PubMed] [Google Scholar]
- 99.Martin FP, Sprenger N, Montoliu I, Rezzi S, Kochhar S, Nicholson JK (2010) Dietary modulation of gut functional ecology studied by fecal metabonomics. J Proteome Res 9:5284–5295 [DOI] [PubMed] [Google Scholar]
- 100.Marcobal A, Yusufaly T, Higginbottom S, Snyder M, Sonnenburg JL, Mias GI (2015) Metabolome progression during early gut microbial colonization of gnotobiotic mice. Sci Rep 5:11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK (2011) Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol 19:349–359 [DOI] [PubMed] [Google Scholar]
- 102.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI (2011) Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 108:6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kumar M, Mathur T, Joshi V, Upadhyay DJ, Inoue SI, Masuda N (2018) Effect of DS-2969b, a novel GyrB inhibitor, on rat and monkey intestinal microbiota. Anaerobe 51:120–123 [DOI] [PubMed] [Google Scholar]
- 104.Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takiishi T, Fenero CIM, Camara NOS (2017) Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers 5:e1373208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ouellette AJ (2011) Paneth cell alpha-defensins in enteric innate immunity. Cell Mol Life Sci 68:2215–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Muniz LR, Knosp C, Yeretssian G (2012) Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol 3:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mantis NJ, Forbes SJ (2010) Secretory IgA: arresting microbial pathogens at epithelial borders. Immunol Investig 39:383–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D (2015) Role of the normal gut microbiota. World J Gastroenterol 21:8787–8803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gill N, Wlodarska M, Finlay BB (2011) Roadblocks in the gut: barriers to enteric infection. Cell Microbiol 13:660–669 [DOI] [PubMed] [Google Scholar]
- 111.Sun M, He C, Cong Y, Liu Z (2015) Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol 8:969–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Akira S, Hemmi H (2003) Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85:85–95 [DOI] [PubMed] [Google Scholar]
- 113.Medzhitov R, Janeway C Jr (2000) Innate immune recognition: mechanisms and pathways. Immunol Rev 173:89–97 [DOI] [PubMed] [Google Scholar]
- 114.Harris G, KuoLee R, Chen W (2006) Role of toll-like receptors in health and diseases of gastrointestinal tract. World J Gastroenterol 12:2149–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22:240–273, Table of Contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Newman MA, Sundelin T, Nielsen JT, Erbs G (2013) MAMP (microbe-associated molecular pattern) triggered immunity in plants. Front Plant Sci 4:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pandey UB, Nichols CD (2011) Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev 63:411–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dicksved J, Schreiber O, Willing B, Petersson J, Rang S, Phillipson M, Holm L, Roos S (2012) Lactobacillus reuteri maintains a functional mucosal barrier during DSS treatment despite mucus layer dysfunction. PLoS One 7:e46399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lin PW, Nasr TR, Berardinelli AJ, Kumar A, Neish AS (2008) The probiotic lactobacillus GG may augment intestinal host defense by regulating apoptosis and promoting cytoprotective responses in the developing murine gut. Pediatr Res 64:511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fukui H (2016) Increased intestinal permeability and decreased barrier function: does it really influence the risk of inflammation? Inflamm Intest Dis 1:135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ren K, Dubner R (2010) Interactions between the immune and nervous systems in pain. Nat Med 16:1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gareau MG, Silva MA, Perdue MH (2008) Pathophysiological mechanisms of stress-induced intestinal damage. Curr Mol Med 8:274–281 [DOI] [PubMed] [Google Scholar]
- 123.Maes M, Twisk FN, Kubera M, Ringel K, Leunis JC, Geffard M (2012) Increased IgA responses to the LPS of commensal bacteria is associated with inflammation and activation of cell-mediated immunity in chronic fatigue syndrome. J Affect Disord 136:909–917 [DOI] [PubMed] [Google Scholar]
- 124.Thomas CM, Hong T, van Pijkeren JP, Hemarajata P, Trinh DV, Hu W, Britton RA, Kalkum M, Versalovic J (2012) Histamine derived from probiotic lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One 7:e31951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, Dougherty DM (2009) L-tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int J Tryptophan Res 2:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B (2007) Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord 98:143–151 [DOI] [PubMed] [Google Scholar]
- 127.Desbonnet L, Garrett L, Clarke G, Bienenstock J, Dinan TG (2008) The probiotic Bifidobacteria infantis: an assessment of potential antidepressant properties in the rat. J Psychiatr Res 43:164–174 [DOI] [PubMed] [Google Scholar]
- 128.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39:372–385 [DOI] [PubMed] [Google Scholar]
- 129.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kebir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Prat A, Antel J, Quintana FJ (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 22:586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, Wild B, Camargo SM, Singer D, Richter A, Kuba K, Fukamizu A, Schreiber S, Clevers H, Verrey F, Rosenstiel P, Penninger JM (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487:477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S (2012) Host-gut microbiota metabolic interactions. Science 336:1262–1267 [DOI] [PubMed] [Google Scholar]
- 132.Pisetsky DS (1996) Immune activation by bacterial DNA: a new genetic code. Immunity 5:303–310 [DOI] [PubMed] [Google Scholar]
- 133.Bird AP (1987) CpG islands as gene markers in the vertebrate nucleus. Trends Genet:342–346 [Google Scholar]
- 134.Kumar M, Khan FG, Sharma S, Kumar R, Faujdar J, Sharma R, Chauhan DS, Singh R, Magotra SK, Khan IA (2011) Identification of mycobacterium tuberculosis genes preferentially expressed during human infection. Microb Pathog 50:31–38 [DOI] [PubMed] [Google Scholar]
- 135.Hergersberg M (1991) Biological aspects of cytosine methylation in eukaryotic cells. Experientia 47:1171–1185 [DOI] [PubMed] [Google Scholar]
- 136.Krieg AM (2006) Therapeutic potential of toll-like receptor 9 activation. Nat Rev Drug Discov 5:471–484 [DOI] [PubMed] [Google Scholar]
- 137.McClure R, Massari P (2014) TLR-dependent Human mucosal epithelial cell responses to microbial pathogens. Front Immunol 5:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Messina JP, Gilkeson GS, Pisetsky DS (1991) Stimulation of in vitro murine lymphocyte proliferation by bacterial DNA. J Immunol 147:1759–1764 [PubMed] [Google Scholar]
- 139.Ballas ZK, Rasmussen WL, Krieg AM (1996) Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J Immunol 157:1840–1845 [PubMed] [Google Scholar]
- 140.O’Hara JR, Feener TD, Fischer CD, Buret AG (2012) Campylobacter jejuni disrupts protective toll-like receptor 9 signaling in colonic epithelial cells and increases the severity of dextran sulfate sodium-induced colitis in mice. Infect Immun 80:1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hooper LV, Littman DR, Macpherson AJ (2012) Interactions between the microbiota and the immune system. Science 336:1268–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thorburn AN, Macia L, Mackay CR (2014) Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40:833–842 [DOI] [PubMed] [Google Scholar]
- 143.Soto M, Herzog C, Pacheco JA, Fujisaka S, Bullock K, Clish CB, Kahn CR (2018) Gut microbiota modulate neurobehavior through changes in brain insulin sensitivity and metabolism. Mol Psychiatry 23:2287–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.van de Wouw M, Schellekens H, Dinan TG, Cryan JF (2017) Microbiota-gut-brain Axis: modulator of host metabolism and appetite. J Nutr 147:727–745 [DOI] [PubMed] [Google Scholar]
- 145.Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH, Zhou Z, Bao Y, Jia W, Nicholson JK, Jia W (2011) The footprints of gut microbial-mammalian co-metabolism. J Proteome Res 10:5512–5522 [DOI] [PubMed] [Google Scholar]
- 146.Gaudet RG, Sintsova A, Buckwalter CM, Leung N, Cochrane A, Li J, Cox AD, Moffat J, Gray-Owen SD (2015) INNATE IMMUNITY. Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 348:1251–1255 [DOI] [PubMed] [Google Scholar]
- 147.Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, Grigg ME, Kastenmayer R, Schwartzberg PL, Belkaid Y (2011) Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34:435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Lar murier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay JM, Langella P, Xavier RJ, Sokol H (2016) CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 22:598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ (2011) The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13:517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504:451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sanderson IR (2004) Short chain fatty acid regulation of signaling genes expressed by the intestinal epithelium. J Nutr 134:2450S–2454S [DOI] [PubMed] [Google Scholar]
- 152.Gaudier E, Jarry A, Blottiere HM, de Coppet P, Buisine MP, Aubert JP, Laboisse C, Cherbut C, Hoebler C (2004) Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am J Physiol Gastrointest Liver Physiol 287:G1168–G1174 [DOI] [PubMed] [Google Scholar]
- 153.Wrzosek L, Miquel S, Noordine ML, Bouet S, Joncquel Chevalier-Curt M, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C, Langella P, Thomas M (2013) Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol 11:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, Pevsner-Fischer M, Shapiro H, Christ A, Harmelin A, Halpern Z, Latz E, Flavell RA, Amit I, Segal E, Elinav E (2015) Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163:1428–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Blacher E, Levy M, Tatirovsky E, Elinav E (2017) Microbiome-modulated metabolites at the Interface of host immunity. J Immunol 198:572–580 [DOI] [PubMed] [Google Scholar]
- 156.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, Ganapathy V (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40:128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H (2011) Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547 [DOI] [PubMed] [Google Scholar]
- 158.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A (2011) Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108:5354–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schluter J, Foster KR (2012) The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol 10:e1001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kostic AD, Howitt MR, Garrett WS (2013) Exploring host-microbiota interactions in animal models and humans. Genes Dev 27:701–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Calatayud M, Dezutter O, Hernandez-Sanabria E, Hidalgo-Martinez S, Meysman FJR, Van de Wiele T (2019) Development of a host-microbiome model of the small intestine. FASEB J 33(3):3985–3996. 10.1096/fj.201801414R:fj201801414R [DOI] [PubMed] [Google Scholar]
- 162.Wang B, Li J, Li Q, Zhang H, Li N (2009) Isolation of adhesive strains and evaluation of the colonization and immune response by lactobacillus plantarum L2 in the rat gastrointestinal tract. Int J Food Microbiol 132:59–66 [DOI] [PubMed] [Google Scholar]
- 163.Yadav M, Verma MK, Chauhan NS (2018) A review of metabolic potential of human gut microbiome in human nutrition. Arch Microbiol 200:203–217 [DOI] [PubMed] [Google Scholar]
- 164.Lea T (2015) Caco-2 cell line. In: Verhoeckx K, Cotter P, Lopez-Exposito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, Wichers H (eds) The impact of food bioactives on health: in vitro and ex vivo models. Springer, Cham (CH), pp 103–111. 10.1007/978-3-319-16104-4_10 [DOI] [PubMed] [Google Scholar]
- 165.Bahrami B, Child MW, Macfarlane S, Macfarlane GT (2011) Adherence and cytokine induction in Caco-2 cells by bacterial populations from a three-stage continuous-culture model of the large intestine. Appl Environ Microbiol 77:2934–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zenhom M, Hyder A, de Vrese M, Heller KJ, Roeder T, Schrezenmeir J (2011) Prebiotic oligosaccharides reduce proinflammatory cytokines in intestinal Caco-2 cells via activation of PPARgamma and peptidoglycan recognition protein 3. J Nutr 141:971–977 [DOI] [PubMed] [Google Scholar]
- 167.Fogh J, Fogh JM, Orfeo T (1977) One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 59:221–226 [DOI] [PubMed] [Google Scholar]
- 168.Pearce SC, Al-Jawadi A, Kishida K, Yu S, Hu M, Fritzky LF, Edelblum KL, Gao N, Ferraris RP (2018) Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol 16:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pham VT, Seifert N, Richard N, Raederstorff D, Steinert R, Prudence K, Mohajeri MH (2018) The effects of fermentation products of prebiotic fibres on gut barrier and immune functions in vitro. PeerJ 6:e5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nepelska M, Cultrone A, Beguet-Crespel F, Le Roux K, Dore J, Arulampalam V, Blottiere HM (2012) Butyrate produced by commensal bacteria potentiates phorbol esters induced AP-1 response in human intestinal epithelial cells. PLoS One 7:e52869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Norouzi Z, Salimi A, Halabian R, Fahimi H (2018) Nisin, a potent bacteriocin and antibacterial peptide, attenuates expression of metastatic genes in colorectal cancer cell lines. Microb Pathog 123:183–189 [DOI] [PubMed] [Google Scholar]
- 172.Brosnahan AJ, Brown DR (2012) Porcine IPEC-J2 intestinal epithelial cells in microbiological investigations. Vet Microbiol 156:229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vergauwen H (2015) The IPEC-J2 cell line. In: Verhoeckx K, Cotter P, Lopez-Exposito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, Wichers H (eds) The impact of food bioactives on health: in vitro and ex vivo models. Springer, Cham (CH), pp 125–134. 10.1007/978-3-319-16104-4_12 [DOI] [PubMed] [Google Scholar]
- 174.Yan H, Ajuwon KM (2017) Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS One 12:e0179586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu HY, Roos S, Jonsson H, Ahl D, Dicksved J, Lindberg JE, Lundh T (2015) Effects of lactobacillus johnsonii and lactobacillus reuteri on gut barrier function and heat shock proteins in intestinal porcine epithelial cells. Phys Rep 3(4):e12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cinova J, De Palma G, Stepankova R, Kofronova O, Kverka M, Sanz Y, Tuckova L (2011) Role of intestinal bacteria in gliadin-induced changes in intestinal mucosa: study in germ-free rats. PLoS One 6:e16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liu Y, Fatheree NY, Mangalat N, Rhoads JM (2010) Human-derived probiotic lactobacillus reuteri strains differentially reduce intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 299:G1087–G1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ren H, Musch MW, Kojima K, Boone D, Ma A, Chang EB (2001) Short-chain fatty acids induce intestinal epithelial heat shock protein 25 expression in rats and IEC 18 cells. Gastroenterology 121:631–639 [DOI] [PubMed] [Google Scholar]
- 179.Lin R, Jiang Y, Zhao XY, Guan Y, Qian W, Fu XC, Ren HY, Hou XH (2014) Four types of Bifidobacteria trigger autophagy response in intestinal epithelial cells. J Dig Dis 15:597–605 [DOI] [PubMed] [Google Scholar]
- 180.Clift MJ, Gehr P, Rothen-Rutishauser B (2011) Nanotoxicology: a perspective and discussion of whether or not in vitro testing is a valid alternative. Arch Toxicol 85:723–731 [DOI] [PubMed] [Google Scholar]
- 181.Bothe MK, Maathuis AJH, Bellmann S, van der Vossen J, Berressem D, Koehler A, Schwejda-Guettes S, Gaigg B, Kuchinka-Koch A, Stover JF (2017) Dose-dependent prebiotic effect of lactulose in a computer-controlled in vitro model of the human large intestine. Nutrients 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Isenmann R, Schwarz M, Rozdzinski E, Marre R, Beger HG (2000) Aggregation substance promotes colonic mucosal invasion of Enterococcus faecalis in an ex vivo model. J Surg Res 89:132–138 [DOI] [PubMed] [Google Scholar]
- 183.Chen HQ, Yang J, Zhang M, Zhou YK, Shen TY, Chu ZX, Zhang M, Hang XM, Jiang YQ, Qin HL (2010) Lactobacillus plantarum ameliorates colonic epithelial barrier dysfunction by modulating the apical junctional complex and PepT1 in IL-10 knockout mice. Am J Physiol Gastrointest Liver Physiol 299:G1287–G1297 [DOI] [PubMed] [Google Scholar]
- 184.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459:262–265 [DOI] [PubMed] [Google Scholar]
- 185.Workman MJ, Gleeson JP, Troisi EJ, Estrada HQ, Kerns SJ, Hinojosa CD, Hamilton GA, Targan SR, Svendsen CN, Barrett RJ (2018) Enhanced utilization of induced pluripotent stem cell-derived Human intestinal Organoids using microengineered chips. Cell Mol Gastroenterol Hepatol 5:669–677.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pearce SC, Coia HG, Karl JP, PantojaFeliciano IG, Zachos NC, Racicot K (2018) Intestinal in vitro and ex vivo models to study host-microbiome interactions and acute stressors. Front Physiol 9:1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rajan A, Vela L, Zeng XL, Yu X, Shroyer N, Blutt SE, Poole NM, Carlin LG, Nataro JP, Estes MK, Okhuysen PC, Maresso AW (2018) Novel segment- and host-specific patterns of enteroaggregative escherichia coli adherence to Human intestinal Enteroids. MBio 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, Zachos NC (2017) A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep 7:45270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zomer-van Ommen DD, Pukin AV, Fu O, Quarles van Ufford LH, Janssens HM, Beekman JM, Pieters RJ (2016) Functional characterization of cholera toxin inhibitors using Human intestinal organoids. J Med Chem 59:6968–6972 [DOI] [PubMed] [Google Scholar]
- 190.Aoki-Yoshida A, Saito S, Fukiya S, Aoki R, Takayama Y, Suzuki C, Sonoyama K (2016) Lactobacillus rhamnosus GG increases toll-like receptor 3 gene expression in murine small intestine ex vivo and in vivo. Benefic Microbes 7:421–429 [DOI] [PubMed] [Google Scholar]
- 191.Pierzchalska M, Panek M, Czyrnek M, Gielicz A, Mickowska B, Grabacka M (2017) Probiotic lactobacillus acidophilus bacteria or synthetic TLR2 agonist boost the growth of chicken embryo intestinal organoids in cultures comprising epithelial cells and myofibroblasts. Comp Immunol Microbiol Infect Dis 53:7–18 [DOI] [PubMed] [Google Scholar]
- 192.Engevik MA, Aihara E, Montrose MH, Shull GE, Hassett DJ, Worrell RT (2013) Loss of NHE3 alters gut microbiota composition and influences Bacteroides thetaiotaomicron growth. Am J Physiol Gastrointest Liver Physiol 305:G697–G711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.O’Callaghan D, Vergunst A (2010) Non-mammalian animal models to study infectious disease: worms or fly fishing? Curr Opin Microbiol 13:79–85 [DOI] [PubMed] [Google Scholar]
- 194.Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A (2011) Bacterial communities of diverse drosophila species: ecological context of a host-microbe model system. PLoS Genet 7:e1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ (2011) Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334:670–674 [DOI] [PubMed] [Google Scholar]
- 196.Iatsenko I, Boquete JP, Lemaitre B (2018) Microbiota-derived lactate activates production of reactive oxygen species by the intestinal NADPH oxidase Nox and shortens drosophila lifespan. Immunity 49:929–942. e5 [DOI] [PubMed] [Google Scholar]
- 197.Trinder M, Daisley BA, Dube JS, Reid G (2017) Drosophila melanogaster as a high-throughput model for host-microbiota interactions. Front Microbiol 8:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF (2011) Evidence for a core gut microbiota in the zebrafish. ISME J 5:1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K (2006) Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297:374–386 [DOI] [PubMed] [Google Scholar]
- 200.Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Ostergren-Lunden G, Petersen LC, Ruf W, Backhed F (2012) Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 483:627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, Liu J, Houpt E, Li JV, Holmes E, Nicholson J, Knights D, Ursell LK, Knight R, Gordon JI (2013) Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339:548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A 108:3047–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Schrumpf E, Kummen M, Valestrand L, Greiner TU, Holm K, Arulampalam V, Reims HM, Baines J, Backhed F, Karlsen TH, Blumberg RS, Hov JR, Melum E (2017) The gut microbiota contributes to a mouse model of spontaneous bile duct inflammation. J Hepatol 66:382–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Spor A, Koren O, Ley R (2011) Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9:279–290 [DOI] [PubMed] [Google Scholar]
- 205.Gordon HA, Pesti L (1971) The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol Rev 35:390–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lee A, Gordon J, Lee CJ, Dubos R (1971) The mouse intestinal microflora with emphasis on the strict anaerobes. J Exp Med 133:339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang B, Yao M, Lv L, Ling Z, Li L (2017) The human microbiota in health and disease. Engineering 3:71–82 [Google Scholar]
- 208.Al Khodor S, Shatat IF (2017) Gut microbiome and kidney disease: a bidirectional relationship. Pediatr Nephrol 32:921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E (2017) Dysbiosis and the immune system. Nat Rev Immunol 17:219–232 [DOI] [PubMed] [Google Scholar]
- 210.Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, Rawls JF, Abdo Z, Fodor AA, Keku TO (2012) Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J 6:1858–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA (2012) Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22:299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD (2013) The gut microbiome modulates colon tumorigenesis. MBio 4:e00692–e00613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, Su L, Li X, Li X, Li J, Xiao L, Huber-Schonauer U, Niederseer D, Xu X, Al-Aama JY, Yang H, Wang J, Kristiansen K, Arumugam M, Tilg H, Datz C, Wang J (2015) Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun 6:6528. [DOI] [PubMed] [Google Scholar]
- 214.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, El-Omar EM, Brenner D, Fuchs CS, Meyerson M, Garrett WS (2013) Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14:207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, Trinchieri G (2017) Microbes and cancer. Annu Rev Immunol 35:199–228 [DOI] [PubMed] [Google Scholar]
- 217.Anonymous (1994) Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum 61:1–241 [PMC free article] [PubMed] [Google Scholar]
- 218.Sears CL, Garrett WS (2014) Microbes, microbiota, and colon cancer. Cell Host Microbe 15:317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Belkaid Y, Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell 157:121–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, Chen Y, Chen H, Hong J, Zou W, Fang JY (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170:548–563.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, Shussman N, Almogy G, Cuapio A, Hofer E, Mevorach D, Tabib A, Ortenberg R, Markel G, Miklic K, Jonjic S, Brennan CA, Garrett WS, Bachrach G, Mandelboim O (2015) Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42:344–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M, Stein E, Vadivelu J, Roslani AC, Malik AA, Wanyiri JW, Goh KL, Thevambiga I, Fu K, Wan F, Llosa N, Housseau F, Romans K, Wu X, McAllister FM, Wu S, Vogelstein B, Kinzler KW, Pardoll DM, Sears CL (2014) Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A 111:18321–18326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, Ivanisevic J, Cho K, Wick EC, Hechenbleikner EM, Uritboonthai W, Goetz L, Casero RA Jr, Pardoll DM, White JR, Patti GJ, Sears CL, Siuzdak G (2015) Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab 21:891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T, Chipashvili O, Hagan T, Walker M, Ramachandran A, Diosdado B, Serna G, Mulet N, Landolfi S, Ramon YCS, Fasani R, Aguirre AJ, Ng K, Elez E, Ogino S, Tabernero J, Fuchs CS, Hahn WC, Nuciforo P, Meyerson M (2017) Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358:1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C (2012) Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338:120–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Uittamo J, Siikala E, Kaihovaara P, Salaspuro M, Rautemaa R (2009) Chronic candidosis and oral cancer in APECED-patients: production of carcinogenic acetaldehyde from glucose and ethanol by Candida albicans. Int J Cancer 124:754–756 [DOI] [PubMed] [Google Scholar]
- 227.Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, Artis D, Garrett WS (2016) Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351:1329–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Plottel CS, Blaser MJ (2011) Microbiome and malignancy. Cell Host Microbe 10:324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Nowak A, Czyzowska A, Huben K, Sojka M, Kuberski S, Otlewska A, Slizewska K (2016) Prebiotics and age, but not probiotics affect the transformation of 2-amino-3-methyl-3H-imidazo[4,5-f]quinoline (IQ) by fecal microbiota - an in vitro study. Anaerobe 39:124–135 [DOI] [PubMed] [Google Scholar]
- 230.Poutahidis T, Cappelle K, Levkovich T, Lee CW, Doulberis M, Ge Z, Fox JG, Horwitz BH, Erdman SE (2013) Pathogenic intestinal bacteria enhance prostate cancer development via systemic activation of immune cells in mice. PLoS One 8:e73933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE (2006) Innate immune inflammatory response against enteric bacteria helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res 66:7395–7400 [DOI] [PubMed] [Google Scholar]
- 232.Yamamoto ML, Maier I, Dang AT, Berry D, Liu J, Ruegger PM, Yang JI, Soto PA, Presley LL, Reliene R, Westbrook AM, Wei B, Loy A, Chang C, Braun J, Borneman J, Schiestl RH (2013) Intestinal bacteria modify lymphoma incidence and latency by affecting systemic inflammatory state, oxidative stress, and leukocyte genotoxicity. Cancer Res 73:4222–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, Thaiss CA, Reuben A, Livny J, Avraham R, Frederick DT, Ligorio M, Chatman K, Johnston SE, Mosher CM, Brandis AM, Fuks G, Gurbatri C, Gopalakrishnan V, Kim M, Hurd MW, Katz M, Fleming J, Maitra A, Smith DA, Skalak M, Bu J, Michaud M, Trauger SA, Barshack I, Golan T, Sandbank J, Flaherty KT, Mandinova A, Garrett WS, Thayer SP, Ferrone CR, Huttenhower C, Bhatia SN, Gevers D, Wargo JA, Golub TR, Straussman R (2017) Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357:1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, Werba G, Zhang K, Guo Y, Li Q, Akkad N, Lall S, Wadowski B, Gutierrez J, Kochen Rossi JA, Herzog JW, Diskin B, Torres-Hernandez A, Leinwand J, Wang W, Taunk PS, Savadkar S, Janal M, Saxena A, Li X, Cohen D, Sartor RB, Saxena D, Miller G (2018) The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 8:403–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Choy ATF, Carnevale I, Coppola S, Meijer LL, Kazemier G, Zaura E, Deng D, Giovannetti E (2018) The microbiome of pancreatic cancer: from molecular diagnostics to new therapeutic approaches to overcome chemoresistance caused by metabolic inactivation of gemcitabine. Expert Rev Mol Diagn. 10.1080/14737159 [DOI] [PubMed] [Google Scholar]
- 236.Greathouse KL, White JR, Vargas AJ, Bliskovsky VV, Beck JA, von Muhlinen N, Polley EC, Bowman ED, Khan MA, Robles AI, Cooks T, Ryan BM, Padgett N, Dzutsev AH, Trinchieri G, Pineda MA, Bilke S, Meltzer PS, Hokenstad AN, Stickrod TM, Walther-Antonio MR, Earl JP, Mell JC, Krol JE, Balashov SV, Bhat AS, Ehrlich GD, Valm A, Deming C, Conlan S, Oh J, Segre JA, Harris CC (2018) Interaction between the microbiome and TP53 in human lung cancer. Genome Biol 19:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Coley WB (1893) Treatment of malignant tumors by repeated inoculation of erysipelas, with a report of 10 cases. Am J Med Sci 105:487–564 [PubMed] [Google Scholar]
- 238.Tang DH, Chang SS (2015) Management of carcinoma in situ of the bladder: best practice and recent developments. Ther Adv Urol 7:351–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Forbes NS (2010) Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer 10:785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bettegowda C, Dang LH, Abrams R, Huso DL, Dillehay L, Cheong I, Agrawal N, Borzillary S, McCaffery JM, Watson EL, Lin KS, Bunz F, Baidoo K, Pomper MG, Kinzler KW, Vogelstein B, Zhou S (2003) Overcoming the hypoxic barrier to radiation therapy with anaerobic bacteria. Proc Natl Acad Sci U S A 100:15083–15088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dang LH, Bettegowda C, Huso DL, Kinzler KW, Vogelstein B (2001) Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A 98:15155–15160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mlynarczyk GS, Berg CA, Withrock IC, Fick ME, Anderson SJ, Laboissonniere LA, Jefferson MA, Brewer MT, Stock ML, Lange JK, Luna KC, Acharya S, Kanuri S, Sharma S, Kondru NC, McCormack GR, Carlson SA (2014) Salmonella as a biological “Trojan horse” for neoplasia: future possibilities including brain cancer. Med Hypotheses 83:343–345 [DOI] [PubMed] [Google Scholar]
- 243.Roy S, Trinchieri G (2017) Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer 17:271–285 [DOI] [PubMed] [Google Scholar]
- 244.Li H, Jia W (2013) Cometabolism of microbes and host: implications for drug metabolism and drug-induced toxicity. Clin Pharmacol Ther 94:574–581 [DOI] [PubMed] [Google Scholar]
- 245.Wilson ID, Nicholson JK (2016) Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 10.1016/j.trsl.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Maurice CF, Haiser HJ, Turnbaugh PJ (2013) Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Haiser HJ, Turnbaugh PJ (2013) Developing a metagenomic view of xenobiotic metabolism. Pharmacol Res 69:21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Bjorkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S (2009) Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One 4:e6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Selwyn FP, Cheng SL, Klaassen CD, Cui JY (2016) Regulation of hepatic drug-metabolizing enzymes in germ-free mice by conventionalization and probiotics. Drug Metab Dispos 44:262–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Selwyn FP, Cheng SL, Bammler TK, Prasad B, Vrana M, Klaassen C, Cui JY (2015) Developmental regulation of drug-processing genes in livers of germ-free mice. Toxicol Sci 147:84–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Yip LY, Chan EC (2015) Investigation of host-gut microbiota modulation of therapeutic outcome. Drug Metab Dispos 43:1619–1631 [DOI] [PubMed] [Google Scholar]
- 252.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ (2016) The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 14:273–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lehouritis P, Cummins J, Stanton M, Murphy CT, McCarthy FO, Reid G, Urbaniak C, Byrne WL, Tangney M (2015) Local bacteria affect the efficacy of chemotherapeutic drugs. Sci Rep 5:14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai RM, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, Trinchieri G, Goldszmid RS (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342:967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gui QF, Lu HF, Zhang CX, Xu ZR, Yang YH (2015) Well-balanced commensal microbiota contributes to anti-cancer response in a lung cancer mouse model. Genet Mol Res 14:5642–5651 [DOI] [PubMed] [Google Scholar]
- 256.Wang Y, Luo X, Pan H, Huang W, Wang X, Wen H, Shen K, Jin B (2015) Pharmacological inhibition of NADPH oxidase protects against cisplatin induced nephrotoxicity in mice by two step mechanism. Food Chem Toxicol 83:251–260 [DOI] [PubMed] [Google Scholar]
- 257.Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther PL, Eberl G, Berard M, Ecobichon C, Clermont D, Bizet C, Gaboriau-Routhiau V, Cerf-Bensussan N, Opolon P, Yessaad N, Vivier E, Ryffel B, Elson CO, Dore J, Kroemer G, Lepage P, Boneca IG, Ghiringhelli F, Zitvogel L (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342:971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Berard M, Nigou J, Opolon P, Eggermont A, Woerther PL, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350:1079–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350:1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, Palmer DC, Boni A, Muranski P, Yu Z, Gattinoni L, Antony PA, Rosenberg SA, Restifo NP (2007) Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest 117:2197–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP (2005) Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res 65:3437–3446 [DOI] [PubMed] [Google Scholar]
- 262.Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, Diefenbach A (2012) Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37:171–186 [DOI] [PubMed] [Google Scholar]
- 263.Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ (2016) Trained immunity: a program of innate immune memory in health and disease. Science 352:aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, Petaccia de Macedo M, Cotechini T, Kumar T, Chen WS, Reddy SM, Sloane RS, Galloway-Pena J, Jiang H, Chen PL, Shpall EJ, Rezvani K, Alousi AM, Chemaly RF, Shelburne S, Vence LM, Okhuysen PC, Jensen VB, Swennes AG, McAllister F, Sanchez EMR, Zhang Y, Le Chatelier E, Zitvogel L, Pons N, Austin-Breneman JL, Haydu LE, Burton EM, Gardner JM, Sirmans E, Hu J, Lazar AJ, Tsujikawa T, Diab A, Tawbi H, Glitza IC et al. (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 539:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359:104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359:91–97 [DOI] [PubMed] [Google Scholar]
- 267.Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, Jiang ZD, Abu-Sbeih H, Sanchez CA, Chang CC, Parra ER, Francisco-Cruz A, Raju GS, Stroehlein JR, Campbell MT, Gao J, Subudhi SK, Maru DM, Blando JM, Lazar AJ, Allison JP, Sharma P, Tetzlaff MT, Wargo JA, Jenq RR (2018) Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med 24:1804–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, No D, Gobourne A, Littmann E, Huttenhower C, Pamer EG, Wolchok JD (2016) Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 7:10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, Boselli L, Routier E, Cassard L, Collins M, Vaysse T, Marthey L, Eggermont A, Asvatourian V, Lanoy E, Mateus C, Robert C, Carbonnel F (2017) Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 28:1368–1379 [DOI] [PubMed] [Google Scholar]
- 270.Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, Koh AY (2017) Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific Human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 19:848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Gharaibeh RZ, Jobin C (2018) Microbiota and cancer immunotherapy: in search of microbial signals. Gut. 10.1136/gutjnl-2018-317220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, Long N, Plodkowski AJ, Arbour KC, Chaft JE, Rouche JA, Zitvogel L, Zalcman G, Albiges L, Escudier B, Routy B (2018) Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol 29:1437–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Yap TW, Gan HM, Lee YP, Leow AH, Azmi AN, Francois F, Perez-Perez GI, Loke MF, Goh KL, Vadivelu J (2016) Helicobacter pylori eradication causes perturbation of the human gut microbiome in young adults. PLoS One 11:e0151893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Fox JG, Wang TC (2007) Inflammation, atrophy, and gastric cancer. J Clin Invest 117:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Atherton JC, Blaser MJ (2009) Coadaptation of helicobacter pylori and humans: ancient history, modern implications. J Clin Invest 119:2475–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song M, Kostic AD, Giannakis M, Bullman S, Milner DA, Baba H, Giovannucci EL, Garraway LA, Freeman GJ, Dranoff G, Garrett WS, Huttenhower C, Meyerson M, Meyerhardt JA, Chan AT, Fuchs CS, Ogino S (2015) Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 10.1136/gutjnl-2015-310101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Lu H, Ren Z, Li A, Zhang H, Jiang J, Xu S, Luo Q, Zhou K, Sun X, Zheng S, Li L (2016) Deep sequencing reveals microbiota dysbiosis of tongue coat in patients with liver carcinoma. Sci Rep 6:33142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ren Z, Li A, Li J, Xu S, Zhang H, Jiang J, Yang J, Luo Q, Zhou K, Zheng S, Li L (2019) Tongue coating microbiome data distinguish patients with pancreatic head cancer from healthy controls AU-Lu, Haifeng. J Oral Microbiol 11:1563409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, Trinchieri G, Pardo-Manuel de Villena F, Yewdell JW, Rehermann B (2017) Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 171:1015–1028.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, Masopust D (2016) Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532:512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Ooijevaar RE, Terveer EM, Verspaget HW, Kuijper EJ, Keller JJ (2019) Clinical application and potential of Fecal microbiota transplantation. Annu Rev Med 70:335–351 [DOI] [PubMed] [Google Scholar]
- 282.Allen-Vercoe E, Petrof EO (2015) Using bugs as drugs: microbial ecosystem therapeutics. Can Commun Dis Rep 45:3–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, Shiota A, Takeshita K, Yasuma-Mitobe K, Riethmacher D, Kaisho T, Norman JM, Mucida D, Suematsu M, Yaguchi T, Bucci V, Inoue T, Kawakami Y, Olle B, Roberts B, Hattori M, Xavier RJ, Atarashi K, Honda K (2019) A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565:600–605. 10.1038/s41586-019-0878-z [DOI] [PubMed] [Google Scholar]