

SUMMARY
The extracellular molecular chaperone heat shock protein 90 (eHSP90) stabilizes protease client the matrix metalloproteinase 2 (MMP2), leading to tumor cell invasion. Although co-chaperones are critical modulators of intracellular HSP90:client function, how the eHSP90:MMP2 complex is regulated remains speculative. Here, we report that the tissue inhibitor of metalloproteinases-2 (TIMP2) is a stress-inducible extracellular co-chaperone that binds to eHSP90, increases eHSP90 binding to ATP, and inhibits its ATPase activity. In addition to disrupting the eHSP90:MMP2 complex and terminally inactivating MMP2, TIMP2 loads the client to eHSP90, keeping the protease in a transient inhibitory state. Secreted activating co-chaperone AHA1 displaces TIMP2 from the complex, providing a “reactivating” mechanism for MMP2. Gene knockout or blocking antibodies targeting TIMP2 and AHA1 released by HT1080 cancer cells modify their gelatinolytic activity. Our data suggest that TIMP2 and AHA1 co-chaperones function as a molecular switch that determines the inhibition and reactivation of the eHSP90 client protein MMP2.
INTRODUCTION
The evolutionarily conserved molecular chaperone heat shock protein-90 (HSP90) is an essential component of the physiological cellular homeostatic machinery in eukaryotes (Schopf et al., 2017). Cytosolic HSP90 interacts with hundreds of proteins that rely on HSP90 for their folding, stability, and activity. HSP90 chaperone function depends on an ordered sequence of dynamic conformational changes, linked to binding and hydrolysis of ATP, that are disrupted by drug occupancy of the ATP pocket (Hessling et al., 2009; Prodromou, 2012). In eukaryotes, the HSP90 conformational cycle is facilitated by a number of proteins termed co-chaperones that directly interact with distinct HSP90 conformational states and serve discrete functions, including assisting in the binding of client proteins to HSP90 (Li et al., 2012a). Co-chaperones also modulate the rate of HSP90-mediated ATP hydrolysis. For example, the activating co-chaperone AHA1 increases the rate of HSP90 ATPase activity, whereas co-chaperone HOP/Sti1 inhibits this activity. HSP90 co-chaperones therefore work in concert to regulate the chaperone cycle and fine-tune the chaperoning of client proteins (Panaretou et al., 2002; Retzlaff et al., 2010; Sahasrabudhe et al., 2017).
Extracellular HSP90 (eHSP90; released or surface bound) binds and chaperones extracellular client proteins such as matrix metalloproteinase 2 (MMP2) (de la Mare et al., 2017; Dong et al., 2016; El Hamidieh et al., 2012; Hance et al., 2012; Li et al., 2012b; Liu et al., 2011; McCready et al., 2014). However, the molecular mechanism of eHSP90 regulation by extracellular co-chaperones and its impact toward the chaperoning and function of an extracellular client protein remain elusive.
In this study, we demonstrate that the endogenous inhibitor of MMPs, the tissue inhibitor of metalloproteinase 2 (TIMP2), is a bona fide extracellular co-chaperone of eHSP90 that inhibits its ATPase activity and decelerates the chaperone cycle (Bourboulia and Stetler-Stevenson, 2010; Brew and Nagase, 2010; Olson et al., 1997). HSP90 was shown to bind to, stabilize, and protect MMP2, increasing the levels of the proteolytically active MMP2 pool in vitro and in vivo (i.e., in the cell-conditioned media [CM] of mammalian cell cultures) (Eustace et al., 2004; Song et al., 2010). Here, we reveal that the functional impact of extracellular co-chaperone TIMP2 on the eHSP90:MMP2 complex is twofold. TIMP2 functions as a disruptor by dissociating MMP2 from eHSP90 and directly inhibiting its proteolytic activity. TIMP2 also functions as a scaffold by loading MMP2 to HSP90, keeping MMP2 in an intermediate inhibitory state in vitro and in CM of fibroblast cells in vivo. We also show that the stress-inducible activating co-chaperone AHA1 is secreted from cells. TIMP2 and eAHA1 compete for binding and displace each other from the eHSP90:MMP2 complex, modulating MMP2 proteolytic activity and in situ gelatinolytic activity of mouse fibroblasts and human HT1080 fibrosarcoma cells. Our results show a mechanism where co-chaperones TIMP2 and AHA1 act competitively in their binding to eHSP90 and as a result directly impact client MMP2 activity and extracellular proteolysis.
RESULTS
TIMP2 Is a Stress-Inducible Protein
Pharmacologic inhibition of HSP90 leads to induction of the cell stress response, which resembles a heat shock stress. Treating HEK293 cells with the HSP90 inhibitor ganetespib (GB) led to a stress response, which was confirmed by the induction of HSP70 (Figure S1A). We also observed a statistically significant 2-fold increase in TIMP2 expression (Figure 1A). The mammalian TIMP family is composed of four members, TIMP1, TIMP2, TIMP3, and TIMP4 (Jackson et al., 2017). No significant changes were observed in TIMP1, TIMP3, and TIMP4 levels, suggesting that the mechanism of transcriptional induction is unique for TIMP2 (Figure S1A). Noticeably, treatment with biotinylated GB (Bio-GB), previously shown to be plasma membrane impermeant (McCready et al., 2014), had no impact on TIMPs or HSP70 expression (Figures S1B and S1C). We next addressed the effect of drug treatment on TIMP2 protein levels. First, we verified that the amount of GB used was not cytotoxic in 24-h drug-treated HEK293 cells (Figure S1D). We confirmed an increase of TIMP2 over 24 h of treatment of HEK293 cells with GB, both in cell extracts and, following normalization to total cellular protein levels (GAPDH loading control), CM (Figure 1B). As expected, the levels of active bona fide HSP90 client, phospho-S473-AKT, were decreased following drug treatment.
Figure 1. TIMP2 Is a Stress-Inducible Protein.
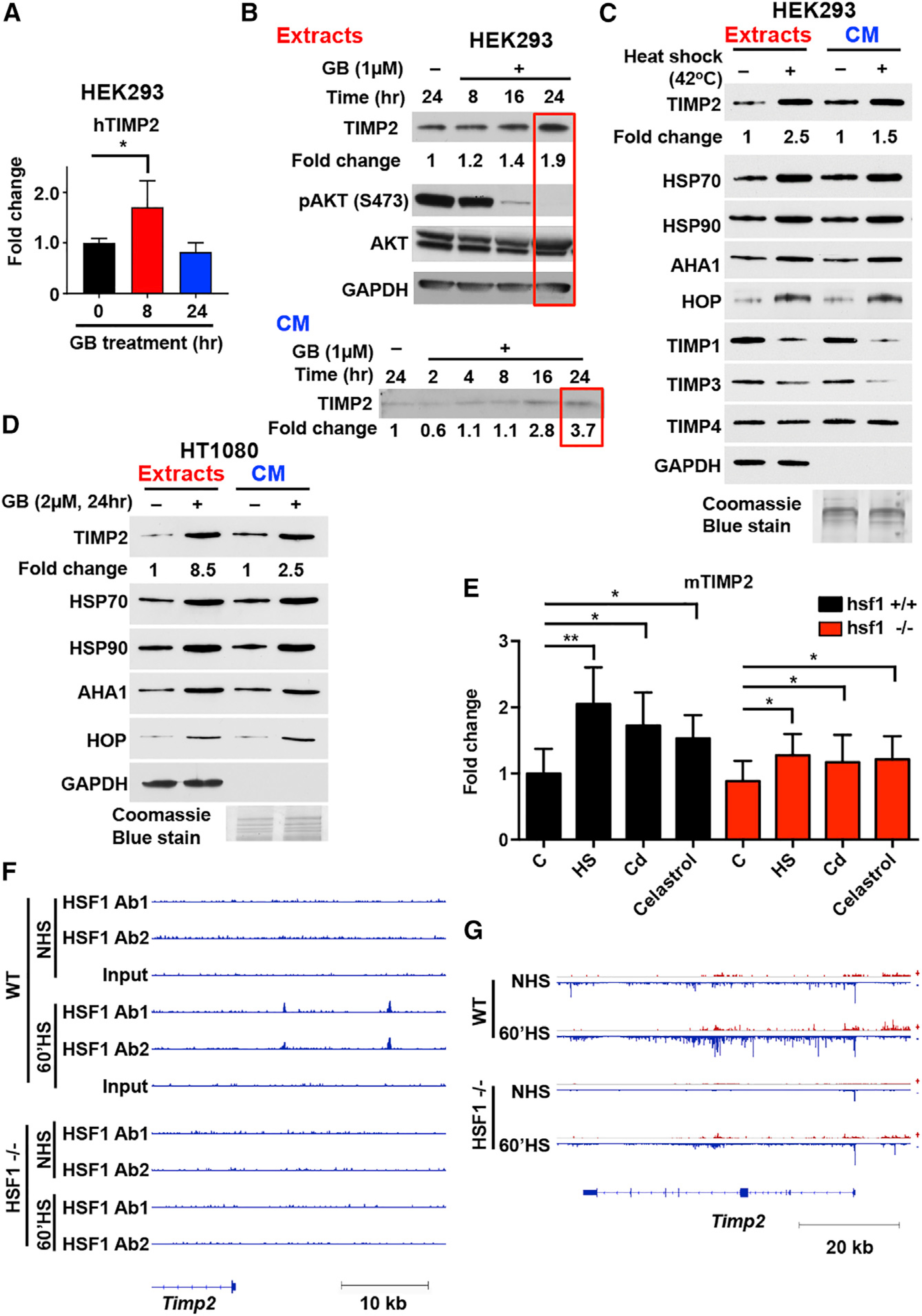
(A) Real-time RT-PCR and transcriptional expression of TIMP2 over housekeeping gene GAPDH. HEK293 cells treated with ganetespib (GB; 1 μM) as indicated. Error bars represent the SEM of n = 3 independent experiments.
(B and C) HEK293 cells were treated with 1 μM GB for the indicated time (hours) (B) or heat shocked (42°C for 1 h) (C).
(D) HT1080 human fibrosarcoma cells were treated with 2 μM GB for 24 h.
(B–D) Proteins were detected in cell extracts and cell-conditioned media (CM) with indicated antibodies (see Key Resources Table). Loading controls were GAPDH (for cell extracts), and Coomassie blue stain (for CM). Fold change was determined following blot analysis with ImageJ. (E) hsf1+/+ and hsf1−/− MEFs were exposed to 1-h heat stress at 42°C (HS), 60 μM CdSO4 for 3 h (Cd), or 4 μM celastrol for 2 h or treated with vehicle (DMSO; C). The mRNA levels of TIMP2 were quantified with qRT-PCR, normalized against RNA18S5, and hsf1+/+ control was set to 1. Error bars represent the SEM of n = 4 independent experiments.
(F) ChIP-seq profile of HSF1 showing read density in the upstream region of TIMP2 gene in hsf1+/+ (WT) and hsf1−/− MEFs grown in control conditions (NHS) or exposed to heat shock for 60 min at 42°C (60’HS). Two different HSF1 antibodies were used for ChIP-seq: custom-made antibody (Ab1) and antibody from Cell Signaling #4356 (Ab2). The data were obtained from Mahat et al. (2016).
(G) PRO-seq profile showing the transcriptional intensity of TIMP2 gene in hsf1+/+ (WT) and hsf1−/− MEF grown in control conditions (NHS) or exposed to heat shock for 60 min at 42°C (60’HS). The data were obtained from (Mahat et al., 2016).
Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S1.
We then examined whether heat shock stress directly impacts TIMP2 protein expression. Heat-shocked HEK293 cells were immunoblotted after normalization to total protein from cell extracts (GAPDH control). Levels of TIMP2 and the stress-inducible proteins Hsp70, AHA1, and HOP (positive controls) increased both in cell extracts and CM, whereas the levels of other TIMPs members remained either unchanged (TIMP4) or decreased (TIMP1 and TIMP3) (Figure 1C). Cancer cells secrete significantly high levels of Hsp90 (Li et al., 2013). Treatment of HT1080 cells with GB confirmed the upregulation of TIMP2, as well as AHA1 and HOP co-chaperones in cancer cells extracts and CM (Figures 1D and S1E).
Heat shock factor 1 (HSF1) is a transcription factor rapidly activated upon cell stress and required for stress-induced heat shock response (Budzyński and Sistonen, 2017; Vihervaara and Sistonen, 2014). Subsequently, we determined the role of HSF1 on TIMP2 response to proteotoxic stress. Wild-type hsf1+/+ and hsf1−/− murine embryonic fibroblasts (MEFs) were untreated (C), heat shocked (HS), or treated with the protein-damaging agents CdSO4 (Cd) or celastrol (Budzyński and Sistonen, 2017). mRNA levels of TIMP2 and HSP70 (HSP1A/B) were quantified by qRT-PCR (Figures 1E and S1F). TIMP2 mRNA was markedly induced (2-fold increase) in stressed hsf1+/+ MEFs, whereas hsf1−/− MEFs displayed 40% lower induction, indicating that TIMP2 is an HSF1 target gene (Figure 1E). Analysis of existing chromatin immunoprecipitation sequencing (ChIP-seq) data on hsf1+/+ and hsf1−/− MEFs, reported by Mahat and co-workers (Mahat et al., 2016), revealed two stress-inducible HSF1-binding sites upstream of the TIMP2 gene (Figure 1F). The precise run-on and sequencing (PRO-seq) data (Mahat et al., 2016) showed a strong transcriptional induction of TIMP2 in heat-shocked hsf1+/+ MEFs, but the induction was less prominent in hsf1−/− MEFs (Figure 1G). Taking together the results of mRNA, ChIP-seq, and PRO-seq analyses, we conclude that the expression of the TIMP2 gene is stress inducible and enhanced by HSF1. Overall, TIMP2 is induced upon pharmacologic inhibition of HSP90 and other proteotoxic stressors that activate HSF1.
TIMP2 Directly Binds to the Middle Domain of eHSP90α
Our data on stress induction of TIMP2 prompted us to examine a possible interaction between HSP90 and TIMP2. Recombinant HSP90α-His6 was first bound to nickel-nitrilotriacetic acid (Ni-NTA) agarose and then incubated with recombinant TIMP2 (Figures 2A and S2A). The addition of 10 ng or 50 ng TIMP2 results in complex formation with HSP90α-His6. Interestingly, high amounts (∼200 ng) of TIMP1, TIMP3, and TIMP4 show minimal interaction, suggesting that TIMP2 has a stronger binding affinity to HSP90α (Figure S2B). We next examined the in vivo interaction between HSP90α and TIMP2. Since HSP90 adopts a variety of functional conformational states in the process of ATP binding and hydrolysis (Schopf et al., 2017), we expressed the wild type (WT) and two HSP90α mutants, D93A (non-ATP bound, which promotes an “open” conformation) and E47A (ATP bound non-hydrolyzing, which promotes a “closed” conformation) (Figure 2B). Following immunoprecipitation (IP) using anti-FLAG M2 affinity gel from CM (Cortes et al., 2018), endogenous TIMP2 was co-immunoprecipitated with eHSP90α, binding stronger to either mutant compared to WT HSP90 (Figure 2B).
Figure 2. The N-Terminal Domain of TIMP2 Interacts with M-Domain of eHSP90α.
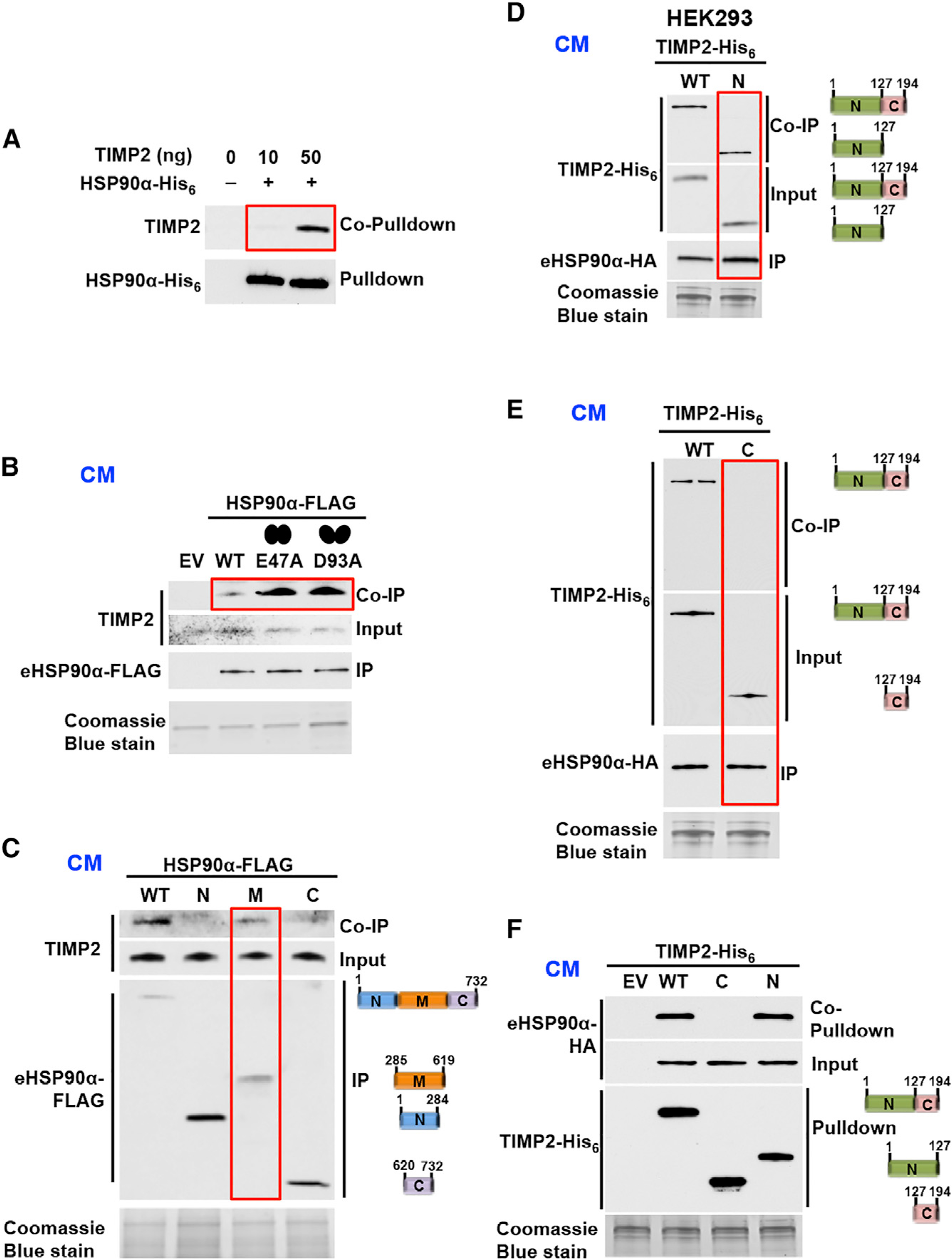
(A) Pull-down of recombinant HSP90α-His6 bound to Ni-NTA agarose followed by co-pull-down of recombinant human TIMP2 (10 ng or 50 ng added).
(B) Immunoprecipitation (IP) of transiently expressed eHSP90α-FLAG, E47A-eHSP90α-FLAG (closed conformation mutant) and D93A-eH-SP90α-FLAG (open conformation mutant) and co-immunoprecipitation (coIP) of endogenous TIMP2 from HEK293 CM.
(C) Interaction of endogenous TIMP2 with full-length, N- (amino), M- (middle), and C- (carboxyl) domains of HSP90α IP from HEK293 CM.
(D and E) TIMP2-His6 (full-length and N-TIMP2- His6) (D) and TIMP2-His6 (full length and C-TIMP2-His6) (E) interactions (coIP) with HSP90α-HA (IP) co-expressed in HEK293 CM.
(F) Empty vector (EV), full-length, C-domain, and N-domain of TIMP2-His6 and HSP90α-HA were co-expressed in HEK293 cells. Immunoblotting was used to detect TIMP2-His6 and co-pull-down eHSP90α-HA from CM.
Coomassie blue stain (loading control for CM). See also Figure S2
HSP90 is composed of N- (amino), M- (middle), and C-(carboxyl) domains (Sahasrabudhe et al., 2017). HSP90 WT and FLAG-tag domains were secreted in HEK293 cells CM and immunoprecipitated as previously shown (Song et al., 2010). It appears that endogenous TIMP2 interacts with the M-domain of eHSP90α (Figure 2C). In similar reciprocal experiments, the N-domain (residues 27–152) or the C-domain (residues 153–220) of TIMP2-His6 was co-expressed with HSP90α -hemagglutinin (HA) in HEK293 cells. Following TIMP2-His6 or eHSP90α -HA precipitation from the CM (Figures 2D–2F) we show that only the N-TIMP2 interacts with eHSP90α. Taken together, the data show that TIMP2 and HSP90α directly interact in vitro and in vivo.
TIMP2 Co-chaperone Is a Potent Inhibitor of eHSP90 Function
HSP90 ATP binding and hydrolysis are coupled to its chaperone function (Panaretou et al., 1998; Schopf et al., 2017). To establish the impact of TIMP2 on HSP90 binding to ATP, we used isothermal titration calorimetry (ITC) for (HSP90α:TIMP2):ATP and HSP90α:ATP (control) protein complexes (Figures 3A and S3A). Full-length recombinant HSP90α and TIMP2 proteins were mixed at stoichiometrically equivalent molar ratios. Consistent with previous studies for both human and yeast HSP90, we measured the HSP90α:ATP dissociation constant (KD) of 270 ± 100 μM (Figure S3A) (McLaughlin et al., 2004; Prodromou et al., 1997). TIMP2 increases HSP90α binding to ATP more than 4-fold in the (HSP90α:TIMP2):ATP complex, with a measured KD of 65 ± 24 μM (Figure 3A). The role of TIMP2 on eHSP90’s ability to bind to ATP was next examined using a TIMP2-deficient (TIMP2−/−) MEF cell line (Wang et al., 2000). We show that eHSP90 binds to agarose-ATP following pull-down of endogenous eHSP90 from CM (Figure 3B). Pretreatment of TIMP2−/− MEFs with TIMP2 significantly increases the binding of eHSP90 to ATP, and TIMP2 also forms a complex with eHSP90 (Figure 3B). In contrast, treatment with same amount of TIMP1 has no effect on the binding of eHSP90 to ATP. Similar experiments using WT MEFs (expressing endogenous TIMP2) confirmed the above data (Figure S3B). Therefore, TIMP2 has the ability to increase HSP90 binding to ATP in vitro and in vivo.
Figure 3. TIMP2 Co-chaperone Inhibits HSP90 ATPase Activity and Increases HSP90 Binding to Both ATP and Drugs.
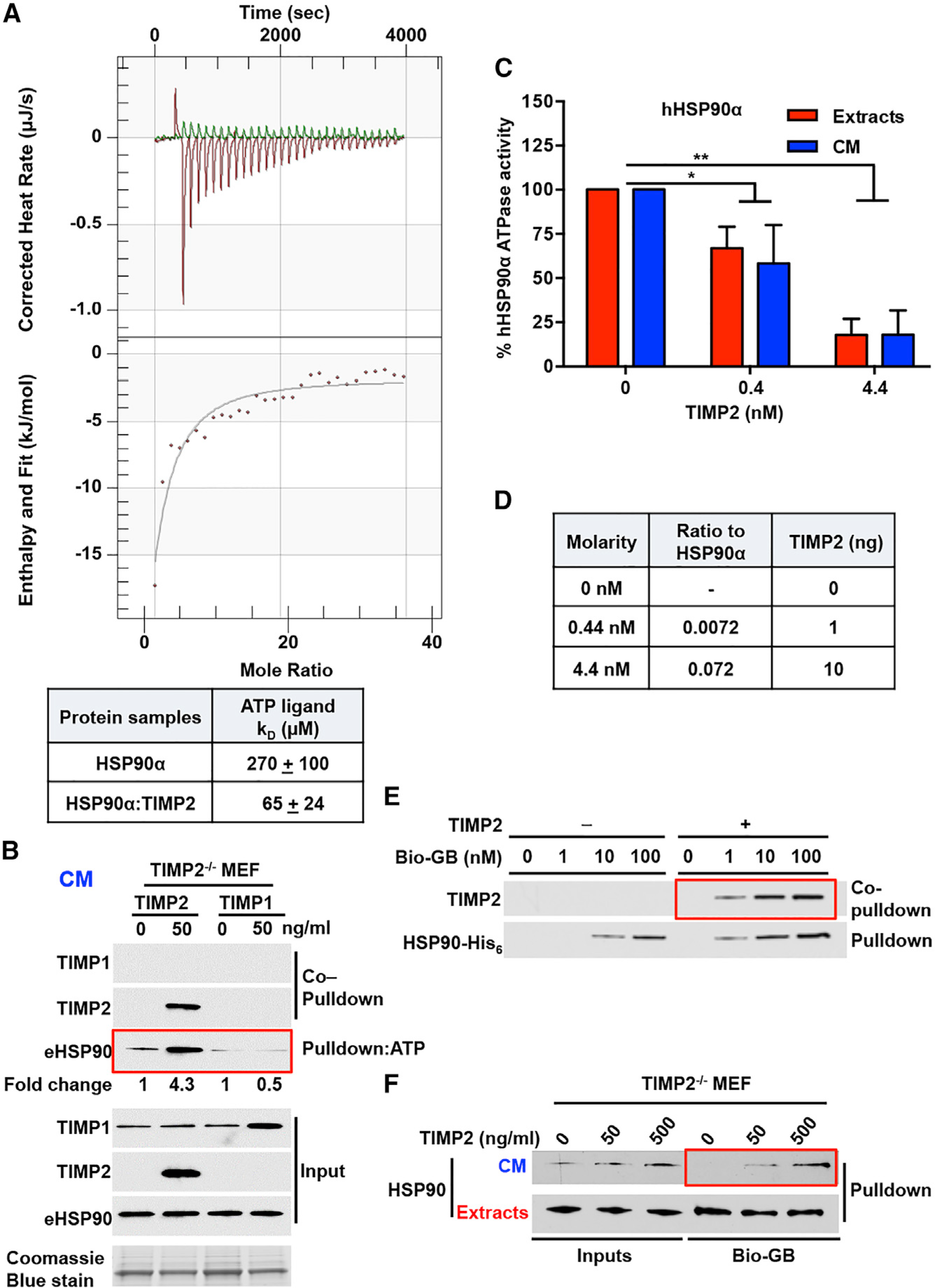
(A) ITC of HSP90-His6:TIMP2-His6 binding to ATP. Corrected heat rate (μJ/s) and enthalpy/fit (kJ/mol) are presented at the top and bottom, respectively. The dissociation constant (KD) is shown in the table for the HSP90-His6:TIMP2-His6 complex and HSP90-His6 alone. Errors represent the SEM of n = 2 independent experiments.
(B) ATP-beads were incubated with TIMP2−/− MEF CM for eHSP90 ATP-pull-down. Co-pull-down of TIMP2 and TIMP1 was performed following addition of each protein. Fold change was determined using ImageJ. Coomassie blue stain was used as a loading control for CM.
(C) ATPase activity of HSP90α purified from HEK293 cell extracts and CM was measured in the absence and presence of different amounts of TIMP2 (nM). Activity (%) was normalized relative to the untreated HSP90. Error bars indicate SEM from n = 3 independent experiments.
(D) Molarity of the proteins in the ATPase assay (Figure 3C), ratio to HSP90α, and total quantity of TIMP2 protein added (ng).
(E) HSP90-bound to the drug was pulled down with streptavidin agarose, and TIMP2 was co-pulled down. Recombinant HSP90α -His6 (100 ng) was incubated with recombinant TIMP2 (10 ng), followed by the addition of different amounts of Bio-GB (1 ng, 10 ng, or 100 ng).
(F) Cell extracts and CM were collected from TIMP2−/− MEFs following the addition of TIMP2 (50 and 500 ng/mL) and incubated with 100 nM of Bio-GB and addition to streptavidin agarose for pull-down experiments.
Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S3.
Given that eHSP90 binds to ATP, we next tested its ability to hydrolyze ATP. HEK293 cells were transfected with HSP90α-FLAG, and cell extracts and CM were collected for HSP90α-FLAG purification. ATPase activity of HSP90α isolated from cell extracts and CM (Figure S3C) was measured using PiPer Phosphate Assay (see STAR Methods and Dunn et al., 2015) (Figures 3C and S3D). Percentage ATPase activity was based on millimoles inorganic phosphate per mole per min (mmol Pi per mol min−1) for HSP90α. Our data show that HSP90 isolated from both CM and cell extracts hydrolyzes ATP (Figure 3C). The addition of TIMP2 significantly inhibited the ATP hydrolysis of HSP90 (both from extracts and CM) at low-nanomolar levels (Figures 3C and 3D). In contrast, TIMP1 did not significantly affect the eHSP90 ATPase activity (Figure S3E). As anticipated, GB potently inhibited HSP90 ATPase activity (Figure S3F). HSP90’s ability to hydrolyze ATP at its N terminus is facilitated by its homodimerization state. Analysis of native forms of mammalian cytosolic HSP90 was previously shown to exist predominantly as a ∼200-kDa homodimer (Minami et al., 1991; Nemoto and Sato, 1998). HEK293 CM, transiently expressing eHSP90α-His6, were analyzed alongside recombinant HSP90α-His6 control in native PAGE (Figure S3G). The data revealed that eHSP90 migrate at ∼200 kDa, confirming that secreted HSP90 exists in a dimeric state (Figure S3G).
We next examined the effects of TIMP2 on HSP90 binding to the N-terminal domain inhibitor GB (Figure 3E). TIMP2 enhances HSP90α binding to 1 nM Bio-GB and leads to the formation of a HSP90α:TIMP2:GB complex (Figure 3E). We also treated TIMP2−/− MEF CM with increasing amounts of TIMP2. The data showed an increase in eHSP90 binding to 100 nM Bio-GB (Figure 3F). Taken together, we show that eHSP90 exists as a dimer, binds to ATP, and has ATPase activity. Our data also show that TIMP2 is a potent inhibitor of HSP90 ATPase activity and increases the affinity of HSP90 to both ATP and N-terminal small-molecule inhibitors.
TIMP2 Co-chaperone Functions as an Adaptor and Disruptor of the HSP90:MMP2 Complex
It has been shown that eHSP90 binding to MMP2 impacts MMP2 stability and results in enhanced levels of active MMP2 (Eustace et al., 2004; Song et al., 2010; Stellas et al., 2010). MMP2 is secreted as a latent enzyme (proMMP2, 72 kDa) that is activated extracellularly (active MMP2, 62 kDa) following proteolytic removal of its N-terminal domain while in association with a plasma-membrane-bound active metalloproteinase (Visse and Nagase, 2003). We found that (100 ng) active-MMP2 and (250 ng) proMMP2 directly bind to HSP90α in vitro (Figures S4A–S4D).
To understand the interplay among HSP90α, MMP2, and TIMP2, we performed a series of in vitro protein-binding experiments described in the flow diagram (Figure 4A). First, active MMP2 was complexed with Ni-NTA-bound HSP90α-His6 (Figures 4B, top, and Figure S4E). The addition of increasing amounts of TIMP2 results in dissociation of MMP2 from HSP90α-His6, while TIMP2 forms a complex with the chaperone (Figure 4B, top). IP of the free MMP2 from the supernatant of the agarose mixture also showed that MMP2 was in complex with TIMP2 (Figure 4B, bottom). Similar results were also obtained using proMMP2 in the complexes (Figures 4C and S4F). The data demonstrate that in this setting, TIMP2 functions as a disruptor of MMP2 from the chaperone, resulting in the formation of two complexes, first with HSP90α and second with the released MMP2 (active and proMMP2) in a dose-dependent manner.
Figure 4. TIMP2 Co-chaperone Functions as an Adaptor and Disruptor of the HSP90-MMP2 Complex.
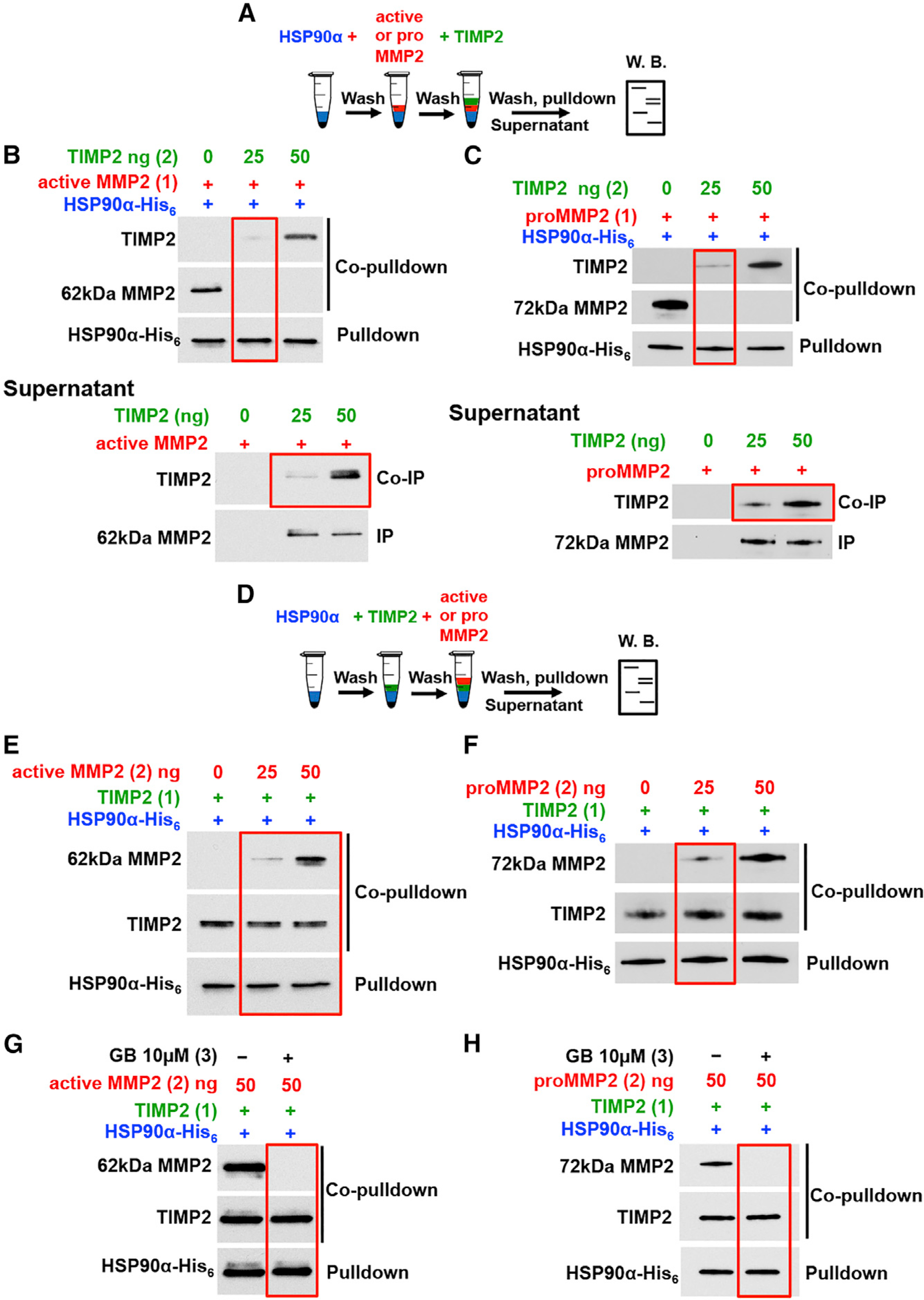
(A) Schematic flowchart of experiments in (B and C). (B) Active MMP2 (100 ng) was added first (1) to Ni-NTA-bound HSP90α -His6 followed by 25ng or 50ng TIMP2 (2). HSP90 pull-down, MMP2 and TIMP2 co-pull-down (top blot). MMP2 was IP and TIMP2 was coIP from the supernatant (bottom blot).
(C) As in (B) with the exception of ProMMP2 (250 ng) was added in place of active MMP2.
(D) Schematic flowchart of experiments in (E and F).
(E) TIMP2 (25 ng) was added first (1) to HSP90α His6 bound to Ni-NTA agarose followed by (2) indicated amounts of active MMP2. Proteins were pull-down and immunoblotted.
(F) As in (E) with the exception of proMMP2 was added in place of active MMP2.
(G) HSP90α-His6 was bound to Ni-NTA agarose, followed by addition of TIMP2 first (1) followed by active MMP2 (2). 10 μM GB was added to the complexes and protein were analyzed by immunoblotting.
(H) As in (G) with the exception of proMMP2 was added in place of active MMP2.
See also Figure S4.
In reciprocal experiments (Figure 4D), TIMP2 was first complexed with HSP90α-His6 bound to Ni-NTA agarose, followed by the addition of client MMP2 (Figures 4E, 4F, S4G, and S4H). To our surprise, the addition of 25 ng active MMP2 (or proMMP2) resulted in the formation of a ternary complex HSP90α-His6:TIMP2:MMP2 (Figures 4E and 4F). The fact that less MMP2 (25 ng and 50 ng) interacts with the HSP90α-His6:TIMP2 complex (Figures 4E and 4F), compared to 100 ng active and 250 ng proMMP2 with HSP90α alone (Figures S4A and S4B), suggests that TIMP2 mediates and promotes binding of MMP2 to the HSP90α-His6:TIMP2 complex. Indeed, in the absence of TIMP2, 25 ng active or proMMP2 does not complex with HSP90α (Figures S4G and S4H). Furthermore, we show a gradual increase of a ternary complex formation HSP90α -His6:TIMP2:MMP2 (Figures 4E and 4F). To gain further evidence that MMP2 makes direct contact with HSP90α in this ternary complex, we added GB (a specific HSP90 inhibitor) to the MMP2:TIMP2:HSP90α -His6. As expected, GB disrupts the interaction between HSP90 and active or proMMP2, as it would if client MMP2 made direct contact with HSP90. Moreover, it does not disrupt TIMP2 from HSP90 (Figures 4G and 4H). These data demonstrate that, in this setting, TIMP2 functions as a scaffolding co-chaperone that loads MMP2 client to HSP90.
TIMP2 and AHA1 Compete for Binding to the HSP90:MMP2 Complex
To determine if a known HSP90 co-chaperone could displace TIMP2 from the HSP90 complex, we first showed that common intracellular co-chaperones, such as AHA1, PP5, and HOP, were secreted from HEK293 cells (Figure 5A). However, p23 and CDC37 co-chaperones were almost undetectable in CM of HEK293 cells (Figure S5A). Endogenous TIMP2 immunoprecipitated from CM of HEK293 cells showed interaction with eHSP90, eHOP, and ePP5, but not eAHA1 (Figure 5A). These data suggested that TIMP2 and AHA1 may not be able to bind to HSP90 simultaneously. Our in vitro data demonstrate that TIMP2 displaces AHA1 from the HSP90α-His6:AHA1 complex (Figures 5B and S5B). Reciprocal experiments also showed that AHA1 releases TIMP2 from HSP90α-His6, allowing the formation of an HSP90α-His6:AHA1 complex (Figure 5C). These data establish that the two co-chaperones, TIMP2 and AHA1, act competitively for binding to HSP90. Interestingly, the M-domain of HSP90 is a binding site for the N-terminal domain of AHA1 (Meyer et al., 2004). To test whether TIMP2 binds to a similar site as AHA1 on HSP90, we utilized the human HSP90α-V411E mutant (yeast HSP82pV391E) that was previously shown to abolish HSP90 and AHA1 complex formation (Meyer et al., 2004; Retzlaff et al., 2010; Woodford et al., 2017). Following IP of WT and mutant HSP90α-V411E-FLAG from HEK293 CM, the data confirm that while both TIMP2 and AHA1 bind to WT HSP90α, their binding to HSP90α-V411E is abolished (Figures 5D and S5C). This indicates that TIMP2 and AHA1 may either share common binding sites on HSP90 M-domain or TIMP2 binds to a conformational epitope of HSP90 disrupted by the HSP90α -V411E mutation.
Figure 5. Extracellular AHA1 Displaces TIMP2 Co-chaperone from the HSP90:MMP2 Complex.
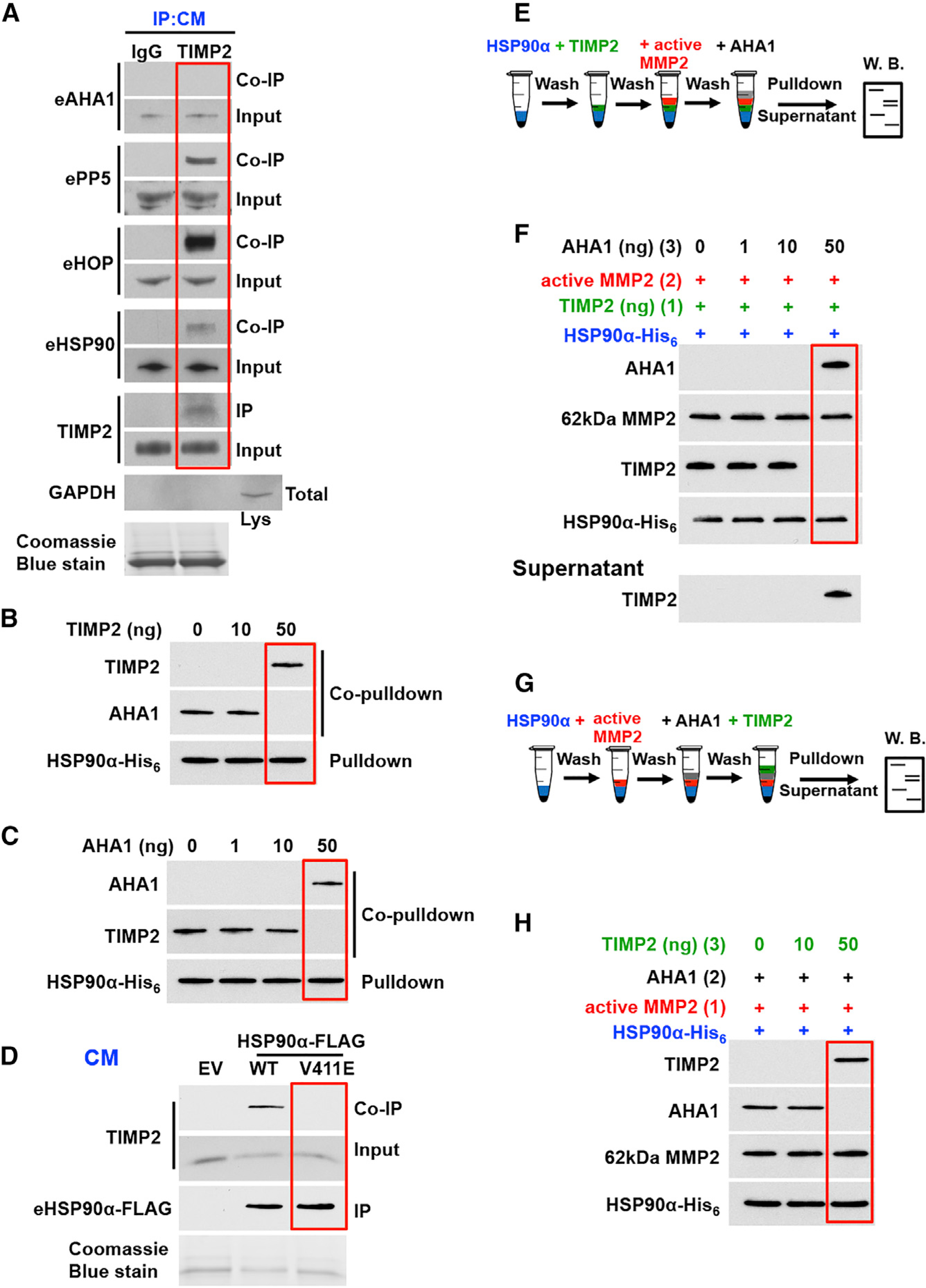
(A) Endogenous TIMP2 (or IgG control) was IP from HEK293 CM, and interaction with eHSP90 and co-chaperones (eAHA1, ePP5 and eHOP) was determined by coIP. GAPDH was used as a control for cell lysis.
(B) HSP90α-His6 (100 ng) was bound to Ni-NTA agarose followed first by the addition of recombinant AHA1 (50 ng) and then by the addition of indicated amounts of TIMP2. Interactions were determined by immunoblotting.
(C) HSP90α-His6 (100 ng) was bound to Ni-NTA agarose followed first by the addition of TIMP2 (50 ng) and then by the addition of increasing amounts of AHA1. Interactions were determined by immunoblotting.
(D) Wild-type and HSP90α-FLAG mutant V411E from HEK293 CM were bound to FLAG affinity agarose and immunoblotted for HSP90 (IP) and TIMP2 (coIP).
(E) Schematic representation of steps involved information of the HSP90α:TIMP2:MMP2 complex followed by the addition of AHA1.
(F) HSP90α -His6 (100 ng) bound to Ni-NTA agarose was followed by the addition of TIMP2 (50 ng). Active MMP2 (50 ng) was added (2) to form the HSP90:TIMP2:MMP2 complex. The indicated amounts of AHA1 (3) were added, followed by pull-down and immunoblotting. Supernatant from the above complexes was analyzed for the presence of TIMP2.
(G) Schematic representation of steps involved in the formation of the HSP90α:AHA1:MMP2 complex, followed by the addition of TIMP2.
(H) HSP90α-His6 (100 ng) bound to Ni-NTA agarose, followed by the addition of active MMP2 (50 ng) (1). AHA1 (50 ng) was added (2) to form the HSP90:MMP2:AHA1 complex. The indicated amounts of TIMP2 (3) were added, followed by pull-down and detection by immunoblotting. Coomassie blue stain was used as a loading control for CM.
See also Figure S5.
Given that TIMP2 modulates the HSP90α:MMP2 complex, we next addressed how TIMP2 and AHA1 influence client binding. First, we analyzed active MMP2 complex with HSP90 in the presence of AHA1 (Figure 5E). The addition of increasing amounts of AHA1 in the formed HSP90α -His6:TIMP2:MMP2 complex results in the dissociation of TIMP2, which is identified in the supernatant. Active MMP2, however, remains in complex with HSP90α and AHA1 (Figure 5F). Similarly, the addition of TIMP2 in the HSP90α-His6:MMP2:AHA1 complex results in the release of AHA1 and the formation of an HSP90α-His6:MMP2:TIMP2 complex (Figures 5G and 5H). Therefore, TIMP2 and eAHA1 compete for binding to the HSP90:MMP2 complex without disrupting the interaction between chaperone and client protein.
Co-chaperone Deficiencies Impact Extracellular Chaperone:Client Complexes
Fibroblasts and other stromal cells are known to secrete components of the extracellular matrix (ECM), as well as significant amounts of MMPs and their endogenous inhibitors (TIMPs) during remodeling of the ECM (Bourboulia and Stetler-Stevenson, 2010; Jackson et al., 2017; Kessenbrock et al., 2010). In addition to HSP90α secretion, AHA1 was reported to co-localize with HSP90 in secretory vesicles and further was detected in cell media (Eustace et al., 2004; Ghosh et al., 2015; Sims et al., 2011). We next questioned whether the in vitro modulation of HSP90:MMP2 complexes by TIMP2 and AHA1 could take place in physiological conditions in vivo.
We utilized gene knockout mouse embryonic fibroblast (MEF) or mouse adult fibroblast (MAF) cell lines in which we exogenously supplemented their CM with physiologically relevant amounts of the missing protein. CM were collected and further analyzed in immunoblotting, IP, enzyme activity, and gelatinolytic assays (Figure 6A). To determine what protein concentrations to add that are physiologically relevant, secreted levels of MMP2, TIMP2, and eAHA1 in CM were estimated using combination of available mouse ELISAs and immunoblotting (Figures S6A and S6B). CM volumes were normalized to total protein from cell extracts (GAPDH control in extracts), and protein levels were also compared to 50 ng/mL recombinant protein as a control (Figure S6B). Following IP of endogenous eHSP90α from CM, we identified extracellular interactions with endogenous MMP2 (Figure 6B), TIMP2 (Figures 6C and 6D), and eAHA1 (Figures 6E and 6F). Supplementing of TIMP2−/− MEFs with increasing amounts of TIMP2 led to evident disruption of endogenous MMP2 from eHSP90α and led to the formation of an eHSP90α:TIMP2 complex (Figure 6B). The addition of pro-and active MMP2 in MMP2−/− MEFs resulted in the formation of eHSP90α:TIMP2:MMP2 ternary complexes (Figures 6C and 6D). We also questioned the competitive binding of eAHA1 and TIMP2 on eHSP90. We found that in the absence of TIMP2 (TIMP2−/− MEF), eAHA1 binding to eHSP90 was stronger (Figure 6E). The addition of 50 ng/mL TIMP2 resulted in a modest decrease of endogenous eAHA1 interaction with eHSP90 to the level of WT MEFs (Figure 6E). Noticeably, the added amount of TIMP2 was similar to the endogenous levels of TIMP2 in WT MEF CM (see Figure 6E, TIMP2 Input). Consistent with the in vitro data, the addition of AHA1 to AHA1−/− MAFs released TIMP2 from the eHSP90:-TIMP2:MMP2 complex (Figure 6F). Our data suggest that the competition and interplay seen between TIMP2 and AHA1 in vitro also occurs in vivo.
Figure 6. The eHSP90:MMP2 Complex Is Regulated by TIMP2/AHA1 Dynamic Interplay.
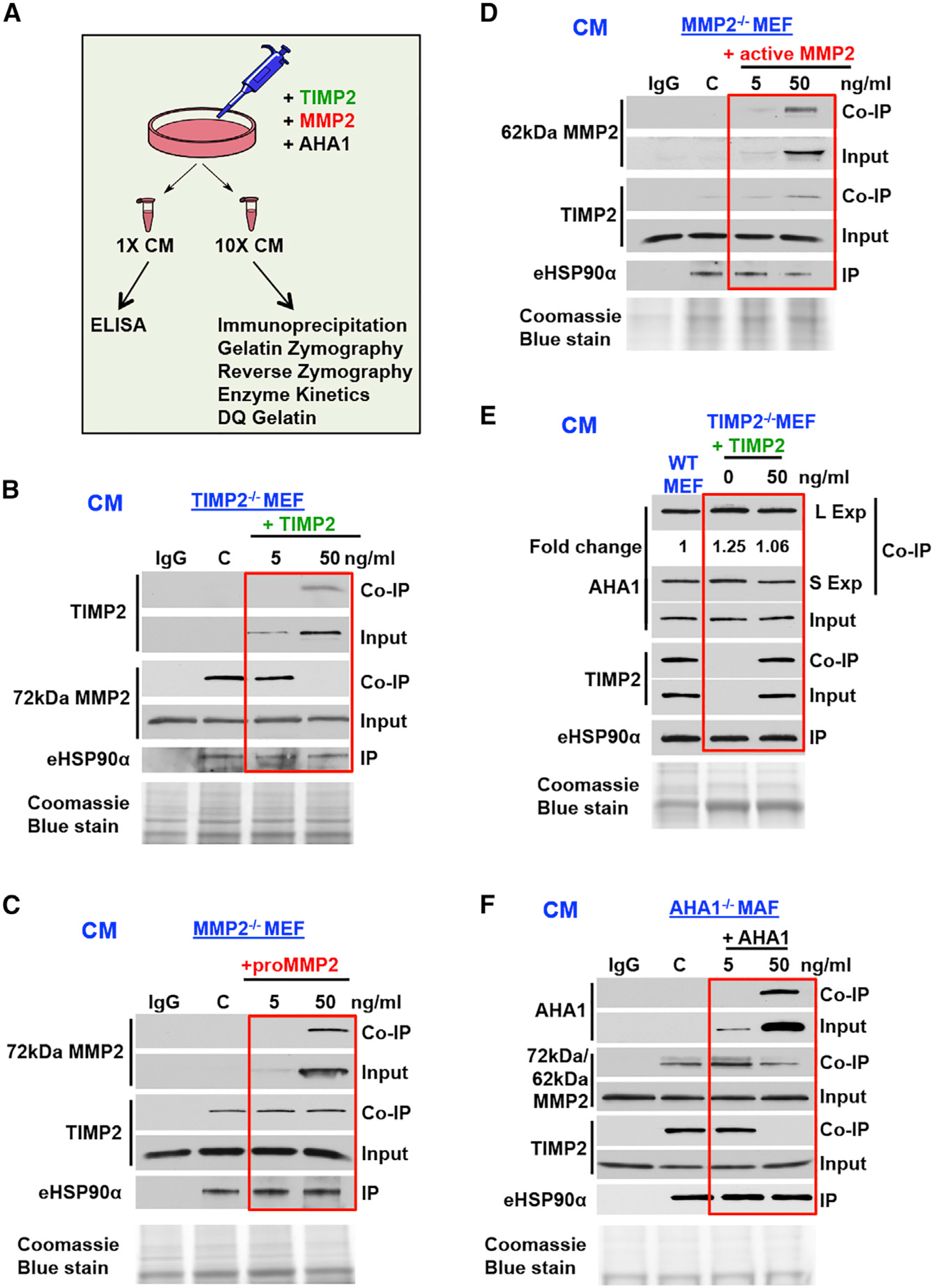
(A) Schematic representation of experiments in Figure 6.
(B) TIMP2−/− MEF cells were supplemented with increasing amounts of TIMP2-His6 (0–50 ng/mL) for 2 h. IP of endogenous eHSP90α (IP) from CM was followed by MMP2 and TIMP2 (coIP).
(C and D) MMP2−/− MEF cells were treated respectively with increasing amounts of 72 kDa (C) or 62 kDa (D) recombinant MMP2 (0–50 ng/mL) for 2 h. IP of endogenous eHSP90α from CM was followed by MMP2 and TIMP2 coIP.
(E) TIMP2−/− MEF cells were treated with 0 and 50 ng/mL TIMP2-His6 for 2 h. eHSP90α IP from TIMP2−/− and WT MEF CM, and analyzed by immunoblot for TIMP2 and AHA1 (coIP). Fold change was determined using ImageJ.
(F) AHA1 MAF−/− cells were treated with increasing amounts of recombinant AHA1 (0–50 ng/mL) for 2 h. IP of endogenous eHSP90α (IP) from CM was followed by MMP2, TIMP2, and AHA1 (coIP). Coomassie blue stain was used as a loading control for CM.
See also Figure S6.
AHA1 and TIMP2 Exert Opposite Functions on MMP2 Activity and Matrix Degradation
To understand how TIMP2 and AHA1 dynamic interplay affects MMP2 activity when in complex with HSP90, we performed fluorometric enzyme kinetic assays (Sánchez-Pozo et al., 2018). HSP90α:active MMP2 complexes (∼1:1 molar ratio) or MMP2 alone was incubated at 4°C, and MMP2 enzymatic activity was determined at 25°C (pH 7.4). Titrating HSP90α against active MMP2 (0.2 nM) does not impact protease activity (Figure S7A). Following titration of TIMP2, the activity of MMP2 alone or in complex with HSP90α was completely repressed (Figure S7B). We next formed the HSP90α -His6:TIMP2:MMP2 ternary complex by adding active MMP2 to the HSP90α-His6:TIMP2 complex. MMP2 activity was reduced by ∼75% when in complex with HSP90α-His6:TIMP2 (Figure S7C). The data suggest that HSP90α, when bound to MMP2, does not directly affect proteolytic activity and is also unable to protect MMP2 from TIMP2-mediated inhibition.
We showed earlier that AHA1 displaces TIMP2 from the HSP90α -His6:TIMP2:MMP2 complex. We therefore tested whether displacement of TIMP2 by AHA1 reactivates MMP2 in complex with HSP90α. Increasing amounts of AHA1 do not affect the activity of MMP2 alone or in complex with HSP90α (Figures S7D–S7F). We then formed the HSP90α -His6:TIMP2:MMP2 complex by performing pull-down experiments as shown earlier (Figures 5E and 5F). AHA1 promoted the “reactivation” of MMP2 in the newly formed HSP90-His6:MMP2:AHA1, showing an average increase in activity of 32% (0–200 ng of AHA1) (Figure 7A). The addition of TIMP2 to the ternary HSP90α-His6:MMP2:AHA1 complex reverses MMP2 activity back to the inhibited state (Figures 7A and S7G). These findings provide strong evidence of an intermediate state of MMP2 activity that is dependent, and tightly modulated by, the interplay between co-chaperones TIMP2 and AHA1 binding to HSP90.
Figure 7. Opposing Effects of the Co-chaperones AHA1 and TIMP2 on MMP2 Activity and Matrix Degradation.
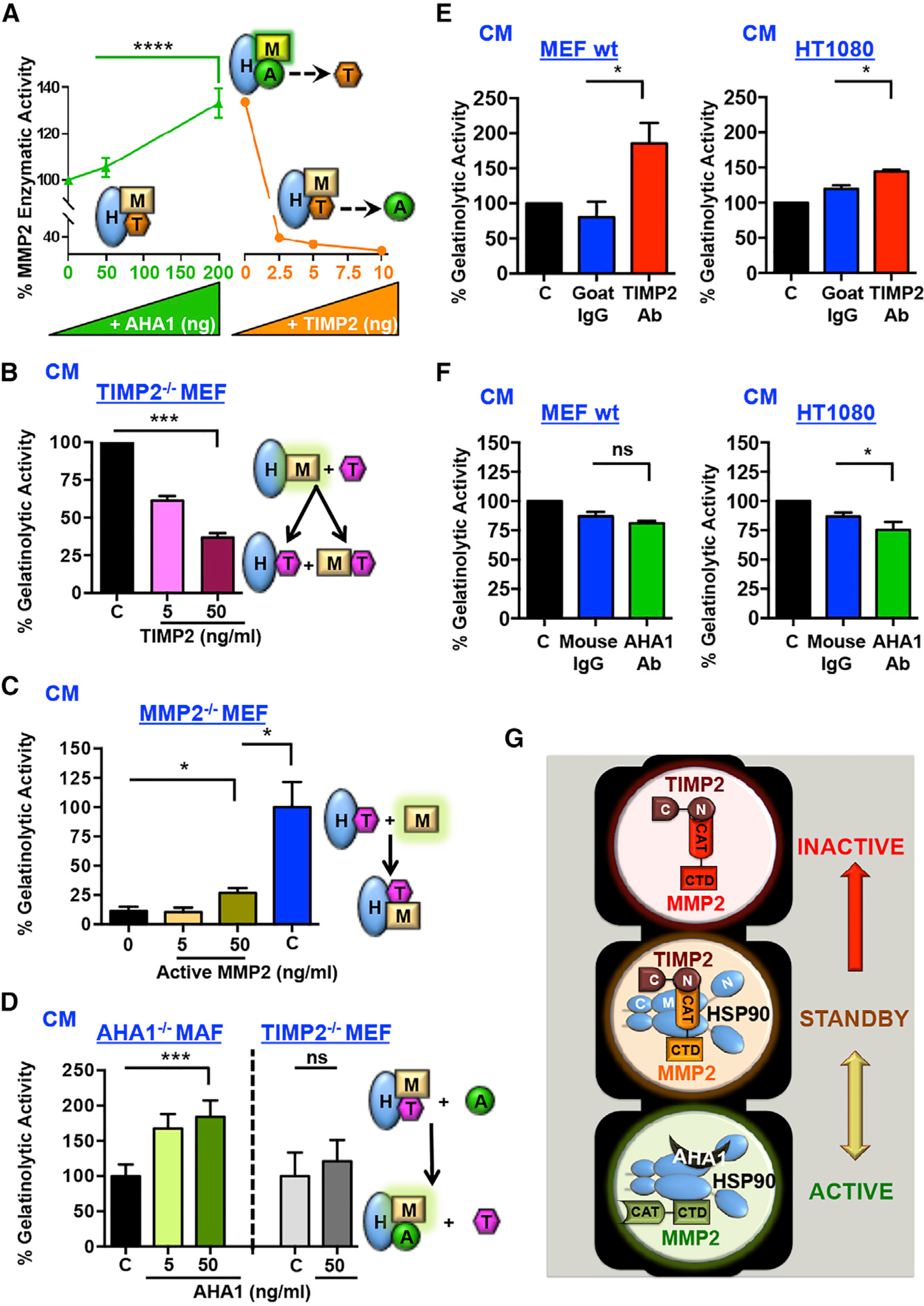
(A) HSP90α:TIMP2:MMP2 complexes were formed on Ni-NTA beads, followed by the addition of the indicated amounts of AHA1 and further addition of increasing amounts of TIMP2. MMP2 activity (%) was determined. Error bars represent the SEM of at least two independent experiments. Statistical significance was determined between control (no AHA1) and 200 ng AHA1
(B) TIMP2−/− MEF cells were treated with increasing amounts (0–50 ng/mL) of TIMP2. Gelatinolysis of CM was measured using DQ fluorescent gelatin. End-point measures were taken. Error bars represent SEM of n = 2 independent experiments.
(C) MMP2−/− MEF cells were treated with increasing amounts (0–50 ng/mL) of 62-kDa active MMP2. Gelatinolytic activity was measured and presented as percentage of 50 ng/mL MMP2. Error bars represent SEM of n = 2 independent experiments.
(D) AHA1−/− MAF or TIMP2−/− MEF cells were left untreated or treated with increasing amounts of recombinant AHA1 (0–50 ng/mL). End-point measurements were taken. Error bars represent SEM of n = 2 independent experiments.
(E and F) MEF WT +/+ and HT1080 human fibrosarcoma cells were treated with anti-TIMP2 (10 μg/mL) or anti-AHA1 (20 μg/mL) antibodies, respectively, or isotype IgG and PBS (C) controls. Gelatinolytic activity in CM was measured using DQ gelatin degradation and plotted as a percentage of untreated control. Error bars represent SEM of n = 3 independent experiments, comparing treated versus IgG controls. ns, non-significant.
(G) Model of the mechanism described in this study. Extracellular molecular chaperone HSP90 (eHSP90) binds directly to its secreted client active protease, MMP2. Co-chaperone TIMP2 facilitates the HSP90:MMP2 complex dissociation, resulting in the formation of a terminally inactivated MMP2 in complex with TIMP2 (inactive, top). TIMP2 binds directly to HSP90, and additional “intermediate” inhibitory complexes can be formed with MMP2 until competing co-chaperone AHA1 switches MMP2 activity back on. The equilibrium between TIMP2 and AHA1 and possibly other unknown extracellular signals or modifications maintain the HSP90:client complexes in a standby mode. CAT, catalytic; CTD, C-terminal domain.
Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S7
To gain further insight of this mechanism in vivo, TIMP2−/− MEF, MMP2−/− MEF, and AHA1−/− MAF CM were treated exogenously with MMP2 or co-chaperones (Figure 6A). Gelatinolytic activity was measured by in situ zymography using a DQ-gelatin fluorescence substrate assay. The activity of endogenously secreted gelatinases, MMP2 and MMP9, and their inhibitors, TIMP2 and TIMP1 respectively, were confirmed by gelatin and reverse zymography (Figures S7H–S7K) in indicated CM. It is worth noting that TIMP2−/− MEF and AHA1−/− MAF CM secrete larger amounts of MMP2 than control WT MEFs, whereas the activity of gelatinase MMP9 is insignificant (Figures S7H and S7I). Similarly, MMP2−/−MEF and AHA1−/− MAF CM also contain high amounts of TIMP2 and TIMP1 and are comparable to WT MEFs. Finally, the added amounts of active MMP2 in MMP2−/− MEFs or TIMP2 in TIMP2−/− MEFs were well within WT MEF protein levels (Figures S7I and S7J).
To test the impact of TIMP2 in a physiologically relevant set, we activated endogenous MMP2 with the addition of APMA (4-aminophenylmercuric acetate), since MEF TIMP2−/− cells secrete endogenous proMMP2 that has no activity (Figure S7H, green border) (Caterina et al., 2000; Wang et al., 2000). APMA-activated MMP2 CM were supplemented with increasing amounts of TIMP2 for 1 h, and CM were collected to measure gelatinolytic activity (Figures 7B and S7L). The addition of 5 ng/mL TIMP2 that does not dissociate MMP2 from HSP90 (Figure 6B) reduced gelatinolytic activity by almost 40%, suggesting TIMP2 inhibited free, HSP90-unbound active MMP2. However, 50 ng/mL TIMP2 dissociates MMP2 from HSP90 and inhibits gelatinolysis even further. We next tested whether the addition of active MMP2 will result in an inhibitory ternary complex. Active MMP2 at 50 ng/mL increases the gelatinolytic activity in CM of MMP2−/− MEF compared to untreated or 5 ng/mL active MMP2 (Figure 7C). However, 50 ng/mL recombinant active MMP2 alone or in MEF TIMP2−/− (control, in the absence of TIMP2) was over 4-fold more proteolytically active, suggesting that the added MMP2 was inhibited by endogenous TIMP2 in the CM.
We earlier showed that AHA1 replaces TIMP2 (Figures 5F and 6F) and activates the HSP90α-His6:TIMP2:MMP2 complex in vitro (Figure 7A). The addition of increasing amounts of AHA1 protein to AHA1−/−MAFs also enhances gelatinolysis (Figure 7D). It is worth noting that control treatments of 50 ng/mL AHA1 alone (Figure S7M) or in TIMP2−/− MEF CM (in the absence of TIMP2) had no significant impact on gelatinolytic activity (Figure 7D). These data suggest that AHA1 reactivates MMP2 in vitro and in vivo through TIMP2 displacement. We further revealed the opposing functions of TIMP2 and AHA1 in physiological settings by treating MEF WT+/+ or HT1080 cells with anti-TIMP2, anti-AHA1, or immunoglobulin G (IgG) control antibodies (Figures 7E and 7F). Indeed, blocking endogenous secreted TIMP2 increases the gelatinolytic activity in both MEF WT and HT1080 cells, whereas blocking endogenous secreted AHA1 has the opposite effect.
DISCUSSION
By definition, co-chaperones are accessory proteins that facilitate functional flexibility and specificity within the HSP90 chaperone machinery (Sahasrabudhe et al., 2017; Schopf et al., 2017). In here, we have identified TIMP2, a ubiquitous and constitutively expressed secretory protein (Bourboulia and Stetler-Stevenson, 2010; Jackson et al., 2017), as a bona fide extracellular co-chaperone of eHSP90. We have demonstrated that TIMP2 is a stress-inducible protein, since heat shock, the HSP90 inhibitor GB, and other proteotoxic stressors upregulate TIMP2 both at the transcriptional and translational levels. Combined with the data-mining analysis of the previous work by Mahat and co-workers (Mahat et al., 2016), these data indicate that TIMP2 is induced upon stress and that its transcription is enhanced in the presence of HSF1. In pathologies that trigger cell stress such as cancer, where it has been well established that eHSP90 and eHsp70 levels are also increased, our findings become highly relevant (De Maio and Vazquez, 2013; Li et al., 2012b; Song et al., 2010; Wong and Jay, 2016). While cytosolic AHA1 is an established stress-inducible activating co-chaperone (Panaretou et al., 2002; Sahasrabudhe et al., 2017), our study is the first to show that, like TIMP2, extracellular AHA1 (eAHA1) is also upregulated upon heat shock and drug treatment. Pharmacologic inhibition of eHSP90 would, therefore, increase TIMP2 and/or eAHA1 levels, which will consequently impact matrix degradation and tumor cell invasiveness.
A required element in cytosolic HSP90 chaperone function is its ATP binding and hydrolysis (Hessling et al., 2009; Prodromou, 2012). Although it has been suggested that eHSP90 may chaperone clients in an ATP-independent manner (Sims et al., 2011; Song et al., 2010), the role of extracellular ATP (eATP) in eHSP90 function is yet to be determined. Upon stress and diseases such as cancer, cells release more ATP, increasing eATP levels (Guzman-Aranguez et al., 2017; Stagg and Smyth, 2010). While TIMP2 is a secretory protein that regulates MMP2 activity, either directly or through eHSP90, in light of our data, we can speculate that eHSP90 in association with TIMP2 has higher affinity toward the limited available amounts of eATP, kick-starting an optimum chaperoning function toward eHSP90 clients. While the extracellular role of AHA1 remains to be uncovered, its secreted levels appear to be critical for the regulation of the eHSP90:MMP2 activity. As MMP/TIMP protein level ratio imbalance alters the ECM composition and facilitates the development of cancer and metastatic disease (Moore and Crocker, 2012), the TIMP2/eAHA1 level ratio may also be an important player in disease progression.
We have shown that the N-domain of TIMP2 interacts with the M-domain of HSP90 and inhibits its ATPase activity. TIMP2 could obstruct the conformational changes in the catalytic loop within the M-domain of HSP90, preventing the release of arginine-400 from its inactivation conformation, therefore inhibiting HSP90. This scenario is further strengthened by the fact that activating co-chaperone AHA1 is also secreted and competes with TIMP2 for binding to eHSP90. Although the V411E mutation may have prevented TIMP2 from binding to HSP90, our data indicate that TIMP2 could occupy the same epitope in the M-domain of HSP90 as the one that AHA1 binds to. This appears to provide TIMP2 with a scaffolding function that loads MMP2 to eHSP90 in order to form a HSP90:TIMP2:MMP2 complex. Based on our findings here and also previously published work (Song et al., 2010), we speculate that the N-domain of TIMP2 binds to the active site (N-domain) of MMP2 while the C-domain of MMP2 interacts with eHSP90. Our claim is further strengthened by the fact that MMP2 in this complex is in a standby inhibitory state, which can be reversed by the co-chaperone AHA1. This leads to displacement of TIMP2 and subsequent reactivation of MMP2 in complex with eHSP90 (Figure 7G).
TIMP2 co-chaperone functions as a disruptor of the MMP2:eHSP90 complex. TIMP2 appears to bind independently to both MMP2 and eHSP90. It is unclear whether TIMP2’s association with MMP2 and eHSP90 occurs sequentially or simultaneously. Irrespective of the order of events, the addition of TIMP2 disrupts this complex and ultimately leads to the inhibition of MMP2 activity. These data become more physiologically relevant as eHSP90 associates with MMP2 and promotes its stability. This extracellular stable protease pool drives pathological processes including cell migration, invasion, and angiogenesis in cancer. We can deduce that upregulation of the inhibitor TIMP2 will suppress the enhanced proteolytic and invasive environment. Recent studies have reported AHA1 in promoting cell migration and invasion (Desjardins et al., 2012; Ghosh et al., 2015). Our observation that eAHA1 promotes matrix degradation indicates that eAHA1 may in fact achieve this through the reactivation of gelatinase MMP2.
In conclusion, eHSP90 appears to utilize two secreted co-chaperones with opposing functions, TIMP2 and AHA1, in order to become a more beneficial chaperone for MMP2. Ultimately, this interplay determines client stability and net activity by co-chaperone-mediated MMP2 client inhibition and reactivation when in complex with HSP90. The question remains whether this mechanism also applies in a similar fashion to other eHSP90 client proteins.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Any further requests or information about reagents or resources should be directed to and will be fulfilled by the Lead Contact; Dr. Dimitra Bourboulia, (bourmpod@upstate.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian Cell Culture
Cultured cell lines: human embryonic kidney (HEK293), hsf1+/+ and hsf1−/− murine embryonic fibroblasts (MEF) (McMillan et al., 1998), WT MEF, TIMP2−/− ras/myc MEF (Wang et al., 2000), MMP2−/− ras/myc MEF, AHA1−/−murine adult fibroblasts (MAF) (Echeverría et al., 2011) and human fibrosarcoma HT1080 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Sigma–Aldrich) supplemented with 10% fetal bovine serum (FBS, Sigma–Aldrich). hsf1+/+ and hsf1−/− MEF were cultured in high glucose DMEM (Sigma) containing 10% FBS (Biowest), 2mM L-glutamine (Sigma), streptomycin (100μg/ml) penicillin (100U/ml) (both from VWR), and supplemented with 1xMEM nonessential amino acid solution (Sigma). HEK293 were acquired from (American Type Culture Collection, ATCC). The TIMP2−/−ras/myc MEF was a generous gift from Prof. Paul Soloway, Cornell University. The MMP2−/− ras/myc MEF was a generous gift from Prof. Chris Overall, The University of British Columbia. The AHA1−/− MAF was a generous gift from Prof. Didier Picard and Dr. Pablo Echeverria, University of Geneva. All cell lines were maintained in a CellQ incubator (Panasonic Healthcare) at 37°C in an atmosphere containing 5% CO2.
METHOD DETAILS
Mammalian Protein Expression
All exogenously expressed proteins were prepared from transiently transfected HEK293 cells. Plasmids (see Key Resources Table) were transfected using TransiiT-2020 Reagent with 2μg plasmid as per manufacturer’s protocol. Cells were washed and serum starved with serum free media. Cell-released proteins generated cell-conditioned media (CM). The experiments performed using cell CM represent our in vivo studies. Cell extracts and CM were collected as listed below.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-rabbit TIMP1 | Abcam | Cat# AB1827; RRID: AB_302627 |
| Anti-rabbit TIMP2 | Cell Signaling | Cat #5738s; RRID: AB_10694774 |
| Anti-rabbit TIMP3 | Cell Signaling Technology | Cat# 5673s; RRID: AB_10694530 |
| Anti-rabbit TIMP4 | NOVUS Biologicals | Cat# AF974; RRID: AB_2205240 |
| Anti-rabbit MMP2 | Cell Signaling Technology | Cat# 13132S; RRID: AB_2798128 |
| Anti-mouse MMP2 | Millipore | Cat# MAB3308; RRID: AB_2235453 |
| Anti-rabbit HSP70 | StressMarq Biosciences | Cat# SPC-103; RRID:AB_2570584 |
| Anti-rabbit AHA1 | Stress Marq Biosciences | Cat# SPC-183D; RRID:AB_2224092 |
| Anti-mouse AHA1 | Stress Marq Biosciences | Cat# SMC-172; RRID:AB_2242422 |
| Anti-rabbit FLAG tag | Thermo Scientific | Cat# PA1–984B; RRID:AB_347227 |
| Anti-mouse 6x-His epitope tag (HIS.H8) | Thermo Scientific | Cat# MA1–21315; RRID:AB_557403 |
| Anti-rat HSP90 (16F1) | Enzo Life Sciences | Cat# ADI-SPA-835; RRID:AB_11181205 |
| Anti-mouse GAPDH (1D4) | Enzo Life Sciences | Cat# ADI-CSA-335; RRID:AB_10617247 |
| Anti-rabbit Phospho-Akt S473 (D9E) | Cell Signaling Technology | Cat# 2289; RRID:AB_2315049 |
| Anti-mouse Akt (2H10) | Cell Signaling Technology | Cat# 2967; RRID:AB_331160 |
| Anti-rabbit HA-Tag (C29F4) | Cell Signaling Technology | Cat# 3724; RRID:AB_1549585 |
| Anti-mouse secondary | Santa Cruz Biotech | Cat# sc-2005; RRID:AB_631736 |
| Anti-rabbit secondary | Santa Cruz Biotech | Cat# sc-2004; RRID:AB_631746 |
| Anti-rat secondary | Santa Cruz Biotech | Cat# sc-2006; RRID:AB_1125219 |
| Mouse IgG1 isotype | R&D Systems | Cat# MAB002; RRID: AB_357334 |
| Goat IgG, isotype | Abcam | Cat# AB37373 |
| Anti-Goat TIMP2 | R&D Systems | Cat# AF971-TM; RRID: AB_355752 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ganetespib | Madrigal Pharma (formerly Synta Pharmaceuticals) | STA-9090 CAS# 888216-25-9 (Ying et al., 2012) |
| Ganetespib, Biotinylated | Madrigal Pharma (formerly Synta Pharmaceuticals) | STA-12–7191 (Ying et al., 2012) |
| Fluorescent Substrate: Dabcyl-GPLGMRGK(5FAM)-NH2 | BioZyme Inc. | Cat# PEPDAB011 |
| MMP-2 (Active) | Sigma-Aldrich | SRP3118 |
| Pro-MMP-2 | Sigma-Aldrich | PF023–5UG |
| TIMP1 | R&D | Q6FGX5 |
| TIMP2, 21.8kDa, E.coli, active | ProSpec | ENZ-782 |
| TIMP2, 21.8kDa | R&D | P16035 |
| TIMP2-His6 | NCI, Dr W. Stetler-Stevenson | (Yamada et al., 2018) |
| TIMP3 | R&D | P35625 |
| TIMP4 | R&D | Q99727 |
| hAHA1 | In House | (Dunn et al., 2015) |
| HSP90His6 | In House | (Woodford et al., 2017) |
| AMP-PNP | Sigma | Cat# 101025470001 |
| Adenosine 5'-triphosphate (ATP) disodium salt hydrate | Sigma | Cat# FLAAS |
| Celastrol | Sigma | Cat# C0869 |
| CdSO4 | Sigma | Cat# 481882 |
| Critical Commercial Assays | ||
| Mirus TransIT-2020 | MirusBio | Cat# MIR5405 |
| PiPer Phosphate Assay | ThermoFisher Scientific | Cat# P22061 |
| Anti-FLAG M2 affinity gel | Sigma-Aldrich | Cat# A2220 |
| Bradford Assay | Bio-Rad | Cat# 5000205 |
| Protein G agarose | ThermoFisher Scientific | Cat# 15-920-010 |
| ATP agarose | Novus Biologicals | Cat# 510–0002 |
| Strep agarose | ThermoFisher Scientific | Cat# 20349 |
| Ni-NTA Agarose | ThermoFisher Scientific | Cat# 88221 |
| iScript cDNA Synthesis Kit | BIO-RAD | Cat# 170–8891 |
| iTaq SYBR GREEN | BIO-RAD | Cat# 172–5121 |
| Purelink RNA mini-kit | Ambion | Cat# 12183025 |
| Total MMP-2 Quantikine ELISA Kit | R&D | Cat# MMP200 |
| Mouse HSP90/Heat shock protein 90 Elisa Kit (Sandwich ELISA) | LSBio | Cat#LS-F21385 |
| Dq Gelatin – EnzChek Gelatinase/Collagenase Assay Kit | Molecular Probes | Cat# D-12054 |
| SensiFAST SYBR lo-ROX kit | Bioline | Cat# BIO-94005 |
| RNeasy mini kit | QIAGEN | Cat# 74136 |
| Pierce LDH Cytotoxicity Assay Kit | ThermoScientific | Cat#88953 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat# CRL-1573 |
| HT1080 | ATCC | Cat# CCL-121 |
| TIMP2 (−/−) MEF | Cornell University Dr. Paul D. Soloway | (Wang et al., 2000) |
| AHA1 (−/−) MAF | University of Geneva, Switzerland Dr. Didier Picard | (Echeverrıá et al., 2011) |
| MMP2 (−/−) MEF | University of British Columbia, Dr. Christopher Overall | N/A |
| MEF WT | Brigham & Womens Hospital, Boston, Dr. David Kwiatkowski | (Woodford et al., 2017) |
| Oligonucleotides | ||
| qRT-PCR Primers | Eurofins Genomics | See Table S1 |
| Recombinant DNA | ||
| pcDNA3-HSP90α-HA | (Woodford et al., 2017) | N/A |
| pcDNA3-HSP90α-His6 | (Woodford et al., 2016) | N/A |
| pcDNA3-HSP90α-Flag | (Woodford et al., 2017) | N/A |
| pcDNA3-HSP90α-E47A | (Woodford et al., 2017) | N/A |
| pcDNA3-HSP90α-D93A | (Woodford et al., 2017) | N/A |
| pcDNA3-HSP90α-V411E | (Woodford et al., 2017) | N/A |
| pcDNA3-C-HSP90-FLAG | (Woodford et al., 2016) | N/A |
| pcDNA3-M-HSP90-FLAG | (Woodford et al., 2016) | N/A |
| pcDNA3-N-HSP90-FLAG | (Woodford et al., 2016) | N/A |
| pcDNA3-TIMP2-His6 | This Work | N/A |
| pcDNA3-N-TIMP2-His6 | This Work | N/A |
| pcDNA3-C-TIMP2-His6 | This Work | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | Graphpad Software | https://www.graphpad.com |
| Nanoanalyse | TA Instruments | https://www.tainstruments.com |
| ImageJ | ImageJ Developers (NIH) | https://imagej.nih.gov/ij/ |
| Integrative Genomics Viewer (IGV) | Broad Institute and the Regents of the University of California | http://software.broadinstitute.org/software/igv |
Immunoblotting and Protein Extraction
Immunoblotting and cellular protein extraction was carried out as previously described in (Dunn et al., 2015). Cell conditioned media (CM) was collected following serum starvation of cells with media containing no supplementing fetal bovine serum (FBS) (Cortes et al., 2018). The CM was centrifuged at 1000rpm for 5–10 mins at 4°C to remove pelleted cells, preventing contamination from cell extracts. CM were concentrated 10x using Amicon Ultra 3K or 10K centrifugal filters (Millipore) according to the manufacturer’s protocol. Proteins were separated by SDS-PAGE, and either stained with Coomassie Blue Stain for loading control, or transferred to nitrocellulose membrane, and detected by immunoblotting with antibodies listed in Key Resources Table.
Ni-NTA Pulldown and Immunoprecipitation
IPs and pulldowns of both in vitro purified proteins, and in vivo cell extracts and conditioned media were performed as described in (Sá nchez-Pozo et al., 2018; Woodford et al., 2017). For immunoprecipitation, cell extracts and CM were incubated with anti-FLAG conjugate beads or anti-HA conjugate beads for 2 hours at 4°C or with HSP90α or TIMP2 antibodies for 1hr at 4°C followed by protein G agarose for 2 hours at 4°C. Pulldowns were prepared following incubation with Ni-NTA agarose or ATP conjugated agarose for 2 hours at 4°C. Pellets were washed 4 times with fresh lysis buffer (20mM HEPES pH7.0, 100mM NaCl, 1mM MgCl2, 0.1% NP40, protease inhibitor cocktail (Roche), and PhosSTOP (Roche)) and eluted in 5x Laemmli buffer. The samples were then boiled and immunoblotted.
RT-PCR
Following serum starvation, HEK293 cells were treated with either 1μM of HSP90 inhibitor Ganetespib, 1μM of biotinylated-Ganetespib or DMSO control. Following 8 or 24 hours treatment, extracts were collected. RNA was extracted using RNA extraction mini-kit, and quantified using NanoQuant (TECAN). 1μg of RNA from each sample was reverse transcribed into cDNA using iScript cDNA synthesis kit (BIO-RAD). RT-qPCR analysis was performed using nucleic acid stain SYBR-GREEN (BIO-RAD) and specific qPCR oligonucleotide primers: GAPDH, TIMP1, TIMP2, TIMP3, TIMP4 and HSP70 control. mRNA expression levels were analyzed over GAPDH control.
Drug Treatment of HEK293 and HT1080 Human Fibrosarcoma
HEK293 and HT1080 fibrosarcoma cells were cultured to a confluency of 70%. Cells were subsequently serum starved and treated with DMSO (negative control) or 1–2 μM Ganetespib. Subsequently cells were cultured for a further 24 hours in unsupplemented DMEM. Cell extracts and conditioned media were collected from cells for immunoblotting.
Heat shock of HEK293 cells
HEK293 cells were heat shocked at 42°C in a water bath for 1 hour. Cell media were replaced with serum free media and incubated at 37°C for 24 hours. CM and cell extracts were collected and analyzed by immunoblotting.
Hsf1, stressors and qRTPCR
For treatments, 0.5×106 hsf1+/+ and hsf1−/− MEF cells were used. Heat shock was conducted in a water bath at 42°C for 1 h. CdSO4 (Sigma), dissolved in sterile water, was used at a concentration of 60 μM for 3 h, while celastrol (Sigma) dissolved in DMSO was used at a concentration of 4 μM for 2 h. A control sample was treated with vehicle (DMSO).
RNA was isolated using an RNeasy mini kit (QIAGEN) according to the manufacturer’s instructions and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Then, one μg of total RNA was reverse transcribed with an iScript kit (Bio-Rad). SensiFAST Probe lo-rox kit and SensiFAST SYBR lo-ROX kit (Bioline) were used for qRT-PCRs that were performed with QuantStudio3 real-time PCR systems (Applied Biosystems). Primers and probes were purchased from Sigma (see Key Resources Table). mRNA expression levels were analyzed over 18S RNA (RNA18S5) control. All reactions were run in triplicate from samples derived from four biological replicates.
Isothermal titration calorimetry
Isothermal titration calorimetry was performed as described (Garnier et al., 2002). TIMP2 was pre- incubated with HSP90 overnight at 4°C prior to ATP injection. Binding of ATP to HSP90α-His6 and TIMP2-His6: HSP90α-His6 was carried out following overnight dialysis in 1 L reaction buffer (20mM Tris 7.4, 100mM NaCl, 5mM MgCl2) using a membrane with a cut off of 12–14 kDa at 4°C to remove any residual ADP or ATP bound to HSP90. Reactions were carried out using Affinity ITC Auto system, TA instruments. Dissociation constant KD was calculated following 30×1μl microinjections of 4mM ATP into a calorimetric cell containing a 10 μM solution of HSP90-His6 or TIMP2-His6:HSP90-His6 from a 200μl syringe at 25°C. The heat of dilution was obtained by injecting the same ATP solution into a calorimetric cell containing either reaction buffer or TIMP2-His6 alone. This resultant baseline was subtracted from titration curves before they were fit using Nanoanalyze, TA Instruments using a single class of site.
ATPase Assay of human HSP90
The ATPase activity of Hsp90 in the presence of potential mediators was performed as described in (Dunn et al., 2015). ATPase activity of human HSP90α with and without recombinant TIMP1 or TIMP2 was measured as described in the PiPer Phosphate Assay Kit instructions for use (Life Technologies). In brief, HSP90α-FLAG was isolated from cell extracts or conditioned media of HEK293 cells transiently transfect with 3μg of HSP90α-FLAG plasmid. The purity of the isolated HSP90 proteins was examined by Coomassie staining of SDS-PAGE gels using GelCode Blue Safe Protein Stain (Thermo Scientific). Triplicate samples of 1μg HSP90α and 10μM ganetespib (GB) or 1ng (0.4nM) or 10ng (4.4nM) TIMP2 or TIMP1 were pre-incubated on ice for 1 hour. Samples were loaded into a 96-well plate and freshly prepared 2x PiPer reaction mixture containing 1mM ATP was added. 96-well plate was then wrapped in foil and incubated at 37°C. Absorbance at 565nm was read on a Tecan infinite M200 Pro plate reader after 60 minutes. Standard curve with linear fit line was created from 0 to 100μM final concentration reactions. ATP turnover was calculated as mmol Pi per mol HSP90α per minute and relative ATPase activity was calculated from those values, with the value of HSP90α alone representing 100% activity.
Biotinylated-drug binding assay
Assay was performed as described in (Dunn et al., 2015). Recombinant HSP90α was incubated with and without recombinant TIMP2 for 1 hour at 4°C. Followed by a further 1 hour incubation with Bio-GB at varying concentrations, the complexes were incubated with streptavidin agarose beads. TIMP2−/− MEF cells were cultured and cell extracts and CM were collected. Extracts and CM were incubated with 100nM Bio-GB for 1 hour at 4°C and further incubated with streptavidin agarose beads. The proteins were then pulldown and immunoblotted.
In vivo TIMP2:eHSP90 ATP pulldown
Wild-type MEF and TIMP2−/− MEF were serum starved and treated with varying concentrations of recombinant TIMP2 or TIMP2 before CM was collected. The CM was then incubated with ATP beads. The proteins were then pulldown and immunoblotted.
In vitro interactions of HSP90, MMP2, TIMP2 and AHA1
100ng of HSP90α-His6 was bound to Ni-NTA agarose, incubated for 1 hour at 4°C and washed 5 times with protein lysis buffer. To form complexes, proMMP2, active MMP2, TIMP2, AHA1 and HSP90 inhibitor Ganetespib were individually added to the beads at varying concentrations (indicated in figure legends). Each time a protein was added it was incubated for 1 hour at 4°C and washed 5 times with protein lysis buffer. The resultant complexes were mixed with protein loading buffer, boiled for 5mins and immunoblotted.
In vivo interaction of MMP2, TIMP2, HSP90, and AHA1
MMP2−/− MEFs and AHA1−/− MAFs were cultured to a confluency of ∼80% in supplemented DMEM. They were subsequently serum-starved in non-supplemented DMEM for 24 hours. Prior to collection, cells were treated with recombinant MMP2 and AHA1 proteins (0–50ng/ml), respectively, for 2 hours. Conditioned media was collected, from cells (alongside cell extracts) and concentrated to 10x. Samples were endogenously immunoprecipitated using anti-HSP9α antibody and resultantly immunoblotted for TIMP2, MMP2, and AHA1 co-immunoprecipitation.
Fluorometric Enzyme Activity Assays
Samples of 62kDa MMP2 were diluted to 25ng/ml in TIMP2 reconstitution buffer (50mM Tris 7.4, 150mM NaCl, 5mM CaCl2, 0.05% Brij-35), and added 1:1 with fluorescent substrate in fluorescent buffer (50mM Tris 7.4, 150mM NaCl, 2mM CaCl2, 5μM Zinc Sulfate), to 96-well optical bottom plate. Fluorescent peptide (Dabcyl-GPLGMRGK(5FAM)-NH2) (in 50mM Tris 7.4, 150mM NaCl, 2mM CaCl2, 5mM ZnSO4) was diluted in DMSO to a concentration of 10mM. Further dilutions were made to 10μM in fluorescent peptide buffer (50mM Tris 7.4, 150mM NaCl, 2mM CaCl2, 5mM ZnSO4). Unless otherwise stated, 0.28nM of HSP90 and 0.2nM of MMP2 were incubated to form HSP90:MMP2 complexes. Prior to dilution, HSP90 or TIMP2 were added to MMP2 and rotated for up to an hour at 4°C.
Alternatively, HSP90-His6 was bound to Ni-NTA beads as described in in-vitro pulldown assays. Following subsequent rotation steps with TIMP2 and MMP2, AHA1 was added at varying concentrations. Following the final rotation, the samples are washed with 0.1% NP40 extraction buffer and finally eluted in TIMP2 reconstitution buffer. 50μl of the slurry was loaded onto a 96-well optical bottom plate. Samples of HSP90α:MMP2 that were incubated at 37°C prior to dilution to 25ng/ml were collected at varying time points up to 1 hour. Fluorescent substrate was then added 1:1 as described above. The plate was loaded into a SpectraMax i3, plate reader (Molecular Devices). Samples were analyzed at an excitation and emission wavelength – 485 and 530nm respectively, every 5 minutes for 30–60 min, at room temperature. Following linear regression, experiments were either modeled to the Michaelis-Menten approach or changes seen in the percentage activity of a MMP2 bound complex in respect to a sole MMP2 control.
Antibody treatments of cells
MEF WT +/+ and HT1080 fibrosarcoma cells, were cultured to a confluency ∼70%–80%. Cells were serum starved in media containing either: no treatment (PBS), IgG (isotype control), or antibody against AHA1 (20 μg/ml) or TIMP2 (10 μg/ml). Conditioned media was collected 24 hours later and concentrated to 10x followed by DQ gelatin degradation assay.
DQ gelatin degradation assay
A DQ-gelatin degradation assay was performed to measure net gelatinolytic activity of 10x cell conditioned media using the EnzCheck® Gelatinase Assay kit according to manufacturer’s instructions (Molecular Probes). To measure the degradation in fluorescence units, a standard curve was generated using different concentrations of active MMP2 (0–250ng/ml) added at 100μl in a black 96-well plate. Serum starved, untreated and treated 10x CM were diluted 1:5 in fluorescent peptide buffer (50mM Tris 7.4, 150mM NaCl, 2mM CaCl2, 5μM ZnSO4) and plated at 100μl onto a 96 well black bottom plate. DQ gelatin substrate was diluted to a 1mg/ml stock in ddH2O followed by subsequent 1:5 dilution in fluorescent peptide buffer (50mM Tris 7.4, 150mM NaCl, 2mM CaCl2, 5μM ZnSO4). Wells were volume up to 200μl with DQ gelatin, 100μg/ml on plate concentration. Plates were covered and incubated for 18 hours at 37°C before analysis at excitation and emission wavelengths of: 486nM and 532nM respectively. End point measures are described in the resultant figures. Graphs and calculations were obtained with Prism 7 (GraphPad Software, Inc.).
Gelatin Zymography
Gelatinase activity of recombinant active MMP2 following stability experiments was determined by gelatin zymography as previously described (Kleiner and Stetler-Stevenson, 1994; Sánchez-Pozo et al., 2018). Following incubation at 37°C, 2ng of active MMP2 alone or in complex with HSP90α were prepared with 5x Laemmli protein loading buffer in non-denaturing non-reducing conditions. Samples were subjected to electrophoresis using 8% acrylamide gels containing 0.1% gelatin. The gels were incubated for 30 min at room temperature in zymogram renaturing buffer (Novex, Invitrogen), 30 min at room temperature in zymogram developing buffer (Novex, Invitrogen), and then transferred to fresh zymogram developing buffer for overnight incubation at 37°C. Gels were then stained with Coomassie Brilliant Blue R-250 (Bio-Rad) and briefly destained in 10% acetic acid, 40% methanol and distilled water. They were imaged using an Epson Perfection V700 scanner. Gelatinase activity was detected as transparent bands on a dark background. Recombinant human proMMP2 was run alongside as a control to confirm the identity of MMP2 in the samples.
Gelatinase activity in CM of TIMP2−/− MEF, MMP2−/− MEF or AHA1−/− MAF following exogenous addition of recombinant TIMP2, MMP2 or AHA1 proteins was determined in 10x CM. Equal amounts of media were analyzed and compared to WT MEF and known amount of recombinant proMMP2 as control.
Reverse Zymography
Reverse gelatin zymography was performed to test TIMP2 inhibitory function toward MMP2 in CM (Sánchez-Pozo et al., 2018). CM were collected from cells, spun down to remove cell pellets and concentrated 10x using Amicon Ultra 3K centrifugal filters (Millipore) according to the manufacturer’s protocol. Equal amounts of CM were run in 15% acrylamide gels containing 0.225% gelatin (Sigma) and 50 ng/ml recombinant proMMP2. The gels were incubated for 2 hours at room temperature in zymogram renaturing buffer, 30 min at room temperature in zymogram developing buffer, and then transferred to fresh zymogram developing buffer for overnight incubation at 37°C. Gels were stained and imaged as described in gelatin zymography. TIMP2 inhibitory activity was detected as dark positive staining bands over a clear background. Recombinant human TIMP2 (Abcam) was run alongside purified TIMP2-His6 mutants as a positive control.
Bioinformatics
ChIP-seq and PRO-seq data (GEO accession number GSE71708) were downloaded from GEO database. Reads from ChIP-seq experiment were mapped to the mouse genome mm10 using Bowtie 2 with default parameters. Aligned reads were converted to bedgraphs using Samtools (sort command) and Bedtools (genomecov command) with default parameters. ChIP-seq peaks were visualized by uploading bedgraphs to Integrative Genomics Viewer (IGV), http://software.broadinstitute.org/software/igv/. PRO-seq data were visualized by uploading existing bigwig files from GEO database to IGV.
LDH Cytotoxicity Assay
Cell cytotoxicity assays were performed using the Pierce LDH Cytotoxicity Assay Kit (ThermoScientific) as described by the manufacturers protocols. To calculate an optimum number of cells to utilize in the assay, varying amounts of cells, 0–10,000 for both HEK293 and HT1080 fibrosarcoma cells in a 96 well plate. Measurements of absorbance were taken at 490nm and 680nm.
We subsequently plated 8,000 cells of both HT1080 and HEK293 cells, in a 96 well plate and followed manufacturers protocol for the chemical-compound mediated cytotoxicity. Cells were cultured for 24hours and subsequently serum starved. Ganetespib 1nM-10 μM or DMSO control were added at different concentrations. Values represent the percentage of 100% maximum LDH release, lysis control.
Native-PAGE Electrophoresis
HEK293 cells were transiently transfected with HSP90α-His6 prior to serum starvation. Following 24 hours culture in unsupplemented DMEM, cell conditioned media was collected. 100X concentrated CM and recombinant human HSP90α expressed in E.coli were separated on Native-PAGE gel. The gel was subsequently transferred to nitrocellulose membrane and Ponceau Stained or immunoblotted as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistics were performed using GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla California USA, https://www.graphpad.com. Statistical significance was ascertained using a simple Student’s t test, multiple comparisons were ascertained using Holm-Sidak correction. Significance was denoted as asterisks in each figure: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Error bars indicate the standard error of the mean (SEM). All band quantification was performed using ImageJ-2, as described (Rueden et al., 2017). Replicates were independent (n = 3) unless otherwise stated in the figure legend.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets nor unique computer codes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Paul Soloway (Cornell University) for the TIMP2−/− ras/myc MEF cell line, Prof. Chris Overall (University of British Columbia) for the MMP2−/− ras/myc MEF cell line, and Prof. Didier Picard and Dr. Pablo Echeverria (University of Geneva) for the AHA1−/− MAF cell line. M.A.B. and S.V.H. were funded by Åbo Akademi University. The laboratory of L.S. is supported by the Academy of Finland, the Sigrid Juseélius Foundation, the Magnus Ehrnrooth Foundation, and Cancer Foundation Finland. This work was supported by the SUNY Upstate Medical University, The Upstate Foundation, the SUNY Research Foundation, and Upstate Breast Cancer Research funds (D.B. and M.M.); a Carol M. Baldwin Breast Cancer Research Fund grant (D.B. and M.M.); and the Associated Medical Schools of New York (D.B.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.045.
REFERENCES
- Bourboulia D, and Stetler-Stevenson WG (2010). Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol 20, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew K, and Nagase H (2010). The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzyński MA, and Sistonen L (2017). Versatile functions of heat shock factors: it is not all about stress. Curr. Immunol. Rev 13, 4–18. [Google Scholar]
- Caterina JJ, Yamada S, Caterina NC, Longenecker G, Holmbäck K, Shi J, Yermovsky AE, Engler JA, and Birkedal-Hansen H (2000). Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2(Timp-2) gene alters proMMP-2 activation. J. Biol. Chem 275, 26416–26422. [DOI] [PubMed] [Google Scholar]
- Cortes S, Baker-Williams AJ, Mollapour M, and Bourboulia D (2018). Detection and Analysis of Extracellular Hsp90 (eHsp90). Methods Mol. Biol. 1709, 321–329. [DOI] [PubMed] [Google Scholar]
- de la Mare JA, Jurgens T, and Edkins AL (2017). Extracellular Hsp90 and TGFβ regulate adhesion, migration and anchorage independent growth in a paired colon cancer cell line model. BMC Cancer 17, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio A, and Vazquez D (2013). Extracellular heat shock proteins: a new location, a new function. Shock 40, 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins F, Delisle C, and Gratton JP (2012). Modulation of the cochaperone AHA1 regulates heat-shock protein 90 and endothelial NO synthase activation by vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol 32, 2484–2492. [DOI] [PubMed] [Google Scholar]
- Dong H, Zou M, Bhatia A, Jayaprakash P, Hofman F, Ying Q, Chen M, Woodley DT, and Li W (2016). Breast cancer MDA-MB-231 cells use secreted heat shock protein-90alpha (Hsp90α) to survive a hostile hypoxic environment. Sci. Rep 6, 20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn DM, Woodford MR, Truman AW, Jensen SM, Schulman J, Caza T, Remillard TC, Loiselle D, Wolfgeher D, Blagg BSJ, et al. (2015). c-Abl mediated tyrosine phosphorylation of Aha1 activates its cochaperone function in cancer cells. Cell Rep. 12, 1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverría PC, Bernthaler A, Dupuis P, Mayer B, and Picard D (2011). An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS ONE 6, e26044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hamidieh A, Grammatikakis N, and Patsavoudi E (2012). Cell surface Cdc37 participates in extracellular HSP90 mediated cancer cell invasion. PLoS ONE 7, e42722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, et al. (2004). Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat. Cell Biol 6, 507–514. [DOI] [PubMed] [Google Scholar]
- Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, Briand C, Makarov AA, Catelli MG, and Peyrot V (2002). Binding of ATP to heat shock protein 90: evidence for an ATP-binding site in the C-terminal domain. J. Biol. Chem 277, 12208–12214. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Shinogle HE, Garg G, Vielhauer GA, Holzbeierlein JM, Dobrowsky RT, and Blagg BS (2015). Hsp90 C-terminal inhibitors exhibit antimigratory activity by disrupting the Hsp90α/Aha1 complex in PC3-MM2 cells. ACS Chem. Biol 10, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Aranguez A, Pérez de Lara MJ, and Pintor J (2017). Hyperosmotic stress induces ATP release and changes in P2X7 receptor levels in human corneal and conjunctival epithelial cells. Purinergic Signal. 13, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hance MW, Dole K, Gopal U, Bohonowych JE, Jezierska-Drutel A, Neumann CA, Liu H, Garraway IP, and Isaacs JS (2012). Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem 287, 37732–37744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessling M, Richter K, and Buchner J (2009). Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol 16, 287–293. [DOI] [PubMed] [Google Scholar]
- Jackson HW, Defamie V, Waterhouse P, and Khokha R (2017). TIMPs: versatile extracellular regulators in cancer. Nat. Rev. Cancer 17, 38–53. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, and Werb Z (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, and Stetler-Stevenson WG (1994). Quantitative zymography: detection of picogram quantities of gelatinases. Anal. Biochem 218, 325–329. [DOI] [PubMed] [Google Scholar]
- Li J, Soroka J, and Buchner J (2012a). The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823, 624–635. [DOI] [PubMed] [Google Scholar]
- Li W, Sahu D, and Tsen F (2012b). Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim. Biophys. Acta 1823, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tsen F, Sahu D, Bhatia A, Chen M, Multhoff G, and Woodley DT (2013). Extracellular Hsp90 (eHsp90) as the actual target in clinical trials: intentionally or unintentionally. Int. Rev. Cell Mol. Biol 303, 203–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yan Z, Huang L, Guo M, Zhang Z, and Guo C (2011). Cell surface heat shock protein 90 modulates prostate cancer cell adhesion and invasion through the integrin-β1/focal adhesion kinase/c-Src signaling pathway. Oncol. Rep 25, 1343–1351. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT (2016). Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol. Cell 62, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCready J, Wong DS, Burlison JA, Ying W, and Jay DG (2014). An impermeant ganetespib analog inhibits extracellular Hsp90-mediated cancer cell migration that involves lysyl oxidase 2-like protein. Cancers (Basel) 6, 1031–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin SH, Ventouras LA, Lobbezoo B, and Jackson SE (2004). Independent ATPase activity of Hsp90 subunits creates a flexible assembly platform. J. Mol. Biol 344, 813–826. [DOI] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, and Benjamin IJ (1998). Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem 273, 7523–7528. [DOI] [PubMed] [Google Scholar]
- Meyer P, Prodromou C, Liao C, Hu B, Roe SM, Vaughan CK, Vlasic I, Panaretou B, Piper PW, and Pearl LH (2004). Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami Y, Kawasaki H, Miyata Y, Suzuki K, and Yahara I (1991). Analysis of native forms and isoform compositions of the mouse 90-kDa heat shock protein, HSP90. J. Biol. Chem 266, 10099–10103. [PubMed] [Google Scholar]
- Moore CS, and Crocker SJ (2012). An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol 180, 12–16. [DOI] [PubMed] [Google Scholar]
- Nemoto T, and Sato N (1998). Oligomeric forms of the 90-kDa heat shock protein. Biochem. J 330, 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MW, Gervasi DC, Mobashery S, and Fridman R (1997). Kinetic analysis of the binding of human matrix metalloproteinase-2 and −9 to tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. J. Biol. Chem 272, 29975–29983. [DOI] [PubMed] [Google Scholar]
- Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, and Pearl LH (1998). ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 17, 4829–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, Singh S, Millson SH, Clarke PA, Naaby-Hansen S, Stein R, et al. (2002). Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 10, 1307–1318. [DOI] [PubMed] [Google Scholar]
- Prodromou C (2012). The ‘active life’ of Hsp90 complexes. Biochim. Biophys. Acta 1823, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, and Pearl LH (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75. [DOI] [PubMed] [Google Scholar]
- Retzlaff M, Hagn F, Mitschke L, Hessling M, Gugel F, Kessler H, Richter K, and Buchner J (2010). Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 37, 344–354. [DOI] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabudhe P, Rohrberg J, Biebl MM, Rutz DA, and Buchner J (2017). The plasticity of the Hsp90 co-chaperone system. Mol. Cell 67, 947–961.e945. [DOI] [PubMed] [Google Scholar]
- Sánchez-Pozo J, Baker-Williams AJ, Woodford MR, Bullard R, Wei B, Mollapour M, Stetler-Stevenson WG, Bratslavsky G, and Bourboulia D (2018). Extracellular phosphorylation of TIMP-2 by secreted c-Src tyrosine kinase controls MMP-2 activity. iScience 1C, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf FH, Biebl MM, and Buchner J (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol 18, 345–360. [DOI] [PubMed] [Google Scholar]
- Sims JD, McCready J, and Jay DG (2011). Extracellular heat shock protein (Hsp)70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 6, e18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Wang X, Zhuo W, Shi H, Feng D, Sun Y, Liang Y, Fu Y, Zhou D, and Luo Y (2010). The regulatory mechanism of extracellular Hsp90alpha on matrix metalloproteinase-2 processing and tumor angiogenesis. J. Biol. Chem 285, 40039–40049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J, and Smyth MJ (2010). Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29, 5346–5358. [DOI] [PubMed] [Google Scholar]
- Stellas D, El Hamidieh A, and Patsavoudi E (2010). Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, and Sistonen L (2014). HSF1 at a glance. J. Cell Sci 127, 261–266. [DOI] [PubMed] [Google Scholar]
- Visse R, and Nagase H (2003). Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res 92, 827–839. [DOI] [PubMed] [Google Scholar]
- Wang Z, Juttermann R, and Soloway PD (2000). TIMP-2 is required for efficient activation of proMMP-2 in vivo. J. Biol. Chem 275, 26411–26415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DS, and Jay DG (2016). Emerging roles of extracellular Hsp90 in cancer. Adv. Cancer Res 129, 141–163. [DOI] [PubMed] [Google Scholar]
- Woodford MR, Dunn DM, Blanden AR, Capriotti D, Loiselle D, Prodromou C, Panaretou B, Hughes PF, Smith A, Ackerman W, et al. (2016). The FNIP co-chaperones decelerate the Hsp90 chaperone cycle and enhance drug binding. Nat. Commun 7, 12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford MR, Sager RA, Marris E, Dunn DM, Blanden AR, Murphy RL, Rensing N, Shapiro O, Panaretou B, Prodromou C, et al. (2017). Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 36, 3650–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Chowdhury A, Schneider JP, and Stetler-Stevenson WG (2018). Macromolecule-network electrostatics controlling delivery of the biotherapeutic cell modulator TIMP-2. Biomacromolecules 19, 1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Du Z, Sun L, Foley KP, Proia DA, Blackman RK, Zhou D, Inoue T, Tatsuta N, Sang J, et al. (2012). Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther 11, 475–484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets nor unique computer codes.