

SUMMARY
Tumor cells encounter a myriad of physical cues upon arrest and extravasation in capillary beds. Here, we examined the role of physical factors in non-random organ colonization using a zebrafish xenograft model. We observed a two-step process by which mammalian mammary tumor cells showed non-random organ colonization. Initial homing was driven by vessel architecture, where greater numbers of cells became arrested in the topographically disordered blood vessels of the caudal vascular plexus (CVP) than in the linear vessels in the brain. Following arrest, bone-marrow- and brain-tropic clones exhibited organ-specific patterns of extravasation. Extravasation was mediated by β1 integrin, where knockdown of β1 integrin reduced extravasation in the CVP but did not affect extravasation of a brain-tropic clone in the brain. In contrast, silencing myosin 1B redirected early colonization from the brain to the CVP. Our results suggest that organ selectivity is driven by both vessel topography and cell-type-dependent extravasation.
INTRODUCTION
Metastasis describes the process whereby cancer cells move from a primary tumor and establish lesions in distinct secondary organs (Nguyen et al., 2009; Obenauf and Massagué, 2015; Steeg, 2016). Metastasis is non-random, with differences in organ targeting (i.e., organ tropism) and clinical latency correlated with initial tumor type in line with Paget’s seed and soil hypothesis (Nguyen et al., 2009; Obenauf and Massagué, 2015; Paget, 1989; Steeg, 2016). Unfortunately, emergent metastatic lesions are often resistant to treatments, even if that treatment was effective in control and eradication of the primary tumor (Correia and Bissell, 2012; Gottesman, 2002; Tanner and Gottesman, 2015). Therefore, understanding how cells target specific organs at the earliest steps of the metastatic cascade, whether differences exist in this targeting, and the factors critical to cell survival following dissemination are critical for developing treatments for metastatic and refractory cancers.
A limited repertoire of isogenic clones exists such that multiple organ-seeking clones were derived from the same parental cell line within the same strain of mouse (Kim et al., 2018; Kusuma et al., 2012; Nguyen et al., 2009; Yoneda et al., 2001; Zhang et al., 2013). From organ-seeking cell line model systems, cell (“seed”) intrinsic differences in genes and signaling pathways that regulate organ specificity have been evaluated (Bos et al., 2009; Kang et al., 2003; Nguyen et al., 2009; Obenauf and Massagué, 2015; Yoneda et al., 2001; Zhang et al., 2013). However, the mechanisms that determine how cells transition from the circulation to successfully colonize the “soil” in distinct organs are not well understood, particularly considering that metastatic lesions are often located in organs that are outside the range of single-cell imaging techniques (Weissleder, 2002).
An important mediator of tumor cell fate that interacts with cell-intrinsic genetics is the microenvironment (Bissell and Hines, 2011; Friedl and Alexander, 2011; Hoshino et al., 2015; Tanner and Gottesman, 2015), and tumor cells along the metastatic cascade encounter different physical microenvironments during capillary arrest and extravasation into organs (Kim and Tanner, 2015; Kumar and Weaver, 2009). One outstanding question is whether organ tropism is initially a passive process whereby disseminated tumor cells (DTCs) arrest within capillary beds due to the layout of the vasculature and size restrictions imposed by blood vessel diameters, or whether selection is instead due to surface markers of the DTCs that allow them to home to specific organs via ligand-receptor interactions between these cells and the luminal walls of the microvasculature (Valastyan and Weinberg, 2011). Following cell arrest, cells must also survive environmental pressures to extravasate into secondary organs.
While steps in early colonization are rare events that are difficult to study using current models (Azevedo et al., 2015), the relatively small size and optical transparency of the larval zebrafish enables imaging of multiple tissues in the same animal at single-cell resolution (Paul et al., 2019; White et al., 2013). The zebrafish is rapidly becoming a model for studying tumor behavior at different stages of the metastatic cascade (Follain et al., 2018; Stoletov et al., 2010, 2007; White et al., 2013), and organs that are frequent sites of metastasis, such as brain and bone marrow niches in human patients, are well conserved between mammals and zebrafish (Meeker and Trede, 2008; Oosterhof et al., 2015). At the cellular level, the zebrafish larval brain shares similarities with the mammalian brain in that the cellular components, for example, microglia, astrocytes, neurons, and blood vessels (Oosterhof et al., 2015), as well as key architectural features, such as a functional blood-brain barrier (Fleming et al., 2013) and choroid vessel, are conserved. The caudal vein plexus (CVP) comprises sinusoidal vessels that surround the caudal hematopoietic tissue (CHT), which is analogous to mammalian fetal liver or bone marrow and harbors hematopoietic stem cells (HSCs), including CD41 positive cells (Murayama et al., 2006). Moreover, recent studies have begun to demonstrate the applicability of the zebrafish model to provide insights relevant to human disease (Fior et al., 2017; Follain et al., 2018; Paul et al., 2019; Roh-Johnson et al., 2017).
Here, we demonstrate that cell line pairs that preferentially target bone marrow and brain niches in mice show similar targeting in the larval fish by 5 days post-injection (dpi) following injection to the circulatory system. We then took advantage of the ability to simultaneously monitor early metastatic events in multiple organs within the same zebrafish larva to examine cell trafficking, arrest, and extravasation of organ-seeking cell lines in the zebrafish brain and CVP. This approach was used to determine where organ selectivity arose and to begin delineating factors influencing non-random organ targeting. We examined both the physical properties of the tissues and the molecular profiles of the organotropic cells and determined that the physical properties of the vessels dictated how cells moved within the vasculature. In addition, greater numbers of cells became arrested within organs with chaotic vessels. This result was observed for all organotropic clones and did not differentiate brain-targeting from bone-marrow-targeting cells. However, differences in extravasation emerged for different cell lines, leading to diverging distributions of brain- or bone-marrow-targeting breast tumor cells in the zebrafish. Proteomic profiling indicated that myosin 1B, an actin-associated molecular motor that has been implicated in metastasis in head and neck cancers (Ohmura et al., 2015; Sherr et al., 1993), was upregulated across multiple brain-tropic clones. Silencing myosin 1B shifted the distribution of cells toward the more chaotic CVP. Conversely, silencing of β1 integrin redirected cell targeting by differentially impacting extravasation in various tissues in a cell-line-dependent manner. Specifically, bone-tropic MDA-MB-231 cell clones, which have greater expression and activation of β1 integrin, showed reduced extravasation throughout the larvae upon silencing of β1 integrin. The extravasation of brain-targeting cells was reduced in the CVP but not in the brain upon integrin inhibition, leading to increased brain targeting. Overall, these data establish zebrafish as a model to study organ-specific patterns of cancer cell dissemination and suggest that both vascular topography and differential extravasation rates contribute to non-random patterns of metastatic spread.
RESULTS
Organ Selectivity during Early Metastasis Is Conserved in a Zebrafish Metastasis Model
The metastatic cascade involves cell invasion or shedding into the circulatory system, arrest in the capillaries of target organs, extravasation into the parenchyma of these organs, and either dormancy or proliferation to establish a secondary lesion (Kim and Tanner, 2015; Kumar and Weaver, 2009). As the key components of the zebrafish brain and hematopoietic niche (in the CVP) have emerged and are functional in larval zebrafish in the first few days after fertilization (Fleming et al., 2013; Goessling and North, 2011; Murayama et al., 2006; Oosterhof et al., 2015), we hypothesized that organotropic cells that target the brain and hematopoietic niche (i.e., bone marrow) in mice would show similar organ targeting in a zebrafish metastasis model.
We injected cancer cell line models of organotropism directly into the circulation of 2 days post-fertilization (dpf) zebrafish with fluorescently labeled vasculature (Figure 1A). We first focused on human MDA-MB-231 brain-tropic (231BR) and bone-marrow-tropic (231BO) cells (Yoneda et al., 2001). In mice, brain-tropic 231BR cells metastasize to the brain at greater rates than parental MDA-MB-231 cells, while bone-marrow-tropic 231BO cells show increased metastasis to the bone marrow (Figure 1B). Fish were imaged at 5 dpi, at an age of 7 dpf, to assess early organ colonization. Brain-tropic and bone-marrow-tropic clones demonstrated different patterns of tissue colonization by 5 dpi (Figures 1C and 1D). As both the brain and CVP of each larva were optically accessible, we classified individual larva based on the presence of cancer cells exclusively in the brain, exclusively in the CVP, or present in both organs (Figure 1E). This classification encompassed quantification of all cells present (i.e., both intravascular and extravascular cells) in a particular organ (Figure 1F). In general, cells were present throughout the larva at 5 dpi, with ~80% of larvae containing cells in both the brain and CVP regardless of cell type (Figure 1E). For both 231BO and 231BR cells, ~20% of larvae contained cells only in the CVP, and no larvae had cancer cells present only in the brain (Figure 1E). We defined a unitless parameter ranging from 0 to 1 based on the relative presence of cells in the brain to the total number of cells in the brain and CVP (Figure 1F). With this unitless parameter, a larva having cells present only in the CVP has a score of 0, while a larva containing cells only in the brain has a score of 1 (Figures 1F and 1G). Larvae with cells in both organs have scores ranging between these values, with the magnitude of the score indicating relative abundance in the brain. Larvae injected with 231BR brain-tropic cells were more likely to have cells present in the brain at a greater abundance than in the CVP (score > 0.5; Figure 1G). Averaging this metric across all larvae examined showed a statistically significant increase in brain homing for 231BR brain-tropic cells (Figure 1H; p = 0.0475, two-tailed unpaired t test with Welch’s correction).
Figure 1. Non-random Targeting of Organ-Tropic Cancer Cell Lines in a Zebrafish Metastasis Model.
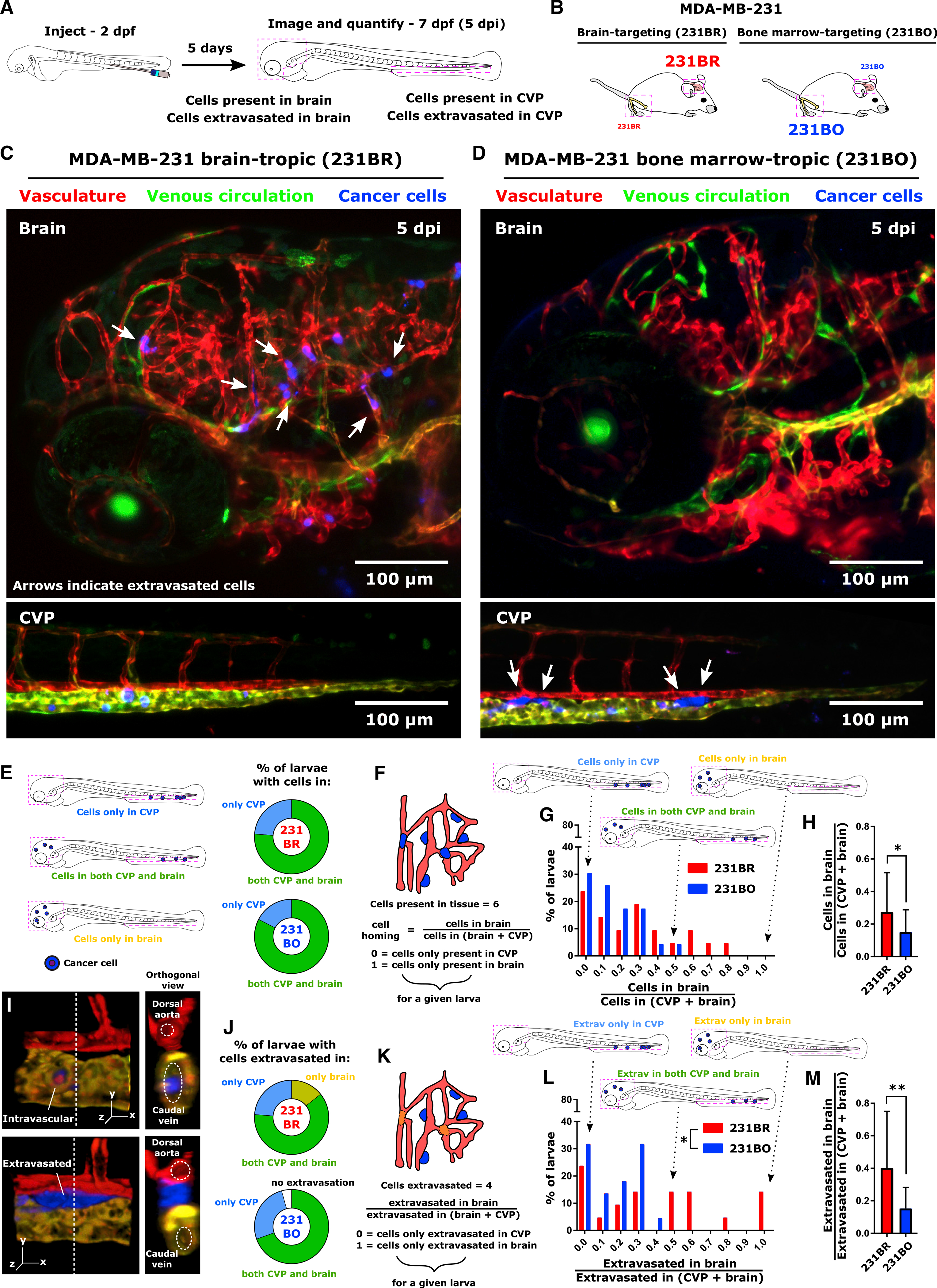
(A) Schematic of experimental setup. Cancer cells were injected into the circulatory system of Tg(flk:mCherry/MRC1a:EGFP) zebrafish at 2 days post-fertilization (dpf) and imaged at 5 days post-injection (dpi; zebrafish age at imaging was 7 dpf). In each larva, both the total number of cells and the number of extravasated cells were counted in the brain and CVP.
(B) Schematic of reported organ colonization of MDA-MB-231 brain-tropic (231BR, red) and bone-marrow-tropic (231BO, blue) subclones in murine models. Font size indicates relative homing within a given organ.
(C and D) Representative images of MDA-MB-231 (C) brain-tropic subclone (231BR) and (D) bone-marrow-tropic subclone (231BO) colonization in the zebrafish brain and CVP at 5 dpi. Arrows indicate extravasated cells. Images are average intensity projections from confocal z stacks. The vasculature is displayed in red, the venous circulation in green, and injected cancer cells in blue. Scales are indicated.
(E) Percentage of larvae with cells present only in the brain, only in the CVP, or in both tissues at 5 dpi. Schematic illustrates these outcomes.
(F) Schematic illustrating assessment of all cells present in a tissue of interest (cell homing).
(G) Distribution of values for cell homing ratio of 231BR and 231BO cells at 5 dpi.
(H) Average ± SD ratio across all larvae of 231BR and 231BO cells present in the brain at 5 dpi to the total cells present in the brain and CVP. *p = 0.0475 by two-tailed unpaired t test with Welch’s correction. For (E), (G), and (H), values were calculated on a per larvae basis for N = 21 larvae injected with 231BR cells (from 398 total cells) and N = 23 larvae injected with 231BO cells (from 433 total cells).
(I) Representative 3D reconstructions from confocal images showing intravascular (top) and extravasated (bottom) cells in the CVP. Dashed line indicates position of orthogonal (yz) slice. In orthogonal views, lumina of neighboring vessels are outlined.
(J) Percentage of larvae with cells extravasated only in the brain, only in the CVP, or in both tissues, or with no extravasated cells, at 5 dpi. Percentages were calculated from N = 21 larvae injected with 231BR cells and N = 23 larvae injected with 231BO cells.
(K) Schematic illustrating assessment of cell extravasation. For extravasation calculations, only extravascular cells were considered. Fish with no extravasated cells were excluded from analysis.
(L) Distribution of values for the ratio of cells extravasated in the brain to total cells extravasated in the brain and CVP for 231BR and 231BO cells at 5 dpi. The distributions of extravasated cells for larvae injected with 231BR and 231BO cells were statistically different (*p = 0.0153, Kolmogorov-Smirnov test).
(M) Average ± SD across all larvae at 5 dpi of extravasated cells in the brain divided by total number of extravasated cells in the brain and CVP for 231BR and 231BO cells. **p = 0.0047 by unpaired two-tailed t test with Welch’s correction. For (L) and (M), values were calculated on a per larva basis for N = 21 larvae with extravasated 231BR cells (from 196 total extravasated cells) and N = 22 larvae with extravasated 231BO cells (from 291 total extravasated cells). See also Figures S1–S3.
We then quantified extravasated cells in both the brain and CVP (Figure 1I). Cells were able to extravasate and survive outside of vessels until at least 5 dpi in both organs (Figures S1A and S1B; Video S1). Extravasated cells often maintained a perivascular position and could be observed both as single cells or as groups of cells (Figure S1B; Video S1). By 5 dpi, a subset of extravasated cells was active in the cell cycle by Ki67 staining, and we did not observe cell expression of the apoptosis marker annexin V (Figure S1C). Larvae injected with brain-tropic 231BR cells were more likely to have extravasated cells exclusively in the brain, while bone-marrow-tropic 231BO cells were only extravasated in the brain if they were also extravasated in the CVP (Figure 1J). Using the aforementioned analysis and scoring larvae based on relative extravasation in the brain compared to the total number of cells extravasated in the brain and CVP (Figure 1K), we noted statistically different distributions of extravasated cells in the larvae based on cell type injected (p = 0.0153, Kolmogorov-Smirnov test), with a relative shift in extravasation to the brain for 231BR cells (Figure 1L). Averaging across all larvae examined, brain-tropic 231BR cells exhibited significantly greater extravasation in the brain relative to their extravasation in the CVP than bone-marrow-tropic 231BO cells (Figure 1M; p = 0.0047, unpaired two-tailed t test with Welch’s correction). Thus, having completed trafficking through the circulation, arrest in the vasculature, and extravasation, the 231 clones demonstrated differences in organ targeting in the zebrafish metastasis model and recapitulated organ targeting observed in mice.
To confirm that organ targeting in zebrafish and murine models was similar for additional cell lines, we injected murine 4T1 brain-tropic (4T1Br4) and bone-marrow-tropic (4T1BM2) cells (Kim et al., 2018; Kusuma et al., 2012), which exhibit weaker patterns of tropism in mice than 231 cells (Figures S2A and S2B). Cell homing was less pronounced in the weakly tropic 4T1 cells than in 231 cells (Figures S2C, S2D, and S2G–S2J), though a higher fraction of larvae injected with bone-marrow-tropic 4T1BM2 cells lacked cells in the brain (Figure S2I). Notably, there was a significant shift in brain extravasation for larvae injected with 4T1Br4 cells compared to those injected with 4T1BM2 cells (Figure S2K; p = 0.0138, chi-square test between distributions of cells; Figures S2L and S2M). Brain-tropic Bt474m1-Br3 cells also had similar organ homing in the zebrafish as has been reported in murine models, though with different dynamics than the other cell line pairs we tested, showing non-random distributions at 1 dpi (Figures S2E, S2F, and S2O–S2Q).
To ensure that cell organ targeting was not due simply to differences in cell survival in the organ parenchyma, we examined the intravital survival of both MDA-MB-231 clones. We co-injected both MDA-MB-231 clones directly into the brain of optically transparent Casper zebrafish (Figure S3A). We determined via both sampling of fish from the injected cohort at different time points (Figures S3A and S3B) and longitudinal imaging of the same fish (Figures S3A and S3C–S3E) that both clones showed similar survival for one week within the brain parenchyma. Consistent with the observation that clones were not differentially cleared from a given tissue, we observed minimal interaction of neutrophils with either MDA-MB-231 clone in the brain and CVP over the first ~14 h post-injection (Video S2) following injection to the circulation. Additionally, at 1 dpi, similar numbers of zebrafish neutrophils were present in the head and tail of larvae injected with either cell type (Figures S3F and S3G), and the distribution of neutrophils between the head and tail was not cell-type dependent (Figures S3G–S3J), indicating that differences in neutrophil recruitment between the cell types were not responsible for organ targeting.
Cell Speed of Organ-Targeting Cell Line Clones Is Identical for a Given Organ Environment
Cell movement within capillaries and arrest within capillary beds are considered to be the first steps dictating transition from the circulation to the parenchyma of secondary organs (Nguyen et al., 2009; Strilic and Offermanns, 2017). We measured the size of zebrafish blood vessels in multiple tissues of 2 dpf zebrafish (Figures 2A–2D). Vessel diameters in the brain and intersegmental vessels (ISVs; nearly linear vessels in the zebrafish tail and trunk) averaged 9.9 ± 2.4 and 10.6 ± 2.6 μm, respectively and ranged from ~5–15 μm in width (Figures 2C and 2D). These values are similar to those measured in human lung and nailbed capillaries and are comparable to those seen in mice, rats, and rabbits (Doerschuk et al., 1993; Kienast et al., 2010; Mathura et al., 2001; Potter and Groom, 1983; Tufto and Rofstad, 1999). Vessel diameters in the CVP were more widely varied, and vessels ranged from ~5–50 μm in width with an average of 16.7 ± 6.8 μm (Figures 2C and 2D). Thus, the vasculature of the zebrafish at this age is analogous to a mammalian capillary bed (Figure 2B).
Figure 2. Cell Speed of Organ-Targeting Cell Line Clones Are Identical for a Given Organ Environment In Vivo.
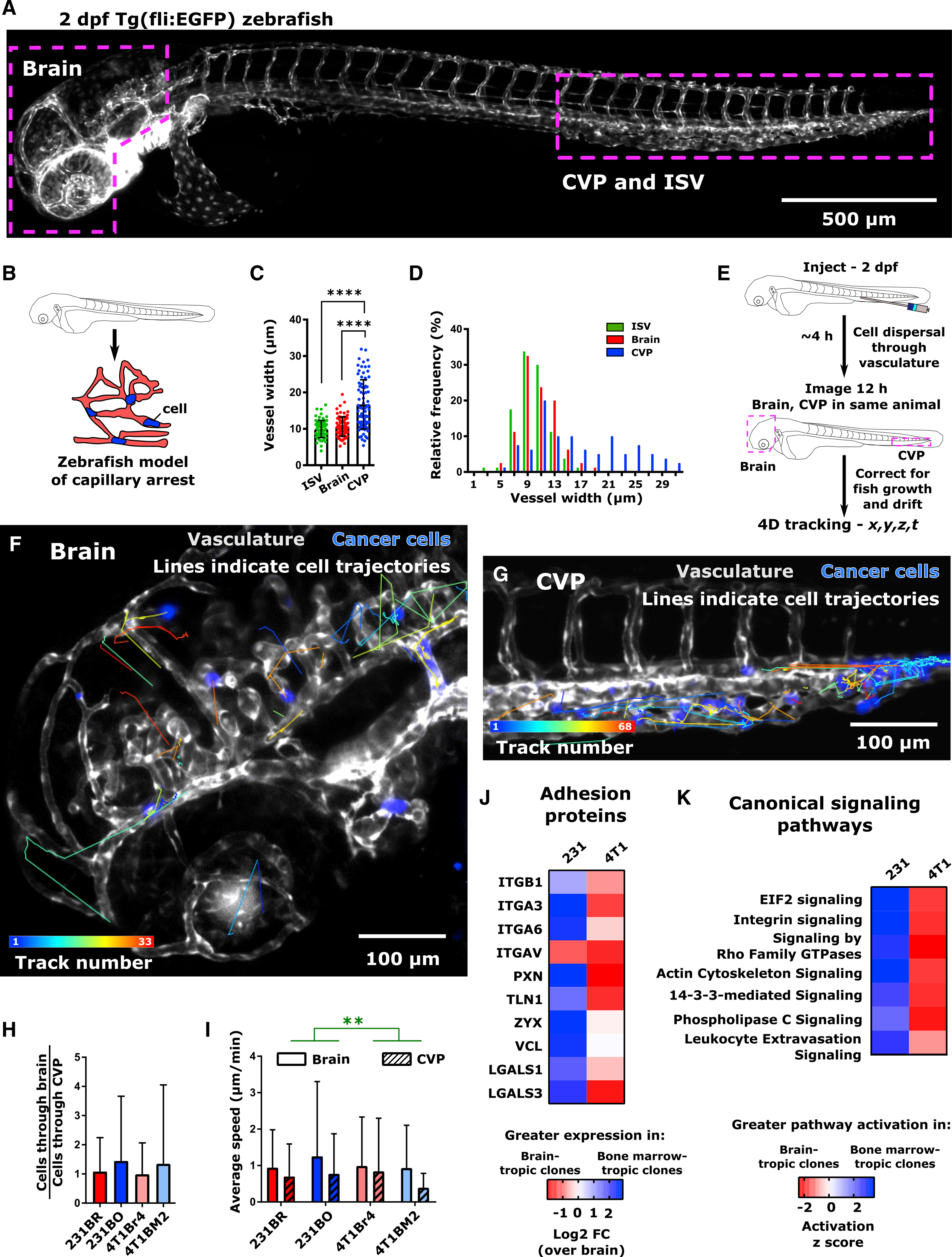
(A) Overview image of 2 days post-fertilization (dpf) Tg(fli1:EGFP) zebrafish. The intersegmental vessels (ISV), caudal vein plexus (CVP), and brain are indicated.
(B) Schematic representation of zebrafish capillary arrest model. The size and architecture of the zebrafish vasculature make it analogous to an organism-scale capillary bed.
(C) Average ± SD and (D) distribution of blood vessel widths in the zebrafish ISV, CVP, and brain. Vessels were measured from four Tg(fli1:EGFP) zebrafish at 2 dpf, 20 vessels per region per fish, to obtain a total of N = 80 vessels at each location. ****p < 0.0001 by Dunn’s multiple comparisons post-test following Kruskal-Wallis test (p < 0.0001).
(E) Schematic of intravascular cell speed experiments.
(F and G) Representative images of cell trajectories within the zebrafish (F) brain and (G) CVP are shown. Trajectories are overlaid on average intensity projection of the t = 12 h image. Tracks are pseudocolored by track number (blue, first track; red, final track). Cells (231BR) are displayed in blue. Zebrafish vasculature and venous circulation are shown in grayscale to facilitate visualization of cell trajectories. Scale bar, 100 μm.
(H) Per larva ratio of cells tracked in the brain to cells tracked in the CVP for both brain-tropic and bone-marrow-tropic cells. Plot displays mean ± SD for this ratio averaged across N = 7 larvae for 231BR cells (310 cells total), N = 10 larvae for 231BO cells (183 cells total), N = 6 larvae for 4T1Br4 cells (139 cells total), and N = 8 larvae for 4T1BM2 cells (96 cells total). Differences were not significant by Dunn’s multiple comparisons test between each cell type following Kruskal-Wallis test (p = 0.5516).
(I) Average ± SD speeds of cells moving through the brain and CVP. Speeds are grouped measurements (231BR cells, N = 91 cells in brain, N = 131 cells in CVP; 231BO cells – N = 46 cells in brain, N = 56 cells in CVP; 4T1Br4 cells – N = 48 cells in the brain, N = 60 cells in the CVP; 4T1BM2 cells – N = 22 cells in the brain, N = 65 cells in the CVP) taken from multiple fish (231Br – 6 larvae; 231BO – 8 larvae; 4T1Br4 – 6 larvae; 4T1BM2 – 8 larvae). **p = 0.0025 by two-way ANOVA, with tissue (F = 9.218, DF = 1) but not cell type as a significant source of variation.
(J) Relative expression of adhesion proteins in 231 and 4T1 bone-marrow-targeting cell clones, relative to expression in brain-targeting clone of each cell line. Heatmap color indicates log2 fold change compared to brain-targeting clone. Protein expression levels were from whole-lysate mass spectrometry, from lysates taken from three independent experiments. Significance levels are not indicated.
(K) Comparison of canonical signaling pathway activation between 231 and 4T1 organ-targeting clones. On heatmap, blue indicates greater pathway activation in bone-marrow-tropic clones and red indicates greater pathway activation in brain-tropic clones by activation Z score in ingenuity pathway analysis (IPA). Pathways were sorted by Z Score using IPA comparison analysis. See also Figure S4.
Having observed non-random organ targeting of brain- and bone-marrow-tropic cells in the zebrafish xenograft model by 7 dpf, we examined the steps of the metastatic cascade from cell dissemination through the circulatory system through extravasation to determine where this selectivity occurred. To determine whether differences in movement through vessels was a precursor to non-random organ targeting, we injected fluorescently labeled 231BR, 231BO, 4T1Br4, or 4T1BM2 cells into the zebrafish circulation and simultaneously imaged cell trafficking in the brain and CVP in three dimensions and over time (Figure 2E). Injection via the posterior cardinal vein led to cell dispersal throughout the vasculature (Figures S4A and S4B). Intravital imaging began from ~4–6 h after injection and continued for the subsequent 12 h (Figure 2E). Because larval zebrafish grow during the imaging period and the vasculature of the larvae thus drifted over time in the reference stage of the microscope, all images were first corrected using 3D image phase-correlation-based translational registration (Parslow et al., 2014), with the vasculature of the fish used as the reference channel (Figure S4C; Video S3). Registration corrected this issue and enabled accurate quantitation of cell speed in vivo (Figure S4D; Video S3).
Tracking of cell movement in the zebrafish brain and CVP (Figures 2F and 2G; Video S4) revealed that on average, an equal number of cells passed through the brain and CVP (normalized per fish, where both organs could be imaged), indicating that these organs were equally exposed to all cell types tested during initial cell dissemination (Figure 2H). Cells moved at moderately higher average speeds within the lumen of vessels in the brain than within the CVP for both 231 and 4T1 cell clones, where the organ of observation but not the cell type was a statistically significant source of variation in cell speed by two-way ANOVA (Figure 2I; p = 0.0025; also see histograms of cell speed in Figures S4E and S4F). Speeds did not differ significantly between cell types within a given organ (Figure 2I). These observed speeds were several orders of magnitude less than the blood flow velocity in larval zebrafish (Watkins et al., 2012), suggesting that cells did not simply move with blood flow. Speeds were similar in a given organ microenvironment for all cell types, regardless of basal levels of expression of proteins associated with cell adhesion (Figure 2J). Patterns of protein expression and canonical signaling pathway expression were not necessarily conserved across organ-targeting cell line pairs, with signaling pathways overexpressed in bone-marrow-targeting cells in the 231 line often regulated in the opposite direction in the 4T1 line (Figures 2J and 2K). Nevertheless, these differences did not affect cell speed in vivo.
To confirm that the similarities in movements observed for tropic clones were not due to a mismatch between human cells and zebrafish stroma, we next asked if the observed cell movements in the zebrafish microenvironment were comparable to how these cells moved in similar geometries in vitro. We fabricated in vitro microchannel mimetics that recreated vessel widths and topographies to compare the basal migration rates of 231BR and 231BO cells in physiologically relevant microenvironments, with microchannels modeling the vascular architecture of the brain and CVP alongside straight microchannels with the same average width as vessels in the brain and CVP (9 μm and 17 μm, respectively; Figures 3A–3C; Video S5). In concert with the in vivo data, both 231BR and 231BO cells migrated through a given microchannel geometry at the same speed when channels were coated with 10 μg/mL fibronectin, indicating that these cells trafficked identically for a given microenvironment (Figure 3D). The majority of cells moved at speeds of less than 1 μm/min both in vivo and in vitro (Figures S4E–S4H). In vitro, the topographical complexity of the brain- and CVP-mimetic environments moderately reduced the average displacement of cells into the microchannels (Figure S4I). To confirm that cell trafficking was not dependent on extracellular matrix (ECM) protein in confinement, we also tested migration in straight microchannels coated with collagen type I, fibronectin, or bovine serum albumin (BSA; Figures 3E and 3F). In these microchannels, though both cell type and channel coating were significant sources of variation by two-way ANOVA, a comparison of cell migration within the geometric in vitro mimetic and in vivo within zebrafish ISVs (Figure 3G) revealed that both 231 cell clones showed similar motilities during confined migration in channels coated with a given mammalian protein (Figure 3H). Cell speeds in vitro were similar to those previously observed with parental MDA-MB-231 cells within confined environments (Balzer et al., 2012; Mathieu et al., 2016).
Figure 3. Organ-Targeting Cell Line Clones Move at Identical Speeds In Vitro for a Given Microenvironment.
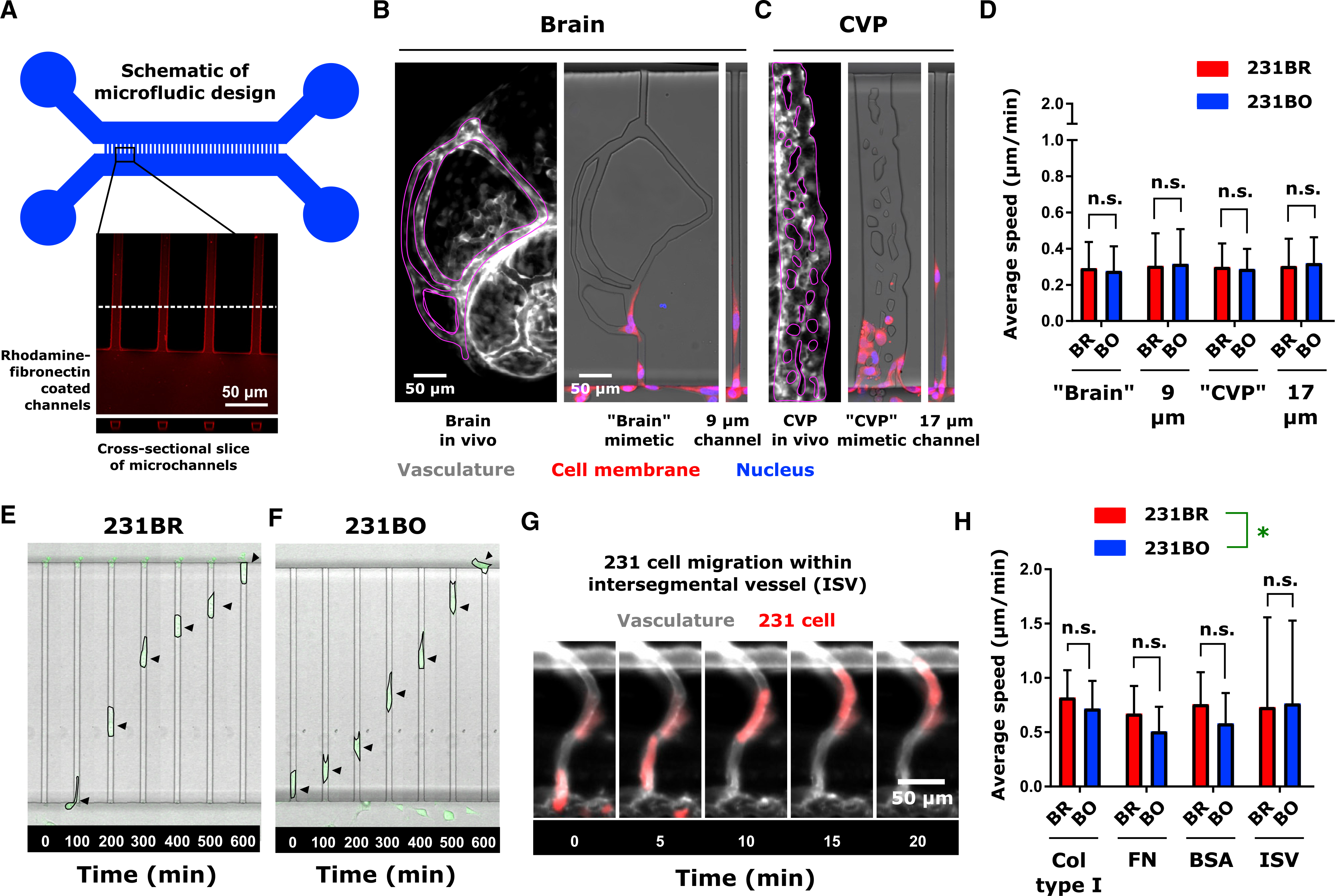
(A) Schematic of microfluidic device. Microchannel inset shows average intensity projection of rhodamine-fibronectin coated microchannels, with a cross-sectional slice to demonstrate the rectangular channel cross section. Microchannel walls were coated with proteins by adsorption, while the interiors of the channels were open.
(B) Vasculature (gray scale) in the zebrafish brain, corresponding “brain” mimetic microchannels, and 9 μm-wide microchannel (average width of vessels in brain; see Figure 2).
(C) Vasculature (gray scale) in the zebrafish CVP, corresponding “CVP” mimetic microchannels, and 17 μm-wide microchannel (average width of vessels in CVP; see Figure 2). In (C) and (D), vasculature images are from 2 dpf Tg(fli1:EGFP) zebrafish, with regions of interest outlined in magenta. Cell membranes are displayed in red and the cell nucleus in blue for in vitro images. Scales are indicated.
(D) Average ± SD speed of cells migrating in microfluidic channels of varying topography. Channels were coated with 10 μg/mL fibronectin. Speeds were calculated from at least 41 cells per condition over 4 biological replicates: 231BR, brain mimetic − N = 41; 231BO, brain mimetic − N = 42; 231BR, 9 μm wide − N = 42; 231BO, 9 μm wide – N = 46; 231BR, CVP mimetic – N = 120; 231BO, CVP mimetic – N = 118; 231BR, 17 μm wide – N = 90; 231BO, 17 μm wide – N = 81. Neither cell type (p = 0.9847, F = 0.0003680, DF = 1) nor microchannel topography (p = 0.3561, F = 1.082, DF = 3) was significant source of variation by two-way ANOVA. Speed differences between cell types were not significant (n.s.) in a given environment by Sidak’s multiple comparisons post-test.
(E and F) Time-lapse images of (E) 231BR cells and (F) 231BO cells migrating through confining microchannels coated with 20-μg/mL rat tail collagen type I. Arrows indicate cell position. For scale, microchannel width, 8 μm.
(G) Representative image of MDA-MB-231 cell (red) migrating through Tg(fli1:EGFP) zebrafish intersegmental vessel (ISV; gray). Arrow indicates cell location. Scale is indicated.
(H) Average speeds (mean ± SD) of cells migrating in microchannels and zebrafish intersegmental vessels. In the microchannels, speeds from at least 52 cells collected over at least 2 biological replicates were measured: 231BR, collagen type I – N = 60 cells, 2 biological replicates; 231BO, collagen type I – N = 60 cells, 2 biological replicates; 231BR, fibronectin – N = 60 cells, 2 biological replicates; 231BO, fibronectin – N = 60 cells, 2 biological replicates; 231BR, BSA – N = 52 cells, 3 biological replicates; 231BO, BSA – N = 57 Cells, 3 biological replicates. Speeds in the intersegmental vessels were measured for N = 38 brain-tropic cells from 6 larvae and N = 14 bone-marrow-tropic cells from 7 larvae. For a given microchannel protein coating and within the ISV, speeds were not significantly different (NS) by Sidak’s multiple comparisons post-test following two-way ANOVA, though cell type (*p = 0.0157, F = 5.888, DF = 1) and ECM coating (p = 0.0115, F = 3.727, DF = 3) were significant sources of variation. See also Figure S4.
Characterization of the Zebrafish Metastatic “Soil”
During transit, cell arrest, and extravasation, cells receive cues from the tissue mechanics of the surrounding blood and lymph vessels and the flow of blood in addition to confinement due to vessel width. We characterized the zebrafish “soil” in the brain and CVP to see how these factors contributed to organ targeting. We employed in vivo optical-trap-based active microrheology (Figure 4A) to measure tissue mechanics (Blehm et al., 2016; Staunton et al., 2017, 2016). Simply, 1-μm-diameter beads embedded in a given material serve as probes to interrogate local mechanical properties. By modulation of the position of the optical tweezer, one can locally apply forces across a wide range of frequencies and measure the bead displacement to calculate the material strain response. The complex modulus and hysteresivity (in terms of phase angle; measurement of viscoelastic parameters) of the zebrafish brain and CVP were nearly identical over a wide range of frequencies at 2 dpf (approximately 50–1,000 Pa; Figures 4B–4D). These values compare well with the mechanical properties of the mammalian brain, mouse mammary fat pad (~5–1,000 Pa), and hydrogels commonly used for 3D culture systems, such as collagen gels (~10–350 Pa), Matrigel mimicking basement membrane (~10–200 Pa), and hyaluronic acid gels mimicking brain ECM microenvironments (~10–200 Pa) (Blehm et al., 2016; Kim et al., 2016; Staunton et al., 2017, 2016). Quantification of the flow speeds of individual blood cells in confocal slices through the CVP and brain using high-speed confocal microscopy revealed that, on average, blood flow rates were identical in the brain and CVP (Figures 4E and 4F; Video S6). Differential expression of proteins available for cell adhesion could also affect cell trafficking. As the endothelial glycocalyx acts as a barrier at the tumor-endothelial cell luminal interface, we next examined if there are differences in the glycocalyx between organs (Reitsma et al., 2007). Albumin is one of the soluble proteins found within and layered on top of proteins that form the endothelial glycocalyx (Reitsma et al., 2007). Visualization using fluorescently labeled albumin revealed that there was a greater intensity of albumin for vessels within the CVP than those found in the brain (Figures 4G and 4H). We also characterized gross protein expression in both the brain and CVP using a panel of histological stains (Figures 4I–4K). Gross protein levels were similar in the brain and CVP, and these levels were similar for larvae between 2–7 dpf and in multiple zebrafish strains.
Figure 4. Characterization of the Zebrafish Metastatic “Soil”.
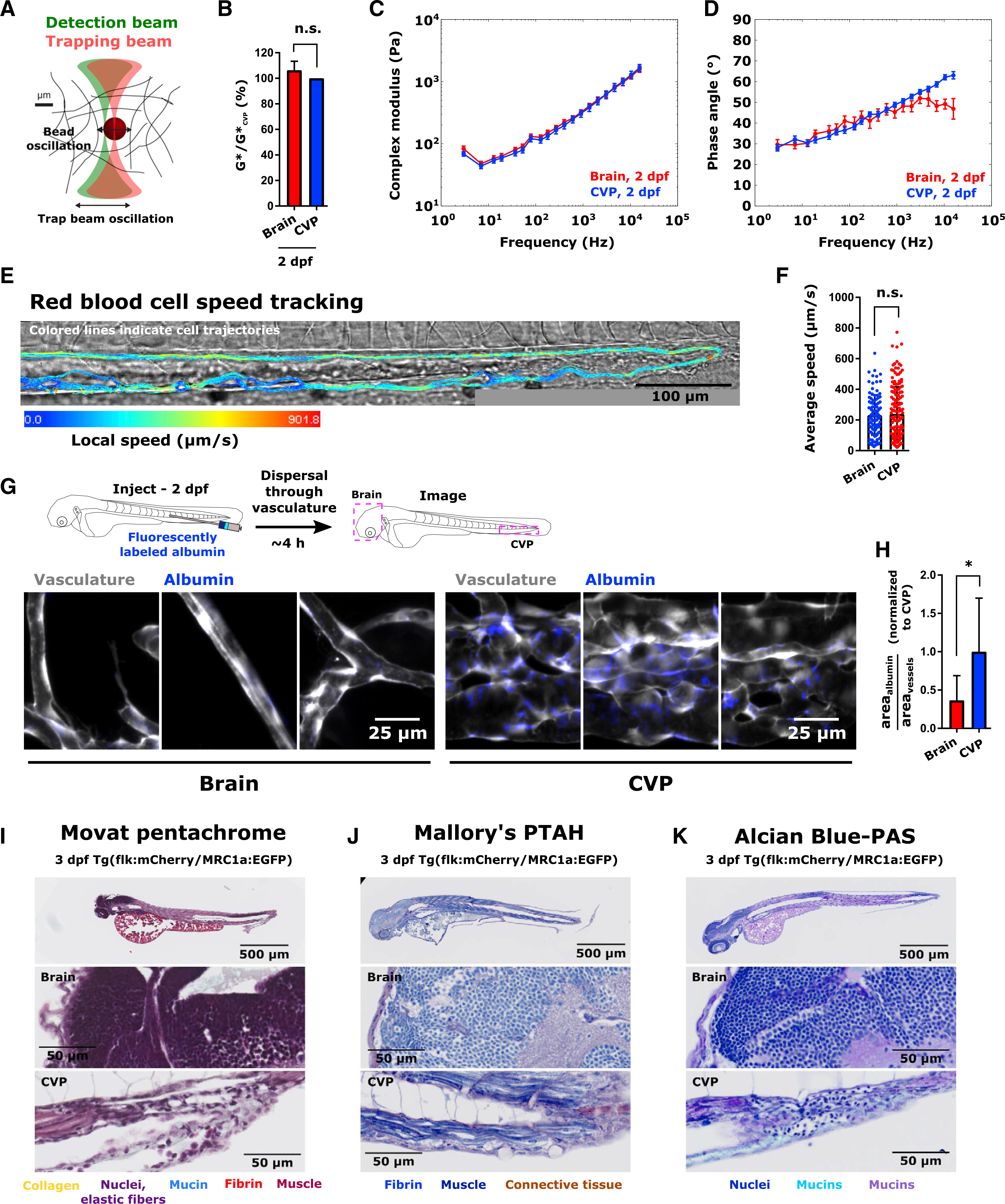
(A) Schematic of optical trap-based microrheology.
(B) Mean ± SD complex modulus in the brain and CVP at 2 dpf, normalized to the mean complex modulus in the CVP on the same day. n.s., not statistically significantly different by Tukey’s honestly significant difference test following two-way ANOVA.
(C) Complex modulus (mean ± SEM) versus frequency curves and (D) phase angle (mean ± SEM) versus frequency curves obtained using optical trap-based microrheology in zebrafish brain and CVP at 2 dpf. Samples were measured in triplicate with at least 30 beads per sample measured.
(E) Red blood cell flow trajectories in CVP of 2 dpf Tg(flk:MCherry/MRC1a:EGFP) larva. Color of trajectory indicates local speed. Scale is indicated.
(F) Mean ± SD red blood cell flow speed in brain and CVP at 2 dpf. Points indicate average speed of an individual cell over the duration of tracking. Bar represents ensemble mean speed. Speeds were measured in the brain and CVP of 4 larvae (N = 102 cells in the brain, N = 160 cells in the CVP, maximum of 40 measurements per larva per tissue of interest). n.s., not significantly different by two-tailed Mann-Whitney test (p = 0.7111).
(G) Schematic and images of fluorescently labeled albumin (blue) accumulation in the glycocalyx of the brain and CVP in 2 dpf Tg(flk:MCherry/MRC1a:EGFP) larvae. Images were acquired ~4 h after albumin injection and are maximum intensity projections of confocal slices. Scale is indicated.
(H) Area coverage of fluorescent albumin as a fraction of total area of vessels in a given region of interest (mean ± SD), normalized to the average ratio in the CVP. In each tissue, 3 images were obtained from 3 larvae (N = 9 per tissue). *p = 0.0306 by unpaired two-tailed t test with Welch’s correction.
(I–K) Histological staining with (I) Movat Pentachrome, (J) Mallory’s PTAH, and (K) Alcian Blue-PAS in 3 dpf Tg(flk:MCherry/MRC1a:EGFP) larvae. Insets in the brain and CVP are shown. Scale is indicated. Color of labels indicate color of corresponding structures in histology images.
Initial Cell Occlusion Is Non-random but Does Not Differ between Organotropic Cell Lines
We next asked if the observed non-random targeting was due to differential occlusion of organ-targeting cells in the brain and CVP. Simply put, does a given tropic cell clone get “stuck” in some tissues but not others, and is that pattern different for a clone that targets another tissue? During cell speed experiments, we noted that all cells examined could either move through and be cleared from both the brain and CVP following injection (Video S4) or could become arrested in zebrafish blood vessels without impeding flow (Videos S4 and S7). We measured the residence time (time cells spend within a given organ) representative of capillary arrest over the first ~12 h after cell injection for larvae with uncompromised blood circulation (Figure 5A). Cells remained in the vessels of the CVP for longer times than in the vessels of the brain during dissemination, where both tissue and cell type were significant sources of variation by two-way ANOVA (Figure 5B). The residence time of both 231BR and 231BO cells was significantly greater in the CVP than in the brain (Figure 5B). Overall, a majority of cells quickly (<3 h) passed through the vasculature and moved out of frame, though a subset of long-resident cells was present in both organs, especially in the CVP (Figures S5A and S5B). We next quantified the fraction of cells occluded after the 12 h tracking period to the total cells passing through each organ for a given cell line. We determined that regardless of cell type injected, approximately 20% of cells passing through the brain became occluded, while this percentage increased to ~40% in the CVP (Figures 5C and 5D). The tissue of observation was a significant source of variation by two-way ANOVA, and a significantly greater fraction of cells became occluded in the CVP than brain for all cells tracked (Figure 5C). Interestingly, cell lines with different patterns of organ targeting did not arrest differently over this period within a given organ. Similar results were observed for the ratio of cell residence time in the brain to that in the CVP and cell occlusion fraction calculated on a per larva basis instead of from the entire pool of cells observed (Figures S5C and S5D). To confirm that initial cell occlusion patterns were largely independent of cell type, we injected inert 10-μm-diameter polystyrene beads to the zebrafish circulation. Residence time and occlusion patterns were identical for the beads as for the cell types tested over the same analysis window (Figures 5B and 5C).
Figure 5. Vessel Topography Drives Cell Residence Time and Occlusion during Initial Dissemination through the Circulatory System.
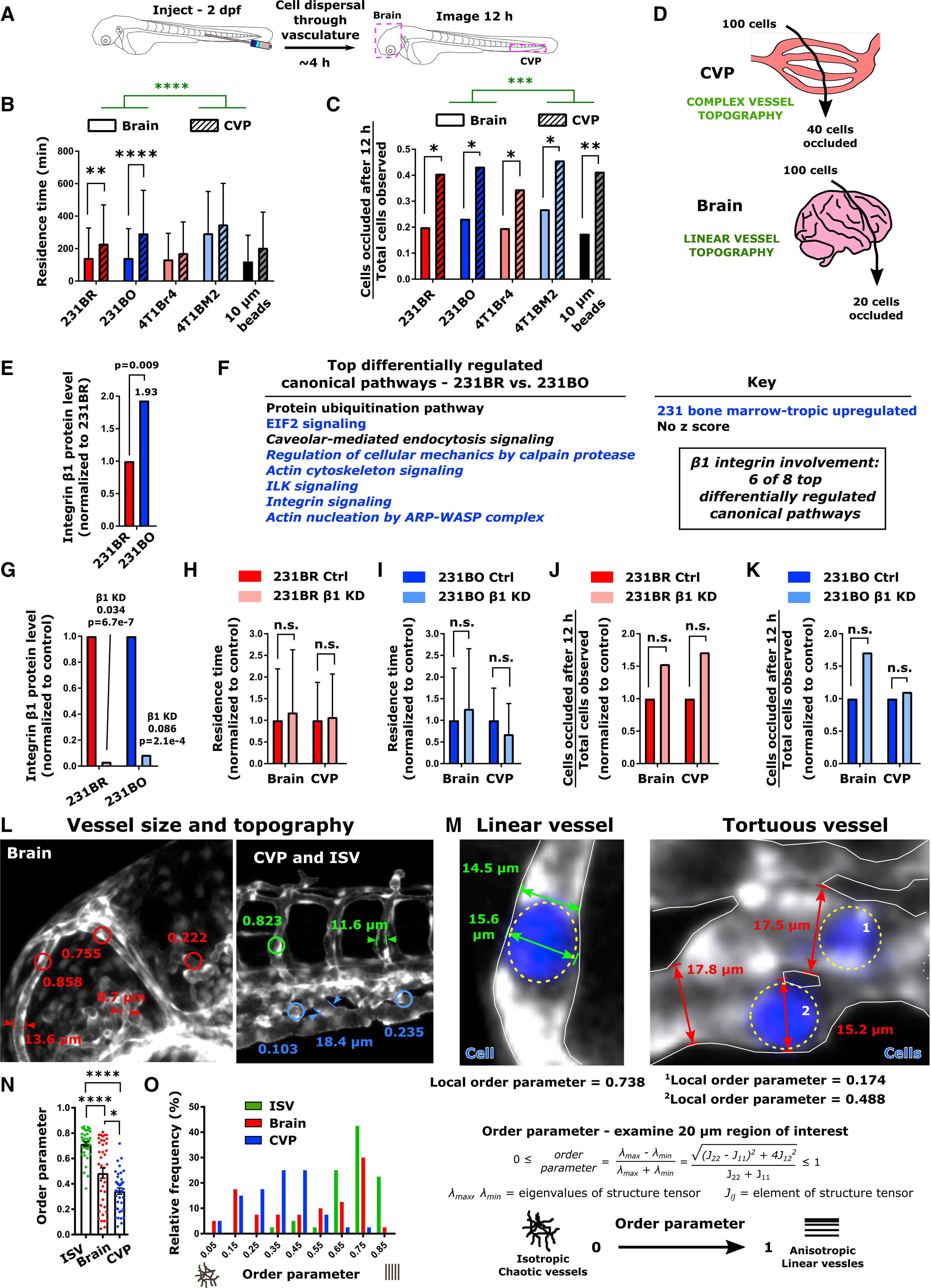
(A) Schematic of experiment. 2 days post-fertilization (dpf) zebrafish were injected with MDA-MB-231 brain-tropic (231BR) or bone-marrow-tropic (231BO) cells, 4T1 brain-tropic (4T1Br4) or bone-marrow-tropic (4T1BM2) cells, or 10-μm-diameter polystyrene beads (beads) in the posterior cardinal vein. Following dispersal through the vasculature, the brain and CVP of each larva were imaged for 12 h via confocal microscopy.
(B) Residence time (mean ± SD) of cells and beads through the brain and CVP. Averages were calculated from the pool of all cells or beads imaged within the indicated organ. Tissue of observation (****p < 0.0001, F = 25.36, DF = 1) and particle/cell type (p < 0.0001, F = 13.65, DF = 4) were significant sources of variation by two-way ANOVA. Additionally, residence times of 231BR and 231BO cells were significantly less in the brain than the CVP (**p = 0.0012; ****p < 0.0001 by Sidak’s multiple comparisons test).
(C) Fraction of cells or beads occluded in the brain and CVP at the end of the 12 h imaging period from the total number of particles observed in each organ over the imaging period. The tissue of observation but not particle/cell type was a significant source of variation by two-way ANOVA (***p = 0.0002, F = 184.2, DF = 1), and occlusion fractions were significantly higher in the CVP than the brain for all particles tested by Sidak’s multiple comparisons test (231BR, *p = 0.0156; 231BO, *p = 0.0172; 4T1Br4, *p = 0.0486; 4T1BM2, *p = 0.0217; beads, **p = 0.0090).
(D) Schematic of cell occlusion in the brain and CVP. Regardless of cell type tested, ~40% of cells observed became occluded in the CVP and ~20% of cells observed became occluded in the brain over the 12 h imaging period tracking initial cell dissemination. For (B) and (C), values were calculated from ensemble data across 7 larvae for 231BR cells (N = 140 cells in the brain, N = 170 cells in the CVP), 10 larvae for 231BO cells (N = 86 cells in the brain, N = 97 cells in the CVP), 6 larvae for 4T1Br4 cells (N = 61 cells in the brain, N = 78 cells in the CVP), 8 larvae for 4T1BM2 cells (N = 26 cells in the brain, N = 70 cells in the CVP), and 6 larvae for polystyrene beads (N = 183 beads in the brain, N = 29 beads in the CVP).
(E) Relative protein expression of integrin β1 in MDA-MB-231 organ-tropic cell lines from mass spectrometry data. Expression in bone-marrow-tropic 231BO cells is normalized to that in brain-tropic 231BR cells. Fold changes in protein level and p values are indicated.
(F) List of the top differentially regulated canonical signaling pathways between MDA-MB-231 brain-tropic (231BR) and bone-marrow-tropic (231BO) cells from pathway analysis of mass spectrometry data. Where possible, the direction of pathway regulation (from IPA Analysis Z score) is indicated (blue, upregulated in 231BO; black, no conclusive direction of regulation). Pathways in which β1 integrin are implicated are italicized.
(G) Knockdown of integrin β1 assessed from mass spectrometry data. β1 integrin levels are normalized within each cell line relative to treatment with control siRNA. Fold changes and p values are indicated.
(H and I) Residence time (average ± SD) of (H) 231BR cells and (I) 231BO cells in the brain and CVP upon β1 integrin silencing, normalized to the average residence time in the same organ for control cells. Integrin β1 silencing was not a significant source of variation by two-way ANOVA (231BR: p=0.4679; 231BO: p=0.8425), and differences between control and knockdown cells in a given organ were not significant (n.s.) by Sidak’s multiple comparisons test (231BR: p=0.7247 in brain, p=0.9413 in CVP; 231BO: p=0.3234 in brain, p=0.1676 in CVP).
(J and K) Fraction of (J) 231BR or (K) 231BO β1 integrin knockdown cells passing through a given organ that became occluded over the 12 h imaging period tracking initial cell dissemination, normalized to the fraction in the same organ for control cells. Fractions were calculated from the pool of all cells imaged in the brain and CVP for a given condition. Integrin β1 silencing was not a significant source of variation by two-way ANOVA (231BR: p=0.0925; 231BO: p=0.4067), and differences between control and knockdown cells in a given organ were not significant (n.s.) by Sidak’s multiple comparisons test (231BR: p=0.1766 in brain and CVP; 231BO: p=0.6480 in brain and CVP). For (H–K), residence times and occlusion fractions were calculated from: 231BR Ctrl – 3 larvae, N = 30 cells in brain, N = 35 cells in CVP; 231BR integrin β1 knockdown – 4 Larvae, N = 94 cells in brain, N = 74 cells in CVP; 231BO Ctrl – 3 larvae, N = 78 cells in brain, N = 70 cells in CVP; 231BO integrin β1 knockdown – 4 larvae, N = 43 cells in head, N = 55 cells in CVP.
(L) Representative vessel width and order parameter measurements in the intersegmental vessels (ISV), Caudal Vein Plexus (CVP), and brain of 2 days post-fertilization (dpf) Tg(fli1:EGFP) zebrafish. Order parameters were measured over 20-μm-diameter circular regions of interest.
(M) Representative images of linear and tortuous zebrafish blood vessels. Cancer cells are shown in blue and outlined by dashed yellow ovals. Representative examples of cell and vessel width measurements are illustrated. Additionally, the local order parameter describing vessel topography as defined is given in the vicinity of each indicated cell.
(N) Average ± SD and (O) distribution of the order parameter characterizing the topography of vessels in the ISV, brain, and CVP over 20-μm-diameter circular regions of interest. A value of 1 indicates aligned structures, while a value of 0 indicates isotropic structures. Vessels were measured from four Tg(fli1:EGFP) zebrafish at 2 dpf, 10 regions per tissue per fish, to obtain a total of N = 40 vessels at each location. *p < 0.05; ****p < 0.0001 by Dunn’s multiple comparisons post-test following Kruskal-Wallis test (p < 0.0001). See also Figure S5.
Integrin β1 has been implicated as one of the key proteins for cell fate decisions on time scales spanning hours-days when cells encounter a new environment (Humphries et al., 2006; Hynes, 2002; Tanner et al., 2012; Weaver et al., 1997). Label-free mass spectrometry of MDA-MB-231 cell lines revealed that integrin β1 was upregulated in bone-marrow-tropic 231BO cells compared to brain-tropic 231BR cells (Figure 5E), and of the canonical signaling pathways differentially regulated between MDA-MB-231 tropic clones from mass spectrometry data, β1 integrin emerged as a key integration point, with 6 of the top 8 differentially regulated canonical signaling pathways involving this integrin (italicized in Figure 5F). We inhibited β1 integrin in the 231 cell line models and investigated the effects on early cell trafficking. We confirmed that β1 integrin was significantly lowered upon knockdown using mass spectrometry (Figure 5G). Silencing β1 integrin prior to injection to the circulation of 2 dpf zebrafish did not significantly affect the residence time of either 231BR or 231BO cells in the brain or CVP compared to cells transfected with a non-targeting control small interfering RNA (siRNA) (Figures 5H, 5I, S5E, and S5F). Additionally, there was no significant change in cell occlusion compared to control in the brain or CVP for either cell type (Figures 5J, 5K, S5G, and S5H). Similar results were obtained when a function-blocking antibody against integrin β1 was used, and no significant difference in the fraction of cells occluded upon antibody treatment was noted (Figures S5I–S5P). In summary, our data indicate that organ targeting cells move and become arrested similarly within a given organ environment (brain or CVP) regardless of the characteristics of the “seed” but that a greater percentage of cells moving through the CVP are arrested compared to the brain.
Topography Drives the Initial Arrest of Disseminated Cells
At the single-cell level, a cell senses its environment using protrusions that may span from a few micrometers (e.g., filopodia) to roughly the diameter of a cell (e.g., blebs and lamellipodia, nuclear confinement). Though vessel sizes are larger in the CVP than the brain on average (Figures 2C and 2D), more cells became arrested in the CVP, suggesting that average vessel size was not the determinant of cell speed or arrest. We thus investigated the topography or architecture of these vessels. We calculated an order parameter describing the tortuosity of the vessel on the length scale corresponding to roughly that of a cell diameter (20 μm), which ranged from values of 0 for isotropic, chaotic vessels to 1 for aligned, linear vessels (Figures 5L and 5M) (Fonck et al., 2009). The order parameter in the highly oriented ISVs dorsal to the CVP was close to 1, while the order parameter was significantly lower in the CVP than both the brain and ISVs. Thus, the ISVs were the most ordered, whereas the CVP vessels had the greatest tortuosity and the brain vessels showed intermediate order (Figures 5N and 5O). Thus, vessel topography on the scale of the cell was systematically varied through these organs. In line with our determination that the microscale ECM protein expression and tumor cell speeds were comparable for both the brain and CVP at the time of cell injection, we reasoned that vessel topography instead of blood flow rates, tissue mechanics, average blood vessel diameter, or cell adhesion protein expression was the primary driver of cell speed during dissemination and initial cell distribution.
Differential Extravasation in the CVP Leads to Organ Selectivity Following Dissemination
Due to differences in the distribution of extravasated cells between brain- and bone-marrow-tropic cells at 5 dpi following similar patterns of dissemination over the first ~12–16 h post-injection, we next asked if cells respond differently to the brain and CVP during extravasation. Extravasation is, by nature, an event that takes place at the endothelial barrier, with arrested cells protruding into the perivascular space, transmigrating through vessels, and establishing themselves in the tissue parenchyma (Figure S6A). We focused on MDA-MB-231 cell clones, which showed distinct distributions of extravasated cells at 5 dpi (Figures 1L and 1M). We analyzed images of a subset of the larvae presented in Figure 1 at 3, 4, and 7 dpf (1, 2, and 5 dpi) to examine the time course of cell extravasation (Figure 6A). The endothelial cells comprising the zebrafish vasculature expressed E-cadherin across this time period, with the greatest amount of staining by immunofluorescence around the peripheries of the blood vessels (Figure S6B).
Figure 6. Differential Extravasation in the Caudal Vein Plexus Leads to Organ Selectivity Following Dissemination.
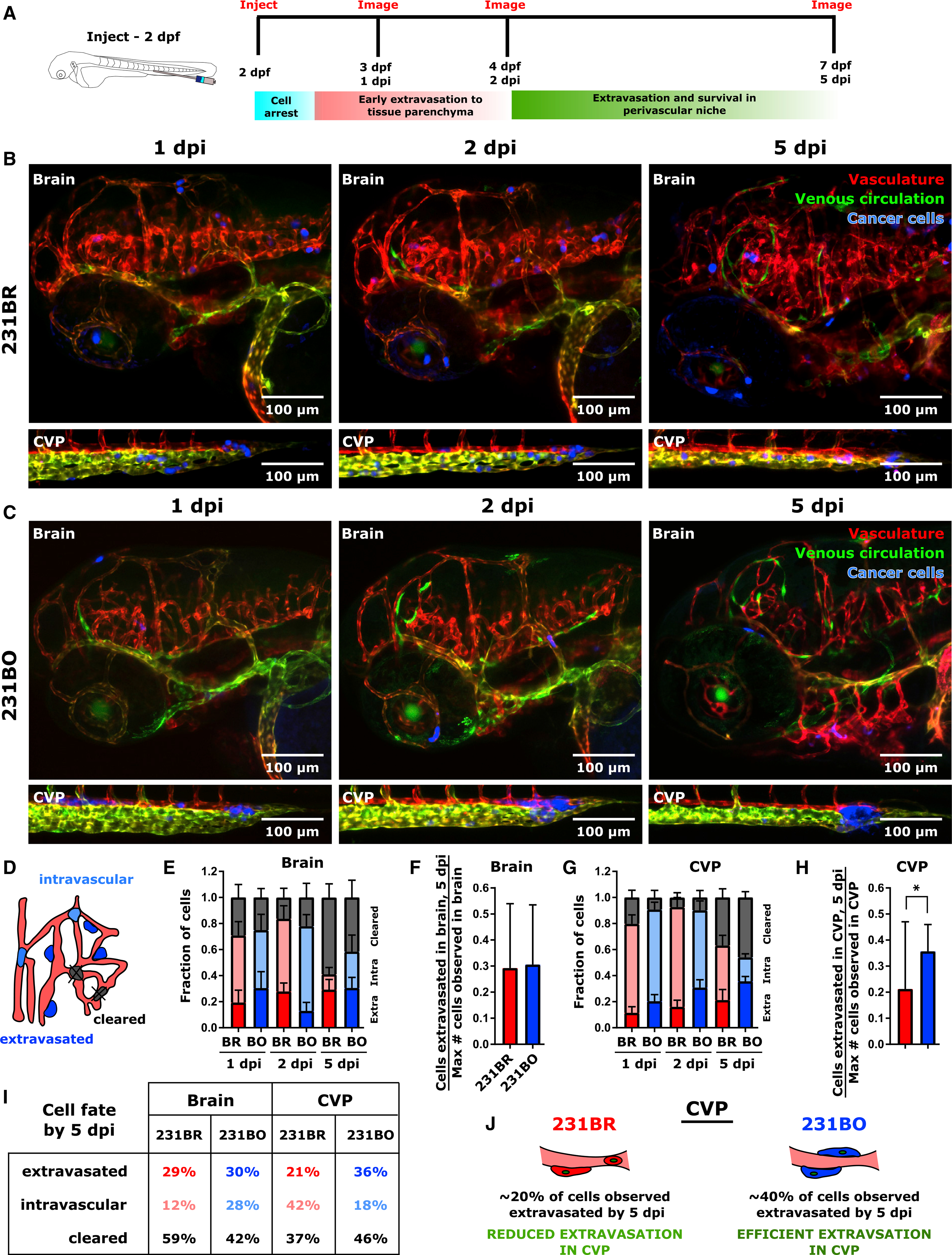
(A) Schematic of experiment. MDA-MB-231 brain-tropic (231BR) or bone-marrow-tropic (231BO) cells were injected to the circulation of Tg(flk:MCherry/MRC1a:EGFP) zebrafish at 2 days post-fertilization (dpf). The brain and caudal vein plexus (CVP) of injected larvae were imaged at 1, 2, and 5 days post-injection (dpi).
(B and C) Representative images of (B) larva injected with 231BR cells and (C) larva injected with 231BO cells at 1, 2, and 5 dpi. Images show the brain and CVP for 1 larva over time for each cell type. Images are average intensity projections from confocal Z stacks. The vasculature is displayed in red, the venous circulation in green, and injected cancer cells in blue. Scales are indicated.
(D) Schematic illustrating cell outcomes after arrival in the vasculature of a target organ. Cells can remain intravascular, extravasate to the parenchyma, or be cleared from the tissue.
(E) Average ± SEM fractions of 231BR and 231BO cells in the brain that are extravasated (bottom portion of bars), intravascular (middle portion of bars), or cleared (top portion of bars) at 1, 2, and 5 dpi.
(F) Average ± SD fraction of the maximum number of cells observed in the brain of a given larvae that were extravasated by 5 dpi. Average is calculated across larvae. Differences were not significant (p = 0.9098 by unpaired t test).
(G) Average ± SEM fractions of 231BR and 231BO cells in the CVP that are extravasated (bottom portion of bars), intravascular (middle portion of bars), or cleared (top portion of bars) at 1, 2, and 5 dpi.
(H) Average ± SD fraction of the maximum number of cells observed in the CVP of a given larvae that were extravasated by 5 dpi. Average is calculated across larvae. *p = 0.0124 by two-tailed Mann-Whitney test.
(I) Table detailing cell fate by 5 dpi in the brain and CVP. Values are averages obtained as in (E–H). In (E–I), values were calculated across N = 10 larvae for 231BR cells and N = 8 larvae for 231BO cells. Average fractions were calculated independently on each day.
(J) Schematic of cell extravasation outcomes in the CVP. Approximately 20% of 231BR cells observed in the CVP at 1 dpi had extravasated by 5 dpi. Extravasation of 231BO cells in the CVP was more efficient, with ~40% of cells observed at 1 dpi extravasating by 5 dpi. See also Figure S6.
Following dissemination, 231BR cells occluded in the CVP were unable to extravasate efficiently and were partially cleared or remained intravascular by 5 dpi (Figure 6B). In contrast, 231BO cells exhibited high rates of extravasation and survival in the CVP (Figure 6C). While extravasation was occasionally observed to occur by 1 dpi (Video S8), we observed that cells generally extravasated at later times (Figures 6B and 6C). To estimate the number of cells that remained intravascular, extravasated, or were cleared from the tissue of interest over time (Figure 6D), we calculated the fraction of cells in each state at each time point as a fraction of the maximum number of cells observed in that organ over the imaging period. In the brain, the percentage of intravascular cells decreased over time, as cells that had not extravasated were cleared (Figure 6E), and ~30% of observed cells extravasated for both 231BO and 231BR cells (Figure 6F). In the CVP, a large percentage of 231BR cells remained intravascular over time (Figure 6G). This led to a significantly lower percentage of 231BR cells (~20%) than 231BO cells (~40%) that were able to extravasate in the CVP (Figure 6H). Over the 5 days of imaging, the total numbers of cells present in the brain plus CVP were similar for larvae injected with 231BR or 231BO cells (Figure S6C). Cell numbers decreased over time, and time but not cell type was a significant source of variation in cell number by two-way ANOVA, consistent with the clearance of intravascular cells over time (Figure S6C).
In summary, cell outcomes by 5 dpi were similar for both cell types in the brain but differed in the CVP (Figure 6I). Thus, differences in extravasation potential in the CVP drove the second phase of organ selectivity for 231BR and 231BO cells (Figure 6J). Decreased extravasation of 231BR cells in the CVP caused an increase in the relative number of 231BR cells in the brain of the zebrafish and led to the observed distributions of extravasated cells (Figures 1L and 1M).
Modulation of Extravasation via Inhibition of β1 Integrin Redirects Organ Targeting
Integrins switch between active and inactive conformations to drive signaling processes (Campbell and Humphries, 2011; Larsen et al., 2006). Having observed that β1 integrin was upregulated in 231BO compared to 231BR cells, we next asked if there is also a difference between brain- and bone-marrow-tropic clones in integrin activation and determined that 231BO cells also showed increased activation of β1 integrin (Figures 7A and 7B). This integrin was found in brain-tropic 231BR cells but remained largely endosomal (Figure 7A). β1 integrin has been shown to be important for extravasation and tumor cell growth (Chen et al., 2016; Hamidi and Ivaska, 2018; Shibue and Weinberg, 2009). Therefore, we probed the effect of siRNA-mediated silencing of β1 integrin expression on cell survival and extravasation in vivo (Figure 7C). We observed that cells with silenced β1 integrin reached the brain and CVP (Figures 5J and 5K) and survived in the larvae (Figures S7A and S7B) at similar rates as cells transfected with non-targeting control siRNA. By 5 dpi, cells with silenced β1 integrin could be found throughout the larvae (Figures S7A and S7C). However, we noted a decrease in the ability of cells to extravasate in the CVP upon β1 integrin knockdown, with a number of cells remaining intravascular (Figure S7A). Following β1 integrin knockdown, extravasation of 231BR cells decreased by ~40% in the CVP but did not change in the brain (Figure 7D). In contrast, 231BO cell extravasation was reduced by ~65% upon knockdown compared to control cell extravasation in both tissues, a reduction that was significant by two-way ANOVA (Figure 7E). Thus, knockdown of β1 integrin in 231BO cells, which have greater endogenous and activated β1 integrin levels than 231BR cells and higher activation of cell integrin and contractility pathways, significantly reduced the ability of cells to extravasate in both the brain and CVP (Figure 7E).
Figure 7. Redirection of Organ Targeting via Knockdown of β1 Integrin or Myosin 1B.
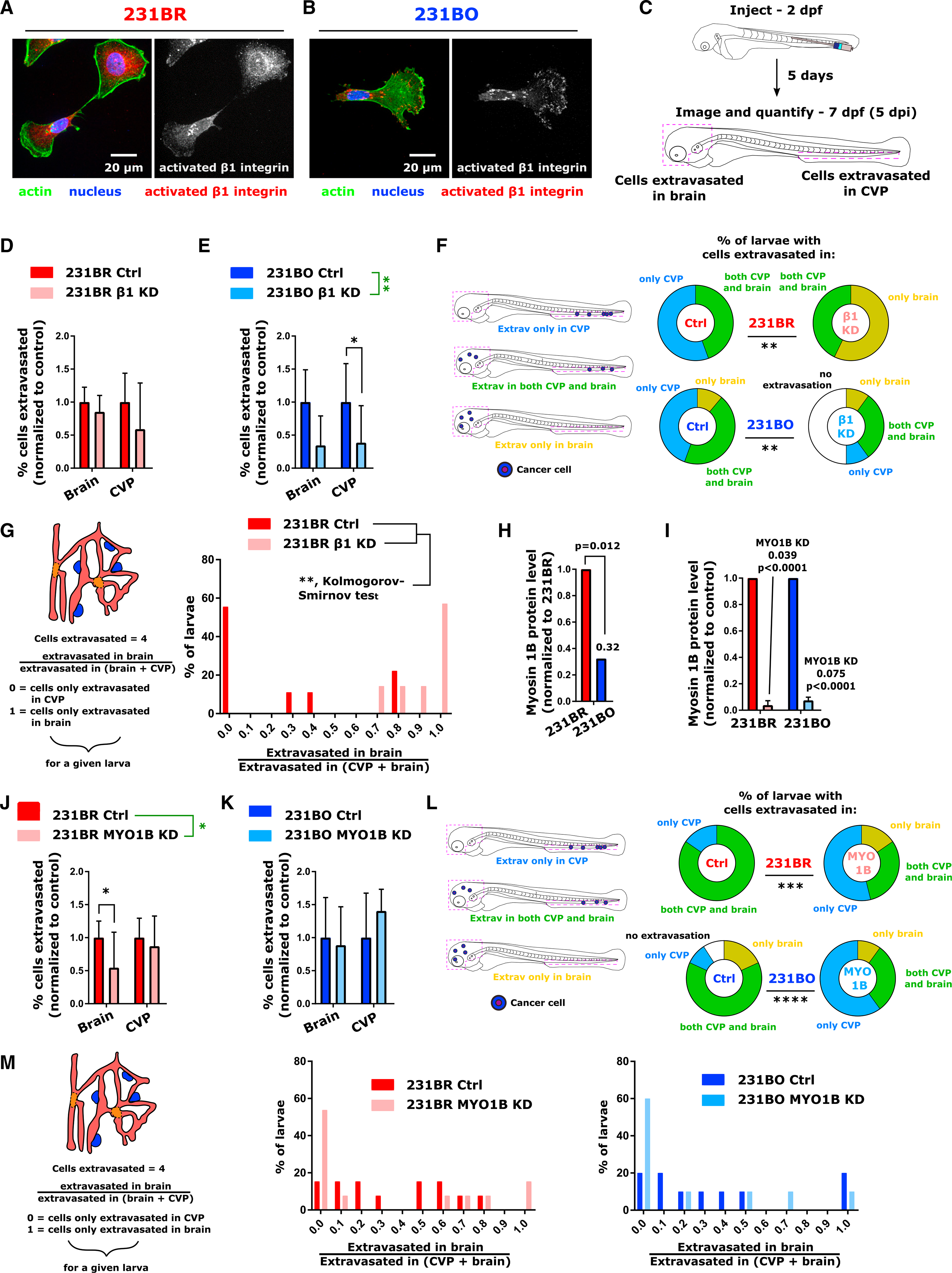
(A and B) Immunofluorescence images of activated β1 integrin in (A) 231BR and (B) 231BO cells. In the left images, actin is displayed in green, the nucleus in blue, and activated β1 integrin in red.
(C) Schematic and outline of siRNA knockdown extravasation experiments. Cells were injected at 2 days post-fertilization (dpf), and extravasation in the brain and caudal vein plexus (CVP) were quantified at 5 days post-injection (dpi).
(D and E) Percentage of (D) 231BR cells and (E) 231BO cells present in the brain or CVP at 5 dpi that had extravasated following transfection with siRNA targeting β1 integrin, normalized to the average extravasation percentage in the same organ for control cells (mean ± SD). Values were averaged from the pool of larvae that had at least one cell present in the brain or CVP. This pool encompassed analysis of: 231BR ctrl cells – N = 9 larvae for CVP, N = 4 larvae for brain; 231BR β1 knockdown cells – N = 6 larvae for CVP, N = 7 larvae for brain; 231BO ctrl cells – N = 9 larvae for CVP, N = 6 larvae for brain; 231BO β1 knockdown cells – N = 9 larvae for CVP, N = 8 larvae for brain. For 231BO cells, knockdown was a significant source of variation (**p = 0.0023, two-way ANOVA, F = 11.24, DF = 1), and extravasation in the CVP was significantly reduced (*p = 0.0398, Sidak’s multiple comparisons test).
(F) Percentage of larvae injected with control or β1 integrin knockdown cells with cells extravasated only in the brain, only in the CVP, or in both tissues, or with no cells extravasated, at 5 dpi. Percentages were calculated from: 231BR ctrl cells – N = 9 larvae; 231BR β1 knockdown cells – N = 7 larvae; 231BO ctrl cells – N = 9 larvae; 231BO β1 knockdown cells – N = 10 larvae. 231BR cells: **p = 0.0034, two-tailed binomial test between fraction of larvae with cells extravasated in head for injection with ctrl or β1 knockdown cells; 231BO cells: **p = 0.0012, chi-square test.
(G) Distribution of values for the ratio of cells extravasated in the brain to total cells extravasated in the brain and CVP for 231BR cells transfected with control siRNA or siRNA targeting β1 integrin. The distributions of extravasated cell locations were statistically different (**p = 0.0052, Kolmogorov-Smirnov test). For (G), values were calculated from the pool of larvae with extravasated cells: 231BR ctrl cells – N = 9 larvae (46 total extravasated cells); 231BR β1 knockdown cells – N = 7 larvae (25 total extravasated cells).
(H) Relative protein expression of myosin 1B in MDA-MB-231 organ-tropic cell lines via mass spectrometry. Expression in bone-marrow-tropic cells (231BO) is normalized to that in brain-tropic cells (231BR), and p value is indicated.
(I) Knockdown of myosin 1B via siRNA assessed from qPCR data. Myosin 1B levels were normalized within each cell line relative to treatment with control siRNA. Fold changes and p values (Sidak’s multiple comparisons test following two-way ANOVA) are indicated.
(J and K) Percentage of (J) 231BR cells and (K) 231BO cells present in the brain or CVP at 5 dpi that had extravasated following transfection with siRNA targeting myosin 1B, normalized to the average extravasation percentage in the same organ for control cells (mean ± SD). Values were averaged from the pool of larvae that had at least one cell present in the brain or CVP. This pool encompassed analysis of: 231BR ctrl cells – N = 13 larvae for CVP, N = 11 larvae for brain; 231BR myosin 1B knockdown cells – N = 12 larvae for CVP, N = 10 larvae for brain; 231BO ctrl cells – N = 10 larvae for CVP, N = 11 larvae for brain; 231BO myosin 1B knockdown cells – N = 9 larvae for CVP, N = 5 larvae for brain. For 231BR cells, knockdown was a significant source of variation (*p = 0.0167, two-way ANOVA, F = 6.220, DF = 1), and extravasation in the brain was significantly reduced (*p = 0.0237, Sidak’s multiple comparisons test).
(L) Percentage of larvae injected with control or myosin 1B knockdown cells with cells extravasated only in the brain, only in the CVP, or in both tissues, or with no cells extravasated, at 5 dpi. Percentages were calculated from: 231BR ctrl cells – N = 13 larvae; 231BR myosin 1B knockdown cells – N = 13 larvae; 231BO ctrl cells – N = 11 larvae; 231BO myosin 1B knockdown cells – N = 10 larvae. ***p = 0.0001; ****p < 0.0001, chi-square test.
(M) Distribution of values for the ratio of cells extravasated in the brain to total cells extravasated in the brain and CVP for 231BR and 231BO cells transfected with control siRNA or siRNA targeting myosin 1B. Values were calculated from the pool of larvae that had at least one extravasated cell: 231BR ctrl cells – N = 13 larvae (132 total extravasated cells); 231BR myosin 1B knockdown cells – N = 13 larvae (92 total extravasated cells); 231BO ctrl cells – N = 10 larvae (164 total extravasated cells); 231BO myosin 1B knockdown cells – N = 10 larvae (101 total extravasated cells). See also Figure S7.
Classification of larvae based on the presence of cells extravasated exclusively in the brain, exclusively in the CVP, or present in both organs reflected these changes in extravasation potential (Figure 7F). Upon β1 integrin knockdown, 231BR cell exclusive targeting of the brain increased, and no larvae with extravasated cells only in the CVP were observed (Figure 7F). In contrast, the decreased extravasation capacity of 231BO cells upon knockdown led to distributions in which half of the larvae examined had cells present in the brain and/or CVP but in which no cells were extravasated, significantly changing the distribution of extravasated cells (Figure 7F). While modulating the extravasation of 231BO cells changed the overall metastatic potential of 231BO cells, knocking down integrin β1 in 231BR cells instead resulted in a statistically significant shift in the distribution of extravasated cells toward the brain compared to control (Figure 7G). In summary, while cells were able to initially seed both the brain and CVP (Figure 5) and inhibition of β1 integrin inhibited the extravasation of both cell types in the CVP, cell-type-specific differences in the effect of β1 integrin knockdown on brain extravasation caused a redistribution of organ targeting in 231BR cells (Figure S7D).
Redirection of Targeting via Myosin 1B Knockdown
While mechanosensing proteins, integrins, and cell contractility proteins were typically upregulated in 231BO cells, we noted that unconventional myosin 1B (MYO1B) was expressed at significantly higher levels (compared to bone-marrow-seeking counterparts) in both 231BR and 4T1Br4 cells on the protein level (Figures 7H and S7E; see also Figure 2J). Myosin 1B serves as a link between the membrane and the actin cytoskeleton, thereby regulating the actin network within growth cones in neurons (Iuliano et al., 2018). We silenced myosin 1B using siRNA and confirmed silencing by qPCR (Figure 7I). Cells were injected into the circulation of 2 dpf zebrafish, and cell targeting was assessed at 5 dpi (Figure S7F). The total number of cells present after knockdown was moderately decreased compared to control cells but did not significantly differ within a given cell line (Figure S7G). While cells were present throughout the larvae of injected fish regardless of knockdown status, a number of cells in the brain remained intravascular upon myosin 1B knockdown (Figures S7F and S7H). Silencing of myosin 1B significantly reduced the percentage of 231BR cells in the brain that were extravasated compared to control without changing extravasation rates in the CVP (Figure 7J). While myosin 1B knockdown of bone-marrow-seeking 231BO cells only slightly reduced extravasation in the brain, it also moderately (though not significantly) increased extravasation rates in the CVP compared to control cells (Figure 7K). This led to a significantly increased percentage of larvae had cells extravasated only in the CVP upon myosin 1B silencing for both cell types (Figure 7L). Single-cell analysis of relative cell distributions at 5 dpi revealed an increase in the percentage of larvae containing extravasated cells present only in the CVP for both cell types upon myosin 1B knockdown (Figure 7M).
DISCUSSION
Tumor cells encounter a myriad of environmental cues along the metastatic cascade before eventual arrest and outgrowth in a secondary organ (Kim and Tanner, 2015; Kumar and Weaver, 2009). While mechanisms of cell escape from a primary tumor have been well characterized (Patsialou et al., 2009; Wyckoff et al., 2007), visualization of the exit from the circulatory system and into secondary organs has been more difficult to achieve. One advantage of the larval zebrafish is the ability to image the whole organism whilst maintaining single-cell resolution not readily achieved in murine systems (Paul et al., 2019; White et al., 2013). Therefore, the zebrafish is an excellent model to study and visualize key aspects of tumor-capillary dynamics and extravasation in multiple tissues and in vivo within the same animal.
Here, we characterize the zebrafish as a tool to begin addressing the role of the physical environment in the earliest stages of organotropic metastasis. In a recent study, homing of patient-derived cells and cell lines for multiple myeloma to the CVP was observed within a few minutes after injection (Sacco et al., 2016), and here, we observed differential targeting for non-hematological cancers. Recent work has highlighted the role of physical forces on cell extravasation in the zebrafish CVP at early time points (1 dpi) (Follain et al., 2018). However, within different organs, tissue architecture, composition, mechanics, and regulatory signaling networks provide cues that span length and time scales to drive the emergence of a metastatic lesion (Kim and Tanner, 2015; Oudin and Weaver, 2016). We therefore built on previous work by simultaneously examining multiple organs and characterizing vessel architecture and tissue mechanics in detail and cell behavior over longer time spans (up to 5 dpi). By monitoring cancer cell dissemination, organ arrest, and extravasation in multiple organs and with multiple organotropic cell types, this system enables determination of key steps in the non-random distribution of cancer cells throughout the organism. This process is illustrated schematically for MDA-MB-231 brain-tropic (231BR) and bone-marrow-tropic (231BO) cells in Figure S8. Vessel topography drove preferential cell occlusion in the CVP over the brain. Once arrested at the vascular interface, differences in extravasation, particularly in the CVP, led to greater relative extravasation of 231BO cells in the CVP (Figures 6 and S8).
Numerous in vitro studies have demonstrated how topographical cues can influence cell motility, invasion, and gene expression (Paul et al., 2017). In the CVP, vessel architecture was comparable to the tortuosity found within bone marrow, which is distinct from the organized linear blood vessels observed in other organs (Kusumbe et al., 2014; Nombela-Arrieta et al., 2013; Ramasamy et al., 2016). In line with Paget’s hypothesis, our results demonstrate that differences in the vessel architecture of the “soil” contribute to the efficacy of the establishment of a distant lesion. However, our results suggest that bone-marrow-tropic and brain-tropic cells do not necessarily arrest at greater frequencies in one organ over another and that aspects of cell dissemination may be independent of the features of the disseminating cells. We confirm that the blood flow rates and mechanical properties of the organ analogs are similar to each other on the day of injection and that the tissue mechanics are similar to 3D hydrogel mimetics (Blehm et al., 2016; Kniazeva et al., 2012; Kotlarchyk et al., 2011; Staunton et al., 2016) to strengthen our assertion that topography is the primary driver of this initial segmentation. While topography played a dominant role in cell arrest over the time period studied here, it is likely that the cell glycocalyx would also contribute to cell survival after arrest or extravasation and thus influence the efficiency of metastasis (Cheung et al., 2011; Li and King, 2012; Mitchell and King, 2014; Paszek et al., 2014). Additionally, differences in the composition of the endothelial glycocalyx in the brain compared to that in the CVP may drive specific cancer cell-adhesion ligand interactions, and the role of the glycocalyx in activating integrins (Paszek et al., 2014) may influence cell extravasation following cell arrest.
Following dissemination, both brain-targeting and bone-marrow-targeting cells arrested in a given microenvironment were exposed to the same cues but extravasated at different rates. Differences in proteins implicated in mechanosensing were observed for the MDA-MB-231 tumor cell “seeds” (Schwartz, 2010). Of these proteins, β1 integrin has been implicated in modulating extravasation for multiple types of tumors using in vivo and in vitro assays (Chen et al., 2016; Stoletov et al., 2010). Our findings suggest that β1-integrin-mediated extravasation is a primary mode of cell exit from the bloodstream in the CVP, in line with previous studies showing that Twist expression regulates β1-integrin-dependent extravasation in the ISVs (Stoletov et al., 2010). However, to our knowledge, the specific role of β1 integrin on cancer cell extravasation in the brain, and whether inhibition of β1 integrin can specifically affect extravasation in one organ differently than in another, has not been tested. In the brain, knockdown of β1 integrin again inhibited extravasation of bone-marrow-tropic 231BO cells, but extravasation of brain-tropic 231BR cells remained at control levels (Figure 7). This phenomenon is reminiscent of T cell but not myeloid cell reliance on β1 integrin for infiltration to the central nervous system in a murine model of experimental autoimmune encephalomyelitis (Bauer et al., 2009). Previous work has demonstrated that tumor cells in confinement are able to migrate efficiently upon inhibition of β1 integrin activity or cell contractility (Balzer et al., 2012; Stroka et al., 2014), and brain-tropic cells may be utilizing these alternate mechanisms for colonization. Myosin 1B, a class I myosin, was expressed at higher amounts in multiple brain-targeting clones. Myosin 1B has been implicated in normal physiology, where it is a widely expressed, single-headed, actin-associated molecular motor in neuronal tissues (Sherr et al., 1993). It is associated with embryonic cell migration, intracellular cargo transport, and filopodia and lamellipodia formation in differentiated cells (Almeida et al., 2011; Iuliano et al., 2018; Salas-Cortes et al., 2005). Its role in cancer is less prevalent. However, a recent report implicated myosin 1B as an important mediator of motility and lymph node metastasis in human head and neck cancers (Ohmura et al., 2015). Here, our data suggest that it may also play a role in organotropism. However, further studies will be needed to confirm this role.
Examination of tumor cell dissemination from the primary site has identified the relative inefficiency of metastatic outgrowth in spite of the presence of circulating tumor cells in the bloodstream (Condeelis and Segall, 2003; Ewald et al., 2011; Friedl and Alexander, 2011; Hirata et al., 2015). In the zebrafish model used here, ~15%–25% of cells injected were present at 5 dpi (Figure S8). This metastatic efficiency—an indicator of how many cells survive dissemination to exit and colonize—is comparable or greater than what has been observed in murine studies (Fidler, 2003). However, while certain aspects of organotropism are likely conserved between zebrafish and murine models, further work will be needed to fully understand how translational tropism results obtained in zebrafish are to human patients and to mechanistically understand why different cell types exhibit distinct extravasation potential in different organs. Many aspects of brain architecture and cell type are conserved in the larval fish at the developmental stage that was used. However, at this stage, osteoclasts are not fully mature; instead, the CVP is analogous to the HSC niche, both of which contain sinusoidal vascular vessels, fibroblastic reticular cell networks, and perivascular hematopoietic stem and progenitor cells (Kusumbe et al., 2014; Murayama et al., 2006; Nombela-Arrieta et al., 2013; Ramasamy et al., 2016). Moreover, the cytokines implicated in maintaining the niche, such as CXCR4-CXCL12, are conserved between zebrafish and mammals (Ma et al., 2011; Murayama et al., 2006). It is worth noting that the results here were obtained in the presence of the innate immune system but before adaptive immunity has emerged (Renshaw and Trede, 2012). Additionally, pre-conditioning of the metastatic niche via integrin-based targeting of primary tumor-derived exosomes has been shown to influence organ targeting and survival of cancer cells in secondary organs (Hoshino et al., 2015). Microenvironmental cues such as tumor-stromal crosstalk are important regulators of disseminated tumor outgrowth (Barnes et al., 2017; Bissell and Hines, 2011; Tanner and Gottesman, 2015), and some but not all of these cues are present in the zebrafish model. Our results suggest that there is sufficient homology to study initial trafficking steps in multiple organs not readily accessible in some studies. Because only ~100 cells are introduced into each zebrafish, this model may be used to further understand how multiple microenvironmental cues may influence organ targeting and establishment of metastatic sites, and, as an extension, drug response (Fior et al., 2017) at the time of diagnosis using patient-derived tissue.
Conclusions
We determined that non-random organ colonization is made possible by a two-step process when mammalian mammary cells were introduced into larval zebrafish. Both organ-targeting clones migrated with similar speeds within the vasculature, but more cells became arrested upon arrival in the topographically disordered blood vessels of CVP than in the linear vessels in the brain, regardless of the cell type tested. Following arrest, bone-marrow- and brain-tropic clones exhibited organ-specific patterns of extravasation. A reduction in extravasation of brain-tropic cells in the CVP led to a relative increase in brain targeting. Proteomic analysis and in vivo screening of brain- and bone-marrow-targeting cells implicated β1 integrin and myosin 1B as important mediators of extravasation in the CVP and brain, respectively (Figure S8). This work emphasizes the importance of the interplay between tissue architecture and cell behavior at the vascular interface to drive preferential colonization of one organ at early stages of metastasis. Our data suggest that the larval fish possesses physiological similarities that may be used to screen potential metastatic sites using patient-derived cells.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kandice Tanner (kandice.tanner@nih.gov). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish Husbandry
Animal studies were conducted under protocols approved by the National Cancer Institute and the National Institutes of Health Animal Care and Use Committee. Zebrafish were maintained at 28.5°C on a 14-hour light/10-hour dark cycle according to standard procedures. Larvae were obtained from natural spawning, raised at 28.5°C, and maintained in fish water (60 mg Instant Ocean© sea salt [Instant Ocean, Blacksburg, VA, Catalog #SS15–10] per liter of DI water). Larvae were checked regularly for normal development. For all experiments, larvae were transferred to fish water supplemented with N-phenylthiourea (PTU; Cat. No. P7629–25G, Millipore Sigma, St. Louis, MO) between 18–22 hours post-fertilization to inhibit melanin formation and maintain optical transparency. PTU water was prepared by dissolving 16 μl of PTU stock (7.5% w/v in DMSO) per 40 ml of fish water. Water was replaced daily. For cancer cell injections, larvae were maintained at 28.5°C until the time of injection (2 dpf) and kept at 33°C following injection. Clutches were divided randomly into experimental groups (e.g., injection with brain-targeting vs. bone marrow-targeting cells) prior to injection. Injected fish were maintained at 33°C until reaching 4 dpf and were then introduced to a larger zebrafish housing system and held in system water at 28.5°C. Regular feeding commenced at 5 dpf. Larvae were collected from the housing system for imaging at 7 dpf. Zebrafish have not completed gonad differentiation by the endpoints used in these experiments.
Zebrafish Lines
For analysis of non-random tissue targeting of MDA-MB-231, Bt474m1, and 4T1 cell lines, the recently described Tg(MRC1a:EGFP) zebrafish (Jung et al., 2017) line was crossed with Tg(flk:mCherry) zebrafish (Wang et al., 2010), as described previously (Venero Galanternik et al., 2017). Transgenics were crossed into a Casper background to avoid pigmentation (White et al., 2008). Stable transgenic lines expressing both fluorophores were used in all experiments. In this transgenic strain, EGFP is expressed under the MRC1a promoter to label, at the larval stage, the venous circulation, while mCherry is expressed under the flk promoter to label the vasculature. This line was also used in experiments in which MDA-MB-231 clones were transfected with integrin β1 siRNA, myosin 1B siRNA, or control siRNA prior to injection, and for whole mount immunofluorescence staining for annexin V after injection of MDA-MB-231 clones. For analysis of the zebrafish vasculature (vessel size and organization), 2 dpf Tg(fli1:EGFP) transgenic fish were used. The Tg(fli1:EGFP) strain (Lawson and Weinstein, 2002) was also used in experiments studying the effects of treatment with β1-integrin function blocking antibodies on initial cell trafficking through the vasculature, and for injection with fluorescent 10 μm-diameter polystyrene beads. In experiments for which cells were stained for expression of Ki67 and in which the response of zebrafish neutrophils to injected cells was assessed, Tg(mpx:EGFP/flk:mCherry) zebrafish (Renshaw et al., 2006; Wang et al., 2010), in which neutrophils express EGFP, were used. Casper zebrafish (White et al., 2008) were used for direct brain injections of MDA-MB-231 clones.
Cell Culture
MDA-MB-231 human bone marrow-tropic (231BO) and brain-tropic (231BR) breast adenocarcinoma cells (Yoneda et al., 2001) were grown and maintained in high glucose (4.5 g/l) DMEM containing 10% FBS, 1% L-glut, and 1% P/S at 37°C. In some cases, cells transformed to express GFP and luciferase (231BR-GFP/Luc) or mKate (231BO-mKate) were used. Bt474m1 and Bt474m1-Br3 human breast cancer cells (Zhang et al., 2013) were maintained in RPMI medium supplemented with 10% FBS, 1% P/S, and 1% L-glut. 4T1Br4 and 4T1BM2 murine breast cancer cells (Kim et al., 2018; Kusuma et al., 2012) were maintained in α-MEM containing sodium pyruvate (Cat. No. 10-022-CV, Corning Mediatech, Corning, NY) and supplemented with 5% FBS, 1% P/S, and 1% L-glut. Cells were passaged upon reaching 70–80% confluency, and media was changed every 2–3 days and 24 h before all zebrafish injections. All cell lines were female.
All cell lines were evaluated for the presence of mycoplasma and tested negative (Genetica DNA Laboratories, Burlington, NC). Cell line STR profiling was performed by Genetica DNA Laboratories. The MDA-MB-231 and Bt474 cell line derivatives had EV≥0.86 compared to parental MDA-MB-231 or Bt474 cells, respectively, and did not match the STR profiles for any other cells in the DSMZ profile database (EV≤0.83) using the DSMZ online STR matching analysis tool (Dirks et al., 2010). 4T1Br4 and 4T1BM2 cells were positive for a mouse marker (PowerPlex16HS kit) and did not have human STR data. 231BR, 231BO, Bt474m1-Br3, and Bt474m1 cells were free of mouse DNA.
METHOD DETAILS
Zebrafish Circulatory Injections
Prior to injection, cells were washed with PBS and detached from the cell culture flask using 10 mM EDTA diluted in PBS for 20 min at 37°C. Cells were resuspended in full growth medium, pelleted, resuspended in growth medium again, and counted. Counted cells were centrifuged at 1000 rpm for 5 min and resuspended at a concentration of 2×106 cells/ml in PBS containing CellTracker Deep Red (Cat. No. C34565, Thermo Fisher Scientific, Waltham, MA) at 1 μM or CellTracker Blue CMAC (Cat. No. C2210, Thermo Fisher Scientific) at 10 μM. Cells were stained with CellTracker at 37°C for 20–30 min. Stained cells were washed twice with PBS, with spins at 2000 rpm for 3 min between washes. After the final spin, cells were resuspended to a concentration of 1 million cells/20 μl in PBS. For integrin-blocking experiments, the final resuspension solution contained 30 μg/ml IgG mouse isotype control (Cat. No. ab91353, abcam, Cambridge, MA) or anti-β1 integrin (abcam, Cat. No. ab24693) antibody. Cells were kept on ice for 1–2 hours during injection, with at least 30 min incubation prior to the first injection. Alternatively, 10 μm-diameter polystyrene beads (crimson fluorescent FluoSpheres, Cat. No. F8831, Thermo Fisher Scientific) were centrifuged and resuspended to 1 million beads/20 μl PBS.
Tricaine stock was prepared by dissolving 400 mg of Tricaine powder (ethyl 3-aminobenzoate methanesulfonate; Cat. No. E10521–50G, Millipore Sigma) with 97.9 ml of deionized water and 2.1 ml of 1 M Tris. Anesthetic fish water was prepared by mixing 4.2 ml of tricaine stock per 100 ml of fish water supplemented with PTU (0.4% buffered tricaine). For injection to the circulation, 2 dpf zebrafish were anesthetized in 0.4% buffered tricaine and oriented in a lateral orientation on an agarose bed. As needed, zebrafish were dechorionated using 2 mg/ml pronase (Millipore Sigma, Cat. No. 10165921001) in fish water prior to anesthetization. A volume of 2–5 nL of the cell or bead suspension at 1 million cells/20 μl (~100–250 cells) were injected directly in the circulation via the posterior cardinal vein using a pulled micropipette. Fish from a given clutch were randomly divided into experimental groups prior to injection. For all experiments involving gene silencing or integrin function blocking, experiments were blocked so that treated and control cells were injected into larvae from the same clutch. Injected fish were screened between 4–20 h post-injection to check for successful dissemination of cells through the circulatory system and were then imaged or housed for later experiments. Fish lacking cells in the circulation were euthanized. Larvae were imaged as described below.
Direct Injections to the Zebrafish Brain Parenchyma
For select experiments, 231BR-GFP/Luc and 231BO-mKate cells were prepared as described above and incubated with 30 μg/ml IgG antibody and mixed in equal parts to obtain a total concentration of 1 million cells/20 μl in PBS. Zebrafish at 2 dpf were prepared for injection as described above. A volume of 2–5 nL of the cell suspension was injected directly to the zebrafish hindbrain using a pulled micropipette. Larvae were imaged as described below.
siRNA Knockdown
Cells were plated at 200,000 cells/well of a 6-well plate in 2.5 ml total growth medium. The following day, cells were transfected with Thermo Fisher Silencer Select siRNA. siRNA was designed to block expression of β1 integrin (Thermo Fisher Scientific, Cat. No. 4390824, siRNA ID s7575), myosin 1B (Thermo Fisher Scientific, Cat. No. 4392420, siRNA ID s8964), or to be used as a negative control (Silencer Select Negative Control No. 1, Thermo Fisher Scientific, Cat. No. 4390843). siRNA was aliquoted and stored at a concentration of 50 μM and stored at −20°C. Prior to transfection, aliquots were diluted 1:10 in Opti-MEM cell culture medium (Thermo Fisher Scientific, Cat. No. 31985070) to reach a concentration of 5 μM. Per well of a 6-well plate, 3 μl of siRNA at 5 μM was mixed with 150 μl Opti-MEM. In a separate tube, 7.2 μl RNAiMAX (Thermo Fisher Scientific, Cat. No. 13778030) was mixed with 150 μl Opti-MEM. Next, 150 μl diluted of the Opti-MEM/siRNA mixture was combined with 150 μl of the Opti-MEM/RNAiMAX mixture and incubated for 5 min at room temperature. To transfect, 250 μl of this mixture was added to cells plated the previous day, directly to the existing medium. The following day, media was replaced with fresh growth media. Two days following transfection, cell lysate or RNA was collected (see below) or cells were used for injection to zebrafish larvae as described above.
Intravital Microscopy of Cell Distributions
Zebrafish larvae were anesthetized and immobilized in a lateral orientation in 1% (w/v) low gelling temperature agarose (Cat. No. A9414–25G, Millipore Sigma) dissolved in fish water. To enable high-resolution confocal imaging of mounted larvae, fish were laterally oriented in coverglass-bottom chamber slides (Nunc Lab-Tek Chambered #1.0 Borosilicate Coverglass slides, Cat. No 155383, Thermo Fisher Scientific). PTU water supplemented with 0.4% buffered tricaine was then added to the imaging chamber to keep the larvae anesthetized over the course of the experiment.
To acquire images of cancer cells and/or neutrophils at single-cell resolution within living zebrafish from 1–5 days post-injection, chamber slides containing the larvae were imaged on Zeiss 710 or 780 laser scanning confocal microscopes. Three-dimensional tile scans of the brain and caudal vein plexus (CVP) were obtained at 1, 2, or 5 dpi to assess cell homing. No overlap was used in tile scans of larvae injected with cancer cells. Images were taken at axial steps of 1 μm and stacked to create three dimensional images. One-photon, confocal 12-bit images were acquired with a Zeiss 20x EC Plan-Apochromat, 0.8 NA objective and a digital zoom of 1, resulting in a field of view of 425.1 μm × 425.1 μm for each tile of the image. Pinhole diameter was set at 90 μm. Samples were simultaneously excited with 488 nm light from an argon laser with a total power of 25 mW, 561 nm light from a solid-state laser with a total power of 20 mW, and 633 nm light from a HeNe633 solid state laser with a total power of 5 mW. Transmitted light was also recorded. Images were taken on two tracks to minimize signal overlap. All lasers were set at or below 8% total power. A beam splitter, MBS 488/561/633, was employed in the emission pathway to delineate the red (band-pass filters ~580–645 nm), green (band-pass filters ~493–574 nm), and far red (band-pass filters ~650–747 nm) channels. The master gain was set at or below 700 for each channel. The zebrafish larvae were maintained at 33°C for the course of imaging. Pixel dwell times of 1.58 ms were used.
Intravital Microscopy of Zebrafish Vasculature
To obtain images of the vasculature of 2 dpf larvae, whole-larva confocal tile scans (15% tile overlap) of 2 dpf Tg(fli1:EGFP) zebrafish were acquired on a Zeiss 780 LSM confocal microscope with a Zeiss 20x EC Plan-Apochromat, 0.8 NA objective and a digital zoom of 1 using an axial step of 1 μm. One-photon, confocal 12-bit images were acquired with a Zeiss 20x EC Plan-Apochromat, 0.8 NA objective and a digital zoom of 1, resulting in a field of view of 425.1 μm × 425.1 μm for each tile of the image. Pinhole diameter was set at 90 μm. Samples were excited with 488 nm light from an argon laser with a total power of 25 mW. Transmitted light was also recorded. Laser power was set at 2%. The master gain was set at or below 700 for each channel. The zebrafish larvae were maintained at 33°C for the course of imaging. Pixel dwell times of 1.58 ms were used.
Intravital Imaging of Cell Trafficking
To image motile cells (or polystyrene beads) over the first 12–16 h following injection to the zebrafish circulation, zebrafish were anesthetized and immobilized as described above approximately 4 h after cell injection. One-photon, confocal, 2-dimensional images were acquired at lateral dimensions of 512 × 512 pixels, which were stacked to acquire 3-dimensional images. Images were obtained on Zeiss 710 or 780 confocal microscopes. Imaging conditions used were:
Tg(flk:mCherry/MRC1a:EGFP) or Tg(flk:mCherry/mpx:EGFP) zebrafish were injected with 231BR, 231BO, 4T1Br4, 4T1BM2, or 231 control or integrin β1 knockdown cells stained with Cell Tracker Deep Red prior to injection. Confocal z-stacks were acquired every 10 minutes for ~90 frames at 2 μm axial steps to image a total depth of ~150 μm. 12-bit images were acquired with a Zeiss 20x EC Plan-Apochromat, 0.8 NA objective and a digital zoom of 0.8, resulting in a field of view of 531.37 μm ×× 531.37 μm. Pinhole diameter was set at 90 μm. Samples were simultaneously excited with 488 nm light from an argon laser with a total power of 25 mW, 561 nm light from a solid-state laser with a total power of 20 mW, and 633 nm light from a HeNe633 solid state laser with a total power of 5 mW. Transmitted light was also recorded. Images were taken on two tracks to minimize signal overlap. All lasers were set at 2% total power. A beam splitter, MBS 488/561/633, was employed in the emission pathway to delineate the red (band-pass filters ~580–645 nm), green (band-pass filters ~493–574 nm), and far red (band-pass filters ~650–747 nm) channels. The master gain was set at or below 700 for each channel. The zebrafish larva was maintained at 33°C for the course of imaging. Pixel dwell times of 1.58 ms were used. Multiple positions were imaged to capture both the brain and tail of a given zebrafish in the same experiment. These imaging conditions were also used in Tg(fli:EGFP) zebrafish following injection of red fluorescent beads to the circulation, with the far red channel omitted.
Tg(fli1:EGFP) zebrafish were injected with 231BR or 231BO cells treated with 30 μg/ml of a function-blocking β1 integrin antibody or IgG control antibody. Cells were stained with CellTracker Deep Red prior to injection. Confocal z-stacks were acquired every 10 minutes for ~90 frames at 2 μm axial steps to image a total depth of ~150 μm. 12-bit images were acquired with a Zeiss Plan-Neofluar 10x/0.30 NA objective and a digital zoom of 1, resulting in a field of view of 850.19 μm × 850.19 μm. Pinhole diameter was set at 100 μm. Samples were simultaneously excited with 488 nm light from an argon laser with a total power of 25 mW and 633 nm light from a HeNe633 solid state laser with a total power of 5 mW. Transmitted light was also recorded. All lasers were set at 2% total power. A beam splitter, MBS 488/633, was employed in the emission pathway to delineate the green (band-pass filters ~493–598 nm) and far red (band-pass filters ~640–747 nm) channels. The master gain was set at or below 700 for each channel. The zebrafish larva was maintained at 33°C for the course of imaging. Pixel dwell times of 1.58 ms were used.
Zebrafish Whole Mount Immunofluorescence Staining
Tg(flk:mCherry/MRC1a:EGFP) zebrafish were injected in the circulation at 2 dpi with CellTracker Blue CMAC-stained 231BR or 231BO cells as described above. At 5 dpi, larvae were fixed overnight at 4°C in 4% paraformaldehyde in PBST (PBS supplemented with 0.1% Tween 20). Alternatively, uninjected Tg(flk:mCherry/MRC1a:EGFP) zebrafish were fixed at 3 dpf or 7 dpf for imaging of zebrafish E-cadherin expression. Fixed larvae were washed three times in PBST and permeabilized with 10 μg/ml Proteinase K (Millipore Sigma, Cat. No. 3115879001) in PBST for 3 h at room temperature. Larvae were washed four times with PBST and blocked for 2 h at room temperature in PBST containing 5% goat serum. Larvae were then incubated in a 1:50 dilution of rabbit polyclonal anti-Ki67 antibody (Millipore Sigma, Cat. No. AB9260), a 1:50 dilution of rabbit polyclonal anti-annexin V antibody (Thermo Fisher Scientific, Cat. No. PA5-27872), a 1:50 dilution of anti-zebrafish Cdh1 antibody (GeneTex, Irvine, CA, Cat. No. GTX125890), or a 1:50 dilution of rabbit IgG isotype control antibody (Thermo Fisher Scientific, Cat. No. 31235; 1:50 dilution from isotype control stock first diluted to 1 mg/ml in PBST). All antibodies were diluted in PBST with 5% goat serum, and larvae were stained at 4°C overnight. Stained larvae were washed quickly in PBST four times, and then washed an additional three times in PBST for 10 min each. Fish were blocked in 5% goat serum in PBST for 2 h at room temperature prior to secondary antibody addition. Larvae were transferred to a 1:100–1:200 (5–10 μg/ml) secondary antibody cocktail containing an AlexaFluor Plus 647 goat anti-rabbit secondary antibody (Thermo Fisher Scientific, Cat. No. A32733) in PBST with 5% goat serum. Fish were stained for 3 h at room temperature, washed three times in PBST, and imaged with a Zeiss 780 confocal microscope.
For control staining of cells on 2D substrates, 231BR or 231BO cells were seeded on ibidi 35 mm μ-Dishes with ibiTreat surfaces (ibidi, Martinsried, Germany, Cat. No. 80136) in full growth medium. The following day, cells were fixed (for Ki67 staining; see fixation and staining details below), or media was replaced with growth medium containing 1 μM vincristine sulfate (Millipore Sigma, Cat. No. V8879-5MG) or DMSO control. After treatment for 24 h, cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, then washed three times with PBST. Cells were permeabilized witih 0.3% Triton X-100 in PBS for 5 min at room temperature and washed three additional times in PBST. Non-specific binding was blocked for 1 h at room temperature with 5% goat serum in PBST. Cells were then stained with 1:500 dilutions of the anti-Ki67, anti-annexin V, or rabbit isotype control antibodies described above diluted in 5% goat serum in PBST at 4°C overnight. Stained cells were washed three times in PBST and stained with a cocktail of 1:200 AlexaFluor Plus 647 goat anti-rabbit antibody, 1:40 AlexaFluor 488 phalloidin (Thermo Fisher Scientific, Cat. No. A12379), and 1 μg/ml Hoechst 33342 (Thermo Fisher Scientific, Cat. No. H3570) in 5% goat serum in PBST for 1 h at room temperature. Cells were washed three times in PBST and imaged.
To image both fixed cells in 2D and stained zebrafish, images were taken with 4 tracks to minimize fluorophore crosstalk. Confocal z-stacks were acquired at 0.5 μm axial steps with the pinhole diameter set at 90 μm. 12-bit images were acquired with a Zeiss 20x EC Plan-Apochromat, 0.8 NA objective. Samples were sequentially excited with 488 nm light from an argon laser with a total power of 25 mW, 561 nm light from a solid-state laser with a total power of 20 mW, 405 nm light from a 30 mW diode laser, and 633 nm light from a HeNe633 solid state laser with a total power of 5 mW. Transmitted light was also recorded. All lasers were set at 2–4% total power. The master gain was set at or below 700 for each channel. Pixel dwell times of 1.58 ms were used. Presented images show single confocal slices in indicated regions (Ki67 and annexin V images) or maximum intensity projections from six confocal slices at the plane of the blood vessels (E-cadherin images). Lookup tables were adjusted to the same minimum and maximum values in the far-red (secondary antibody) channel in each image.
Microchannel Experiments
Master molds for the microchannel devices were fabricated from SU-8 on silicon wafers using standard photolithographic techniques for dual-height structures. Two chrome-glass contact masks were designed using L-Edit (Mentor Graphics, Wilsonville, OR) or AutoCAD (Autodesk, San Rafael, CA) and purchased from Front Range Photomask (Lake Havasu City, AZ). Four-inch silicon wafers (NOVA Electronic Materials, Flower Mound, TX) were first cleaned with acetone, isopropanol, and water, then dehydrated for 10 minutes on a 200°C hotplate. Three layers of SU-8 photoresist (Microchem, Newton, MA) were used to pattern the devices, with minor modifications of the manufacturer suggested protocols. First, an adhesion layer of SU-8 2000.5 was spun on the wafer using a single wafer spin coater (Model WS-650S-8NPP/LITE, Laurell Technologies, North Wales, PA), softbaked, flood-exposed with the mask aligner (Model 208, Optical Associates, Inc., San Jose, CA), and post-exposure baked. Second, the microchannel layer was patterned using the first chrome-glass mask with SU-8 2005 to make an array of 7 μm high, 8 μm wide channels; the SU-8 2005 layer was softbaked, exposed, and post-exposure baked. Both the SU-8 2000.5 and the SU-8 2005 were prepared in our laboratory by dilution of SU-8 2050 with SU-8 thinner (Microchem) to obtain the desired percent solids as per the manufacturer product sheets.
The large fluid channels were patterned with SU-8 2100 as received, using the second chrome-glass mask. Solvent edge bead removal was performed during the spinning step to ensure good contact between the mask and resist, and alignment marks were used to ensure the small microchannels were centered in the gap of the larger channels. For the thick layer, a long-pass filter (PL-360-LP, Omega Optical, Brattleboro, VT) was used during exposure to prevent overexposure of the top of the resist layer. After this final layer was softbaked, exposed, and post-exposure baked, the pattern was developed with SU-8 developer, rinsed thoroughly, and hardbaked at 150°C for two minutes. The templates were then treated with silane vapor (tridecafluoro-1,1,2,2-tetrahydrooctyl trichlorosilane (Gelest, Morrisville, PA, Cat. No. SIT8174.0) for one hour to reduce adhesion during use.
Final microfluidic devices were formed using replica molding with polydimethylsiloxane (PDMS; Sylgard 184 kit, Dow Corning, Midland, MI, USA) at a ratio of 10 parts prepolymer to 1 part crosslinker. The PDMS components were mixed, poured over the patterned wafer, and degassed. PDMS devices were polymerized at 85°C for ~2 h before removal from the master mold. Devices were then cut, and fluid access ports were punched with 4 mm biopsy punches (Miltex 69031–05, Electron Microscopy Sciences, Hatflield, PA).
PDMS devices were cleaned with ethanol and DI water. Devices and two-well coverglass-bottom chamber slides (Nunc Lab-Tek Chambered #1.0 Borosilicate Coverglass slides; Cat. No 155380, ThermoFisher Scientific) were exposed to oxygen plasma for 30 sec. PDMS devices were then irreversibly bound to the slides and filled with PBS. Device walls were coated with 20 μg/ml rat tail collagen type I (BD Biosciences, San Jose, CA, Cat. No. 354236), 10 μg/ml fibronectin from human plasma (Millipore Sigma, Cat. No. FC010), or 1% BSA (Millipore Sigma, Cat. No. A7906-100G) by adsorption for 1 h at 37°C, 5% CO2. All proteins were dissolved from stock concentrations in PBS. Devices were washed with PBS following removal of the protein solution.
To examine cell migration in microchannels of varying topography, devices were coated with 10 μg/ml human fibronectin. 231BR or 231BO cells were detached with 10 mM EDTA in PBS containing 1 μg/ml Hoechst 33342 for 20 min at 37°C, 5% CO2. Cells were resuspended in growth medium, centrifuged for 5 min at 1000 rpm, resuspended in growth medium, and counted. Cells were then incubated in a solution of 1 μM CellTracker Deep Red in PBS for 20 min at 37°C. After staining, cells were washed once in PBS and resuspended to 500,000 cells/ml in serum containing medium. To examine cell migration in straight 8 μm-wide, 10 μm-tall microchannels coated with a panel of extracellular matrix proteins, 231BR and 231BO cells transformed to express green fluorescent protein (GFP) and luciferase (Luc), hereafter referred to as 231BR-GFP/Luc and 231BO-GFP/Luc, respectively, were washed with PBS, detached with 0.25% trypsin-EDTA, and resuspended in serum containing medium at 1×106 cells/ml.
For all devices, cells were introduced to the device by adding 20 μl of the cell seeding solution to the device inlet well, and the device was placed in the incubator for 15 min for cell attachment. If cell inlet flow stopped during this period, some of the seeding solution was moved to the other cell well to re-initiate flow. Following seeding, devices were washed by adding serum free medium to each well and incubating the device for 30 min. Devices were then washed three times using medium with serum. After the final wash, medium with serum was added to all wells, with an additional 2.5 ml/well of serum containing medium added to each well of the chamber slide to keep the device immersed and minimize evaporation during imaging. Thus, cells migrated in the device in the absence of flow and chemical gradients.
To obtain live cell images of migrating cells, devices were mounted in a stage top incubator on a Zeiss 780 LSM confocal microscope ~2 h after the final device wash. Devices were maintained at 37°C, 5% CO2 and imaged every 10 min for up to 12 h. Imaging was performed as described below:
Microchannels of varying topography – A Zeiss 20x EC Plan-Apochromat, 0.8 NA objective and a digital zoom of 0.8 was used to capture a field of view of 607.28 μm × 607.28 μm. Multiple positions were used to obtain fields of view encompassing the entire device. Samples were simultaneously excited with 633 nm light from a HeNe laser with a total power of 5 mW and 405 nm light from a diode laser with a total power of 30 mW to capture 2048 pixel × 2048 pixel, 12 bit, 2 line mean averaged images with the pinhole width set a 181 μm. Lasers were set at 2% of total power, and detection gain was kept below 700. Four biological replicates were run for each cell type.
Straight microchannels coated with panel of ECM proteins – A Zeiss Plan-Neofluar 10x/0.30 NA objective and a digital zoom of 1 was used to capture a field of view of 850.19 μm × 850.19 μm. Multiple positions were imaged to obtain fields of view encompassing the entire device. Samples were excited with 488 nm light from an argon laser with a total power of 25 mW to capture 2048 pixel × 2048 pixel, 16 bit, 2 line mean averaged images with the pinhole width set at 90 μm. The laser was set at 2% of total power. Biological duplicates were run for each combination of cell type and ECM protein, with the exception of BSA, where three devices were analyzed for each cell type to obtain a sufficient number of cells in microchannels.
Optical Tweezer-Based Microrheology
For all experiments, at 24 hours post fertilization (hpf), larvae were transferred to fish water supplemented with PTU to inhibit melanin formation for increased optical transparency. Larvae were then returned to the incubator at 28.5°C and checked for normal development. Zebrafish larvae at 2 dpf were anesthetized using 0.4% buffered tricaine. Polystyrene beads, 1 micron in diameter resuspended in PBS with a final cell concentration of 1 × 107 beads/ml, were injected directly into the organ of interest as previously described (Blehm et al., 2016; Staunton et al., 2017). Larvae were then anesthetized using 0.4% buffered tricaine and embedded in a lateral orientation in 1% low melting point agarose and allowed to polymerize in with cover glass (no. 1.5 thickness) as previously described (Blehm et al., 2016). Fish water supplemented with tricaine was added to the agarose hydrogel for the entire time of data acquisition. For complete experimental details, see (Blehm et al., 2016; Staunton et al., 2017, 2016).
Our home-built setup consists of a 1064 nm trapping beam steered by an acousto-optic deflector to oscillate the trap at frequency ω and a stationary 975 nm detection beam that is coupled into and colocated with the trap with a dichroic before being sent into the backport of an inverted microscope with a long working distance water objective and a high NA condenser. Telescope lenses conjugate the optical plane at the acousto-optic deflector (AOD) to the back aperture of the condenser, which is placed in Kohler illumination after the object is focused in the specimen place. Above the condenser, the detection beam is relayed to a quadrant photodiode (QPD) for back focal plane interferometric position detection. Each bead was positioned precisely in the center of the trap by scanning it through the detection beam in three dimensions using a piezo nanopositioning stage while recording the voltages from the QPD. The V-nm relation, β, of the QPD was calibrated in situ by fitting the central linear region of the detector response to scanning the bead through the detection beam in the direction of the oscillations, giving β in V/nm. A second QPD recorded the position of the trapping laser to find the relative phase lag between the bead and trap oscillations. The optical trap stiffness k was determined in situ for each measured bead using the active-passive calibration method (Fischer and Berg-Sørensen, 2007). The stiffness was determined from: the real part of the active power spectrum, , with and the Fourier transforms of the time series of the positions of the trapping laser and the driven bead, respectively, recorded while the trap was oscillating; and the passive power spectrum, , where is the Fourier transform of the time series of the undriven bead’s thermally fluctuating position while the trap was held stationary. Each bead was subjected to fourteen consecutive 2 s pulses, with the trap alternately oscillating or stationary. Amplitude of oscillations was set to 20 nm. Laser power was set to 100 mW at the microscope back port. Only probes at distances exceeding ~30 μm away from the cover slip surface were measured, to minimize drag in consideration of Faxen’s law (Neuman and Block, 2004). Experiments were controlled using custom LabVIEW programs. Samples were measured in triplicate with at least 30 beads per sample measured.
Histological Staining
Uninjected Tg(flk:mCherry/MRC1a:EGFP) larvae were fixed using 4% paraformaldehyde in PBST overnight at 4°C at 2, 3, and 7 dpf. Fixed larvae were stored in 70% ethanol prior to processing for histology. Paraffin embedding and sectioning and staining with Movat pentachrome, Mallory’s PTAH, or Alcian Blue-PAS was performed by Histoserv, Inc. (Germantown, MD). Stained sections were imaged on a Zeiss Axio Scan.Z1 slide scanner, or evaluated by a pathologist.
Red Blood Cell Flow Imaging
Images of red blood cells flowing in the zebrafish CVP or brain were obtained on a Nikon A1R resonant scanning confocal microscope following immobilization of 2 dpf Tg(flk:mCherry/MRC1a:EGFP) or Tg(flk:mCherry/mpx:EGFP) zebrafish as described above. One z slice was obtained to maximize scan speed with a confocal pinhole size of 19.16 μm. The scan area was limited to 1024 pixels × 128 pixels to obtain a 12-bit image of 880.6 μm × 110.1 μm. Images were acquired for 15 seconds at a rate of 114.8 frames per second for a total of 1722 images. Images were acquired using a Nikon APO LWD 20x, 0.95 NA objective. Samples were simultaneously excited with 488 nm and 561 nm light, and transmitted light was also collected. Images were acquired from three Tg(flk:mCherry/MRC1a:EGFP) larvae and one Tg(flk:mCherry/mpx:EGFP) larvae.
Imaging of Albumin in the Endothelial Glycocalyx
Albumin from bovine serum, Alexa Fluor 647 conjugate (BSA-647, Thermo Fisher Scientific, Cat. No. A34785) was diluted in PBS to 100 μg/ml. BSA-647 was injected into the circulation of 2 dpf Tg(flk:mCherry/MRC1a:EGFP) zebrafish as described above. Approximately 4 h following injection, larvae were anesthetized, mounted in agarose as described above, and imaged to assess BSA-647 accumulation. Three-dimensional scans of the brain CVP were taken at axial steps of 0.5 μm and stacked to create three dimensional images. One-photon, confocal 12-bit images were acquired with a Zeiss 20x EC Plan-Apochromat, 0.8 NA objective and a digital zoom of 4, resulting in a field of view of 106.07 μm × 106.07 μm for each tile of the image. Pinhole diameter was set at 36.2 μm. Samples were simultaneously excited with 488 nm light from an argon laser with a total power of 25 mW, 561 nm light from a solid-state laser with a total power of 20 mW, and 633 nm light from a HeNe633 solid state laser with a total power of 5 mW. Transmitted light was also recorded. Images were taken on two tracks to minimize signal overlap. All lasers were set at 2% total power. A beam splitter, MBS 488/561/633, was employed in the emission pathway to delineate the red (band-pass filters ~580–645 nm), green (band-pass filters ~493–574 nm), and far red (band-pass filters ~650–747 nm) channels. The master gain was set at or below 700 for each channel. The zebrafish larvae were maintained at 33°C for the course of imaging. Pixel dwell times of 1.58 ms were used. Three regions in both the brain and CVP were imaged for three larvae.
Mass Spectrometry
For proteomic analysis of brain- and bone marrow-tropic MDA-MB-231 or 4T1 cells, cells were washed with PBS and detached from cell culture flasks using 10 mM EDTA for 20 min at 37°C. Cells were resuspended in full growth medium, pelleted, resuspended in growth medium again, and counted. Cells were then centrifuged at 1000 rpm for 5 min and resuspended in PBS containing CellTracker Deep Red at 1 μM at a concentration of 2×106 cells/ml and incubated at 37°C for 20 min to match the preparation procedure used for zebrafish injections. Stained cells were washed twice with PBS, with spins at 2000 rpm for 3 min between washes. After the final spin, cells were resuspended to a concentration of 1 million cells/20 μl in PBS. For the final resuspension, cells were in PBS containing 30 μg/ml IgG. Cells were incubated with the antibody for 30 min, diluted to a concentration of 1e6 cells/ml in serum free media, and seeded in 6-well plate wells (300,000 cells/well). Additional serum free media was added to bring the total volume in each well to 2 ml. Cells were incubated for 8 h at 37°C, 5% CO2 prior to lysate collection. This cell preparation method was analogous to cell preparation for injection to the zebrafish.
For siRNA-mediated gene knockdown experiments, cells were prepared and transfected as described above. Lysate was collected at 2 days post-transfection as described above, without staining or detaching cells prior to lysis.
Protein lysates were prepared by adding 40 μl of lysis buffer containing 1 part Calbiochem 539131 Protease inhibitor cocktail set 1 (Cat. No. 539131, Calbiochem, Millipore Sigma), 1 part phosphatase inhibitor cocktail 2 (Cat. No. P5726, Millipore Sigma), 1 part phosphatase inhibitor cocktail 3 (Cat. No. P0044, Millipore Sigma), and 100 parts M-PER Mammalian Protein Extraction Reagent (Cat. No. 78501, Thermo Fisher Scientific) to each well of a seeded 6-well plate. Cells were scraped, and lysate was collected. Lysate was sonicated 20 sec, spun down 5 min at 1600 rpm, and supernatant stored at −80°C prior to mass spectrometry. Lysates were prepared over three separate days to obtain biological triplicates.
Cell lysates (250 μg each) were digested with trypsin using the filter-aided sample preparation (FASP) protocol as previously described with minor modifications (Wiśniewski et al., 2009). Lysates were first reduced by incubation with 10 mM DTT at 55 °C for 30 min. Each lysate was then diluted with 8 M urea in 100 mM Tris-HCl (pH 8.5 [UA]) in a Microcon YM-10 filter unit and centrifuged at 14,000 × g for 30 min at 20°C. The lysis buffer was exchanged again by washing with 200 μL UA. The proteins were then alkylated with 50 mM iodoacetamide in UA, first incubated for 6 min at 25 °C and then excess reagent was removed by centrifugation at 14,000 × g for 30 min at 20°C. Proteins were then washed 3 × 100 μL 8 M urea in 100 mM Tris-HCl (pH 8.0) (UB). The remaining urea was diluted to 1 M with 100 mM Tris-HCl pH 8 and then the proteins were digested overnight at 37°C with trypsin at an enzyme to protein ratio of 1:100 w/w. Tryptic peptides were recovered from the filter by first centrifugation at 14,000 × g for 30 min at 20°C followed by washing of the filter with 50 μL 0.5 M NaCl. The peptides were acidified and desalted on a C18 SepPak cartridge (Waters, Milford, MA) and dried by vacuum concentration (Labconco, Kansas City, MO). Dried peptides were fractionated by high pH reversed-phase spin columns (Thermo Fisher Scientific). The peptides from each fraction were lyophilized, and dried peptides were solubilized in 4% acetonitrile and 0.5% formic acid in water for mass spectrometry analysis. Each fraction of each sample was separated on a 75 μm × 15 cm, 2 μm Acclaim PepMap reverse phase column (Thermo Fisher Scientific) using an UltiMate 3000 RSLCnano HPLC (Thermo Fisher Scientific) at a flow rate of 300 nL/min followed by online analysis by tandem mass spectrometry using a Thermo Orbitrap Fusion mass spectrometer. Peptides were eluted into the mass spectrometer using a linear gradient from 96% mobile phase A (0.1% formic acid in water) to 35% mobile phase B (0.1% formic acid in acetonitrile) over 240 minutes. Parent full-scan mass spectra were collected in the Orbitrap mass analyzer set to acquire data at 120,000 FWHM resolution; ions were then isolated in the quadrupole mass filter, fragmented within the HCD cell (HCD normalized energy 32%, stepped ± 3%), and the product ions analyzed in the ion trap.
Assessment of Gene Knockdown by qPCR
Cells were seeded and transfected as described above. At 2 days post-transfection, RNA was isolated using TRIzol (Thermo Fisher Scientific, Cat. No. 15596018) according to the manufacturer’s instructions. Briefly, 0.4 ml Trizol per well was added directly to cells plated in 6-well plates after aspirating media to lyse cells. Homogenized lysate was mixed with 0.2 ml chloroform per ml of TRIzol and centrifuged for 15 minutes at 12,000 × g at 4 °C. The upper aqueous phase containing RNA was mixed with an equal volume of 100% ethanol and processed with the PureLink RNA Mini Kit (Thermo Fisher Scientific, Cat. No. 12183020). The mixture was processed through the kit Spin Cartridge by centrifugation at 12,000 × g for 1 minute, washed once with 700 μl per tube of Wash Buffer I, washed twice with 500 μl per tube of Wash Buffer II, and eluted in RNase-free water. The resulting RNA concentration was assessed via Nanodrop. Biological duplicates were collected for each sample.
cDNA was synthesized using SuperScript IV VILO Master Mix (Thermo Fisher Scientific, Cat. No. 11755050). For each sample, 10 μl of RNA at 100 ng/μl was combined with 4 μl of SuperScript IV VILO Master Mix or No RT Control, and 6 μl nuclease-free water was added to bring the total volume to 20 μl. Solutions were mixed and incubated at 25°C for 10 minutes, 50°C for 10 minutes, and 85°C for 5 minutes. The synthesized cDNA was used for qPCR. For each well of a 96-well reaction plate, 10 μl TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific, Cat. No. 4444557) was mixed with 2 μl of the synthesized cDNA, 7 μl nuclease-free water, and 1 μl of the TaqMan probe. Each biological sample was run in technical triplicates. The FAM-MGB TaqMan probes used were: GAPDH (Thermo Fisher Scientific, Cat. No. 4331182, Assay ID Hs04420697_g1) and MYO1B (Thermo Fisher Scientific, Cat. No. 4331182, Assay ID Hs00362654_m1). Gene expression assays were performed on an Applied Biosystems QuantStudio 3 (Thermo Fisher Scientific).
Activated Integrin β1 Immunofluorescence
231BR or 231BO cells were seeded on MatTek dishes (MatTek, Ashland, MA) coated with 100 μg/ml collagen type I and spread prior to fixation. All fixation and permeabilization steps were performed at 37°C. Cells were permeabilized and fixed in 4% paraformaldehyde in cytoskeletal buffer (CBS; 10 mM MES, 138 mM KCl, 2 mM EGTA, 3 mM MgCl2 plus 5% sucrose) for 15 minutes. Cells were rinsed 3 times in PBS++ and permeabilized with 0.5% Triton X-100 in CBS for 5 minutes. Cells were rinsed 5 times over 40 minutes with PHEM+glycine (60 mM PIPES, 2 mM HEPES, 10 mM EGTA, 2 mM MgCl2, 100 mM glycine, pH 6.9). Non-specific binding sites were blocked with 20% donkey serum in PHEM+glycine buffer for 1 hour. Cells were then rinsed 3 times with PHEM+glycine over 30 minutes. A primary antibody detecting activated integrin β1 (purified rat anti-mouse CD29, Clone 9EG7, BD Biosciences, Cat. No. 553715) was diluted in PHEM+glycine with 10% donkey serum to 10 μg/ml and incubated for 45 minutes. The secondary antibody (Jackson ImmunoResearch) was diluted in PHEM+glycine with 10% donkey serum and incubated for 25 minutes.
Stained cells were imaged using a CSU-X1 spinning disk confocal (Yokogawa, Tokyo, Japan) attached to a Zeiss Axiovert 200M microscope with a 63X Apochromat objective (NA 1.4) oil immersion objective (Zeiss). An LMM5 laser merge module (Spectral Applied Research, Ontario, Canada) equipped with 405 nm (100 mW), 488 nm (100 mW), 561 nm (50 mW), and 642 nm (100 mW) diode lasers provided excitation wavelengths. The primary dichroic (405/488/561/640) and accompanying emission filters were from Semrock (Rochester, NY). Images were captured at 16-bits using a 512 backthinned EM CCD camera (Photometrics). X, Y, and Z positioning was performed with a MS-2000 Z-piezo stage from ASI (Eugene, OR).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of Cell Numbers and Extravasated Cells in the Zebrafish Brain and CVP
Fish injected with cells were screened for health and successful dissemination of cells throughout the circulatory system prior to incubation at 33°C (2–4 dpf) and 28.5°C (4–7 dpf). At 5 dpi, fish were randomly selected from the husbandry tank, anesthetized, mounted in agarose, and imaged as described above. Larvae were imaged only if blood flow was visually confirmed in both the brain and CVP.
From the confocal images, cells were counted as being in the CVP if they were within or proximal to the cardinal artery and caudal vein plexus, from the first 15 intersegmental vessels to the end of the cardinal artery. Cells were counted as being in the brain if they were within the first 700 μm from the most distal location of arteries in the head. Cells in the heart sac, Duct of Cuvier, and aortic arches were not counted as being in the brain. In the CVP, cells that were present at the injection site were excluded from analysis. The minimum size of particles in the deep red channel to be counted as a cell was 10 μm × 10 μm, as determined by the size of the particle at the z slice of its largest diameter in Fiji. Cells were counted as extravasated if they were outside of the vessel lumen, judged by 3D volume reconstructions in Fiji.
To determine whether cells homed to the brain or CVP, the total number of cells (including intravascular and extravascular cells) in the brain and CVP as described above were counted for each larva. For a given larva, the ratio of cells in the brain to the total number of cells counted in the brain and CVP was calculated. All fish with at least one cell present were included in analysis. Cells were counted from: N=21 fish for 231BR cell injections (398 total cells); N=23 fish for 231BO cell injections (433 total cells); N=28 fish for 4T1Br4 cell injections (316 total cells); N=25 fish for 4T1BM2 cell injections (153 total cells); N=7 fish for Bt474m1-Br3 cell injections (53 total cells); N=11 fish for Bt474m1 cell injections (64 total cells); N=9 fish for injection of 231BR cells transfected with control siRNA in association with integrin β1 knockdown (68 total cells); N=7 fish for 231BR integrin β1 knockdown injections (49 total cells); N=9 fish for injection of 231BO cells transfected with control siRNA in association with integrin β1 knockdown (52 total cells); N=10 fish for 231BO integrin β1 knockdown injections (80 total cells); N=13 fish for injection of 231BR cells transfected with control siRNA in association with myosin 1B knockdown (196 total cells); N=13 fish for 231BR myosin 1B knockdown injections (135 total cells); N=11 fish for injection of 231BO cells transfected with control siRNA in association with myosin 1B knockdown (238 total cells); and N=10 fish for 231BO myosin 1B knockdown injections (129 total cells). Cells were counted in larvae at 5 dpi (231BR, 231BO, 4T1Br4, 4T1BM2, and gene silencing experiments using 231 cells) or 1 dpi (Bt474m1-Br3, Bt474m1). For a given organotropic cell line pair, the average ratio of cells in the brain to total number of cells counted was compared by unpaired two-tailed t test (Bt474m1), unpaired two-tailed t test with Welch’s correction for populations with significantly different variances (MDA-MB-231), or Mann-Whitney test if distributions did not pass D’Agostino and Pearson normality test (4T1). The distributions of these values were also compared using the Kolmogorov-Smirnov test. Additionally, the distribution of cells throughout each larva was scored according to whether cells were found only in the brain, only in the CVP, or in both the brain and CVP. The percentage of larvae fitting each category were plotted and compared by chi-square test. To further study the effect of siRNA-mediated knockdown of β1 integrin or myosin 1B on MDA-MB-231 cell survival, the total number of control and knockdown cells present at 5 dpi were counted as described above from the same pool of larvae. Totals were normalized to the number of control cells present for a given cell line and gene silenced. The effect of knockdown on the total number of cells present for a given gene was compared by two-way ANOVA on these normalized values, with differences between knockdown and control cells for a given cell type further analyzed by Sidak’s multiple comparisons test.
To study topography-mediated extravasation of MDA-MB-231 and 4T1 cells in the brain and CVP, the distribution of extravasated cells throughout each larva was scored according to whether no extravasated cells were present, or whether cells were extravasated only in the brain, only in the CVP, or in both the brain and CVP. All fish with at least one cell present were included in analysis. Cells were counted from: N=21 fish for 231BR cell injections (398 total cells); N=23 fish for 231BO cell injections (433 total cells); N=28 fish for 4T1Br4 cell injections (316 total cells); N=25 fish for 4T1BM2 cell injections (153 total cells); N=9 fish for injection of 231BR cells transfected with control siRNA in association with integrin β1 knockdown (68 total cells); N=7 fish for 231BR integrin β1 knockdown injections (49 total cells); N=9 fish for injection of 231BO cells transfected with control siRNA in association with integrin β1 knockdown (52 total cells); N=10 fish for 231BO integrin β1 knockdown injections (80 total cells); N=13 fish for injection of 231BR cells transfected with control siRNA in association with myosin 1B knockdown (196 total cells); N=13 fish for 231BR myosin 1B knockdown injections (135 total cells); N=11 fish for injection of 231BO cells transfected with control siRNA in association with myosin 1B knockdown (238 total cells); and N=10 fish for 231BO myosin 1B knockdown injections (129 total cells). Bt474m1-Br3 and Bt474m1 cells were not typically extravasated at 1 dpi and were therefore excluded from this analysis. The distributions of larvae fitting into these categories for a given pair of organotropic clones, or of a given clone with gene silencing and matched control, were compared by chi-square test, with one exception. For 231BR cells transfected with non-targeting control siRNA or siRNA targeting integrin β1, distributions were compared by two-tailed binomial test between the percentages of larvae with cells extravasated in the head following injection with either control or integrin β1 knockdown cells.
Additionally, the number of cells that had extravasated in the brain and CVP were counted for each larva. For a given larva, the ratio of cells extravasated in the brain to the total number of cells extravasated in the brain and CVP was calculated. Zebrafish in which no extravasation had occurred in either organ were excluded from this analysis. This sample encompassed larvae from N=21 fish for 231BR cell injections (196 total extravasated cells); N=22 fish for 231BO cell injections (291 total extravasated cells); N=27 fish for 4T1Br4 cell injections (258 total extravasated cells); N=23 fish for 4T1BM2 cell injections (130 total extravasated cells); N=9 fish for injection of 231BR cells transfected with control siRNA in association with integrin β1 knockdown (46 total extravasated cells); N=7 fish for 231BR integrin β1 knockdown injections (25 total extravasated cells); N=9 fish for injection of 231BO cells transfected with control siRNA in association with integrin β1 knockdown (41 total extravasated cells); N=5 fish for 231BO integrin β1 knockdown injections (31 total extravasated cells); N=13 fish for injection of 231BR cells transfected with control siRNA in association with myosin 1B knockdown (132 total extravasated cells); N=13 fish for 231BR myosin 1B knockdown injections (92 total extravasated cells); N=10 fish for injection of 231BO cells transfected with control siRNA in association with myosin 1B knockdown (164 total extravasated cells); and N=10 fish for 231BO myosin 1B knockdown injections (101 total extravasated cells). The population average extravasation ratio between isogenic clones was analyzed using unpaired two-tailed t test with Welch’s correction for populations with significantly different variances (MDA-MB-231) or Mann-Whitney test if distributions did not pass D’Agostino and Pearson normality test (4T1). The distributions of these values were also compared using the Kolmogorov-Smirnov test.
To study the effect of siRNA-mediated knockdown of β1 integrin or myosin 1B on MDA-MB-231 cell extravasation, the percentages of cells present in the brain or CVP at 5 dpi that had extravasated were also calculated for a given larvae and organ. Average percentages in a given organ upon integrin β1 or myosin 1B knockdown were normalized to the corresponding average control cell percentages in that same organ. For a given organ, these percentages were calculated when at least one cell was present in that organ at 5 dpi. This encompassed the following pool of larvae: control 231BR cells associated with integrin β1 knockdown – N=9 larvae for CVP (53 total cells), N=4 larvae for brain (15 total cells); 231BR integrin β1 knockdown cells – N=6 larvae for CVP (17 total cells), N=7 larvae for brain (32 total cells); control 231BO cells associated with integrin β1 knockdown – N=9 larvae for CVP (27 total cells), N=6 larvae for brain (25 total cells); 231BO integrin β1 knockdown cells – N=9 larvae for CVP (54 total cells), N=8 larvae for brain (26 total cells); control 231BR cells associated with myosin 1B knockdown – N=13 larvae for CVP (133 total cells), N=11 larvae for brain (63 total cells); 231BR myosin 1B knockdown cells – N=12 larvae for CVP (96 total cells), N=10 larvae for brain (39 total cells); control 231BO cells associated with myosin 1B knockdown – N=10 larvae for CVP (189 total cells), N=11 larvae for brain (16 total cells); 231BO myosin 1B knockdown cells – N=9 larvae for CVP (113 total cells), N=5 larvae for brain (26 total cells). For a given cell type and genetic intervention, normalized values were compared by two-way ANOVA with Sidak’s multiple comparisons test between cells within a given tissue.
Finally, to analyze cell survival in the brain parenchyma, larvae co-injected with 231BR-GFP/Luc and 231BO-mKate directly in the brain parenchyma were imaged. Cell survival was assessed by two methods. For population sampling of co-injected cells, a subset of fish was imaged at 1 dpi prior to being euthanized (N=10 larvae at 1 dpi, 520 total cells counted). At 7 dpi, a second subsampling from the injected pool of fish was imaged (N=6 larvae at 7 dpi, 142 total cells counted). The ratio of 231BR:231BO cells present from population subsamplings was analyzed by unpaired two-tailed t test following D’Agostino and Pearson normality test. Alternatively, for timecourse analysis, injected larvae were imaged at 1, 2, and 7 dpi, and total cell numbers were counted on each day from N=4 larvae.
Quantification of Neutrophils in the Head and Tail
Tg(flk:mCherry/mpx:EGFP) zebrafish at 2 dpf were injected with MDA-MB-231 brain-tropic (231BR) or bone marrow-tropic (231BO) cells as described above were imaged on the night of injection and at 1 dpi. Images were oriented along the horizontal axis and then cropped. Images in the zebrafish tail were cropped vertically to a height of 512 pixels (425.10 μm) and horizontally to encompass the tail and 15 most posterior ISVs in Fiji. Images of the zebrafish head were cropped to a size of 844×722 pixels (width × height; 700.74 μm × 599.45 μm) from the most anterior blood vessel in the head. Confocal z stacks were maximum intensity projected, and the channel showing zebrafish neutrophils was processed in Fiji. Images were auto threshold using the Huang method and automatic Fiji binarization threshold. Binarized images were run through the Fiji watershed algorithm, and the number of particles present between the sizes of 20–10000 μm2 were counted. Larvae in which background fluorescence intensity was too high to obtain accurate counts were discarded from further analysis. Average neutrophil counts in the head and tail were calculated for N=7 larvae injected with brain-tropic cells (total of 255 neutrophils counted in the head and 421 neutrophils counted in the tail) and N=5 larvae injected with bone-tropic cells (total of 360 neutrophils counted in the head and 490 neutrophils counted in the tail). Neutrophil numbers were analyzed by two-way ANOVA with Sidak’s multiple comparisons post-test comparing cell types within each tissue. For this population, the average ratio of neutrophils in the head to total neutrophils counted in the head and tail was also calculated on a per larva basis. This ratio was analyzed between larvae injected with brain- and bone marrow-tropic cells using an unpaired two-tailed t test.
Quantification of Vessel Size and Topography
Average intensity projections of the entire larvae were made in Fiji. The line tool in Fiji was used to obtain widths of 20 vessels per fish in four fish in the brain, CVP, and ISV for a total of N=80 vessels at each location. To characterize vessel topographical complexity, the circle tool was used to draw 20-μm-diameter circular regions of interest. This region of interest was randomly placed over vessels in the brain, CVP, or ISV, and the Orientation J plugin in Fiji was used to measure the coherency (defined here as the order parameter) at each region of interest and is described in detail elsewhere (Fonck et al., 2009; Püspöki et al., 2016). The order parameter ranged from a value of 0 for isotropic distributions to 1 for aligned regions. For each region of interest size and tissue (brain, CVP, or ISV), 10 regions were measured for 4 fish, giving a total number of N=40 regions. Vessel widths in each region were compared using Dunn’s multiple comparisons test following Kruskal-Wallis test after distribution did not pass D’Agostino and Pearson normality test. Order parameters in each region were compared using the Kruskal-Wallis test with Dunn’s multiple comparisons test.
Intravital Cancer Cell Tracking
Time-lapse microscopy images were exported to Fiji for analysis. Images were only analyzed if blood flow was visually confirmed in both the brain and CVP. To adjust for fish growth during imaging, images were first registered using the Correct 3D Drift plugin (Parslow et al., 2014), with the vasculature of the fish used as a topographical reference. For registration, the multi-time scale computation, subpixel drift correction, and edge enhancement options were enabled. Cancer cells were tracked in 3D in registered images over a 12 h imaging window from the registered image stacks using the TrackMate plugin for Fiji (Tinevez et al., 2017). Images were segmented in the cancer cell fluorescence channel in each frame using a Laplacian of Gaussian (LoG) detector with a 15 μm estimated particle diameter. A 10 μm estimated particle diameter was used to track injected fluorescent beads. An initial threshold of 1.0 was set, and the sub-pixel localization and median filter options in TrackMate were activated. Segmentation was then further refined by manual adjustment of the threshold to minimize detection of background objects. Segmented objects were linked from frame to frame with a Linear Assignment Problem (LAP) tracker with 30 μm maximum frame-to-frame linking distance. Tracks were visually inspected for completeness and accuracy over the entire acquisition period and were manually edited to ensure that point-to-point tracks were generated for the entire time that a cell was in the field of view.
Cell tracks (x,y,t,z) were exported to MATLAB for analysis. For each cell, frame-to-frame speed was calculated by dividing the displacement of the cell by the time interval between frames. An average speed for that cell over the course of imaging was then calculated by averaging these frame-to-frame speeds, and that average speed was used in subsequent plots and calculations. Speeds were only calculated for those cells remaining in frame for at least 4 frames (30 min). Reported speeds are grouped measurements (231BR – 91 cells in brain, 131 cells in CVP; 231BO – 46 cells in brain, 56 cells in CVP; 4T1Br4 – 48 cells in the brain, 60 cells in the CVP; 4T1BM2 – 22 cells in the brain, 65 cells in the CVP) made from multiple fish (231BR – 6 larvae; 231BO – 8 larvae; 4T1Br4 – 6 larvae; 4T1BM2 – 8 larvae). Speeds were compared by two-way ANOVA with Sidak’s multiple comparisons post-test between cell types in each tissue. This is the same method used for tracking cells in the intersegmental vessels in vivo, where cells were tracked in the ~12 h following injection to the circulation of 2 dpf Tg(fli1:EGFP) zebrafish (231BO – 14 cells in 7 larvae; 231BR – 38 cells in 6 larvae). In these calculations, N was equal to the total number of cells observed for each cell type and tissue. Histograms of cell speed distributions were also prepared.
To calculate the relative number of cells through the brain to the CVP, the ratio of cells observed in the brain to cells observed in the CVP was calculated for each fish. These ratios were averaged across all larvae for a given condition, with N=7 larvae for 231BR cells (310 cells total), N=10 larvae for 231BO cells (183 cells total), N=6 larvae for 4T1Br4 cells (139 cells total), and N=8 larvae for 4T1BM2 cells (96 cells total). Distributions were analyzed using D’Agostino and Pearson normality test and were not normally distributed. This ratio for each cell type was then compared using Kruskal-Wallis test with Dunn’s multiple comparisons test between each cell type.
To calculate cell and bead residence time in the brain and CVP, the number of frames that cells were tracked were simply counted and multiplied by the imaging interval (10 min) for observations made during the total analysis period, which was set at 72 frames. Cells that were in frame for only one frame were not considered. Residence time values were calculated from data across: 7 larvae for 231BR cells (N=140 cells in the brain, N=170 cells in the CVP), 10 larvae for 231BO cells (N=86 cells in the brain, N=97 cells in the CVP), 6 larvae for 4T1Br4 cells (N=61 cells in the brain, N=78 cells in the CVP), 8 larvae for 4T1BM2 cells (N=26 cells in the brain, N=70 cells in the CVP), 6 larvae for polystyrene beads (N=183 beads in the brain, N=29 beads in the CVP); 3 larvae for 231BR control cells associated with integrin β1 knockdown (N=30 cells in the brain, N=35 cells in the CVP); 4 larvae for 231BR integrin β1 knockdown cells (N=94 cells in the brain, N=74 cells in the CVP); 3 larvae for 231BO control cells associated with integrin β1 knockdown (N=78 cells in the brain, N=70 cells in the CVP), 4 larvae for 231BO integrin β1 knockdown cells (N=43 cells in the brain, N=55 cells in the CVP); 5 larvae for 231BR cells treated with IgG antibody (N=207 cells in the brain, N=159 cells in the CVP); 4 larvae for 231BR cells treated with anti-integrin β1 antibody (181 cells in the brain, 296 cells in the CVP); 5 larvae for 231BO cells treated with IgG antibody (N=78 cells in the brain, N=139 cells in the CVP); and 3 larvae for 231BO cells treated with anti-integrin β1 antibody (26 cells in the brain, 77 cells in the CVP). All experiments with 231BR cells, 231BO cells, 4T1Br4 cells, 4T1BM2 cells, 231BR control and integrin β1 knockdown cells, and 231BO control and integrin β1 knockdown cells were performed in Tg(flk:mCherry/MRC1a:EGFP) zebrafish injected at 2 dpf. Experiments with polystyrene beads, 231BR cells treated with IgG or anti-integrin β1 antibodies, or 231BO cells treated with IgG or anti-integrin β1 antibodies were performed in Tg(fli:EGFP) zebrafish injected at 2 dpf.
Average residence times of untreated 231BR cells, 231BO cells, 4T1Br4 cells, 4T1BM2 cells, and beads were calculated from the ensemble of cells grouped across larvae for a given cell type and tissue to obtain population average residence times. Values were compared using two-way ANOVA with Sidak’s multiple comparisons test comparing residence time for a given cell type within each tissue. Histograms of cell residence times were also generated. In these calculations, N was equal to the total number of cells observed for each cell type and tissue. Additionally, the ratio of average residence time in the brain to that in the CVP was calculated on a per larvae basis. The ratio of average residence time in the brain to average residence time in the CVP was calculated for N=7 larvae for 231BR cells, N=10 larvae for 231BO cells, N=6 larvae for 4T1Br4 cells, N=8 larvae for 4T1BM2 cells, and N=6 larvae for polystyrene beads. Distributions were analyzed using D’Agostino and Pearson normality test and were not normally distributed. This ratio was compared across cell types using Kruskal-Wallis test with Dunn’s multiple comparisons post-test.
To calculate the normalized residence times of cells upon silencing or blocking of integrin β1, the ensemble averaged residence time for a given condition was divided by the ensemble averaged residence time of the associated control cells within the same organ. These normalized values were compared by two-way ANOVA with inhibition and tissue of observation as factors, and normalized residence times between control and inhibited cells in a given organ were further compared by Sidak’s multiple comparisons test. In these calculations, N was equal to the total number of cells observed in a given organ for each cell type and treatment. Histogramas of non-transformed residence time data is also presented.
To calculate the fraction of cells observed that were occluded after 12 h of timelapse imaging, the number of cells that were tracked for residence time calculations was totaled. The number of cells in frame at the end of the 12 h tracking period was divided by this total. Running totals were made across all larvae imaged (N=7 larvae for 231BR cells, N=10 larvae for 231BO cells, N=6 larvae for 4T1Br4 cells, N=8 larvae for 4T1BM2 cells, and N=6 larvae for polystyrene beads) in each tissue. This resulted in the occlusion fractions summarized in Table S1. Data were analyzed by two-way ANOVA with Sidak’s multiple comparisons test comparing the occlusion fractions in each tissue for a given cell type. The final occlusion fraction (one value for each cell type and tissue) was used in calculations.
Additionally, the occlusion fractions in each organ were calculated on a per larva basis. For each larva (N=7 larvae for 231BR cells, N=10 larvae for 231BO cells, N=6 larvae for 4T1Br4 cells, N=8 larvae for 4T1BM2 cells, and N=6 larvae for polystyrene beads), the occlusion fraction as described above was calculated in the brain and in the CVP. These fractions were averaged across all larvae for a given organ and cell type. Data were compared by two-way ANOVA with Sidak’s multiple comparisons test comparing occlusion in each tissue for a given cell type.
To study the effect of integrin β1 knockdown or function blocking on cell arrest, the total number of cells present at the final frame (12 h) across larvae was divided by the total number of cells observed in all fish larvae for a given condition (e.g., 231BR cells with knockdown of integrin β1). In each organ, this fraction was normalized by dividing by the arrest fraction observed for all cells of the same type treated with the corresponding non-targeting siRNA or control IgG antibody in a given organ. Cells that were in frame for only one frame were not considered. Normalized values were compared by two-way ANOVA with organ and integrin β1 inhibition as factors. Within a given organ, control and inhibited cells were further compared using Sidak’s multiple comparisons test. Occlusion fractions were calculated from: 3 larvae for 231BR control cells associated with integrin β1 knockdown, 4 larvae for 231BR integrin β1 knockdown cells, 3 larvae for 231BO control cells associated with integrin β1 knockdown, 4 larvae for 231BO integrin β1 knockdown cells, 5 larvae for 231BR cells treated with IgG antibody, 4 larvae for 231BR cells treated with anti-integrin β1 antibody, 5 larvae for 231BO cells treated with IgG antibody, and 3 larvae for 231BO cells treated with anti-integrin β1 antibody. This resulted in the occlusion fractions summarized in Table S2.
Additionally, cell occlusion fractions were calculated on a per larvae basis. For each larva (N=3 larvae for 231BR control cells associated with integrin β1 knockdown, N=4 larvae for 231BR integrin β1 knockdown cells, N=3 larvae for 231BO control cells associated with integrin β1 knockdown, N=4 larvae for 231BO integrin β1 knockdown cells, N=5 larvae for 231BR cells treated with IgG antibody, N=4 larvae for 231BR cells treated with anti-integrin β1 antibody, N=5 larvae for 231BO cells treated with IgG antibody, and N=3 larvae for 231BO cells treated with anti-integrin β1 antibody), the occlusion fraction as described above was calculated in the brain and in the CVP. For a given organ, cell type, and treatment, these fractions were averaged across all larvae. In each organ, the resulting arrest fraction was then normalized by the average arrest fraction in the same organ of the corresponding control cells. Normalized data were compared by two-way ANOVA, with organ and inhibition of integrin β1 as factors. Within a given organ, average cell arrest fractions between control and inhibited cells were further compared using Sidak’s multiple comparisons test. Code used for calculating the number of occluded cells is included in Methods S1.
Calculation of Cell Speed in Microchannels
Migrating cells were tracked within the microchannels. To track cells, the fluorescence channels of each image were first preprocessed to better visualize cell morphology. In Fiji, the background was subtracted using the rolling ball method with a radius of 50 pixels. Images were then subjected to a Gaussian blur with sigma=2 pixels, following by an unsharp mask at a radius of 2 pixels and a mask value of 0.60. Processed images were saved as tiff files and tracked using TrackMate (Tinevez et al., 2017). For GFP-positive cells, images were segmented in the GFP channel using the Downsample LoG detector with a 40 μm estimated particle diameter, threshold of 0.1, and downsampling factor of 2. For Hoechst-stained cells, images were segmented in the Hoechst channel using the LoG detector with a 15 μm estimated particle diameter and threshold of 0.1. During segmentation, the median filtering and subpixel localization options were selected. In all experiments, the entrance and exit of the microchannels was manually identified, and tracking was confined to the regions where cells were fully within channels. Only tracks containing at least 12 points were retained for analysis. After initial track assignment using a LAP tracker, tracks were manually checked and updated for completeness and accuracy. Tracking was discontinued if a cell reached the end of a microchannel or exited the base of a microchannel, encountered a cell in the channel traveling in the opposite direction, or was in contact with a preceding cell in the microchannel. Dividing cells were excluded from analysis.
Cell tracks (x,y,t,z) were exported to MATLAB for analysis. For each combination of cell type, microchannel topography, and ECM protein, a maximum of 30 tracks from each biological replicate were randomly selected, and an average speed for a given cell was obtained by averaging the frame-to-frame speed (calculated by dividing the displacement of the cell by the time interval between frames) over the first 30 frames for which the cell was tracked. The displacements of the cells were calculated by measuring the straight-line distance from the first point in the track to the final point in the track. For BSA coated devices and most replicates of experiments with varying microchannel topography, fewer cells entered the microchannels, and fewer than 30 cells were used per biological repeat in some instances. Average speeds and displacements were grouped from each biological replicate to obtain an ensemble of cell speeds and displacements. Ensembles were averaged to obtain a mean cell migration speed or displacement for a given condition. Average speeds and displacements between cell types in microchannels of varying topography were analyzed by two-way ANOVA, with speeds between cell types in a given microchannel geometry compared by Sidak’s multiple comparisons post-test. Average speeds between cell lines for a given ECM coating and in the ISV were analyzed by two-way ANOVA with Sidak’s multiple comparisons post-test. Summaries of the number of cells analyzed for each condition are included in Tables S3 and S4. Code used for analysis is included in Methods S1.
Optical Trap Data Analysis
Samples were measured in triplicate with at least 30 beads per sample measured. Data were analyzed using custom MATLAB programs. Trajectories of bead positions together with β, k, and the bead’s mass m and radius a were used to calculate the complex modulus as a function of frequency, G*(ω), of each bead’s surrounding microenvironment. The complex modulus, G*(ω), can be broken down into components, with , where the real part, G’(ω), is the elastic component and the imaginary part, G”(ω), is the viscous component. The complex modulus, G*( ω), is calculated as , where the friction relaxation spectrum is related by the equation to the active power spectrum , with and the Fourier transforms of the time series of the positions of the trapping laser and the driven bead respectively, recorded while the trap is oscillating. Code used for analysis is included in Methods S1.
Active microrheology summary data are presented as mean complex modulus, normalized to the mean complex modulus of the CVP at 2 dpf, and averaged across frequency (mean ± standard deviation). Data normalized to the complex modulus of the CVP were analyzed by two-way ANOVA with Tukey’s honestly significant difference post-test. Data are also represented as mean complex modulus and phase angle vs. frequency from 3 Hz to 15 kHz.
Red Blood Cell Speed Tracking
Images of red blood cell flow were acquired in the brain and CVP from three Tg(flk:mCherry/MRC1a:EGFP) larvae and one Tg(flk:mCherry/mpx:EGFP) larvae. To isolate images of moving objects (red blood cells), average intensity projections of the brightfield channel over time were taken for each image. This average projection was subtracted from the original image to remove stationary objects. This method does not provide a comprehensive profiling of blood flow in all regions of a given tissue but provides a snapshot of flow through a confocal slice. Red blood cells were segmented from the resultant image using the Yen Auto Threshold method in Fiji across the image stack. Background noise was removed by running the Fiji Despeckle command. Processed images were tracked in Fiji using the TrackMate plugin (Tinevez et al., 2017). Objects were identified using the LoG detector with a 4 μm estimated particle diameter and threshold of 1. During object identification, the median filtering and subpixel localization options were selected. Tracks were assembled from identified objects using the LAP tracker. Only tracks containing at least 12 points were retained for analysis. Cell tracks (x,y,t,z) were exported to MATLAB for analysis. A maximum of 31 points per track were retained to calculate cell speed. For each larva, a maximum of 40 tracks from each organ (brain or CVP) were randomly selected, and an average speed for a given cell was obtained by averaging the frame-to-frame speed (calculated by dividing the displacement of the cell by the time interval between frames) over time for which the cell was tracked. Tracking resulted in measurement of N=102 cells in the brain and N=160 cells in the CVP. Information on the samples used for red blood cell tracking is provided in Table S5. Code used for analysis is included in Methods S1.
Glycocalyx Albumin Intensity Quantification
Confocal images from regions in both the brain and CVP were acquired for three Tg(flk:mCherry/MRC1a:EGFP) larvae at 2 dpf. For each image, maximum intensity projections were made in the vasculature (mCherry) and BSA-647 channels. The total area occupied by the vessels in a given image was then calculated by first segmenting the vasculature image using the Triangle Auto Threshold method in Fiji and measuring the total segmented area with the Analyze Particles function in Fiji, with a minimum size of 2 and objects on the edges of images counted. The total area occupied by BSA-647 in a given image was calculated by segmenting the BSA-647 image using the Yen Auto Threshold method in Fiji and measuring the total segmented area with the Analyze Particles function, with no size restrictions and objects on the edges of images again counted. For a given image, the ratio of the area occupied by BSA-647 was normalized by dividing by the total area occupied by the vessels. Values were then normalized to the average value of this ratio in the CVP. Averages across N=9 images in both the brain and CVP were compared by unpaired two-tailed t test with Welch’s correction.
Longitudinal analysis of Cells in the Brain and CVP
A subset of the larvae injected with 231BR and 231BO cells and used to examine cell homing and extravasation at 5 dpi were imaged at 1, 2, and 5 dpi. The brain and CVP were imaged, and cells were counted and classified as described above. Images were obtained for N=10 larvae injected with 231BR cells and N=8 larvae injected with 231BO cells.
For cells in the CVP for each larva, the maximum total number of cells (intravascular plus extravasated) observed over the three days of imaging was calculated. At each time point, the number of intravascular cells and number of extravasated cells was divided by this maximum number to obtain the fraction of intravascular and extravasated cells, respectively. For each day, if the sum of these fractions was less than one, the remainder of cells were classified as “cleared.” The fraction of cells in each state was averaged across larvae for each cell type, and averages were calculated independently on each day. The same procedure was followed in the brain. Comparison of the average fraction of cells observed in the brain that had extravasated by 5 dpi was made using an unpaired two-tailed t test following D’Agostino and Pearson normality test. Comparison of the average fraction of cells observed in the CVP that had extravasated by 5 dpi was made using the Mann-Whitney test after distributions did not pass D’Agostino and Pearson normality test.
Additionally, the total number of cells present in the brain plus CVP of each fish was counted from this same pool of larvae on each day. The average total number of cells present on a given day was averaged across all larvae injected with each cell type. Values were compared by two-way ANOVA, with both time and cell type as factors. The number of 231BR and 231BO cells present on a given day were further compared by Sidak’s multiple comparisons test.
Proteomic Analysis from Mass Spectrometry
The mass spectrometry data were analyzed and label-free quantitation performed using MaxQuant version 1.5.7.4 (Cox et al., 2014; Cox and Mann, 2008) with the following parameters: variable modifications - methionine oxidation and N-acetylation of protein N-terminus; static modification – cysteine carbamidomethylation; first search was performed using 20 ppm error and the main search 10 ppm; maximum of two missed cleavages; protein and peptide FDR threshold of 0.01; min unique peptides 1; match between runs; label-free quantitation, with minimal ratio count 2. Proteins were identified using a Uniprot human database from November 2016 (20,072 entries). Statistical analysis was performed using Perseus version 1.5.6.0 (Tyanova et al., 2016). After removal of contaminant and reversed sequences, as well as proteins that were only quantified in one of the three replicate experiments, the label-free quantitation values were base 2 logarithmized and missing values were imputed from a normal distribution of the data. Statistically significant differences were assigned using a two-way t-test with a p-value cut-off of 0.05. Hierarchical clustering was also performed in Perseus. Fold changes were calculated relative to protein expression in 231BR or 4T1Br4 cells from the average base 2 logarithmic differences across biological replicates. Fold changes in select proteins associated with cell adhesion and cell contractility are plotted. Proteomics data following analysis in Perseus is presented in Table S6.
For analysis of signaling pathways that were differentially regulated between the cell types, the annotated list of base 2 logarithmized values and p-values was imported to Ingenuity Pathway Analysis (IPA, QIAGEN, Hilden, Germany). For comparison of 231BR and 231BO cells incubated with the IgG antibody prior to lysate collection, proteins were filtered to retain those with a p-value less than 0.05. This retained 477 of the total 2180 mapped proteins returned from Perseus analysis for 231 cells. For comparison of 4T1Br4 and 4T1BM2 cells incubated with the IgG antibody prior to lysate collection, proteins were filtered to retain those with a p-value less than 0.05. This retained 308 of the total 1831 mapped proteins returned from Perseus analysis for 4T1 cells. The top differentially regulated pathways between 231BR and 231BO cells are listed. Pathways that involved β1 integrin are displayed in italic text. The activation pattern of each pathway was assessed using the built-in IPA z-score analysis. Pathways are displayed in black if no activity pattern was available.
To identify potential regulators of brain metastasis, the list of proteins from 231 and 4T1 clones incubated with the IgG antibody prior to lysate collection were filtered to retain those proteins that were significantly different (p<0.05) between brain- and bone marrow-targeting clones for a given cell line. Those lists were then compared across 231 and 4T1 cells to retain those proteins that were also differently regulated in the same direction for both cell pairs. From this list, myosin 1B was the only protein that was upregulated in both brain-tropic clones.
To compare canonical signaling pathways between 231 and 4T1 cell line clones, the Comparison Analysis function in IPA was used. Analysis data sets in IPA generated for 231 and 4T1 cells incubated with IgG antibody prior to lysate collection and filtered for significance as described above were compared. Canonical pathways were sorted using the IPA z-score function, and activation levels were visualized based on the activation z-score.
Gene Expression Analysis by qPCR
Gene expression was normalized to that of GAPDH for each cell type and siRNA status. For cells transfected with siRNA targeting myosin 1B, fold change was calculated from the mean ΔΔcT of three technical triplicates, as indicated by the manufacturer (Applied Biosystems, “Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR”). For cells transfected with control siRNA, expression of myosin 1B was set to 1. Results from two biological replicates were averaged to obtain the mean and standard deviation fold change upon siRNA transfection.
Software Used for Statistical Analysis
Statistical calculations were carried out in MATLAB (optical trap microrheology data), Perseus version 1.5.6.0 (mass spectrometry data), or GraphPad Prism 7 (other data; GraphPad Software, La Jolla, CA). Details on statistical tests are given in each section.
DATA AND CODE AVAILABILITY
The analyzed datasets generated during this study are available at Mendeley Data under https://doi.org/10.17632/rbckhhkczj.1. Corresponding files of cell trajectories used in cell speed, residence time, and occlusion experiments are available at Mendeley Data under the same DOI. Raw proteomics data is available on ProteomeXchange with accession number PXD014345 (ProteomeXchange: PXD014345). Analyzed proteomics data generated in this study is included in the Supplemental Information (Table S6). Code used to analyze cell trajectories in zebrafish and in microfluidic devices is included in this paper as Supplemental Information, along with code used to analyze optical trap microrheology data (Methods S1). Raw microscopy data are several terabytes in size and are available from the corresponding author on request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal IgG1, kappa [B11/6] isotype control | abcam | Cat#ab91353 |
| Mouse monoclonal anti-integrin beta1 antibody [P5D2] | abcam | Cat#ab24693; RRID: AB_448230 |
| Rabbit polyclonal anti-Ki67 | Millipore Sigma | Cat#AB9260; RRID: AB_2142366 |
| Rabbit polyclonal anti-annexin V | Thermo Fisher | Cat#PA5-27872; RRID: AB_2545348 |
| Rabbit polyclonal anti-Cdh1 | GeneTex | Cat#GTX125890; RRID: AB_11167551 |
| Rabbit IgG isotype control | Thermo Fisher | Cat#31235; RRID: AB_243593 |
| Rat anti-mouse CD29, clone 9EG7 | BD Biosciences | Cat#553715; RRID: AB_395001 |
| Alexa Fluor Plus 647 goat anti-rabbit secondary antibody | Thermo Fisher | Cat#A32733; RRID: AB_2633282 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Instant Ocean Sea Salt | Instant Ocean | Cat#SS15-10 |
| N-phenylthiourea (PTU) | Millipore Sigma | Cat#P7629-25G |
| Tricaine | Millipore Sigma | Cat#E01521-50G |
| Low gelling temperature agarose | Millipore Sigma | Cat#A9414-25G |
| Proteinase K | Millipore Sigma | Cat#3115879001 |
| CellTracker Deep Red | Thermo Fisher | Cat#C34565 |
| CellTracker Blue CMAC | Thermo Fisher | Cat#C2110 |
| 10 μm-diameter FluoSpheres | Thermo Fisher | Cat#F8831 |
| Albumin from bovine serum, Alexa Fluor 647 conjugate | Thermo Fisher | Cat#A34785 |
| Opti-MEM cell culture medium | Thermo Fisher | Cat#31985070 |
| RNAiMAX | Thermo Fisher | Cat#13778030 |
| Protease inhibitor cocktail set 1 | Millipore Sigma | Cat#539131 |
| M-PER Mammalian Protein Extraction Reagent | Thermo Fisher | Cat#78501 |
| Phosphatase inhibitor cocktail 2 | Millipore Sigma | Cat#P5726 |
| Phosphatase inhibitor cocktail 3 | Millipore Sigma | Cat#P0044 |
| TRIzol | Thermo Fisher | Cat#15596018 |
| SuperScript IV VILO Master Mix | Thermo Fisher | Cat#11755050 |
| TaqMan Fast Advanced Master Mix | Thermo Fisher | Cat#4444557 |
| SU-8 2050 | Microchem | http://microchem.com/Prod-SU82000.htm |
| SU-8 2100 | Microchem | http://microchem.com/Prod-SU82000.htm |
| SU-8 thinner | Microchem | http://microchem.com/Prod-SU82000.htm |
| Tridecafluoro-1,1,2,2-tetrahydrooctyl trichlorosilane | Gelest | Cat#SIT8174.0 |
| Polydimethylsiloxane (PDMS), Sylgard 184 | Dow Corning | Cat#1673921 |
| Rat tail collagen type I | BD Biosciences | Cat#354236 |
| Fibronectin from human plasma | Millipore Sigma | Cat#FC010 |
| Bovine serum albumin | Millipore Sigma | Cat#A7906-100G |
| Hoechst 33342, trihydrochloride, trihydrate | Thermo Fisher | Cat#H3570 |
| Alexa Fluor 488 phalloidin | Thermo Fisher | Cat#A12379 |
| Critical Commercial Assays | ||
| PureLink RNA Mini Kit | Thermo Fisher | Cat#12183020 |
| Deposited Data | ||
| Proteomics datasets for 231BR, 231BO, 4T1Br4, 4T1BM2, 231BR ctrl siRNA, 231BR integrin beta1 siRNA, 231BO ctrl siRNA, and 231BO integrin beta1 siRNA cells | This paper | ProteomeXchange, accession number: PXD014345; Table S6 |
| Datasets used to generate plots | This paper | Mendeley Data, https://doi.org/10.17632/rbckhhkczj.1 |
| Cell and bead trajectory files | This paper | Mendeley Data, https://doi.org/10.17632/rbckhhkczj.1 |
| Experimental Models: Cell Lines | ||
| Human: MDA-231BR cells | Yoneda et al., 2001 | N/A |
| Human: MDA-231BO cells | Yoneda et al., 2001 | N/A |
| Human: Bt474m1 cells | Zhang et al., 2013 | N/A |
| Human: Bt474m1-Br3 cells | Zhang et al., 2013 | N/A |
| Murine: 4T1Br4 | Kim et al., 2018 | N/A |
| Murine: 4T1BM2 | Kusuma et al., 2012 | N/A |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: Tg(flk:mCherry/MRC1a:EGFP), Casper background | Jung et al., 2017; Venero Galanternik et al., 2017; Wang et al., 2010; White et al., 2008 | ZFIN: ZDB-PUB-100826-21; ZDB-PUB-170517-11; ZDB-PUB-170412-9; ZDB-FISH-150901-6638 |
| Zebrafish: Tg(flk:mCherry/mpx:EGFP) | Wang et al., 2010; Renshaw et al., 2006 | ZFIN: ZDB-PUB-100826-21; ZDB-FISH-150901-23930 |
| Zebrafish: Tg(fli:EGFP) | Lawson and Weinstein, 2002 | ZFIN: ZDB-FISH-150901-3654 |
| Zebrafish: Casper | White et al., 2008 | ZFIN: ZDB-FISH-150901-6638 |
| Oligonucleotides | ||
| Silencer Select siRNA: ITGB1, Assay ID s7575 | Thermo Fisher | Cat#4390824 |
| Silencer Select siRNA: MYO1B, Assay IDs8964 | Thermo Fisher | Cat#4392420 |
| Silencer Select siRNA: Negative Control No. 1 | Thermo Fisher | Cat#4390843 |
| FAM-MGB TaqMan Probe: MYO1B, Assay ID Hs00362654_m1 | Thermo Fisher | Cat#4331182 |
| FAM-MGB TaqMan Probe: GAPDH, Assay ID Hs04420697_g1 | Thermo Fisher | Cat#4331182 |
| Software and Algorithms | ||
| MaxQuant version 1.5.7.4 | Cox et al., 2014; Cox and Mann, 2008 | http://www.coxdocs.org/doku.php?id=maxquant:start |
| Perseus version 1.5.6.0 | Tyanova et al., 2016 | http://www.coxdocs.org/doku.php?id=perseus:common:download_and_installation |
| Ingenuity Pathway Analysis, Version 46901286 | QIAGEN | https://www.qiagen.com/us/shop/analytics-software/biological-data-tools/ingenuity-pathway-analysis/#orderinginformation |
| Fiji | N/A | https://fiji.sc/ |
| OrientationJ | Fonck et al., 2009; Püspöki et al., 2016 | http://bigwww.epfl.ch/demo/orientation/ |
| Correct 3D Drift plugin | Parslow et al., 2014 | https://imagej.net/Correct_3D_Drift |
| TrackMate plugin | Tinevez et al., 2017 | https://imagej.net/TrackMate |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| MATLAB code for analysis of cell trajectories and optical trap data | This paper | This paper (Methods S1) |
| AutoCAD | Autodesk | https://www.autodesk.com/products/autocad/overview |
| L-Edit | Mentor Graphics | https://www.mentor.com/tannereda/l-edit |
| Other | ||
| Thermo Orbitrap Fusion Mass Spectrometer | Thermo Fisher | N/A |
| UltiMate 3000 RSLCnano HPLC | Thermo Fisher | Cat#ULTIM3000RSLCNANO |
| Applied Biosystems QuantStudio 3 | Thermo Fisher | N/A |
| Nunc Lab-Tek Chambered #1.0 Borosilicate Coverglass | Thermo Fisher | Cat#155383, Cat#155380 |
| 780 LSM confocal microscope | Zeiss | N/A |
| 10x Plan-Neofluar, 0.3 NA objective | Zeiss | N/A |
| 20x EC Plan-Apochromat, 0.8 NA objective | Zeiss | N/A |
| 63x Apochromat, 1.4 NA objective | Zeiss | N/A |
| A1R confocal microscope | Nikon | N/A |
| Zeiss Axio Scan.Z1 slide scanner | Zeiss | N/A |
| Four-inch silicon wafers | NOVA Electronic Materials | http://www.novawafers.com/ |
| Spin coater | Laurell Technologies | Model#WS-650S-8NPP/LITE |
| Model 208 mask aligner | Optical Associates, Inc. | N/A |
| Long-pass filter | Omega Optical | Cat#PL-360-LP |
| Optical tweezer setup | Blehm et al., 2016 | N/A |
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute. The MDA-MB-231 clones were a kind gift of Professor Toshiyuki Yoneda (Indiana University-Purdue University). The Bt474m1 clones were a kind gift from Professor Dihua Yu (University of Texas, MD Anderson Cancer Center). The 4T1 clones were a kind gift of Professor Normand Pouliot (Olivia Newton-John Cancer Research Institute, Melbourne, Australia). Transgenic zebrafish lines used for cancer cell injections were kindly provided by Dr. Brant Weinstein (NICHD, NIH). We thank Dan Castranova, Jian Liu, and Blake Carrington for useful discussions. We thank Victoria Hoffman, Division of Veterinary Resources, Office of the Director, NIH for assistance with pathology. We would like to thank Susan Garfield, Michael Kruhlak, and Langston Lim, CCR Confocal Microscopy Core Facility, Laboratory of Cancer Biology and Genetics, NCI for use of the core microscopes. We would also like to thank Nikon microscopy for demo use of an A1R confocal microscope.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2019.07.005.
REFERENCES
- Almeida CG, Yamada A, Tenza D, Louvard D, Raposo G, and Coudrier E (2011). Myosin 1b promotes the formation of post-Golgi carriers by regulating actin assembly and membrane remodelling at the trans-Golgi network. Nat. Cell Biol 13, 779–789. [DOI] [PubMed] [Google Scholar]
- Azevedo AS, Follain G, Patthabhiraman S, Harlepp S, and Goetz JG (2015). Metastasis of circulating tumor cells: favorable soil or suitable biomechanics, or both? Cell Adhes. Migr 9, 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzer EM, Tong Z, Paul CD, Hung WC, Stroka KM, Boggs AE, Martin SS, and Konstantopoulos K (2012). Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 26, 4045–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes JM, Przybyla L, and Weaver VM (2017). Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci 130, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Brakebusch C, Coisne C, Sixt M, Wekerle H, Engelhardt B, and Fässler R (2009). Beta1 integrins differentially control extravasation of inflammatory cell subsets into the CNS during autoimmunity. Proc. Natl. Acad. Sci. USA 106, 1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, and Hines WC (2011). Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med 17, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blehm BH, Devine A, Staunton JR, and Tanner K (2016). In vivo tissue has non-linear rheological behavior distinct from 3D biomimetic hydrogels, as determined by AMOTIV microscopy. Biomaterials 83, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ID, and Humphries MJ (2011). Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MB, Lamar JM, Li R, Hynes RO, and Kamm RD (2016). Elucidation of the roles of tumor integrin beta1 in the extravasation stage of the metastasis cascade. Cancer Res. 76, 2513–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung LS, Raman PS, Balzer EM, Wirtz D, and Konstantopoulos K (2011). Biophysics of selectin-ligand interactions in inflammation and cancer. Phys. Biol 8, 015013. [DOI] [PubMed] [Google Scholar]
- Condeelis J, and Segall JE (2003). Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 3, 921–930. [DOI] [PubMed] [Google Scholar]
- Correia AL, and Bissell MJ (2012). The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat 15, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dirks WG, MacLeod RAF, Nakamura Y, Kohara A, Reid Y, Milch H, Drexler HG, and Mizusawa H (2010). Cell line cross-contamination initiative: an interactive reference database of STR profiles covering common cancer cell lines. Int. J. Cancer 126, 303–304. [DOI] [PubMed] [Google Scholar]
- Doerschuk CM, Beyers N, Coxson HO, Wiggs B, and Hogg JC (1993). Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J. Appl. Physiol 74, 3040–3045. [DOI] [PubMed] [Google Scholar]
- Ewald AJ, Werb Z, and Egeblad M (2011). Dynamic, long-term in vivo imaging of tumor-stroma interactions in mouse models of breast cancer using spinning-disk confocal microscopy. Cold Spring Harb. Protoc 2011, pdb.top97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler IJ (2003). The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 3, 453–458. [DOI] [PubMed] [Google Scholar]
- Fior R, Póvoa V, Mendes RV, Carvalho T, Gomes A, Figueiredo N, and Ferreira MG (2017). Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 114, E8234–E8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, and Berg-Sørensen K (2007). Calibration of trapping force and response function of optical tweezers in viscoelastic media. J. Opt. A: Pure Appl. Opt 9, S239–S250. [Google Scholar]
- Fleming A, Diekmann H, and Goldsmith P (2013). Functional characterisation of the maturation of the blood-brain barrier in larval zebrafish. PloS One 8, e77548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follain G, Osmani N, Azevedo AS, Allio G, Mercier L, Karreman MA, Solecki G, Garcia Leòn MJ, Lefebvre O, Fekonja N, et al. (2018). Hemodynamic forces tune the arrest, adhesion, and extravasation of circulating tumor cells. Dev. Cell 45, 33–52.e12. [DOI] [PubMed] [Google Scholar]
- Fonck E, Feigl GG, Fasel J, Sage D, Unser M, Rüfenacht DA, and Stergiopulos N (2009). Effect of aging on elastin functionality in human cerebral arteries. Stroke 40, 2552–2556. [DOI] [PubMed] [Google Scholar]
- Friedl P, and Alexander S (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009. [DOI] [PubMed] [Google Scholar]
- Goessling W, and North TE (2011). Hematopoietic stem cell development: using the zebrafish to identify the signaling networks and physical forces regulating hematopoiesis. Methods Cell Biol. 105, 117–136. [DOI] [PubMed] [Google Scholar]
- Gottesman MM (2002). Mechanisms of cancer drug resistance. Annu. Rev. Med 53, 615–627. [DOI] [PubMed] [Google Scholar]
- Hamidi H, and Ivaska J (2018). Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, and Sahai E (2015). Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries JD, Byron A, and Humphries MJ (2006). Integrin ligands at a glance. J. Cell Sci 119, 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687. [DOI] [PubMed] [Google Scholar]
- Iuliano O, Yoshimura A, Prospéri MT, Martin R, Knölker HJ, and Coudrier E (2018). Myosin 1b promotes axon formation by regulating actin wave propagation and growth cone dynamics. J. Cell Biol 217, 2033–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HM, Castranova D, Swift MR, Pham VN, Venero Galanternik M, Isogai S, Butler MG, Mulligan TS, and Weinstein BM (2017). Development of the larval lymphatic system in zebrafish. Development 144, 2070–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, and Massagué J (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549. [DOI] [PubMed] [Google Scholar]
- Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WEF, Goldbrunner R, Herms J, and Winkler F (2010). Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med 16, 116–122. [DOI] [PubMed] [Google Scholar]
- Kim J, Staunton JR, and Tanner K (2016). Independent control of topography for 3D patterning of the ECM microenvironment. Adv. Mater. Weinheim 28, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Tanner K (2015). Recapitulating the tumor ecosystem Along the metastatic cascade using 3D culture models. Front. Oncol 5, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Redvers RP, Chi LH, Ling X, Lucke AJ, Reid RC, Fairlie DP, Martin ACBM, Anderson RL, Denoyer D, et al. (2018). Identification of brain metastasis genes and therapeutic evaluation of histone deacetylase inhibitors in a clinically relevant model of breast cancer brain metastasis. Dis. Model. Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeva E, Weidling JW, Singh R, Botvinick EL, Digman MA, Gratton E, and Putnam AJ (2012). Quantification of local matrix deformations and mechanical properties during capillary morphogenesis in 3D. Integr. Biol. (Camb) 4, 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlarchyk MA, Shreim SG, Alvarez-Elizondo MB, Estrada LC, Singh R, Valdevit L, Kniazeva E, Gratton E, Putnam AJ, and Botvinick EL (2011). Concentration independent modulation of local micromechanics in a fibrin gel. PloS One 6, e20201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, and Weaver VM (2009). Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuma N, Denoyer D, Eble JA, Redvers RP, Parker BS, Pelzer R, Anderson RL, and Pouliot N (2012). Integrin-dependent response to laminin-511 regulates breast tumor cell invasion and metastasis. Int. J. Cancer 130, 555–566. [DOI] [PubMed] [Google Scholar]
- Kusumbe AP, Ramasamy SK, and Adams RH (2014). Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M, Artym VV, Green JA, and Yamada KM (2006). The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr. Opin. Cell Biol 18, 463–471. [DOI] [PubMed] [Google Scholar]
- Lawson ND, and Weinstein BM (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol 248, 307–318. [DOI] [PubMed] [Google Scholar]
- Li J, and King MR (2012). Adhesion receptors as therapeutic targets for circulating tumor cells. Front. Oncol 2, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Zhang J, Lin HF, Italiano J, and Handin RI (2011). The identification and characterization of zebrafish hematopoietic stem cells. Blood 118, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu E, Paul CD, Stahl R, Vanmeerbeeck G, Reumers V, Liu C, Konstantopoulos K, and Lagae L (2016). Time-lapse lens-free imaging of cell migration in diverse physical microenvironments. Lab Chip 16, 3304–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathura KR, Vollebregt KC, Boer K, De Graaff JC, Ubbink DT, and Ince C (2001). Comparison of OPS imaging and conventional capillary microscopy to study the human microcirculation. J. Appl. Physiol. (1985) 91, 74–78. [DOI] [PubMed] [Google Scholar]
- Meeker ND, and Trede NS (2008). Immunology and zebrafish: spawning new models of human disease. Dev. Comp. Immunol 32, 745–757. [DOI] [PubMed] [Google Scholar]
- Mitchell MJ, and King MR (2014). Physical biology in cancer. 3. The role of cell glycocalyx in vascular transport of circulating tumor cells. Am. J. Physiol. Cell Physiol 306, C89–C97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama E, Kissa K, Zapata A, Mordelet E, Briolat V, Lin HF, Handin RI, and Herbomel P (2006). Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity 25, 963–975. [DOI] [PubMed] [Google Scholar]
- Neuman KC, and Block SM (2004). Optical trapping. Rev. Sci. Instrum 75, 2787–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, and Massagué J (2009). Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284. [DOI] [PubMed] [Google Scholar]
- Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, Park SY, Lu J, Protopopov A, and Silberstein LE (2013). Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Biol 15, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauf AC, and Massagué J (2015). Surviving at a distance: organ specific metastasis. Trends Cancer 1, 76–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmura G, Tsujikawa T, Yaguchi T, Kawamura N, Mikami S, Sugiyama J, Nakamura K, Kobayashi A, Iwata T, Nakano H, et al. (2015). Aberrant myosin 1b Expression Promotes Cell Migration and lymph node Metastasis of HNSCC. Mol. Cancer Res 13, 721–731. [DOI] [PubMed] [Google Scholar]
- Oosterhof N, Boddeke E, and van Ham TJ (2015). Immune cell dynamics in the CNS: learning from the zebrafish. Glia 63, 719–735. [DOI] [PubMed] [Google Scholar]
- Oudin MJ, and Weaver VM (2016). Physical and chemical gradients in the tumor microenvironment regulate tumor cell invasion, migration, and metastasis. Cold Spring Harb. Symp. Quant. Biol 81, 189–205. [DOI] [PubMed] [Google Scholar]
- Paget S (1989). The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 8, 98–101. [PubMed] [Google Scholar]
- Parslow A, Cardona A, and Bryson-Richardson RJ (2014). Sample drift correction following 4D confocal time-lapse imaging. JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, et al. (2014). The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, and Condeelis JS (2009). Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 69, 9498–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CD, Devine A, Bishop K, Xu Q, Wulftange WJ, Burr H, Daly KM, Lewis C, Green DS, Staunton JR, et al. (2019). Human macrophages survive and adopt activated genotypes in living zebrafish. Sci. Rep 9, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CD, Mistriotis P, and Konstantopoulos K (2017). Cancer cell motility: lessons from migration in confined spaces. Nat. Rev. Cancer 17, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter RF, and Groom AC (1983). Capillary diameter and geometry in cardiac and skeletal muscle studied by means of corrosion casts. Microvasc. Res 25, 68–84. [DOI] [PubMed] [Google Scholar]
- Püspöki Z, Storath M, Sage D, and Unser M (2016). Transforms and operators for directional BioImage analysis: a survey. Adv. Anat. Embryol. Cell Biol 219, 69–93. [DOI] [PubMed] [Google Scholar]
- Ramasamy SK, Kusumbe AP, Schiller M, Zeuschner D, Bixel MG, Milia C, Gamrekelashvili J, Limbourg A, Medvinsky A, Santoro MM, et al. (2016). Blood flow controls bone vascular function and osteogenesis. Nat. Commun 7, 13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, and oude Egbrink MG (2007). The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 454, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw SA, Loynes CA, Trushell DMI, Elworthy S, Ingham PW, and Whyte MKB (2006). A transgenic zebrafish model of neutrophilic inflammation. Blood 108, 3976–3978. [DOI] [PubMed] [Google Scholar]
- Renshaw SA, and Trede NS (2012). A model 450 million years in the making: zebrafish and vertebrate immunity. Dis. Models Mech 5, 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh-Johnson M, Shah AN, Stonick JA, Poudel KR, Kargl J, Yang GH, di Martino J, Hernandez RE, Gast CE, Zarour LR, et al. (2017). Macrophage-dependent cytoplasmic transfer during melanoma invasion in vivo. Dev. Cell 43, 549–562.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A, Roccaro AM, Ma D, Shi J, Mishima Y, Moschetta M, Chiarini M, Munshi N, Handin RI, and Ghobrial IM (2016). Cancer cell dissemination and homing to the bone marrow in a zebrafish model. Cancer Res. 76, 463–471. [DOI] [PubMed] [Google Scholar]
- Salas-Cortes L, Ye F, Tenza D, Wilhelm C, Theos A, Louvard D, Raposo G, and Coudrier E (2005). Myosin Ib modulates the morphology and the protein transport within multi-vesicular sorting endosomes. J. Cell Sci 118, 4823–4832. [DOI] [PubMed] [Google Scholar]
- Schwartz MA (2010). Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol 2, a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr EH, Joyce MP, and Greene LA (1993). Mammalian myosin I alpha, I beta, and I gamma: new widely expressed genes of the myosin I family. J. Cell Biol 120, 1405–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, and Weinberg RA (2009). Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 106, 10290–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunton JR, Blehm B, Devine A, and Tanner K (2017). In situ calibration of position detection in an optical trap for active microrheology in viscous materials. Opt. Express 25, 1746–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunton JR, Vieira W, Fung KL, Lake R, Devine A, and Tanner K (2016). Mechanical properties of the tumor stromal microenvironment probed in vitro and ex vivo by in situ-calibrated optical trap-based active microrheology. Cell. Mol. Bioeng 9, 398–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS (2016). Targeting metastasis. Nat. Rev. Cancer 16, 201–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, and Klemke R (2010). Visualizing extravasation dynamics of metastatic tumor cells. J. Cell Sci 123, 2332–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletov K, Montel V, Lester RD, Gonias SL, and Klemke R (2007). High-resolution imaging of the dynamic tumor cell vascular interface in transparent zebrafish. Proc. Natl. Acad. Sci. USA 104, 17406–17411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strilic B, and Offermanns S (2017). Intravascular survival and extravasation of tumor cells. Cancer Cell 32, 282–293. [DOI] [PubMed] [Google Scholar]
- Stroka KM, Jiang H, Chen SH, Tong Z, Wirtz D, Sun SX, and Konstantopoulos K (2014). Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner K, and Gottesman MM (2015). Beyond 3D culture models of cancer. Sci. Transl. Med 7, 283ps289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner K, Mori H, Mroue R, Bruni-Cardoso A, and Bissell MJ (2012). Coherent angular motion in the establishment of multicellular architecture of glandular tissues. Proc. Natl. Acad. Sci. USA 109, 1973–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: an open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Tufto I, and Rofstad EK (1999). Interstitial fluid pressure and capillary diameter distribution in human melanoma xenografts. Microvasc. Res 58, 205–214. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Valastyan S, and Weinberg RA (2011). Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venero Galanternik MV, Castranova D, Gore AV, Blewett NH, Jung HM, Stratman AN, Kirby MR, Iben J, Miller MF, Kawakami K, et al. (2017). A novel perivascular cell population in the zebrafish brain. eLife 6, e24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kaiser MS, Larson JD, Nasevicius A, Clark KJ, Wadman SA, Roberg-Perez SE, Ekker SC, Hackett PB, McGrail M, et al. (2010). Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development 137, 3119–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SC, Maniar S, Mosher M, Roman BL, Tsang M, and St Croix CM (2012). High resolution imaging of vascular function in zebrafish. PLoS One 7, e44018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, and Bissell MJ (1997). Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol 137, 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R (2002). Scaling down imaging: molecular mapping of cancer in mice. Nat. Rev. Cancer 2, 11–18. [DOI] [PubMed] [Google Scholar]
- White R, Rose K, and Zon L (2013). Zebrafish cancer: the state of the art and the path forward. Nat. Rev. Cancer 13, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, Bourque C, Dovey M, Goessling W, Burns CE, et al. (2008). Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, and Mann M (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, Wang Y, Lin EY, Li J. f., Goswami S, Stanley ER, Segall JE, Pollard JW, and Condeelis J (2007). Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 67, 2649–2656. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Williams PJ, Hiraga T, Niewolna M, and Nishimura R (2001). A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res 16, 1486–1495. [DOI] [PubMed] [Google Scholar]
- Zhang S, Huang W-C, Zhang L, Zhang C, Lowery FJ, Ding Z, Guo H, Wang H, Huang S, Sahin AA, et al. (2013). SRC family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res. 73, 5764–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The analyzed datasets generated during this study are available at Mendeley Data under https://doi.org/10.17632/rbckhhkczj.1. Corresponding files of cell trajectories used in cell speed, residence time, and occlusion experiments are available at Mendeley Data under the same DOI. Raw proteomics data is available on ProteomeXchange with accession number PXD014345 (ProteomeXchange: PXD014345). Analyzed proteomics data generated in this study is included in the Supplemental Information (Table S6). Code used to analyze cell trajectories in zebrafish and in microfluidic devices is included in this paper as Supplemental Information, along with code used to analyze optical trap microrheology data (Methods S1). Raw microscopy data are several terabytes in size and are available from the corresponding author on request.