

Abstract
Gene essentiality estimation is a popular empirical approach to link genotypes to phenotypes. In humans, essentiality is estimated based on loss-of-function (LoF) mutation intolerance, either from population exome sequencing (in vivo) data or CRISPR-based in vitro perturbation experiments. Both approaches identify genes presumed to have detrimental consequences on the organism upon mutation. Are these genes constrained by having key cellular/organismal roles? Do in vivo and in vitro estimations equally recover these constraints? Insights into these questions have important implications in generalizing observations from cell models and interpreting disease risk genes. To empirically address these questions, we integrate genome-scale datasets and compare structural, functional and evolutionary features of essential genes versus genes with extremely high mutational tolerance. We found that essentiality estimates do recover functional constraints. However, the organismal or cellular context of estimation leads to functionally contrasting properties underlying the constraint. Our results suggest that depletion of LoF mutations in human populations effectively captures organismal-level functional constraints not experimentally accessible through CRISPR-based screens. Finally, we identify a set of genes (OrgEssential), which are mutationally intolerant in vivo but highly tolerant in vitro. These genes drive observed functional constraint differences and have an unexpected preference for nervous system expression.
INTRODUCTION
Understanding the patterns and phenotypic consequences of genetic alterations is a fundamental problem in evolution and development (1–4). A popular empirical approach to link genotypes to phenotypes is by estimating the degree of essentiality of a gene. A gene is considered ‘essential’ if it is required to sustain life in cells or whole organisms, and this requirement is often estimated by experimental perturbations (5,6). The study of essential genes was originally conducted on prokaryotes, due to their accessibility to genetic manipulation. More recently, however, gene essentiality has been estimated in multicellular eukaryotes, including mammals (7). Despite the absolute character of the ‘essential’ gene denomination, data from multiple studies in model organisms have shown strong context dependency: genes are required or not for survival depending on environmental conditions and developmental stages (5,8,9).
The advent of sequencing technologies and gene editing techniques enabled the estimation of gene essentiality in humans (6). The problem has been addressed following two approaches. On one hand, systematic testing of gene silencing effects on human cell cultures identifies genes that affect cell viability or optimal fitness upon perturbation (10–13). On the other hand, population-level statistical estimates of unexpected mutational depletion identifies genes presumed to be subjected to functional constraints (14). Both approaches aim at ranking genes according to their effect on the organism (or cell) upon loss-of-function mutations. However, given the context dependency of gene essentiality, and the differences in the organizational level at which the effects of genotypic changes are assessed, the parallels of the two types of essentiality approximations are unclear.
In vitro screens of mutation tolerance identify genes with an immediate effect on cell proliferation and viability; consequently, the corresponding essentiality estimates depend on the specific cell line and culture conditions being tested. Furthermore, cell culture experiments do not capture developmental and functional constraints intrinsic to the organism. In vitro estimation of gene essentiality is thus inevitably tailored to cell viability. On the other hand, ‘in vivo’ measures of mutational tolerance estimated from population-level genetic variation score genes according to the prevalence/depletion of loss-of-function (LoF) mutations. Genes under mutational constraint are assumed to be consistent with a scenario where purifying selection filters out protein-altering mutations with detrimental effects, thus eluding fixation within the population. In this sense, in vivo estimates of mutational tolerance are considered a proxy for the effect of mutations on organismal fitness. Such effect, in turn, mirrors to some extent the notion of essentiality in the context of population dynamics (5). Both estimation types (in vitro and in vivo) have been discussed within the context of human gene essentially, nonetheless (6). Hereafter we use the terms cellular viability (CV) and organismal fitness (OF) to refer to the context in which human gene essentiality is estimated: by means of in vitro perturbation experiments (CV), or in vivo population-based mutation tolerance estimates (OF).
Notably, in both the CV and the OF context, a subset of mutational intolerant genes has been identified, leading to the idea of defining an ‘essential genome’ containing genes that do not tolerate mutations, and a ‘dispensable genome’ including mutation-tolerant genes (6,14). Intolerant genes (essential) are commonly of interest due to their potential detrimental effect on phenotype and disease association; however, highly tolerant genes (nonessential) might be relevant for evolvability, due to the plasticity they confer to the system at longer time-scales—for example, as sources of cryptic genetic variation (4) or possible editable links that integrate subsystems (15). Hereafter we will use the terms tolerant and intolerant to refer to human nonessential or essential genes as estimated by the degree of LoF mutation tolerance.
Despite the potential functional relevance of tolerant and intolerant genes, an understanding of the molecular determinants that discriminate between the two groups has been only partially explored for humans (6). Moreover, an understanding of the dependency of molecular determinants of gene essentiality on the differences between the operational context of estimation (CV, in vitro versus OF, in vivo) is lacking. To address these problems, here we systematically defined groups of human tolerant and intolerant genes and performed an integrative and comparative analysis of the structural, functional, and evolutionary features associated with gene essentiality. We analyzed the particularities and commonalities between genes that show extreme (in)tolerance to LoF mutation in a given context: CV, OF or both (Figure 1).
Figure 1.

Overview. (A) Mutational tolerance scores used to categorize human (in)tolerant genes. (B) Consensus mutational tolerance score derivation (mean rank distribution) and corresponding (in)tolerant gene sets. (C–F) Features considered as potential determinants of mutational constraint and gene essentiality, including structural and functional features (C), evolutionary (D), protein functional characterization (E) and expression enrichment (F).
MATERIALS AND METHODS
Gene essentiality
Human gene essentiality estimations based on measures of tolerance to LoF mutations were taken from (6). Estimates include the following scores based on the Exome Aggregation Consortium (ExAC) sample of 60 706 human exomes (14): residual variation intolerance score (RVIS) (16), EvoTol (17), missense Z-score (18), LoFtool (19), probability of haploinsufficiency (Phi) (20), probability of loss-of-function intolerance (pLI) (14) and selection coefficient against heterozygous loss-of-function (shet) (21). Scores based on cell culture perturbation-based experiments include data from KBM7, Raji, Jiyoye, HCT116 and K562 cell lines (12); the KBM7 cell line (10), and RPE1, GBM514, HeLa and DLD1 cell lines (22).
Intrinsic structural disorder
Disorder predictions for each protein in the human proteome were generated at residue resolution using IUPred (23). A gene intrinsic disorder score was calculated by averaging the predicted residue scores over the corresponding protein. Scores range from 0 to 1, with higher scores indicating a higher propensity toward intrinsic disorder.
Haploinsufficiency
A predictive genome-wide haploinsufficiency score (GHIS) was obtained from (24).
Gene expression specificity
Reference RNA-seq data for human tissues was downloaded from the Genotype-Tissue Expression project (GTEx.v7) (25) (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5214/), and the Human protein atlas (HPA) (26) (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2836/). The GTEx dataset includes 53 tissues profiled from 961 donors. The HPA dataset includes 32 tissues profiled from 122 control subjects. For both datasets the median expression over replicates was considered as the expression value of the tissue. Expression breath values for each gene were calculated as the fraction of tissues in which the gene is expressed, using an arbitrary cut-off value of 2 RPKM to determine expression. Expression specificity was measured using the Tau statistic on the same tissue-median matrix as computed in (27). Briefly, the Tau statistic is calculated as follows:
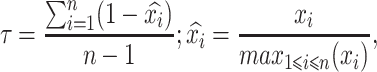 |
where  is the expression of gene x in tissue i and n is the number of tissues. From this definition, Tau varies from 0 to 1, with ubiquitously expressed genes having 0 value and extremely specific genes a value of 1.
is the expression of gene x in tissue i and n is the number of tissues. From this definition, Tau varies from 0 to 1, with ubiquitously expressed genes having 0 value and extremely specific genes a value of 1.
Protein classifications
Proteins were classified as transcription factors (TF), transporters, receptors, enzymes, peptidase, kinase, cancer-related, and RNA-binding proteins (RBP) based on combined curated annotations extracted from the Human Protein Atlas (https://www.proteinatlas.org/humanproteome/proteinclasses) (26), TF reference in (28), RBPs reference from (29), and transporters and receptors reported in (30).
Evolutionary conservation
Comprehensive gene homology information for each human gene with respect to 187 species was extracted from Ensembl comparative genomics resources (31). Only one-to-one orthology relationships were considered to build a binary gene-species matrix. A gene conservation index was calculated for each human gene as the fraction of species having a corresponding ortholog (31). Gene duplication data were extracted from (32,33).
Developmental annotations
Developmental expression classes and developmental process gene annotations were downloaded from the Online Gene Essentiality database (OGEE.v2) at (http://ogee.medgenius.info/downloads/) (33).
Mutational tolerance gene group definition
Mutational tolerance groups were defined based on consensus tolerance scores estimated by averaging gene ranks across available tolerance measures (OF, n = 6; and CV, n = 10 measures). For all measures, as reported in (6), values increase with the degree of intolerance to mutation: intolerant genes have high values. Tolerant genes were defined as the bottom 20% genes in the consensus score rank (lowest constraint) and Intolerant genes as the top 20% (highest constraint). The choice of cut-off values captures extreme values from a long-tailed distribution, which approximates the cut-off proposed in (14) to define the widely used LoF-intolerance metric pLI (cut-off pLI>0.9). Four additional subgroups were defined based on the patterns of overlap between intolerant and intolerant groups (Figure 2C): consistent tolerant genes (n = 714 genes classified as tolerant in both conditions), consistent intolerant genes (n = 771 genes classified as intolerant in both conditions), organismal intolerant but cellular tolerant genes (OI-CT) (n = 567 genes classified as OF intolerant and CV tolerant), and cellular intolerant but organismal tolerant genes (CI-OT) (n = 351 genes classified as OF tolerant and CV intolerant).
Figure 2.

Mutational tolerant and intolerant groups definition. (A) Correlation plot of mutational tolerance measures, adapted from (6). (B) Mutational tolerance measures mean rank distribution. Tolerant and intolerant gene sets are defined as the bottom and top 20%, respectively. (C) Venn diagram of the defined gene sets. Group intersections are highlighted to represent tolerant (714 genes) and intolerant (771) genes found consistently in both OF and CV groups, and inconsistent genes found in contradictory groups depending on the context.
Analysis of gene set aggregation and centrality in the PPI network
A reference human protein-protein interaction (PPI) network was obtained from (34). Briefly, the network is based on experimentally supported protein–protein interactions from different sources that through a stringent orthology mapping scheme recover 625 641 interactions among 17 530 human proteins.
The degree to which a set of genes is aggregated forming a neighborhood within the PPI network was quantified using three complementary approaches: (i) estimating the deviation of the size of the subnetwork produced by genes within the set and their interactions (module size) from expectation, (ii) estimating the strength of association among genes within the set by clustering enrichment and (iii) contrasting observed pairwise gene network distances with expectation. Subgraph module size was calculated by counting the number of nodes (Sc) and edges (Cc) of the largest connected subgraph formed by proteins belonging to a given gene set. Clustering enrichment was measured using spatial analysis of network association, as implemented in SANTA (35). Pairwise shortest distance between every protein pair was measured using the igraph R package (36). The distance distribution of each gene set was characterized by calculating its minimum (Ds) and mean (Dsm) distances. Network centrality was calculated using three complementary measures: degree, betweenness and coreness. These measures were quantified for every node using the igraph R package (36). For each gene set and aggregation statistic, enrichment was calculated by estimating the deviation of the observed gene set average from that expected in a distribution obtained from 10 000 randomly sampled gene sets of the same size. Deviation was quantified with a z-score.
Network structural robustness analysis
Network robustness was characterized by measuring the effect on network structure of targeted removal of nodes according to mutational tolerance ranking and estimating its deviation from random expectation. Network structural response was assessed by calculating the number of nodes (Sf) and edges (Cf) in the perturbed largest connected component after removing a fraction (f) of nodes relative to the unperturbed measures. For each measure, random expectation was estimated by removing fractions from 0.01 to 0.99 of randomly selected nodes 10 000 times.
Functional enrichment and protein class distribution
Over-representation of gene functional features (GHIS, ID, expression specificity, expression breadth, earliest stage expression and developmental process annotation) was estimated by contrasting the gene set average and the random expectation obtained from measuring the given feature in 10 000 randomly sampled same-sized gene sets. Deviation in protein class distribution among gene sets was assessed by quantifying the deviation of the percentage of genes belonging to each protein class from its random expectation as estimated from 10 000 randomly sampled same-sized gene sets.
Gene set evolutionary analysis
Starting from a binary gene-species matrix based on one-to-one orthology relationships, species were classified by taxonomic group resulting in: Archaea (21 species), Bacteria (99 species), Protozoa (16 species), Fungi (8 species), Plants (9 species), Invertebrates (24 species) and Vertebrates (10 species). Percentage of orthologs was calculated for each taxonomic group and mutational tolerance gene group. Orthology relationships were extracted from homologies reported in Ensembl Compara v101 (37).
Tissue specific and expression enrichment
Patterns of preferential expression across tissues were assessed using transcriptomic data (RNA-seq) from the Genotype-Tissue Expression Project (GTEx) (51 tissues) (25) and from the Human Protein Atlas (HPA) (32 tissues) (26). Gene expression enrichment was estimated by pairwise differential expression analysis across all tissue pairs. Differential expression was calculated using voom and lmFit functions from the limma package in R (38). Expression enrichment scores are defined as the sum of the differential expression coefficients across comparisons, discarding genes with a Bonferroni corrected P-value >0.05. Tissue specificity of mutational tolerance groups was estimated by quantifying per tissue and gene groups the deviation from random expectation of expression enrichment scores. Random expectation was estimated by randomly sampling same-sized gene sets 10 000 times.
Gene set enrichment analysis
Gene ontology enrichment analysis was performed using DAVID (https://david.ncifcrf.gov/summary.jsp) (39,40).
GWAS trait associated genes enrichment
Genome-wide association studies (GWAS) data were downloaded from the NHGRI-EBI Catalog of human genome-wide association studies (41). Significant associations and mapped genes as reported by the Catalog were for the following traits were considered: cancer (EFO_1000654), type 2 diabetes (T2D, EFO_0001360), Alzheimer disease (AD, EFO_1001870), amyotrophic lateral sclerosis (ALS, EFO_0001357), Parkinson’s disease (PD, EFO_0002508), schizophrenia (EFO_0004609), major depression (EFO_0009854), cognition (EFO_0005229), intelligence (EFO_0004337) and bipolar disorder (BP, EFO_0009963). Over-representation of trait associated genes among the (in)consistency mutational tolerance classes was measured using Fisher’s exact test as implemented in the R package SuperExactTest (42). Gene set enrichment analysis was performed using fgsea package (43) using GWAS associated genes, mutational tolerance mean ranking scores for OF and CV measures, and the rank difference between OF and CV measures as input. A functional classification of the top/bottom 15 genes in the rank difference distribution associated with any trait was defined using information from Uniprot (44).
Code and data availability
Code and data to reproduce results and all figures are available through GitHub: https://github.com/jlcaldu/Gene-essentiality-analysis.
RESULTS
Context-dependent mutational tolerance categorises human genes
To define genes with extreme (in)tolerance to detrimental mutation, as estimated from patterns of mutational depletion in exome sequencing data (OF) or fitness effects in CRISPR-based cell culture perturbation experiments (CV), we first calculated for each gene and context a consensus tolerance score by averaging gene ranks across available tolerance measures (OF, n = 6; and CV, n = 10 measures). The high pairwise correlation (average Pearson correlation = 0.59, 0,42; OF, CV) between individual measures within each context justifies the use of the proposed consensus score (Figure 2A). We then defined a set of mutation-intolerant (tolerant) genes based on the distribution of consensus tolerance scores. We used the 80th percentile of the distribution as arbitrary cut-off value, a choice that captures the extreme values observed in the long tailed distribution of the measures, and which approximates the cut-off proposed in (14) to define the widely used LoF-intolerance metric pLI (cut-off pLI>0.9). In addition, we defined a contrasting, similar-sized set of mutation-tolerant (intolerant) genes by selecting the 20% bottom ranked genes of the consensus score distribution (Figure 2B). The size of the resulting gene sets are 3028 genes for both intolerant and tolerant OF groups and 3139 genes for both intolerant and tolerant CV groups. Exploring the intersection between gene sets, we identified 714 tolerant and 771 intolerant genes with consistent tolerance behavior across contexts. In contrast, we identified 918 inconsistent genes, whose tolerance behavior depends on the context (OF/CV) (Figure 2C).
Structural and functional constraints predict mutational tolerance classes
We next tested whether the different gene groups are distinctively associated with network structural, and with functional molecular properties. Following previous studies that point to a central role of essential genes in the interactome (45–48), we first asked if (in)tolerant genes have contrasting positions in the interactome and whether such pattern is consistent in genes affecting both organismal and cellular fitness. From a network perspective, in the context of the present study, we hypothesized that (i) mutational tolerance estimation may allow to identify evidence of a core constrained neighborhood within the human interactome, which is separated from a more peripheral, scattered layer formed by mutationally tolerant genes and (ii) given the central role of the intolerant neighborhood, perturbations affecting the corresponding genes are more likely to confer structural fragility to the entire system.
By measuring network features associated with node centrality and aggregation (Figure 3A, see Materials and Methods section), we confirmed that irrespective of the context of estimation, intolerant genes are aggregated and central in the interactome (P values < 0.001, two-sided z test); while tolerant genes consistently show the opposite behavior: loose aggregation and peripheral positioning (Figure 3C and Supplementary Figure S1). To test the vulnerability of the interactome to perturbations targeting tolerant or intolerant genes, we analyzed the network's behavior as a function of the progressive removal of nodes in decreasing order of mutational intolerance score (Materials and Methods section). This analysis further confirmed that there is a strong association between mutational patterns and the global structural properties of the interactome, revealing that intolerant gene removal produces a higher structural damage than random node removal (Figure 3B).
Figure 3.

Structural and functional constraints. (A) Network features measured in the PPI. (B) Structural robustness of the PPI after random removal of nodes (gray lines) and directed removal of genes ranked by mutational intolerance score (inset shows the pattern around the 20% of nodes removal, corresponding to the intolerant gene sets). (C) Deviation (z score) of every network feature. (D) Deviation (z score) of the functional features measured for every gene set (significance: P < 0.05 = n.s., 0.001 < P < 0.05 = *, 0.0001 < P <0.001 = **, P < 0.0001 = ***).
Our results suggest that global structural properties of the interactome are related to mutational tolerance classes. We reasoned that molecular properties suggestive of functional constraint might discriminate tolerance groups as well. By analyzing the degree of estimated haploinsufficiency (GHIS), protein intrinsic disorder (ID), expression breadth and specificity, we similarly found contrasting patterns between tolerant and intolerant genes. Intolerant genes are more likely to be haploinsufficient, to have more intrinsic disorder in protein structure, and to be more broadly expressed across tissues; while tolerant genes show exact opposite behavior (Figure 3D and Supplementary Figure S1). Together, our results confirm that mutationally tolerant and intolerant genes can be consistently discriminated by features indicative of structural and functional constraints, and that this property is independent of the context in which tolerance is estimated (OF or CV).
Evolutionary history of tolerance gene classes
Gene essentiality and interactome centrality have been previously related to evolutionary conservation, with a tendency for topologically central and essential genes to be conserved (evolutionarily old) (49,50). We analyzed whether the tolerance gene groups identified here similarly have contrasting evolutionary conservation patterns, and whether associations are consistent in genes affecting organismal or cellular fitness. We analyzed two features of evolutionary conservation: gene orthology and paralogy.
First, we evaluated the degree of gene conservation by calculating a gene conservation index (CI) that measures the proportion of species in which a human gene has a one-to-one ortholog (Figure 4A). We considered a total of 187 species from 7 taxonomic groups (archaea, bacteria, protozoa, fungi, plants, invertebrates and vertebrates) (Materials and Methods section). Intolerant genes are significantly more conserved than tolerant genes in both OF and CV contexts (pval < 0.0001, two-sided z test) (Figure 4B and Supplementary Figure S2). Notably, however, the conservation of intolerant genes that affect cell viability is considerably higher than that of genes affecting organismal fitness. To further explore the difference in conservation, we calculated the proportion of genes having a one-to-one ortholog by taxonomic and tolerance group (Figure 4C). This analysis revealed a clear difference between CV and OF gene groups. In particular, CV intolerant genes are more represented in every taxonomic group except for vertebrates, while the behavior of OF intolerant genes does not deviate from random expectation, presenting only a marginal enrichment among Plants and Fungi and depletion in Bacteria (Figure 4C). This result demonstrates deep conservation of intolerant genes affecting cellular viability in humans, possibly reflecting the relevance of such genes in core cell-autonomous functions.
Figure 4.

Genes evolutionary features. (A), Presence of gene orthologs among taxa, each row shows the presence (black) of an ortholog in the given taxon. (B) Deviation of mean gene C.I. (C) Percent of genes with an ortholog in each taxon (random expectation is shown in black). (D) Deviation in the number of singleton and duplicated genes per set. (significance: P < 0.05 = n.s., 0.001 < P < 0.05 = *, 0.0001 < P <0.001 = **, P < 0.0001 = ***).
We next analyzed the association between tolerance groups and copy number variation, considering the number of gene duplication events represented in each gene group. The number of duplication events is consistently depleted among intolerant genes in both OF and CV. In contrast, tolerant genes show over-representation of duplication events, but only in the OF context. These results are consistent with a scenario in which paralogs might be buffering phenotypic effects of gene deletion (51) (Figure 4D). The evolutionary pattern of reduced duplication events in intolerant genes is also consistent with a reduction in gene family size distribution for intolerant relative to tolerant genes (Supplementary Figure S2). Together, these results confirm that there is a marked difference in the evolutionary history of tolerant versus intolerant genes. Within tolerant classes, we also found differences between genes that affect cell viability (CV) or organismal fitness (OF): CV intolerant genes are evolutionarily older than OF genes, and only OF but not CV tolerant genes tend to keep multiple gene copies in evolutionary history, suggesting that only organismal but not cellular fitness captures a role for paralogs on phenotypic buffering—organismal.
Molecular classes predict context-dependent mutational tolerance
Our previous results revealed differences in the evolutionary history of tolerance gene classes that affect CV or OF, suggesting that the deep conservation of intolerant genes with effect in cellular viability possibly stems from their role in core unicellular functions. To further explore this association and unravel the differences between OF and CV gene sets, we analyzed the differential over-representation of tolerance gene groups within protein classes and gene ontology terms.
We again found differences in the protein class distribution of (in)tolerant genes, depending on the context of influence CV or OF (Figure 5A). Protein kinases, receptors, transcription factors (TF) and cancer associated proteins show an unexpected contrasting pattern in CV versus OF context, with enrichment of OF intolerant genes but depletion of CV intolerant genes, and a reverse pattern for tolerant genes: enrichment for CV and depletion for OF. Thus, these four protein categories, which together have a key role in developmental processes and associated signaling pathways, tend as a group to not tolerate LoF mutations in the human population, yet are not strongly required for human cell viability.
Figure 5.

Protein class distribution and gene ontology term enrichment. (A) Percent of genes belonging to each protein class distribution, dashed horizontal lines indicate the random expectation, deviation from expectation is shown on top of each bar (significance: P < 0.05 = n.s., 0.001 < P < 0.05 = *, 0.0001 < P <0.001 = **, P < 0.0001 = ***). (B) Top five enriched terms from functional cluster enrichment (DAVID) for each gene set.
Gene ontology enrichment analysis further supports the difference between tolerance groups depending on CV or OF context, with distinct over-represented terms (Figure 5B). Consistent with the previous result, OF intolerant genes show over-representation of gene ontology terms related to development and cell communication, such as transcription regulation, kinases and synapse. CV intolerant genes, on the contrary, show over-representation of core cellular processes related to cell energetics, replication, transcription and translation. Consistent with contrasting functional properties of mutational gene tolerance classes depending on context, human genes that tolerate LoF mutations in cell culture are over-represented in processes related to cell adhesion and communication, i.e. in processes that do not tolerate mutations in the human population context (OF) (Figure 5B).
Contrasting tissue-specificity and developmental activity of (in)tolerant genes
While global network structural and molecular functional properties provide evidence of consistent strong functional constraint on genes that do not tolerate LoF mutations in either human populations (OF context) or in human culture experiments (CV context); more detailed analyses of evolutionary history, protein classes and gene ontology terms suggest that the two contexts (OF and CV) capture distinct functional roles of intolerant genes in the organism. To further explore the hypothesis of contrasting functional constraints, we gathered and interrogated data informative of developmental involvement and tissue-specific expression.
First, we evaluated the distribution of developmental stages in which genes are first expressed. Intolerant genes are expressed earlier in development than tolerant genes in both OF and CV contexts (Figure 6A), with at least 98% of the genes already expressed in prenatal stages. On the contrary, tolerant genes are depleted in prenatal stages and preferentially expressed after birth. This similar pattern of early expression is consistent with the high involvement of both TF-mediated specification, cell-attachment and core cellular replication in embryogenesis and organogenesis. However, when considering curated gene sets involved in specific developmental processes, we found a contrasting pattern between OF and CV gene sets, consistent with previous results. In OF context, intolerant genes are over-represented in every developmental category, while tolerant genes are depleted in all categories. In sharp contrast, in CV context, tolerant genes are over-represented in developmental processes, while intolerant genes are depleted (Figure 6B). This result further supports the view that genes intolerant of LoF mutations in the human population are preferentially involved in organismal development, and that the same constraint is not captured by in vitro screens of gene essentiality.
Figure 6.

Gene set enrichment in tissue-specific expression, temporal stage expression and developmental processes. (A) Cumulative distribution of gene set percentage of genes first expressed by developmental stage. (B) Number of genes per gene set associated with a developmental process. (C) Expression enrichment deviation by tissue, tissues are ordered according to rank log ratio. Tissues highlighted with a star are part of the central nervous system (significance: P < 0.05 = n.s., 0.001 < P < 0.05 = *, 0.0001 < P < 0.001 = **, P < 0.0001 = ***).
In addition to developmental-stage associations, we next explored whether (in)tolerant gene classes of CV or OF context recover distinct preferential behavior in adult tissues. We used RNA-seq data from the Genotype-Tissue Expression (GTEx) project (25) to analyze patterns of tissue-specific expression. First we performed gene expression specificity analysis to compute for each tissue and gene a quantitative measure of specific expression relative to all other tissues (Materials and Methods section). We then used the specificity values to estimate the degree to which a gene tolerance group shows unexpectedly high preferential expression in a given tissue relative to random expectation. We performed these calculations independently for each tissue and tolerance group, and for each context (CV or OF). We found both common and particular patterns of behavior among OF and CV contexts. We found consistent opposite behavior in tissue preference in tolerant versus intolerant genes: in both contexts tissues with preferential expression of intolerant genes show depleted preferential expression of tolerant genes, and vice versa (Figure 6C).
Next, to contrast the tissue-preference behavior of tolerance groups in each context, we ordered the tissues by their relative preference to preferentially express intolerant versus tolerant genes. We measured this preference by the ratio of how each tissue ranks in intolerant versus tolerant preferential expression. Using this approach, tissues that tend to preferentially express intolerant genes and not tolerant genes appear on top (Figure 6C). Notably, this analysis uncovered a contrasting behavior between OF and CV contexts: tissues from the central nervous system as a group (n = 12 brain regions) show the largest relative preference of intolerant gene expression in the OF context and the least preference in the CV context. In other words, we found that the adult human brain tends to preferentially express genes that do not tolerate LoF mutations in the human population while preferentially repressing both OF tolerant genes and genes required for cell viability (CV intolerant genes) (Figure 6C). The other tissues do not show any clear pattern distinguishing CV and OF measures. We corroborated the reproducibility of these results by using independent gene expression reference data form the human protein atlas (26) (Supplementary Figure S3).
Genes with inconsistent mutational tolerance behavior
The contrasting patterns found in OF versus CV gene groups suggests that genes with an inconsistent tolerance behavior across contexts might be driving the observed functional differences. To test this hypothesis and to identify specific genes that capture the differential functional constraints accessible through population-based versus CRISPR-bases essentiality estimations, we defined (in)consistency mutational tolerance classes and repeated all association analyses for the new groups (Figure 7A). We identified a group of 567 genes that do not tolerate LoF mutations in human populations, but that are not required for survival in human cells (organismal intolerant but cellular tolerant genes, OI-CT). Similarly, we identified a group of 351 genes that are cellular intolerant but organismal tolerant (CI-OT). Consistent with our previous results, OI-CT genes include major TF regulators of early cell lineage specification (e.g. SOX1, PAX6 and OLIG1), members of signaling pathways regulating these TFs (NOTCH1, NOTCH3, SMAD1), and genes encoding proteins relevant for noncell autonomous physiological integration (e.g. ion channels CACNA1C, CLCN3, GABRA1) (Supplementary Data S1).
Figure 7.

Consistent and inconsistent subgroups analyses. (A) Subgroups definitions based on the overlap between OF and CV gene sets. (B) Ontology term enrichment for each subgroup. (C) Protein class distribution enrichment. (D) Percent of orthologs in each taxon, color of the column indicates subgroup, black column shows the expected values. (E) Percent of genes that only have orthologs among vertebrates. (F) Enrichment of gene presence in developmental processes. (G) Brain tissues expression enrichment (significance: P < 0.05 = n.s., 0.001 < P < 0.05 = *, 0.0001 < P < 0.001 = **, P < 0.0001 = ***).
As expected, association analyses revealed clear differences in these two inconsistent groups (OI-CT and CI-OT), in particular with respect to categories with contrasting behavior in OF versus CV gene sets. OI-CT genes are associated with gene ontology terms related to transcriptional regulation and neuronal communication, while CI-OT genes are associated with unicellular functions (Figure 7B). Similarly, protein classes with contrasting behavior in OF versus CV (i.e., TFs, receptors and kinases) are highly over-represented in OI-CT (Figure 7C). Notably, the same enrichment patterns are not observed when considering (in)tolerant genes with consistent behavior in both OF and CV contexts.
We similarly identified discrepancies in the evolutionary history of the new gene groups. OI-CT genes have less orthologs than expected within every taxonomic group except for Vertebrates, suggesting that many of these genes emerged relatively late in evolution, in pair with the emergence of vertebrates (Figure 7D). To further explore this observation, we calculated the number of genes in each tolerance gene subgroup that have one-to-one orthologs only within vertebrates, and not in other taxonomic groups. This analysis confirmed that >80% of OI-CT genes are exclusive to the vertebrate branch, in sharp contrast with both consistently intolerant genes and with CI-OT genes (Figure 7E). Lastly, OI-CT genes are also highly over-represented within every developmental process considered (Figure 7F) and are preferentially expressed in adult human brain tissues (Figure 7G). The evolutionary and functional patterns associated with the OI-CT group suggests that the genes in this group are relevant for organismal physiology.
Mutational intolerant genes nonessential for cell survival are associated with brain disease and cognitive traits
Because OI-CT genes are evolutionarily novel, involved in development, preferentially expressed in the brain, and do not tolerate deleterious mutations in the human population; we hypothesized that their function might be associated with phenotypic traits characteristic of humans. To test this hypothesis, we compiled genes associated with cognitive traits (cognition and intelligence), psychiatric disorders (schizophrenia, depression and bipolar disorder BD) and neurodegenerative diseases (Alzheimer’s disease [AD], amyotrophic lateral sclerosis [ALS] and Parkinson’s disease [PD]). These and related traits and diseases have been considered either of particular relevance for human biology or human-specific (52–54). For contrast, we included cancer and type 2 diabetes (T2D), diseases not directly associated with cognitive traits. In support of our hypothesis, we found that every disease gene set is over-represented in the OI-CT genes, with the psychiatric/cognitive traits having higher fold enrichment (FE > 2 for every gene set) than the other traits (Figure 8A). In almost every other tolerant class disease associated genes are underrepresented, the only exception being consistently tolerant genes, where cancer genes are over-represented. This pattern is consistent with the relevance of OI-CT gene function in organismal physiology, and in cognitive functions and human brain neurophysiology in particular. We further explored these associations by investigating more globally the degree to which large differences in the degree to which genes tolerate detrimental mutation in the human population versus in cell culture conditions tend to recover physiologically relevant genes. To this end, we scored every gene by the rank difference between its OF and CV tolerance scores and tested whether GWAS trait associated genes tend to have unexpectedly large rank differences (Figure 8B and C). We found that, indeed, GWAS genes have large OF-CV rank differences, with stronger enrichment than that obtained when considering OF or CV tolerance scores alone, with the latter lacking any significant association. Among GWAS traits, cognitive and psychiatric traits showed the highest association with OF-CV rank differences, indicating that genes associated with these traits are particularly intolerant of detrimental mutation in the human population and yet nonessential for cell survival (Figure 8C). To examine whether gene function might help explain such a pattern, we looked more closely at the GWAS genes having the most extreme top/bottom OF-CV rank differences (Figure 8D). Genes with high OF and low CV ranking, and thus mutationally constrained in humans but not in cells, tend to be involved in neurodevelopment and signal transduction, and to function as ion channels; processes relevant for multicellular communication and proper brain structure/function. On the other extreme, genes with high CV and low ranking, and thus mutationally constrained in cells but not in humans, are associated with cell autonomous functions like cell replication and transcription. This evidence demonstrates that genes with contrasting mutational tolerance behavior are indeed associated with cognitive traits, which are biologically relevant functions with a more recent evolutionary history.
Figure 8.

Subgroups association with psychiatric disorders and cognitive traits. (A) Over-representation of GWAS associated genes and (in)consistent tolerance groups. Fold enrichment (FE) of observed over expected overlap. Gene sets are colored according to the trait classification. (B) Cumulative sum of genes belonging to each GWAS set, genes sorted by the difference in OF and CV mutational tolerance rank. (C) Normalized enrichment score (NES) from gene set enrichment analysis (GSEA) performed test over-representation of GWAS genes with respect to CV, OF, rank difference scores. (D) Top/bottom 15 genes with extreme difference in OF and CV mutational tolerance scores and their traits and functional annotations. (significance: P > 0.05 = n.s., 0.001< P <0.05 = *, 0.0001< P <0.001 = **, P < 0.0001 = ***).
DISCUSSION
We examined the degree to which measures that rank human genes according to their degree of tolerance to LoF mutations capture functional constraints. We considered tolerance estimations based on either in vivo exome-based population data or in vitro CRISPR-based perturbation experiments. To interpret evidence of differences in functional constraint in essential versus mutational tolerant genes, we integrated genome-wide data related to gene function, including structural, functional and evolutionary features. Our results indicate that intolerant genes (i) form a core network neighborhood in the human protein interactome, (ii) are enriched in molecular properties suggestive of functional constraint, (iii) are evolutionarily conserved and (iv) show preferential expression in specific tissues and developmental stages (Figure 9). The molecular and network properties that consistently discriminate intolerant from tolerant genes suggest that essentiality estimates based on mutational tolerance inference do recover functional constraints, irrespective of estimation context (OF or CV). However, we also found differences in the discriminatory properties of genes depending on whether their tolerance to mutation was estimated at the organismal or cellular level.
Figure 9.

Results summary. Enrichment patterns of the main features found associated with genes essentiality. Top panel: Structural and functional features distinctive of tolerant/intolerant genes irrespective of the context. Bottom panel: Distinctive features that show a divergent pattern depending on the context in which mutational tolerance is defined.
Consistent with previous observations (6,14), we found that structural network properties consistently discriminate tolerance/essentiality classes. Intolerant genes are central and localized in the human interactome, while tolerant genes are dispersed in the periphery. This relative organization predicts preferential vulnerability of the cell to intolerant gene failure, a principle that we confirmed by simulated network perturbation analysis. Both centrality and perturbation results provided results consistent with the hypothesis of a dominant role of intolerant genes in influencing cell behavior. Molecularly, intolerant genes also show properties often associated with gene functional relevance. We observe a significant tendency (FDR<0.01) for intolerant genes to be haploinsufficient, to have structural disorder and to be broadly expressed. In contrast, tolerant genes show under-representation of these properties. These observations further support the functional relevance of intolerant genes, as similar to broadly expressed (55), intrinsically disordered proteins are highly pleiotropic given their structural flexibility and interaction promiscuity (56), while dosage alteration of haploinsufficient genes is similarly prone to be detrimental (57).
From an evolutionary perspective, we also observe a clear distinction between tolerant and intolerant genes, consistent with previous reports (48,50). Genes intolerant to LoF mutations have an older evolutionary history, with deep one-to-one orthology across species. Notably, we found that this evolutionary pattern is accentuated in intolerant genes estimated at the cellular-level compared to those measured at the organismal-level, suggesting that CV intolerant genes are more prone to be involved in basic cell-autonomous processes shared among all taxa, with prominent presence in unicellular organisms. In contrast, OF intolerant genes are either not enriched in unicellular groups (Archaea and Protozoa) or are underrepresented in Bacteria, suggesting that these genes emerged more recently in evolution and acquired a central role at a higher level of multicellular organization.
The idea that the context at which mutational tolerance is estimatimated discriminates genes operating at different levels of organization is reinforced when considering the differences we found in molecular classes among tolerance groups. The ontology terms associated with CV intolerant genes (e.g. mitochondria, RNA processing, ribonucleoprotein and cell cycle) relate to core functions required for cell survival, such as cellular metabolism and replication. On the contrary, CV tolerant genes relate to intercellular adhesion and communication (glycoprotein, cell junction and protein kinase). In sharp contrast, OF intolerant genes are enriched in functional features key to multicellularity, such as organismal development and cell–cell communication, as evidenced by over-representation of transcriptional regulators, synapse genes, kinases and receptors. These results suggest that OF measures recover functional constraints stemming from multicellularity and organismal regulation, a property not readily captured by CV estimations. Consistent with this view, by considering curated gene annotations for developmental processes, we found that genes involved in developmental processes are enriched for OF intolerant genes and CV tolerant genes, and depleted in OF tolerant genes and CV intolerant genes.
Contrasting behaviors also manifest in tissue-specificity. We found that OF intolerant and CV tolerant genes are preferentially expressed in the adult human brain, in contrast with the underexpression of both OF tolerant genes and genes required for cell viability (CV intolerant genes). The over-representation of OF intolerant genes in the adult brain requires a careful explanation, considering additional functions of these pleiotropic genes in the organism and close relatives, and potential reasons why they might be structurally constrained, a study beyond the scope of the current paper. From the current results, we speculate that, in addition to multicellular functional constraints, the depletion of LoF mutations estimated in human populations might capture constraints stemming from functional properties of species-specific relevance, such as higher cognition and associated traits grounded on the complexified human brain (54).
Although essentiality estimates from both cellular and organismal contexts do recover functional constraints, we found that a subgroup of 567 genes estimated as essential at the organismal level, yet nonessential at the cellular level, is responsible for the contrasting functional patterns found between OF and CV intolerant genes. These genes, which we refer to as (OrgEssential), are enriched in developmental processes, transcriptional regulation and neuronal communication and are preferentially expressed in the human brain. Furthermore, these genes are also associated with cognitive and psychiatric traits, underscoring the functional relevance of human-specific constraints recovered only from the organismal level of mutational intolerance. Despite being evolutionary younger than other essential genes, sharing one-to-one orthologs mainly with Vertebrates, OrgEssential genes seem to have developed a central role in the organism, providing an example of how during evolution novel genes can acquire essential properties by acting at levels of biological organization beyond core cell functionality.
Supplementary Material
ACKNOWLEDGEMENTS
J.C.P. is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowship 446988 from CONACYT.
Contributor Information
Jose Luis Caldu-Primo, Instituto de Ecología, Universidad Nacional Autónoma de México, Cd. Universitaria, CDMX., 04510, México; Centro de Ciencias de la Complejidad (C3), Universidad Nacional Autónoma de México, Cd. Universitaria, CDMX., 04510, México.
Jorge Armando Verduzco-Martínez, Departamento de Biología Celular y Genética, Facultad de Ciencias Biológicas, Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Nuevo León, 66400, México.
Elena R Alvarez-Buylla, Instituto de Ecología, Universidad Nacional Autónoma de México, Cd. Universitaria, CDMX., 04510, México; Centro de Ciencias de la Complejidad (C3), Universidad Nacional Autónoma de México, Cd. Universitaria, CDMX., 04510, México.
Jose Davila-Velderrain, MIT Computer Science and Artificial Intelligence Laboratory, Cambridge, MA 02139, USA; Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Consejo Nacional de Ciencia y Tecnología (CONACYT) [46988 to J.C.P]. Funding for open access charge: Universidad Nacional Autónoma de México (UNAM-DGAPA-PAPIIT http://dgapa.unam.mx/index.php/impulso-a-la-investigacion/papiit) [IN211721 to E.R.A.B.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mayr E. The determinants and evolution of life. The evolution of living systems. Proc. Natl. Acad. Sci. USA. 1964; 51:934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dobzhansky T., Levene H.. Genetics of natural populations; proof of operation of natural selection in wild populations of Drosophila pseudoobscura. Genetics. 1948; 33:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waddington C.H. Canalization of development and the inheritance of acquired characters. Nature. 1942; 150:563–565. [Google Scholar]
- 4. Gibson G., Dworkin I.. Uncovering cryptic genetic variation. Nat. Rev. Genet. 2004; 5:681–690. [DOI] [PubMed] [Google Scholar]
- 5. Zhan T., Boutros M.. Towards a compendium of essential genes - From model organisms to synthetic lethality in cancer cells. Crit. Rev. Biochem. Mol. Biol. 2016; 51:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartha I., di Iulio J., Craig Venter J., Telenti A. Human gene essentiality. Nat. Rev. Genet. 2017; 19:51–62. [DOI] [PubMed] [Google Scholar]
- 7. Rancati G., Moffat J., Typas A., Pavelka N.. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018; 19:34–49. [DOI] [PubMed] [Google Scholar]
- 8. Chen P., Wang D., Chen H., Zhou Z., He X.. The nonessentiality of essential genes in yeast provides therapeutic insights into a human disease. Genome Res. 2016; 26:1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu G., Yong M.Y.J., Yurieva M., Srinivasan K.G., Liu J., Lim J.S.Y., Poidinger M., Wright G.D., Zolezzi F., Choi H.et al.. Gene essentiality is a quantitative property linked to cellular evolvability. Cell. 2015; 163:1388–1399. [DOI] [PubMed] [Google Scholar]
- 10. Blomen V.A., Májek P., Jae L.T., Bigenzahn J.W., Nieuwenhuis J., Staring J., Sacco R., van Diemen F.R., Olk N., Stukalov A.et al.. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015; 350:1092–1096. [DOI] [PubMed] [Google Scholar]
- 11. Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M.. Identification and characterization of essential genes in the human genome. Science. 2015; 350:1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang T., Wei J.J., Sabatini D.M., Lander E.S.. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014; 343:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang T., Yu H., Hughes N.W., Liu B., Kendirli A., Klein K., Chen W.W., Lander E.S., Sabatini D.M.. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell. 2017; 168:890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirschner M.W., Gerhart J.C., Norton J.. The Plausibility of Life: Resolving Darwin’s Dilemma. 2005; New Haven, Connecticut: Yale University Press. [Google Scholar]
- 16. Petrovski S., Wang Q., Heinzen E.L., Allen A.S., Goldstein D.B.. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013; 9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rackham O.J.L., Shihab H.A., Johnson M.R., Petretto E.. EvoTol: a protein-sequence based evolutionary intolerance framework for disease-gene prioritization. Nucleic Acids Res. 2015; 43:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnström K., Mallick S., Kirby A.et al.. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014; 46:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fadista J., Oskolkov N., Hansson O., Groop L.. LoFtool: a gene intolerance score based on loss-of-function variants in 60 706 individuals. Bioinformatics. 2017; 33:471–474. [DOI] [PubMed] [Google Scholar]
- 20. Bartha I., Rausell A., McLaren P.J., Mohammadi P., Tardaguila M., Chaturvedi N., Fellay J., Telenti A.. The characteristics of heterozygous protein truncating variants in the human genome. PLoS Comput. Biol. 2015; 11:e1004647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cassa C.A., Weghorn D., Balick D.J., Jordan D.M., Nusinow D., Samocha K.E., O’Donnell-Luria A., MacArthur D.G., Daly M.J., Beier D.R.et al.. Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat. Genet. 2017; 49:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S.et al.. High-Resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015; 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 23. Dosztanyi Z., Csizmok V., Tompa P., Simon I.. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005; 21:3433–3434. [DOI] [PubMed] [Google Scholar]
- 24. Steinberg J., Honti F., Meader S., Webber C.. Haploinsufficiency predictions without study bias. Nucleic Acids Res. 2015; 43:e101–e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carithers L.J., Ardlie K., Barcus M., Branton P.A., Britton A., Buia S.A., Compton C.C., DeLuca D.S., Peter-Demchok J., Gelfand E.T.et al.. A novel approach to High-Quality postmortem tissue Procurement: The GTEx project. Biopreserv. Biobank. 2015; 13:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S.et al.. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010; 28:1248–1250. [DOI] [PubMed] [Google Scholar]
- 27. Kryuchkova-Mostacci N., Robinson-Rechavi M.. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017; 18:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 175:598–599. [DOI] [PubMed] [Google Scholar]
- 29. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Southan C., Sharman J.L., Benson H.E., Faccenda E., Pawson A.J., Alexander S.P.H., Buneman O.P., Davenport A.P., McGrath J.C., Peters J.A.et al.. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 2016; 44:D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herrero J., Muffato M., Beal K., Fitzgerald S., Gordon L., Pignatelli M., Vilella A.J., Searle S.M.J., Amode R., Brent S.et al.. Ensembl comparative genomics resources. Database. 2016; 2016: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen W.-H., Minguez P., Lercher M.J., Bork P.. OGEE: an online gene essentiality database. Nucleic Acids Res. 2012; 40:D901–D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen W.-H., Lu G., Chen X., Zhao X.-M., Bork P.. OGEE v2: an update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res. 2017; 45:D940–D944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li T., Wernersson R., Hansen R.B., Horn H., Mercer J., Slodkowicz G., Workman C.T., Rigina O., Rapacki K., Stærfeldt H.H.et al.. A scored human protein–protein interaction network to catalyze genomic interpretation. Nat. Meth. 2017; 14:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cornish A.J., Markowetz F.. SANTA: quantifying the functional content of molecular networks. PLoS Comput. Biol. 2014; 10:e1003808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gabor C., Nepusz T.. The igraph software package for complex network research. InterJ. Complex Syst. 2006; 1695:1–9. [Google Scholar]
- 37. Vilella A.J., Severin J., Ureta-Vidal A., Heng L., Durbin R., Birney E.. EnsemblCompara Genetrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009; 19:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang D.W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 40. Huang D.W., Sherman B.T., Zheng X., Yang J., Imamichi T., Stephens R., Lempicki R.A.. Extracting biological meaning from large gene lists with DAVID. Curr. Protoc. Bioinform. 2009; 27:1–13. [DOI] [PubMed] [Google Scholar]
- 41. Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E.et al.. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019; 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang M., Zhao Y., Zhang B.. Efficient test and visualization of Multi-Set intersections. Sci. Rep. 2015; 5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A.. Fast gene set enrichment analysis. 2021); bioRxiv doi:01 February 2021, preprint: not peer reviewed 10.1101/060012. [DOI]
- 44. UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zotenko E., Mestre J., O’Leary D.P., Przytycka T.M. Why do hubs in the yeast protein interaction network tend to be Essential: Reexamining the connection between the network topology and essentiality. PLoS Comput. Biol. 2008; 4:e1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Batada N.N., Hurst L.D., Tyers M.. Evolutionary and physiological importance of hub proteins. PLoS Comput. Biol. 2006; 2:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu H., Greenbaum D., Lu H.X., Zhu X., Gerstein M.. Genomic analysis of essentiality within protein networks. Trends Genet. 2004; 20:227–231. [DOI] [PubMed] [Google Scholar]
- 48. Fraser H.B. Evolutionary rate in the protein interaction network. Science. 2002; 296:750–752. [DOI] [PubMed] [Google Scholar]
- 49. Fraser H.B. Modularity and evolutionary constraint on proteins. Nat. Genet. 2005; 37:351–352. [DOI] [PubMed] [Google Scholar]
- 50. Hahn M.W., Kern A.D.. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol. Biol. Evol. 2005; 22:803–806. [DOI] [PubMed] [Google Scholar]
- 51. Gu Z., Steinmetz L.M., Gu X., Scharfe C., Davis R.W., Li W.-H.. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003; 421:63–66. [DOI] [PubMed] [Google Scholar]
- 52. Burns J.K. An evolutionary theory of schizophrenia: cortical connectivity, metarepresentation, and the social brain. Behav. Brain Sci. 2004; 27:831–855. [DOI] [PubMed] [Google Scholar]
- 53. Varki A., Geschwind D.H., Eichler E.E.. Human uniqueness: genome interactions with environment, behaviour and culture. Nat. Rev. Genet. 2008; 9:749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Geschwind D.H., Rakic P.. Cortical evolution: judge the brain by its cover. Neuron. 2013; 80:633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Watanabe K., Stringer S., Frei O., Umićević Mirkov M., de Leeuw C., Polderman T.J.C., van der Sluis S., Andreassen O.A., Neale B.M., Posthuma D.. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019; 51:1339–1348. [DOI] [PubMed] [Google Scholar]
- 56. Wright P.E., Dyson H.J.. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015; 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang N., Lee I., Marcotte E.M., Hurles M.E.. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010; 6:e1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code and data to reproduce results and all figures are available through GitHub: https://github.com/jlcaldu/Gene-essentiality-analysis.