

Abstract
Chinese hamster ovary (CHO) cells are widely used for producing biopharmaceuticals, and engineering gene expression in CHO is key to improving drug quality and affordability. However, engineering gene expression or activating silent genes requires accurate annotation of the underlying regulatory elements and transcription start sites (TSSs). Unfortunately, most TSSs in the published Chinese hamster genome sequence were computationally predicted and are frequently inaccurate. Here, we use nascent transcription start site sequencing methods to revise TSS annotations for 15 308 Chinese hamster genes and 3034 non-coding RNAs based on experimental data from CHO-K1 cells and 10 hamster tissues. We further capture tens of thousands of putative transcribed enhancer regions with this method. Our revised TSSs improves upon the RefSeq annotation by revealing core sequence features of gene regulation such as the TATA box and the Initiator and, as exemplified by targeting the glycosyltransferase gene Mgat3, facilitate activating silent genes by CRISPRa. Together, we envision our revised annotation and data will provide a rich resource for the CHO community, improve genome engineering efforts and aid comparative and evolutionary studies.
INTRODUCTION
Chinese hamster ovary (CHO) cells are the predominant mammalian system for large-scale production of clinical therapeutic proteins (1). They are valued for their high growth rate (2), ease of genetic manipulation and ability to properly fold, assemble and produce complex post-translationally modified proteins that are not immunogenic in humans (3). As of 2018, 84% of FDA approved monoclonal antibodies were produced in CHO cells (1) and by sales in 2020, 5 out of the top 10 drugs are CHO-derived recombinant proteins (4). Optimizing CHO cells to increase production quantity and quality has been a priority for efforts to reduce the costs of biopharmaceuticals. Over the past few decades, these optimization efforts have progressed from engineering the media and bioreactors to transgene codon sequence and more recently, cell engineering and synthetic biology (5,6).
Genome sequencing efforts for CHO cells and the Chinese hamster (7–9) have been fundamental for studying and engineering CHO cells. In particular, they enabled systematic identification of genes associated with improved cell performance and product quality (10–16). Furthermore, the sequences enabled the implementation of CHO cell engineering using tools including transcription activator-like effector nucleases (TALENs, (17), RNA-directed DNA methylation (RdDM), (18), CRISPR-Cas9 (19)) and others for genetic screens and the targeted inhibition or activation of genes (6,20). However, the Chinese hamster genome annotation remains far from complete, especially for the approximately 50% of genes that are silenced in CHO cells, including many needed for producing more human-like proteins (21). CRISPR activation (CRISPRa (20)) and other genetic engineering methods could be instrumental to improve therapeutic protein production. However, to engineer gene expression, knowledge of the underlying regulatory elements is critical.
Recruitment of the RNA Polymerase II pre-initiation complex (RNAPII) by CRISPRa or blocking of the RNAPII by CRISPR inhibition (CRISPRi) and promoter editing (22,23) require knowledge of the polymerase’s native transcription start site (TSS). Unfortunately, the vast majority of TSSs in the Chinese hamster RefSeq annotation were predicted computationally (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/) and may not correspond to the actual start sites in vivo. While previous work annotated 6,547 TSSs using steady-state 5′RNA ends by cap analysis gene expression (CAGE) in CHO cells (24), the data and annotation are not publicly available. Consequently, current inaccuracies in the annotation of the Chinese hamster genome and its TSSs present a major hurdle for targeted engineering of gene expression in CHO cells.
To remedy this issue, we generated multiple complementary experimental data types to accurately capture nascent transcription start sites (TSSs) at single nucleotide resolution [5′GRO-seq (25), csRNA-seq (26), GRO-seq (27)], expressed genes (ribosomal RNA-depleted RNA-seq), small RNAs [sRNA-seq (26)] and open chromatin (ATAC-seq (28)). To more comprehensively define regulatory elements in CHO cells, including for silenced genes, we interrogated not only CHO K1 cells but also 10 tissues and bone marrow derived macrophages (BMDMs) from Chinese hamsters of the original colony where CHO cells were derived (29). Through this work, we developed a comprehensive compendium of Chinese hamster gene expression, including genes, enhancers, unstable divergent transcripts, diverse non-coding RNAs and their respective TSSs. Given their importance in deploying CRISPRa, we further analyzed the TSSs of protein-coding genes. These data enabled us to accurately annotate the TSS or TSSs of 15 308 protein-coding genes and 3034 non-coding RNAs. Notably, for 13 037 (85% of observed genes) genes, all detected TSSs were revised by >10 base pairs (bp) from the nearest NCBI RefSeq TSS, and 2607 (17%) by >150 bp. To demonstrate the accuracy and functionality of our revised TSS annotation we activated the dormant Mgat3 (β‐1,4‐mannosyl‐glycoprotein 4‐β‐N‐acetylglucosaminyltransferase) in CHO cells by CRISPRa (30) using a novel identified TSS. In addition to accurate TSSs, the data generated provide insights into the DNA motifs and transcriptional regulatory pathways underlying tissue specificity in hamsters. Together, we envision our data and revised TSS annotation for the Chinese hamster will provide a rich resource for the CHO community, facilitate integrating the Chinese hamster into comparative studies, and improve engineering and manipulation for optimizing the production of therapeutic recombinant proteins in CHO cells.
MATERIALS AND METHODS
Sample preparation
Female Chinese hamsters (Cricetulus griseus) were generously provided by George Yerganian (Cytogen Research and Development, Inc.) and housed at the University of California San Diego animal facility on a 12h/12h light/dark cycle with free access to normal chow food and water. All animal procedures were approved by the University of California San Diego Institutional Animal Care and Use Committee in accordance with University of California San Diego research guidelines for the care and use of laboratory animals. None of the used hamsters were subject to any previous procedures and all were used naively, without any previous exposure to drugs. Euthanized hamsters were quickly chilled in a wet ice/ethanol mixture (∼50/50), organs were isolated, placed into Trizol LS, flash frozen in liquid nitrogen and stored at -80°C for later use. CHO-K1 cells were grown in F-K12 medium (GIBCO-Invitrogen, Carlsbad, CA, USA) at 37°C with 5% CO2.
Bone marrow-derived macrophage (BMDM) culture
Hamster bone marrow-derived macrophages (BMDMs) were generated as detailed previously in (31). Femur, tibia and iliac bones were flushed with DMEM high glucose (Corning), red blood cells were lysed, and cells cultured in DMEM high glucose (50%), 30% L929-cell conditioned laboratory-made media (as source of macrophage colony-stimulating factor (M-CSF)), 20% FBS (Omega Scientific), 100 U/ml penicillin/streptomycin+L-glutamine (Gibco) and 2.5 μg/ml Amphotericin B (HyClone). After 4 days of differentiation, 16.7 ng/ml mouse M-CSF (Shenandoah Biotechnology) was added. After an additional 2 days of culture, non-adherent cells were washed off with room temperature DMEM to obtain a homogeneous population of adherent macrophages which were seeded for experimentation in Nunc Cell Culture dishes (Thermo Scientific) overnight in DMEM containing 10% FBS, 100 U/ml penicillin/streptomycin+L-glutamine, 2.5 μg/ml Amphotericin B and 16.7 ng/ml M-CSF. For Kdo2-Lipid A (KLA), activation, macrophages were treated with 10 ng/ml KLA (Avanti Polar Lipids) for 1 hour.
RNA-seq
RNA was extracted from organs that were homogenized in Trizol LS using an Omni Tissue homogenizer. After incubation at RT for 5 min, samples were spun at 21 000 g for 3 min, supernatant transferred to a new tube and RNA extracted following manufacturer’s instructions. Strand-specific total RNA-seq libraries from ribosomal RNA-depleted RNA were prepared using the TruSeq Stranded Total RNA Library kit (Illumina) according to the manufacturer-supplied protocol. Libraries were sequenced 100 bp paired-end to a depth of 29.1–48.4 million reads on an Illumina HiSeq2500 instrument.
csRNA-seq protocol
Capped small RNA-sequencing was performed identically as described in (26). Briefly, total RNA was size selected on 15% acrylamide, 7 M UREA and 1× TBE gel (Invitrogen EC6885BOX), eluted and precipitated over night at -80°C. Given that the RIN of the tissue RNA was often as low as 2, essential input libraries were generated to facilitate accurate peak calling (26). csRNA libraries were twice cap selected prior to decapping, adapter ligation and sequencing. Input libraries were decapped prior to adapter ligation and sequencing to represent the whole repertoire of small RNAs. Samples were quantified by Qbit (Invitrogen) and sequenced using the Illumina NextSeq 500 platform using 75 cycles single end.
Global run-on nuclear sequencing protocol
Nuclei from hamster tissues were isolated as described in (32). Hamster BMDM and CHO nuclei were isolated using hypotonic lysis [10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2] with 0.1% and 0.5% IGEPAL, respectively. Nuclei were flash frozen and later 0.5–1 × 106 nuclei in 200 μl GRO-freezing buffer [50 mM Tris-HCl pH 7.8, 5 mM MgCl2, 40% Glycerol] were used in reactions with 3x NRO buffer [15 mM Tris-Cl pH 8.0, 7.5 mM MgCl2, 1.5 mM DTT, 450 mM KCl, 0.3 U/μl of SUPERase In, 1.5% Sarkosyl, 366 μM ATP, GTP (Roche) and Br-UTP (Sigma Aldrich) and 1.2 μM CTP (Roche, to limit run-on length to ∼40 nt)] as described in (33). Run-on reactions were stopped, purified and GRO-seq and 5′GRO-seq libraries generated exactly as described in (31). BrU enrichment was performed using a BrdU Antibody (Sigma B8434-200 μl Mouse monoclonal BU-33) coupled to Protein G (Dynal 1004D) beads. For each sample, 3 × 20 μl of Protein G beads were washed twice in DPBS+0.05% Tween 20 (DPBS+T) and then the antibody coupled in a total volume of 1 ml DPBS+T under gentle rotation. About 1 μl of antibody was used per 8 μl of beads. Samples were amplified for 14 cycles, size selected for 160–250 bp and sequenced on an Illumina NextSeq 500 using 75 cycles single end.
Assay for transposase-accessible chromatin sequencing (ATAC-seq) protocol
Approximately 150 k nuclei in 22.5 μl GRO freezing buffer (isolated as described for GRO-seq above) were mixed with 25 μl 2× DMF buffer [66mM Tris-acetate (pH = 7.8), 132 K-Acetate, 20 mM Mg-Acetate, 32% DMF] and tagmented using 2.5 μl DNA Tn5 (Nextera DNA Library Preparation Kit, Illumina) added. The mixture was incubated at 37°C for 30 min and subsequently purified using the Zymogen ChIP DNA purification kit (D5205) as described by the manufacturer. DNA was amplified using the Nextera Primer Ad1 and unique Ad2.n barcoding primers using NEBNext High-Fidelity 2× PCR MM for 8 cycles. PCR reactions were purified using 1.5 volumes of SpeedBeads in 2.5 M NaCl, 20% PEG8000, size selected for 140–240 bp fragments and sequenced using the Illumina NextSeq 500 platform using 75 cycles single end. This size range was selected to enrich for nucleosome-free regions.
CRISPRa
CRISPRa was carried out as previously described in (6). Briefly, guide RNAs (gRNAs) were designed in a region proximal to our new revised TSS for Mgat3 (NCBI GeneID: 100689076) and prioritized based on off-targets/proximity to the TSS. Target sequences and gRNA oligos are listed in Supplementary Tables S2 and S3, respectively. gRNAs were transfected along with a dCas9 VPR fusion plasmid [VPR-dCas9 (addgene #134601)] into mutant CHO-S cells carrying knockouts of Mgat4a,4b and 5, St3gal3,4 and 6, B3gnt2, Sppl3 and Fut8 in biological triplicates. Non-targeting gRNAs were transfected with (NT-gRNA) and without VPR-dCas9 (NT-Cas9) as controls. Two days after transfection, cells were harvested to assess activation via qRT-PCR (in technical triplicate) and N-glycan analysis. Transcript levels were normalized to the mean of Hprt and Gnb1 and relative expression levels were calculated using the 2–ΔΔCt method (34).
Glycan quantification
N-Glycans were fluorescently labeled and quantified via LC-MS as described previously in (35). Briefly, the supernatant was concentrated using Amicon® Ultra‐4 Centrifugal Filter Units. Secretome proteins were fluorescently labeled with GlycoWorks RapiFluor‐MS N‐Glycan Kit (Waters, Milford, MA). N‐linked glycan analysis was performed by LC‐MS using a Waters Acquity Glycan BEH Amide 130 Å, 2.1 mm × 150 mm, 1.7 μm column (Waters, Milford) with a Thermo Ultimate 3000 HPLC with the fluorescence detector coupled on‐line to a Thermo Velos Pro Iontrap MS (run in positive mode) and a separation gradient of 30–43% buffer. The amount of N‐glycan was measured by integrating the areas under the normalized fluorescence spectrum peaks with Thermo Xcalibur software (Thermo Fisher Scientific) giving normalized, relative glycan quantities.
RNA-seq processing
Sequence data for all RNA-seq (ribosomal-depleted RNA-seq, csRNA-seq, 5′GRO-seq, sRNA-seq, GRO-seq), data were quality controlled using FastQC (v0.11.6. Babraham Institute, 2010), and cutadapt v1.16 (36) was used to trim adapter sequences and low quality bases from the reads. Reads were aligned to the Chinese hamster genome assembly PICR and annotation GCF_003668045.1, part of the NCBI Annotation Release 103. Sequence alignment was accomplished using the STAR v2.5.3a aligner (37) with default parameters. Reads mapped to multiple locations were removed from analysis.
ATAC-seq processing
Sequence data for ATAC-seq was processed using the ENCODE ATAC-seq pipeline (https://github.com/kundajelab/atac_dnase_pipelines). The reads were trimmed using cutadapt v1.9.1. Reads were aligned using Bowtie2 v2.2.4 (38) to the same Chinese hamster genome. Peaks were called using MACS2 v2.1.0 (39) with a P-value of 0.01 and replicates were merged using irreproducible discovery rate (IDR) (40) of 0.1. The fold-change value is the number of normalized counts over the local background, taken as a 10 000 bp surrounding region.
Detecting TSSs
To call TSS peaks, the Homer (41) version 4.10 TSS pipeline was used with the command ‘findPeaks -style tss’ (http://homer.ucsd.edu/homer/ngs/tss/index.html). Briefly, fragment lengths are set to 1, and 150 bp regions significantly enriched with fragments above the local genomic background region, as well as 2-fold above the input data (GRO-seq and sRNA-seq). FDR correction of 0.01 across peaks in each sample was used. The samples are then merged together into our initial, putative experimental TSSs. Additionally, the total RNA-seq were used to call TSSs as stable if reads are identified between -100 and +500 bp upstream of the TSS.
Sample peaks were merged using the mergePeaks command in Homer. If samples have overlapping peaks, they are combined into one, where the start position is the minimum start position and the end is maximum end position. When merging the replicate peak expression in the same biological sample, the average counts per million (CPM) was used.
Revised promoter annotation
To annotate protein-coding TSSs, a distance threshold from the original annotations was enforced. Ultimately, we retained TSSs that are within -1000 bp and +1000 bp from the initial reported TSS. Additionally, TSSs found in introns, coding sequences, and opposite strand TSSs (divergent transcripts) found in the TSS region were removed (Supplementary Figure S1). There were two annotations used in this study to provide gene promoter landmarks, one from the NCBI RefSeq Annotation 103 release using the PICR genome, and the other with both NCBI’s annotation and a proteogenomics annotation (doi:10.7303/syn17037372) that used RNA-seq, proteomics and Ribo-seq to refine gene mappings (42).
When samples are merged together, the TSSs that are merged may be offset by a few bp. Our revised annotation TSS location is assigned as the CHO TSS location if there is one present, or the location of the TSS in the sample which had the highest expression in CPM. An additional annotation integrating promoter TSSs found in either annotation is also reported.
The annotation provided (Supplementary Data S2 and S3) includes the chromosome, start position (0-based index similar to bed format), strand, position, corresponding gene name, corresponding transcript, comma-separated list of biosamples that express the TSS, and a confidence score signifying the TSS having 2 CPM in at least 2 5′GRO-seq and/or csRNA-seq experiment.
Distal TSSs
Distal TSSs (dTSSs), or intergenic TSSs, were defined as being >1000 kilobase pairs (kbp) away from an annotated gene (ncRNA and protein-coding).
RNA-seq/TSS-seq comparison
To compare RNA-seq to TSS-seq, we used 1558 CHO samples of different lines that were a combination of in-house and public samples (see Supplementary Table S4 for accession IDs). These were quantified and converted into transcript per kilobase gene per million mapped reads (TPM) using Salmon with default parameters (43).
Read histograms
For Figure 2A and B, Homer annotatePeaks.pl with the -hist command was used to construct the histogram with a bin size of 1 bp, and the CPM per TSS was calculated. We restrict the maximum number of tags to count per nucleotide to 3 to prevent high-expressing TSSs from saturating the signal.
Figure 2.
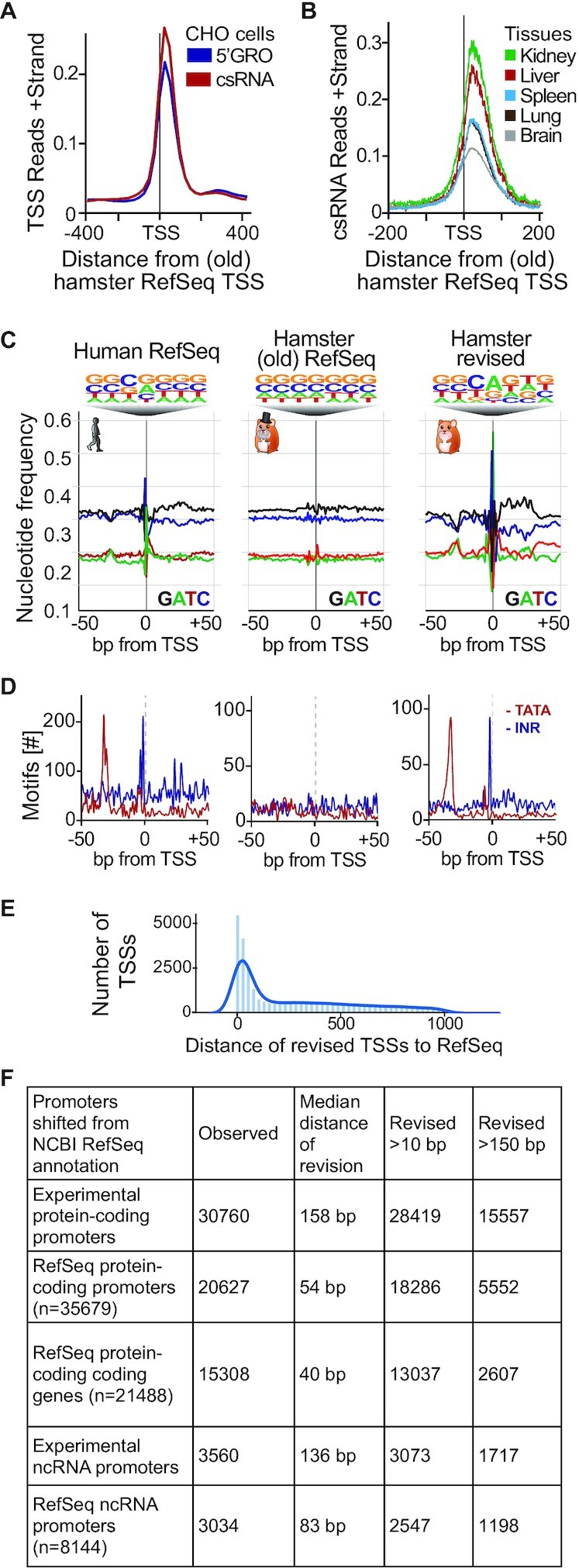
An experimental realignment of TSS annotation for the Chinese hamster uncovers expected genomic elements. A comparison of our TSSs to Chinese hamster RefSeq annotation GCF_003668045.1 (A andB) Average normalized CPM around protein-coding reference TSSs. (A) Comparison of experimentally defined TSSs from CHO cells by 5′GRO-seq and csRNA-seq relative to the RefSeq annotation. (B) Comparison of experimentally defined TSSs from representative tissues relative to the RefSeq annotation. (C) Nucleotide frequency plots of TSSs and their relative information content in Human RefSeq, Chinese hamster RefSeq, and our revised Chinese hamster annotation. (D) Frequency of positional core promoter elements: the TATA box and the Initiator that are commonly found at -30 and +1, relative to the TSS. (E) Frequency of distance between revised TSSs observed and the nearest RefSeq TSS. (F) Summary of total protein-coding and non-distal ncRNA TSSs observed and their distances to RefSeq TSSs.
Motif analysis
Motif analysis of the core promoter elements the Initiator element and the TATA-Box seen in Figure 2 were done using FIMO of the MEME Suite 5.0.2 package with default parameters (44), scanned across a 150 bp window centered on the TSS. Position weight matrix scores of the motifs are summed across all TSSs and converted into a log2-likelihood ratio score for each motif with respect to each sequence position and then converts these scores to P-values, with a cutoff of 0.0001.
For motif analysis in Figure 3, the promoter regions were -300 bp to +100 bp downstream of each TSS using Homer command ‘findMotifsGenome.pl’ with parameters ‘-size -300,100 -len 6,8,10’. For each sample, protein-coding TSSs with log2 CPM of 2 standard deviations above the mean were taken as enriched promoters. The background chosen was randomly selected GC-controlled regions. The negative logeP-value of the top 3 enriched motifs from each sample are taken and the TFs were clustered based on their enrichment P-values.
Figure 3.
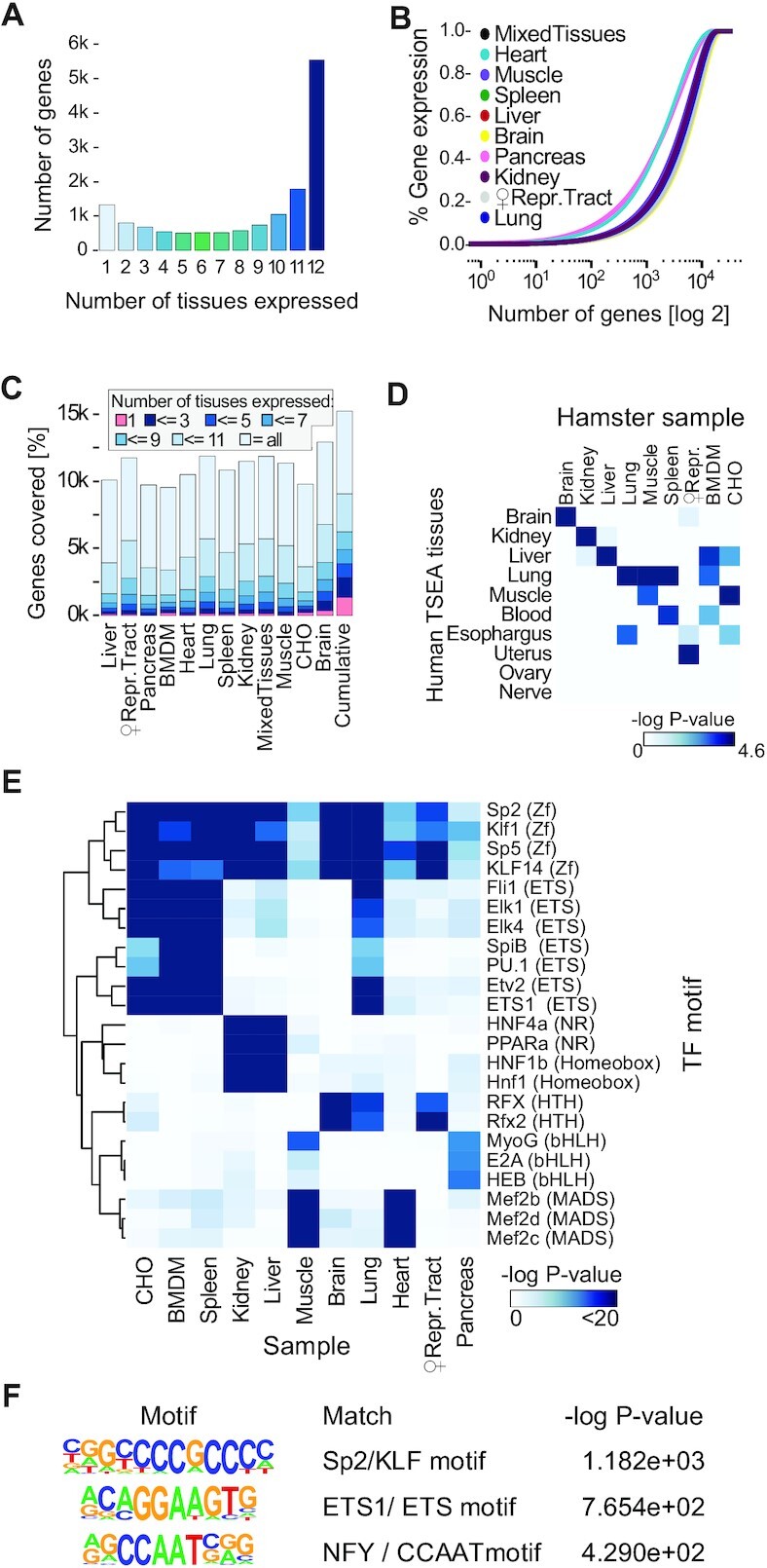
Composition of diverse tissue-specific Chinese hamster transcriptomes. (A) Experimentally detected genes and the number of tissues wherein they were confidently expressed, as defined by csRNA-seq and 5′GRO-seq. (B) Cumulative plot of the distribution of transcript abundance as defined by RNA-seq in various tissues. The transcriptome of highly specialized tissues such as the heart or the pancreas is more dominated by the high expression of a small set of specific RNAs than those of complex tissues such as the brain. (C) Comparison of gene expression distributions across tissues as defined by csRNA-seq and 5′GRO-seq. (D) Tissue-specific gene enrichment analysis (TSEA) comparing the gene expression patterns of our samples as defined by csRNA-seq and 5′GRO-seq to orthologous human pre-defined tissue-specific genes. -loge (P-value) values are shown. (E andF) Motif analysis with Homer. Significance of hypergeometric enrichment of the motifs shown as -loge (P-value). (E) Transcription factor motifs (top 3 per sample) enriched in TSSs for each tissue highlight conservation and factors involved in maintaining tissue-specific expression patterns. (F) Transcription factor motifs enriched in all protein-coding gene-associated TSSs in the revised TSS annotation.
Tissue-specific gene enrichment analysis (TSEA)
TSEA was done using the webserver http://genetics.wustl.edu/jdlab/tsea/. This performs enrichment analysis using Fisher’s Exact test, and the Benjamini-Hochberg corrected log P-values were used for Figure 3D. Unique genes for each sample were defined as only one sample having an observed promoter in that gene. Homologous genes to the human set in TSEA were taken using gene names.
GlycoGene database
Human glycosylation genes and their associated enzyme classes were taken from the ‘Enzymatic Activity’ section of the GlycoGene Database (45). Homologous genes were taken as described above.
RESULTS
Nascent 5′ RNA sequencing across hamster tissues enables accurate reannotation of RNA start sites at single nucleotide resolution
Algorithms predicting gene annotations rely on highly conserved features such as protein domains (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/). Consequently, although gene exon regions are commonly assigned correctly, the annotations of their TSSs, and each associated promoter, are often inaccurate, as these features evolve rapidly and can relocate to non-homologous regions (46). To correctly annotate the TSS of protein genes and non-coding RNAs (e.g. pri-miRNAs, lncRNAs and snoRNAs), it is necessary to experimentally determine these features. We therefore captured [5′GRO-seq (25), csRNA-seq (26)], active transcription [GRO-seq (27)], expressed genes (ribosomal RNA-depleted RNA-seq), small RNAs [sRNA-seq (26)], and open chromatin [ATAC-seq (28)] in CHO-K1 cells as well as ten tissues and bone marrow derived macrophages (BMDMs) in female hamsters from Dr George Yerganian, representing the original colony from which CHO cells were derived in 1957 (47) (Figure 1A; Supplementary Figure S1A and Table S1). Unlike RNA-seq, 5′GRO-seq and csRNA-seq provide accurate TSSs of stable transcripts such as mRNAs (Figure 1B) or ncRNAs but also unstable RNAs such as enhancers RNAs (Figure 1C) (48,49) at single nucleotide resolution. Even for highly expressed genes, such as the Eukaryotic Translation Elongation Factor 1 Alpha (Eef1a1), RNA-seq and related methods that capture the complete transcriptome have limited information about the exact location where genes start and often fall short in the detection of the TSSs for less abundant transcripts (Figure 1B). Capturing the TSSs of nascent transcripts further helps to avoid potential false-positive 5′ends caused by RNA processing or recapping of cytosolic (steady-state) mRNAs (50).
Figure 1.
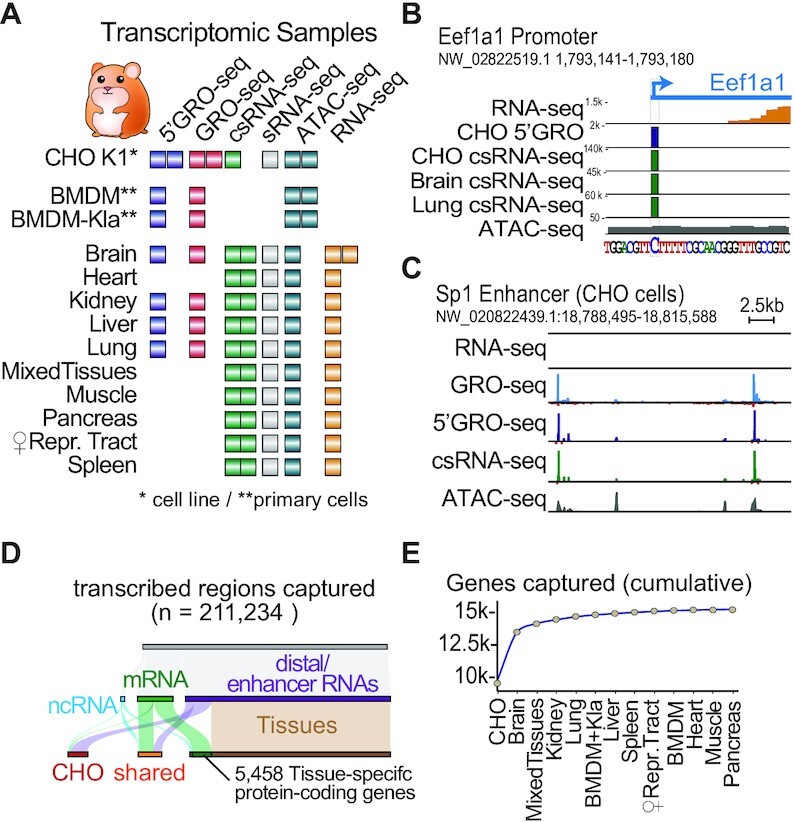
A Chinese hamster Transcriptome Atlas. (A) Overview of datasets generated to identify transcription start sites. * Denote cell lines, ** denote primary cells. (B andC) IGV viewer of data. Units are in counts per million (CPM) (B) Example transcription start site at single-nucleotide resolution as defined by 5′GRO-seq and csRNA-seq (using GRO-seq and sRNA-seq as input, respectively) of the focused Eukaryotic Translation Elongation Factor 1 Alpha (Eef1A1) promoter in CHO cells and diverse tissues. Brain RNA-seq reads are shown in orange. (C) Example of unstable transcription start sites of enhancer RNAs that are poorly detected by conventional RNA-seq at the Sp1 ‘super enhancer’ locus in CHO cells. Note: Raw IGV browser visualization data are provided in Supplementary Figure S3. (D) Number of TSSs captured, grouped by TSS type and samples detected in (E). Cumulative plot across all samples of protein-coding genes with a TSS detected by csRNA-seq and/or 5′GRO-seq enrichment over GRO-seq and/or csRNA-seq. Sorted by taking CHO as the first sample, followed by hamster tissues.
As the primary goal of this study was the determination of confident TSSs, we employed two independent nascent TSS methods, csRNA-seq and 5′GRO-seq. However, while csRNA-seq accurately captures TSSs from total RNA, 5′GRO-seq requires several million purified nuclei, which was not feasible for some tissues. Using csRNA-seq allowed us to expand our analysis across more diverse hamster tissues. In addition, we employed GRO-seq and small RNA-seq (sRNA-seq) data as a background control (also known as input) to boost the confidence of TSS calls by 5′GRO-seq and csRNA-seq, respectively (Supplementary Figure S1B). Next, we integrated ATAC-seq to filter TSSs that mapped outside of open chromatin regions (Supplementary Figure S4A). Finally, as nascent TSS methods detect both stable and unstable TSSs, we used conventional ribosomal RNA-depleted RNA-seq to assign TSSs as stable if RNA-seq coverage was detected between -100 and +500 base pairs from the TSS (26). Integrating these multiple independent data sets also enabled an intrinsic quality control metric and highlighted the confidence of captured TSSs. For example, the correlation among 5′GRO-seq replicates and between 5′GRO-seq and csRNA-seq were highly consistent in their position and expression strength (Pearson correlation of r = 0.96 and r = 0.88, respectively, Supplementary Figure S2). A list of the 71 datasets generated in this study is provided in Supplementary Table S1. These data capture over 210 000 transcribed regions at single-nucleotide resolution (Figure 1D, Supplementary Figure S3A–C, Supplementary Data S1) and provide a comprehensive view of the hamster transcriptome. The majority of these regions (n = 154 736) mark putative distal regulatory elements (sometimes referred to as ‘enhancers’ for simplicity (49)) and unstable divergent transcripts, two common hallmarks of mammalian gene expression (27,51), as well as 3560 non-coding RNAs (Figure 1D). Importantly for protein engineering, we focus on the detected TSSs that mark the promoter or promoters of a cumulative 15 308 RefSeq protein-coding genes captured and their revised promoter TSSs (Figure 1E, Supplementary Data S2). Functional gene groups that were less covered by our data include those associated with olfaction, taste, the male sex organ (testis), development and the adaptive immune system (Supplementary Figure S3 D,E). Together, our experimental data provide accurate TSSs for 72% of annotated hamster protein-coding genes and 3034 non-coding RNAs. We additionally leverage promoter TSSs predicted by a recent proteogenomics annotation (42) to detect and revise additional promoters (Supplementary Data S3).
Realignment of NCBI Chinese hamster RefSeq TSSs exposes key features of transcription
Genome annotations are an essential part of many sequencing and bioinformatic analyses and TSSs provide the foundation for accurate annotation of 5′ ends. We therefore tested the rigor of our experimentally determined protein-coding TSSs and our revised annotation using a number of independent measures. First, we evaluated the relationship of our revised TSSs to the Chinese hamster RefSeq TSSs (GCF_003668045.1). TSSs called by either 5′GRO-seq or csRNA-seq displayed similar distributions (Figure 2A). However, both experimentally determined TSSs displayed a clear offset from the RefSeq annotation. A comparable offset was also observed for protein-coding TSSs measured in diverse tissues (Figure 2B). To further explore these differences we next plotted the proximate DNA nucleotide frequency distributions for both RefSeq and our revised TSS. Basal transcription factors often bind core promoter elements to recruit and position the RNAP II transcription complex which preferentially initiates on purines (52–54). These nucleotide preferences are clearly visible when analyzing the human RefSeq (GRCh38) annotation and in our revised hamster annotation, but not in the current Chinese hamster RefSeq annotation (Figure 2C). In addition to the increased information content in the TSS-proximate nucleotide frequencies, the TATA box and Initiator (Inr) core promoter elements (55–57), were found at the expected -30 and +1 bp positions respectively in the human RefSeq and in our revised hamster annotations, but not the old RefSeq annotation (Figure 2D).
Next, we utilized published epigenetic chromatin states from CHO samples (58) which revealed a striking enrichment of our revised TSSs in the ‘active promoter’ category, highlighting that our experimental CHO dataset is consistent with prior published CHO chromatin states (Supplementary Figure S4B). On the contrary, both our revised promoter TSSs and the NCBI Refseq TSSs fell into more quiescent states, suggesting these regions are near silenced CHO genes. Lastly, we integrated 1558 CHO RNA-seq samples (21,59–61) to assess potential false positive and false negative TSSs in our revised annotation. Genes where we failed to experimentally detect a TSS showed little to no expression across the CHO RNA-seq datasets while those where we captured a CHO TSS were consistently expressed (Supplementary Figure S5), suggesting a low false discovery rate.
Overall, the distance of protein-coding TSSs to the nearest RefSeq TSS varied widely (Figure 2E), with a median distance of 158 bp (Figure 2F). Notably, RefSeq promoters (that represent different transcript isoforms) with a detectable TSS were revised by a median of 54 bp, and 5552 of the promoter TSSs were revised by >150 bp. When we look further at the smallest revision across the promoter TSS’ of each gene, the median distance is 40 base pairs. Importantly, 13 037 were revised >10 bp, and 2607 >150 bp (Figure 2E and F). ncRNAs TSSs also varied, and had a median distance of 83 bp, and 1198 revised by >150 bp (Figure 2F). In summary, these observations provide an independent validation for our revised annotation and stress the importance of experimental TSS data for accurate genome annotations.
Tissue-specific TSS and gene expression patterns in the Chinese hamster
Capturing the protein-coding TSSs across tissues and cell lines revealed that about 1/3 of annotated genes were ubiquitously expressed, while only a comparatively small number of genes were tissue-specific (Figure 3A). Using ribosomal-depleted RNA-seq to measure the steady-state transcriptome highlights the variation of gene expression across tissues (Figure 3B). The number of genes with detected mRNA were 9596 and 9850 in bone marrow-derived macrophages (BMDMs, pooled rested and stimulated conditions) and CHO cells, respectively. Meanwhile, the number spanned from 9782 genes with measured mRNA in the pancreas to 13 007 in the brain (Figure 3B). The number of tissue-specific genes is related to the tissues' degree of specialization and the number of different cell types found within the tissue, but also affected by high abundance transcripts that can hinder detection of less abundant ones (62). In the pancreas, for example, much of transcription is directed towards expressing secretory enzymes such as chymotrypsinogen or carboxypeptidase (63), while in the brain, a higher diversity of transcripts are expressed (64,65).
Of the genes for which TSSs were confidently detected, 40% were expressed in all 12 tissues or cell types and another 19% were found in 11 samples. Approximately 8% of captured genes were unique to a tissue or cell type which increased to 18% and 25% for genes expressed in <3 or <5 samples, respectively (Figure 3C). The genes underlying these tissue-specific gene expression signatures in hamsters at large resembled those of analogous human tissues, as determined by Tissue-Specific Gene Enrichment Analysis (TSEA (66), Figure 3D). Capturing the TSS of a given gene across multiple tissues provided an additional control, in addition to our use of two distinct methods for TSS detection. Nevertheless, for many conserved genes (in which at least one TSS was detected in each sample), the respective promoters detected differed among tissues (Supplementary Figure S6D). Additionally, within conserved promoters, there were small but slight shifts of the called tissue TSS from the revised TSS annotation (Supplementary Figure S6E), together showing a remarkable diversity in 5′ ends. This finding highlights regulatory plasticity as a critical factor to maintain gene expression in distinct cell types.
To gain insights into the underlying regulatory program, we next probed the promoters of tissue-specific genes’ promoters for differentially enriched transcription factor binding motifs. To do this, we used Homer to scan for known motifs 300 bp upstream to 100 bp downstream of TSSs unique to a sample (see Materials and Methods section). The top 3 enriched motifs from each sample and their enrichment values are shown in Figure 3E. We found key regulators or lineage determining transcription factors with preferential expression and binding sites for each tissue such as RFX factors for the brain (67), HNF1 (68) and PPARα factors for the kidney and liver (69,70) or the MADS-box transcription factors Mef2b,c and d for the heart and muscle (Figure 3E) (71,72). Closely related tissues, such as muscle and heart or liver and kidney, displayed a combination of shared and unique factors, which also became apparent for other tissues when more motifs were integrated into the analysis. This observation is in line with the hypothesis that tissue-specific regulatory pathways arise by tinkering with existing pathways, rather than complete innovation (73,74) of regulatory elements needed. On the other hand, ubiquitously expressed genes were enriched for the binding motifs of strong, ubiquitous activators such as SP2/KLF family members (75), ETS factors or NFY (Figure 3F). Together, these findings argue that a comparatively large fraction of genes, including ubiquitous transcription factors, ensure the cell's vital core programs, while a smaller number of genes effectively facilitates specialization. Moreover, our identification of tissue-specific genes and transcription factors enriched in their promoters are consistent with other mammals, further validating our revised TSSs and RNA-seq data.
Profiling diverse hamster tissues identifies TSSs for important, but silenced genes in CHO cells
While CHO cells are exceptional protein production hosts, many genes that could improve product quality or quantity lay dormant. Indeed, about 50% of genes, including many that contribute to important human post-translational modifications, are silent (21). We detected TSSs for only 46% of all protein-coding genes in CHO cells (Figure 1E). Integrating our TSSs from ten tissues and macrophages (76) confidently defined TSSs from an additional 5458 protein-coding genes. In addition, we identified alternative promoters responsible for transcript isoforms for 55% of the RefSeq annotated protein-coding promoters (Supplementary Figure S6C). Our revised TSS annotation provides multiple promoters per gene along with additional promoters uncharacterized in RefSeq (Supplementary Figure S6A and B). This isoform annotation is important as it facilitates the tailored expression of protein isoforms that can exhibit differential activity or distinct functions (77,78). This characterization of >15 k protein-coding genes and >20 k annotated promoters provide the necessary foundation for ongoing efforts to optimize drug production in CHO cells through engineered activation of dormant genes. Given that most protein therapeutics are glycosylated, and the glycans can impact drug safety, efficacy and half-life (79), we next specifically investigated glycosylation-related genes in the context of our updated annotation (Figure 4A). When examining CHO homologues of curated human glycosylation enzymes, we detected dozens of TSSs across diverse classes of glycosylation enzyme genes (Figure 4A). Together, these new annotations should open up new possibilities for engineering gene expression programs, such as glycosylation in CHO cells.
Figure 4.
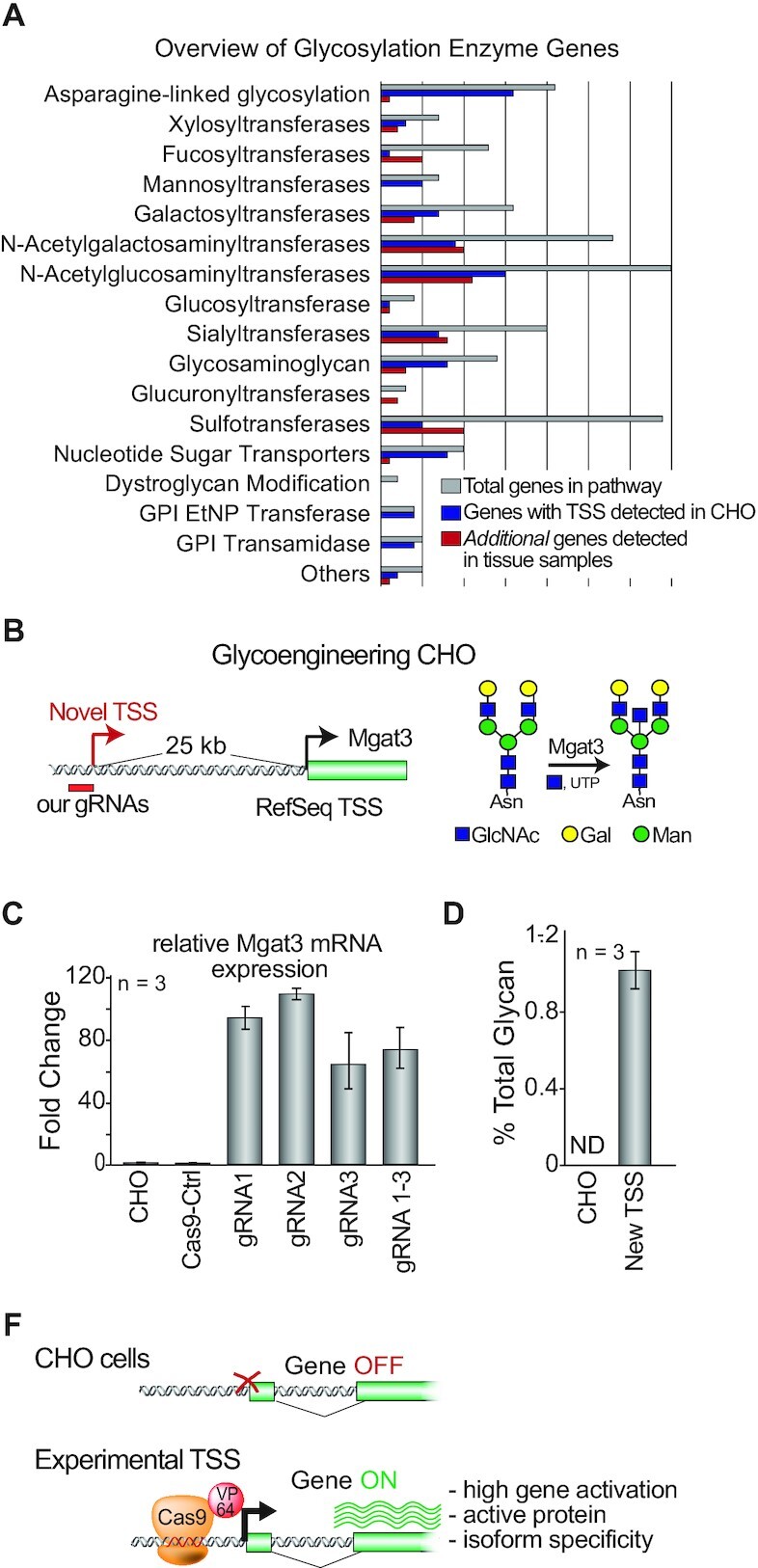
Experimentally measured TSSs facilitates genome engineering to humanize glycosylation. (A) List of human glycosylation enzyme classes detected in our samples as defined by 5′GRO-seq/csRNA-seq in the Chinese hamster. The number of genes expressed in CHO cells (blue) and additional genes for which experimental TSSs were discovered in our tissue samples (red) are shown. (B) Overview of the RefSeq TSS targeted by guide RNAs with CRISPRa to induce Mgat3 expression in CHO cells. The Mgat3-encoded glycosyltransferase catalyzes the addition of bisecting N‐acetylglucosamines on glycoproteins, but is silenced in CHO cells. (C) Quantitative RT-PCR measurement of Mgat3 expression in CHO cells and upon activation by the three designed gRNAs using our new TSSs. As a control, the cells were transfected with NT-gRNA (gRNA-Ctrl) or NT-gRNA and VPR-dCas9 (Cas9-Ctrl). (E) Comparison of the levels of bisecting N‐acetylglucosamines in secretome following CRISPRa. As a control, the cells were transfected with NT-gRNA (gRNA-Ctrl). (E) Overview: Experimental TSS facilitates efficient engineering of Mgat3 in an upstream promoter.
TSS detected in upstream promoter facilitates CRISPR activation of the dormant gene Mgat3 in CHO
To test the feasibility of genome engineering based on our revised annotations we aimed to activate Mgat3 (Figure 4B), which is naturally dormant in CHO cells (80) using a novel identified alternative promoter stable TSS that is 25,481 bp upstream of the promoter previously used for Mgat3 activation (6). Mgat3 is required for bisecting N‐acetylglucosamines which play an important role in regulating complex glycosylation maturation and impact antibody effector function (81,82) and is hence well studied in both humans and CHO cells.
CRISPR/Cas9 enables rapid and cost‐effective genome editing, gene inhibition (CRISPRi), and activation (CRISPRa) without altering the native DNA sequence (19,20,22). However, the success of these and similar precise genome engineering approaches depends on accurate gene annotations (18,83). Given that Mgat3 has previously been targeted by CRISPRa (6), we used this experimental system to show that the gene can be activated by targeting an experimentally identified promoter, even when located >25 kb away from the RefSeq gene TSS. To activate Mgat3 we designed three CRISPR guide RNAs (gRNAs) complementary to the DNA sequence near our alternative TSS (Figure 4C). CRISPRa resulted in a mean of 94-,109-, and 64-fold upregulation of Mgat3 using the three different gRNAs individually (n = 3 samples each), and 73-fold for a mixture of the 3, as measured by qRT-PCR (Figure 4D). To test if the activation of Mgat3 transcripts impacts glycan synthesis, we measured the relative abundance of glycans on the secretome. This analysis revealed that while undetectable in control cells, 1.08% of glycans were bisecting N‐acetylglucosamines after Mgat3 activation (Figure 4E). Together, these data show the use of our revised annotation for genome engineering. With our newly reported TSSs for 15 308 genes across >30 000 detected promoters, we anticipate further usage of these TSSs for cell line engineering.
DISCUSSION
In this study we measured and analyzed the coding and non-coding RNA in the Chinese hamster genome using steady state and nascent RNA sequencing experiments for diverse hamster tissues and cell lines. Through this we were able to comprehensively map TSSs for >70% of annotated Chinese hamster genes and non-coding RNAs, including many genes normally silenced in CHO cells. Importantly, these experimentally determined TSS enabled us to realign current RefSeq TSSs, which were predominantly computationally predicted and often inaccurate. Unlike the previous RefSeq TSS, our revised TSSs annotations display expected DNA nucleotide frequency features such as the Initiator motif or the TATA box in the core promoter. Furthermore, we demonstrated that accurate TSSs and knowledge of alternative promoters can be used to activate a silenced gene of interest using CRISPRa. Through this we present a resource to guide genome editing and genomic analysis of CHO cells.
Here, we captured 30 760 nascent protein-coding TSSs corresponding to 15 308 genes, along with 3560 ncRNAs (lncRNA, miRNA, snRNA, snoRNA and tRNA), and 176 914 distal peaks (enhancer RNAs etc.). This resource provides rich information for precise cell engineering. Furthermore, including diverse hamster tissues helps in efforts to fine tune existing CHO gene regulatory programs, as well as activate genes or pathways naturally encoded in the Chinese hamster genome but dormant in CHO cells. Our TSSs are a prerequisite for the design and testing of gRNAs and eventually, an effective gRNA library for the activation of diverse Chinese hamster genes by CRISPRa (Figure 4F). It can also complement existing data on epigenetic markers of CHO cells in efforts to find endogenous promoters that avoid silencing seen with common viral promoters or harness endogenous regulatory circuits involved in ER stress or cold shock (84).
Our transcriptomic datasets also provide a comprehensive resource for future research and discovery. In addition to our gene-centric atlas of Chinese hamster TSSs reported here, our data cover a plethora of transcriptomic features that remain to be explored including miRNAs, pri-miRNAs and well over a 100 k putative distal regulatory elements that are commonly referred to as enhancers (Supplementary Data S2). Although beyond the focus of this manuscript, this extensive, transcript stability-independent resource of TSSs could also aid to improve our understanding of how gene expression is regulated in hamsters and how tissue-specific regulatory programs emerged. While a key advantage of CRISPRa is the ability to activate desired genes independent of tissue-specific transcription factors, future engineering efforts may be more tailored towards adjusting transcriptional programs, rather than one or a few specific genes. For example, our definition of transcription factors that were highly enriched in the promoters of tissue-specific genes provides a first step to advance our understanding of which and why specific genes or pathways are silent in CHO cells. Improved knowledge of how gene regulatory networks function in hamsters may ultimately allow to predict how activation of one gene impacts the hamster regulome and to eventually fine-tune desired regulatory programs, rather than individual genes (85). Going beyond capturing TSSs, our data also contain maps of open chromatin, as defined by ATAC-seq, nascent transcription, as defined by GRO-seq, and mature RNAs, as defined by ribosomal RNA-depleted RNA-seq for CHO cells, hamster macrophages and diverse hamster tissues that were primarily used in this study as a critical input for the identification of high-confidence TSSs. Our data thus also provide a rich resource for future studies and enable the integration of the Chinese hamster into comparative or evolutionary studies, for example, as an outgroup to mice (31).
In summary, our data have enabled the development of a compendium of experimentally defined TSSs and transcriptomic features from multiple tissues and cell types from the same hamster colony from which CHO cells were generated. Our revised annotation shows considerable improvement over the current RefSeq by several measures including agreement with published RNA-seq datasets, TSS information content as well as core promoter motifs. More broadly, these findings emphasize the importance of refined TSS mapping methods such as 5′GRO-seq/GROcap or csRNA-seq for accurate annotation of a gene’s 5′ end. The TSS is a landmark in gene regulation and its accuracy becomes imperative in an era of genetic engineering. We further envision that our data and annotation will provide a rich resource for the CHO community and beyond as the Chinese hamster is further included in comparative and evolutionary studies. At its core, the improved TSSs map will aid CHO gene engineering efforts aiming to improve the quality and quantity of desired recombinant proteins and ultimately reduce drug manufacturing costs.
DATA AVAILABILITY
All sequencing data are submitted to the Gene Expression Omnibus (GEO) with GEO ID GSE159044. The Supplementary Data provided is also uploaded to Synapse (synapse.org), with ID syn22969187. This includes our revised protein-coding promoter TSS annotation, in which each of TSS has an associated RefSeq transcript and gene association. This is done for both NCBI RefSeq (Supplementary Data S2) and with RefSeq in conjunction with the proteogenomics annotation reported in (42) (Supplementary Data S3). Open-chromatin regions merged across samples are provided on synapse as a bed file as well.
In addition, we release all our TSSs detected (Supplementary Data S1), which include distal TSSs (putative enhancer regions, divergent transcripts), as well as non-coding RNA promoter TSSs and protein-coding TSSs, along with the CPM from each tissue per TSS and the respective TSS locations of the tissue if it expressed that TSS. This will allow researchers studying regulatory elements to have easy access to a comprehensive TSS dataset.
Supplementary Material
ACKNOWLEDGEMENTS
We like to thank Marten A. Hoeksema for culturing BMDMs.
Dedication: The authors dedicate this work to Dr. George Yerganian (1924–2019), who provided the hamsters for this study, and for the original CHO cells in 1957.
Contributor Information
Isaac Shamie, Novo Nordisk Foundation Center for Biosustainability at UC San Diego 92093, USA; Bioinformatics and Systems Biology Program, University of California, San Diego 92093, USA.
Sascha H Duttke, Department of Medicine, University of California, San Diego 92093, USA.
Karen J la Cour Karottki, Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800, Denmark; Department of Pediatrics, University of California, San Diego 92093, USA.
Claudia Z Han, Department of Cellular and Molecular Medicine, University of California, San Diego 92093, USA.
Anders H Hansen, Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800, Denmark; National Biologics Facility, Technical University of Denmark, 2800, Denmark.
Hooman Hefzi, Novo Nordisk Foundation Center for Biosustainability at UC San Diego 92093, USA; Department of Pediatrics, University of California, San Diego 92093, USA; Department of Bioengineering, University of California, San Diego 92093, USA.
Kai Xiong, Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800, Denmark.
Shangzhong Li, Novo Nordisk Foundation Center for Biosustainability at UC San Diego 92093, USA; Department of Bioengineering, University of California, San Diego 92093, USA.
Samuel J Roth, Department of Medicine, University of California, San Diego 92093, USA; Bioinformatics and Systems Biology Program, University of California, San Diego 92093, USA.
Jenhan Tao, Department of Cellular and Molecular Medicine, University of California, San Diego 92093, USA; Bioinformatics and Systems Biology Program, University of California, San Diego 92093, USA.
Gyun Min Lee, Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800, Denmark; Department of Biological Sciences, KAIST, 34141, South Korea.
Christopher K Glass, Department of Cellular and Molecular Medicine, University of California, San Diego 92093, USA.
Helene Faustrup Kildegaard, Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800, Denmark.
Christopher Benner, Department of Medicine, University of California, San Diego 92093, USA.
Nathan E Lewis, Novo Nordisk Foundation Center for Biosustainability at UC San Diego 92093, USA; Department of Pediatrics, University of California, San Diego 92093, USA; Department of Bioengineering, University of California, San Diego 92093, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Institutes of Health/National Institute of General Medical Sciences [K99GM135515 to S.H.D]; National Institutes of Health [AI135972, GM134366 to C.B.]; Novo Nordisk Foundation [NNF10CC1016517, NNF20SA0066621 to N.E.L., A.H.H.; NNF16OC0021638 to H.F.K.]; Cancer Research Institute Irvington Postdoctoral Fellowship Program (to C.Z.H.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Walsh G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018; 36:1136–1145. [DOI] [PubMed] [Google Scholar]
- 2. Golabgir A., Gutierrez J.M., Hefzi H., Li S., Palsson B.O., Herwig C., Lewis N.E.. Quantitative feature extraction from the Chinese hamster ovary bioprocess bibliome using a novel meta-analysis workflow. Biotechnol. Adv. 2016; 34:621–633. [DOI] [PubMed] [Google Scholar]
- 3. Jenkins N., Parekh R.B., James D.C.. Getting the glycosylation right: implications for the biotechnology industry. Nat. Biotechnol. 1996; 14:975–981. [DOI] [PubMed] [Google Scholar]
- 4. Urquhart L. Top companies and drugs by sales in 2020. Nat. Rev. Drug Discov. 2021; 20:253. [DOI] [PubMed] [Google Scholar]
- 5. Popp O., Moser S., Zielonka J., Rüger P., Hansen S., Plöttner O.. Development of a pre-glycoengineered CHO-K1 host cell line for the expression of antibodies with enhanced Fc mediated effector function. MAbs. 2018; 10:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karottki K.J. la C., Hefzi H., Xiong K., Shamie I., Hansen A.H., Li S., Pedersen L.E., Li S., Lee J.S., Lee G.M.et al.. Awakening dormant glycosyltransferases in CHO cells with CRISPRa. Biotechnol. Bioeng. 2020; 117:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu X., Nagarajan H., Lewis N.E., Pan S., Cai Z., Liu X., Chen W., Xie M., Wang W., Hammond S.et al.. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011; 29:735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brinkrolf K., Rupp O., Laux H., Kollin F., Ernst W., Linke B., Kofler R., Romand S., Hesse F., Budach W.E.et al.. Chinese hamster genome sequenced from sorted chromosomes. Nat. Biotechnol. 2013; 31:694–695. [DOI] [PubMed] [Google Scholar]
- 9. Rupp O., MacDonald M.L., Li S., Dhiman H., Polson S., Griep S., Heffner K., Hernandez I., Brinkrolf K., Jadhav V.et al.. A reference genome of the Chinese hamster based on a hybrid assembly strategy. Biotechnol. Bioeng. 2018; 115:2087–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ritter A., Rauschert T., Oertli M., Piehlmaier D., Mantas P., Kuntzelmann G., Lageyre N., Brannetti B., Voedisch B., Geisse S.et al.. Disruption of the gene C12orf35 leads to increased productivities in recombinant CHO cell lines. Biotechnol. Bioeng. 2016; 113:2433–2442. [DOI] [PubMed] [Google Scholar]
- 11. Laux H., Romand S., Nuciforo S., Farady C.J., Tapparel J., Buechmann-Moeller S., Sommer B., Oakeley E.J., Bodendorf U.. Degradation of recombinant proteins by Chinese hamster ovary host cell proteases is prevented by matriptase-1 knockout. Biotechnol. Bioeng. 2018; 115:2530–2540. [DOI] [PubMed] [Google Scholar]
- 12. Tang D., Subramanian J., Haley B., Baker J., Luo L., Hsu W., Liu P., Sandoval W., Laird M.W., Snedecor B.et al.. Pyruvate Kinase Muscle-1 Expression Appears to Drive Lactogenic Behavior in CHO Cell Lines, Triggering Lower Viability and Productivity: A Case Study. Biotechnol. J. 2019; 14:1800332. [DOI] [PubMed] [Google Scholar]
- 13. Kim C.L., Lee G.M.. Improving recombinant bone morphogenetic protein-4 (BMP-4) production by autoregulatory feedback loop removal using BMP receptor-knockout CHO cell lines. Metab. Eng. 2019; 52:57–67. [DOI] [PubMed] [Google Scholar]
- 14. Kol S., Ley D., Wulff T., Decker M., Arnsdorf J., Schoffelen S., Hansen A.H., Jensen T.L., Gutierrez J.M., Chiang A.W.T.et al.. Multiplex secretome engineering enhances recombinant protein production and purity. Nat. Commun. 2020; 11:1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiong K., Marquart K.F., la Cour Karottki K.J., Li S., Shamie I., Lee J.S., Gerling S., Yeo N.C, Chavez A., Lee G.M.et al.. Reduced apoptosis in Chinese hamster ovary cells via optimized CRISPR interference. Biotechnol. Bioeng. 2019; 116:1813–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. la Cour Karottki K.J., Hefzi H., Li S., Pedersen L.E., Spahn P., Ruckerbauer D., Bort J.H., Thomas A., Lee J.S.et al.. A metabolic CRISPR-Cas9 screen in Chinese hamster ovary cells identifies glutamine-sensitive genes. Metab. Eng. 2019; 66:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christian M., Cermak T., Doyle E.L., Schmidt C., Zhang F., Hummel A., Bogdanove A.J., Voytas D.F.. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010; 186:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gallego-Bartolomé J., Liu W., Kuo P.H., Feng S., Ghoshal B., Gardiner J., Zhao J.M.-C., Park S.Y., Chory J., Jacobsen S.E.. Co-targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell. 2019; 176:1068–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doudna J.A., Charpentier E.. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 20. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A.et al.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hefzi H., Ang K.S., Hanscho M., Bordbar A., Ruckerbauer D., Lakshmanan M., Orellana C.A., Baycin-Hizal D., Huang Y., Ley D.et al.. A consensus genome-scale reconstruction of chinese hamster ovary cell metabolism. Cell Syst. 2016; 3:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chavez A., Tuttle M., Pruitt B.W., Ewen-Campen B., Chari R., Ter-Ovanesyan D., Haque S.J., Cecchi R.J., Kowal E.J.K., Buchthal J.et al.. Comparison of Cas9 activators in multiple species. Nat. Methods. 2016; 13:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horlbeck M.A., Gilbert L.A., Villalta J.E., Adamson B., Pak R.A., Chen Y., Fields A.P., Park C.Y., Corn J.E., Kampmann M.et al.. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. 2016; 5:e19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jakobi T., Brinkrolf K., Tauch A., Noll T., Stoye J., Pühler A., Goesmann A.. Discovery of transcription start sites in the Chinese hamster genome by next-generation RNA sequencing. J. Biotechnol. 2014; 190:64–75. [DOI] [PubMed] [Google Scholar]
- 25. Lam M.T.Y., Cho H., Lesch H.P., Gosselin D., Heinz S., Tanaka-Oishi Y., Benner C., Kaikkonen M.U., Kim A.S., Kosaka M.et al.. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013; 498:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duttke S.H., Chang M.W., Heinz S., Benner C.. Identification and dynamic quantification of regulatory elements using total RNA. Genome Res. 2019; 29:1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Puck T.T., Cieciura S.J., Robinson A. Genetics of somatic mammalian cells. J. Exp. Med. 1958; 108:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Durocher Y., Butler M.. Expression systems for therapeutic glycoprotein production. Curr. Opin. Biotechnol. 2009; 20:700–707. [DOI] [PubMed] [Google Scholar]
- 31. Link V.M., Duttke S.H., Chun H.B., Holtman I.R., Westin E., Hoeksema M.A., Abe Y., Skola D., Romanoski C.E., Tao J.et al.. Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell. 2018; 173:1796–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hetzel J., Duttke S.H., Benner C., Chory J.. Nascent RNA sequencing reveals distinct features in plant transcription. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12316–12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duttke S.H.C., Lacadie S.A., Ibrahim M.M., Glass C.K., Corcoran D.L., Benner C., Heinz S., Kadonaga J.T., Ohler U.. Human promoters are intrinsically directional. Mol. Cell. 2015; 57:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 35. Karottki K.J. la C., Hefzi H., Xiong K., Shamie I., Hansen A.H., Li S., Pedersen L.E., Li S., Lee J.S.et al.. Awakening dormant glycosyltransferases in CHO cells with CRISPRa. Biotechnol. Bioeng. 2020; 117:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10. [Google Scholar]
- 37. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Q., Brown J.B., Huang H., Bickel P.J.. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011; 5:1752–1779. [Google Scholar]
- 41. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li S., Cha S.W., Heffner K., Hizal D.B., Bowen M.A., Chaerkady R., Cole R.N., Tejwani V., Kaushik P., Henry M.et al.. Proteogenomic annotation of chinese hamsters reveals extensive novel translation events and endogenous retroviral elements. J. Proteome Res. 2019; 18:2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bailey T.L., Johnson J., Grant C.E., Noble W.S.. The MEME Suite. Nucleic Acids Res. 2015; 43:W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Togayachi A., Dae K.-Y., Shikanai T., Narimatsu H.. Taniguchi N., Suzuki A., Ito Y., Narimatsu H., Kawasaki T., Hase S.. A Database System for Glycogenes (GGDB). Experimental Glycoscience: Glycobiology. 2008; Tokyo: Springer Japan; 423–425. [Google Scholar]
- 46. Young R.S., Hayashizaki Y., Andersson R., Sandelin A., Kawaji H., Itoh M., Lassmann T., Carninci P.FANTOM Consortium FANTOM Consortium Bickmore W.A.et al.. The frequent evolutionary birth and death of functional promoters in mouse and human. Genome Res. 2015; 25:1546–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wurm F. CHO Quasispecies—implications for manufacturing processes. Processes. 2013; 1:296–311. [Google Scholar]
- 48. Field A., Adelman K.. Evaluating enhancer function and transcription. Annu. Rev. Biochem. 2020; 89:213–234. [DOI] [PubMed] [Google Scholar]
- 49. Halfon M.S. Studying transcriptional enhancers: the founder fallacy, validation creep, and other biases. Trends Genet. 2019; 35:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Affymetrix ENCODE Transcriptome Project and Cold Spring Harbor Laboratory ENCODE Transcriptome Project Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature. 2009; 457:1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seila A.C., Calabrese J.M., Levine S.S., Yeo G.W., Rahl P.B., Flynn R.A., Young R.A., Sharp P.A.. Divergent transcription from active promoters. Science. 2008; 322:1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smale S.T., Kadonaga J.T.. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003; 72:449–479. [DOI] [PubMed] [Google Scholar]
- 53. Danino Y.M., Even D., Ideses D., Juven-Gershon T.. The core promoter: at the heart of gene expression. Biochim. Biophys. Acta. 2015; 1849:1116–1131. [DOI] [PubMed] [Google Scholar]
- 54. Haberle V., Stark A.. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018; 19:621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grosveld G.C., Shewmaker C.K., Jat P., Flavell R.A.. Localization of DNA sequences necessary for transcription of the rabbit β-globin gene in vitro. Cell. 1981; 25:215–226. [DOI] [PubMed] [Google Scholar]
- 56. Vo Ngoc L., Cassidy C.J., Huang C.Y., Duttke S.H.C., Kadonaga J.T.. The human initiator is a distinct and abundant element that is precisely positioned in focused core promoters. Genes Dev. 2017; 31:6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smale S.T., Baltimore d. The ‘initiator’ as a transcription control element. Cell. 1989; 57:103–113. [DOI] [PubMed] [Google Scholar]
- 58. Feichtinger J., Hernández I., Fischer C., Hanscho M., Auer N., Hackl M., Jadhav V., Baumann M., Krempl P.M., Schmidl C.et al.. Comprehensive genome and epigenome characterization of CHO cells in response to evolutionary pressures and over time. Biotechnol. Bioeng. 2016; 113:2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Wijk X.M., Döhrmann S., Hallström B.M., Li S., Voldborg B.G., Meng B.X., McKee K.K., van Kuppevelt T.H., Yurchenco P.D., Palsson B.O.et al.. Whole-Genome Sequencing of Invasion-Resistant Cells Identifies Laminin α2 as a Host Factor for Bacterial Invasion. mBio. 2017; 8:e02128-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chiang A.W.T., Li S., Kellman B.P., Chattopadhyay G., Zhang Y., Kuo C.-C., Gutierrez J.M., Ghazi F., Schmeisser H., Ménard P.et al.. Combating viral contaminants in CHO cells by engineering innate immunity. Sci. Rep. 2019; 9:8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Singh A., Kildegaard H.F., Andersen M.R.. An online compendium of CHO RNA-Seq data allows identification of CHO cell line-specific transcriptomic signatures. Biotechnol. J. 2018; 13:e1800070. [DOI] [PubMed] [Google Scholar]
- 62. Yu N.Y.-L., Hallström B.M., Fagerberg L., Ponten F., Kawaji H., Carninci P., Forrest A.R.R.The FANTOM Consortium The FANTOM Consortium Hayashizaki Y., Uhlén M.et al.. Complementing tissue characterization by integrating transcriptome profiling from the Human Protein Atlas and from the FANTOM5 consortium. Nucleic Acids Res. 2015; 43:6787–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Danielsson A., Pontén F., Fagerberg L., Hallström B.M., Schwenk J.M., Uhlén M., Korsgren O., Lindskog C.. The human pancreas proteome defined by transcriptomics and antibody-based profiling. PLoS One. 2014; 9:e115421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hawrylycz M.J., Lein E.S., Guillozet-Bongaarts A.L., Shen E.H., Ng L., Miller J.A., van de Lagemaat L.N., Smith K.A., Ebbert A., Riley Z.L.et al.. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012; 489:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cardoso-Moreira M., Halbert J., Valloton D., Velten B., Chen C., Shao Y., Liechti A., Ascenção K., Rummel C., Ovchinnikova S.et al.. Gene expression across mammalian organ development. Nature. 2019; 571:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dougherty J.D., Schmidt E.F., Nakajima M., Heintz N.. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 2010; 38:4218–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sugiaman-Trapman D., Vitezic M., Jouhilahti E.-M., Mathelier A., Lauter G., Misra S., Daub C.O., Kere J., Swoboda P.. Characterization of the human RFX transcription factor family by regulatory and target gene analysis. BMC Genomics. 2018; 19:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rey-Campos J., Chouard T., Yaniv M., Cereghini S.. vHNF1 is a homeoprotein that activates transcription and forms heterodimers with HNF1. EMBO J. 1991; 10:1445–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tremblay M., Sanchez-Ferras O., Bouchard M.. GATA transcription factors in development and disease. Development. 2018; 145:dev164384. [DOI] [PubMed] [Google Scholar]
- 70. Berger J., Moller D.E.. The mechanisms of action of PPARs. Annu. Rev. Med. 2002; 53:409–435. [DOI] [PubMed] [Google Scholar]
- 71. Lin Q., Schwarz J., Bucana C., Olson E.N.. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997; 276:1404–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Black B.L., Olson E.N.. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 1998; 14:167–196. [DOI] [PubMed] [Google Scholar]
- 73. Sanetra M., Begemann G., Becker M.-B., Meyer A.. Conservation and co-option in developmental programmes: the importance of homology relationships. Front. Zool. 2005; 2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gregory T.R., Ryan Gregory T.. The evolution of complex organs. Evol. Educ. Outreach. 2008; 1:358–389. [Google Scholar]
- 75. Kadonaga J.T., Courey A.J., Ladika J., Tjian R.. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science. 1988; 242:1566–1570. [DOI] [PubMed] [Google Scholar]
- 76. Raetz C.R.H., Garrett T.A., Reynolds C.M., Shaw W.A., Moore J.D., Smith D.C. Jr, Ribeiro A.A., Murphy R.C., Ulevitch R.J., Fearns C.et al.. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 2006; 47:1097–1111. [DOI] [PubMed] [Google Scholar]
- 77. Spain M.M., Caruso J.A., Swaminathan A., Pile L.A.. Drosophila SIN3 isoforms interact with distinct proteins and have unique biological functions. J. Biol. Chem. 2010; 285:27457–27467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reyes A., Huber W.. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018; 46:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Solá R.J., Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs. 2017; 24:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stanley P., Sundaram S., Tang J., Shi S.. Molecular analysis of three gain-of-function CHO mutants that add the bisecting GlcNAc to N-glycans. Glycobiology. 2005; 15:43–53. [DOI] [PubMed] [Google Scholar]
- 81. Umaña P., Jean-Mairet J., Moudry R., Amstutz H., Bailey J.E.. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999; 17:176–180. [DOI] [PubMed] [Google Scholar]
- 82. Schachter H. Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochem. Cell Biol. 1986; 64:163–181. [DOI] [PubMed] [Google Scholar]
- 83. Marx N., Grünwald-Gruber C., Bydlinski N., Dhiman H., Nguyen L.N., Klanert G., Borth N.. CRISPR-based targeted epigenetic editing enables gene expression modulation of the silenced beta-galactoside alpha-2,6-sialyltransferase 1 in CHO cells. Biotechnol. J. 2018; 13:1700217. [DOI] [PubMed] [Google Scholar]
- 84. Nguyen L.N., Baumann M., Dhiman H., Marx N., Schmieder V., Hussein M., Eisenhut P., Hernandez I., Koehn J., Borth N.. Novel promoters derived from chinese hamster ovary cells via in silico and in vitro analysis. Biotechnol. J. 2019; 14:e1900125. [DOI] [PubMed] [Google Scholar]
- 85. Edros R., McDonnell S., Al-Rubeai M.. The relationship between mTOR signalling pathway and recombinant antibody productivity in CHO cell lines. BMC Biotech. 2014; 14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data are submitted to the Gene Expression Omnibus (GEO) with GEO ID GSE159044. The Supplementary Data provided is also uploaded to Synapse (synapse.org), with ID syn22969187. This includes our revised protein-coding promoter TSS annotation, in which each of TSS has an associated RefSeq transcript and gene association. This is done for both NCBI RefSeq (Supplementary Data S2) and with RefSeq in conjunction with the proteogenomics annotation reported in (42) (Supplementary Data S3). Open-chromatin regions merged across samples are provided on synapse as a bed file as well.
In addition, we release all our TSSs detected (Supplementary Data S1), which include distal TSSs (putative enhancer regions, divergent transcripts), as well as non-coding RNA promoter TSSs and protein-coding TSSs, along with the CPM from each tissue per TSS and the respective TSS locations of the tissue if it expressed that TSS. This will allow researchers studying regulatory elements to have easy access to a comprehensive TSS dataset.