

Abstract
The Gq-coupled P2Y6 receptor (P2Y6R) is a component of the purinergic signaling system and functions in inflammatory, cardiovascular and metabolic processes. UDP, the native P2Y6R agonist and P2Y14R partial agonist, is subject to hydrolysis by ectonucleotidases. Therefore, we have synthesized UDP/CDP analogues containing a stabilizing α,β-methylene bridge as P2Y6R agonists and identified compatible affinity-enhancing pyrimidine modifications. A distal binding region on the receptor was explored with 4-benzyloxyimino cytidine 5′-diphosphate analogues and their potency determined in a calcium mobilization assay. A 4-trifluoromethyl-benzyloxyimino substituent in 25 provided the highest human P2Y6R potency (MRS4554, 0.57 μM), and a 5-fluoro substitution of the cytosine ring in 28 similarly enhanced potency, with >175- and 39-fold selectivity over human P2Y14R, respectively. However, 3-alkyl (31 – 33, 37, 38), β-D-arabinofuranose (39) and 6-aza (40) substitution prevented P2Y6R activation. Thus, we have identified new α,β-methylene bridged N4-extended CDP analogues as P2Y6R agonists that are highly selective over the P2Y14R.
Keywords: P2Y6 receptor, G protein-coupled receptor, structure-activity relationship, pyrimidine nucleotides, calcium mobilization.
Graphical Abstract
Among the eight metabotropic P2Y receptors (P2YRs) are four subtypes that respond to uracil mono- and dinucleotides.1 The human (h) Gq protein-coupled P2Y2 receptor (P2Y2R) and P2Y4R are potently activated by UTP, but UDP is ~900- and ~160-fold weaker, respectively.2 However, UDP acts as an agonist and partial agonist at the P2Y6R (Gq-coupled) and P2Y14R (Gi-coupled), respectively.1 The P2Y14R is potently activated by endogenous UDP-sugars, principally UDP-glucose,1 and the P2Y6R is also activated by certain synthetic nucleoside 5′-triphosphate and dinucleoside triphosphate analogues.2,3 UTP is inactive at the hP2Y6R (EC50 >10 μM) and P2Y14R, while UMP does not activate any hP2YR.1-6 Thus, it is logical to compare and contrast the P2Y6R and P2Y14R due to their common endogenous agonist UDP, even though they are linked to different second messenger pathways.7–9
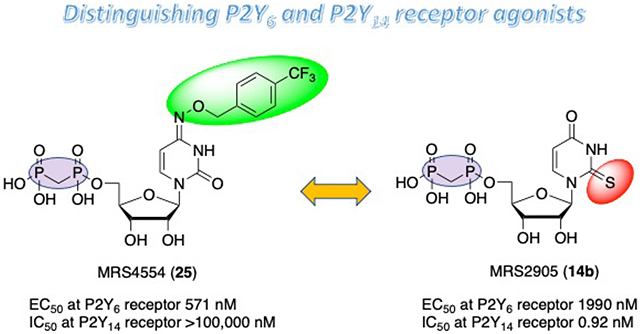
We focus here on the structure-activity relationship (SAR) of pyrimidine mononucleotides as P2Y6R agonists (e.g., 1 – 10, Chart 1) and demonstrate the ability to distinguish the P2Y6R and P2Y14R subtypes with analogues of UDP 1 or CDP that contain a 5′-α,β-methylene diphosphonate. Among known P2Y6R selective UDP analogues, 5-iodo-UDP 2, 5-methoxy-UDP 3, 3-phenylacetyl-UDP 5 and MRS4383 6 are 5′-diphosphates that tend to be hydrolyzed in biological tissue by ectonucleotidases.2,3,10 These enzymes include nucleoside triphosphate diphosphohydrolase-1 (NTPDase1, CD39), 5′-nucleotidase (CD73, hydrolyzes 5′-monophosphates) and other ectonucleotidases.10,11 A 5′-α,β-methylene diphosphonate group in nucleotide analogues, e.g. 9 and 10, lends greater, but not absolute, stability toward enzymatic hydrolysis compared to the native 5′-diphosphate and is compatible with P2Y6R activation (Table 1) 3-5,10,12 We have also reported 5′-α,β-methylene diphosphonates that potently activate the P2Y14R, such as MRS2905 14b (2-thiouridine-5′-O-(α,β-methylene)diphosphate, the structure shown in Table 2 footnote).9 In addition to phosphonate bridges, another means of increasing the resistance to hydrolytic degradation is to include an α-borano-diphosphate moiety as in potent P2Y6R agonist 4.13 Dinucleotide P2Y6R agonists are also more stable toward hydrolysis by CD39 and CD73 in tissues than 5′-diphosphate agonists such as UDP.10 However, dinucleotides are cleaved by ectonucleotide pyrophosphatase/phosphodiesterases (ENPPs).10
Chart 1.
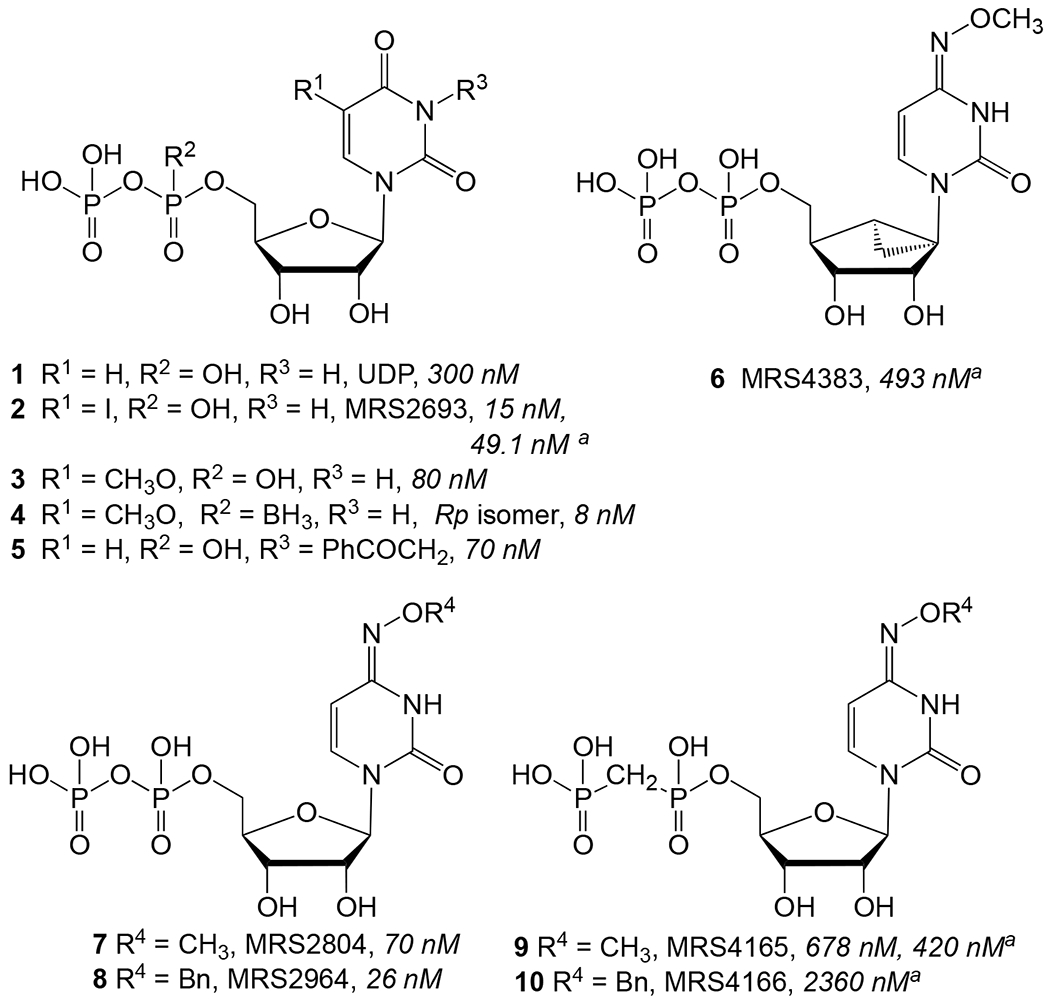
Structures of known P2Y6R agonists that are UDP analogues.3,4 The reported potency in activation of PLC (EC50), is shown in italics after each compound, unless noted (“a” indicates EC50 in nM in the calcium mobilization assay).
Table 1.
Potencies of various uridine 5’-diphosphate (UDP) and α,β-methylene-UDP analogues in comparison to known UDP derivatives for activation of the human P2Y6R, as determined in an assay of calcium mobilization, unless noted.a
 | ||||
|---|---|---|---|---|
| Compound | x = | Y = | Z = | P2Y6R, EC50, μM (Ca2+, unless noted) |
| 5′-Diphosphates | ||||
| 1 UDP | H | H | O | 0.0416±0.007 |
| 2 | I | H | O | 0.0491±0.0195, 0.015±0.002b |
| 11 | H | H | NOCH3 | 0.493±0.128 |
| 12 | H | H | NNHCOOCH3 | 1.54f |
| 13 | H | H | NOCH2C6H5 | 0.793±0.149 |
| 5′-α,β-Methylene diphosphonates | ||||
| 14ac | H | H | O | 0.70±0.11b |
| 15c | I | H | O | 0.0491±0.0195,c 0.13±0.02b |
| 16c | F | H | O | 0.203 ± 0.030c |
| 17c | Cl | H | O | 0.097 ± 0.011 |
| 18c | Br | H | O | 0.104 ± 0.010 |
| 9 | H | H | NOCH3 | 0.42±0.08d |
| 10 | H | H | NOCH2C6H5 | 2.36±0.73d |
| 19 | H | H | NOCH2C6H5-3-F | 2.70 ± 0.78 |
| 20 | H | H | NOCH2C6H5-3-Cl | 1.45 ± 0.22 |
| 21 | H | H | NOCH2C6H5-3-CF3 | 1.91 ± 0.47 |
| 22 | H | H | NOCH2C6H5-3-CO2CH3 | 0.902 ± 0.05 |
| 23 | H | H | NOCH2C6H5-4-Cl | 1.32 ± 0.08 |
| 24 | H | H | NOCH2C6H5-4-CH3 | 5.12 ± 0.28 |
| 25c | H | H | NOCH2C6H5-4-CF3 | 0.571 ± 0.196 |
| 26 | H | H | NOCH2C6H5-4-CO2CH3 | 1.67 ± 0.34 |
| 27 | H | H | NOCH2C6H5-4-SF5 | 1.59 ± 0.09 |
| 28c | F | H | NOCH2C6H5 | 0.549 ± 0.070 |
| 29c | CH3 | H | NOCH2C6H5 | 1.09 ± 0.32 |
| 30c | H | H | N4-CO-Ph | 1.39 ± 0.223c |
| Inactive (% activation at 3 μM of <10%) | ||||
| 31c | H | CH2CH3 | O | 3.5% |
| 32c | H | (CH2)2CH3 | O | 2.7% |
| 33c | H | CH2C6H5 | O | 5.3% |
| 34c | F | - | - | 5.7% |
| 35c | I | - | - | 3.4% |
| 36c | CH3 | - | - | 3.0% |
| 37c | H | CH3 | NOCH2C6H5 | 3.6%c |
| 38c | H | CH2CH3 | NOCH2C6H5 | 5.7% |
| Other inactives (% activation at 3 μM of <10%) | ||||
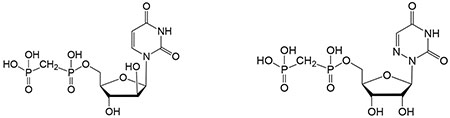
| 39c | Structure shown in footnotee | 2.4% | ||
| 40c | Structure shown in footnotee | 5.2% | ||
Agonist potencies reflect P2Y6R-dependent calcium mobilization in 1321N1 human astrocytoma cells stably expressing the human P2Y6R. Potencies are presented in the form of EC50 values, which represent the concentration of agonist at which 50% of the maximal effect on calcium mobilization is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments, unless noted, with each molecule.
EC50 values from Maruoka et al. (PLC assay).4
All syntheses for indicated compounds were reported in Junker et al., as well as the P2Y6R Ca2+ assay results for compound 30.33
Reported in Toti et al. (Ca2+ assay).3
Structures of 39: 40:
n = 1 (tested once).
Table 2.
Potency of α,β-methylene analogues of UDP and CDP at the human P2Y6R and P2Y14R.a
| Compound | hP2Y6R EC50 ± SEM (μM) or % |
hP2Y14R IC50 ± SEM (μM) |
|---|---|---|
| 14a | 0.339b | 0.011b |
| 14bc | 1.99b | 0.00092b |
| 16 | 0.203 ± 0.030 | 0.362 ± 0.090 |
| 17 | 0.097 ± 0.011 | 16.6 ± 10.3 |
| 20 | 1.45 ± 0.22 | >100 |
| 23 | 1.32 ± 0.08 | >100 |
| 25 | 0.571 ± 0.196 | >100 |
| 28 | 0.549 ± 0.070 | 21.5 ± 11.1 |
| 30 | 1.39 ± 0.223 | 6.66 ± 4.74 |
FLIPR (Ca2+) assay in hP2Y6R-1231N1 astrocytoma cells, expressed as EC50 or % activation at 3 µM, unless noted. Fluorescent antagonist was used as a tracer for flow cytometry of hP2Y14R-CHO cells, unless noted.
Potency in a phospholipase C assay of hP2Y6R activation (Maruoka et al., 2010)4 or in a cAMP assay of hP2Y14R activation (Das et al., 2010).9
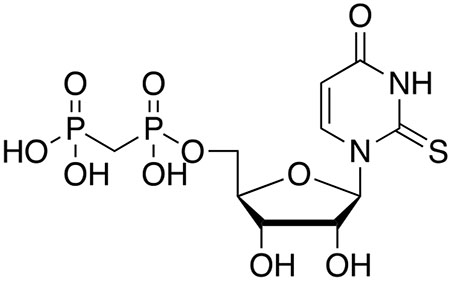
Structure of 14b:
P2Y6R antagonists have broader envisioned therapeutic use than P2Y6R agonists, for treating cancer, inflammation, neurodegeneration, and diabetes.14-22 However, P2Y6R agonists are also proposed for neurodegeneration, glaucoma, asthma, and other conditions.13,23-27 A nucleoside precursor prodrug of a P2Y6R agonist entered a Phase 1b clinical trial for Alzheimer’s disease to target microglial cells (ClinicalTrials.gov: NCT02254369, NCT02386306).26,27 Unlike other Gq-coupled P2YRs, P2Y6R counteracts apoptosis in various cell lines and tissue,3 and it also has vascular effects, including both vasoconstriction and vasodilation,28 and synergizes with the hypertensive effects of angiotensin II.21,22 P2Y6R agonists that might be relatively stable and suitable for use as in vivo pharmacological probes are also needed. Previously, we reported that UDP-related mononucleotides and Up3U-related dinucleotide analogues that contained a rigidified, carbocyclic ribose moiety, i.e. a South (S)-bicyclo[3.1.0]hexane ((S)-methanocarba) ring system as in methoxyimino derivative MRS4383 6, retained P2Y6R agonist potency and efficacy with high selectivity.3,17 The ability of MRS4383 to stimulate PLC activity in mouse adipocytes disappeared in adipocytes derived from P2Y6R-knockout mice, demonstrating its selectivity.17 However, the (S)-methanocarba modification was not compatible with a stability-enhancing α,β-methylene 5′-diphosphonate.3
Here, we have modified the pyrimidine ring 4-position for extension in the form of (aryl)alkoxyimino moieties, in the ribose series of α,β-methylene 5′-diphosphonates (Table 1). A chemically stable N4-methoxyimino or N4-benzyloxyimino modification of the pyrimidine ring in UDP/CDP and α,β-methylene-UDP/CDP analogues was shown to be permissive for P2Y6R activation.3,4,29 Thus, an (aryl)alkoxyimino moiety binds in a P2Y6R region that is not highly sterically constrained, likely facing the extracellular side based on the freedom of chain extension at that position.29 Here, we have empirically explored effects on P2Y6R of ring substitutions of the N4-benzyloxyimino moiety and other modifications. Since the Gq protein pathway activates phospholipase C (PLC) β and subsequent calcium mobilization, we have used for ligand characterization a Ca2+ mobilization assay in an astrocytoma cell line stably expressing the hP2Y6R4,30,31 To demonstrate selectivity against the hP2Y14R, we have used a fluorescent binding assay in Chinese hamster ovary (CHO) cells stably expressing the receptor.34
The pyrimidine nucleotides (Table 1) were synthesized as shown in Schemes 1 and 2. Synthesis of compounds 10 and 19 – 27 required the preparation of various aryl-substituted benzyloxyamines (42 – 51). These benzyloxyamines were produced following the procedure already reported,30 starting from the corresponding substituted benzyl bromides. The synthesis of compounds 14a, 14b, 15 – 18, 25, and 28 – 40 was reported.9,33
Scheme 1.
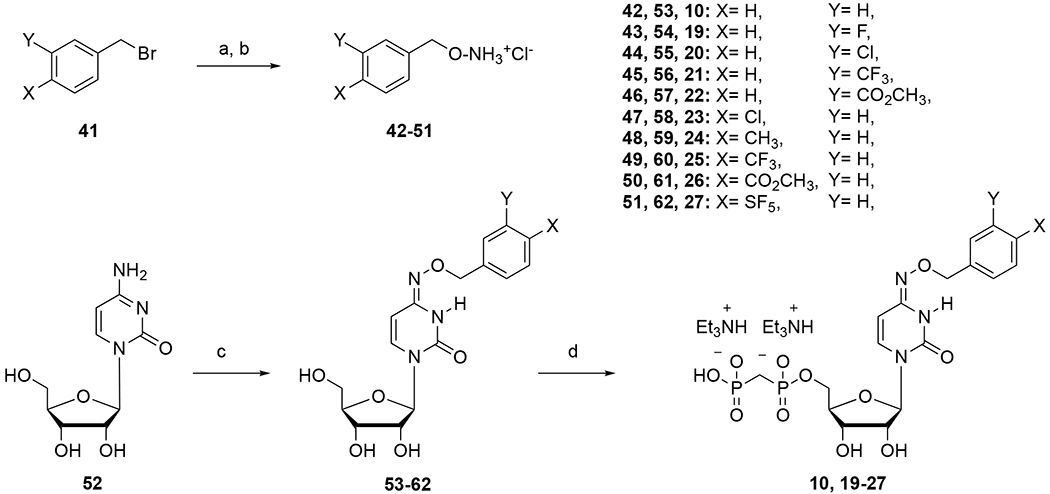
Reagents and conditions: Synthesis of the modified uridine/cytidine nucleotides. Reagents and conditions: a. (Boc)2NOH, DBU, DMF, 50 °C, 2 h; b. HCl, 4 N in dioxane, rt, 12 h; c. desired benzyl-hydroxylamine (53 – 62), pyridine, 80 °C, 12 h; d. Methylenebis(phosphonic dichloride), (CH3O)3PO, 0 °C, 3 h.
Scheme 2.
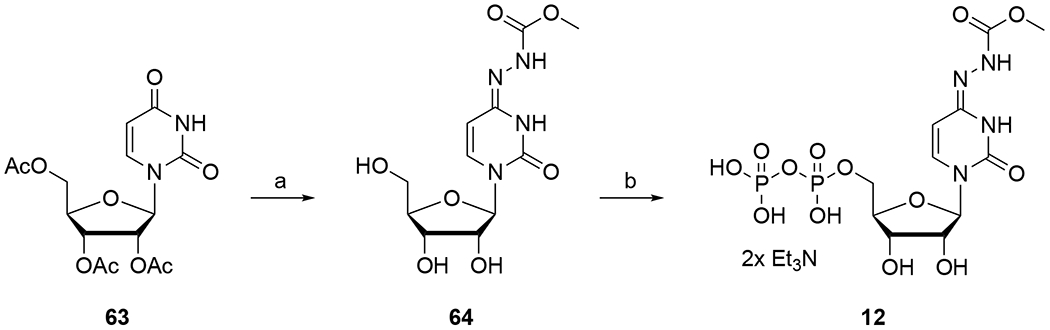
Synthesis of the cytidine nucleotide 12. Reagents and conditions: a. (i) dioxane, DMAP, TEA, 2,4,6-triisopropylbenzenesulfonyl chloride, rt, 18 h; (ii) NH2NHCOOMe, rt, 18 h; (iii) 1 N NH3 in methanol, rt, 18 h, 6%; b. (i) PO(OMe)3, Proton Sponge®, POCl3, 0 °C, 2 h; (ii) DMF, 2[Bu3NH]+HPO42−, 0 °C → rt, 15 min, 5.3%.
The reaction of cytidine (52) with the desired substituted benzyloxyamine (42 – 51) in pyridine at 80 °C, gave the desired intermediates (53 – 62) in good yields. 5′-O-Phosphonylation of the latter was performed using methylenebis(phosphonic dichloride) in trimethyl phosphate to obtain the desired final compounds 10, 19-27 in acceptable yields (Scheme 1).35
Compound 12 was designed to probe if replacement of the O of the hydroxylamine moiety with N was possible. To prepare compound 12, the oxygen at the 4-position of pyrimidine base of protected uridine (63) was activated using 2,4,6-triisopropylbenzenesulfonyl chloride and then reacted with methyl hydrazinecarboxylate (Scheme 2). This intermediate was unstable to purification by silica-gel chromatography; hence the acetates were deprotected without purification by treating with dilute ammoniacal methanol to give 64 in low isolated yields. Compound 64 was converted to corresponding diphosphate 12 under standard phosphorylation conditions by employing phosphoryl oxychloride and tributylammonium pyrophosphate in low yields.
The P2Y6R potency of the nucleotide derivatives was determined in concentration-response assays of calcium mobilization, using fluorescence-based calcium mobilization in 1321N1 human astrocytoma cells expressing the human P2Y6R, as was previously reported.3 This functional data is only indirectly comparable to data previously reported by us and others using a PLC assay.2,4 It is to be noted that EC50 values of UDP analogues in the calcium assay often underestimate their potency when compared to P2Y6R-induced PLC stimulation.3,36 Representative concentration-response curves are shown in Figure 1.
Figure 1.
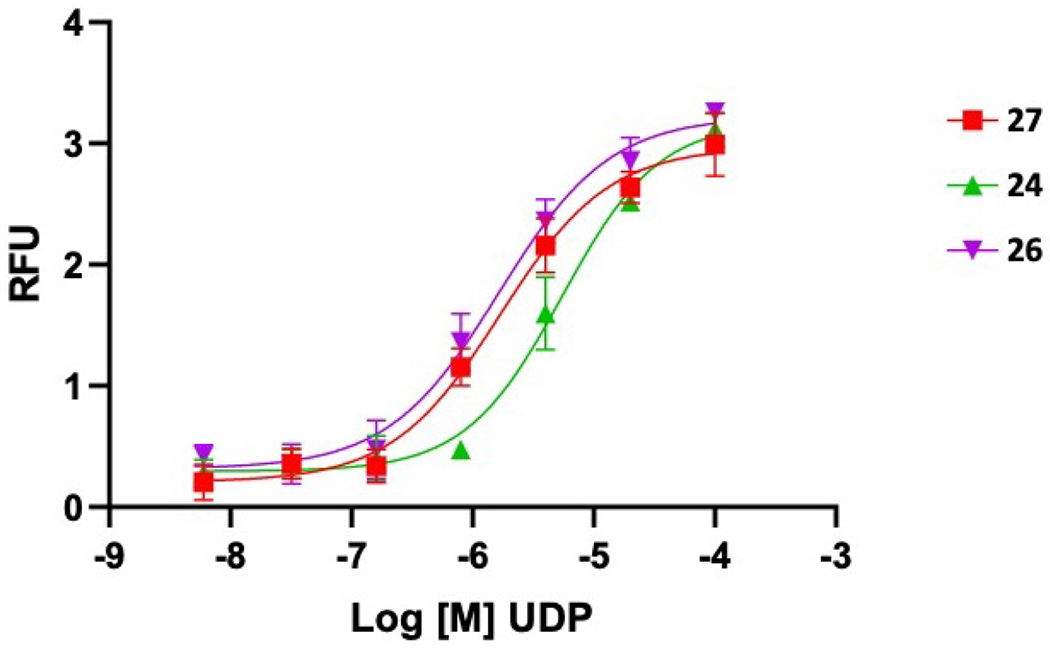
Representative concentration-response curves for P2Y6R activation in a calcium mobilization assay, using 1321N1 cells expressing the human P2Y6R (EC50 values (µM): 24, 5.47; 26, 1.65; 27, 1.69).
All of the compounds synthesized here contained a α,β-methylene bridge, while 14 – 30 and 37, 38 additionally contained an alkyloxyimino-pyrimidine nucleobase. Several of the compounds were previously reported by us as intermediates and as P2Y6R agonists in the synthesis of CD73 inhibitors.33 Selected analogues were also measured in a fluorescent P2Y14R binding assay by flow cytometry of whole CHO cells expressing the hP2Y14R to establish selectivity (Table 2).
Reference compounds 1, 2, 9, 10, and 15 are included in Table 1.3,4,36 The P2Y6R potency of UDP 1 (EC50 0.0416 μM) was reduced 12- to 19-fold with 4-imino-oxyalkyl modifications in 11 and 13. Nevertheless, we sought to explore the interaction of bound to a nucleotide agonist with this distal region of the receptor, especially in combination with a stabilizing methylene phosphonate. Previously, considerable P2Y6R agonist potency was observed upon 4-imino-oxyalkylaryl substitution of UDP.4
Although the N4-methoxy α,β-methylene analogue 9 (0.42 μM) was more potent than the corresponding N4-benzyloxy analogue 10 (2.36 μM), we examined functional group substitution of the phenyl ring of 10. 3-Substituted benzyloxo groups (19 – 22) displayed EC50 values in the range of 0.9–2.7 μM, while 4-substituted benzyloxo groups (23 – 27) displayed EC50 values in the range of 0.57–2.3 μM. Notably, the IC50 values for hP2Y14R binding of compounds 20, 23 and 25 were >100 μM, indicating a high degree of selectivity. A 4-trifluoromethyl-benzyl substituent in 25 provided the highest potency (0.57 μM), and a 5-fluoro substitution of the cytosine ring in 28 similarly enhanced potency, with >175- and 39-fold selectivity over P2Y14R, respectively. N4-Benzoyl derivative 30 was a potent P2Y6R agonist (1.39 μM), but nonselective. A 4-pentafluorosulfanyl group (SF5) group37 in 27 maintained P2Y6R potency. 5-Methyl substitution of the cytosine ring in 29 led to an EC50 of 1.09 μM, i.e. slightly more potent than 10. 3-Alkyl or aryl alkyl (31 – 33, 37, 38), β-D-arabinofuranose 39, 6-aza 40 substitution, as previously synthesized,33 was shown here to completely prevent P2Y6R activation. The 5-substituted cytosine derivatives 34–36 in the α,β-methylene series were inactive at P2Y6R, in contrast to closely related, potent uracil derivatives (15, 16). Curiously, alkylation of N3-position of N4-benzyloxy analogues (37, 38) led to a complete loss of P2Y6R activity. 5-Fluoro-5′-αβ-methylene-UDP 16 was nonselective between P2Y6R and P2Y14R (affinity 0.2–0.3 μM).
P2Y6 agonist 26 and 27 and P2Y14 agonist 14b were chosen as representative UDP analogues containing a stabilizing α,β-methylene bridge for the determination of off-target activities at 46 receptors, channels and transporters (Supporting information) by the Psychoactive Drug Screening Program (PDSP).38 14b showed no significant (i.e. >50% binding inhibition at 10 μM) off-target activities. At 10 μM, 26 inhibited binding at the D3 dopamine receptor (85%), and 27 inhibited binding at the σ1 receptor (59%). Nevertheless these nucleotides are non-promiscuous in binding.
N4-Benzyloxy-CDP analogues containing a stabilizing α,β-methylene bridge were prepared and evaluated as P2Y6R agonists. The reactions between cytidine and alkoxy amines afforded single condensation products, which we represent with a Z-exo C-N double bond, consistent with previous reports, including X-ray crystallographic structures obtained for model compounds.39,40 Considerable freedom of substitution of the benzyl ring is present, while substitution of the pyrimidine C5 position (halo, methyl) but not the N3 position (alkyl) is compatible with P2Y6R activation. SAR studies of several other P2YRs have revealed that substitution of the phosphate moiety of agonists with an α,β-methylene or β,γ-methylene bridge greatly reduces affinity. For example, at the P2Y1R, agonist activity is lost in α,β-methylene-ATP.41 However, β,γ-methylene-or β,γ-dihalomethylene analogues of ATP are potent P2Y12R antagonists.42 A previous study comparing α,β-methylene bridged UDP analogues showed a tendency toward P2Y14R selectivity in comparison to P2Y6R.9 Here, we have demonstrated that a wide range of α,β-methylene-CDP derivatives are suitable as selective P2Y6R agonists, but that is dependent on the presence of a N4-benzyloxyimino group. The potency may not be as high as α,β-methylene-UDP derivatives, but these CDP derivatives have high P2Y6R selectivity compared to the P2Y14R. Additional P2Y6R agonists are needed to explore P2Y6R physiological effects and interactions of the P2Y6R with other proteins, such as the angiotensin 1 (AT1) receptor. This significance of increased P2Y6R mRNA expression as a signature of human tumors needs to be explored.43 We have not tested these P2Y6R agonists in vivo. Still, because of the phosphonate group, they might display a longer duration of action than various UDP analogues that have been used in vivo and isolated in tissue studies.17,28
In this study, inhibition of 5′-nucleotidase (CD73) was not measured. If present, this activity could be a caveat during the interpretation of pharmacological data using nucleoside/nucleotide ligands of purinergic receptors. Some of the compounds included here were found to inhibit CD73 by Junker et al.33 Using a rat CD73 radiometric assay with [3H]AMP as a substrate,44 with a substrate concentration of 5 μM AMP, the following IC50 values (μM or % inhibition at 1 μM) were determined: 9, 0.257; 10, 0.112; 14a, 1.83; 14b, 7%; 15, 0.162; 16, >1; 25, 0.030; 28, 0.085; 29, 0.321.
Within the P2YR family, only P2Y1R and P2Y12R experimental structures are available.1 Thus, current structural hypotheses for P2Y6R ligand recognition are necessarily based on homology modeling,3,45 which needs refinement. Expanding the range of agonist SAR can be informative for GPCR modeling, and P2Y6R agonist SAR is already more highly advanced than its antagonist SAR. By analogy, we effectively used molecular dynamics (MD) simulation to recapitulated P2Y14R agonist SAR, based on homology to the P2Y12R, and this homology modeling approach later led to the effective discovery of potent P2Y14R antagonists.46,47 Although P2Y1R and P2Y6R belong to the same Gq-coupled subfamily of P2YRs, their similarity and identity of sequence are only 41% and 28%, respectively 45 Thus, a new P2Y6R homology model would be strengthened and validated by extensively simulating the binding of known ligands, perhaps including the present agonists.
In conclusion, we have synthesized, and evaluated as P2Y6R agonists in a calcium mobilization assay, various UDP/CDP analogues containing a stabilizing α,β-methylene bridge. A putative distal binding region of the receptor surrounding a 4-benzyloxyimino cytidine group was explored with aryl substitution. A 4-trifluoromethyl-benzyloxyimino substituent in 25 provided the highest potency (MRS4554, 0.57 μM), and a 5-fluoro substitution of the cytosine ring in 28 similarly enhanced potency, with >175- and 39-fold selectivity over P2Y14R, respectively. Thus, we have expanded the range of P2Y6R agonists, and explored the SAR of pyrimidine nucleoside 5′-α,β-methylene diphosphonates that are highly selective over P2Y14R. The distinguishing features are an N4-extension in the CDP analogues in the P2Y6R agonists reported here versus a 2-thio group in uracil-containing P2Y14R-selective agonists, such as MRS2905 14b.
Supplementary Material
Highlights.
P2Y6 receptor (P2Y6R) agonists proposed for neurodegeneration, glaucoma, asthma, and other conditions.
We have synthesized and evaluated various UDP/CDP analogues, containing a stabilizing α,β-methylene bridge, in a calcium mobilization assay.
We identified new P2Y6R agonists that are highly selective over P2Y14R.
The distinguishing features are an N4-extension in CDP analogues as P2Y6R agonists versus a 2-thio group in uracil-containing P2Y14R-selective agonists.
Acknowledgements
Mass spectral measurements were performed by John Lloyd (NIDDK). This work was supported by the NIDDK Intramural Research Program (ZIADK031116). Financial support of Anna Junker by the Deutsche Forschungsgemeinschaft (German Research Foundation, JU 2966/1-1 and JU 2966/2-1) is gratefully acknowledged. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (Contract #HHSN-271-2008-00025-C) for screening data (https://kidbdev.med.unc.edu).
Abbreviations:
- CDI
carbonyldiimidazole
- DCC
dicyclohexylcarbodiimide
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- GPCR
G protein-coupled receptor
- PDSP
Psychoactive Drug Screening Program
- PLC
phospholipase C
- RMSD
root-mean-square deviation
- SAR
structure activity relationship
- TBAP
tetrabutylammonium dihydrogen phosphate
- TEA
triethylamine
- TEAB
tetraethylammonium tetrafluoroborate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- UDP
uridine 5′-diphosphate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information
Supplementary data (chemical synthesis, characterization data, HPLC purification and purity analysis procedures, pharmacological assays and spectral data associated with this article) can be found, in the online version.
References and notes
- 1.Jacobson KA, Delicado EG, Gachet C, et al. , Br. J. Pharmacol 2020;177:2413–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Tayeb A, Qi A, Nicholas RA, et al. J. Med. Chem 2006;49 (24):7076. [DOI] [PubMed] [Google Scholar]
- 3.Toti KS, Jain S, Ciancetta A, et al. Med. Chem. Commun 2017;8:1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maruoka H, Barrett MO, Ko H, et al. J. Med. Chem 2010;53:4488–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ko H, Carter RL, Cosyn L, et al. Bioorg. Med. Chem 2008; 16:6319–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko H, Fricks I, Ivanov AA, et al. J. Med. Chem 2007;50:2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazarowski ER, Harden TK. Mol. Pharmacol 2015;88:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harden TK, Sesma JI, Fricks IP, et al. Acta Physiol (Oxf). 2010;199(2):149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das A, Ko H, Burianek LE, et al. J. Med. Chem 2010;53:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zimmermann H. Biochem. Pharmacol 2021;187:114322. [DOI] [PubMed] [Google Scholar]
- 11.Bhattarai S, Pippel J, Scaletti E, et al. J. Med. Chem 2020;63:2941–2957. [DOI] [PubMed] [Google Scholar]
- 12.Tomczyk M,Mierzejewska P, Slominska EM, et al. Nucleosides Nucleotides Nucleic Acids 2018;37(12):709–716. [DOI] [PubMed] [Google Scholar]
- 13.Jacob TF, Singh V, Dixit M, et al. Purinergic Signal. 2018;14:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oliveira-Giacomelli Á, Albino CM, de Souza HDN, et al. Front. Cell. Neurosci 2019;13:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia RA, Yan M, Search D, et al. PLoS ONE 2014;9(10):e111385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Placet M, Arguin G, Molle CM, et al. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 2018;1864:1539–1551. [DOI] [PubMed] [Google Scholar]
- 17.Jain S, Pydi SP, Toti KS, et al. Proc. Natl. Acad. Sci. USA 2020;117(48):30763–30774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salem M, El Azreq M-A, Pelletier J, et al. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 2019;1865(10):2595–2605. [DOI] [PubMed] [Google Scholar]
- 19.Fanning LB, Garofalo D, Boyce JA. J. Allergy Clin. Immunol 2015;135(2):AB63. [Google Scholar]
- 20.Chetty A, Sharda A, Warburton R, et al.J. Asthma Allergy 2018;11:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura A, Sunggip C, Oda S, et al. Pharmacol. Therap 2017;180:113–128. [DOI] [PubMed] [Google Scholar]
- 22.Nishimura A, Sunggip C, Tozaki-Saitoh H. Sci. Signal 2016;9(41l):ra7. [DOI] [PubMed] [Google Scholar]
- 23.Shimoda K, Nishimura A, Sunggip C, et al. Sci. Rep 2020;10(1):13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neher JJ, Neniskyte U, Hornik T, et al. Glia 2014;62(9):1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan H, Xie R, Xu J, et al. Sci. Rep 2017;7(1):2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haydon PG, Lee J. PCT Int. Appl WO 2014160502 A1 20141002, 2014. [Google Scholar]
- 27.Haydon PG, Lee J. US 2018/0085386 A1 [Google Scholar]
- 28.Wihlborg AK, Balogh J, Wang L, et al. Circ. Res 2006;98:970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jayasekara PS, Barrett MO, Ball CB. et al. Med. Chem. Commun 2013;4:1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robaye B, Boeynaems J-M, Communi D. Eur. J. Pharmacol 1997;329(2):231–236. [PubMed] [Google Scholar]
- 31.Ginsburg-Shmuel T, Haas M, Schumann M, et al. J. Med. Chem 2010;53(4):1673–1685. [DOI] [PubMed] [Google Scholar]
- 32.Junker A, Balasubramanian R, Ciancetta A, et al. J. Med. Chem 2016;59:6149–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junker A, Renn C, Dobelmann C, et al. J. Med. Chem 2019;62:3677–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayasekara PS, Jacobson KA. Synth. Commun 2014;44:2344–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalek M, Jemielity J, Stepinski J, et al. Tetrahedron Lett. 2005;46:2417–2421. [Google Scholar]
- 36.Ko H, Carter RL, Cosyn L, et al. Bioorg. Med. Chem 2008;16:6319–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sowaileh MF, Hazlitt RA, Colby DA. ChemMedChem 2017;12:1481. [DOI] [PubMed] [Google Scholar]
- 38.Besnard J, Ruda GF, Setola V, et al. Nature 2012;492:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maruoka H, Jayasekara MPS, Barrett MO, et al. J. Med. Chem 2011;54:4018–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Meervelt L Acta Crystallogr. 1991;C47:2635–2637. [Google Scholar]
- 41.Burnstock G, Fischer B, Maillard M, et al. Drug Devel. Res 1994;31:206–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Springthorpe B, Bailey A, Barton P, et al. Bioorg. Med. Chem. Lett 2007;17:6013–6018. [DOI] [PubMed] [Google Scholar]
- 43.Sriram K, Moyung K, Corriden R, et al. PLoS Biol. 2019;17(11):e3000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freundlieb M, Zimmermann H, Müller CE. Anal. Biochem 2014;446:53–58. [DOI] [PubMed] [Google Scholar]
- 45.Neumann A, Müller CE, Namasivayam V. WIREs Comput Mol Sci. 2020;10:e1464. [Google Scholar]
- 46.Trujillo K, Paoletta S, Kiselev E, et al. Bioorg. Med. Chem 2015;23:4056–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung YH, Salmaso V, Wen Z, et al. J. Med. Chem 2021;64(8):5099–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.