

Abstract
The purinergic signaling system includes membrane-bound receptors for extracellular purines and pyrimidines, and enzymes/transporters that regulate receptor activation by endogenous agonists. Receptors include: adenosine (A1, A2A, A2B and A3) and P2Y (P2Y1 P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14) receptors (all GPCRs), as well as P2X receptors (ion channels). Receptor activation, especially accompanying physiological stress or damage, creates a temporal sequence of signaling to counteract this stress and either mobilize (P2Rs) or suppress (ARs) immune responses. Thus, modulation of this large signaling family has broad potential for treating chronic diseases. Experimentally determined structures represent each of the three receptor families. We focus on selective purinergic agonists (A1, A3), antagonists (A3, P2Y14), and allosteric modulators (P2Y1, A3). Examples of applying structure-based design, including the rational modification of known ligands, are presented for antithrombotic P2Y1R antagonists and anti-inflammatory P2Y14R antagonists and A3AR agonists. A3AR agonists are a potential, nonaddictive treatment for chronic neuropathic pain.
Keywords: drug discovery, molecular modeling, nucleosides, nucleotides, receptors
Graphical Abstract

1. Introduction
G protein-coupled receptors (GPCRs) remain important targets for drug discovery, and current approaches to GPCR drug discovery are structure-based.[1] Many well-established drugs that act at GPCRs, either directly at the receptor, or indirectly by modulating the concentration of their endogenous GPCR ligands, are clearly among the most highly prescribed pharmaceuticals for diverse therapeutic areas.[2] Six out of 24 New Drug Applications (NDAs) approved by the US Food and Drug Administration (FDA) in 2019 were for GPCR ligands.[3] All of the newly FDA-approved GPCR ligands, including an A2A adenosine receptor (AR) antagonist (by a resubmitted application after prior disapproval),[4] are for neurological indications. One of the advantages of developing drugs for GPCRs and other transmembrane receptors is their cell surface location. Thus, achieving cell permeability, which is challenging for many compound classes such as peptides, nucleosides, and nucleotides when directed toward intracellular targets, is a less stringent factor for GPCR ligands. Nevertheless, these ligands must possess favorable pharmacokinetic properties to ensure their distribution and localization at specific action sites.
A major part of this review focuses on the specific research efforts of our own team.
1.1. GPCR structural advances
The drugs introduced in previous decades that act at GPCRs were found mainly by empirical methods of probing structure–activity relationships (SAR), often inspired by natural products. However, modern ligand discovery for GPCRs is largely structure-based, and knowledge of GPCR structures is rapidly accruing, beginning with the structures of bovine rhodopsin (2000) and later (2007) the β2-adrenergic receptor.[1,5] Hundreds of new structures determined in recent years have greatly aided this effort. As of March 2020, 419 structures have been determined for 86 different GPCRs, most of which are members of the rhodopsin-like Class A family.[6]
Table 1 shows current approaches used for the study of GPCRs, encompassing both biophysical approaches, novel assay methods, and sophisticated ligand design. Biophysical methods used to study GPCR structures include X-ray crystallography, X-ray free-electron laser (XFEL), cryogenic electron microscopy (cryo-EM), and NMR.[7] Also, advances in the computational modeling of membrane-bound proteins like GPCRs have provided an interface for medicinal chemists to predict the effects of ligand structural changes on the GPCR binding and activation processes.[8–10] The derived theoretical modeling enhances the utility of the crystallographic structures generated toward predictions for novel ligands, beyond the ligands that are present in the X-ray structures. Medicinal chemists can now predict the effects of chemical modifications of known ligands to explore SAR prior to analogue synthesis. Also, methods for receptor stabilization by multi-site mutagenesis and processing a redesigned receptor protein have greatly facilitated this technology.[7,11] The availability of an increasing number of structures has led to significant acceleration in GPCR drug design using computational methods.
Table 1.
Current approaches to GPCR structure-based drug discovery and references for representative examples pertaining to the purinergic receptors.
| Approach | Example purine receptors | Refs. |
|---|---|---|
| I. Structure-based drug design (SBDD): | ||
| A. Structure determination | ||
| X-ray crystallography | A1,A2A,P2Y1,P2Y12 | [9,30–36,42,66] |
| X-ray free-electron laser (XFEL) | A2A | [87] |
| NMR spectroscopy | A2A | [67,68] |
| Cryo-electron microscopy | A1 | [37] |
| B. Computational modeling[a] | ||
| In silico library screening | A2A | [44–48] |
| Building on known ligands | A2A, A2B | [49,88] |
| Artificial intelligence, machine learning | – | [89] |
| II. Screening/characterizing drug action: | ||
| Fluorescent ligands | A1,A2A,A3,P2Y14 | [23,63,64] |
| Residence time | A2A, A3 | [79,90] |
| Mass spectroscopy | A2A | [91] |
| III. Allostery: | ||
| Within a GPCR monomer[b] | P2Y1, A3 | [59,92,93] |
| Between multimeric GPCR protomers[c] | A2A | [95,96] |
| IV. Novel GPCR modulators: | ||
| Biased agonists | A1, A3 | [93,96,97] |
| Intracellular signaling/ compartmentalization | – | [98] |
| Monoclonal antibodies | A3 | [99] |
| Polypharmacology | A3 | [100] |
| Photopharmacology | A2A | [101] |
Allosteric binding of ligands outside the canonical (orthosteric) binding region in the center of the 7 transmembrane (7TM) helical bundle of rhodopsin-like (Family A) GPCRs has challenged our former concept of where small-molecule ligands might bind to GPCRs.[8] In addition to the canonical binding site, allosteric sites have been discovered all around the protein scaffold. Positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) of GPCRs are increasingly attractive as drug targets, as they promise to be spatially and temporally more selective in the body than orthosteric GPCR ligands (i.e. acting at the same site as the endogenous activators).[1,12]
2. Purinergic GPCRs
Molecules that subserve purinergic signaling, that is, adenosine, inosine, ATP, and various other adenine and uracil nucleotides, are ubiquitously present in living tissues, and their receptors are present in some combination in nearly every cell.[13] Purinergic receptors include both GPCRs (ARs and P2Y receptors (P2YRs) for purine and pyrimidine nucleotides) and ligand-gated P2X receptors (Figure 1).[13] A temporal sequence of signaling, which is involved in physiology and pathology, regulates the innate immune system and many other processes in responding to challenges. Multiple enzymes and transporters in the purinergic system, such as adenosine kinase (ADK, removes intracellular adenosine), adenosine deaminase (ADA, converts adenosine into inosine), CD39/CD39L1 (nucleoside triphosphate diphosphohydrolases 1/2, NTPDase1/2, form extracellular AMP) and CD73 (ecto-5′-nucleotidase, forms extracellular adenosine), and equilibrative nucleoside transporter 1 (ENT1 in the SLC29 family, effecting communication between intra- and extracellular adenosine pools), control the endogenous agonist levels. Medicinal chemists are developing selective agonists, antagonists, and allosteric modulators of these proteins for therapeutic and diagnostic applications in humans.[14] Also, inhibitors of the enzymes that process extracellular nucleosides and nucleotides and of the transporters and channels that link intracellular and extracellular levels of these molecules, indirectly modulate signaling at ARs, P2YRs, and P2XRs. Our laboratory and others have long been involved in developing ligands for various purinergic signaling proteins. For example, our selective ligands of various P2Y receptors (P2Y1, P2Y2, P2Y6, P2Y13, and P2Y14) and highly potent and selective ligands for the A3AR and A2BAR are used widely as probes (Figure 2) to explore the biological role of these receptors.[14–22]
Figure 1.

Early signal transduction by purinergic GPCRs that respond to extracellular nucleotides (P2YRs) and to adenosine (ARs). Enzymes, transporters, channels and cell damage control the level of endogenous agonists, which act in an autocrine or paracrine fashion. Pathways downstream of the G proteins leading to various second messengers, kinases and gene transcription are not shown. P2XRs, which respond rapidly to ATP and are ligand-gated cation channels, consisting of 7 monomer subtypes forming active trimers, are not shown. Various sources of extracellular nucleotides are indicated, in addition to synaptic release from neurons and astrocytes. Ectonucleotidase CD39 is an integral membrane protein and hydrolyzes both ATP and ADP to AMP, while CD73 is anchored through glycosylphosphatidyl inositol (GPI) and acts only on AMP to produce adenosine.
Figure 2.
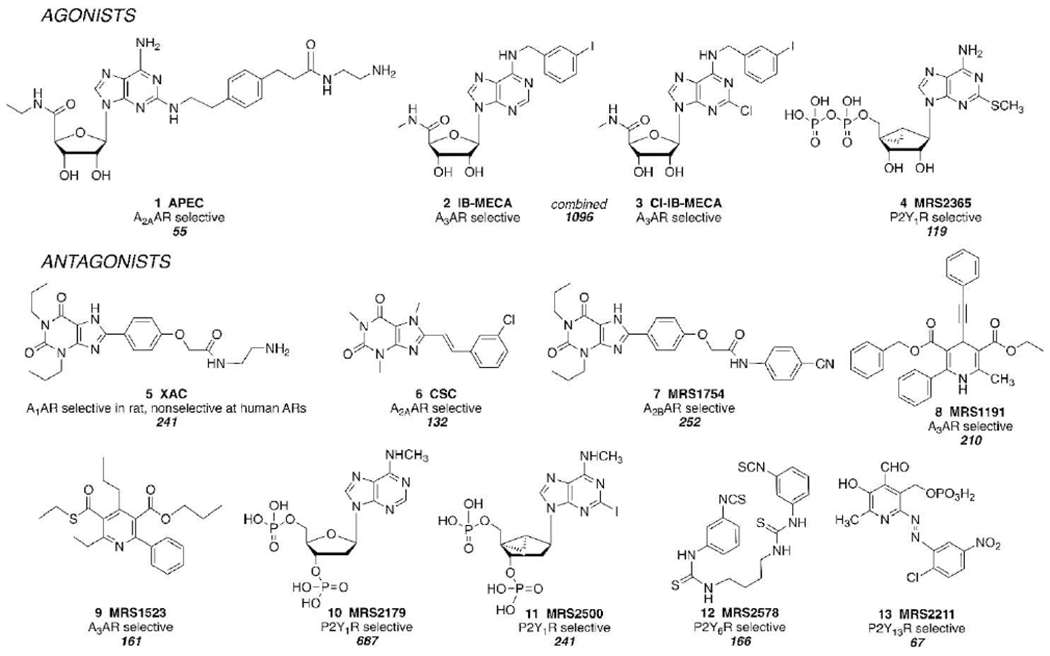
Representative ligand toolbox of purinergic receptor agonists and antagonists that were developed by Jacobson et al. through empirical probing of GPCR SAR, from 1985 (XAC) to 2004 (MRS2578). The citation numbers are shown below each description of selectivity (according to SciFinder, https://scifinder.cas.org/, accessed May 22, 2020).
Despite enormous efforts devoted to the synthesis of new AR and P2YR ligands (Figures 2 and 3) and their relevance to disease biology, most of the clinical trials of agonists and antagonists have failed and few drugs are already approved.[16] Adenosine itself and A2AAR agonist regadenoson 16 (Figure 3) are approved for coronary stress imaging.[16] Adenosine was first approved in 1989 for the emergency treatment of paroxysmal supraventricular tachycardia (PSVT) by activating the A1AR.[16] Theophylline and other nonselective xanthine antagonists have been used for asthma treatment.[19] In addition, an NDA resubmission for selective A2A adenosine receptor (AR) antagonist istradefylline (Nourianz) 15 as a cotherapy with levodopa/carbidopa for increasing “on-time” in Parkinson’s disease resulted in its being the first selective AR antagonist to be FDA-approved.[4] Among P2YRs, only P2Y12R antagonists are clinically approved (as antithrombotic drugs), although a P2Y2R agonist is approved in Japan and Korea for dry eye treatment.[15]
Figure 3.
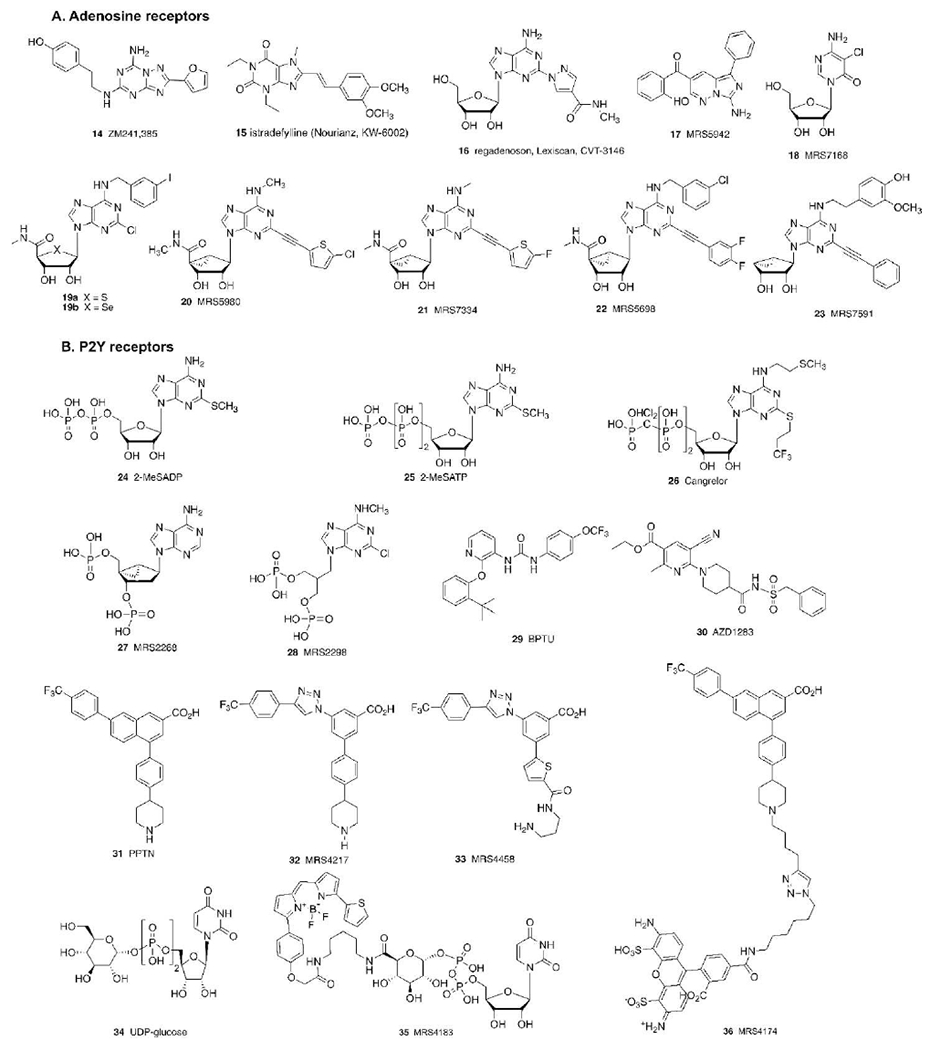
Various other purine receptor ligands discussed in the text, including both compounds discovered by empirical methods and designed by structure-based approaches. Most of the compounds shown are GPCR antagonists, but some agonists (16, 18, 19-22, 24, 25, 34, 35) are included. Compound 23 is a partial agonist selective for the A3AR. Cangrelor is an approved i.v. antithrombotic drug, while BPTU and AZD1283 are experimental antithrombotics. 35 and 36 are high affinity P2Y14R fluorescent probes.
We explore both the medicinal chemistry and pharmacology of purinergic signaling through four inter-related project areas: structures, drug candidates, biological understanding, and receptor probes. Widely used receptor probes (a ligand toolbox, with representative agonists and antagonists 1–13 shown in Figure 2) were first made by empirically probing the SAR of purine receptors, and most are first of their type or designed for expanded utility in receptor characterization. For example, potent AR antagonist XAC 5 and agonist APEC 1 were designed as functionalized congeners,[23,24] such that they could be coupled to carrier or reporter moieties without loss of receptor affinity. In 1985 the first biotin conjugates of any GPCR ligand were synthesized from these amino congeners.[23,24] This toolbox has aided in hundreds of studies in the purinergic field, including pharmacological probing of the role of each receptor, that have enabled clinical trials. IB-MECA 2 and CI-IB-MECA 3 have progressed from probe molecules to advanced clinical trials.[16,25,26] MRS1754 7, MRS2578 12, and MRS2211 13 were the first antagonists reported for the A2BAR, P2Y6R, and P2Y13R, respectively.[22,27] Dihydropyridine MRS1191 8 and pyridine MRS1523 9, both selective A3AR antagonists, are chemically optimized versions of screening hits identified through binding performed in the Jacobson lab from a manually collected chemical library prior to the commercial availability of libraries. We are also working on inhibitors of ADK (to increase intracellular and, indirectly, extracellular adenosine levels) and CD73 (to prevent the formation of adenosine from nucleotides, useful as co-therapy in cancer immunotherapy).[28,29]
2.1. Structure-guided discovery of purinergic GPCR ligands
Advanced methods have been used by various laboratories to determine AR and P2YR structures (Table 1).[1,8,30–37] We have collaborated in this effort with Ray Stevens at the University of Southern California and his colleagues. Furthermore, we and others have employed the receptor coordinates in structure-based drug design (SBDD) of new ligands.[1,8,9,38] At least 57 purinergic GPCR structures are reported in the public Protein Data Bank:[39] 52 for ARs and 5 for P2YRs.[5] Thus, ~14% of all GPCR structures so far obtained are for purinergic receptors [as of March 2020]. Therefore, the purinergic GPCRs, especially the human (h) A2AAR, are well-represented and serve as prototypes for applying new experimental and computational methods. High-resolution X-ray and cryo-EM structures of ARs and X-ray structures of P2YRs facilitate the rational design of ligands, either by modification of known agonists and antagonists or by virtual screening to discover novel chemotypes.
The first AR structure determined was for the A2AAR in complex with a synthetic antagonist ZM241385 14 (Figure 3), which is still the most commonly used antagonist co-crystallized in A2AAR structures.[30] The A2AAR was also the first GPCR for which agonist-bound structures were determined without a G protein or its equivalent present.[31,32] Along with the β2 adrenergic receptor, the A2AAR often exemplifies how the X-ray and cryo-EM structures are informative concerning the structure-function relationship of receptor binding and activation.[1] In fact, experimental structures of the active A2AAR bound to a mini-Gs protein have been released.[40,41] Recently, a second AR subtype, A1AR, has been structurally elucidated, with the determination of its inactive[36,42] and Gi/o protein-bound active states.[37] The structures of two P2YRs, determined by our lab in collaboration with Ray Stevens, along with colleagues Beili Wu and Qiang Zhao (Shanghai Inst. Materia Medica), contained many unanticipated features that challenged our previous concepts of conserved structural characteristics of Family A GPCRs. For example, the first case of a GPCR ligand that binds entirely outside the seven helical bundle, in contact with the phospholipid bilayer was the P2Y1R antagonist BPTU 29 (Figure 3).[35,43] Also, the “conserved” disulfide bond between the second extracellular loop (EL2) and TM3 seems dynamic in the P2Y12R structure, with different ligands binding to either oxidized (closed, with nucleotide analogues of ATP and ADP) or reduced (open, with non-nucleotide antagonist AZD1283 30) forms.[33,34]
Now SBDD is the standard for GPCR drug discovery in general and, specifically, for purine receptors.[1,8,9] The particular approach taken to discover new ligands for a given receptor depends on the available data and current state of known ligands for that receptor. For example, novel AR chemotypes can be discovered using in silico screening and biophysical mapping.[44–51] Otherwise, known ligands can be modified rationally to increase affinity and selectivity at a particular receptor subtype.[17,18,21] There is no “one size fits all” approach. Rather, the approaches used are tailored to known characteristics of each receptor target and its ligands. Three separate examples of using GPCR structures to guide ligand design are presented below: P2Y1R antagonists, P2Y14R antagonists, and A3AR agonists.
3. P2Y1R Receptor Structures Used To Analyze Known Agonist and Antagonist SAR
The P2Y1R is representative of the Gq-coupled first subfamily of five P2YRs, termed P2Y1-like and comprising P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11Rs. There is a strong justification for designing novel P2Y1R antagonists. The initial focus on P2Y1R antagonists as potential antithrombotic drugs since 1999[52] has expanded to encompass their application to neurodegenerative conditions. Notably, P2Y1R antagonists have shown efficacy in models of Alzheimer’s disease.[53,54] We have characterized in detail the interaction of many antagonists with the receptor, an effort that can contribute to the rational modification of known ligands.
The most potent reported antagonist of this receptor is the bis-phosphate nucleotide MRS2500 11 (Figure 2), which contains a sterically locked bicyclic ring in place of ribose.[35] The [3.1.0]bicyclohexane ring system, also called methanocarba, maintains this pseudoribose moiety in either a south (S) or north (N) conformation, depending on the site of cyclopentane-cyclopropane ring fusion. The (N)-methanocarba structure bridges the 4′ and 6′ carbons with a cyclopropyl ring, while in the (S)-methanocarba structure (not shown) a bridging methylene is present between the 1′ and 6′ carbons. The (N)-methanocarba modification is clearly the P2Y1R-preferred one, and its incorporation in place of the ribose tetrahydrofuryl group enhances both affinity and selectivity at this receptor. A potent (N)-methanocarba antagonist MRS2500 has become a useful tool for defining the role of this receptor in vitro and in vivo. When injected i.v. in the mouse, MRS2500 is sufficiently stable to phosphate group hydrolysis to allow studies of in vivo P2Y1R antagonism within a span of 30 min or longer.[55] Studies in pharmaceutical companies have demonstrated the feasibility of using P2Y1R antagonists, including MRS2500, as antithrombotic agents with the advantage of less bleeding liability compared to use of P2Y12R antagonists.[56]
Figure 4 shows an overlay of two separately determined P2Y1R X-ray structures with orthosteric and allosteric antagonists.[35] Two disparate antagonist sites were found on the P2Y1R – and both were surprising. Neither corresponds to the canonical ligand binding site of Family A GPCRs. In fact, before the release of the X-ray structure, site-directed mutagenesis (SDM) of P2Y1R was not successful in predicting the position of MRS2500 binding.[57] Nucleotide binding and activation were found to depend on amino acid residues deeper in the binding site than the site of MRS2500 11 in the P2Y1R structure. In hindsight, these residues could be important for the receptor’s overall structural integrity. Then, structural determination, supported by SDM, computational modeling and comparing antagonism in different signaling pathways, distinguished the two distinct sites for MRS2500 and BPTU 29 [35,57–59] MRS2500 binds the receptor in a region exposed to the polar, extracellular environment, instead of within a deep pocket within the transmembrane bundle. The adenine scaffold is involved in a bidentate H-bond with Asn2836.58 through the N6H and N7, and the 2-iodo substituent forms a halogen bond with the Cys42N-ter carbonyl.
Figure 4.

A superposition of separate X-ray structures of the hP2Y1R in complex with MRS2500 11 and BPTU 29.[35] These inactive receptor conformations are nearly identical, even though the ligands bind to disparate sites on the protein. The MRS2500-bound structure (PDB ID: 4XNW) is shown in light gray (ligand in lime) and the BPTU-bound structure (PDB ID: 4XNV) in dark gray (ligand in violet). Specific stabilizing interactions are indicated by dashed lines.
Both phosphate groups are required for high-affinity interaction with the P2Y1R, consistent with the MRS2500-P2Y1R structure.[35,57] Upon removal of the 5′-phosphate of MRS2500 leaving 5′-OH, the affinity is reduced ~2000-fold, and the parent nucleosides in this bisphosphate series are completely inactive. Thus, the specific interactions of each phosphate moiety are critical, which was confirmed by SDM of cationic and H-bonding residues. The 3′-phosphate makes salt-bridges with Lys46N-ter and Arg195EL2 and H-bonds with Tyr1102.63 and Tyr3037.32; while the 5′-phosphate interacts with Arg3107.39 through a salt-bridge and with Thr205EL2 and Tyr3067.35 through H-bonds. The involvement of Lys46N-ter, Arg195EL2, and Tyr3037.32 in stabilizing the 3′-phosphate was confirmed by the selective binding affinity reduction of the Lys46Ala, Arg195Ala and Tyr303Phe-mutated receptors for MRS2500, but not for the agonist 2-MeSADP, lacking the phosphate at the 3′ position. Differently, the Tyr306Phe mutation affected both MRS2500 and 2-MeSADP, both presenting a 5′ phosphate moiety.
Allosteric binding of diarylurea BPTU was undetected prior to the X-ray structure of its P2Y1R complex, and an early report of this hydrophobic antagonist series envisioned the binding site in the canonical TM region.[58] The allosteric antagonist is positioned between TM2 and TM3, facing the phospholipid bilayer, and exclusively makes two H-bonds with the receptor, in particular between the urea NHs and the backbone O lone pairs of Leu1022.55. Following the appearance of an unambiguous allosteric site in the BPTU-P2Y1R complex structure, BPTU’s allostery was demonstrated pharmacologically by its increasing the rate of agonist radioligand dissociation.[35,35] SDM to replace the small Ala1062.59 with the bulkier Trp/Phe/Leu to sterically prevent binding of the pyridine ring of BPTU, removed the effect of BPTU to allosterically inhibit [3H]2-MeSADP binding and to increase its dissociation rate.[35] Concerning the question of how BPTU binding prevents receptor activation, it was noted that other GPCRs undergo a relative movement of TM3 with respect to TM2, which would be blocked in P2Y1R by BPTU.[35,43]
Later, the allosteric effects of BPTU were revealed in greater detail pharmacologically by comparing antagonism of full and partial agonists at different P2Y1R signaling pathways.[59] We have compared the action of these two P2Y1R antagonists on full and partial agonists – and different second messenger functional assays have also uncovered the different modes of action. Results for BPTU indicate that with partial agonists an insurmountable antagonism is seen. In cases where there is signal amplification, that is, readouts that are further down the signaling cascade from the G protein itself, such as ERK1/2 stimulation mediated by either Gq or β-arrestin2, BPTU antagonized full agonists in a surmountable fashion.[59] In the past, the presence of surmountable antagonism was taken as evidence that the binding is competitive. As expected, the orthosteric antagonist MRS2500 always produces surmountable antagonism. However, in some cases BPTU shows surmountable antagonism – leading to the incorrect conclusion that its binding might be competitive. Thus, antagonism by urea BPTU can appear to be either surmountable or insurmountable, although it binds only in the allosteric site.
The first step in validating a modeling approach for a given GPCR is to try to recapitulate the known ligand SAR. In a study of molecular dynamics (MD) simulation, Ciancetta and Jacobson compared the P2Y1R recognition of known ligands, that is, 100 nucleotides (mostly antagonists) and 46 non-nucleotide arylurea antagonists.[57] That was accomplished by first docking and then refining with MD the P2Y1R complexes with these two classes of ligands. The MRS2500 (PDB ID: 4XNW[35]) and BPTU-bound (PDB ID: 4XNV[35]) P2Y1R X-ray structures were employed for docking the nucleotide and non-nucleotide ligands, respectively, after removal of the crystallographic ligands. Focusing on the nucleotide ligands, their docking poses generally agreed with the crystallographic binding mode of MRS2500, suggesting that the site for MRS2500, although more external than the canonical GPCR binding site, could be also a site for nucleotide agonist binding. The endocyclic N7 and the exocyclic N6H of modeled, bound nucleotides were engaged in H-bonds with Asn2836.58, the N6-alkyl substituent, with methyl as the favored alkyl group, was hosted in a small hydrophobic pocket between TM6 and TM7. The C2 substituent occupied a hydrophobic cavity defined by residues of the N-terminus and EL3 and expanding toward the extracellular environment, with long hydrophobic solvent-exposed moieties more disfavored than polar ones, which are instead able to interact with polar residues in EL2 and N-term. The P2Y1R affinity of antagonists containing extended 2-alkynyl chains bearing polar groups was rationalized by the predictions of specific distal interactions with the receptor. The most affinity-enhancing C2 substitution was provided by an iodine atom, by interacting through a halogen-bond with the backbone of Cys42N-ter. The unfavored C8-substitution was rationalized by its effect in disfavoring the anti-glycosidic bond, which was instead observed in the X-ray structure and maintained during the MD simulations.
The MD simulations allowed the recognition modes of key ligands to be analyzed in detail, considering each of the recognition elements.[38,57] The docking poses of the 3′,5′-bisphosphate antagonist MRS2500 and closely related bisphosphate agonist MRS2268 27 (Figure 3), and of the 5′-diphosphate agonist 2-MeSADP 24, to the hP2Y1R were subjected to MD simulation. The MD trajectories were analyzed by computing the RMSD of the protein Cα-carbon atoms and of the ligand barycenter and by evaluating the stability of the ligand-receptor interaction patterns over time. This MD analysis also allowed a determination of the favored protonation state for receptor-bound MRS2500. It was the triple anion, in which the upward facing 3′-phosphate is doubly charged, and the downward facing 5′-phosphate is singly charged. This result agrees intuitively with the relative location of these two groups, with the 3′-phosphate being more external and in contact with multiple cationic functional groups in the ELs.[57]
Considering the pattern of interactions, the bidentate H-bond with Asn2836.58 remained stable during the simulations of bound agonists 2-MeSADP and MRS2268 but was less persistent for antagonist MRS2500.[57] P2Y1R docking alone failed to explain the functional activity of MRS2268, which differs from the antagonist in the absence of N6-methyl and 2-iodo substitution, but the following agonist signatures were identified using MD: MRS2268, but not MRS2500: 1) had a persistent bidentate H-bond with Asn2836.58; 2) established an additional H-bond with the sidechain of Asn2997.28; and allowed more water molecules to diffuse into the cavity. Two arginine residues, Arg195EL2 and Arg3107.39, contributing to the binding of the 3′-phosphate and 5′-phosphate groups, respectively, in the crystallographic pose of MRS2500, had a different behavior in the case of 2-MeSADP: the β-phosphate was engaged in salt-bridges with the two arginine residues, but only transiently with Arg195EL2. This is consistent with SDM, with Arg310Ala mutation abolishing 2-MeSADP affinity and Arg195Ala not affecting it. MD simulations also confirmed the preference for the (N) puckering of the ribose of 2-MeSADP, which was previously determined empirically using the bicyclic methanocarba substitutions: the ribose ring (N) conformation was indeed prevalent during the MD simulation of the complex between P2Y1R and 2-MeSADP.[57]
Thus, computational studies derived from the antagonist-bound P2Y1R X-ray structures are able to explain empirical observations with a wide range of reported agonist and antagonist ligands. The ability of MD to recapitulate the SAR of two ligand classes establishes it as a likely predictive approach. Thus, future SAR studies have gained a means of rationally designing new ligands.
4. P2Y12R Receptor Structures Used To Design New P2Y14R Antagonists
The P2Y12R, a clinically validated drug target for antithrombotic agents, belongs to the Gi-coupled subfamily of P2YRs, which consists of three receptors, also including the ADP-activated P2Y13R and the UDP-glucose (UDPG 34) activated P2Y14R. There are currently three orally administered P2Y12R antagonist drugs, ticagrelor and prodrugs clopidogrel and prasugrel, that are approved (structures not shown).[15] In addition, there is a nucleotide antagonist, cangrelor 26, that is delivered by infusion. We have established a theoretical model of ligand recognition at the P2Y14R and used the gained insights to select proposed molecules for synthesis.[18,21] Iterative cycles of modeling and synthesis have greatly expanded the number of potent antagonists of this receptor.
The P2Y14R has a specific distribution, function, and growing number of agonist and antagonist ligands (Figure 3).[18,21] It is widely distributed in placenta, spleen, bone marrow, thymus, stomach, intestine, adipose tissue, lung, and brain. It has a role in immune and inflammatory responses, renal inflammation, mast cell degranulation, smooth muscle contraction, and gastric function. P2Y14R antagonists may eventually be used in treating asthma, as human P2Y14R gene variants were found to correlate with the disease.[60] Lazarowski and colleagues used a transgenic mouse model of cystic fibrosis (CF) to demonstrate that antagonist PPTN 31 reduced neutrophil-mediated lung inflammation and detected elevated UDPG in human CF sputum samples.[61] P2Y14R is expressed on human neutrophils, and its activation by UDPG stimulates chemokinesis. Also, P2Y14R on mast cells promotes the release of inflammatory mediators.[19a] Breton and colleagues have studied the role of P2Y14R in renal intercalated cells and found P2Y14R antagonist PPTN to be useful in controlling inflammation.[19b] Thus, P2Y14R antagonists might have a role in acute kidney injury.
P2Y12R structures are representative of the Gicoupled second subfamily of three P2YRs, and the structures do not closely resemble the P2Y1R structures. Three P2Y12R X-ray structures have been determined, two structures bound to the agonists 2-MeSADP 24 (PDB ID: 4PXZ[33]) and 2-MeSATP 25 (PDB ID: 4PY0[33]) and one structure bound to the antagonist AZD1283 30 (PDB ID: 4NTJ[34]). The two ligand classes differ dramatically in their P2Y12R recognition.[33,34] There are two subpockets in the P2Y12R binding site, and only one of the subpockets of this bifurcated binding site is ligand-occupied. Both AZD1283 and the nucleoside portion of the nucleotide agonists bind in subpocket “1”, and the nucleotides’ phosphate chains wrap around the barrier separating the two subpockets but do not fill the second pocket. As concerning nucleotide agonists, the orientation of adenosine is roughly flipped in the P2Y12R (with the nucleobase binding deeper than the ribose moiety) compared to its orientation in A2AAR. The purine aromatic scaffold is engaged in a π-π interaction with Tyr1053.33, the ribose 2′ and 3′ hydroxy groups interact with His1875.36, Lys179EL2, Thr163EL2 and Cys973.25, and the phosphate groups interact with numerous polar and charged residues. There is greater P2Y12R structural plasticity compared to other GPCRs having multiple X-ray structures. The largely positively charged P2Y12R extracellular loops fold around nucleotide ligands through charge complementarity with the phosphate moiety, and the conserved disulfide bond between TM3 and EL2 is dynamic. In fact, in the antagonist-bound structure, the disulfide bridge between Cys973.25 and Cys175EL2 is not present. Moreover, helices TM6 and TM7 are moved outward as compared to the agonist-bound structures, in order to accommodate AZD1283 that spans from TM3 to TM7 making a π-π interaction with Tyr1053.33 similarly to the agonists.
Our primary interest is not the development of P2Y12R antagonists, which is highly advanced in the pharmaceutical industry, rather the design of ligands for homologous receptor subtypes. Given the availability of P2Y12R X-ray structures, we were able to derive a homology model of the closely related (45% sequence identity) hP2Y14R and apply that model to ligand design.[10] We had previously introduced numerous P2Y14R agonists, and now the availability of a close template for P2Y14R homology modeling allows us to both retrospectively analyze agonist binding and to prospectively guide antagonist design.
We have used an agonist-bound hP2Y12R X-ray structure (PBD ID: 4PXZ[33]) as a template to build a structural model of P2Y14R to predict agonist binding.[62] We proposed binding contacts of UDPG in the bifurcated binding site of P2Y14R, by analogy to the P2Y12R binding site. The hypothetical orientation of UDP is similar to that of 2-MeSADP and 2-MeSATP at P2Y12R binding site, with the base, in the predicted anti conformation, involved in a π-π interaction with Tyr1023.33, the ribose 2′ and 3′ hydroxy groups interacting with His1845.35, Lys176EL2 and Cys943.25, and the phosphates with Arg2536.55, Tyr2566.58, Gln2606.62, Lys27773.5, and Arg2747.32. Differently from P2Y12R, where subpocket “2” is unoccupied, in our P2Y14R model the UDPG glucose moiety conveniently occupies subpocket “2” (Figure 5A), thus finally identifying a putative biological function for this half of the binding site, i.e. glucose recognition. Following this UDPG binding mode, we have even identified putative H-bonds of the glucose moiety with polar residues in this subpocket, like Lys772.60, Asp812.64, Asn903.21, Ile170EL2, Arg2747.32, Glu2787.36, with the importance of each glucose-OH group of UDPG probed using F substitution.[62] The degree of glucose OH coordination in our homology model for UDPG binding roughly corresponded to the affinity loss upon F substitution of each OH. There is a close parallel in both SAR and putative interactions with conserved residues for agonist binding to both P2Y12R and P2Y14R. All indications, from empirical SAR and its docked pose in our model,[62] pointed to the flexibility of substitution of the glucose moiety, since it is the most externally exposed portion on the receptor-bound UDPG molecule. Thus, we utilized this moiety as a point for chain attachment for the design of P2Y14R agonist functionalized congeners.[62,63] The carboxylic acid of UDP-glucuronic acid was used as an attachment point for linker extension through amide formation. This attachment point was also used to design an agonist fluorescent probe MRS4183 35, potentially useful for drug discovery.[63]
Figure 5.

A) Docking pose of UDP-glucose 34 (lime) at the hP2Y14R (light gray) structure, obtained by homology modeling based on the P2Y12R X-ray structure.[62] Two subpockets were found in the P2Y14R putative orthosteric binding site, occupied by the UDP (subpocket 1) and glucose (subpocket 2) moieties. B) Docking pose of the antagonist PPTN 31 (violet) at the P2Y14R model (gray).[21]
The application of P2YR structures to P2Y14R antagonists presented additional challenges. The prototypical P2Y14R antagonist is PPTN, a 2-naphthoic acid derivatized with a 4-(trifluoromethyl)-phenyl at position 7 and a 4-(piperidin-4-yl)-phenyl at position 4.[21a] The ligand does not resemble the native agonist in any obvious manner. The aryl carboxylic acid and the aliphatic secondary amine groups appended to a large multicyclic hydrophobic moiety provide an amphipathic character to the compound, which presents poor water solubility. There is a need for a bioisosteric replacement of the naphthalene moiety, which contributes to the dominant hydrophobicity and insolubility of PPTN.[21a]
Although targeting new antagonists, we used the MD-refined model built using the nucleotide agonist-bound P2Y12R as a starting template, as in the previously described study. We chose the agonist-bound template because extracellular regions were largely defined in this structure in contrast to the antagonist-bound P2Y12R (PDB ID: 4NTJ[34]). It also contained the intact conserved disulfide bridge between TM3 and EL2.[21a] Moreover, an attempted docking of PPTN to a P2Y14R model based on the antagonist-bound P2Y12R template resulted in poor score poses, so this model was discarded.[64] Instead, we employed the docking pose of PPTN obtained at the 2-MeSADP—P2Y12R-based P2Y14R model to devise a new fluorescently labeled antagonist as a screening tool.[21a,64] There is no high-affinity radioligand for the P2Y14R, although a relatively weak-binding radiolabeled agonist has been used for this purpose. Thus, it was of great interest to design fluorescent P2Y14R ligands, both agonists and antagonists. We chain modified PPTN through its piperidine nitrogen atom, which was the most solvent-exposed functionality in our PPTN pose (Figure 5B). We installed an N-hexynyl group, to which an azide-functionalized fluorophore, Alex Fluor488, was coupled by a click reaction. The resulting compound, MRS4174 36, is a bitopic ligand of exceptionally high affinity in a functional assay.[64] The greatly increased affinity was predicted to be a result of specific polar interactions of charged groups on the fluorophore with the receptor’s EL region, which anchored the conjugate. The availability of fluorescent P2Y14R ligands provided binding test capacity and paved the way to the design and optimization of new P2Y14R ligands, which is an iterative process involving design through modeling, synthesis, and evaluation of new compounds. Having a potent and selective fluorescent P2Y14R antagonist MRS4174 for compound screening by flow cytometry was particularly valuable in light of an unsuccessful attempt to use [3H]PPTN as a radioligand for binding assays.[18] The same site at the distal piperidine N of PPTN, predicted by modeling to be in a sterically permissive P2Y14R region facing the extracellular space, could also serve as a polyethylene glycol) (PEG) conjugation site to increase aqueous solubility while retaining binding ability.[18]
The first challenge that we addressed was the bioisosteric substitution of naphthalene. A computational pipeline comprising three phases was established to compare different potential bioisosteric replacements: i) a bioisostere search, ii) compound screening, and iii) hit selection.[21a] The bioisostere search process involved docking runs followed by MD simulations to identify linking groups able to suitably replace half of the naphthalene ring while preserving the ligand – receptor interactions observed for PPTN. First, the PPTN-bound hP2Y14R homology model was MD-refined, and the PPTN was removed from the binding site. Among the potential naphthalene bioisosteres considered, two analogues, an alkyne or a triazole separating two phenyl moieties, were oriented comparably to the parent PPTN by P2Y14R docking, but not following MD simulation. The triazole, MRS4217 32, was predicted to bind with greater stabilization than the alkyne and does in fact, with an IC50 of 32 nM, which is far more potent. This triazole maintained a π-cation interaction with Arg2536.55, and the carboxylate group was coordinated by three polar amino acid side chains: Lys772.60, Lys2777.35, and Tyr1023.33. Importantly, MRS4217 had an only fivefold lower affinity than PPTN (IC50 6 nM, by fluorescent binding assay) and had a clogP of 4.64 compared to 6.18 for PPTN. Various triazole-analogues bearing different aryl groups were compared by computational modeling to select most promising candidates to synthesize. This provided a SAR highlighting a general preference for the para substitution of the aryl group with bulky hydrophobic substituents. The compound found to have the best affinity, MRS4217, contains the same optimized p-CF3-phenyl substitution as PPTN.
The next iteration of SBDD used a strategy to modify functionality and cyclic structures on the aryl group attached to the core benzene ring of MRS4217, and its aliphatic terminal amino group by trying to dock various combinations of these components.[21b] Visual inspection after docking identified analogues that maintained the important P2Y14R polar interactions with the aryl-COOH and hydrophobic interactions of p-CF3-Ph. The MD refinement phase of selected compounds was accelerated using High Throughput MD (HTMD). Docking and MD were used to compare structures and predict that five-membered rings in place of the other p-phenyl group of PPTN would bind appropriately to the P2Y14R, with thiophene maintaining a π-cation interaction with Arg2747.32. An amide linker extending from the thiophene was predicted to be better than a sulfonamide. Comparing different lengths of the alkyl amino spacer chain, the three-methylene linker appeared optimal, both in the computational analysis and in the affinity of the synthesized compounds. This process of MD simulation and chemical synthesis resulted in alkyl amino congener MRS4458 33 (IC50 169 nM), which had a c log P of 0.84.
Thus, P2Y14R modeling has provided a means of analyzing the structural basis for binding of known agonists and antagonists, as well as rationally extending the SAR of both ligand classes. Many, but not all of the predicted outcomes for novel antagonists related to PPTN have been confirmed through synthesis and affinity determination. One outcome of the structure-based approach to P2Y14R ligands is the discovery of high affinity functionalized congeners in both ligand classes, bearing fluorophores or other conjugated moieties. These fluorescent probes, particularly the antagonist MRS4174, are now useful tools for characterization of P2Y14R ligand binding.[18,21]
5. Design of AR Agonists and Their Receptor Interactions
The general role of adenosine as a neuromodulator is in allostasis of an organism, rather than homeostasis.[65] Thus, knockout (KO) of each AR subtype is nonlethal and well-tolerated, for the most part. Indeed, a mouse line lacking all four ARs had a very similar phenotype to the corresponding WT mice.[65] In the mouse, adenosine agonists are potent hypothermic agents, which is paralleled by their reduction of open field locomotor activity. Each of the four AR subtypes has a role in inducing hypothermia in the mouse, each by a distinct mechanism and receptor location.
The medicinal chemistry of the ARs is much further advanced than that of the P2YRs.[16,22] In the 1960s to early 1980s, N6-(aryl)alkyl and 2-substituted adenosines were discovered as potent agonists for the only two known AR subtypes during that period, A1 and A2A.[16,19,23,24] 8-Arylxanthines were found to be much more potent than the naturally occurring alkylxanthine antagonists, for example, caffeine and theophylline.[23] Functionalized congeners of adenosine and theophylline were introduced in the 1980s by Jacobson et al. as a general, conceptual approach to conjugating GPCR ligands for the purpose of providing receptor probes as pharmacological tools.[23] Also, the approach was considered a possible route to the “development of new therapeutic agents having favorably altered potency, selectivity (including targeted delivery), or duration in vivo.”[24] This approach anticipated bitopic ligands of GPCRs, in which one portion of a tethered ligand can bind to extracellular regions or in an outer vestibule, and a vestibule was later shown to exist in the A2AAR.[23,24,66]
Currently, 49 A2AAR structures have been deposited in the PDB, with 40 antagonist-bound and nine agonist-bound complexes; two antagonist-bound and one agonist-bound structures are available for A1AR.[6] However, no experimental structure has been released so far for A3AR, but theoretical structures can be obtained through homology modeling for SBDD purposes.
5.1. Current approaches to AR ligand discovery
In silico screening with A2AAR inactive structures has been used by Carlsson et al., Katritch et al. and others to discover diverse chemotypes as AR antagonists.[29,44–46] We have collaborated with Jens Carlsson (Uppsala Univ.) on various AR modeling and ligand design projects, resulting in novel antagonists, such as 17, and agonists, such as mixed A1/A3AR agonist 18. Carlsson and co-workers also screened chemical libraries using the agonist-bound A2AAR structure, but the search returned mainly antagonists.[47] Only one of two known classes of non-nucleoside agonists, 2-amino-3-cyanopyridines, was also found as hits, but no novel agonist chemotypes. This appears to be because of the high stringency of spatial requirements for AR interactions of agonists.
An improved method for the discovery of AR agonists was to recognize that nucleosides were the best scaffold for virtual searches.[48] However, the number of nucleosides in existing chemical libraries, at least in 2016, was limited to several hundred. Instead, Carlsson and co-workers used the approach of condensing the nucleobase with the ribose moiety first virtually, and then experimentally (synthesis and receptor binding) only for the highest ranking hits. There were ~7000 nucleobases from which to select hits, which greatly improved the chemical diversity. This led to the discovery of novel nucleosides as AR agonists, for example compound 18 (Figure 3). The discovery and application of A3AR agonists is described below.
5.2. Allosteric coupling and microswitches
How does the activation signal propagate through the receptor protein? How to relate structural dynamics to function? Eddy et al. used NMR to study these A2AAR conformational changes by following signals generated from Gly and Trp residues, which were like beacons distinctly separated from other resonances.[67] Propagation of conformational change follows a path that is detectable using NMR. Extrinsic Trp residues could be substituted in the A2AAR sequence strategically at positions that do not interfere with ligand binding or signaling but act as NMR sensors.[68] Using NMR it was possible to distinguish agonist vs. antagonist binding at A2AAR, and the conformational effects of a G protein mimic were also studied. White et al. provided X-ray structures of mutant A2AAR, to propose how signals are transmitted between two domains of the receptor.[69] Ciancetta et al. adopted a MD approach to predict conformational signatures of A3AR activation without a bound ligand by using molecular models of pharmacologically validated constitutively active mutant receptors.[70] Thus, modeling methods have been used in combination with experimental data to predict limited conformational changes involved in GPCR activation, even in the absence of an active state structure with a G protein present.
5.3. Discovery, optimization and therapeutic potential of A3AR agonists
Recently, our focus has been on the discovery of A3AR agonists with computational modeling playing a major role in both understanding the pharmacodynamic properties of known ligands and the further exploration of SAR. Historically, the first A3AR agonists were adenosine derivatives that contained a combination of N6-benzyl and 5′-small N-alkyl carboxamido substituents, that is, the combination of already known adenosine modifications, but in combination, and with optimized substitution, produced moderate A3AR selectivity.[16] Thus, the prototypical agonists IB-MECA 2 and Cl-IB-MECA 3 became widely used pharmacological probes and advanced to Phase 3 and 2 clinical trials, respectively, for autoimmune inflammatory diseases including rheumatoid arthritis and psoriasis (2) and liver conditions including hepatocellular carcinoma and nonalcoholic steatohepatitis (3).[16,25,26] The subsequently developed SAR of A3AR agonists has now been analyzed in great detail and a current survey recently published.[71]
Since the initial A3AR cloning in several species,[72,73] our lab has remained active in the design of A3AR agonists, leading eventually to novel therapeutic approaches for treating chronic diseases.[25,26,71] The two components of adenosine, nucleobase and ribose, have been explored separately for modifications that greatly increase A3AR selectivity. Suitably functionalized adenine derivatives are AR antagonists, while a ribose or pseudoribose moiety is required for AR activation by nucleoside derivatives. The nucleobase can be considered the “address” portion of the molecule, determining largely the subtype to which the molecule favors, and the ribose contains the “message” to activate the receptor.
The lack of an experimental A3 AR structure might be viewed as a limitation for SBDD, but the increasing number of A2A and A1 AR structures provide good homology modeling templates. The agonist-bound structures provide insights into the typical pattern of interactions that agonists engage with residues surrounding the orthosteric site that are either conserved within the AR family or particular for a given subtype. We have both analyzed the binding modes of known agonists and predicted synthetic targets that were ultimately found to enhance affinity, for example, at the A3AR.
A focus on sterically constrained substitutes for the ribose ring identified the (N)-methanocarba modification (present in 20-23) as a general substitution that maintains an A3AR-preferred conformation and is compatible with AR activation and suitable for combination with other A3AR-enhancing modifications to further increase A3AR selectivity.[71] The preference for the ribose ring (N)-conformation in the AR-bound nucleosides has now been confirmed with the experimental structures of agonist- bound A2A and A1ARs.[74] The ribose ring substitution with a (N)-methanocarba ring system thus has the effect of constraining the agonists in a receptor-preferred conformation. Why this constraint is most effective for the A3AR is unclear. However, it does increase both affinity and selectivity at this subtype without reducing efficacy to activate the receptor. A side-by-side comparison of (N)-methanocarba and ribose equivalents confirmed that the (N)-methanocarba derivatives displayed significantly higher affinity and selectivity at the A3AR. A 5-fluorothien-2-yl analogue MRS7334 21 was identified as one of the most potent and selective (~40000-fold) A3AR agonists.[17] A structure-based approach comparing the behavior of MRS7334 21 and its ribose analogue (not shown) through MD simulations showed a higher variability of the ribose puckering over time, suggesting an entropic advantage when the ribose is substituted with a (N)-methanocarba ring system.[17]
The replacement of the ribose ring O of Cl-IB-MECA with S and Se, that is, 19a and 19b, is tolerated with only minor changes in A3AR affinity.[75] This was surprising because the Se analogue of Cl-IB-MECA 3 has a south conformation in its pure crystalline state, but in the AR binding site a north conformation is required, according to multiple A1AR and A2AAR structures. The A3AR contains most of the conserved H-bonding residues surrounding the ribose and adenine moieties. Therefore, we must conclude that to maintain conserved H-bonds of selenoadenosines in A3AR binding - the forces of interaction with the protein overcome the tendency to adopt a (S) conformation.
C2 extension in combination with the (N)-methanocarba modification achieves > 10000-fold selectivity.[17,76] This unusually high selectivity can be used to interrogate biological systems more effectively than the prototypical A3AR agonists IB-MECA and Cl-IB-MECA, which already have demonstrated examples of activating other AR subtypes, depending on the system studied and concentration.[71] How do these rigid features reconcile with the receptor structure? Receptor structural plasticity was proposed, with a displacement of TM2 and an outward movement from the TM bundle, that was not possible in the A2AAR due to the constraints from 3 disulfide bonds in ELs. This was accomplished by employing hybrid models, using an opsin, β2-adrenergic[77] or A1 adenosine[78] receptor structure for TM2, and an A2AAR structure for the rest of the receptor. This would permit docking of nucleoside ligands to the binding site while retaining the conserved agonist contacts observed in A1AR and A2AAR structures. The conserved features in the hybrid models are analogous to conserved nucleoside interactions in A1AR and A2AAR structures and include: a bidentate H-bond of N6.55 with the ligands′ N7 and N6H; a π-π stacking between the base aromatic scaffold and a phenylalanine in EL2; and two H-bonds between the ligands′ 2′ and 3′ hydroxy groups and the receptor’s H7.43 and S7.42, respectively (Figure 6). TM2 plasticity also enabled the introduction of bulky fluorophores in high affinity A3AR agonists. As for A1AR, a 1-deaza modification of adenine is favorable for A3AR affinity, but not 3-deaza or 7-deaza modifications because these two nitrogen atoms are engaged in conserved H-bonding.[48]
Figure 6.

Pose of MRS7591 23 at the A) human and B) mouse A3AR hybrid models, built using hA2AAR and hA1AR X-ray (for TM2) structures.[78] A) Last frame of a 30-ns MD simulation starting from the docking pose of MRS7591 (lime) at the hA3AR model (light gray). B) Last frame of a 30-ns MD simulation starting from the docking pose of MRS7591 (violet) at the mA3AR model (gray).
We successfully predicted likely sites on the A3AR agonist scaffold in which a permanently charged sulfonate group would enhance affinity, in order to prepare a tool compound that would not diffuse across biological membranes.[76] Guided by molecular modeling, a charged sulfonate group was strategically placed at the meta position of a C2-phenylethynyl group in MRS5841 (not shown) to maintain A3AR agonist ~nanomolar affinity and selectivity at both h and mA3ARs.
Also, we are able to reduce the A3AR efficacy by truncation or rigidifying the ribose moiety (5′-position) to provide low efficacy partial agonists or antagonists. Conversion of full agonists into partial agonists is particularly facile at the A3AR compared to other ARs. Truncation or rigidification of the ribose 5′ group, e.g. CH2OH or CONHCH3 for potent analogues, has been shown to reduce the A3AR efficacy, consistent with the ribose moiety requiring specific polar interactions with the receptor in its active state. However, this truncation often lowers mouse (m) A3AR affinity. To compensate for this loss, we noted that by strategically designing the N6 group of truncated analogues we could access polar groups that are particularly prominent in mA3AR ELs. Following MD simulation of agonists bound to hA3AR and mA3AR homology models, nucleoside synthesis confirmed the enhanced mA3AR affinity in a series of partial agonists bearing H-bonding (OH/OMe) groups at a distal point on a hydrophobic N6−2-phenylethyl substituent.[78] MD simulations of a 4′ truncated (N)-methanocarba nucleoside MRS7591 23 (Figure 3) have predicted its distal polar interaction at both h and mA3AR models (Figure 6). This was a means of using modeling-derived insights to maintain interspecies A3AR affinity.
Signaling bias and receptor residence time have been examined for rigidified A3AR agonists.[79] A rigid C2 extension, which we proposed to outwardly displace TM2, is a source of signaling bias in addition to increasing A3AR vs A1/A2AAR affinity. In collaboration with Lauren May (Monash University), we reported that some A3AR agonists containing rigid C2 extensions have a signaling bias for cell survival, compared to other downstream signaling readouts.[96] However, bias was not evident in comparing Gregion and β-arrestin pathways.[96]
Our prototypical A3AR agonists, IB-MECA (piclodenoson) and Cl-IB-MECA (namodenoson), have reached advanced clinical trials.[25,26] Perhaps the most exciting new potential clinical application of A3AR agonists is in the treatment of chronic neuropathic pain.[20,80] The history of adenosine’s involvement in pain began with the A1AR and A2AAR, and ironically A3AR agonists were dismissed. A1AR agonists and allosteric enhancers relieve pain. The receptors on primary afferents decrease the release of excitatory neurotransmitters in the dorsal root ganglions/dorsal horn. There were at least three unsuccessful attempts to implement A1AR activation as a pain treatment in clinical trials.[20] In addition to the undesired side effects of A1AR agonists (bradycardia, hypothermia), there was a lack of clinical efficacy.[16] Curiously, acupuncture was shown to involve the antinociceptive effects of endogenous adenosine by activating the A1AR.[80]
Early reports on A3AR’s role in pain were contradictory, and some review papers noted that “A3AR cause pain”.[20] However, it is now clear that A3AR agonists effectively reduce or prevent the development of chronic neuropathic pain. In collaboration with Daniela Salvemini at St. Louis University, we have studied >100 A3AR agonists in the mouse chronic constriction injury (CCI) pain model, which has been predictive of the clinical efficacy of later approved pain drugs.[81,82] As a phenotypic screen, this in vivo assay has directed the SAR exploration as a principal criterion along with binding affinity/selectivity. We have employed this in vivo phenotypic screen, in which the A3AR agonists are administered orally, to provide a window on both pharmacodynamic and pharmacokinetic factors. The synthesized adenosine derivatives were preselected to satisfy the binding site criteria. Although most of the compounds tested had very high A3AR affinity (roughly nanomolar), the precise binding affinity did not correlate well with in vivo efficacy and duration of action. Our leading candidates for use in chronic pain display favorable absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) properties. One such molecule, MRS5980 21 has good drug-like properties. These highly A3AR-selective agonists overcome the problem of cardiovascular side effects and other “toxicities” of earlier generations of A1AR and A2AAR agonists.[16]
Highly selective, rigidified agonists MRS5980 20 and MRS5698 22 have demonstrated efficacy in diverse pain models and with greater molar potency than several other widely used pain treatments. Comparison of their efficacy in the CCI model with the inactivity of MRS5698 and other agonists in the A3AR KO mouse confirms that activation of this receptor is the sole mechanism. The protective effects of A3AR agonists, even at a subthreshold dose, in chronic pain are also additive with the effects of known pharmacological agents used clinically for this condition. The protective effect of A3AR agonists did not desensitize, at least over 1 week of repeated or constant (delivered through implanted Alzet minipump) administration. It is very promising that A3AR agonists coadministered with morphine make the chronically dosed opioid safer and more efficacious.[83]
Not only do exogenously administered A3AR agonists reduce chronic pain, but there is a similar effect of endogenous adenosine acting at A3AR when its concentration is elevated by the administration of an inhibitor of intracellular adenosine kinase (ADK).[82] Also, unlike opioid pain treatments, there is no inherent reward of A3AR agonists as indicated in a conditioned place preference test in which rats spontaneously self-administer MRS5698 only when they are nerve-injured.[82] Moreover, A3AR agonists lessen the desensitization to opioids when used in subthreshold dose.
There are three possible sites of action for A3AR agonists in controlling pain: peripheral sensory afferent neurons, the spinal cord dorsal horn, and the brain. Mechanisms of A3AR-induced nociception were studied in the spinal cord, and the protection was found to be associated with normalization of imbalances in cytokine and glutamate signaling and with the reduction of nitro-oxidative pathways. The protection by A3AR agonists is IL-10 dependent, and the protection by Cl-IB-MECA and MRS5980 correlates with a block of N-type calcium channels in the spinal cord, that is, the site of action of a conotoxin inhibitor now used clinically for intense pain.[84,85] The action in the spinal cord is GABAA dependent (bicuculine, but not antagonists of opioids or cannabinoid receptors, block the A3AR effects). In the CNS, A3AR in the rostral ventromedial medulla (RVM) activates descending antinociceptive pathways.[82] A3AR agonists control chemotherapy-induced pain and bone metastasis pain in rodent models.[82,84] Oxaliplatin increases ADK in the spinal cord, which leads to activation of the nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3 (NLRP3) inflammasome, and this imbalance is attenuated by MRS5698.[84] With this multiplicity of mechanisms, the exploration of more specific A3AR is well justified, and we continue to apply structure-based methods to that end.
We have also introduced hA3AR PAMs having a 1H-imidazo [4,5-c]quinolin-4-amine scaffold.[86] The enhancement of A3AR agonists action was fully apparent at dog, rabbit and hA3ARs, but was barely detectable at mA3AR.
6. Conclusions
New structures of purinergic GPCRs have been determined and used in the design of novel ligands, including high-affinity fluorescent probes as tools for drug discovery. The P2YR structures had many unanticipated features, such as a dynamic disulfide bridge and unusually high conformational differences between agonist-bound and antagonist-bound states for P2Y12R. The P2Y1R has disparate binding sites for orthosteric and allosteric antagonists, and the presumed orthosteric site for nucleotides does not match the canonical Family A GPCR binding site. The first step in validating a GPCR modeling approach is the recapitulation of known SAR, which was done for the P2Y1R with docking and MD of 146 ligands. The analysis was consistent with mutagenesis results. We have used a structure-based design approach to discover a new triazole-based scaffold for hP2Y14R antagonists. The computation pipeline led us to design and synthesize new tools for understanding P2Y14R pharmacology. Structure-guided design approaches were used to enhance the affinity and selectivity of known A3AR agonists to achieve >10000-fold selectivity. Efficacy, in in vivo chronic pain models, of these highly selective agonists increased confidence in the assigned mechanism compared to use of previous moderately selective A3AR agonists. Their high potency and oral efficacy in vivo in chronic neuropathic pain models suggested they might be clinically useful drug candidates. Thus, structure-guided design has advanced the medicinal chemistry of adenosine and P2Y receptors, which have untapped potential for treatment of chronic diseases.
Acknowledgements
We thank the NIDDK Intramural Research Program for funding (ZIADK31116, ZIADK31117, ZIADK31126).
Biographies
Kenneth Jacobson obtained a B.A. from Reed College, a Ph.D. in chemistry from the University of California, San Diego, and completed postdoctoral studies in organic chemistry at the Weizmann Institute. He is currently John W. Daly Distinguished Scientist and Chief of the Molecular Recognition Section, Laboratory of Bioorganic Chemistry at the National Institute of Diabetes and Digestive and Kidney Diseases. He is interested in medicinal chemistry of G protein-coupled receptors, in particular receptors for adenosine and for purine and pyrimidine nucleotides, and has introduced many of the widely used purinergic receptor pharmacological probes, including several compounds in clinical trials for inflammatory and liver diseases.

Veronica Salmaso earned a Master’s Degree in chemistry and pharmaceutical techniques and a Ph.D. in molecular sciences-pharmaceutical sciences, under the supervision of Prof. Stefano Moro at the University of Padova. She is currently a postdoctoral fellow in K.A.J.’s group at NIH, exploring computational chemistry in a multidisciplinary team, with the goal to develop new purinergic receptors agonists and antagonists (P2Y receptors and adenosine receptors). Her scientific interests include rational drug design of G protein-coupled receptors (GPCRs) agonists and antagonists, with expertise in docking, virtual screening, homology modeling and molecular dynamics.

Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Congreve M, de Graaf C, Swain NA, Tate CG, Cell 2020, 181, 81–91. [DOI] [PubMed] [Google Scholar]
- [2].Fuentes AV, Pineda MD, Nagulapalli Venkata KC, Pharmacy (Basel) 2018, 6, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Novel Drug Approvals for 2019. Accessed: Dec. 29, 2019: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2019.
- [4].Chen JF, Cunha R, Purinergic Signal. 2020, 16, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC, Science 2007, 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Experimentally solved GPCR structures. Accessed: June 20, 2020: http://costanziresearch.blogspot.com/p/table.html.
- [7].Congreve M, de Graaf C, Swain NA, Tate CG, Cell 2020, 181, 81–91. [DOI] [PubMed] [Google Scholar]
- [8].Deflorian F, Mason JS, Bortolato A, Tehan BG, Impact of Recently Determined Crystallographic Structures of GPCRs on Drug Discovery. In Structural Biology in Drug Discovery, Renaud J-P (Ed.). 2020, doi: 10.1002/9781118681121.ch19. [DOI] [Google Scholar]
- [9].Jazayeri A, Andrews SP, Marshall FH, Chem. Rev 2017, 117, 21–37. [DOI] [PubMed] [Google Scholar]
- [10].Salmaso V, Jacobson KA, Biomolecules 2020, 10, 812, doi: 10.3390/biom10060812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Popov P, Peng Y, Shen L, Stevens RC, Cherezov V, Liu Z-J, Katritch V, ELife, 2018, 7, e34729. 10.7554/eLife.34729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gregory KJ, Bridges TM, Gogliotti RG, Stauffer SR, Noetzel MJ, Jones CK, Lindsley CW, Conn PJ, Niswender CM, ACS Pharmacol. Transl. Sci 2019, 2,442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Antonioli L, Blandizzi C, Pacher P, Haskó G, Pharmacol. Rev 2019, 71, 345–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dal Ben D, Antonioli L, Lambertucci C, Spinaci A, Fornai M, D’Antongiovanni V, Pellegrini C, Blandizzi C, Volpini R, Exp. Opin. Drug Disc 2020, 1–17. [DOI] [PubMed] [Google Scholar]
- [15].Müller CE, Baqi Y, Namasivayam V, Methods Mol. Biol 2020, 2041, 45–64. [DOI] [PubMed] [Google Scholar]
- [16].Jacobson KA, Tosh DK, Jain S, Gao ZG, Frontiers Cell. Neurosci 2019, 13, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tosh DK, Salmaso V, Rao H, Campbell R, Bitant A, Gao ZG, Auchampach JA, Jacobson KA, ACS Med. Chem. Lett 2020, DOI: 10.1021/acsmedchemlett.9b00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jung YH, Yu J, Wen Z, Salmaso V, Karcz TP, Phung NB, Chen Z, Duca S, Bennett JM, Dudas S, Cook DN, Salvemini D, Gao ZG, Jacobson KA, J. Med. Chem 2020, 63, 9563–9589, DOI: 10.1021/acs.jmed-chem.0c00745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Gao ZG, Jacobson KA, Front. Pharmacol 2017, 8, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Battistone MA, Mendelsohn AC, Spallanzani RG, Allegretti AS, Liberman RN, Sesma J, Kalim S, Wall SM, Bonventre JV, Lazarowski ER, Brown D, Breton S, J. Clin. Invest 2020, 130, 3734–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jacobson KA, Giancotti LA, Lauro F, Mufti F, Salvemini D, Pain 2020, 161, 1425–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Junker A, Balasubramanian R, Ciancetta A, Uliassi E, Kiselev E, Martiriggiano C, Trujillo K, Mtchedlidze G, Birdwell L, Brown KA, Harden TK, Jacobson KA, J. Med. Chem 2016, 59, 6149–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yu J, Ciancetta A, Dudas S, Duca S, Lottermoser J, Jacobson KA, J. Med. Chem 2018, 61, 4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Conroy S, Kindon N, Kellam B, Stocks MJ, J. Med. Chem 2016, 59, 9981–10005. [DOI] [PubMed] [Google Scholar]
- [23].Jacobson KA, Bioconjugate Chem. 2009, 20, 1816–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jacobson KA, Kirk KL, Daly JW in Physiology and Pharmacology of Adenosine and Adenine Nucleotides (Ed.: Paton D) Taylor and Francis, London, 1988, pp. 27–38. [Google Scholar]
- [25].Fishman P, Cohen S, Clin. Rheumatol 2016, 35, 2359–2362. [DOI] [PubMed] [Google Scholar]
- [26].Fishman P, Cohen S, Itzhak I, Amer J, Salhab A, Barer F, Safadi R, Int. J. Mol. Med 2019, 44, 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Borea PA, Gessi S, Merighi S, Vicenzi F, Varani K, Physiol. Rev 2018, 98,1591–1625. [DOI] [PubMed] [Google Scholar]
- [28].Junker A, Renn C, Dobelmann C, Namasivayam V, Jain S, Losenkova K, Irjala H, Duca S, Balasubramanian R, Chakraborty S, Börgel F, Zimmermann H, Yegutkin GG, Müller CE, Jacobson KA, J. Med. Chem 2019, 62, 3677–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Toti K, Osborne D, Ciancetta A, Boison D, Jacobson KA, J. Med. Chem 2016, 59, 6860–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC, Science 2008, 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC, Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG, Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang K, Zhang J, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q, Nature 2014, 509, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Müller CE, Yang H, Cherezov V, Katritch V, Han GW, Jacobson KA, Stevens RC, Wu B, Zhao Q, Nature, 2014, 509, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, Han GW, Liu H, Cherezov V, Katritch V, Jiang H, Stevens RC, Jacobson KA, Zhao Q, Wu B, Nature, 2015, 520, 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Glukhova A, Thal DM, Nguyen AT, Vecchio EA, Jörg M, Scammells PJ, May LT, Sexton PM, Christopoulos A, Cell 2017, 168, 867–877. [DOI] [PubMed] [Google Scholar]
- [37].Draper-Joyce CJ, Khoshouei M, Thal DM, Liang Y-L, Nguyen ATN, Furness SGB, Venugopal H, Baltos J-A, Plitzko JM, Danev R, Baumeister W, May LT, Wootten D, Sexton PM, Glukhova A, Christopoulos A, Nature 2018, 558, 559–563. [DOI] [PubMed] [Google Scholar]
- [38].Jespers W, Oliveira A, Prieto-Díaz R, Majellaro M, Åqvist J, Sotelo E, Gutiérrez-de-Terán H, Molecules 2017, 22, 1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Carpenter B, Nehmé R, Warne T, Leslie AGW, Tate CG, Nature 2016, 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].García-Nafría J, Lee Y, Bai X, Carpenter B, Tate CG, eLife 2018, 7, e35946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cheng RKY, Segala E, Robertson N, Deflorian F, Doré AS, Errey JC, Fiez-Vandal C, Marshall FH, Cooke RM, Structure 2017, 25, 1275–1285. [DOI] [PubMed] [Google Scholar]
- [43].Chan HCS, Li Y, Dahoun T, Vogel H, Yuan S, Trends Biochem. Sci 2019, 44, 312–330. [DOI] [PubMed] [Google Scholar]
- [44].Katritch V, Jaakola VP, Lane JR, Lin J, IJzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R, J. Med. Chem 2010, 53, 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lenselink EB, Beuming T, van Veen C, et al. J. Comput.-Aided Mol. Des 2016, 30, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Carlsson J, Yoo L, Gao ZG, Irwin J, Shoichet B, Jacobson KA, J. Med. Chem 2010, 53, 3748–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rodríguez D, Gao ZG, Moss SM, Jacobson KA, Carlsson J, J. Chem. Inf. Model 2015, 55, 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rodríguez D, Chakraborty S, Warnick E, Crane S, Gao ZG, O’Connor RO, Jacobson KA, Carlsson J, ACS Chem. Biol 2016, 11, 2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Langmead CJ, Andrews SP, Congreve M, Errey JC, Hurrell E, Marshall FH, Mason JS, Richardson CM, Robertson N, Zhukov A, Weir M, J. Med. Chem 2012, 55, 1904–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Katritch V, Kufareva I, Abagyan R, Neuropharmacology 2011, 60, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Congreve M, Andrews SP, Doré AS, et al. , J. Med. Chem 2012, 55, 1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Léon C, Hechler B, Freund M, Eckly A, Vial C, Ohlmann P, Dierich A, LeMeur M, Cazenave JP, Gachet C, J. Clin. Invest 1999, 104, 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Reichenbach N, Delekate A, Breithausen B, Keppler K, Poll S, Schulte T, Peter J, Plescher M, Hansen JN, Blank N, Keller A, Fuhrmann M, Henneberger C, Halle A, Petzold GC, J. Exp. Med 2018, 215, 1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Simões AP, Silva CG, Marques JM, Pochmann D, Porciúncula LO, Ferreira S, Oses JP, Beleza RO, Real JI, Köfalvi A, Bahr BA, Lerma J, Cunha RA, Rodrigue RJ, Cell Death Dis. 2018, 9, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bird JE, Wang X, Smith PL, Barbera F, Huang C, Schumacher WA, J. Thromb. Thrombolysis 2012, 34, 199–207. [DOI] [PubMed] [Google Scholar]
- [56].Wong PC, Watson C, Crain EJ, J. Thromb. Thrombolysis 2016, 41, 514–521. [DOI] [PubMed] [Google Scholar]
- [57].Ciancetta A, Paoletta S, Jacobson KA, J. Chem. Inf. Model 2017, 57, 3104–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].a) Chao H, Turdi H, Herpin TF, Roberge JY, Liu Y, Schnur DM, Poss MA, Rehfuss R, Hua J, Wu Q, Price LA, Abell LM, Schumacher WA, Bostwick JS, Steinbacher TE, Stewart AB, Ogletree ML, Huang CS, Chang M, Cacace AM, Arcuri MJ, Celani D, Wexler RR, Lawrence RM, J. Med. Chem 2013, 56, 1704–1714; [DOI] [PubMed] [Google Scholar]; b) Qiao JX, Wang TC, Hiebert S, Hu CH, Schumacher WA, Spronk SA, Clark CG, Han Y, Hua J, Price LA, Shen H, Chacko SA, Everlof G, Bostwick JS, Steinbacher TE, Li YX, Huang CS, Seiffert DA, Rehfuss R, Wexler RR, Lam PYS, ChemMedChem 2014, 9, 2327–2343. [DOI] [PubMed] [Google Scholar]
- [59].Gao ZG, Jacobson KA, Mol. Pharmacol 2017, 92, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ferreira MAR, Jansen R, Willemsen G, Penninx B, Bain LM, Vicente CT, Revez JA, Matheson MC, Hui J, Tung JY, Baltic S, Le Souëf P, Montgomery GW, Martin NG, Robertson CF, James A, Thompson PJ, Boomsma DI, Hopper JL, Hinds DA, Werde RB, Phipps S, J. Allergy 2017, 139, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sesma JI, Weitzer CD, Livraghi-Butrico A, Dang H, Donaldson S, Alexis NE, Jacobson KA, Harden TK, Lazarowski ER, Purinergic Signal. 2016, 12, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Trujillo K, Paoletta S, Kiselev E, Jacobson KA, Bioorg. Med. Chem 2015, 23, 4056–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kiselev E, Balasubramanian R, Uliassi E, Brown KA, Trujillo K, Katritch V, Hammes E, Stevens RC, Harden TK, Jacobson KA, Bioorg. Med. Chem. Lett 2015, 25, 4733–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kiselev E, Barrett M, Katritch V, Paoletta S, Weitzer CD, Hammes E, Yin AL, Zhao Q, Stevens RC, Harden TK, Jacobson KA, ACS Chem. Biol 2014, 9,2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Xiao C, Liu N, Jacobson KA, Gavrilova O, Reitman ML, PLoS Biol. 2019, 17, e3000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun B, Bachhawat P, Chu ML-H, Wood M, Ceska T, Sands ZA, Mercier J, Lebon F, Kobilka TS, Kobilka BK, Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Eddy MT, Lee MY, Gao ZG, White KL, Didenko T, Horst R, Audet M, Stanczak P, McClary KM, Han GW, Jacobson KA, Stevens RC, Wüthrich K, Cell 2018, 172, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Eddy MT, Gao ZG, Mannes P, Patel N, Jacobson KA, Katritch V, Stevens RC, Wüthrich K, J. Am. Chem. Soc 2018, 140, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].White KL, Eddy MT, Gao ZG, Han GW, Lian T, Deary A, Patel N, Jacobson KA, Katritch V, Stevens RC, Structure 2018, 26, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ciancetta A, Rubio P, Lieberman DI, Jacobson KA, J. Comput.-Aided Mol. Des 2019, 33, 983–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jacobson KA, Tosh DK, Gao ZG, Yu J, Suresh RR, Rao H, Romagnoli R, Baraldi PG, Tabrizi MA in The Receptors, Vol. 34: The Adenosine Receptors (Ed.: Varani K), Springer, 2018, pp. 169–198. [Google Scholar]
- [72].Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O, Proc. Natl. Acad. Sci. USA 1992, 89, 7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG, Proc. Natl. Acad. Sci. USA 1993, 90,10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Salmaso V, Jacobson KA, Nucleosides Nucleotides Nucleic Acids 2020, 39,322–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yu J, Zhao LX, Park J, Lee HW, Sahu P, Cui M, Moss S, Hammes E, Warnick G, Gao ZG, Noh M, Choi S, Ahn HC, Choi J, Jacobson KA, Jeong LS, J. Med. Chem 2017, 60, 3422–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].a) Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski E, Auchampach JA, Salvemini D, Jacobson KA, J. Med. Chem 2014, 57, 9901–9914; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Paoletta S, Tosh DK, Finley A, Gizewski E, Moss SM, Gao ZG, Auchampach JA, Salvemini D, Jacobson KA, J. Med. Chem 2013, 56, 5949–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA, J. Med. Chem 2012, 55, 4847–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tosh DK, Salmaso V, Rao H, Bitant A, Fisher CL, Lieberman DI, Vorbrüggen H, Reitman ML, Gavrilova O, Gao ZG, et al. , J. Med. Chem 2020, 63, 4334–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Xia L, Kyrizaki A, Tosh DK, van Duijl TT, Roorda JC, Jacobson KA, IJzerman AP, Heitman LH, Biochem. Pharmacol 2018, 153, 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Goldman N, Chen M, Fujita T, Xu Q, Peng W, Liu W, Jensen TK, Pei Y, Wang F, Han X, Chen JF, Schnermann J, Takano T, Bekar L, Tieu K, Nedergaard M, Nat. Neurosci. 2010, 13, 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bennett GJ, Xie YK, Pain 1988, 33, 87–107. [DOI] [PubMed] [Google Scholar]
- [82].Janes K, Symons-Liguori AM, Jacobson KA, Br. J. Pharmacol 2016, 173, 1253–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Doyle TM, Largent-Milnes TM, Chen Z, Staikopoulos V, Esposito E, Dalgarno R, Fan C, Tosh DK, Cuzzocrea S, Jacobson KA, Trang T, Hutchinson MR, Bennett GJ, Vanderah TW, Salvemini D, J. Pharmacol. Exp. Ther 2020, DOI: 10.1124/jpet.120.000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wahlman C, Doyle T, Little JW, Luongo L, Janes K, Chen Z, Espostio E, Tosh DK, Cuzzocrea S, Jacobson KA, Salvemini D, Pain 2018, 159, 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Coppi E, Cherchi F, Fusco I, Failli P, Vona A, Dettori I, Gaviano L, Lucarini E, Jacobson KA, Tosh DK, Salvemini D, Ghelardini C, Pedata F, Di Cesare Mannelli L, Pugliese AM, Pain 2019, 160, 1103–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Du L, Gao ZG, Paoletta S, Wan TC, Barbour S, van Veldhoven JP, IJzerman AP, Jacobson KA, Auchampach JA, Purinergic Signal. 2018, 14,59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Batyuk A, Galli L, Ishchenko A, Han GW, Gati C, Popov PA, Cherezov V, Sci. Adv 2016, 2, e1600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mallo-Abreu A, Prieto-Diaz R, Jespers W, Azuaje J, Majellaro M, Velando C, García-Mera X, Caamaño O, Brea J, Loza MI, Gutiérrez-de-Terán H, Sotelo E, J. Med. Chem 2020, 63, 7721–7739. [DOI] [PubMed] [Google Scholar]
- [89].Raschka S, Kaufman B, Machine learning and AI-based approaches for bioactive ligand discovery and GPCR-ligand recognition. arXiv preprint arXiv:2001.06545. 2020. January 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Potterton A, Husseini FS, Southey MWY, Bodkin MJ, Heifetz A, Coveney PV, Townsend-Nicholson A, J. Chem. Theory Comput. 2019, 15,3316–3330. [DOI] [PubMed] [Google Scholar]
- [91].Lu Y, Qin S, Zhang B, Dai A, Cai X, Ma M, Gao ZG, Yang D, Stevens R, Jacobson KA, Wang MW, Shui W, Analyt. Chem 2019, 91, 8162–8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Du L, Gao ZG, Paoletta S, Wan TC, Barbour S, van Veldhoven JP, IJzerman AP, Jacobson KA, Auchampach JA, Purinergic Signal. 2018, 14,59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Baltos JA, Gregory KJ, White PJ, Sexton PM, Christopoulos A, May LT, Biochem. Pharmacol 2016, 99, 101–112. [DOI] [PubMed] [Google Scholar]
- [94].Federico S, Lassiani L, Spalluto G, Pharmaceuticals 2019, 12, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dix AV, Moss SM, Phan K, Hoppe T, Paoletta S, Kozma E, Gao ZG, Durell SR, Jacobson KA, Appella DH, J. Am. Chem. Soc 2014, 136, 12296–12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].a) Baltos JA, Paoletta S, Nguyen ATN, Gregory KJ, Tosh DK, Christopoulos A, Jacobson KA, May L, Mol. Pharmacol 2016, 90, 12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Storme J, Tosh DK, Gao ZG, Jacobson KA, Stove CP, Biochem. Pharmacol. 2018, 158, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Baltos JA, Vecchio EA, Harris MA, Qin CX, Ritchie RH, Christopoulos A, White PJ, May LT, Biochem. Pharmacol. 2017, 135, 79–89. [DOI] [PubMed] [Google Scholar]
- [98].Shchepinova MM, Hanyaloglu AC, Frost GS, Tate EW, Curr. Opin. Chem. Biol. 2020, 56, 98–110. [DOI] [PubMed] [Google Scholar]
- [99].Therapeutic Uses of A3 Adenosine Receptor Antibodies, Fishman P (Can-Fite Biopharma Ltd.), WO/2007/063539, 2007. [Google Scholar]
- [100].Yu J, Ahn S, Kim HJ, Lee M, Ahn S, Kim J, Jin SH, Lee E, Kim G, Cheong JH, Jacobson KA, Jeong LS, Noh M, J. Med. Chem. 2017, 60, 7459–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Taura J, Nolen EG, Cabré G, Hernando J, Squarcialupi L, López-Cano M, Jacobson KA, Fernández-Dueñas V, Ciruela F, J. Controlled Release 2018, 283,135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]