

Abstract
Focal adhesion kinase (FAK) is both a non-receptor tyrosine kinase and an adaptor protein that primarily regulates adhesion signalling and cell migration, but FAK can also promote cell survival in response to stress. FAK impacts on a diverse range of cancer cell functions mediated by both its kinase and scaffolding functions. It is commonly overexpressed in cancer and has been considered a high value druggable target for therapy, with multiple FAK kinase inhibitors currently in development. Substantial evidence has emerged implying that it is as clinical combination strategies that FAK targeting will be most effective, so as to reverse failure of chemotherapies or targeted therapies and enhance efficacy of immune-based treatments of solid tumours. Here we discuss the recent pre-clinical evidence that implicates FAK in anti-cancer therapeutic resistance, leading to the view that FAK kinase inhibitors will have their greatest utility as combination therapies in stratified contexts.
Introduction
Focal adhesion kinase (FAK; gene name PTK2) is a non-receptor tyrosine kinase that classically transduces signalling from cell adhesions to regulate multiple biological cellular functions, including cell survival, migration and invasion of cancer cells (Box 1) – activities of FAK that have been reviewed extensively1,2. Briefly, engagement of transmembrane integrin receptors to extracellular matrix recruits FAK to sites where integrins cluster, termed focal adhesions [G]. FAK does not interact with integrins directly, but instead binds to the membrane and to other focal adhesion proteins such as paxillin and talin through its C-terminal FAT domain3. Once recruited to focal adhesions, FAK becomes catalytically active in a multi-step process. Initially, inactive FAK forms dimers that interact with phosphatidylinositol 4,5-bisphosphate-rich membranes that dissociates the autoinhibitory interaction between the FERM (4.1 protein, ezrin, radixin, moesin) [G] and kinase domains and, exposes the autophosphorylation site tyrosine 397 (Y397) for trans-autophosphorylation4. Once phosphorylated, FAK acts as a molecular scaffold and recruits SRC kinase(s) to further phosphorylate FAK on Y576 and Y577 in the activation loop4. Autophosphorylation of FAK on Y397 is a major target of its catalytic activity and as this is a key step in its priming for activation, as well as for subsequent signalling from focal adhesions, it makes it an attractive target for anti-cancer therapy. FAK also acts as an adaptor protein at focal adhesions, which can be independent of its kinase activity, to recruit other proteins to link through to the actin cytoskeleton (Box 1).
Text Box 1.
Focal Adhesion Kinase (FAK) was first discovered as a heavily tyrosine phosphorylated protein located at focal adhesions, the points of cellular plasma membranes that link to extracellular matrix (ECM) via transmembrane receptors, typically integrin heterodimers1,2,119–121. Upon integrin-ECM engagement, FAK clusters and is activated in a complex series of increasingly well-documented events involving lipid binding, conformational switches, tyrosine phosphorylation (and perhaps other post-translational) events, culminating in full catalytic activation. FAK is also an adaptor protein, and multiple domains, including the so-called FERM domain47, are involved in the binding of key cellular protein partners and their co-recruitment into larger heteromeric protein complexes. From integrin-induced nascent focal complexes or more stable, larger focal adhesions, FAK signals into the cell interior, so controlling adhesion, migration, cancer cell invasion, survival and proliferation. FAK expression is often increased in solid epithelial cancers and is required for tumour development that results in malignant behaviour1,2,9–11.
In regard of FAK’s adaptor functions, these have long been recognised47; specifically, at cell-ECM adhesion sites, FAK is involved in scaffolding molecular complexes that link to actin and adhesion regulatory networks, so promoting cellular dynamics and enhanced migration and cancer cell invasion113,114. More recently, it has become clear that FAK also scaffolds transcriptional regulatory complexes in the nucleus16,46, underlining the importance of FAK’s adaptor functions in cell biology. However, as yet these protein-protein interactions have not been directly targeted for therapeutic purposes, but it is likely that some of FAK’s vital scaffolding interactions are regulated by its kinase activity via protein conformational switches114.
As mentioned above, FAK not only resides at peripheral focal adhesions, but it can also be found in the nucleus of cancer cells15,16,46,122. Nuclear localisation of FAK is evident from biochemical fractionation studies and may be a function of cellular response to stress. FAK functions in the nucleus by scaffolding transcriptional regulators, including p53 in some scenarios15, and influencing the transcription and expression of biologically important target genes, such as chemokines that influence the tumour microenvironment and anti-tumour immunity16,45.
Little is yet known about why and how FAK translocates to the nucleus or whether the nuclear pools of FAK overlap with those at peripheral focal adhesions. One hypothesis is that FAK is part of stress-sensing and -response machinery at the cell periphery, and that upon receipt of specific cues in the cellular environment, FAK moves to the nucleus to perform functions that buffer cells against stress-induced cell death. This may be part of (perhaps multiple) mechanisms by which elevated FAK expression and activity contribute to tumour cell survival, including under unfavourable conditions, such as after chemotherapy treatment37. It is equally possible that FAK’s biological activity, including its kinase and/or scaffolding activity at cell adhesions is important.
FAK is rarely mutated in cancer, most frequently occurring in cancers arising from the uterus (9.24% of samples), colon (7.25%) and liver (3.92%) with simple somatic mutations resulting in a missense, stop or frameshift5. In addition to the low frequency, mutations in the PTK2 gene encoding FAK show no preference for the position or functional domain. However, FAK expression is very commonly increased or ‘gene-amplified’ in a number of cancers1,2, with ovarian (including high grade serious ovarian carcinoma; HGSOC), lung squamous cell neoplasms, oesophagus and uveal melanoma tumours displaying >20% of samples with an increased copy number of FAK and is predictive of poor patient outcome (TCGA Research Network)5–8.
As first shown by Agochiya et al. in 19999, many cell lines derived from invasive epithelial tumours have increased copies PTK2, located at chromosome 8q24.3. Frequent gains or amplifications of 8q24 in tumours10,11 include increased copies of the MYC gene12, an important oncogene in many cancers13. The amplification of this chromosomal region has thus been suggested to be MYC-associated. Yet, in some tumours, such as head and neck squamous cell carcinoma, PTK2 gains occur more frequently than MYC, and it cannot therefore be assumed that it is always, or only, elevated MYC contributing to cancer phenotypes6–8.
FAK activation in tumours occurs via the well described mechanisms following engagement of integrin-mediated cell adhesions or, for example by activating mutations in heterotrimeric G-proteins [G] in uveal melanoma or mutations of RHOA [G] in diffuse gastric cancer (see below); both of these activate RHOA and FAK-dependent focal adhesion signalling7,14. FAK also has what are considered non-canonical roles in cancer cells that are experiencing ‘cellular stress’, and nuclear functions have been described to regulate p53 and cytokine expression15,16.
PYK2 is a closely related paralogue of FAK sharing ~48% amino acid similarity and shares analogous structural domain organisation and many protein binding partners2,17. Unlike FAK, PYK2 displays copy number variation losses in cancer, most frequently in ovarian (17.4% of cases), prostate (14.2%) and breast (8.21%)13. FAK and PYK2 can have redundant roles, for example PYK2, like FAK, is overexpressed in a mutant-KRAS model of PDAC where it regulates Wnt-β-catenin signalling during intestinal tumorigenesis, PDAC formation and maintenance18. However, FAK and PYK2 can also perform distinct roles, for example PYK2 does not localise to focal adhesions19,20 and PYK2 activation inhibits cell cycle progression17,21. FAK knockout or pharmacological inhibition can increase PYK2 expression or phosphorylation in both normal and cancer cells22–24. As regards PYK2, FAK kinase inhibitors fall into two categories: FAK-specific inhibitors (e.g. GSK2256098) or dual FAK/PYK2 inhibitors (e.g. VS-4718 or PF-573228). Both types of FAK inhibitors have been tested clinically and it is not clear whether using FAK or dual FAK/PYK2 kinase inhibitors would provide any specific differences in clinical efficacy.
Despite the accumulated evidence that FAK plays important roles in cancer progression18,25–29, single agent FAK kinase inhibitor clinical trials have only occasionally resulted in stabilisation of disease30–33. Given the genetic, cellular and stromal complexity of advanced solid tumours, it is perhaps not surprising monotherapies that target single signal transduction proteins, or pathways, are often unsuccessful; exceptions to this occur when single agents target known molecular drivers of disease, such as the use of BRAF inhibitors in BRAF-mutant melanoma34. Melanoma frequently (~60%) harbour activating mutations (V600E) in the BRAF protein and although the successful development of pharmacological inhibitors, such as vemurafenib that specifically targets this mutated form of BRAF, have performed well clinically, patients often relapse due to a number of now well-documented genetic and non-genetic resistance mechanisms35,36.
FAK inhibition has recently been identified as a potential strategy to overcome adaptive resistance to chemo-37,38, radio-26,39–41, or targeted therapies, including the use of BRAF inhibitors in BRAF-mutant cancers42–44, or therapies that target the immune microenvironment16,38,45,46. There are now excellent examples of biological scenarios in which FAK is a key mediator of therapeutic resistance, including for both tumour cell survival signalling mechanisms and for influence over the tumour microenvironment [G], or both. Indeed, it could be argued that FAK inhibitors used as a monotherapy may be likely to supress the more classical adhesion and migration roles of FAK as primary effects, which is perhaps unlikely to have a major impact on the outcomes of patients with advanced cancers; an aspiration of combination therapeutics would therefore be to also specifically target the roles of FAK in buffering therapeutic stress and so trigger cancer cell death or immune response. Therefore, there is growing interest in how best to use FAK kinase inhibitors as therapeutic combinations in clinical trials. Here, we review recent insights that highlight FAK’s role in regulating inherent and acquired resistance to anti-tumour therapies.
Combination therapies targeting FAK
The critical role of autophosphorylation in FAK activation has led to the development and clinical testing of a number of therapies that target FAK’s catalytic activity; defactinib, IN10018, VS-4718, GSK2256098 and PF-5732281,2,47. Of these FAK kinase inhibitors, two are currently under clinical evaluation in a number of phase I/II combination trials (see Table 1 and 2). The clinical development of VS-4718 has been discontinued and GSK2256098 currently is not in active clinical trials. Defactinib (VS-6063, PF-04554878) and IN10018 (BI 853520) both exhibit FAK kinase inhibition in vitro at low nanomolar concentrations (0.4–0.6 nM and 1 nM, respectively)32,48. Defactinib was developed as a second generation FAK inhibitor, and, in addition to FAK, inhibits 9 other kinases in vitro IC50 < 1 μM49,50. By contrast, IN10018 is a more specific FAK kinase inhibitor, co-targeting only four other kinases (of 262 tested) with an in vitro IC50 < 1 μM48. IN10018 is selective for FAK (PYK2 IC50 = 2 – 50 μM48) while defactinib inhibits PYK2 at low nanomolar concentrations (IC50 ranging from 0.632 to 423.4 nM49,50).
Table 1.
Kinase selectivity of current clinical FAK inhibitors.
| Compound | IC50 values for in vitro kinase inhibition [nM] (assay type) | # kinases with IC50 < 1 μM | Reference | |
|---|---|---|---|---|
| FAK | PYK2 | |||
| IN10018 (BI 853520) | 1 (DELFIA) 38.1 (Z-LYTE™) |
>50,000 (DELFIA) 2000 (Z-LYTE™) |
4 | 48 |
| Defactinib (VS-6063) (PF-04554878) | 0.47 (Kinobeads) 0.6 (Kinase assay) |
423 (Kinobeads) 0.6 (Kinase assay) |
9 | 32,49 |
Table 2.
Active clinical trials with FAK inhibitor combinations.
| Compound | Clinical trials | Cancer type | Combination agents | Molecular targets |
|---|---|---|---|---|
| Defactinib (VS-6063) (PF-04554878) |
NCT02546531 (Phase I)93 |
Solid Tumours / Advanced Solid Tumours Pancreatic Cancer | Pembrolizumab / gemcitabine | PD-1 |
|
NCT02758587 (Phase I/IIA)90 |
Non-Small Cell Lung Carcinoma, Mesothelioma, Pancreatic Neoplasms | Pembrolizumab | PD-1 | |
|
NCT03287271 (Phase I/II)67 |
Ovarian Cancer | Paclitaxel and carboplatin | - | |
|
NCT03727880 (Phase II)91 |
Pancreatic Ductal Adenocarcinoma | Pembrolizumab | PD-1 | |
|
NCT03875820 (Phase I)124 |
Advanced RAS mutant solid tumours; Non-Small Cell Lung Carcinoma, Low Grade Serous Ovarian Cancer, Colorectal Cancer, other RAS mutant solid tumours. | VS-6766 | MEK/RAF | |
|
NCT04201145 (Phase Ia-Ib)92 |
Malignant Pleural Mesothelioma | Pembrolizumab | PD-1 | |
|
NCT04620330 (Phase II)126 |
Non-Small Cell Lung Cancer with a KRAS Activating Mutation | VS-6766 | MEK/RAF | |
|
NCT04625270 (Phase II)125 |
Ovarian Cancer | VS-6766 | MEK/RAF | |
|
NCT04331041 (Phase II)129 |
Pancreatic Cancer | Radiotherapy | - | |
| IN10018 (BI 853520) |
NCT04109456 (Phase Ib)127 |
Metastatic uveal melanoma or cutaneous NRAS-mutant melanoma | Cobimetinib | MEK |
As mentioned above, FAK kinase inhibitors have performed poorly to date when tested as single agents for the treatment of cancer30–33. However, FAK is clearly an important signalling ‘hub’ through which cancer cells buffer stress following treatment with chemo-37,38, radio-39–41, or targeted therapies42–44. FAK is therefore a rational target for clinical combinations, where co-targeting may reveal vulnerabilities that are not evident from using FAK inhibitors, or specific therapeutics, alone - especially as FAK kinase inhibitors are orally administered and well tolerated clinically2,30–32. However, this requires strong, scientifically-evidenced combination hypotheses and clear strategies for patient stratification, without which combination therapy approaches are likely to fail. Early combination trials in patients with advanced or refractory ovarian cancer resulted in partial responses to the combination of defactinib and paclitaxel2,51, while early results from a trial of the FAK-specific inhibitor, GSK2256098 in combination with the MEK inhibitor, trametinib in patients with advanced pancreatic cancer showed no activity, with only one patient (out of 11 evaluated) exhibiting stable disease52. In addition, in patients with advanced solid tumours with mitogen-activated protein kinase (MAPK) pathway activation, including mesothelioma, a combination of GSK2256098 and trametinib failed to show any improvement over GSK2256098 monotherapy, with the best response being disease stabilization in 13 patients (38%)53. Analysis of NF2 (also called Merlin)-negative mesothelioma patients in this trial, which was hypothesized to predict sensitivity to FAK inhibitors in vitro54, also revealed no clinical benefit from GSK2256098 and trametinib combination53.
Hence, a greater understanding of how FAK signalling interacts with druggable oncogenic drivers [G], and FAK’s role in adaptive and acquired resistance to anti-cancer therapies, is required to improve stratification; in particular, identifying the tumour types and patients that may benefit from specific FAK kinase inhibitor combinations will be enormously helpful. Therefore, we now describe preclinical studies that provide some rationale for use of FAK inhibitor combinations because they highlight where FAK plays an important role in the survival and growth of a particular cancer after therapeutic intervention; in turn, each of these preclinical studies have led to clinical trials focussed on using FAK inhibitors (defactinib or IN10018) in combination with RAF/MEK inhibitors, anti-PD-1 immunotherapy, and chemo- or radiotherapy (see Table 2). We select key exemplars of FAK-mediated control of resistance to therapy that represent exciting co-targeting opportunities.
FAK mediates resistance to therapy
FAK connections to chemotherapy resistance.
More than any other cancer, FAK is amplified in HGSOC, the subtype of ovarian cancer accounting for the greatest mortality55. Importantly, the expression of FAK mRNA is concordant with PTK2 gains, and this is associated with tumour progression and poorer HGSOC patient prognosis2,37. In HGSOC, the copy number of FAK corresponds to FAK transcript levels, while such an association is not true for MYC56,57. As we now know that cellular roles for FAK extend beyond acting to promote cell adhesion and motility, HGSOC represents a good cancer system in which to dissect connections between FAK and its role in malignancy.
Standard of care for HGSOC patients is treatment with cytoreductive surgery followed by carboplatin (to induce DNA damage) and paclitaxel (to stabilize microtubules) chemotherapy to kill residual tumour cells58,59. In contrast to immunohistochemical staining of pancreatic cancer, which displays high levels of FAK tyrosine phosphorylation in the stromal cells [G] surrounding tumours60,61, HGSOC is characterized by elevated FAK tyrosine phosphorylation within the tumour cells themselves37. Although basic research62 and clinical studies63 have examined the role of FAK in promoting paclitaxel resistance in HGSOC, the role of FAK in mediating platinum chemotherapy resistance is less well understood. In paired patient tumour samples taken prior to, and after, several cycles of carboplatin and paclitaxel chemotherapy, FAK tyrosine phosphorylation was elevated further in non-necrotic residual tumour cells37. In HGSOC models in vivo, FAK Y397 phosphorylation – a surrogate for activity - increased upon sublethal cisplatin treatment of platinum-resistant tumour cells. Interestingly, cisplatin, but not paclitaxel, increased FAK Y397 phosphorylation in anchorage-independent tumourspheres in vitro37. Since platinum-induced cell stress can activate FAK, it has been suggested that paradoxical FAK activation may function to permit acquired platinum tumour resistance37.
Few ovarian tumour models exist to study the impact of copy number alterations on tumour state. In this regard, murine ID8 cells are immortalized ovarian epithelial cells that form slow growing tumours64, but isolation and expansion of early ascites ID8 cells led to isolation of spontaneously aggressive counterparts that readily form tumours in mice65. These cells contained chromosome gains encompassing several amplicons in common with HGSOC37. One region of interest included murine chromosome 15qA1-D3, which contained the MYC, FAK and RECQL4 genes, a region orthologous to human 8q24.3 that is often amplified in epithelial cancers. Supporting the notion that this may be one important driver of tumour malignancy, exome sequencing did not reveal de novo oncogenic somatic mutations, and these tumour cells were therefore termed KMF, denoting gains in the genes encoding Kras, Myc, and FAK. The KMF variants of ID8 cells exhibit more aggressive tumour growth characteristics, enhanced tumoursphere-forming capability in vitro, elevated FAK Y397 phosphorylation, increased β-catenin transcriptional activity, augmented detoxifying enzyme expression (such as aldehyde dehydrogenase) associated with stem-like properties and greater intrinsic resistance to cisplatin-mediated cytotoxicity compared to the parental ID8 cells37 (Fig. 1). As β-catenin is linked to HGSOC cisplatin resistance66, and FAK signalling to β-catenin is an adaptive chemo-resistance pathway in BRAF mutated colon cancer42, this pathway may promote resistance to radiation and other chemotherapies in HGSOC18,26,41. A central role for FAK in promoting stemness-associated genes suggests that FAK inhibitors could complement classical chemotherapy by targeting populations of cells that do not respond to treatment37. This rationale underlies the ROCKIF trial (see Table 2; NCT0328727167) in which FAK inhibitors are being used to re-sensitize platinum-resistant ovarian cancer disease by targeting stem-like cells in the tumour compartment.
Fig 1. FAK mediates resistance to therapy in high grade serious ovarian cancer.
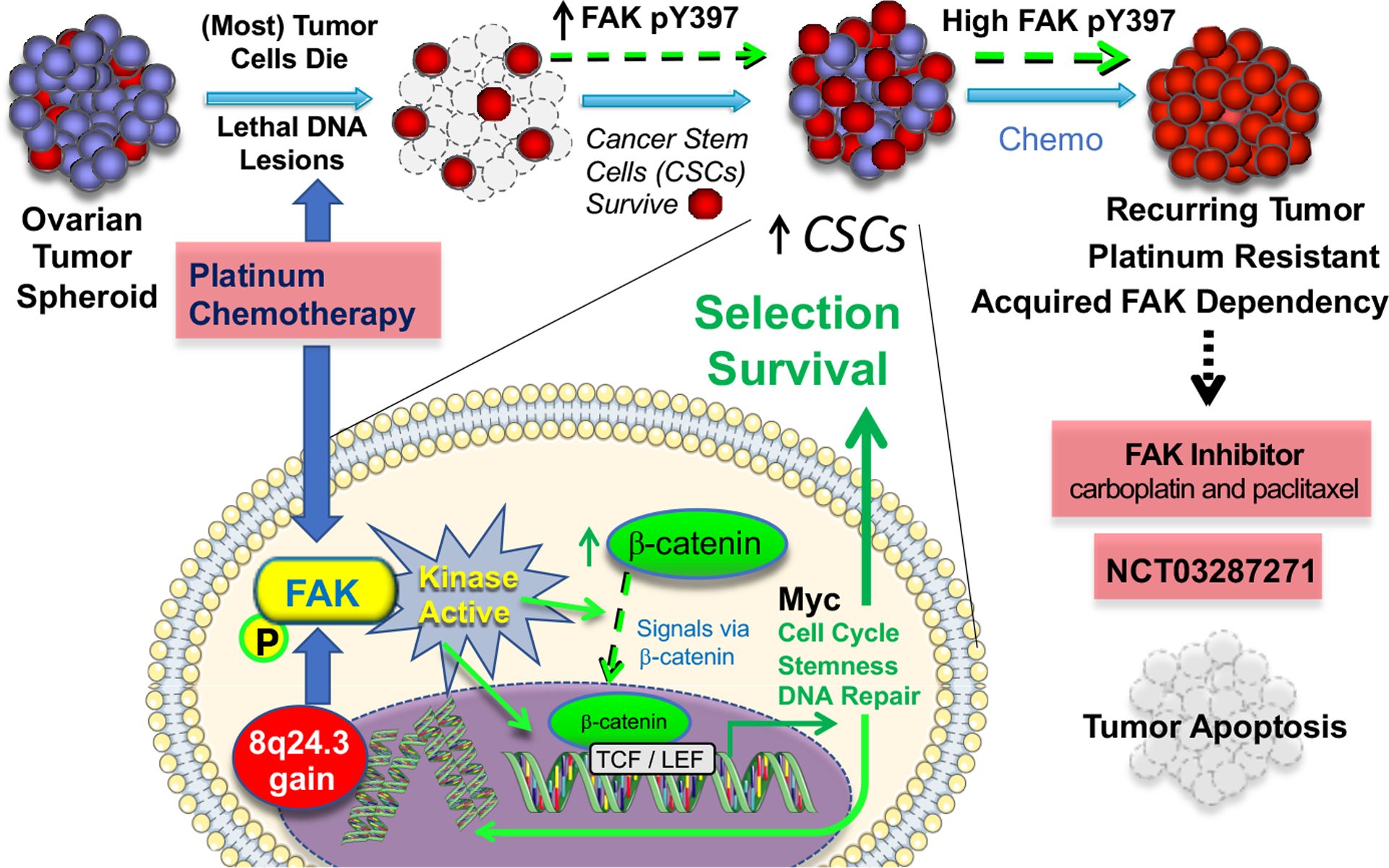
Model of chromosome 8q24.3 gain encoding the gene (PTK2) for focal adhesion kinase (FAK), FAK activation in tumourspheres surviving platinum chemotherapy, and the survival of cancer stem cells (CSCs) in high grade serous ovarian cancer (HGSOC). Gene breakage, gains, or losses are common drivers of HGSOC phenotypes. Over 70% of patient tumours contain gains at both PTK2 and MYC loci within chromosome 8q24. FAK signalling sustains tumoursphere growth, CSC survival, and enhances platinum resistance. Although the mechanisms of FAK activation in tumourspheres by increased expression or chemotherapy stress remains to be determined, intrinsic FAK activity is needed for β-catenin-associated increases in mRNA levels of MYC, cell cycle, pluripotency, and DNA repair gene expression. Spontaneous and induced cellular platinum resistance is associated with high levels of FAK tyrosine phosphorylation and the acquired dependence on FAK activity for platinum-resistant cell survival in culture. As the combination of a small molecule FAK inhibitor and cisplatin trigger platinum-resistant tumour apoptosis, a clinical trial67 is testing the combination of FAK inhibition, carboplatin, and paclitaxel for recurrent platinum-resistant HGSOC for which no approved treatments are available.
By exploiting the murine KMF model of HGSOC to evaluate genetic alterations and therapeutic opportunities, pharmacological and genetic approaches were used to delineate the role of FAK in both intrinsic and acquired resistance to platinum chemotherapy. In KMF cells, transcriptomic and bioinformatic analyses of KMF FAK-null, FAK-reconstituted, and TCGA HGSOC patient tumours revealed 135 shared targets that were associated with chemoresistance, stemness, and the regulation of cell metabolism. All were induced by the expression of an active form of β-catenin within cells lacking FAK37. Among these, many were related to DNA repair processes, such as DNA Repair and DNA Replication. Kinase-dependent FAK signalling was also linked to increased expression of Hippo pathway [G] components37. In uveal melanoma, FAK activates YAP by MOB1 phosphorylation, also resulting in Hippo pathway inhibition7 (see below). Although the activation of Wnt-β-catenin signalling by FAK may represent an important adaptive signalling response to stress68, it is clear that FAK activity is more complex, since active β-catenin over-expression was sufficient to induce chemoresistance, yet paradoxically insufficient to rescue FAK-null tumour growth defects in mice in the KMF HGSOC model.
Bioinformatic analyses of FAK mRNA transcripts that are elevated in HGSOC and associated with decreased relapse-free survival have identified a set of 36 genes that exhibit a significant change in tumours that display elevated FAK mRNA37. Within this set, none were documented oncogenes or tumour suppressor [G] genes listed in the COSMIC database. Of the 36 genes, 25 were upregulated and 11 were downregulated; many of the upregulated transcripts were products of genes on chromosome 8q that were amplified, like FAK, and another six elevated transcripts were products of genes on other chromosomes (ST6GALNAC5, SPON1, PTGER3, KRT14, NRP2, and ATP10A). Amongst the FAK-downregulated genes associated with poor patient outcome was Brain-Expressed X-linked protein-1 (BEX1)69, which is thought to function as a tumour suppressor; a potential inverse linkage to FAK remains under investigation37.
The most genetically enriched signalling activities in HGSOC are pathways that modulate lipid metabolism70. This may reflect the metastatic tropism of HGSOC cells to lipid-rich secondary sites, such as omentum71. However, HGSOC cells are also highly enriched in copies of genes regulating cell adhesion, extracellular matrix and FAK signalling pathways. Among HGSOC tumours with PTK2 gene amplification, there are also frequently gains in a number of stemness-related genes, including SOX2, SOX9, and OCT3/437. SOX9 is linked to both Wnt signalling and MAPK activation72. Analysing time to recurrence and stemness-associated genes that are gained in HGSOC revealed two genes, DUSP1 and IER5, whose protein products may cooperate with FAK and are significantly elevated in patients with decreased overall survival37. Importantly, both are linked to chemoresistance; DUSP1 via its modulation of the MAPK signalling pathway, and IER5 via its key role in DNA repair. The amplification of the genes for DUSP1 and IER5, coincident with FAK, may prove useful as HGSOC biomarkers for rapid disease recurrence or progression, and could be useful co-targets in this lethal gynaecologic disease.
FAK and the RAS/RAF/MEK pathway.
Activation of the RAS/RAF/MEK signalling pathway, in particular by mutation of the genes encoding RAS or RAF proteins, is very frequent pathway amongst common cancers. FAK is activated following inhibition of the RAS/RAF/MEK pathway in several pre-clinical tumour models (Fig. 2)42,44,73 and this has also been observed analysis of patient tumours74,75. FAK signalling can be negatively regulated by the RAS/RAF/MEK pathway; in mutant RAS expressing fibroblasts, ERK-mediated phosphorylation of FAK on S910 provides feedback leading to FAK inactivation by dephosphorylation and focal adhesion turnover during migration76. Loss of this negative regulation may partially explain the activation of FAK in cancer cells following RAS/RAF/MEK pathway blockade.
Fig 2. Molecular targets for FAK inhibitor combination therapy.
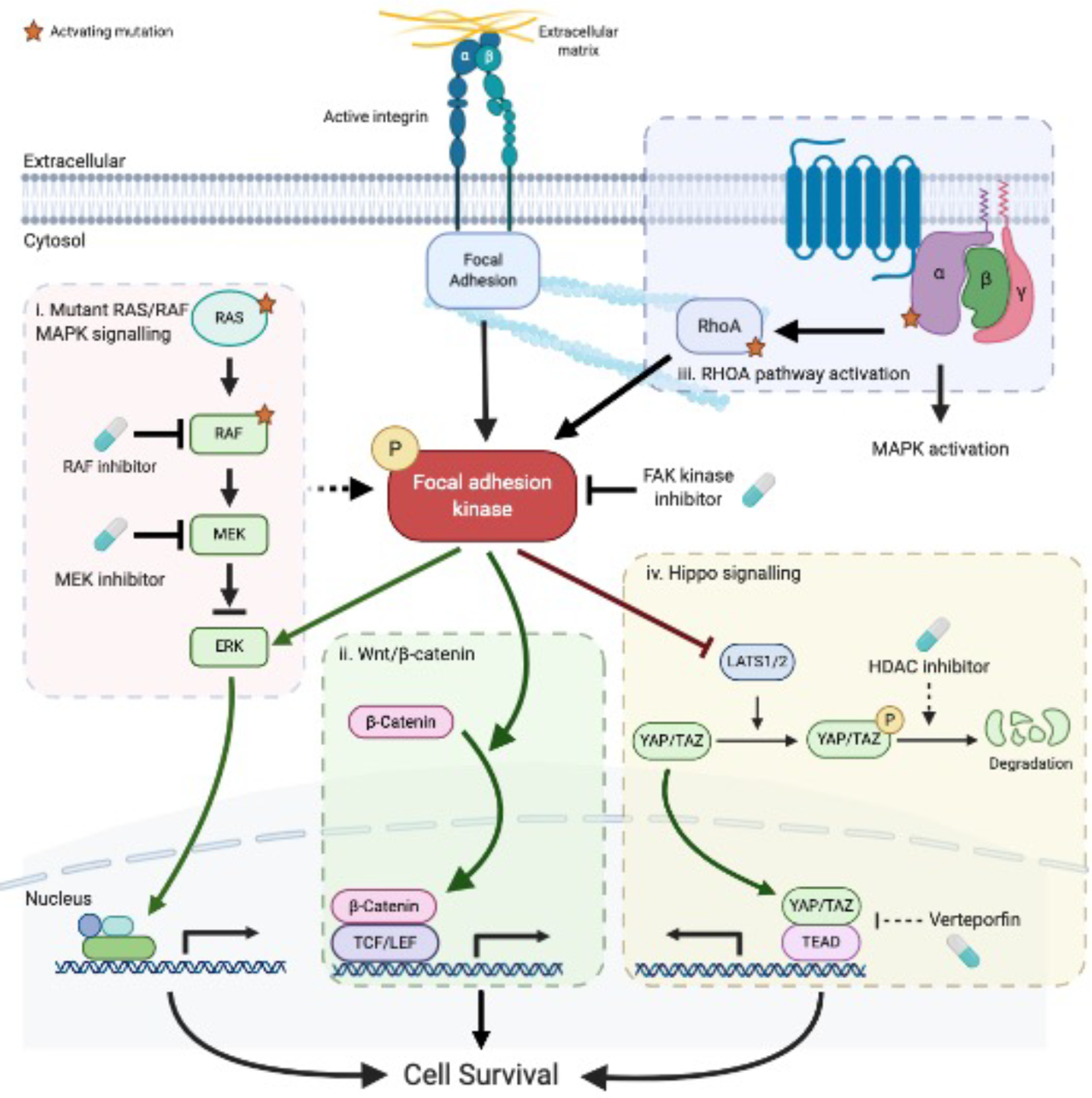
Focal adhesion kinase (FAK) supports myriad oncogenic processes. (i) Activation of the RAS/RAF/MEK signalling cascade is a common oncogenic driver in many tumour types and can be activated by mutations in a tumour type specific manner. KRAS mutations at amino acids G12 or G13 are for example commonly found in colorectal and pancreatic cancer which results in defective GAP-mediated GTP hydrolysis and constitutive activation123. In melanoma, BRAF frequently has an activating phosphomimetic mutation at amino acid V600E36. In RAS or RAF mutant cancer cells, blockade of the RAS/RAF/MEK pathway using either RAF or MEK inhibitors activates FAK and promotes cell survival by reactivation of ERK signalling42,44,75. The combination of FAK (defactinib) and dual RAF/MEK (VS-6766) inhibitors is being investigated in clinical trials in patients with melanoma, non-small cell lung carcinoma, low grade serous ovarian cancer, colorectal cancer, and other RAS mutant solid tumours124–126. (ii) In mutant-BRAF colorectal cancer cells, MAPK pathway blockade activates FAK in a β1-integrin and SRC independent way and promotes Wnt/β-catenin signalling and survival42. (iii) The small GTPase RHOA regulates the actin cytoskeleton. Activation of the RHOA signalling pathway by either gain-of-function mutation of RHOA and inactivation of the tumour suppressor CDH1 in diffuse gastric cancer activates FAK and subsequent YAP, PI3K and β-catenin signalling14. Alternatively, activating mutations in Gαq subunits (GNAQ or GNA11) of heterotrimeric G-proteins in uveal melanoma activates FAK via the RHOA signalling pathway to support YAP signalling and tumour growth7. (iv) Activated FAK in diffuse gastric cancer and uveal melanoma alleviates the negative regulation of YAP by LAST1/2. As mutant GNAQ/GNA11 signalling in uveal melanoma also activates the MAPK pathway, the combination of FAK (IN10018) and MEK (cobimetinib) inhibition is being tested in a clinical trial127. FAK activity can promote the nuclear translocation of YAP and combinations of FAK inhibitors with inhibitors of YAP expression, for example Histone deacetylase (HDAC) inhibitors85, or transcriptional activity88,89 may be needed to reinforce inhibition of oncogenic YAP signalling.
In preclinical models of melanoma and colorectal cancer with mutant BRAF, treatment with RAF inhibitors (dabrafenib, GDC-0879 or vemurafenib) or MEK inhibitors (trametinib) induces a rapid (within hours) paradoxical activation of FAK42–44. This can occur via tumour cell intrinsic mechanisms or cell extrinsic mechanisms; for example, treatment of mice bearing melanoma tumours with vemurafenib activates melanoma associated fibroblasts (MAF), which remodels the extracellular matrix into a cancer cell protective environment44 (see below). In melanoma cells following vemurafenib treatment, FAK is activated as part of a c-Jun/FAK/SRC pathway that promotes de-differentiation of cells that express the low-affinity nerve growth factor receptor, and these melanoma cells are tolerant to BRAF-V600E inhibition. Co-targeting of BRAF (with vemurafenib) and FAK (with the dual FAK/PYK2 kinase inhibitors PF-562271 or defactinib), impairs the acquisition of this “vemurafinib tolerant” state and induces cell death43. In BRAF-V600E mutant colorectal cell lines, BRAF inhibitors activate the Wnt/β-catenin pathway via activation of FAK, independently of β1-integrin and SRC, and vemurafenib and PF-562271 together reduce HT-29 tumour growth in vivo. Interestingly, ‘reinforced’ inhibition of RAF/MEK signalling by targeting BRAF-V600E (with vemurafenib) and MEK (with trametinib) in a triple combination with the FAK inhibitor PF-562271 robustly blocks HT-29 tumour growth42.
In preclinical models of non-small cell lung carcinoma (NSCLC), KRAS-mutant cells depend on FAK for survival, and pharmacological inhibition with the dual FAK/PYK2 kinase inhibitors PF-562,271 or VS-4718, inhibits tumour growth40. However, patients with KRAS-mutant NSCLC treated with the FAK/PYK2 inhibitor defactinib as a single agent showed only modest effects, in keeping with the likely requirement to co-target the FAK and KRAS pathways33. Preliminary results from a Phase I clinical trial reports FAK activation in KRAS-G12V mutant NSCLC tumours from patients treated with the dual RAF/MEK inhibitor VS-6766 (formally CH5126766 or RO5126766)74 and potential clinical activity of defactinib in combination with VS-6766 in the NSCLC and low grade serous ovarian cancer cohorts and which has led to two Phase II clinical trials (see Table 2)75. Following pharmacological inhibition of RAF and/or MEK signalling, FAK activation again provides survival signalling. Therefore, FAK activation in patients with tumours harbouring RAS/BRAF mutations treated with RAF and/or MEK inhibitors may be susceptible to clinical combinations that include a FAK kinase inhibitor.
FAK is involved in oncogenic YAP signalling?
FAK forms part of the “consensus integrin adhesome” [G] and so sits at the core of an extracellular matrix-focal adhesion signalling network77 (Box 1) that transduces mechanical cues into transcriptional responses by mechanisms that include the cytoplasmic to nuclear translocation of the transcriptional co-activators YAP and TAZ78,79. YAP/TAZ ‘activation’ correlates with grade, state of metastasis, and poor outcome in breast, lung, liver, pancreatic, and skin cancer80, with YAP/TAZ signalling being important in both the tumour cell and stromal compartments79,81. The dysregulation of Hippo signalling, and YAP in particular, is widely implicated in resistance to anti-cancer therapies; one study in melanoma demonstrated that shRNA-mediated knockdown of YAP is synthetically lethal with BRAF inhibitors in BRAF-V600E melanoma cells82. In addition, diffuse gastric cancer has gain-of-function mutations of the RHOA [G] gene that synergises with inactivation of the tumour suppressor CDH1 and drives FAK-dependent activation of YAP signalling14, while uveal melanoma has both amplification of FAK and activating mutations in heterotrimeric G-protein Gαq subunits (GNAQ and GNA11) that induce pro-tumorigenic YAP signalling via the TRIO/RHOA/FAK pathway7 (Fig. 2). In both of these preclinical models, FAK-dependent YAP activation is nullified by dual FAK/PYK2 inhibitors, VS-4718 or PF-573228, and tumour growth is inhibited7,14. Uveal melanoma, unlike cutaneous melanoma, do not display activating mutations in BRAF, but activating mutations in GNAQ/GNA11 proteins stimulate the RAS/RAF/MEK pathway and combined inhibition of YAP and RAS/RAF/MEK pathways suppress tumour growth83. Targeting FAK (and therefore YAP where YAP activation is FAK-dependent) and MEK in metastatic uveal and cutaneous melanoma is the subject of a combination clinical trial using the FAK kinase (IN10018) and MEK (cobimetinib) inhibitors (see Table 2).
In mouse skin, YAP and its paralogue TAZ are required for skin homeostasis and wound healing; indeed, conditional genetic deletion of FAK in mouse skin blocks YAP nuclear translocation following inflammation induced by chemical treatment with 12-O-Tetradecanoylphorbol-13-acetate (TPA)84. YAP is strongly localised to the nucleus of mouse skin papillomas and squamous cell carcinomas (SCCs) induced by 7, 12-dimethylbenzanthracene (DMBA)-TPA-induced chemical carcinogenesis and pharmacological inhibition of FAK using PF-573228 inhibits the accumulation of nuclear YAP84. Therefore, FAK appears to be crucial for the activation of YAP/TAZ signalling during chemically-induced skin tumour development and this is consistent with conditional deletion of FAK in the same model inhibiting SCC tumour progression25. Using SCC cells derived from this model expressing a FAK kinase mutation to mimic a kinase inhibitor, a chemical-genetic phenotypic screen identified FAK-dependent reorganisation of the actin cytoskeleton following treatment with histone deacetylase (HDAC) inhibitors, such as vorinostat85. Combination of the dual FAK/PYK2 inhibitor VS-4718 with HDAC inhibitors synergistically inhibited the growth of mutant H-Ras-driven SCC tumours85 by cooperatively inhibiting YAP nuclear localisation and expression. In vitro, HDAC inhibitors reduce YAP expression in many cancer cell lines86, and while HDAC inhibitors have failed to live up to their potential as single anti-cancer agents, in part due to toxicity, they are being investigated in combination clinical trials where it may be possible to lower their doses87. Whether the clinical use of FAK inhibitors to block the nuclear translocation of YAP can be effective in combination with inhibitors of YAP expression, for example using HDAC inhibitors85,86 (Fig. 2), or its transcriptional activity (using verteporfin88 or bromo domain inhibitors89), remains to be seen (Fig 2).
FAK activation by the microenvironment
In addition to FAK’s signalling role in cancer cells, there is also abundant evidence that FAK is regulated by, and regulates, the tumour microenvironment, including the tumour immune cell compliment and anti-tumour immunity. We discuss a number of contexts by which FAK inhibitors may modulate the tumour microenvironment to influence responses to therapy.
FAK regulates adaptive resistance to targeted therapy in melanoma
In a mouse model of BRAF-mutant melanoma, tumour cells display a heterogeneous response to treatment with the BRAF inhibitor vemurafinib44; intra-vital imaging demonstrated that tumour areas with a low stromal density are sensitive to the BRAF inhibitor, while areas with high stromal density are more resistant. In this case, resistance is mediated by activation of melanoma-associated fibroblasts (MAFs) in response to vemurafanib. Activated MAFs lay down, and remodel, extracellular matrix resulting in a tumour environment with increased rigidity, creating protective microenvironmental ‘sanctuaries’ where tumour cells are no longer sensitive to the BRAF inhibitor. Extracellular matrix-driven resistance is distinct from other types of resistance where signalling networks are reprogramed to bypass mutant-BRAF signalling, leading to reactivation of MAPK signalling, for example by switching to other RAF isoforms or (re)activation of receptor tyrosine kinase signalling35. The MAF-driven adaptive resistance mechanism is rapid as cells respond within hours to vemurafenib treatment to begin remodelling the extracellular matrix; such ‘protected’ tumour cells can then provide the foundation for eventual relapse and tumour regrowth. In this case, resistance is dependent on a β1 integrin-FAK signalling axis in the melanoma cells that is negated by the use of FAK kinase inhibitors PF-573228, PF-562271, and FAKi14; melanoma cells isolated from high stromal environments can be rendered sensitive again, demonstrating the importance of the modified tumour microenvironment (Fig. 3). Elevated extracellular matrix deposition and/or stiffness can activate YAP signalling79, highlighting commonality of FAK signalling in different mechanisms that cancer cells use to escape therapeutic responses. FAK (IN10018) and MEK (cobimetinib) inhibitors are being investigated in a clinical trial of patients with metastatic melanoma (see Table 2). Whilst the study by Hirata et al.44 was in a preclinical model of melanoma, we assume the role of the microenvironment in modulating FAK-dependent survival signalling in the tumour niche is not specific to melanoma and it may play a part in resistance mechanisms in multiple different cancers.
Fig 3. FAK regulates adaptive resistance to targeted therapy in melanoma.
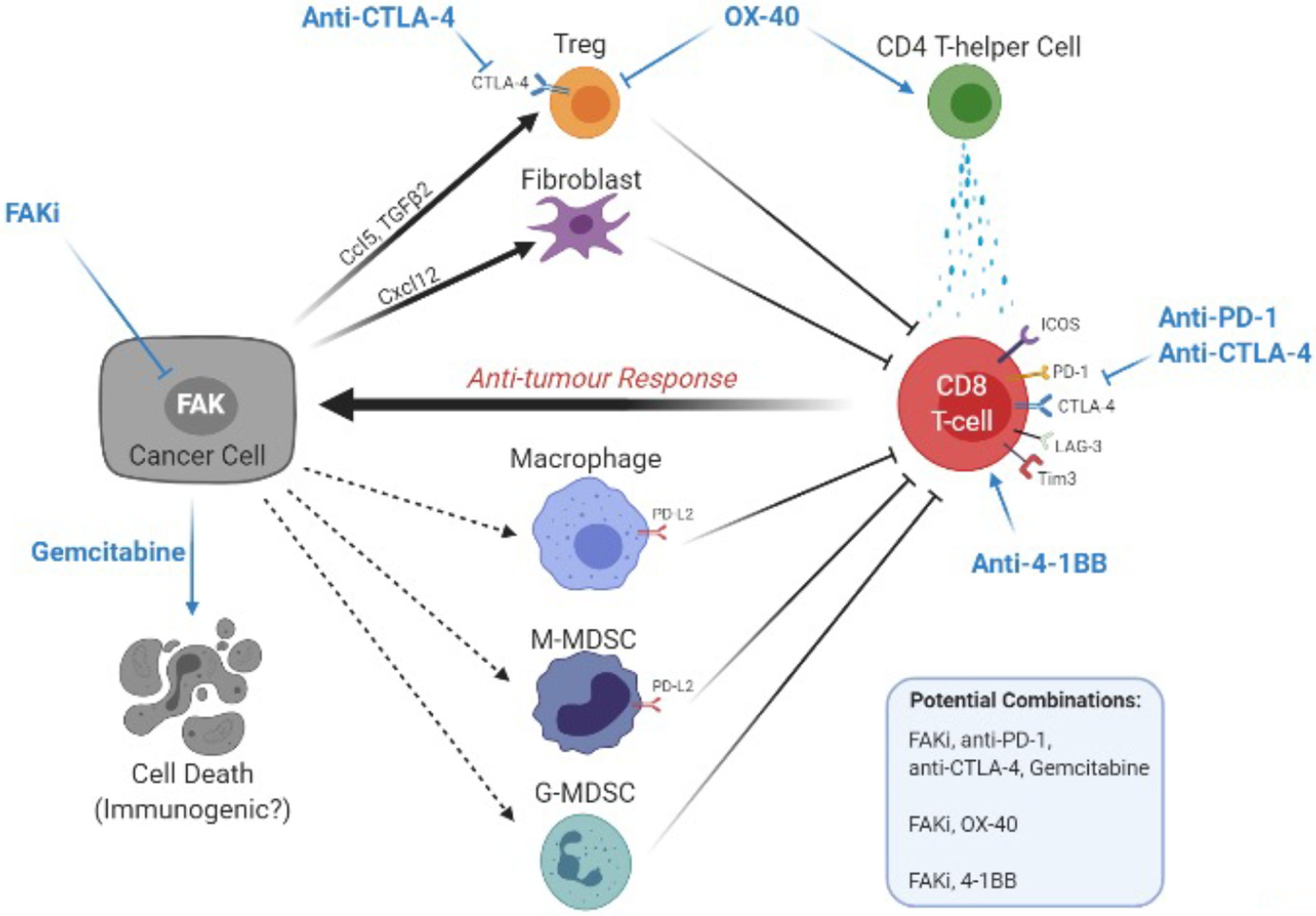
(A) In melanoma, BRAF is commonly mutated (~60% of tumours) and its oncogenic signalling can be blocked with molecularly targeted therapy such as vemurafenib34. BRAF inhibitors block mutant BRAF signalling and inhibit the pro-survival signalling via ERK to induce cancer cell apoptosis. (B) In tumour areas with a high stromal density, melanoma cells become rapidly resistant to vemurafenib treatment by reactivating ERK signalling via activation of β1-integrin receptors by the extracellular matrix deposited and remodelled by the activation of melanoma associated fibroblasts (MAFs). Activation of β1-integrin activates focal adhesion kinase (FAK) and SRC which reactivates ERK-dependent pro-survival signalling, bypassing mutant BRAF signalling and mitigating the effect of BRAF inhibitors. (C) BRAF inhibitor resistance in melanoma cells driven by stromal remodelling of the extracellular matrix can be targeted with FAK inhibitors to re-sensitise tumour cells to BRAF inhibitors.
FAK regulates the tumour immune microenvironment
Activation of the anti-tumour immune response has shown great promise for the treatment of a number of cancers. Tumour cells evade immune-mediated killing through numerous mechanisms, including downregulation of antigen processing or presentation pathways, expression and secretion of immune-regulatory molecules such as immune checkpoint ligands and suppressive cytokines, and the assembly of a highly immune-suppressive microenvironment. Overcoming these mechanisms of immune suppression is central to many therapeutic strategies aimed at stimulating, or reinvigorating, a natural anti-tumour immune response; FAK is emerging as a promising target in this context. In the chemical carcinogenesis mouse model of SCC described above, FAK-depletion from malignant cells or treatment of tumours with the small molecule FAK/PYK2 inhibitor VS-4718 resulted in complete immune-mediated tumour regression and lasting immunological memory16. FAK-dependent expression of chemokine ligand 5 (Ccl5) and the cytokine transforming growth factor β2 (TGFβ2) in SCC cells was found to increase the intra-tumoral density of Regulatory T-cells [G] (Tregs), thereby shifting the balance of Tregs to cytotoxic CD8+ T lymphocytes in favour of immune evasion. This was a kinase-dependent function of nuclear FAK in SCC cells that required association of FAK with the multi-functional cytokine interleukin-33, enabling FAK to functionally interact with a network of chromatin modifiers and transcriptional regulators linked to genes encoding pro-inflammatory chemokines, including Ccl516,46 (Fig. 4). Nuclear FAK seems to accumulate in response to cellular stress15 and in the SCC study it was not present in the relevant normal cell counterparts, namely skin keratinocytes16, implying that FAK inhibitors may offer improved disease-specific immune modulation when compared with some more direct immune-targeted therapies.
Fig 4. FAK mediates protective effects of the tumour immune microenvironment; combination opportunities.
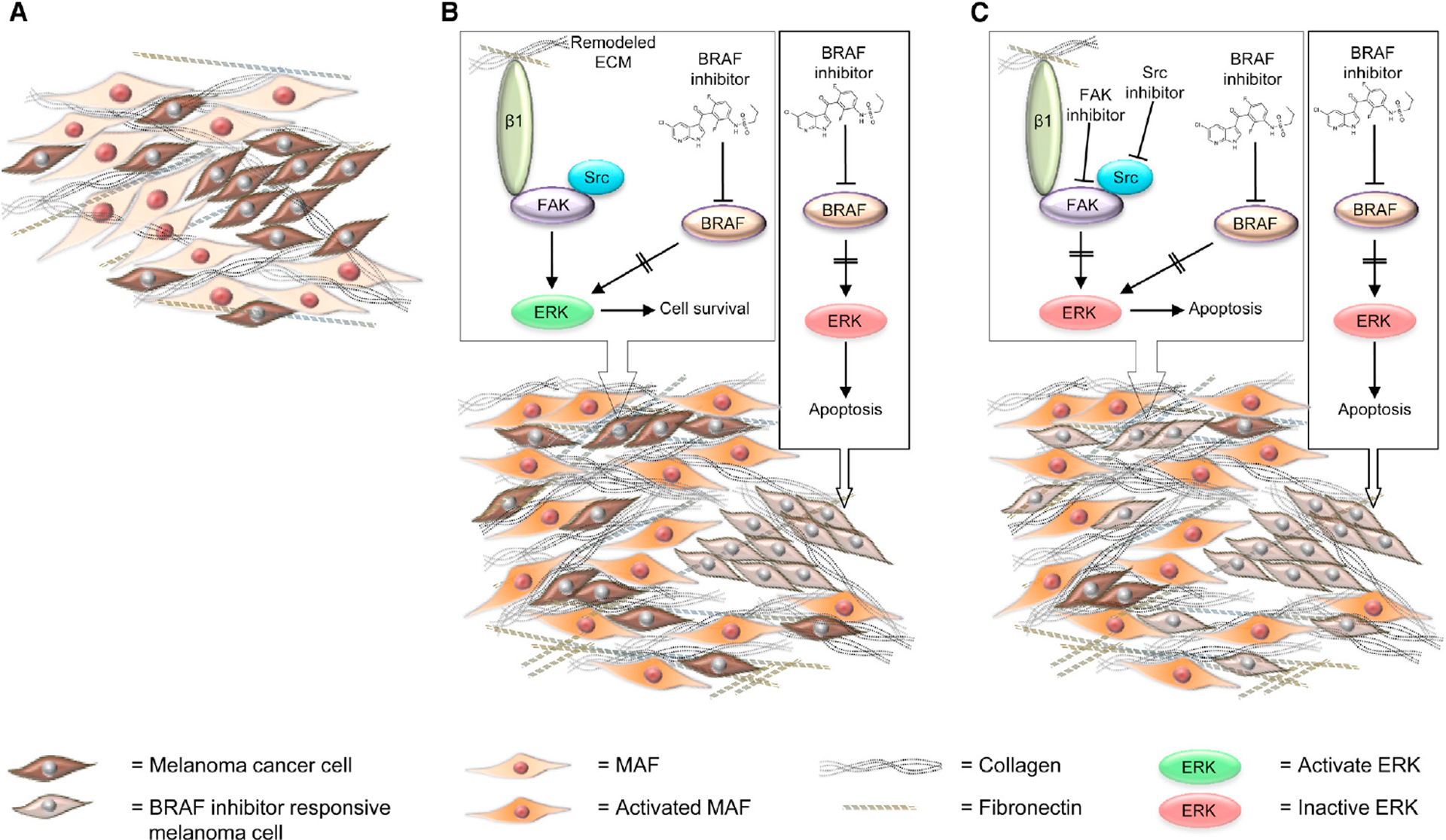
Focal adhesion kinase (FAK) inhibition can modulate the cellular and molecular composition of the immuno-suppressive TME. FAK-dependent expression of Ccl5 and transforming growth factor (TGF)β2 has been shown to impact Treg numbers in squamous cell carcinoma (SCC) tumours16, while Cxcl12 has been linked to promoting pancreatic fibroblast proliferation38. In some cases, FAK inhibition has also been shown to result in a decrease in macrophages, M-MDSCs and G-MDSCs in tumours38,128. These cell types can also act to supress anti-tumour CD8+ T-cell activity. Therefore, a decrease in their abundance is likely to contribute to the enhanced anti-tumour activity of FAK inhibitors in combination with immunotherapies. The mechanisms through which FAK inhibition can regulate macrophages, M-MDSCs and G-MDSCs remain to be defined (dashed arrows). For example, it is not known whether FAK-dependent cancer cell paracrine signalling leads to the recruitment of these cell types into tumours, or whether FAK inhibitors can act directly on these cell types to impact abundance / function. FAK inhibition can also lead to a decrease in PD-L2 expression on some of these cell types45, the mechanisms for which remain to be defined.
FAK inhibition can lead to upregulation of ICOS on CD8 T-cells and reduced co-expression of the exhaustion markers programmed cell death protein 1 (PD-1), LAG-3 and Tim3, likely enhancing the cytolytic activity of CD8 T-cells.
Targeting FAK can sensitise mouse pancreatic tumours to a combination of gemcitabine, anti-PD-1 and anti-CTLA-4. Anti-PD-1 and anti-CTLA-4 target mechanisms of T-cell exhaustion, but anti-CTLA-4 can also have effects on Tregs. The role of gemcitabine is less clear; Gemcitabine alone does not drive immunogenic cell death but whether this is altered when used in combination with a FAK inhibitor (such as has been reported with other targeted therapies) is not known.
FAK inhibition can enhance the anti-tumour efficacy of either anti-OX-40 or anti-4-1BB antibodies in mouse models of SCC and pancreatic cancer. FAK inhibition can increase expression of ICOS on effector CD8+ T-cells and this plays an important role in the efficacy of a FAK inhibitor in combination with either OX-40 or 4-1BB. FAK inhibition can also result in downregulation of PD-L2 on multiple cell types within the tumour microenvironment and this may further contribute to responses to combination treatment with anti-OX-40.
There are four clinical trials testing FAK inhibitor (defactinib) in combination with anti-PD-1 antibody (pembrolizumab) in patients with pancreatic and mesothelioma cancers90–93.
Several studies have now shown that FAK inhibitors can modulate mechanisms of immune suppression in cancer, sensitising mouse cancer models to immunotherapies38,45. Indeed, treatment of genetically engineered (p48-Cre;KrasG12D;Trp53flox/+) and transplantable mouse models of pancreatic cancer with the dual FAK/PYK2 kinase inhibitor VS-4718 is able to overcome resistance to a combination of immunotherapy (anti-programmed cell death protein 1 [G] (PD-1) + anti-cytotoxic T-lymphocyte-associated protein 4 [G] (CTLA-4)) and chemotherapy (gemcitabine), resulting in improved overall survival38. This synergistic activity is underpinned by reprogramming of the fibrotic and immune-suppressive microenvironment in response to the FAK inhibitor and is, at least in part, dependent on FAK driven paracrine signalling between malignant cells and the surrounding microenvironment.
The FAK-specific inhibitor IN10018 can also promote anti-tumour immunity against transplantable mouse models of SCC and breast cancer that express CD80. CD80 is a ligand for both the immune stimulatory T-cell co-receptor CD28 and the inhibitory receptor CTLA-4. IN10018 reduces the intra-tumoral frequency of CTLA-4+ immune cells, promoting an anti-tumour immune response against SCC tumours that is dependent on both CD80 and CD2845. In mouse models of SCC and pancreatic cancer that lack malignant cell CD80 expression, FAK inhibition combined with activating antibodies targeting either the inducible T-cell costimulatory receptor OX-40 or 4-1BB also results in robust anti-tumour immunity, and in SCC this can induce complete tumour regression. In addition to decreasing the intra-tumoral frequency of Tregs in SCC tumours, IN10018 treatment results in broad downregulation of the immune checkpoint ligand programmed death ligand 2 (PD-L2) in the tumour microenvironment and elevated expression of the immune costimulatory molecule ICOS on effector CD8+ T-cells. Elevated expression of ICOS on CD8+ T-cells is important for the enhanced efficacy of both IN10018 + OX-40 or IN10018 + 4-1BB, while PD-L2 regulation may contribute to the efficacy of IN10018 in combination with OX-4045.
Thus, FAK regulates multifaceted immune evasion programmes in multiple tumour contexts that can impact the efficacy of anti-tumour T-cell responses (Fig. 4). These studies have led to the first clinical trials testing FAK inhibitors in combination with immunotherapies. Defactinib is currently being studied in combination with the PD-1 receptor inhibitor, pembrolizumab (lambrolizumab, brand name Keytruda) in patients with pancreatic, NSCLC and mesothelioma cancers90–92, and in combination with both pembrolizumab and gemcitabine in patients with advanced pancreatic cancer93 (see Table 2).
FAK is at the early stages of investigation as an immune oncology target and many questions remain regarding the mechanisms that underpin the immune modulatory activity of FAK and dual FAK/PYK2 inhibitors. The majority of research to-date has focussed on the role of FAK in malignant cells (Box 1). However, both FAK and PYK2 are expressed in a range of cell types present within the tumour microenvironment and dual inhibition of FAK/PYK2 function in these cells may also be important or modulate positive or negative effects. For example, FAK is expressed and phosphorylated in human T-cells94 where it can be found in complex with the T-cell Receptor (TCR)95. CD4/CD8 coreceptors recruit the SRC family kinase LCK to the peptide MHC:TCR complex where it can phosphorylate tyrosine residues in the immunoreceptor tyrosine-based activation motifs of CD3, the ζ chains of the TCR complex and ZAP7096. The TCR complex must be sufficiently phosphorylated in order to initiate a cellular response. Using Jurkat T-cells and activated human peripheral blood CD4+ T-cells, FAK depletion using micro-RNAs sensitised T-cells to low-dose TCR stimulation, resulting in enhanced TCR signalling, cytokine production and expression of the activation marker CD6995,97. FAK is required to promote association of CSK with LCK at the TCR-CD4 complex, resulting in inhibition of LCK activity, implying that FAK may act as a rheostat to prevent inappropriate T-cell activation. These findings support the hypothesis that targeting FAK in T-cells may sensitise them to low affinity tumour antigens, thereby helping to promote anti-tumour immunity. FAK can also interact with and phosphorylate the adaptor protein linker for the activation of T-cells (LAT) on Y171 to promote dissociation of T-cell–dendritic cell conjugation and T-cell motility98. Prolonged T-cell–dendritic cell conjugation is important for optimal activation of naïve T-cells99,100, suggesting that FAK inhibitors could have benefits in this regard also. By contrast though, the dual FAK/PYK2 inhibitor PF-562,271 can impair ZAP70 phosphorylation, CD4+ T-cell activation and interaction of T-cells with antigen presenting cells (APCs)101. However, genetic depletion of FAK in OTII CD4+ T-cells resulted in only modest differences in adhesion to ICAM-1 and conjugation to APCs at higher doses of soluble ovalbumin peptide stimulation, suggesting that the action of PF-562,271 could not be explained solely by inhibition of FAK. PYK2 is also expressed by human T-cells and the relative roles of FAK and PYK2 in regulating T-cell biology may not be functionally redundant97, raising the possibility that FAK-specific and dual FAK/PYK2 kinase inhibitors may elicit different immune modulatory activities that would influence outcomes in patients. Further work is required to reach a consensus on the relative functions of FAK and PYK2 in regulating T-cell biology that is relevant to the anti-tumour T-cell response, since this may have important consequences for how we develop FAK and FAK/PYK2 kinase inhibitors as combination immunotherapies moving forward.
The necessary cautionary tale!
While the overwhelming body of evidence supports the conclusion that FAK kinase inhibitors may be beneficial in the treatment of cancer, a small number of studies have found that FAK loss in some specific cell types can contribute to enhanced tumour progression and potentially metastasis in pre-clinical models. For example, FAK deletion in haematopoietic cells can increase the incidence of liver metastasis in the RIP-tag2 mouse model of pancreatic cancer and liver, lung and bone metastasis following tail vein injection of B16 melanoma cells into mice102. FAK depletion in pericytes is reported to promote angiogenesis and tumour growth in mouse models of lung, melanoma and pancreatic cancer103, while FAK depletion or expression of non-phosphorylatable FAK-Y397F in endothelial cells inhibits metastasis, tumour angiogenesis, growth, and enhances sensitivity to chemotherapy104–107. FAK deletion in cancer-associated fibroblasts (CAFs) can alter malignant cell metabolism in a way that is linked to tumour growth in mouse models of lung and pancreatic cancer108; by contrast, FAK depletion in malignant cells can down-regulate glycolysis109 and so the situation is complex. In patients’ PDAC tumour samples, elevated levels of FAK Y397 are observed in CAFs61, and in mouse models of PDAC and breast cancer, targeting of FAK in the stromal compartment by either genetic or pharmacological kinase inhibitors prevents metastasis61,110. However, prolonged pharmacological inhibition of FAK in PDAC models eventually leads to stromal depletion that is associated with drug resistance111. Transient pharmacological inhibition of the RHOA effector ROCK [G] in pancreatic tumour stroma is sufficient to sensitise tumours to chemotherapy and reduce metastasis112 and such a treatment strategy could be adopted in concert with FAK inhibitors to prevent the development of drug resistance. Therefore, there are complex preclinical data that imply FAK’s role in distinct cell types in tumours can contribute to a greater or lesser extent to the overall tumour microenvironment and therapeutic responses. On this basis, it is hard to predict how the anti-tumour, and potentially pro-tumour, effects of FAK inhibitors will be integrated in patients to produce particular responses. This awaits the outcomes of on-going and future, likely combination, clinical trials – but it is noteworthy that, as yet, there are no reports that FAK inhibitors are toxic or promote tumour progression.
6. Conclusions.
Preclinical and clinical evaluation of molecular targeted therapies against cancer driver pathways shows that tumours almost always display inherent or acquired resistance. Solid tumours consist of a complex heterogeneous mixture of cancer cells, immune cell populations, and stromal cells, and for this reason, combination therapies are mostly required for durable responses. As a key coordinator of cellular responses to environmental cues and stresses, including therapeutic interventions, FAK is an attractive common target supporting a myriad oncogenic processes and resistance mechanisms. Indeed, FAK is frequently pivotal in ‘cellular mitigation’ against stress, including therapeutic stress. FAK is therefore likely to be a more effective target when inhibitors are used in the context of combination therapies, especially if tumour cells rely on anchorage-dependent signalling initiated from the microenvironment. Aside from FAK activation itself8, robust biomarkers are required to identify patients with tumours whose survival and growth are driven by chemo-protective FAK-dependent signalling, whether by intrinsic or extrinsic mechanisms, that could guide the best use of FAK inhibitors for clinical use. We do not dismiss the potential for future targeting of FAK’s protein-protein scaffolding functions, as these play a vital role in cancer cell biology113–115, especially if tumour-specific adaptor functions are defined. For example, proteolysis targeting chimera (PROTAC)-mediated degradation of FAK may also be considered as a useful therapeutic strategy that would simultaneously remove both FAK’s catalytic and adaptor functions116–118.
Acknowledgements
Margaret Frame is supported by a Cancer Research UK Programme Award (C157/A24837). David Schlaepfer and Dwayne Stupack are supported by funding from the National Institute of Health (R01 CA247562 and R01 CA254342). Alan Serrels is supported by a Cancer Research UK Career Development Award (C39669/A25919), has received research funding from Boehringer Ingelheim to work on IN10018 (then BI-853520), and is on the scientific advisory board of InxMed in relation to the development of IN10018.
Glossary
- Consensus integrin adhesome
The proteins that make up the core cell adhesion machinery of integrin adhesion complexes.
- Cytotoxic T-lymphocyte-associated protein 4 (CTLA4)
A protein expressed by Tregs that functions as an immune check point to inhibit the immune response.
- FERM (4.1 protein, ezrin, radixin, and moesin) domain
A protein domain that is involved in localising proteins to the plasma membrane.
- Focal adhesion
A points of cellular plasma membranes that link to extracellular matrix via transmembrane receptors, typically integrin heterodimers.
- Heterotrimeric G-proteins
A GTPase complex made up of three subunits α,β, and γ that links transmembrane receptors to intracellular signalling pathways.
- Hippo pathway
A signalling pathway that controls organ size by regulating cell proliferation and apoptosis that can be dysregulated in cancer.
- Oncogenic drivers
Genes which when mutated are responsible for the initiation and maintenance of cancer.
- Programmed cell death protein 1 (PD-1)
A protein expressed on the surface of cells that inhibits the activation of the immune system.
- Regulatory T-cells (Treg)
A sub-population of T-cells that suppress the immune response.
- RHOA
A small GTPase primarily associated with regulating the actin cytoskeleton.
- ROCK (Rho-associated protein kinase)
a serine-threonine kinase downstream effector of RHOA involved in the formation of actin stress fibres.
- Stromal cells
Connective tissue cells such as fibroblasts that support the other cells of that organ.
- Tumour suppressor
A gene that negatively regulates cell proliferation and whose loss of function are a significant step in the development of cancer.
- Tumour microenvironment
The environment found within a tumour encompassing the tumour, immune and stromal cells, extracellular matrix, and blood vessels.
References:
- 1.Lee BY, Timpson P, Horvath LG & Daly RJ FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther 146, 132–149, doi: 10.1016/j.pharmthera.2014.10.001 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Sulzmaier FJ, Jean C & Schlaepfer DD FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14, 598–610, doi: 10.1038/nrc3792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lietha D et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell 129, 1177–1187, doi: 10.1016/j.cell.2007.05.041 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Acebron I et al. Structural basis of Focal Adhesion Kinase activation on lipid membranes. EMBO J, e104743, doi: 10.15252/embj.2020104743 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grossman RL et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med 375, 1109–1112, doi: 10.1056/NEJMp1607591 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaveh F et al. A systematic comparison of copy number alterations in four types of female cancer. BMC Cancer 16, 913, doi: 10.1186/s12885-016-2899-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng X et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 35, 457–472 e455, doi: 10.1016/j.ccell.2019.01.009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes FAK as a therapeutic target for the treatment of uveal melanoma with oncogenic mutations in GNAQ heterotrimeric G-proteins.
- 8.Kim YH et al. FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer. Cancers (Basel) 11, doi: 10.3390/cancers11091288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agochiya M et al. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene 18, 5646–5653, doi: 10.1038/sj.onc.1202957 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Gorringe KL et al. Copy number analysis identifies novel interactions between genomic loci in ovarian cancer. PLoS One 5, e11408, doi: 10.1371/journal.pone.0011408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramakrishna M et al. Identification of candidate growth promoting genes in ovarian cancer through integrated copy number and expression analysis. PLoS One 5, e9983, doi: 10.1371/journal.pone.0009983 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goode EL et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet 42, 874–879, doi: 10.1038/ng.668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez-Vega F et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e310, doi: 10.1016/j.cell.2018.03.035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H et al. Gain-of-Function RHOA Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer. Cancer Discov, doi: 10.1158/2159-8290.CD-19-0811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim ST et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell 29, 9–22, doi: 10.1016/j.molcel.2007.11.031 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrels A et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of antitumor immunity. Cell 163, 160–173, doi: 10.1016/j.cell.2015.09.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the was the first to show that nuclear FAK promotes a suppressive immune environment.
- 17.Naser R, Aldehaiman A, Diaz-Galicia E & Arold ST Endogenous Control Mechanisms of FAK and PYK2 and Their Relevance to Cancer Development. Cancers (Basel) 10, doi: 10.3390/cancers10060196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao C et al. FAK/PYK2 promotes the Wnt/beta-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3beta. Elife 4, doi: 10.7554/eLife.10072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lulo J, Yuzawa S & Schlessinger J Crystal structures of free and ligand-bound focal adhesion targeting domain of Pyk2. Biochem Biophys Res Commun 383, 347–352, doi: 10.1016/j.bbrc.2009.04.011 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Thul PJ et al. A subcellular map of the human proteome. Science 356, doi: 10.1126/science.aal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Zhao J, Zheng C & Guan J Pyk2 and FAK differentially regulate progression of the cell cycle. J Cell Sci 113 (Pt 17), 3063–3072 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Lim Y et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol 180, 187–203, doi: 10.1083/jcb.200708194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weis SM et al. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol 181, 43–50, doi: 10.1083/jcb.200710038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan H & Guan JL Compensatory function of Pyk2 protein in the promotion of focal adhesion kinase (FAK)-null mammary cancer stem cell tumorigenicity and metastatic activity. J Biol Chem 286, 18573–18582, doi: 10.1074/jbc.M110.200717 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLean GW et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev 18, 2998–3003, doi: 10.1101/gad.316304 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashton GH et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev Cell 19, 259–269, doi: 10.1016/j.devcel.2010.07.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahlou H et al. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci U S A 104, 20302–20307, doi: 10.1073/pnas.0710091104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo M et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res 69, 466–474, doi: 10.1158/0008-5472.CAN-08-3078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slack-Davis JK, Hershey ED, Theodorescu D, Frierson HF & Parsons JT Differential requirement for focal adhesion kinase signaling in cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Ther 8, 2470–2477, doi: 10.1158/1535-7163.MCT-09-0262 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soria JC et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann Oncol 27, 2268–2274, doi: 10.1093/annonc/mdw427 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Shimizu T et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 77, 997–1003, doi: 10.1007/s00280-016-3010-1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones SF et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs 33, 1100–1107, doi: 10.1007/s10637-015-0282-y (2015). [DOI] [PubMed] [Google Scholar]
- 33.Gerber DE et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 139, 60–67, doi: 10.1016/j.lungcan.2019.10.033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapman PB et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364, 2507–2516, doi: 10.1056/NEJMoa1103782 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luke JJ, Flaherty KT, Ribas A & Long GV Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 14, 463–482, doi: 10.1038/nrclinonc.2017.43 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Nazarian R et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977, doi: 10.1038/nature09626 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz Osterman CJ et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 8, doi: 10.7554/eLife.47327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that Wnt/beta-catenin signalling activated by anchorage independent activation of FAK supports platinum chemoresistance in ovarian cancer.
- 38.Jiang H et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860, doi: 10.1038/nm.4123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that FAK inhibition in PDAC tumours can overcome an immunosuppressive fibrotic tumour microenvironment and render tumours responsive to immuno- and chemotherapy.
- 39.Skinner HD et al. Proteomic Profiling Identifies PTK2/FAK as a Driver of Radioresistance in HPV-negative Head and Neck Cancer. Clin Cancer Res 22, 4643–4650, doi: 10.1158/1078-0432.CCR-15-2785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang KJ et al. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin Cancer Res 22, 5851–5863, doi: 10.1158/1078-0432.CCR-15-2603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams KE, Bundred NJ, Landberg G, Clarke RB & Farnie G Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 33, 327–341, doi: 10.1002/stem.1843 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Chen G et al. Wnt/β-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Molecular Cancer Therapeutics 17, 806–813, doi: 10.1158/1535-7163.mct-17-0561 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fallahi-Sichani M et al. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol Syst Biol 13, 905, doi: 10.15252/msb.20166796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirata E et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27, 574–588, doi: 10.1016/j.ccell.2015.03.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study used intravital imaging to identify how molecularly targeted therapy can activate the tumour stroma to rapidly promote a drug tolerant environment and protect melanoma via activation of FAK.
- 45.Canel M et al. T-cell co-stimulation in combination with targeting FAK drives enhanced antitumor immunity. Elife 9, doi: 10.7554/eLife.48092 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serrels B et al. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci Signal 10, doi: 10.1126/scisignal.aan8355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frame MC, Patel H, Serrels B, Lietha D & Eck MJ The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol 11, 802–814, doi: 10.1038/nrm2996 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Hirt UA et al. Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype. Oncogenesis 7, 21, doi: 10.1038/s41389-018-0032-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klaeger S et al. The target landscape of clinical kinase drugs. Science 358, doi: 10.1126/science.aan4368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang J et al. Drug Target Commons: A Community Effort to Build a Consensus Knowledge Base for Drug-Target Interactions. Cell Chem Biol 25, 224–229 e222, doi: 10.1016/j.chembiol.2017.11.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel MR et al. Phase 1/1b study of the FAK inhibitor defactinib (VS-6063) in combination with weekly paclitaxel for advanced ovarian cancer. Journal of Clinical Oncology 32, 5521–5521, doi: 10.1200/jco.2014.32.15_suppl.5521 (2014). [DOI] [Google Scholar]
- 52.Aung KL et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma (PDAC) (MOBILITY-002 Trial, NCT02428270). Journal of Clinical Oncology 36, 409–409, doi: 10.1200/JCO.2018.36.4_suppl.409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mak G et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br J Cancer 120, 975–981, doi: 10.1038/s41416-019-0452-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shapiro IM et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci Transl Med 6, 237ra268, doi: 10.1126/scitranslmed.3008639 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siegel RL, Miller KD & Jemal A Cancer statistics, 2020. CA Cancer J Clin 70, 7–30, doi: 10.3322/caac.21590 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615, doi: 10.1038/nature10166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ducie J et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat Commun 8, 990, doi: 10.1038/s41467-017-01217-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matulonis UA et al. Ovarian cancer. Nat Rev Dis Primers 2, 16061, doi: 10.1038/nrdp.2016.61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JM, Minasian L & Kohn EC New strategies in ovarian cancer treatment. Cancer 125 Suppl 24, 4623–4629, doi: 10.1002/cncr.32544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laklai H et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med 22, 497–505, doi: 10.1038/nm.4082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zaghdoudi S et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Molecular Medicine n/a, e12010, doi: 10.15252/emmm.202012010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies stromal activation of FAK as an independent prognostic marker for disease free survival in pancreatic cancer and highlights the potential of targeting the tumour stroma for the treatment of cancer.
- 62.Kang Y et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. Journal of the National Cancer Institute 105, 1485–1495, doi: 10.1093/jnci/djt210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.US National Library of Medicine. ClinicalTrials.gov. <http://www.clinicaltrials.gov/ct2/show/NCT01778803> (2013). [DOI] [PubMed]
- 64.Walton J et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res 76, 6118–6129, doi: 10.1158/0008-5472.CAN-16-1272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ward KK et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clinical & experimental metastasis 30, 579–594, doi: 10.1007/s10585-012-9562-5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagaraj AB et al. Critical role of Wnt/beta-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget 6, 23720–23734, doi: 10.18632/oncotarget.4690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT03287271> (2017). [DOI] [PubMed]
- 68.Kolev VN et al. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget 8, 51733–51747, doi: 10.18632/oncotarget.18517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kazi JU, Kabir NN & Ronnstrand L Brain-Expressed X-linked (BEX) proteins in human cancers. Biochim Biophys Acta 1856, 226–233, doi: 10.1016/j.bbcan.2015.09.001 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Delaney JR et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat Commun 8, 14423, doi: 10.1038/ncomms14423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lengyel E, Makowski L, DiGiovanni J & Kolonin MG Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 4, 374–384, doi: 10.1016/j.trecan.2018.03.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jana S et al. SOX9: The master regulator of cell fate in breast cancer. Biochem Pharmacol 174, 113789, doi: 10.1016/j.bcp.2019.113789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim MH et al. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J 35, 462–478, doi: 10.15252/embj.201592081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shinde R et al. Abstract CT143: Phase I study of the combination of a RAF-MEK inhibitor CH5126766 and FAK inhibitor defactinib in an intermittent dosing schedule with expansions in KRAS mutant cancers. Cancer Research 80, CT143–CT143, doi: 10.1158/1538-7445.Am2020-ct143 (2020). [DOI] [Google Scholar]
- 75.Verastem Oncology. Addressing RAS Pathway Blockade & Resistance. VS-6766 & Defactinib Combination Data in KRAS Mutant Solid Tumors. Investor Conference Call and Webcast., <https://investor.verastem.com/static-files/93835009-c4b6-4818-9075-16cb64237930> (2020). [Google Scholar]
- 76.Zheng Y et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell 35, 11–25, doi: 10.1016/j.molcel.2009.06.013 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Horton ER et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 17, 1577–1587, doi: 10.1038/ncb3257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study defines a core machinery that is required for integrin-based focal adhesions in cells attached to fibronectin.
- 78.Kim NG & Gumbiner BM Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol 210, 503–515, doi: 10.1083/jcb.201501025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zanconato F, Cordenonsi M & Piccolo S YAP and TAZ: a signalling hub of the tumour microenvironment. Nat Rev Cancer 19, 454–464, doi: 10.1038/s41568-019-0168-y (2019). [DOI] [PubMed] [Google Scholar]
- 80.Zanconato F, Cordenonsi M & Piccolo S YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803, doi: 10.1016/j.ccell.2016.05.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zanconato F, Battilana G, Cordenonsi M & Piccolo S YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol 29, 26–33, doi: 10.1016/j.coph.2016.05.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin L et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 47, 250–256, doi: 10.1038/ng.3218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H et al. YAP/TAZ Activation Drives Uveal Melanoma Initiation and Progression. Cell Rep 29, 3200–3211 e3204, doi: 10.1016/j.celrep.2019.03.021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elbediwy A et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 143, 1674–1687, doi: 10.1242/dev.133728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dawson JC et al. A Synergistic Anticancer FAK and HDAC Inhibitor Combination Discovered by a Novel Chemical-Genetic High-Content Phenotypic Screen. Mol Cancer Ther 19, 637–649, doi: 10.1158/1535-7163.MCT-19-0330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han H et al. Hippo signaling dysfunction induces cancer cell addiction to YAP. Oncogene 37, 6414–6424, doi: 10.1038/s41388-018-0419-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suraweera A, O’Byrne KJ & Richard DJ Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front Oncol 8, 92, doi: 10.3389/fonc.2018.00092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu-Chittenden Y et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26, 1300–1305, doi: 10.1101/gad.192856.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zanconato F et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med 24, 1599–1610, doi: 10.1038/s41591-018-0158-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT02758587> (2016). [DOI] [PubMed]
- 91.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT03727880> (2018). [DOI] [PubMed]
- 92.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT04201145> (2019b). [DOI] [PubMed]
- 93.US National Library of Medicine. ClinicalTrials.gov. <http://www.clinicaltrials.gov/ct2/show/NCT02546531> (2015). [DOI] [PubMed]
- 94.Whitney GS et al. Human T and B lymphocytes express a structurally conserved focal adhesion kinase, pp125FAK. DNA Cell Biol 12, 823–830, doi: 10.1089/dna.1993.12.823 (1993). [DOI] [PubMed] [Google Scholar]
- 95.Chapman NM, Connolly SF, Reinl EL & Houtman JC Focal adhesion kinase negatively regulates Lck function downstream of the T cell antigen receptor. J Immunol 191, 6208–6221, doi: 10.4049/jimmunol.1301587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Courtney AH et al. A Phosphosite within the SH2 Domain of Lck Regulates Its Activation by CD45. Mol Cell 67, 498–511 e496, doi: 10.1016/j.molcel.2017.06.024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chapman NM & Houtman JC Functions of the FAK family kinases in T cells: beyond actin cytoskeletal rearrangement. Immunol Res 59, 23–34, doi: 10.1007/s12026-014-8527-y (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raab M et al. LFA-1 activates focal adhesion kinases FAK1/PYK2 to generate LAT-GRB2-SKAP1 complexes that terminate T-cell conjugate formation. Nat Commun 8, 16001, doi: 10.1038/ncomms16001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gett AV, Sallusto F, Lanzavecchia A & Geginat J T cell fitness determined by signal strength. Nat Immunol 4, 355–360, doi: 10.1038/ni908 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Iezzi G, Karjalainen K & Lanzavecchia A The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 8, 89–95, doi: 10.1016/s1074-7613(00)80461-6 (1998). [DOI] [PubMed] [Google Scholar]
- 101.Wiemer AJ et al. The focal adhesion kinase inhibitor PF-562,271 impairs primary CD4+ T cell activation. Biochem Pharmacol 86, 770–781, doi: 10.1016/j.bcp.2013.07.024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Batista S et al. Haematopoietic focal adhesion kinase deficiency alters haematopoietic homeostasis to drive tumour metastasis. Nat Commun 5, 5054, doi: 10.1038/ncomms6054 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Lechertier T et al. Pericyte FAK negatively regulates Gas6/Axl signalling to suppress tumour angiogenesis and tumour growth. Nat Commun 11, 2810, doi: 10.1038/s41467-020-16618-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tavora B et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. ature 514, 112–116, doi: 10.1038/nature13541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tavora B et al. Endothelial FAK is required for tumour angiogenesis. EMBO Mol Med 2, 516–528, doi: 10.1002/emmm.201000106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pedrosa AR et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and −861. Cancer Res 79, 4371–4386, doi: 10.1158/0008-5472.CAN-18-3934 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Jean C et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol 204, 247–263, doi: 10.1083/jcb.201307067 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Demircioglu F et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat Commun 11, 1290, doi: 10.1038/s41467-020-15104-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang J et al. Focal adhesion kinase-promoted tumor glucose metabolism is associated with a shift of mitochondrial respiration to glycolysis. Oncogene 35, 1926–1942, doi: 10.1038/onc.2015.256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu HJ, Hao M, Yeo SK & Guan JL FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene 39, 2539–2549, doi: 10.1038/s41388-020-1162-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang H et al. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 69, 122–132, doi: 10.1136/gutjnl-2018-317424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vennin C et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med 9, doi: 10.1126/scitranslmed.aai8504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schoenherr C et al. Ambra1 spatially regulates Src activity and Src/FAK-mediated cancer cell invasion via trafficking networks. Elife 6, doi: 10.7554/eLife.23172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Serrels B et al. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol 9, 1046–1056, doi: 10.1038/ncb1626 (2007). [DOI] [PubMed] [Google Scholar]
- 115.Luo M et al. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res 73, 5591–5602, doi: 10.1158/0008-5472.CAN-13-1351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gao H et al. FAK-targeting PROTAC as a chemical tool for the investigation of non-enzymatic FAK function in mice. Protein Cell 11, 534–539, doi: 10.1007/s13238-020-00732-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cromm PM, Samarasinghe KTG, Hines J & Crews CM Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J Am Chem Soc 140, 17019–17026, doi: 10.1021/jacs.8b08008 (2018). [DOI] [PubMed] [Google Scholar]
- 118.Popow J et al. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J Med Chem 62, 2508–2520, doi: 10.1021/acs.jmedchem.8b01826 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Guan JL & Shalloway D Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature 358, 690–692, doi: 10.1038/358690a0 (1992). [DOI] [PubMed] [Google Scholar]
- 120.Schaller MD et al. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci U S A 89, 5192–5196, doi: 10.1073/pnas.89.11.5192 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hanks SK, Calalb MB, Harper MC & Patel SK Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci U S A 89, 8487–8491, doi: 10.1073/pnas.89.18.8487 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Canel M et al. Nuclear FAK and Runx1 Cooperate to Regulate IGFBP3, Cell-Cycle Progression, and Tumor Growth. Cancer Res 77, 5301–5312, doi: 10.1158/0008-5472.CAN-17-0418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hobbs GA, Der CJ & Rossman KL RAS isoforms and mutations in cancer at a glance. J Cell Sci 129, 1287–1292, doi: 10.1242/jcs.182873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT03875820> (2019a). [DOI] [PubMed]
- 125.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT04625270> (2020c). [DOI] [PubMed]
- 126.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT04620330> (2020b). [DOI] [PubMed]
- 127.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT04109456> (2019c). [DOI] [PubMed]
- 128.Stokes JB et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther 10, 2135–2145, doi: 10.1158/1535-7163.MCT-11-0261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.US National Library of Medicine. ClinicalTrials.gov. <https://www.clinicaltrials.gov/ct2/show/NCT04331041> (2020a). [DOI] [PubMed]