

Abstract

The chiral oxazoline motif is present in many ligands that have been extensively applied in a series of important metal-catalyzed enantioselective reactions. This Review aims to provide a comprehensive overview of the most significant applications of oxazoline-containing ligands reported in the literature starting from 2009 until the end of 2018. The ligands are classified not by the reaction to which their metal complexes have been applied but by the nature of the denticity, chirality, and donor atoms involved. As a result, the continued development of ligand architectural design from mono(oxazolines), to bis(oxazolines), to tris(oxazolines) and tetra(oxazolines) and variations thereof can be more easily monitored by the reader. In addition, the key transition states of selected asymmetric transformations will be given to illustrate the features that give rise to high levels of asymmetric induction. As a further aid to the reader, we summarize the majority of schemes with representative examples that highlight the variation in % yields and % ees for carefully selected substrates. This Review should be of particular interest to the experts in the field but also serve as a useful starting point to new researchers in this area. It is hoped that this Review will stimulate both the development/design of new ligands and their applications in novel metal-catalyzed asymmetric transformations.
1. Introduction
Ligands containing a chiral oxazoline are some of the most successful, versatile, and commonly used ligand classes in asymmetric catalysis due to their ready accessibility, modular nature, and applicability in a wide range of metal-catalyzed transformations.
The vast majority of these ligands are formed in short, high yielding synthesis from readily available chiral β-amino alcohols. Therefore, the stereocenter controlling the enantioselectivity of the metal-catalyzed process resides α- to the oxazolinyl nitrogen donor and, as a result, is in close proximity to the metal active site to directly influence the asymmetry induced in the reaction.
Since the first report of chiral oxazoline-based ligands in asymmetric catalysis in 1986, an enormous range of ligands containing one, two, three or four oxazolines incorporating various heteroatoms, additional chiral elements and other specific structural features have been subsequently developed with great success in a significant range of asymmetric reactions. SciFinder data for the years 2009 to 2019 confirm the significance of research related to the term “oxazoline” (Figure 1).
Figure 1.

SciFinder data on oxazoline-related research for the years 2009 to 2019.
This review reports on the use of such ligands in homogeneous metal-catalyzed asymmetric synthesis since 2009, when the area was last reviewed by us.1 This review does not cover the application of such ligands in heterogeneous systems or in the synthesis of polymers. We cover, in our view, the most significant applications of oxazoline-containing ligands reported in the literature until the end of 2018. This review will be structured in the same manner as our 20042 and 2009 reviews as we classify ligands, not by the reaction to which their metal complexes have been applied, but by the nature of the denticity, chirality and donor atoms involved. As a result, the continued development of ligand architectural design can be more easily monitored. In addition, the key transition states of selected asymmetric transformations employing metal complexes of oxazoline ligands will be given to illustrate the features that give rise to high levels of asymmetric induction. As a further aid to the reader, we summarize the majority of schemes with representative examples which highlight the variation in % yields and % ees for carefully selected substrates.
2. Mono(oxazoline) Ligands
2.1. Mono(oxazoline) P,N-Ligands (Phosphinooxazoline)
2.1.1. Phosphinooxazoline Ligands with One Stereocenter
Phosphinooxazoline (PHOX) is a popular class of bidentate ligand in which the chiral oxazoline moiety is solely responsible for asymmetric induction in product formation (Figure 2).3−6 In this section, various PHOX ligands (1–17) and their applications in a broad range of asymmetric transformations will be discussed. This section is further divided into subsections based on the type of reactions the metal complexes of PHOX ligands have been used for.
Figure 2.
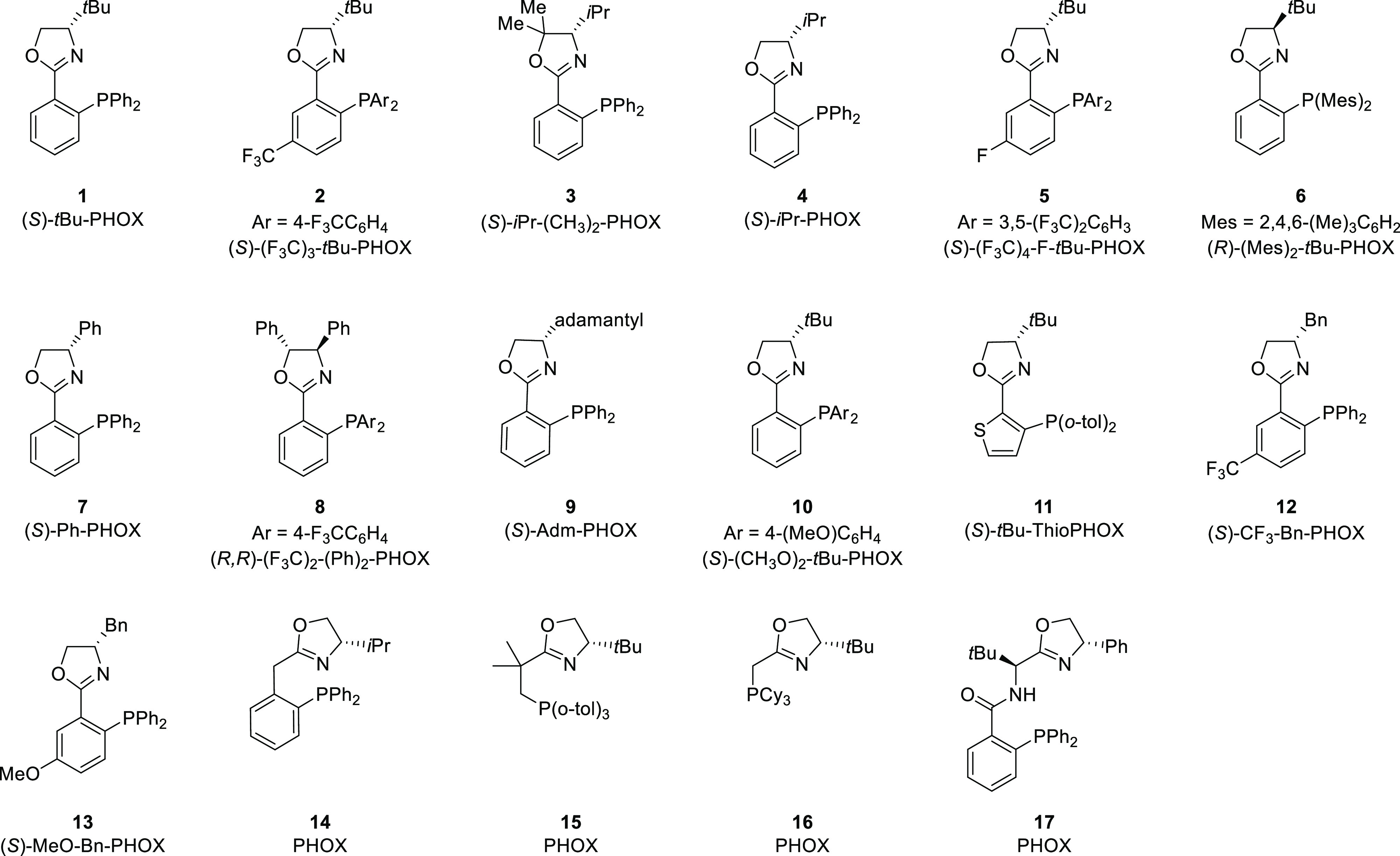
Various phosphinooxazoline (PHOX) ligands
2.1.1.1. Applications of PHOX in Asymmetric Allylation
In late 2004, Stoltz and Trost independently developed a Pd-catalyzed decarboxylative asymmetric allylic alkylation (DAAA) reaction using tBu-PHOX (1) and Trost-type ligands, respectively (Scheme 1).7,8 The generally accepted mechanism of the DAAA involves the coordination of the Pd(0) complex to the allyl moiety leading to oxidative addition, followed by the loss of CO2 to generate the Pd enolate 18-Y, which then attacks the intermediate allyl group to form the enantioenriched product 19 and regenerate the Pd(0) catalyst.9
Scheme 1. Decarboxylative Asymmetric Allylic Alkylation (DAAA).

Since the seminal report from Stoltz, the scope and application of PHOX ligand in DAAA has been expanded to different substrate classes and also in natural products synthesis.
In 2008, Stoltz described a concise and versatile strategy for the preparation of the natural products, cyanthiwigin B, F and G by using a key double stereoablative decarboxylative asymmetric allylic alkylation of bis(β-keto ester) 20 (Scheme 2).10,11 Their strategy involved the synthesis of the central six-membered ring, cyclohexadione 21, with a special emphasis on the early installation of two of the most critical stereocenters of the cyanthiwigin framework in good yield with a high level of stereoselectivity (4.4:1 dr and 99% ee). The major diastereomer of cyclohexadione (R,R)-21-A was subsequently converted for the efficient synthesis of cyanthiwigin B (24-A), F (24-B), and G (24-C). It is worthwhile noting that the observed selectivity in the double decarboxylative allylation with the (S)-tBu-PHOX (1) ligand was excellent given the fact that the reaction begins with a complicated mixture of racemic and meso-diastereomers which leads to several possible stereochemical outcomes, and pathways.
Scheme 2. Catalytic Asymmetric Synthesis of Tricyclic Cyanthiwigin Framework.

The key double catalytic enantioselective alkylation was troublesome on a large scale due to the poor solubility of the catalyst which required low reaction concentrations (0.01 M). This problem was improved significantly in their subsequent report wherein the enantioselective alkylation was carried out on a 10 g scale furnishing the desired diketone (R,R)-21-A in 94% yield, 4.2:1 dr and 99% ee.12
Stoltz extended DAAA to seven membered cyclic β-keto-allyl ester (25) to generate allylated products (26), which were further employed in the asymmetric synthesis of densely functionalized acylcyclopentenes (28), valuable intermediates for the synthesis of natural products, in excellent yields (up to 99%) and high enantioselectivities (up to 92% ee) (Scheme 3).13 The synthesis of 28 from 25 involves a Pd-catalyzed decarboxylative asymmetric allylic alkylation followed by a two-carbon ring contraction.
Scheme 3. Asymmetric Synthesis of Densely Functionalized Acylcyclopentenes.

Later in 2013, Stoltz reported a Pd-catalyzed enantioselective α-alkylation to cyclobutanones (30). The palladium complex of an electron-deficient (S)-(CF3)3-tBu-PHOX (2) ligand demonstrated excellent catalytic activity to afford α-quaternary cyclobutanones in good to excellent yields and enantioselectivities (Scheme 4).14 A wide variety of substituents were compatible at both the α-keto and 2-allyl positions especially considering the presence of highly electrophilic cyclobutanones. Furthermore, chiral cyclobutanones were converted into dialkyl γ-lactams, dialkyl γ-lactones, α-quaternary cyclopentanones, and quaternary [4.5]-spirocycles.
Scheme 4. Enantioselective α-Alkylation of Cyclobutanones.

In 2013, Lupton reported a Pd-catalyzed decarboxylative asymmetric allylation for the enantioselective synthesis of carbazolone and indolone heterocycles (32) (Scheme 5).15 A variety of carbazolones and indolones containing a quaternary carbon center were synthesized in excellent yields (up to 98%) and enantioselectivities (up to 94% ee). Moreover, the application of this methodology was shown in the synthesis of (+)-kopsihainanine (33).
Scheme 5. Catalytic Decarboxylative Asymmetric Allylation of Carbazolones and Indolones.

In 2013, Shao reported the Pd-catalyzed enantioselective decarboxylative allylic alkylation of carbazolone heterocycles (34) (Scheme 6).16 A variety of highly functionalized chiral carbazolones (35) featuring an α-quaternary carbon center were synthesized in good yields (up to 93%) with high levels of enantioselectivity (up to 97% ee).
Scheme 6. Catalytic Decarboxylative Asymmetric Allylation of Carbazolones.

Using this methodology, a catalytic asymmetric strategy for the synthesis of (+)-kopsihainanine A (33) and (−)-aspidospermidine (38) was accomplished from a common intermediate (Scheme 7).
Scheme 7. Catalytic Asymmetric Synthesis of (−)-Aspidospermidine and (+)-Kopsihainanine A.

Stoltz in 2013 reported a Pd-catalyzed decarboxylative allylic alkylation of novel allyl ester substrates (39, 41, and 43) to probe the influence of enolate electronics and the role of α-functionality on the selectivity (Scheme 8A–C).17 It was observed that the high enantioselectivities obtained with imides and lactams are due to both electronic and steric effects associated with the α-substituent, and the enolate electronics alone contribute comparatively less to the stereochemical outcome of the reaction.
Scheme 8. Decarboxylative Asymmetric Allylic Alkylation.

In 2012, Stoltz reported the highly enantioselective Pd-catalyzed decarboxylative allylic alkylation of readily available lactams (45) to form 3,3-disubstituted pyrrolidinones, piperidinones, caprolactams and structurally related lactams (46) (Scheme 8D).18 This method was employed for the catalytic asymmetric synthesis of key intermediates previously used for the construction of Aspidosperma alkaloids quebrachamine and rhazinilam.
In 2015, Stoltz reported the synthesis of α-tertiary piperazin-2-ones (48) by a Pd-catalyzed decarboxylative allylic alkylation (Scheme 8E).19 This method employed a more electron-rich Pd catalyst, [Pd2(pmdba)3] and an electron-deficient PHOX ligand to afford products in good to excellent yields and enantioselectivities. Additionally, this method also allows for the synthesis of α-secondary piperazin-2-ones in modest to excellent yields and good to excellent enantioselectivities. A variety of substituents at nitrogen and also at the stereocenter were tolerated under the reaction conditions employed.
In 2015, Stoltz developed a method to synthesize α,α-disubstituted N-heterocyclic carbonyl compounds (50), by using the well-established DAAA reaction (Scheme 8F).20 Various heterocycles including morpholinone, thiomorpholinone, oxazolidin-4-one, 1,2-oxazepan-3-one, and 1,3-oxazinan-4-one performed well to furnish carbonyl products with fully a substituted stereocenter at the α-position. The presence of an electron-deficient N-substituent was required for high reactivity and enantioselectivity.
Guillou reported a method to access enantioenriched spiroimines (54) using Pd-catalyzed decarboxylative allylic alkylation (Scheme 9).21 A variety of cyclic ketones (52) having α-allyl, propyl or butyl azido groups were synthesized in moderate to good enantioselectivity which, upon isomerization of the allyl group followed by a [3 + 2]-cycloaddition of the azidoalkene, afforded a variety of chiral spiroimines.
Scheme 9. Enantioselective Synthesis of Spiroimines.

In 2016, Stoltz reported the first catalytic asymmetric total synthesis of (−)-goniomitine (59) starting from indole in 11 steps with an 8% overall yield (Scheme 10).22 The important step during this synthesis involves the Pd-catalyzed decarboxylative allylic alkylation of rationally designed heteroaryl bromide 55 furnishing product in 83% yield and 96% ee. Additionally, the formal synthesis of (+)-aspidospermidine (57) and (−)-quebrachamine (58) was also demonstrated.
Scheme 10. Catalytic Asymmetric Total Synthesis of (−)-Goniomitine, (+)-Aspidospermidine, and (−)-Quebrachamine.

In 2018, Stoltz reported a strategy to construct fluorine-containing tetra-substituted stereocenters by the enantioselective Pd-catalyzed decarboxylative allylic alkylation of various carbonyl compounds (60) (Scheme 11).23 An electron-deficient (S)-(CF3)3-tBu-PHOX (2) ligand was optimal and this reaction showed significant substituent tolerance as a variety of five- and six-membered ketones and lactams (60) afforded the corresponding products with good yields and enantioselectivities.
Scheme 11. Enantioselective Synthesis of Fluorine-Containing Tetra-Substituted Stereocenters.

In 2010, Stoltz devised a Pd-catalyzed highly enantio- and diastereoselective α-alkylation cascade protocol (Scheme 12A).24 This method allowed for the installation of vicinal quaternary and tertiary stereocenters in a single step. Cyclohexanone based β-keto-ally esters 62 and arylidene malononitrile 63 were suitable substrates and the desired products 64 were formed in up to 99% ee and with a > 20:1 dr. Small alkyl groups (R) were tolerated best in the reaction, although the use of electron-donating substituents on the arylidene malononitrile gave rise to lower yields. The installation of three stereocenters was also attempted by employing α-nitrile esters 65 (Scheme 12B). However, using a palladium complex of the chiral ligand (CF3)3-t-Bu-PHOX (2) in the reaction gave a poor yield of 15% and 64% ee. The reaction proceeds via the decarboxylation of β-keto-allyl ester (62) to generate a Pd-enolate of cyclohexanone (62-X), followed by a conjugate addition to a prochiral activated Michael acceptor which generates densely functionalized molecules (64) that possess a quaternary stereocenter next to a tertiary center (Scheme 12C).
Scheme 12. Enantioselective Decarboxylative Enolate Alkylation Cascade.

In 2008, Paquin detailed a Pd-catalyzed enantioselective decarboxylative allylic alkylation of fluorinated allyl enol carbonates (67) (Scheme 13).25 They discovered an important and unusual effect of the L/Pd ratio on the enantioselectivity, whereby a L/Pd ratio of 1:1.67 (and as low as 1:4) is required to afford the α-fluoroketones (68) in high enantioselectivity (up to 94% ee).
Scheme 13. Enantioselective Decarboxylative α-Allylation of Fluorinated Allyl Enol Carbonates.

In 2008, Stoltz devised a concise enantioselective strategy for the syntheses of elatol (72) and (+)-laurencenone B (71) (Scheme 14).26 The key Pd-catalyzed decarboxylative enantioselective alkylation was employed to access sterically encumbered enantioenriched vinylogous esters.
Scheme 14. Enantioselective Synthesis of Elatol and (+)-Laurencenone B.

In 2011, Stoltz completed the catalytic enantioselective total synthesis of (+)-liphagal (76) in 19 overall steps from commercially available starting materials (Scheme 15).27 The key step involves a Pd-catalyzed decarboxylative allylic alkylation of enol carbonate 73 to furnish tetra-substituted ketone 74 in 87% yield and 92% ee. This intermediate was elaborated to bicycle 75 in a two-step sequence which was further used for the completion of the first total synthesis of (+)-liphagal.
Scheme 15. Catalytic Enantioselective Total Synthesis of (+)-Liphagal.

In 2011, Stoltz developed a formal synthesis of (+)-hamigeran B 79 starting from tetralone 77 using a key Pd-catalyzed enantioselective decarboxylative allylic alkylation (Scheme 16).28 The other key reaction involves a Ru-mediated cross-metathesis with methyl vinyl ketones and a CuH-mediated domino conjugate reduction-cyclization.
Scheme 16. Formal Synthesis of (+)-Hamigeran B.

In 2018, Zhang reported a catalytic asymmetric synthesis of (−)-cephalotaxine 82 using a Pd-catalyzed Tsuji allylation (Scheme 17).29 The synthesis involved 15 steps starting from homopiperonylamine with a 6.2% overall yield. The key allyl enol carbonate precursor 80-B was prepared from the classic Hanaoka’s pyrrolobenzazepine intermediate (80-A).
Scheme 17. Catalytic Asymmetric Synthesis of (−)-Cephalotaxine.

In 2012, Hanessian reported a new approach for the synthesis of tekturma (85), a drug used in the treatment of hypertension. This approach involves a modified Stoltz Pd-catalyzed decarboxylative asymmetric allylation (Scheme 18).30 A remarkable effect of BHT (2,6-di-tBu-p-cresol) as a protic additive on the reaction time and enantioselectivity was discovered. The alkyl aryl enol carbonate (83-A) was converted into intermediate, (R)-α-isopropyl allyl aryl ketone (84-A) in 90% yield and 90% ee which was used for the synthesis of tekturna 85.
Scheme 18. Application of DAAA for Synthesis of Tekturma.

In 2018, Stoltz reported the first enantioselective Pd-catalyzed decarboxylative allylic alkylation of fully substituted acyclic enol carbonates (86) providing linear α-quaternary ketones (87) (Scheme 19).31 High yields and enantiomeric excesses of the product were achieved using the electron-deficient ligand, (S)-(CF3)3-tBu-PHOX (2). Previous reports in Pd-catalyzed allylic alkylation suggested differing selectivities with different enolate geometries but here it was found that the enolate geometry of the starting material has no adverse effect as the same enantiomer of the product was obtained with the same level of selectivity regardless of the starting ratio of enolates. It was postulated that a dynamic kinetic resolution of the two enolate geometries occurs in the reaction possibly due to facile equilibration between O-bound and C-bound Pd enolates in the presence of the electron-deficient PHOX ligand.
Scheme 19. DAAA of Fully Substituted Acyclic Enol Carbonates.

In 2008, Stoltz developed a Pd/(S)-tBu-PHOX-catalyzed asymmetric alkylation of simple dioxanone derivatives (88) to obtain enantioenriched tetra-substituted dioxanone 89 possessing allyl group at α-position (Scheme 20).32 The enantioenriched tetra-substituted dioxanone subsequently was transformed into the enantioenriched α-hydroxyketones, acids, and esters via a three-step sequence which involved acetonide cleavage catalyzed by p-toluenesulfonic acid (TsOH·H2O), oxidative cleavage with periodic acid (HIO4) and finally methylation of the acid moiety. Additionally, this procedure allowed a catalytic enantioselective formal synthesis of (−)-quinic acid, a useful chiral building block.
Scheme 20. DAAA of Dioxanone Derivatives.

Paquin reported a new member of the PHOX family, 5,5-(dimethyl)-iPr-PHOX (3), having parallel reactivity to (S)-(CF3)3-tBu-PHOX (2) with the key advantage of being easily accessible as both enantiomers (Scheme 21).33 The application of ligand was carried out in the enantioselective allylation of fluorinated silyl enol ether (92) and the enantioselective Heck reaction between 2,3-dihydrofuran (94) and phenyl triflate (95). It was observed that the new ligand’s reactivity was as good as (S)-tBu-PHOX (1) and better than the (S)-iPr-PHOX (4), which lacks the gem-dimethyl at C-5, thus demonstrating the beneficial effect of the substituents at C-5.
Scheme 21. Enantioselective Allylation of Fluorinated Silyl Enol Ether and Heck Reaction.

Riant reported a cooperative dual catalysis based on a Pd(0)/Cu(I) system to generate quaternary carbon stereocenters (Scheme 22).34 In this domino process, the first reaction involves a 1,4 reduction of cyclic enones using Cu(I) hydride to generate the Cu enolate species in situ which further reacts with the π-allyl-Pd complex to form the desired allylation product in an asymmetric fashion. The role of KOtBu as an additive was crucial to increase the enantioselectivity. This method showed tolerance toward alkyl and benzyl groups at the α-position of enones furnishing products in comparable yields and enantioselectivities. The presence of the phenyl group at the α-position of enones was also tolerated to give the expected products in good yield, but unfortunately with low levels of enantioselectivity.
Scheme 22. Asymmetric Cooperative Dual Catalysis of α-Substituted Cyclic Enones.

Tunge reported a method for the asymmetric allylic alkylation of α-tetralones via deacylative allylation using Pd(0) and (S)-tBu-PHOX (1) ligand (Scheme 23).35 This method uses a readily available cyclic ketone pro-nucleophile (100) and allylic alcohol (101) as the allyl source. The reaction involves a retro-Claisen cleavage which results in the formation of both reactive intermediates, the active nucleophile (alkoxide) and electrophile (π-allyl). It was observed that the starting acetyl tetralone (100) must be used in slight excess (1.05 equiv) compared to the alkoxide as it had a detrimental effect on ee. The possible explanation for this is that excess alkoxide competes with the enolate for coordination to Pd which interferes with the inner sphere mechanism for allylation as reported by Stoltz.36
Scheme 23. Asymmetric Deacylative Allylation.

After Tunge’s initial report, Aponick extended this intermolecular Tsuji allylation to enol acetates (Scheme 24).37 The enol acetate substrate (103) is advantageous over the Tunge type 1,3-diketones as they can be easily prepared. This intramolecular allylation version facilitates rapid diversification of the α-quaternary stereocenter-containing products. The method tolerates a wide range of functional groups on the allylic part and the enol acetate of both aromatic and aliphatic ketones. The reaction proceeds with enantioselectivity up to 96% ee and the selectivity has a strong dependence on concentration in intermolecular acyl-transfer reactions as the lower concentrations gives products with higher enantioselectivity. The present protocol is better for 2-substituted allyl alcohols compared to simple allyl alcohol substrates.
Scheme 24. Intermolecular Tsuji Allylation of Enol Acetates.

Stoltz disclosed an elegant approach toward the synthesis of the polycyclic norditerpenoid ineleganolide (Scheme 25).38 A key Pd/(S)-tBu-PHOX (1)-catalyzed enantioselective allylic alkylation was employed to synthesize ketone 108, bearing a chloro-substituted allyl moiety and a methyl group at the α-position, in 82% yield and 92% ee. Ketone 108 underwent a series of diastereoselective transformations to provide a 1,3-cis-cyclopentenediol 109 in 91% yield over five steps. This diol served as a building block for the synthesis of ineleganolide (110).
Scheme 25. Synthesis of Polycyclic Norditerpenoid Ineleganolide.

In 2014, Guo and Chen reported a method for the asymmetric synthesis of acyclic α-carbonyl tertiary alkyl fluorides (113) using an enantioselective Tsuji-Trost reaction of racemic acyclic α-fluorinated ketones (111) (Scheme 26).39 The enantioselectivity for the acyclic α-fluoro ketones (113) products formed ranged from 60 to 90% ee. The good (Z)-configuration of in situ generated enolate intermediate and the presence of a fluorine atom in the starting material had a positive effect on the enantioselectivity observed. It is well documented that cyclic ketones give a better level of enantioselectivity compared to acyclic ketones due to the low selectivity of acyclic ketones toward the formation of an E/Z mixture of the Pd enolate in situ.
Scheme 26. Asymmetric Synthesis of Acyclic α-Carbonyl Tertiary Alkyl Fluorides.

Malcolmson developed a Pd/tBu-PHOX (1)-catalyzed enantioselective intermolecular addition of aliphatic amines (115) with acyclic 1,3-dienes (116) (Scheme 27).40 The chiral allylic amines (117) were synthesized in up to 93% ee. The electron-deficient phosphine ligand not only leads to a more active catalyst but also was important to achieve high levels of enantioselectivity. Mechanistic details revealed that the diene stereochemistry has a strong effect on site selectivity as (Z)-phenylbutadiene (116-B) under optimal conditions furnished a dramatically higher proportion of the 1,4-hydroamination product (118) instead of an expected 1,2-hydroamination product (117-A) (1:2 ratio) compared to the reaction of the (E)-isomer (116-A) which also proceeded with reduced enantioselectivity. It was also noted that both hydroamination products are formed exclusively as the (E)-isomer, indicating that olefin isomerization is faster than amine attack. Additionally, 1,2-hydroamination (117-A) formed the same enantiomer from either the (E)- or (Z)-isomer of the starting 1,3-butadiene. This catalytic system is highly efficient for promoting regio-and enantioselective additions of amines to terminal 1,3-dienes, although internal dienes fail to react.
Scheme 27. Enantioselective Hydroamination of Acyclic 1,3-Dienes.

After a year, Malcolmson extended their enantioselective hydroamination strategy to the challenging 1,4-disubstituted acyclic 1,3-dienes (119) (Scheme 28).41 The success of this method was largely due to a Pd-PHOX catalyst with a noncoordinating BArF4 counteranion, triethylamine as an additive and a nonpolar reaction medium. The variety of aryl/alkyl-disubstituted dienes (119) reacted with several aliphatic amines, primary anilines and indoline to generate allylic amines (120) possessing different α-alkyl groups in good yields and selectivities (up to 78% yield, >98:2 rr, 97% ee). Interestingly, secondary amines, such as piperidine and pyrrolidine, were unreactive under the optimal reaction conditions despite having greater nucleophilicity, perhaps due to the higher pKa values of their conjugate acids compared to the more electron-deficient morpholine, piperazines, and tetrahydroisoquinoline (THIQ) which reacts remarkably well in this reaction. Mechanistic details indicated that the hydroamination is reversible as the ee of the product was found to diminish over time.
Scheme 28. Enantioselective Hydroamination of 1,4-Disubstituted Acyclic 1,3-Dienes.

Malcolmson employed a catalytic system of Pd-PHOX for the enantioselective intermolecular hydroalkylation of acyclic 1,3-dienes with Meldrum’s acid derivatives and other activated carbon pro-nucleophiles (Scheme 29).42 A variety of aryl- and alkyl-substituted dienes (116) react with different β-dicarbonyl-like nucleophiles (122) to generate allylic stereogenic centers at the carbonyl’s β-position in up to 96% yield and 95% ee. Similar to hydroamination (Scheme 28), here the use of triethylamine is essential as it acts as a base which upon deprotonation of carbonyl nucleophile would generate the corresponding ammonium enolate and also its protonated source, might potentially act as an acid source for Pd–H formation within the catalytic cycle. Additionally, it is important to note that the reaction is highly site-selective as addition occurs across the diene’s terminal olefin.
Scheme 29. Enantioselective Hydroalkylation of Acyclic 1,3-Diene.

Wolf presented a regio-, diastereo-, and enantioselective allylation of 3-fluorinated oxindoles (124) using catalytic amounts of Pd and (S)-tBu-PHOX (1) (Scheme 30).43 This method offers a synthesis of 3,3-disubstituted fluorooxindole alkaloids (126) possessing vicinal chiral centers. This protocol was effective with allylic acetates which carried only one aryl terminus and C–C bond formation occurs at less hindered sites irrespective of the origin of the acetyl group.
Scheme 30. Enantioselective Allylation of 3-Fluorinated Oxindoles.

You reported a stereoselective synthesis of tetrahydrofuro-benzofurans (129) and tetrahydrofurobenzothiophenes by the dearomatization of nitrobenzofurans (127) and nitrobenzo-thiophenes, respectively using a Pd/tBu-PHOX (1)-catalyzed formal [3 + 2] cycloaddition (Scheme 31).44 The electronic and steric factors on the phosphine of the PHOX ligand were crucial for the success of this reaction. The products with vicinal stereogenic carbon centers were obtained in good to excellent diastereoselectivity (13/1 to >20/1 dr), and enantioselectivity (75–94% ee).
Scheme 31. Enantioselective [3 + 2] Cycloaddition with 2-Nitrobenzofurans.

You also extended this methodology to the 3-nitroindole substrates (130) (Scheme 32). A stereodivergent synthesis of tetrahydrofuroindoles (131) is reported through a diastereoselective and enantioselective dearomative formal [3 + 2] cycloaddition by using catalytic Pd and PHOX ligand.45 The polarity of the solvent was key to obtain high levels of diastereoselectivity. The use of toluene furnished tetrahydrofuroindole products in good to excellent yields (70–99%), diastereoselectivity (87:13 → 95:5 dr), and enantioselectivity (70–88% ee) whereas acetonitrile yielded another diastereomer in good to excellent yields (75–98%) and stereoselectivity (78:22–93:7 dr and 86–98% ee). Mechanistic studies suggested that the origin of the diastereodivergency was due to the different rate-limiting steps in different solvents, thereby leading to a reversal of stereocontrol.
Scheme 32. Enantioselective [3 + 2] Cycloaddition with 3-Nitroindoles.

In 2014, Takemoto achieved a catalytic enantioselective total synthesis of (−)-aurantioclavine (134) in 16 steps starting from 3-nitro-2-iodophenol (Scheme 33).46 The key step involves intramolecular asymmetric amination of allylic carbonate (132), which has a TsNHR group, using Pd and (S)-tBu-PHOX (1) which proceeded smoothly to furnish a seven-membered heterocycle (133) in 89% yield and 92% ee. This asymmetric allylic amination reaction is a powerful tool to construct a chiral azepinoindole skeleton.
Scheme 33. Catalytic Enantioselective Total Synthesis of (−)-Aurantioclavine.

Kozlowski established a first catalytic, enantioselective Meerwein–Eschenmoser Claisen rearrangement using Pd/t-Bu-PHOX (1) complex (Scheme 34).47 This method allowed the synthesis of a range of oxindoles (136) bearing a quaternary stereocenter. The counterion on Pd(II) was crucial in line with catalyst coordination to the β-amidoester substrate (135), the SbF6 and BF4 complexes provided faster rates and greater turnover than the corresponding perchlorates, triflates, and halides. The Pd/tBu-PHOX (1) combination provided the best enantioselectivity with the smaller C3 methyl ester (R1) and the larger C2′ groups (R2). Deuterium labeling experiments suggested a Lewis acid-catalyzed mechanism and excluded the possibility of π-allyl cation chemistry. In the proposed stereochemical model (137), substrate β-amidoester (135) shows two-point coordination from both oxygens to the chiral Pd complex.
Scheme 34. Enantioselective Meerwein–Eschenmoser Claisen Rearrangement.

Kozlowski presented the first Pd/tBu-PHOX (1)-catalyzed, enantioselective Saucy–Marbet Claisen rearrangement to generate enantiopure allenyl oxindoles (139) bearing a quaternary stereocenter at C3 from propargyl-substituted indole derivatives (138) (Scheme 35).48 Alkyne substrates possessing ortho-substituted aryl groups at R2 gave high levels of enantioselectivity and rapid conversion (up to 96% ee). However, alkynes with larger groups like trimethylsilyl (TMS), tert-butyl, or N-methyliminodiacetic acid (MIDA) boronate were less successful. The alkyne geometry significantly alters the topology of the rearrangement transition state. Additionally, the ester functionality interacts with larger alkyne terminal substituents and destabilizes the less-favorable reaction pathway. Furthermore, improved results were achieved with nonaryl alkynes using Binap and its congeners.
Scheme 35. Enantioselective Saucy–Marbet Claisen Rearrangement.

2.1.1.2. Application of PHOX in Decarboxylative Asymmetric Protonation (DAP)
PHOX ligands have also been extensively employed in the Pd-catalyzed decarboxylative asymmetric protonation (DAP) to generate products with a tertiary stereocenter adjacent to a carbonyl group.49
Stoltz reported a Pd/tBu-PHOX (1)-catalyzed DAP of racemic α-aryl-β-ketoallyl esters (141) (Scheme 36).50 The reaction generated α-alkylated cyclic ketones (142) in excellent yields and enantioselectivities (up to 97% yield and 92% ee) with Meldrum’s acid as the proton source.
Scheme 36. Enantioselective Decarboxylative Protonation of α-Aryl-β-ketoallyl Esters.

In 2014, Shao reported an enantioselective synthesis of C3-substituted chiral carbazolones (142-A) by using Pd/PHOX-catalyzed decarboxylative protonation of α-alkyl-β-keto allyl esters (141-A) (Scheme 37).51 The use of methyl 2-cyclopentanonecarboxylate (143) as the proton donor was found to be more effective compared to the normally used Meldrum’s acid. A range of C3-monosubstituted chiral carbazolones (142-A) carrying various valuable functionalities such as, nitrile, ester, amine and azide were accessed in excellent yields (up to 95%) with good enantioselectivities (up to 92% ee). The application of this methodology was demonstrated in the synthesis of a key pentacyclic intermediate of the naturally occurring (−)-aspidofractinine (146).
Scheme 37. Enantioselective Synthesis of C3-Substituted Chiral Carbazolones.

Guiry reported the first catalytic asymmetric synthesis of isoflavanones (148) containing an α-aryl substituent by using enantioselective decarboxylative protonation of α-aryl-β-ketoallyl ester (147) using Meldrum’s acid as the proton source (Scheme 38).52 It was observed that this method is influenced by both the sterics and the electronics on the α-aryl ring of the isoflavanone. The substrate containing coordinating methoxy substituents at both ortho-positions seems to be essential to achieve optimal enantioselectivity. This method allowed them to synthesize highly sterically hindered α-aryl ketones with enantioselectivities of up to 92%.
Scheme 38. Asymmetric Synthesis of Isoflavanones.

In their following report, Guiry developed a Pd-catalyzed decarboxylative asymmetric protonation of α-aryl-β-ketoallyl esters (149) to generate a tertiary α-aryl isoflavanones (150-A) using formic acid as a proton source (Scheme 39).53 It is interesting to note that, a switch in the preferred enantiomer formed was observed with formic acid as the proton source compared to their earlier work using Meldrum’s acid as the proton source. It is one of the rare examples of an asymmetric protonation wherein dual stereocontrol was observed with the same chiral ligand but with a different achiral proton donor. The Pd-enolate formed after decarboxylation of α-aryl-β-ketoallyl esters (149) was protonated by Meldrum’s acid. Remarkably, the rates of the reactions are significantly different as the Meldrum acid-mediated reaction completes in 30 min, whereas the formic acid reaction takes up to 10 h which suggests the necessity for a carbopalladation to occur and subsequent quenching by formic acid. Alternatively, some precoordination of formic acid to the chiral Pd–enolate complex may result in an inner-sphere-type protonation to a different face of the enolate than with Meldrum’s acid.
Scheme 39. Synthesis of Tertiary α-Aryl Isoflavanones.

In the same year, Guiry extended the methodology for the synthesis of a series of tertiary α-aryl cyclopentanones (152) and cyclohexanones (154) from α-aryl-β-keto allyl esters (151 and 153) in moderate to good levels of enantioselectivity (Scheme 40).54 Similar to earlier methods, good levels of enantioselectivity were achieved for substrates containing aryl groups with mono- and di-ortho-substitution.
Scheme 40. Asymmetric Synthesis of Tertiary α-Aryl Cyclopentanones and Cyclohexanones.

Guiry further studied a scope of the decarboxylative asymmetric protonation with indanone based α-aryl-β-keto allyl ester substrates (155) (Scheme 41).55 As seen with other cyclic ketones, both enantiomers of a series of tertiary α-aryl-1-indanones (156) were accessible by simply switching the achiral proton source. In this example of dual stereocontrol, enantioselectivities up to 92% ee (R) and 94% ee (S) were achieved using formic acid and Meldrum’s acid, respectively.
Scheme 41. Synthesis of Tertiary α-Aryl-1-indanones.

2.1.1.3. Application of PHOX in Asymmetric Hydrogenation
Bolm and Hou independently reported the asymmetric hydrogenation of α-substituted enones (Schemes 42 and 43).
Scheme 42. Asymmetric Hydrogenation of α-Substituted Enones.

Scheme 43. Asymmetric Hydrogenation of α-Substituted Enones.

Bolm discovered a simple and highly efficient asymmetric synthesis of α-substituted ketones (158) from α-substituted enones (157) (Scheme 42).56 Both acyclic and cyclic enones underwent smooth catalytic enantioselective hydrogenations using the Ir complex of tBu-PHOX (1) furnishing α-substituted ketones in high yields and enantioselectivities while tolerating a wide variety of substituents.
Hou reported an Ir-catalyzed asymmetric hydrogenation of α-substituted-α,β-unsaturated ketones (157) to generate a chiral center at the α-position of ketones (158) (Scheme 43).57 Different ketone substrates possessing exocyclic alkenes furnished products in high enantioselectivity, even under ambient pressure of hydrogen.
Ding reported an enantioselective synthesis of cyclohexyl-fused chiral spirobiindane derivative (161) via a sequence of an Ir-catalyzed asymmetric hydrogenation of α,α′-bis(arylidene)ketones (159), followed by an asymmetric spiroannulation of hydrogenated chiral ketones (160) promoted by TiCl4 (Scheme 44).58 A variety of cyclohexyl-fused chiral spirobiindanes (161) were synthesized in high yields and excellent stereoselectivities (up to >99% ee). The asymmetric synthesis of cyclohexyl-fused spiroindanediol (1S,2S,2′S)-162, was carried out in >99% ee and 67% overall yield over four steps without chromatographic purification. The (1S,2S,2′S)-162 was then used to access chiral monodentate phosphoramidites 164 and tridentate phosphorus-aminopyridine 164 and the applications of these ligands were shown in various metal-catalyzed enantioselective reactions including hydrogenation, hydroacylation, and [2 + 2] reactions. The catalytic performances of these ligands were very similar to the corresponding well-established spirobiindane backbone based privileged ligands.
Scheme 44. Asymmetric Hydrogenation of α,α′-Bis(arylidene)ketones.

Pfaltz discovered an asymmetric hydrogenation of enamines (165) catalyzed by cationic Ir complexes derived from tBu-PHOX (1) (Scheme 45).59 The level of enantioselectivites observed in these hydrogenations greatly depend on the substitution pattern at the double bond and the nitrogen atom. An N-aryl or N-benzyl group seems to have a beneficial effect on the enantioselectivity (>90% ee), whereas enantioselectivities for the hydrogenation of cyclic and acyclic 1,2-disubstituted enamines were lower.
Scheme 45. Asymmetric Hydrogenation of Enamines.

Pfaltz applied an Ir-catalyzed hydrogenation for the stereoselective and flexible synthesis of long-chain polydeoxypropionates (Scheme 46).60,61 The Ir complex of chiral PHOX-ligand 6 catalyzed the hydrogenation of 167 with excellent levels of diastereoselectivity. The benzoate anti-167 were obtained with 96:4 diastereoselectivity and >95% isolated yield. Interestingly the opposite diastereofacial selectivity was obtained when the mesityl group on the phosphine ligand was changed to an o-tolyl or a cyclohexyl. Hydrogenated benzoate 168 was further utilized to complete the synthesis of the glycolipid components (+)-phthioceranic acid (169) and (+)-hydroxyphthioceranic acid (170).
Scheme 46. Stereoselective Hydrogenation of 167.

Hou reported an Ir-catalyzed asymmetric hydrogenation of unfunctionalized alkenes (171), α,β-unsaturated esters (157), allyl alcohols, α,β-unsaturated ketones (157), and ketimines (173) using benzylic substituted PHOX ligand 14 (Scheme 47).62 A wide range of substrates were successfully reduced to the corresponding chiral products (172, 158, and 174) with high conversions and high ee values (>99% conversion and up to 99% ee). The reaction of (Z)- and (E)-alkenes gave rise to enantiomeric products in almost the similar enantiomeric excess catalyzed by the same Ir-PHOX 14 complex. The reported P,N-ligands have a six-membered ring with Ir, whereas ligand 14 which is derived from a benzylic substituted P,N-ligand has a seven-membered ring. The increased tether length is more flexible and is believed to have a better chance to fit the steric demands of various substrates. The slightly larger bite angle of ligand was confirmed by X-ray analysis of the corresponding Ir complex.
Scheme 47. Asymmetric Hydrogenation of Unfunctionalized Alkenes.

Pfaltz reported a further Ir-catalyzed asymmetric hydrogenation of unfunctionalized and functionalized alkenes (175) using a new P,N-ligand 15 introduced from their group (Scheme 48).63 The application of their methodology was demonstrated in the synthesis of the antitumor natural product, (R)-(+)-7-demethyl-2-methoxycalamenene (178), a superior approach to an earlier published route with respect to the number of steps and the overall yield (4 steps, 21% overall yield and 98% ee). The catalytic system was also extended to the asymmetric hydrogenation of allylic alcohols, α,β-unsaturated esters and imines which resulted in the corresponding products in good to excellent ees (up to 96%).
Scheme 48. Asymmetric Hydrogenation of Unfunctionalized Alkenes.

Pfaltz described a cationic Ir/chiral P,N-ligand 16 system for the efficient asymmetric hydrogenation of 2-substituted N-protected indoles (179) (Scheme 49).64 The Ir catalyst employed is highly air and moisture sensitive. Various indoles with N-Boc-, N-acetyl-, and N-tosyl protecting groups were hydrogenated in excellent yields and enantioselectivities. The influence of N-protection on the reactivity and enantiomeric excess was lessened by employing the right combination of the catalytic system.
Scheme 49. Asymmetric Hydrogenation of 2-Substituted N-Protected Indoles.

Kuwano developed an Ir-catalyzed enantioselective hydrogenation of isoxazolium triflates (181 and 183) to furnish enantioenriched 4-isoxazolines (182) or isoxazolidines (184) (Scheme 50).65 The hydrogenation of 5-arylisoxazolium triflates (181) exclusively provided the corresponding 4-isoxazolines (182) with good enantioselectivities (up to 90% ee) whereas 5-alkylated isoxazolium triflates (183) selectively furnished cis-3,5-disubstituted isoxazolidines (184) with ees up to 78%. The enantioselectivity was strongly affected by the hindrance from the 5-substituent on the isoxazole ring. Interestingly, the commonly occurred and undesired N–O bond cleavage was not observed under the reaction conditions employed.
Scheme 50. Enantioselective Hydrogenation of Isoxazolium Triflates.

2.1.1.4. Application of PHOX in Addition of Arylboronic Acids to Alkyne, Allene, or Imine
Lam reported a Ni/(R)-Ph-PHOX (7)-catalyzed reaction of arylboronic acids with alkynes (185, 187, and 189) followed by the enantioselective cyclization of an alkenyl Ni species onto a tethered electrophilic trap such as ketones or enones (Scheme 51).66 The success of this domino cyclization is reliant upon the formal anti-carbonickelation of the alkyne, which is suggested to occur by the reversible E/Z-isomerization of an alkenylnickel species.
Scheme 51. Enantioselective Domino Cyclization of Tethered Alkynes.

Furthermore, an intramolecular reaction between 1-phenyl-1-butyne (192) and 2-formylphenylboronic acid (191) using (S,Sp)-tBu-FcPHOX (288) (discussed in detail in section 2.3) furnished the expected indene 193 in 81% yield and 87% ee via a syn-arylnickelation of the alkyne followed by cyclization of the resulting alkenylnickel species onto the aldehyde (Scheme 52). Interestingly, the same chiral ligand, (S,Sp)-tBu-FcPHOX (288), earlier furnished a product via anti-arylnickelation. The ability to synthesize enantioenriched products from either a syn- or anti-carbometallative cyclization further suggests that reversible E/Z-isomerization of the alkenylnickel species is operative and also demonstrates the adaptive power of this Ni-based catalytic system.
Scheme 52. Intramolecular Reaction between 1-Phenyl-1-butyne and 2-Formylphenylboronic Acid.

Lam reported a Ni/(R)-Ph-PHOX (7)-catalyzed desymmetrizing arylative cyclization for the enantioselective synthesis of chiral cyclopent-2-enones (196) by using alkynyl bis(2,2,2-trifluoroethyl) malonates (194) and arylboronic acids (195) (Scheme 53).67 A wide variety of cyclopent-2-enones (196) containing a quaternary stereocenter were synthesized in good yields and excellent enantioselectivities (up to 98% and 94% ee). This cyclization was promoted by the reversible E/Z-isomerization of the alkenylnickel species formed during the reaction.
Scheme 53. Desymmetrizing Arylative Cyclization of Alkynyl Bis(2,2,2-trifluoroethyl) Malonates.

Lam developed a Ni-catalyzed highly diastereoselective annulation between 2-acetylphenylboronic acid (197) or 2-formylarylboronic acid and activated allenes (198) to furnish 3-methyleneindanols (199) (Scheme 54).68 Preliminary results using the chiral phosphinooxazoline ligand, (R,R)-(CF3)2-(Ph)2-PHOX (8) furnished the product in 76% yield and good stereoselectivities (19:1 dr and 74% ee).
Scheme 54. Diastereoselective Annulations between 2-Acetylphenylboronic Acid and Allene.

Cheng reported the synthesis of chiral 1-aminoindenes (202/203) through an intramolecular reaction between o-imodoylarylboronic acids or aryl halides or pseudohalides (200) with alkynes (201) using a Co/chiral PHOX-catalyzed enantioselective [3 + 2] annulation reaction (Scheme 55).69 This reaction achieved a controlled synthesis of both enantiomers of variety of 1-aminoindenes (202/203) in high yields and good to excellent enantioselectivities (up to 98% yield and 98% ee). The unsymmetrical alkynes reacted regioselectively affording single stereoisomers in high yield and excellent ee values. The role of ZnCl2 was found to be crucial as it dramatically enhanced the yield without any negative effect on enantioselectivity. The configuration of the desired product is dominated and controlled by the steric bulk of the substituent attached to the nitrogen-adjacent carbon on the oxazoline ring. As demonstrated in the proposed model, the phenyl group of complex 203-A forces the R group to be far away from the oxazoline moiety and leads the subsequent intramolecular addition of imine from the Re-face, while the isopropyl group of complex 202-A leads to Si-face insertion.
Scheme 55. Enantioselective [3 + 2] Annulation Reaction.

Lam and co-workers earlier discovered that the Ni/(R)-Ph-PHOX (7) complex is highly effective in promoting enantioselective anticarbometallative cyclizations of alkynyl electrophiles with arylboronic acids.66−68 They hypothesized that Ni/(R)-Ph-PHOX (7) complex could also promote a catalytic enantioselective intramolecular 1,4-allylation of substrates containing an allene tethered to an electron-deficient alkene. Considering these facts Lam 2018 described the enantioselective Ni-catalyzed arylative desymmetrization of allenyl cyclohexa-2,5-dienones (204-A) by using arylboronic acids (205) (Scheme 56).70 A variety of hexahydroindol-5-ones and hexahydrobenzofuran-5-ones with three contiguous stereocenters were synthesized in moderate to good yield with high diastereo- and enantioselectivities. The proposed mechanism begins with the Ni/(R)-Ph-PHOX (7)-catalyzed addition of an arylboronic acid to an allenyl cyclohexa-2,5-dienone (204). The resulting allylnickel species 204-X would undergo intramolecular 1,4-allylation to give nickel enolate 204-Y, which upon protonation would liberate the Ni(II) catalyst and a cis-fused hexahydroindol-5-one or hexahydrobenzofuran-5-one (206).
Scheme 56. Enantioselective Ni-Catalyzed Desymmetrization of Allenyl Cyclohexa-2,5-dienones.

In 2016, Hayashi reported a Pd-catalyzed asymmetric arylation of fluoroalkyl-substituted 2-quinazolinone derivatives (207) with arylboronic acids using (S)-iPr-PHOX (4) ligand (Scheme 57).71 A variety of trifluoromethylated and perfluoromethylated 2-quinazoline (208) products possessing quaternary carbon stereocenters were synthesized in excellent enantioselectivities (>99% ee).
Scheme 57. Pd-Catalyzed Asymmetric Arylation.

Later, Zhou employed a Pd-catalyzed enantioselective addition of arylboronic acids to five- and six-membered cyclic α-ketiminophosphonates (209/211) using (S)-tBu-PHOX (1) ligand (Scheme 58).72 The method provided an elegant and efficient route to access chiral α-aminophosphonates (210/212) possessing a quaternary carbon stereocenter in high yields and excellent enantioselectivities (up to 99.9% ee). The use of low catalytic loading and a wide substrate scope are the highlights of this arylation methodology.
Scheme 58. Enantioselective Addition of Arylboronic Acids to Cyclic α-Ketiminophosphonates.

Chen reported a Pd-catalyzed enantioselective addition of arylboronic acids to cyclic iminosulfates (213) using an adamantyl-substituted phosphinooxazoline ligand 9 (Scheme 59).73 This enantioselective arylation tolerates a wide variety of arylboronic acids as well as cyclic iminosulfates to provide cyclic sulfamidates (214) in high yields with excellent ees (up to 97% yield and up to 99% ee). The methodology was applied to synthesize verubecestat (217), a compound under clinical evaluation for the treatment of Alzheimer’s disease.
Scheme 59. Enantioselective Addition of Arylboronic Acids to Cyclic Iminosulfates.

2.1.1.5. Application of PHOX in Asymmetric Borylation of Alkenes
In 2011, Mazet developed an Ir-catalyzed regio- and enantioselective hydroboration of terminal olefins (218) using the tBu-PHOX (1) ligand (Scheme 60).74 The reaction is highly dependent on sterics as changing the R group from methyl to ethyl or cyclohexyl or by replacing the phenyl ring with an o-tolyl group radically decreased the enantioselectivity of the expected hydroboration products.
Scheme 60. Enantioselective Hydroboration of Terminal Olefins.

2.1.1.6. Application of PHOX in Asymmetric α-Arylation
Buchwald reported a Pd-catalyzed α-arylation of aldehydes (220) forming all-carbon-substituted asymmetric centers in high yields and enantioselectivities (Scheme 61).75 Generally, substrates with α-aryl substituents gave rise to products with higher optical purity than these with α-alkyl analogues. The efficiency of the method is excellent for substrates forming five-membered rings, but it dropped significantly for substrates forming a six-membered ring; tetrahydronaphthalene derivatives were prepared in moderate to good yields with moderate levels of enantioselectivity.
Scheme 61. Enantioselective α-Arylation of Aldehydes.

2.1.1.7. Application of PHOX in Asymmetric [2 + 2] Cycloaddition
In 2018, Rajanbabu reported a Co/(R)-Ph-PHOX (7)-catalyzed [2 + 2] cycloaddition between 1,3-enynes (222) and ethylene followed by an enantioselective hydrovinylation of the resulting vinylcyclobutene (223) to give highly functionalized cyclobutanes (224) with an all-carbon quaternary stereocenter, as the (E)-isomer (Scheme 62).76 The reaction proceeds in a tandem fashion to form three highly selective C–C bonds in one pot using a single chiral Co catalyst. The use of an activator, Et2AlCl, was important to promote this cycloaddition reaction. The cycloaddition initiates with an oxidative dimerization of ethylene and the enyne (222) in the coordination sphere of an activated Co(I) species to afford a metallacyclopentene 225-A, which after reductive elimination provides cyclobutene 223. This diene 223 further undergoes oxidative dimerization with ethylene to deliver the metallacycloheptene 225-B. Sterically congested 225-B, (Z)-allylcobalt(III)-hydride undergoes (Z)/(E)-isomerization through an η3-(allyl) intermediate 225-C, which upon β-hydrogen elimination and reductive elimination finally leads to the (E)-224 isomer as the major product. The ratio of (Z)- and (E)-isomers depended on the nature of the R substituent and the nature of the ligand used.
Scheme 62. Asymmetric [2 + 2] Cycloaddition between 1,3-Enynes and Ethylene.

2.1.1.8. Application of PHOX in Asymmetric Heck Reaction
Overman and Wrobleski reported a synthesis of (+)-minifiensine (228), which involves an asymmetric Heck-cyclization of dienyl aryl triflate 226 using a catalytic amount of Pd and (S)-tBu-PHOX (1) as the key step to provide dihydrocarbazole 227 in 99% ee (Scheme 63).77 It is noteworthy that microwave heating (170 °C) was employed to accelerate this catalytic transformation.
Scheme 63. Asymmetric Heck-Cyclization of Dienyl Aryl Triflate 226.

In 2009, Guiry reported a Pd-catalyzed intermolecular asymmetric Heck reaction between 2,3-dihydrofuran (94) and a range of triflates using HetPHOX ligands (Scheme 64).78 The HetPHOX ligand derived from tert-leucinol and di-o-tolylphosphine, (S)-(o-tol)2-tBu-ThioPHOX (11) proved the most effective affording ee values of up to 96, 95, and 94% in the phenylation, cyclohexenylation (using triflate (229)), and naphthylation, respectively, of 2,3-dihydrofuran.
Scheme 64. Intermolecular Asymmetric Heck Reaction.

Zhu reported an asymmetric reductive Heck reaction using a catalytic amount of Pd, (S)-tBu-PHOX (1) and diboron-H2O as a hydride source (Scheme 65).79 This method was applied to synthesize a library of enantioenriched oxindoles (232) possessing a C3-quaternary stereocenter in excellent yields and enantioselectivities. Alkenes possessing aryl and alkyl groups at R2 were compatible. The use of D2O instead of H2O as a D-donor enabled the synthesis of CH2D substituted oxindoles. The catalytic cycle initiates with oxidative addition of the tBu-PHOX (1) ligated Pd(0) to aryl triflate 231, followed by intramolecular carbopalladation, generating a cationic alkyl-Pd(II) intermediate 231-A which reacts with B2(OH)4 and H2O in a series of steps to afford alkyl-Pd(II)-H species 231-E Finally, reductive elimination from 231-E provides the desired product 232 with simultaneous regeneration of the Pd(0)/tBu-PHOX (1) catalyst.
Scheme 65. Asymmetric Reductive Heck Reaction.

Tong developed a Pd-catalyzed asymmetric vinylborylation of (Z)-1-iodo-diene (233) with B2Pin2 using (S)-CF3–Bn-PHOX (12) (Scheme 66).80 This method allowed them to synthesize a variety of 3,3-disubstituted tetrahydropyridines (234) derivatives in good yields and high enantioselectivities. The choice of the tether between vinyl iodide and the alkene had a strong effect on the reaction outcome, as a tether with a strong coordinating atom with Pd had a positive effect and this protocol was limited to the synthesis of a 6-membered cyclic product. This reaction begins by the oxidative addition of Pd(0) to vinyl iodide followed by a Ag-mediated transmetalation with B2Pin2 to form a vinyl-Pd(II)-Bpin species which, upon intramolecular enantioselective carbopalladation followed by reductive elimination, furnished the expected product. Mechanistic studies revealed that the transmetalation step occurs before the alkene insertion, which was complementary to the previous understanding of a similar reaction.
Scheme 66. Asymmetric Vinylborylation.

In 2017, Tong developed an asymmetric reductive Heck cyclization of (Z)-1-iodo-1,6-dienes (235) using Pd2(dba-OMe)3 and (S)-MeO-Bn-PHOX (13) as the ligand (Scheme 67).81 This method provided a facile access to chiral quaternary tetrahydropyridines (236) with good to excellent yields and enantioselectivities. Additionally, this method tolerates susceptible β-hydrogen elimination and a substrate bearing a trisubstituted alkene. The linker between the vinyl iodides and alkenes as well as substituent R1, R2, and R3 had a strong influence on the yield and enantioselectivity of product.
Scheme 67. Asymmetric Reductive Heck Cyclization.

2.1.1.9. Application of PHOX in Asymmetric Alkylation
In 2018, Bisai reported a Cu-catalyzed asymmetric alkylation of 3-hydroxy-2-oxindole (237) with a variety of malonates (238) using the tBu-PHOX (1) ligand (Scheme 68).82 The presence of an indole moiety at C3 is critical for success and no reaction occurred when indole was replaced with an alkyl group. This observation suggested in situ formation of a highly reactive intermediate 237-A followed by an enantioselective malonate addition. The mechanistic studies confirmed that the reaction is reversible in nature and the involvement of a Cu(II)-complex with a distorted trigonal bipyramid (TBP) geometry.
Scheme 68. Asymmetric Alkylation of 3-Hydroxy-2-oxindole.

The application of the method was illustrated in the formal total synthesis of (−)-folicanthine (242) by synthesizing a C2-symmetric dimeric 2-oxindole 241 possessing an all-carbon quaternary stereocenter in 5 steps (Scheme 69).
Scheme 69. Synthesis of (−)-Folicanthine.

2.1.1.10. Application of PHOX in Desymmetrizing Cross-Coupling
Rovis reported a Rh/tBu-PHOX (1)-catalyzed cross-coupling between sp3 organozinc reagents and 3,5-dimethylglutaric anhydride (243) (Scheme 70).83 A variety of syn-deoxypolypropionates (244) were synthesized in excellent yields and good to excellent enantioselectivities. This catalytic system worked well with alkyl and benzyl zinc reagents possessing various functionalities, however it was less efficient with phenyl and isopropyl zinc reagents.
Scheme 70. Desymmetrizing Cross-Coupling.

2.1.1.11. Application of PHOX in Asymmetric Conjugate Addition
Jung reported a Cu-catalyzed asymmetric conjugate addition of alkylzinc reagents to nitroalkenes (245) and cyclic enones by using the newly synthesized P,N-ligand 16 (Scheme 71).84 A variety of chiral nitroalkanes (247) and β-alkyl cyclic ketones were synthesized in good yields and good enantioselectivity (up to 92% yield and 95% ee). Computational studies suggested that the chiral substituents in the oxazoline moiety of P,N-ligand 16 are not crucial for the selectivity whereas the other alkyl substituent in the bridging moiety is involved in the steric repulsion during the transition state.
Scheme 71. Asymmetric Conjugate Addition.

Metal complexes of PHOX ligands with one stereocenter (1–17) have been extensively studied in the past decade. These complexes have been successfully applied in various asymmetric transformations. Palladium complexes were excellent for (I) decarboxylative allylic alkylation and protonation, (II) Heck reaction, (III) hydroamination and alkylation of diene, (IV) [3 + 2] cycloaddition, (V) Meerwein-Eschenmoser and Saucy-Marbet Claisen rearrangement, (VI) aryl boronic acid addition to various imines. Iridium complexes were mostly utilized in hydrogenation of diverse types of alkenes and imines wheras nickel complexes showed excellent results when employed in annulation reactions between aryl boronic acids and alkynes, allenes and imines. Cobalt complexes were specifically successful in (I) intramolecular reactions between o-imodoylarylboronic acids or aryl halides or pseudohalides with alkynes, and (II) [2 + 2] cycloadditions between 1,3-enynes and ethylene. Copper complexes were successfully employed in (I) asymmetric alkylations of 3-hydroxy-2-oxindole, and (II) asymmetric conjugate additions of alkylzinc reagents to nitroalkenes while rhodium complexes found their application in desymmetrizations of 3,5-dimethylglutaric anhydride with alkyl zinc bomides. It is evident from the literature covered in this section of the review that PHOX ligands with only one stereocenter are adaptable with various metals and hence researchers should seek to continue to expand their utilization in asymmetric catalysis.
2.1.2. Phosphinooxazoline Ligands with Stereoaxis or Stereocenter
This section deals with the application of phosphinooxazoline ligands (248a–m) possessing a stereoaxis or stereocenter in metal-catalyzed enantioselective reactions (Figure 3).
Figure 3.

Phosphinooxazoline ligands with a stereocenter or stereoaxis.
2.1.2.1. BiphPHOX Ligands
BiphPHOX ligands (248a–b) possess a chiral oxazoline ring and exist as an equilibrium mixture of diastereomers in solution due to rotation around the internal bond of the biphenyl groups. Interestingly, when these ligands coordinate to Pd or Ir, only one of two possible diastereomeric complexes are formed. The Zhang group extensively utilized the metal-complexes of BiphPHOX ligands (248a–b) in the Ir-catalyzed asymmetric hydrogenation of variably substituted olefins and in the Ni-catalyzed arylation of cyclic aldimines and ketimines.
In 2013, Zhang reported the first asymmetric hydrogenation of α-alkylidene succinimides (249a) using an Ir/(S)-iPr-BiphPHOX (248a) complex (1 mol %) to afford hydrogenated products (249b) in excellent yields (>99%) and enantioselectivities (up to 99% ee) under 20 bar H2 (Scheme 72).85 The reactions performed under reduced catalytic loading (0.05 mol %) and reduced pressure (1 bar) were successful although at the expense of a prolonged reaction time. The iPr substituent on the oxazoline ring had a strong impact on the enantioselectivity (99% ee). The α-alkylidene succinimide (249a) with (E)-configuration was essential for the success of hydrogenation whereas the N-protecting group did not affect the yield and enantioselectivity of the reaction.
Scheme 72. Asymmetric Hydrogenation of α-Alkylidene Succinimides.

In a subsequent study, Zhang described the first asymmetric hydrogenation of unfunctionalized exocyclic C=C bonds (250) with Ir and In-BiphPHOX (248b) delivering the expected chiral 1-benzyl-2,3-dihydro-1H-indene products (251) in up to 98% ee (Scheme 73).86 The use of coordinating solvents like THF and dioxane dramatically decreased the conversion by deactivating the Ir catalyst while the additive acetate ion plays a crucial role in improving the enantioselectivity.
Scheme 73. Asymmetric Hydrogenation of Unfunctionalized Exocyclic Alkenes.

Afterward, Zhang reported the asymmetric hydrogenation of substituted 2H-chromenes and substituted benzo[e][1,2]oxathiine 2,2-dioxides (252) (Scheme 74).87 A variety of 2H-chromenes possessing 3-aryl/alkyl substituents were hydrogenated to the corresponding chiral 3-substituted chromanes (253) in high yields (92–99%) with excellent enantioselectivities (>99% ee). The 4-phenyl substituent on 2H-chromenes also gave the corresponding product in excellent yield but with poor enantioselectivity. Interestingly, benzo[e][1,2]oxathiine 2,2-dioxides with phenyl and methyl substituents produced the desired products in excellent yields but with contrasting enantioselectivity for phenyl (94% ee) and methyl substituents (27% ee).
Scheme 74. Asymmetric Hydrogenation of Substituted 2H-Chromenes and Benzo[e][1,2]oxathiine 2,2-Dioxides.

Similarly, Zhang extended their catalytic system for the asymmetric hydrogenation of 3-substituted 2,5-dihydropyrroles (254) and 3-substituted 2,5-dihydrothiophene 1,1-dioxides (256) (Scheme 75).88 The hydrogenation of 3-substituted 2,5-dihydrothiophene 1,1-dioxides was more challenging compared to the 3-substituted 2,5-dihydropyrroles and hence, a higher temperature and hydrogen pressure were required to achieve full conversions.
Scheme 75. Asymmetric Hydrogenation of 3-Substituted 2,5-Dihydropyrroles and 2,5-Dihydrothiophene 1,1-Dioxides.

Later on, Zhang employed iPr-BiphPHOX (248a) in a Ni(II)-catalyzed asymmetric addition of arylboronic acids to cyclic aldimines and ketimines (258) furnishing products with excellent yields (up to 99%) and enantioselectivities (>99% ee) (Scheme 76).89 Ligand iPr-BiphPHOX (248a) coordinates with Ni(II) to form a complex with (S)-axial chirality, while the complex with (R)-axial chirality was disfavored because of the steric hindrance of the iPr group and the anion coordinated to Ni(II). Crystallographic studies confirmed the (S)-configuration and tetrahedral geometry of the Ni(II) complex. Interestingly, DFT calculations suggested that in solution the configuration of the Ni(II) complex changes from tetrahedral to planar.
Scheme 76. Asymmetric Arylation of Cyclic Aldimines and Ketimines.

Ligand 248c is architecturally very similar to BiphPHOX (248a–b) and it has been applied in the asymmetric hydrogenation of unsaturated acids.
In 2017, Zhang developed a series of highly modular phosphinooxazoline ligands from readily available (S)-(+)-2-phenylglycinol. These ligands were utilized in the Ir-catalyzed asymmetric hydrogenation of α,β-unsaturated carboxylic acids (260) to provide chiral α-substituted carboxylic acids (261) (up to 97% ee, 98% yield, 2000 TON) (Scheme 77).90 Ligand 248c possessing a 3,5-(tBu)2C6H3 substituent on the oxazoline was found to be optimal. Changing the 3,5-(tBu)2C6H3 substituent to a phenyl or a tBu drastically reduced the reactivity and enantioselectivity in hydrogenation. The role of Et3N as an additive was crucial as the reactivity dropped dramatically without it.
Scheme 77. Asymmetric Hydrogenation of α,β-Unsaturated Acids.

2.1.2.2. SIPHOX Ligands
SIPHOX ligands (248d–g) have been employed in the Ir-catalyzed asymmetric hydrogenation of olefins, Pd-catalyzed Narasaka-Heck cyclization, Pd-catalyzed enantioselective formal [6 + 4] cycloaddition of vinyl oxetanes with azadienes and Ni-catalyzed enantioselective arylation of cyclic N-sulfonyl imines.
Zhou reported the asymmetric hydrogenation of α,β-unsaturated carboxylic acids (262) possessing tetra-substituted olefins using Ir and chiral P,N-ligands (248d) based on the spiro-backbone (Scheme 78).91 This method offers a direct approach to chiral carboxylic acids possessing an α-stereocenter having aryl, alkyl, aryloxy, and alkyloxy substituents. The substituents on both phosphorus (3,5-di-t-butylphenyl) and the oxazoline ring of 248d had a strong effect on the outcome of the reaction. A chiral induction model was proposed which showed that the (R)-aryloxy or (R)-alkyloxy-β,β-dimethyl acrylic acids 262 prefer to coordinate to catalyst (Sa,S)-248d with Re-facial selectivity and consequently generates products 263 preferably with (S)-configuration.
Scheme 78. Asymmetric Hydrogenation Reaction of α,β-Unsaturated Carboxylic Acids.

In 2013, Zhou extended the Ir-catalyzed enantioselective hydrogenation strategy to 1,1-diarylethenes (264) and 1,1-dialkylethenes (267) by using a carboxylic acid as a directing group (Scheme 79).92 The carboxylic acid group is essential for the success of this method as no reaction was observed without it or with the corresponding ester. The diphenylethene substrates having a carboxylic acid group either at the meta- or para-position could not be hydrogenated under these reaction conditions. Importantly, no reaction occurred in the absence of the base triethylamine. These experiments revealed that the carboxylic acid reacts with a base and acts as an anchor by coordinating with the Ir catalyst. This enables the catalyst to discriminate between the prochiral faces of the substrates and catalyze their hydrogenation with high levels of enantioselectivity.
Scheme 79. Enantioselective Hydrogenation.

In 2017, Bower developed a Pd-catalyzed highly enantioselective Narasaka-Heck cyclization starting from oxime esters with tethered trisubstituted alkenes (269) using Spinol-derived chiral ligands 248f (Scheme 80).93 This method provides access to dihydropyrrole derivatives (270) with a nitrogen-containing stereocenter in up to 86% yield and 90% ee. It is important to note that the alkene geometry is criticial for reaction success, as cyclization with the (Z)-isomer proceeds with considerably lower ees and yields compared to the corresponding (E)-isomer. Additionally, alkenes with 1,2-disubstitution resulted in a mixture of dihydropyrrole and pyrrole products due to competing β-hydride elimination. The proposed reaction mechanism suggested the enantioselective migratory insertion of alkenes into Pd–N bond was the key step.
Scheme 80. Enantioselective Narasaka–Heck Cyclization.

Zhao reported a highly enantioselective synthesis of benzofuran/indole-fused ten-membered heterocycles (273) via a formal [6 + 4] cycloaddition of vinyl oxetanes (272) with azadienes (271) (Scheme 81A).94 A wide range of benzofuran- and indole-fused heterocycles were accessed in excellent yields and enantioselectivities. The use of aryl- and alkynyl-substituted vinyl oxetanes as substrates was successful whereas an alkyl-substituted vinyl oxetane failed to furnish the desired product, possibly due to the difficulty in the formation of the Pd-π-allyl intermediate. Furthermore, a unique fragmentation of these ten-membered heterocycles (273) was achieved under Lewis acid catalysis to furnish a thermodynamically more favorable six-membered ring (274). Additionally, this method was also employed to generate nine-membered heterocycles (276) by reacting azadiene (271a) with vinyl epoxide (275) via a formal [5 + 4] cycloaddition reaction (Scheme 81B).
Scheme 81. Enantioselective Formal [6 + 4] Cycloaddition of Vinyl Oxetanes with Azadienes.

Lin reported a Ni-catalyzed enantioselective arylation of cyclic N-sulfonyl imines (258) with arylboronic acids (Scheme 82).95 A newly developed chiral phosphinooxazoline ligand, (Ra,S,S)-248h, based on the hexamethyl-1,1′-spirobiindane backbone (HMSI-PHOX), which is a derivative of SIPHOX, provided optically active amines in high yields (up to 94%) and enantioselectivities (up to 99% ee).
Scheme 82. Enantioselective Arylation of Cyclic N-Sulfonyl Imines.

2.1.2.3. SpinPHOX Ligands
SpinPHOX ligands (248i–k) have been employed in the Ir-catalyzed asymmetric hydrogenation of α-aryl-β-substituted acrylic acids, exocyclic α,β-unsaturated carbonyl compounds, and 3-ylidenephthalides.
Ding reported the enantioselective hydrogenation of a series of α-aryl-β-substituted acrylic acids (260) by using an Ir-(R,S)-SpinPHOX (248i) based catalytic system (Scheme 83).96 A series of biologically interesting α-aryl acetic acids (261) were synthesized in excellent yields and enantioselectivity (up to 96% ee). The addition of triethylamine as a basic additive was essential for both conversion and enantioselectivity due to the poor solubility and potential impact of the carboxylate group on the catalysis. Additionally, hydrogenation of the (Z)-isomer with (R,S)-SpinPHOX (248i) was not as successful as the conversion to products was too low. It was found that the chirality at the spiro backbone of (R,S)-SpinPHOX (248i) had a significant impact on the asymmetric induction of the reaction. The (R)-configuration in the spiro backbone and an (S)-configuration on the oxazoline component was found to be the matched ligand whereas the corresponding diastereomeric ligand (S,S)-SpinPHOX gave lower conversion and enantioselectivity for the opposite enantiomer.
Scheme 83. Enantioselective Hydrogenation α-Aryl-β-Substituted Acrylic Acids.

The Ir/SpinPHOX (248j) catalytic system was extended to the enantioselective hydrogenation of six- and seven-membered exocyclic α,β-unsaturated carbonyl compounds (277) with a focus on the challenging lactam derivatives (Scheme 84).97 A series of optically active carbonyl compounds (278) such as lactams, lactones and cyclic ketones with an α-chiral carbon stereocenter was prepared in excellent enantiomeric excess (up to 97%). An excellent stereoselectivity was observed when either (R,S)-248j or (S,S)-248j were employed in the reaction and opposite enantiomers were formed preferentially, which indicated that the spiro chirality of the ligand primarily controls the sense of asymmetric induction while the chirality of the oxazoline moiety might have some influence on the level of enantioselectivity. Additionally, the application of this methodology was shown by synthesizing an ε-aminocaproic acid derivative (280) and the nonsteroidal anti-inflammatory drug loxoprofen (282).
Scheme 84. Enantioselective Hydrogenation of Exocyclic α,β-Unsaturated Carbonyl Compounds.

In 2018, Ding reported the enantioselective hydrogenation of 3-ylidenephthalides (283) by using an Ir/SpinPHOX (248k) catalyst (Scheme 85).98 This method provides a straightforward approach to a wide variety of 2-substituted chiral phthalides (284) in high yields with excellent enantioselectivities (up to >99% yield and 98% ee). The application of this methodology was demonstrated in the synthesis of chiral drugs and natural products such as (R)-chuangxinol (284a), (R)-typhaphthalide (284b), and (S)-3-n-butylphthalide (NBP) (284c), a constituent of celery seed oil.
Scheme 85. Enantioselective Hydrogenation of 3-Ylidenephthalides.

In 2018, Teng reported a new rigid phosphinooxazoline ligand (248l,m) possessing a chiral spiro core and it was applied in Pd-catalyzed asymmetric allylic alkylation and amination with ees up to 99% (Scheme 86).99,100
Scheme 86. Pd-Catalyzed Asymmetric Allylic Alkylation and Amination.

To conclude this section, metal-complexes of PHOX ligands with Stereoaxis or Stereocenter (248a–m) have been employed in various asymmetric transformations. Iridium complexes were successful in the hydrogenation of a variety of alkenes whereas nickel complexes were excellent when used in the addition of aryl boronic acids to imines. Palladium complexes found their application in (I) Narasaka-Heck cyclization, (II) [6 + 4] cycloadditions of vinyl oxetanes with azadienes, and (III) allylic alkylation and amination.
2.1.3. Phosphinooxazoline Ligands with a Stereoplane
Ferrocene phosphinooxazoline (FcPHOX) ligand (other acronyms include FOXAP, Phosferrox, FOX) is the major contributor in this category of P,N-ligands (Figure 4). This ligand was first reported by Richards and Uemura in 1993 for Pd-catalyzed asymmetric allylic alkylation and since then has been employed in a myriad of metal-catalyzed asymmetric transformations.101
Figure 4.

FcPHOX ligands.
2.1.3.1. FcPHOX Ligands for Asymmetric Cycloaddition Reactions
In 2014, Wang developed a Cu/(S,Sp)-tBu-FcPHOX (288)-catalyzed asymmetric inverse-electron-demand aza-Diels–Alder reaction of indoles (301) with in situ formed azoalkenes (Scheme 87).102 This method provided access to a variety of biologically important [2,3]-fused indoline tetrahydropyridazine heterocycles (303) in good yields (up to 97%) with high regioselectivity and diastereoselectivity (>20:1 dr), and excellent enantioselectivity (up to 99% ee). The success of this reaction lies with the coordination of the chiral Cu/FcPHOX (288) complex to the in situ formed azoalkene.
Scheme 87. Asymmetric Inverse-Electron-Demand aza-Diels–Alder Reaction of Indoles.

Later, Wang successfully employed azoalkenes in an unprecedented Cu/(S,Sp)-iPr-FcPHOX (287)-catalyzed asymmetric 1,3-dipolar [3 + 4] cycloaddition with nitrones (304) to generate rapid access to biologically important 1,2,4,5-oxatriazepanes (305) in excellent regioselectivity and enantioselectivity (Scheme 88).103 Interestingly, the [3 + 2] cycloadducts which could have potentially been formed were not observed. Alkyl-substituted nitrones resulted in only the racemic products and ortho-substituted aryl hydrazones and alkyl α-chloro-N-acyl hydrazone were not suitable substrates for this reaction. A proposed transition state 306 depicts that the in situ generated azoalkene coordinates with Cu through nitrogen and oxygen. The bulky isopropyl group of the oxazoline ring blocks the backside of the coordinated azoalkene, and hence the nitrone approaches the R group to avoid unfavorable steric congestion between the diphenylphosphine group in the chiral ligand and the R group of the nitrone.
Scheme 88. Asymmetric 1,3 Dipolar [3 + 4] Cycloaddition with Nitrones.

Deng reported the first Cu/(S,Sp)-Ph-FcPHOX (289)-catalyzed asymmetric [3 + 3] cycloaddition of azomethine ylides (308-A) with 2-indolylnitroethylenes (307) which afforded the highly substituted tetrahydro-γ-carboline derivatives (309) in moderate to high yields, and excellent levels of stereoselectivity (up to >98:2 dr, > 99% ee) (Scheme 89).104 This reaction is highly chemoselective toward the formation of the [3 + 3] cycloadduct over the [3 + 2] cycloadduct (up to 94:6 ratio). An alkyl-substituted azomethine ylide afforded the [3 + 2] cycloadduct in 87% yield instead of the expected [3 + 3] cycloadduct, presumably due to the less-reactive nature of the aliphatic imine. Because of the steric effects of the bulkier phenyl group in the oxazoline ring and the diphenylphosphine group of the ligand, the reaction is proposed to involve Si-face attack of nucleophilic complex 310, generated in situ from the chiral Cu/FcPHOX (289) and azomethine ylide (308-A).
Scheme 89. Asymmetric [3 + 3] Cycloaddition of Azomethine Ylides with 2-Indolylnitroethylenes.

During the Michael addition, the carbanion generated adjacent to the nitro group is stable enough at lower temperature and therefore suppresses the Mannich cyclization which gives the [3 + 2] cycloaddition product. Also, similar levels of stereoselectivity were observed for both the [3 + 3] and [3 + 2] cycloadducts suggesting that the Michael addition should be the crucial step for asymmetric induction.
Following their earlier work, Deng described the first example of the Ag(I)/(S,Sp)-Ph-FcPHOX (289)-catalyzed regioselective and stereoselective [3 + 3] annulation of ketone-derived azomethine ylides (311) with 2-indolylethylenes (312) (Scheme 90).105 This tandem method involved a Michael addition followed by a BF3·Et2O-promoted Friedel–Crafts reaction. Like their earlier report, the traditional [3 + 2] cycloaddition was prevented by using sterically hindered ketone-derived azomethine ylides (>20:1 rr). A wide variety of highly substituted tetrahydro-γ-carboline derivatives (313) were obtained in high yields (up to 99%) with excellent stereoselectivities (up to >20:1 dr, up to 99% ee). Remarkably, the stereochemistry for the [3 + 3] annulation of the ketone-derived azomethine ylides was different from that of aldehyde-derived azomethine ylides although the same ligand Ph-FcPHOX (289) was used. This was due to the favored Re-face attack of the nucleophilic ketone-derived azomethine ylide to 2-indolylethylenes because of the steric effect generated by the two bulky phenyl groups in the nucleophile.
Scheme 90. Regioselective and Stereoselective [3 + 3] Annulation of Ketone-Derived Azomethine Ylides with 2-Indolylethylenes.

In 2014, Guo reported a synthetic method to access chiral azacyclic nucleoside analogues (316) via a Cu/(S,S,Rp)-diPh-FcPHOX (290)-catalyzed highly exo- and enantioselective 1,3-dipolar cycloaddition of azomethine ylides (308-A) with β-nucleobase substituted acrylates (315) (Scheme 91).106 A variety of azacyclic nucleoside analogues (316) were synthesized in high yields (up to 99%), excellent exo- (up to 93:7) and enantioselectivities (>99% ee). The use of dipolarophiles including pyrimidine-, benzimidazole-, imidazole-, benzotriazole-, and indole-substituted acrylates, afforded the desired pyrrolidine derivatives with excellent results. It is important to note that the (E)-geometry of β-nucleobase acrylate is essential since the reaction of the (Z)-isomer of β-nucleobase acrylate 315 furnished the expected products in low conversion along with poor enantioselectivity.
Scheme 91. Highly exo- and Enantioselective 1,3-Dipolar Cycloaddition of Azomethine Ylides with β-Nucleobase Substituted Acrylates.

In 2016, Zhou developed the first catalytic asymmetric synthesis of pyrrolidines (318-A) possessing a trifluoromethylated quaternary stereogenic center at the C-3 position of the pyrrolidine ring using the Cu/FcPHOX (291)-catalyzed 1,3-dipolar [3 + 2] cycloaddition between azomethine ylides (308-B) and β-trifluoromethyl β,β-disubstituted nitroalkenes (317) (Scheme 92).107 This exo-selective method provided different pyrrolidine derivatives (318-A) with high diastereoselectivities (up to >98:2 dr) and excellent enantioselectivities (up to >99.9 ee). Azomethine ylides bearing an alkyl-substituent and an ortho-hydroxy substituent on the aromatic ring failed to give any product. Additionally, azomethine ylides derived from alanine did not react under this catalytic protocol.
Scheme 92. [3 + 2] Cycloaddition between Azomethine Ylides and β-Trifluoromethyl β,β-Disubstituted Nitroalkenes.

The proposed transition state 319 shows that the position of the substituents on the aromatic ring of azomethine ylides affects the result through their steric impact. This rationalizes the higher enantioselectivities observed for ortho-substituted azomethine ylides than the corresponding para- and meta-analogues whereas the substituents on the aromatic rings of β-trifluoromethyl nitroalkenes had a limited effect on the enantioselectivity of the reaction.
In 2017, Liu and Zhang reported a Cu/(S,Sp)-iPr-FcPHOX (287)-catalytic asymmetric Michael addition of ketone-derived azomethine ylides (311-A) to β-trifluoromethyl β,β-disubstituted enones (320-A) and subsequent hydrolytic cyclization to generate 1-pyrrolines (321) (Scheme 93).108 This method offers a facile entry to highly functionalized 1-pyrrolines (321) bearing two contiguous stereocenters in excellent stereoselectivities (up to >20:1 dr, 98% ee). Remarkably, the addition of trace amounts of water is essential for the chemoselective formation of 1-pyrrolines, rather than pyrrolidines (formed via direct 1,3-dipolar cycloaddition). The addition of 6 equiv of water changed the ratio between 1-pyrroline to pyrrolidine from 9:1 to >100:1. Similar levels of chemoselectivity were achieved when ethanol was used instead of water.
Scheme 93. Asymmetric Michael Addition of Ketone-Derived Azomethine Ylide to β-Trifluoromethyl β,β-Disubstituted Enones.

In 2017, Deng reported the first example of a Cu/(S,S)-Bn-FcPHOX (292)-catalyzed 1,3-dipolar cycloaddition of azomethine ylides (323) with β-phthaliminoacrylate esters (322) to generate pyrrolidine β-amino esters (324) in high yields with excellent diastereo- and enantioselectivities (up to 98%, > 20:1 dr, > 99% ee) (Scheme 94).109 It was noted that less reactive aliphatic-substituted azomethine ylides and azomethine ylides (323) derived from alanine were well tolerated in the reaction furnishing the corresponding pyrrolidines (324) in excellent stereoselectivities.
Scheme 94. 1,3-Dipolar Cycloaddition of Azomethine Ylides with β-Phthaliminoacrylate Esters.

In 2018, Yang and Deng reported the first example of a highly efficient and stereoselective synthesis of chiral bicyclic 3-azabicyclo[3.1.0]hexanes (326) using a Cu/(S,S)-Ph-FcPHOX (289)-catalyzed asymmetric [3 + 2] cycloaddition of azomethine ylides (308) with trisubstituted cyclopropenes (325) (Scheme 95).110 With this new desymmetrization process, the asymmetric construction of 3-azabicyclo[3.1.0]hexane derivatives (326) possessing five contiguous stereogenic centers and two bridgehead quaternary stereogenic centers was achieved in high yields (up to 99%) and enantioselectivities (up to 99% ee). A variety of functionalities (CO2R, CN, CONMe2) on the cyclopropane ring and aliphatic-substituted azomethine ylides were well tolerated.
Scheme 95. Stereoselective Synthesis of Chiral Bicyclic 3-Azabicyclo[3.1.0]hexanes.

In 2018, Liu and Zhang reported an unprecedented ligand-controlled regiodivergent Cu(I)/chiral P,N-ligand (287/328)-catalyzed asymmetric intermolecular [3 + 2] cycloaddition of α-substituted iminoesters (323) with β-fluoromethyl β,β-disubstituted enones (320) (Scheme 96).111 This novel method allowed for the enantioselective regiodivergent synthesis of pyrrolidines (327 A-B) bearing two adjacent quaternary stereocenters or two discrete quaternary stereocenters, in high yields (up to >99%), and high regio- (up to >20:1 rr), diastereo- (up to >20:1 dr), and enantioselectivity (up to >99% ee). Mechanistic studies provided insights into the ligand controlled origins of the regioselective control of the cycloaddition. The phosphorus and nitrogen atoms of (S,S)-iPr-FcPHOX (287), remain coordinated to the Cu(I) throughout the whole catalytic process, and 327-A was obtained as the main product due to a combination of the electron distribution across the complex, steric hindrance effects, and a π–π interaction between the two aryl rings of the iminoester 323 and enone 320. In contrast ligand 328 acts as a pseudobidentate ligand. The formation of an O–Cu bond with the carbonyl oxygen atom of the enone 320 and dissociation of the amine from the Cu(I) center occurs which makes ligand 328 monodentate and results in switching the regioselectivity of the reaction to form product 327-B.
Scheme 96. Asymmetric Intermolecular [3 + 2] Cycloaddition of α-Substituted Iminoesters.

In 2016, Zhao developed an unprecedented formal [3 + 2] cycloaddition of p-quinone methides (333-A) with vinyl epoxides (334) under Pd/(S,Sp)-iPr-FcPHOX (287) catalysis (Scheme 97).112 A wide range of spiro[4.5]decanes (335) were obtained in high efficiency and stereoselectivity. It is noteworthy that the bulky t-butyl substituents on the para-quinone methide structure were important for the high diastereoselectivity and enantioselectivity of this catalytic transformation. Changing the t-butyl to the smaller isopropyl or methyl groups led to a drastically reduced dr and ee of the reaction (up to 5:1 dr, up to 30% ee). Importantly, a variety of aryl and alkyl-substituted p-quinone methides were tolerated as substrates.
Scheme 97. Formal [3 + 2] Cycloaddition of p-Quinone Methides.

In 2010, Murakami reported a Ni/(S,Sp)-iPr-FcPHOX (287)-catalyzed denitrogenative annulation of 1,2,3-benzotriazin-4(3H)-ones (336) with allenes (337-A) (Scheme 98). A variety of substituted 3,4-dihydroisoquinolin-1(2H)-ones (338), found in a wide variety of plant alkaloids and bioactive compounds, were synthesized in high regio- and enantioselectivity (up to 99% yield and 97% ee).113 This process tolerates a variety of functionalities and worked well with sterically and electronically different N-aryl substituents. The regioselectivity was significantly affected by the sterics of the allene substituent as changing the hexyl group to a cyclohexyl group led to a drop in regioselectivity from 98:2 to 73:27, whereas the enantioselectivity remained unaltered.
Scheme 98. Denitrogenative Annulation of 1,2,3-Benzotriazin-4(3H)-ones with Allenes.

In 2010, Murakami developed a Ni/(S,Sp)-iPr-FcPHOX (287)-catalyzed enantioselective cycloaddition between isocyanate (339) and allene (337-B) (Scheme 99).114 This process involves the [2 + 2 + 2] cycloaddition of two molecules of isocyanate and one molecule of allene, providing an efficient access to enantiomerically enriched dihydropyrimidine-2,4-diones (340). This method is limited to aryl isocyanates since various alkyl isocyanates failed to undergo this cycloaddition. Surprisingly, electron-rich aryl isocyanates furnished products with higher regioselectivity compared to electron-deficient aryl isocyanates. A mechanistic cycle involves intermolecular Ni(0) oxidative cyclization between the allene (337-B) and isocyanate (339), followed by the addition of one more molecule of isocyanate to generate a zwitterionic π-allylnickel species (339-III) which, upon cyclization at the more substituted carbon of the allyl moiety, affords product 340 along with Ni(0).
Scheme 99. Enantioselective Cycloaddition between Isocyanate and Allene.

In 2011, Matsubara developed a decarbonylative cycloaddition of phthalic anhydride (341) with allenes (337-C) to give δ-lactone derivatives (342) (Scheme 100A).115 The reaction proceeds via asymmetric insertion of a carbon–carbon double bond into a carbon–oxygen bond. The use of (S,Sp)-iPr-FcPHOX (287) afforded chiral δ-lactone derivatives (342) in moderate yields and enantioselectivities of up to 81%. This decarbonylation was also extended to thiophthalic anhydride (343) and N-pyrrole-substituted phthalimide (345), which furnished the corresponding chiral cycloadducts (344/346) in high yield and enantioselectivities of up to 87% and 82% ee, respectively (Scheme 100B,C).
Scheme 100. Decarbonylative Cycloaddition of Phthalic Anhydride with Allenes.

In 2017, Sarlah reported a concise synthesis of (+)-pancratistatin (353) and (+)-7-deoxypancratistatin (352) from benzene (347-A) using a dearomative 1,2-transcarboamination approach in six and seven steps in 19% and 12% overall yields, respectively (Scheme 101).116 The key step was to install the first two vicinal stereocenters which involved visible light-promoted para-cycloaddition of the benzene with the N–N arenophile, N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 348), to generate the MTAD-benzene cycloadduct 349. The cycloadduct 349 was further reacted with aryl Grignard reagent 350 using a catalytic amount of Ni/(R,Rp)-iPr-FcPHOX (287) to generate the desired dearomatized product 351-A in 75% yield as a single diastereoisomer (trans) with high enantioselectivity (96% ee) on a decagram scale. The diene intermediate 351-A was further used to complete the synthesis of the (+)-pancratistatin core.
Scheme 101. Synthesis of (+)-Pancratistatin and (+)-7-Deoxypancratistatin.

The cycloadduct 349 comprises bis-allylic bridgehead positions bearing an electron-deficient urazole and hence it is prone for oxidative addition. Mechanistically, the catalytic process involves anti-π-coordination of the diene to the metal complex, oxidative addition, transmetalation to generate a symmetric η5-complex which, upon enantiodiscrimination of the enantiotopic termini of the cyclohexadienyl system, yields the product enantioselectively.
In 2018, Sarlah applied their novel dearomative trans-1,2-carboamination strategy to a range of aromatic precursors (347) including naphthalene and a series of Grignard reagents using an air-stable Ni(acac)2 and (R,Rp)-iPr-FcPHOX (287) to obtain diene products (351) with exclusive 1,2-trans selectivity, and high enantioselectivity (Scheme 102).117 Interestingly, the methodology was also feasible with different vinyl Grignard reagents which allowed for the enantioselective installation of an alkene moiety.
Scheme 102. Dearomative trans-1,2-Carboamination.

In subsequent studies, Sarlah developed a Pd/(S,Sp)-tBu-FcPHOX (288)-catalyzed enantioselective ring-opening reaction of the cycloadduct derived from naphthalene (347-B) and MTAD using lithium enolates derived from ketone (354) (Scheme 103).118 The method is highly step- and atom-economical and delivers products with exclusive syn-1,4-selectivity and high enantioselectivity (up to 94% ee). Compared to the Ni-catalyzed process, the present protocol is limited to specific substrates. Additionally, enantioselective ring-opening with ester-derived lithium enolates was also achieved by using a [Pd(allyl)Cl]2 and (R)-DTBM-SEGPHOS combination.
Scheme 103. Enantioselective Ring-Opening Reaction.

The complementary selectivity observed in Pd and Ni catalysis is likely due to the result of the inner-sphere delivery of the Grignard reagent in the case of cationic Ni η5-complex 356 and outer sphere attack of the enolate on Pd η3-intermediate 357 (Scheme 104).
Scheme 104. Mechanistic Details.

2.1.3.2. FcPHOX Ligands for Asymmetric 1,4- and 1,2-Addition
In 2011, Zajac reported an enantioselective reaction between glycine derivatives (311) and α,β-unsaturated ketones (358) using a Cu complex of (S,Sp)-iPr-FcPHOX (287) ligand (Scheme 105).119 The chiral Michael adducts (359) formed were directly transformed into the cyclized product upon acid/base workup. The use of acrylonitrile, methyl acrylate or phenylvinyl sulfone as Michael acceptors afforded cycloaddition products exclusively instead of the expected chiral Michael addition adducts. The mechanistic studies performed showed that structure 360 retains a distorted tetrahedral geometry around Cu. It is believed that the phenyl groups of the diphenylphosphine and the imine in 311 occupy a coplanar conformation due to possible π-stacking, with the ester t-butyl group underneath the complex. This orientation requires the electrophilic, α,β-unsaturated ketone (358) to approach the nucleophile from the Si-face and eclipse the phenyl groups.
Scheme 105. Enantioselective Reaction between Glycine Derivatives and α,β-Unsaturated Ketones.

In 2010, Hou reported a Cu-catalyzed asymmetric Michael addition of glycine derivatives (311-A) to nitroalkenes (247) using a derivative of the (S,Sp)-iPr-FcPHOX (287-A) ligand (Scheme 106).120 This method provided a route to prepare a variety of β-substituted-α,γ-diaminobutyric acid derivatives (361) in high diastereo- and enantioselectivities (up to 98% de, 98% ee). The ortho-substituted nitrostyrene derivatives provided much lower enantioselectivities (76 to 89% ee) with the (S,Sp)-iPr-FcPHOX ligand. FcPHOX 289 with a P-bound pyrrole group possessing strong π-acceptor and weak σ-donor properties dramatically improved the enantioselectivity (up to 93% ee) for this class of substrates.
Scheme 106. Asymmetric Michael Addition of Glycine Derivatives to Nitroalkenes.

In 2009, Pu reported a Cu/(S,Sp)-tBu-FcPHOX (288)-catalyzed enantioselective synthesis of 4-alkylidenylglutamic acid derivatives (364) from the reaction of a glycinate Schiff base (311-A) with activated alkyl- and aryl-substituted allylic acetates (363) (Scheme 107).121 The presence of electron-withdrawing groups was essential, as no product was formed when glycine t-butyl ester (311-A) reacted with simple allyl acetate. This suggested that the reaction proceeds via a tandem conjugate addition–elimination pathway.
Scheme 107. Enantioselective Synthesis of 4-Alkylidenylglutamic Acid Derivatives.

In 2014, Lin reported the Cu/(S,Sp)-iPr-FcPHOX (287)-catalyzed asymmetric borylation of β-substituted α-dehydroamino acid derivatives (365) using B2Pin2 to furnish enantioenriched syn- and anti-β-boronate-α-amino acid derivatives (366) in a 1:1 ratio (Scheme 108).122 The double bond geometry in the β-phenyl-substituted substrate was very important since the use of the (E)-isomer did not afford any product under these reaction conditions. Changing an aryl group to an alkyl group in the starting alkene afforded the product in high yield but with poor enantioselectivity (<30%) for both regioisomers.
Scheme 108. Asymmetric Borylation of β-Substituted α-Dehydroamino Acid Derivatives.

In 2016, Deng described a Cu/(S,Sp)-Ph-FcPHOX (289)-catalyzed highly enantioselective catalytic 1,6-conjugate addition of glycine Schiff bases (311) to p-quinone methides (333) (Scheme 109).123 A series of p-quinone methides (333) derived from aryl aldehydes reacted with glycine Schiff bases to provide nonracemic β-Ar,Ar′-α-amino acid derivatives (367) with two contiguous stereogenic centers in high yields with excellent diastereoselectivities (up to 99:1) and enantioselectivities (up to >99% ee).
Scheme 109. Enantioselective 1,6-Conjugate Addition of Glycine Schiff Bases.

In 2013, Zhang developed a Cu/(S,Sp)-tBu-FcPHOX (288)-catalyzed regio- and enantioselective 1,4-conjugate addition reaction of alkyl Grignard reagents to α,β,γ,δ-unsaturated ketones (368) (Scheme 110).124 A series of γ,δ -unsaturated ketones (369) were obtained in high yields with high enantioselectivity (up to 92% yield and 96% ee). The increase in the steric hindrance of the Grignard reagent had a negative impact on enantioselectivity as well as on reaction yield. The mechanism is proposed to involve the formation of π-complex 368-I between Cu–ligand, MeMgBr and substrate 368-A. Later, Cu(III) σ-complex 368-II forms via oxidative addition, which is stabilized by the diconjugated system. This stability accounts for the preference of the 1,4-selectivity over 1,6-selectivity. Finally, the reductive elimination of 368-II delivers Cu-FcPHOX and the 1,4-addition product 369.
Scheme 110. Regio- and Enantioselective 1,4-Conjugate Addition Reaction of Alkyl Grignard Reagents.

In 2018, Deng developed the Cu(I)/(S,Sp)-Ph-FcPHOX (289)-catalyzed diastereo- and enantioselective Mannich reaction of glycine Schiff bases (311-B) with isatin N-Boc ketimines (370) (Scheme 111).125 A wide range of 3-substituted 3-aminooxindole compounds (371) were obtained in high yields (up to 98%) with excellent enantioselectivities (>99% ee in most cases) and moderate to high diastereoselectivities (up to 97:3 dr). The reactivities and stereoselectivities of the Mannich reaction were greatly reliant on the steric hindrance of the ester groups employed. The methyl and ethyl glycine esters were most suitable, whereas the diastereoselectivities decreased when isopropyl or benzyl esters were tested. The t-butyl glycine ester Schiff base was found to be unreactive. The N-protection of isatin is crucial as no reaction occurred with isatin possessing a free NH. The imine derived from N-methyl isatin was found to be the best substrate whereas changing to either an N-benzyl or an N-acetyl isatin produced disappointing diastereoselectivities.
Scheme 111. Diastereo- and Enantioselective Mannich Reaction of Glycine Schiff Bases.

2.1.3.3. FcPHOX Ligands for Asymmetric Allylic Alkylation
In 2013, Zhang reported a Pd/(S,Sp)-iPr-FcPHOX (287)-catalyzed asymmetric allylic amination of 4-aryl-1,3-dioxolan-2-one (373) to synthesize chiral β-aryl-α,β-unsaturated amino alcohols (375) (Scheme 112).126 A wide variety of chiral amino alcohols (374) were obtained in good to excellent yields (up to 92%) and enantioselectivities (up to 98% ee) under mild reaction conditions.
Scheme 112. Asymmetric Allylic Amination of 4-Aryl-1,3-dioxolan-2-ones.

In 2017 and 2018, Zhang and Wang independently published a series of papers based on a Pd/Cu dual catalyst system for the asymmetric α-allylation of Schiff base activated amino acids (Scheme 113).127−133
Scheme 113. Asymmetric α-Allylation of Schiff Base Activated Amino Acids.

Zhang reported a Pd/Cu cooperative catalytic system for the asymmetric α-allylation of Schiff bases derived from imine esters (323) (Scheme 114).127 A dimeric tBu-RuPHOX (296) was found to be a common ligand of choice. A wide variety of α,α-dialkyl α-amino acid derivatives (378) were synthesized in high yields and with excellent enantioselectivities (up to >99% ee). The method was easily extended to the asymmetric allylation of a range of small peptides. The catalytic system utilizes chiral complexes of Cu and Pd with the chiral ligand, tBu-RuPHOX (296), and the use of the same ligand on both metals avoids the possible problem of ligand exchange and also makes it practically simple. The catalytic cycle involves the simultaneous generation of a five-membered N,O-bidentate metalated nucleophilic species 323-A between Cu and the Schiff base and the electrophilic π-allyl Pd intermediate 376-II. This intermediate further reacts to give allylated product in high selectivity. Both studies results suggested that the rigid structure of the five-membered N,O-bidentate metalated azomethine ylide 323-A enables the asymmetric induction from the chiral ligand (Scheme 113).
Scheme 114. Cu/Pd-Catalyzed Asymmetric α-Allylation of Schiff Base.

Zhang found that the aldehyde-derived glycine esters (323) always furnished bisallylated products. This limitation was solved in the subsequent studies involving asymmetric allylation of ketone-derived prochiral glycine ester and amide derivatives (311) using the catalytic Cu/Pd cooperative system (Scheme 115).128 This time ligand (S,Sp)-tBu-RuPHOX (294) performed better compared to the earlier investigated dimeric ligand tBu-RuPHOX (296). A range of α-substituted α-amino acid derivatives (379) were efficiently synthesized in high yields and with excellent enantioselectivities under mild conditions (up to 98% yield and 99% ee).
Scheme 115. Cu/Pd-Catalyzed Asymmetric α-Allylation of Schiff Bases.

Later, Zhang expanded the scope of this asymmetric allylation to 1-pyrroline-5-carboxylic esters, cyclic imino esters (380) to generate a variety of 3,4–2H-pyrrole (381) derivatives bearing a quaternary stereogenic center in high yields and excellent enantioselectivities (up to 98% yield and >99% ee) (Scheme 116).129 Similar to earlier studies, mechanistic investigations uncovered that the synergistic action of the two chiral metal complexes is responsible for its high reactivity and excellent enantioselectivity; the steric hindrance and electronic factors of the electrophiles and the nucleophiles are crucial for the formation of the linear products.
Scheme 116. Asymmetric Allylation to 1-Pyrroline-5-carboxylic Esters.

In a recent report, Zhang introduced a synergistic Ir/Cu-catalyzed allylic alkylation of imine esters to construct a range of α,α-disubstituted α-amino acids (377-A–D) bearing two vicinal stereocenters with excellent levels of enantio- and diastereoselectivity (up to >99% ee and >20:1 dr) (Scheme 117).130 The (S,Sp)-iPr-RuPHOX (295) ligand on Cu and Feringa’s chiral phosphoramidite ligands (382-A) on Ir were necessary for the success of the reaction. Significantly, the two chiral catalysts allowed complete control over the absolute and relative configuration, affording all stereoisomers of the desired products. This methodology was applied to synthesize dipeptides and analogues of bioactive molecules bearing vicinal stereocenters in a stereodivergent manner. The mechanism is proposed to involved the generation of a five-membered N,O-bidentate metalated nucleophilic species (323-I) using the Cu/iPr-RuPHOX (295) complex followed by interception of the in situ formed reactive allyl Ir intermediate (376-I) to generate enantiopure α,α-disubstituted α-amino acid derivatives (377) bearing vicinal stereocenters (Scheme 113). It is important to note that the well-defined geometry of intermediates 323-I and 376-I allows for the control of the configurations of both stereocenters.
Scheme 117. Ir/Cu-Catalyzed Allylic Alkylation of Imine Esters.

At the same time, Wang independently reported a Pd/Cu dual catalyst system for the asymmetric α-allylation of Schiff bases derived from imine esters (323) to synthesize a variety of α,α-dialkyl α-amino acid derivatives (383) in high yields and with excellent enantioselectivities (up to 97% yield, > 99% ee) (Scheme 118).131 Similar to Zhang’s report, the presence of Cu and Pd is indispensable to this reaction as no product was observed in the absence of either. Wang’s catalytic system involves Cu(I) and chiral ligand 287 which in situ generates the chiral complex (323-A) from the Schiff base of imine ester (Scheme 116). Unlike Zhang’s report, the achiral ligand Ph3P was used with Pd to generate the achiral π-allylpalladium intermediate 376-II (Scheme 113). Besides this, the 31P NMR spectroscopic analysis confirmed that either negligible or no ligand scrambling was observed from Cu to Pd. This observation confirmed that the rigid structure of the five-membered N,O-bidentate chiral Cu complex 323-A controls the stereoselectivity of the allylation.
Scheme 118. Pd/Cu-Catalyzed Allylic Alkylation of Imine Esters.

Wang further employed this synergistic Cu/Pd catalytic system for the stereoselective α-allylation of both acyclic (311-A) and cyclic ketimine esters (384) (Scheme 119).132 The combination of the Cu/(S,Sp)-iPr-FcPHOX (287) complex and an achiral palladium complex delivered an array of biologically important α-allyl α-amino acid derivatives (386) and 2H-pyrrole derivatives (385) in excellent yields and enantioselectivities (up to 99% yield, >99% ee).
Scheme 119. Stereoselective α-Allylation of Acyclic and Cyclic Ketimine Esters.

Like Zhang, Wang established a stereodivergent synergistic Cu/Ir catalyzed α-allylation of aldimine esters to generate a series of α-amino acid derivatives (377) possessing vicinal stereocenters in high yield and excellent stereoselectivity (Scheme 120).133 For the success of reaction, the use of (S,Sp)-iPr-FcPHOX (287) ligand on Cu and Feringa’s chiral phosphoramidite ligands 382-B on Ir was necessary. All four stereoisomers of 377 were accessed in good yield (up to 96%) with excellent enantioselectivity (>99% ee) and exclusive diastereoselectivity (>20:1 dr) from the same set of starting materials with the current dual Cu/Ir catalysis by simply selecting a pairwise combination of two chiral catalysts. This outcome suggested that the two distinct metal catalysts exert almost absolute control over the corresponding stereogenic centers, respectively. Additionally, the dual catalysts can be prepared in situ from a mixture of Cu(MeCN)4BF4, [Ir(cod)Cl]2, (S,Sp)-iPr-FcPHOX (287), and (R,R,R)-382-B in a one-pot protocol to furnish the expected allylated products (377) in comparable levels of yield and stereoselectivity.
Scheme 120. Stereodivergent Synergistic Cu/Ir Catalyzed α-Allylation of Aldimine Esters.

2.1.3.4. FcPHOX Ligands for Asymmetric Intramolecular Arylcyanation/Diarylation of Alkenes
In 2008, Nakao described an intramolecular arylcyanation of alkenes to synthesize a range of synthetically interesting nitriles possessing a benzylic quaternary carbon stereocenter (Scheme 121).,134 The combination of Ni(cod)2 along with the chiral ligand, (R,R)-iPr-FcPHOX (287) and the Lewis acid, AlMe2Cl, efficiently formed product 388 in 96% ee and 88% yield, which is a key intermediate in the subsequent synthesis of (−)-esermethole (389). Later in 2010, the scope of the methodology was expanded by synthesizing a variety of enantiomerically enriched 3,3-disubstituted indoline derivatives.135 It was observed that the electron density on the benzene ring slightly affected the enantioselectivity, possibly through altering the ability of the nitrogen or olefin to coordinate to Ni. Surprisingly, aryl–halogen bonds were tolerated whereas activation of C–CN bonds was achieved exclusively. Additionally, the size of the N-substituents significantly affected both the chemical yield and enantioselectivity as the bulkier substituents on the nitrogen slow down oxidative addition of the Ar–CN bond as well as coordination of the double bond to Ni. Mechanistic studies revealed that the AlMe2Cl promoted η2-nitrile complex formation by coordinating with the cyano nitrogen.
Scheme 121. Asymmetric Intramolecular Arylcyanation of Alkenes.

An enantioselective diarylation of activated alkenes was reported by Kong wherein structurally distinguishable aryl bromides react together via a domino Heck cyclization/reductive cross coupling process using a catalytic amount of Ni/(S,S)-iPr-FcPHOX (287) (Scheme 122).136 This method allowed direct access to various bis-heterocycles 392 bearing an all-carbon quaternary center in moderate to good yields (up to 81%) with excellent enantioselectivity (up to 99% ee). Experimental evidence suggested that (i) no intermediacy of arylboronic reagent instead B2Pin2 acts as a coreducing agent; (ii) the enantioselective-determining step of the reaction is the migratory insertion, and it should be irreversible; (iii) the cyclization process is not the turnover-limiting step. A plausible mechanism of the reaction involved two reaction pathways. Initial oxidative addition of aryl bromide 391 to the Ni(0) species followed by an intramolecular carbonickelation results in the formation of a σ-alkyl-Ni(II)Br species 392-A. In pathway A, 392-A was then reduced by stoichiometric Zn/Pin2B2 to generate σ-alkyl-Ni(I)Br species 392-B, which further undergoes a second oxidative addition with aryl bromide 391 to form the σ-alkyl-Ni(III)ArBr species 392-C. Finally, reductive elimination of 392-C affords the desired product 392 and regenerates the Ni(0) catalyst upon reduction with Zn/Pin2B2. Pathway B involves transmetalation between both Ni(II) centers 392-A and ArNi(II)Br to form the σ-alkyl-Ni(II)ArBr species 392-D, which generates product 392 following reductive elimination.
Scheme 122. Enantioselective Diarylation of Activated Alkenes.

2.1.3.5. FcPHOX Ligands for Ir-Catalyzed Asymmetric Hydrogenation
In 2009, Hou developed an asymmetric hydrogenation of α,β-unsaturated amides (393) using an Ir/(S,Sp)-tBu-FcPHOX (288) catalytic system to produce amides (394) with an α-chiral center in high yield and excellent enantioselectivity (Scheme 123).137 The presence of hydrogen on the amide nitrogen is imperative for the enantioselectivity of the reaction. Interestingly, amides having an aliphatic substituent at the β-position were also suitable substrates.
Scheme 123. Asymmetric Hydrogenation of α,β-Unsaturated Amides.

In 2017, Zhang developed a Ir/(S,Sp)-tBu-RuPHOX (294)-catalyzed asymmetric hydrogenation of 5,6-dihydropyrazin-2(1H)-ones (395) to synthesize chiral piperazin-2-ones (396) in excellent yields and with moderate to high ees (up to 97% yield, up to 94% ee) (Scheme 124).138 This asymmetric hydrogenation was easily extended to protected and unprotected amide substrates.
Scheme 124. Asymmetric Hydrogenation of 5,6-Dihydropyrazin-2(1H)-ones.

Zhang reported the Ir-catalyzed asymmetric hydrogenation of simple ketones (397) and exo-α,β-unsaturated cyclic ketones (399) using a novel planar chiral ferrocene phosphinooxazoline ligand 297 (Scheme 125).139 A variety of ketones (397/399) were converted into their corresponding secondary alcohols (398/400) in up to 98% yield and 99% ee. The Ir-297 complex was very stable and displayed high catalytic activity (0.005 mol %, S/C = 20 000) during the asymmetric hydrogenation of acetophenone on a 4.80 g scale providing alcohol product in 95% yield and 96% ee. Deuterium labeling studies confirmed that the reaction proceeds with hydrogen gas rather than via transfer hydrogenation with the alcoholic solvent.
Scheme 125. Asymmetric Hydrogenation of Ketones.

Zhang, Dong and Lan reported a novel modular electron-donating tridentate ligand having a chiral ferrocenylphosphine motif linked to the oxazoline unit through a secondary amine (Figure 4). The highly active and air-stable ferrocene aminophosphooxazoline (f-amphox, 298) ligands were applied in the Ir-catalyzed asymmetric hydrogenation of various ketones to generate alcohol derivatives in excellent yields and enantioselectivities (Scheme 126-131).140−146
Scheme 126. Asymmetric Hydrogenation of Methylketones.

Scheme 131. Asymmetric Hydrogenation of Halogenated Ketones.

The initial application of this Ir-f-amphox catalyst in the asymmetric hydrogenation of simple ketones (401) provided the corresponding secondary alcohols (402) in excellent enantioselectivities (up to 99.9% ee) and activities (up to 1 000 000 TON) (Scheme 126).140 It was observed that the (Sc,Sc,RFc) configuration of the f-amphox is very critical for enantioselectivity and reactivity.
Later, various α-hydroxy ketones (403) were converted to the corresponding chiral vicinal 1,2-diols (404) with excellent outcomes (TON up to 1 000 000, up to 99% yield, >99% ee) (Scheme 127).141
Scheme 127. Asymmetric Hydrogenation of α-Hydroxy Ketones.

Similarly, the asymmetric hydrogenation of α-amino ketones (405) afforded the corresponding chiral 1,2-amino alcohols (406) in excellent activities and enantioselectivities (TON was up to 500 000, >99% conversion, >99% ee). The hydrogenated product possessing 3-hydroxy functionality on benzene ring is the key intermediate of (S)-phenylephrine, an enantiomer of an important α-adrenergic receptor agonist (Scheme 128).142
Scheme 128. Asymmetric Hydrogenation of α-Amino Ketones.

Asymmetric hydrogenation of a series of racemic α-amino β-unfunctionalized ketones (407) via a dynamic kinetic resolution (DKR) process allowed for the construction of a series of chiral 1,2-amino alcohols (408) bearing vicinal stereocenters with excellent results (all products >99% ee and >99:1 dr, TON up to 100 000). The success of this DKR was due more to the base-mediated rapid racemization of the substrate than hydrogenation. As expected, no reaction occurred without a base. The catalytic asymmetric hydrogenation with the process of DKR provided a powerful synthetic strategy for the synthesis of a key chiral intermediate of the preclinical antitumor agent (S,S)-R116010 (Scheme 129).143
Scheme 129. Asymmetric Hydrogenation of α-Amino Ketones via Dynamic Kinetic Resolution.

Further, the asymmetric hydrogenation of a wide range of prochiral α-, β-, γ-, and δ-keto amides (409/411) was achieved to deliver a range of chiral hydroxy amides (410/412) with excellent results (TON was up to 100 000, >99% conversion and >99% ee). A DFT study suggested that noncovalent interactions between the ligand and substrate play an important role in achieving high enantioselectivity (Scheme 130A,B).144,145
Scheme 130. Asymmetric Hydrogenation of Ketoamides.

Finally, the asymmetric hydrogenation of prochiral halogenated ketones (413) was also achieved to produce chiral halohydrins (414) with excellent results (TON up to 20 000, up to 99% yield and >99% ee) (Scheme 131).146
Mechanistic studies for the asymmetric hydrogenation of acetophenone indicated that the formation of intermediate 415 is kinetically and thermodynamically favored (Scheme 132). Then, the hydrogenation of acetophenone with intermediate 415 takes place via transition state 416 which forms the major product 402. Studies showed that in 415 the right-hand side is occupied by the ligand’s cyclopentadiene moiety and an equatorial phenyl group, which blocks the right-hand side during hydrogenation and hence acetophenone is thought to approach the Ir-center of 415 by placing the larger group at the unencumbered left-hand side.
Scheme 132. Mechanistic Studies for Asymmetric Hydrogenation of Acetophenone with f-Amphox.

2.1.3.6. Asymmetric Allylation with 1,1′-P,N-Ferrocene Ligand
In 2008, Hou developed a Pd-catalyzed asymmetric allylic alkylation of acyclic amides (417) using the 1,1′-P,N-ferrocene ligand (299) (Scheme 133).147 Among the various ligands tested, (Sc,Sphos,Sa)-299 afforded the allylated products in high yields (up to 99%) with excellent enantioselectivities (up to 93% ee). The nature of the substituents on the amide nitrogen and the use of allyl acetate instead of the corresponding carbonate or phosphonate had a significant influence on the efficiency and enantioselectivity of the reaction. The application of (Sc,Sphos,Sa)-299 without a stereogenic center in the oxazoline decreased the yield (65%) and ee of the product (55%) suggesting its importance. Also, the product configuration was determined by the stereochemistry of the Binol component rather than that of the phosphorus atom. The presence of a lithium counterion and the exact amount of base was crucial for the success of the reaction. In the proposed stereochemical model, the Binol subunit and the nucleophile occupies the same side of the π-allyl complex which favors Re face attack to provide the product with (R)-configuration.
Scheme 133. Asymmetric Allylic Alkylation of Acyclic Amides.

Later in 2009, Hou reported the highly efficient kinetic resolution of 2,3-dihydro-2-substituted 4-quinolones (421) using a Pd/(S,Rphos,R)-300-catalyzed asymmetric allylic alkylation (AAA) (Scheme 134).148 This method provided the allylated products, 2,3-disubstituted 2,3-dihydro-4-quinolones (424), with trans-selectivity in 37–48% yields and 83–93% ee along with 37–47% yields of recovered starting materials (423) in 87–99% ee (S-factor of 40–145). It is noteworthy that this method incorporates chiral centers at both α- and β-positions of the ketone. The nitrogen substituent of 421 also played a role in the reaction, as for example, when the acetyl group of 4-quinolone was replaced by H or Boc, both the allylated product and recovered substrates were obtained in lowered ee. The application of the methodology was demonstrated in the synthesis of pyrrolo[3,2-c]quinoline (425), a core structural motif of biologically active Martinella alkaloids.
Scheme 134. Kinetic Resolution of 2,3-Dihydro-2-substituted 4-Quinolones.

In 2012, Hou reported a Pd/(Sc,Rphos,Ra)-299-catalyzed asymmetric allylic alkylation (AAA) between allylic carbonate (376) and nitromethane (Scheme 135).149 Among the various ligands tested, (Sc,Rphos,Ra)-299 afforded branched allylated products (426) in high yields with excellent regio- and enantioselectivities. The synthetic application of the method was shown by preparing (R)-baclofen 427, an antispasmodic and (R)-rolipram, an anti-inflammatory agent and antidepressant.
Scheme 135. Asymmetric Allylic Alkylation of Nitromethane.

In 2014, Hou reported a Pd/(Sc,Rphos,Ra)-299-catalyzed asymmetric allylic alkylation of α-fluoro-alkylphosphonates (428) with monosubstituted allylic substrates (376) to afford allylated products (429) with two chiral centers in high regio-, diastereo- and enantioselectivities (Scheme 136).150 The reaction was highly diastereoselective for all allyl substrates tested possessing either electron-donating or electron-withdrawing substituents on the phenyl ring whereas the regioselectivity was sensitive to the substituent on the aryl group of the carbonates (376). The reaction of cinnamyl carbonate furnished the expected product with >20:1 branch to linear ratio, whereas any other substituent on the phenyl ring of carbonates (376) lowered the b/l ratio to 7:1. Additionally, highly functionalized products with two adjacent stereogenic centers and three functional groups were easily elaborated to more complex products.
Scheme 136. Asymmetric Allylic Alkylation of α-Fluoro-alkylphosphonates.

To conclude this section, metal-complexes of PHOX ligands (287–300) with a stereoplane have been employed in various asymmetric cycloaddition reactions. Copper and nickel complexes were extensively employed in (I) [4 + 2], [4 + 3], [3 + 3], [2 + 2 + 2] cycloadditions, (II) 1,2- and 1,4-additions. Copper complexes in combination with palladium were excellent for asymmetric α-allylation of Schiff base activated amino acids. Nickel complexes were also used in intramolecular arylcyanation and diarylation of alkenes whereas iridium complexes were successful in alkene, imine and ketone hydrogenations.
2.1.4. Phosphinooxazoline with Phosphorus Bonded to N- and O-Atom
This section deals with the application of phosphino-oxazoline ligands with phosphorus bonded to N- and O- atom (430a–k) in Ir-catalyzed asymmetric hydrogenations. The phosphino-oxazoline ligands discussed in this section are shown in Figure 5.
Figure 5.

Phosphino-oxazoline ligands with Phosphorus Bonded to N- and O- Atom.
The Andersson group reported a series of papers on the applications of P,N-ligands of type 430a–d in the Ir-catalyzed asymmetric hydrogenation of various types of olefins (Scheme 137).
Scheme 137. Ir-Catalyzed Asymmetric Hydrogenation of Olefins.

In 2008, Andersson described the Ir-catalyzed asymmetric hydrogenation of a variety of di- and trisubstituted enol phosphinates (431) using ligand 430a (Scheme 138).151,152 A range of olefin substrates possessing both aromatic and aliphatic substituents afforded hydrogenated products (432) with excellent enantioselectivities (up to >99% ee). Trisubstituted enol phosphinates were hydrogenated in better yields and selectivities than the related 1,1-disubstituted compounds. More importantly, enol phosphinates were stable toward degradation under the hydrogenation reaction conditions employed.
Scheme 138. Ir-Catalyzed Asymmetric Hydrogenation of Di- and Trisubstituted Enol Phosphinates.

In 2009, the first Ir-catalyzed asymmetric hydrogenations of vinyl boronates (433) was developed using ligand 430a/b under low catalyst loadings (0.5 mol %) and pressure (as low as 1 bar) (Scheme 139).153 For most of the vinyl boronates, the selectivity was either decreased or even reversed with decreased hydrogen pressure suggesting the possibility of different mechanisms at play at different concentrations of hydrogen. Vinyl boronates having aromatic or polarizing groups gave good results (98% ee), whereas aliphatic substituents on vinyl boronates were less successful as they gave hydrogenated products with <50% ee.
Scheme 139. Ir-Catalyzed Asymmetric Hydrogenations of Vinyl Boronates.

Andersson subsequently employed ligand 430b for the Ir-catalyzed asymmetric hydrogenation of cyclic allylic amines (435) thereby synthesizing chiral piperidines (436) in good to excellent enantioselectivities (Scheme 140).154
Scheme 140. Ir-Catalyzed Asymmetric Hydrogenation of Cyclic Allylic Amines.

In 2011, Andersson reported the enantioselective synthesis of β,β-disubstituted aldehydes (438) using the Ir-catalyzed asymmetric isomerization of (E)- and (Z)-trisubstituted allylic alcohols (437) (Scheme 141).155 The Ir complex of the bulkier ligand 430b was successfully employed in the isomerization of a broad range of primary allylic alcohols. The yield of product was significantly impacted by the size of the substituent on the allyl alcohols. (E)-Cinnamyl alcohols with Cy and iPr substituents reacted smoothly (86% yield, >99% ee and 88% yield, >99% ee). For ethyl and methyl substituents, the enantioselectivities were only marginally affected (97% ee and 91% ee), but the aldehyde product was recovered in poor yields (21% and <5%). Compared to the (E)-trisubstituted allylic alcohols, the catalytic system was less sensitive to steric effects in the isomerization of (Z)-trisubstituted allylic alcohols.
Scheme 141. Asymmetric Isomerization of (E)- and (Z)-Trisubstituted Allylic Alcohols.

Later in 2012, the same catalytic system was extended to the asymmetric hydrogenation of α,β-unsaturated esters (439) (Scheme 142).156 A wide variety of ester substrates (439) with both aromatic and aliphatic substituents on the prochiral carbon were hydrogenated with excellent enantioselectivities (up to 99%).
Scheme 142. Asymmetric Hydrogenation of α,β-Unsaturated Esters.

More recently, Andersson reported a highly stereoselective synthesis of chiral fluorine-containing compounds (442) with two vicinal stereogenic centers via the asymmetric hydrogenation of tetra-substituted vinyl fluorides (441) (Scheme 143).157 The Ir complex of the newly developed ligand (430d) was determined to be the most effective catalyst. Several aromatic, aliphatic, and heterocyclic substrates with a variety of functional groups were tolerated to provide chiral fluoroalkanes in excellent yield, diastereoselectivity, and enantioselectivity. Moreover, this catalytic hydrogenation system substantially overcomes the problem of defluorination.
Scheme 143. Asymmetric Hydrogenation of Tetra-Substituted Vinyl Fluorides.

Riera and Verdaguer developed a novel ligand, MaxPHOX (430e–g), which contains three stereogenic centers that can be introduced from three separate and simple building blocks. Initially, Ir-complexes of MaxPHOX-445a were applied in the asymmetric hydrogenation of cyclic enamides derived from α- and β-tetralones (443) (Scheme 144A).158,159 The enantioselectivities observed with Ir-MaxPHOX were much better than those with Ru and Rh catalysts. It was noticed that the enantioselectivity was pressure dependent as reducing the hydrogen pressure to 3 bar increased the enantioselectivity. The X-ray crystallographic study of Ir-445a complex showed that the six-membered metallacycle adopts a boat-like conformation. The phosphorus atom and the bulky t-butyl group on the oxazoline ring are syn to each other on the same face of the metallacycle. In addition to this the catalytic activity observed in coordinating solvents like EtOAc and MeOH suggested bidentate binding of the substrate to the cationic Ir-445a complex. It was believed that the directing amide group binds to an axial position away from the bulky t-butyl group, while the alkene binds equatorially trans to phosphorus. Finally, the asymmetric induction dependence on the hydrogen pressure indicated that hydrogen is involved in the enantioselectivity-determining step.
Scheme 144. Ir-MaxPHOX Catalyzed Asymmetric Hydrogenation.

A further application of Ir-MaxPHOX-445b complexes was shown in the asymmetric hydrogenation of N-aryl imines (Scheme 144B).160 Ir-MaxPHOX-445b possessing an iPr group both on the oxazoline ring and in the backbone with a P-stereogenic center was capable of efficient imine hydrogenation. The chirality on phosphorus has a significant impact on the catalyst activity as the non-P-stereogenic equivalent of Ir-MaxPHOX-445b gave rise to lower selectivity. Also, the nature of the counterion considerably affects the reaction outcome; smaller counterions (e.g., BF4) reduced the enantiomeric excess and slowed down the reaction.
Subsequently, the catalytic system was also employed in the synthesis of the antibiotic (R)-sarkomycin methyl ester (449) (Scheme 144C).161 The key step was an unprecedented asymmetric isomerization of cyclic enone 446 catalyzed by the Ir-MaxPHOX-445c complex resulting in the formation of exocyclic enamine in good yield (73%) and excellent enantioselectivity (99% ee). The exocyclic enamine 447 was formed because of stabilization due to the conjugation and formation of a strong intramolecular hydrogen bond. The exclusive formation of the enamide (Z)-isomer 447 also supported strong hydrogen bonding stabilization. The exocyclic enamine was subsequently reduced, the amine deprotected followed by spontaneous elimination to provide the desired antibiotic 449 in 45% yield and 98% ee.
In 2013, Sigman developed a novel phosphoramidite ligand, Phos-PrOx (430h) and applied it in the Ir-catalyzed asymmetric hydrogenation of 1,1-diarylalkenes (450) (Scheme 145).162 The presence of a coordinating group on the meta-position of one of the phenyl rings was essential for the success of the reaction. It was proposed that the 3,5-dimethoxy substitution acts as a directing group by precoordinating the substrate alkenes to Ir. This coordination orients one alkene face toward Ir, poising the substrate for subsequent alkene coordination and hydrogenation to deliver 1,1-diarylmethines in moderate to good levels of enantioselectivity.
Scheme 145. Ir-Catalyzed Asymmetric Hydrogenation of 1,1-Diarylalkenes.

Pfaltz developed the first Ir-catalyzed asymmetric hydrogenation of α,β-unsaturated nitriles (452) using the chiral P,N-ligand, ThrePHOX (430i) and N,N-diisopropylethylamine (DIPEA) (Scheme 146).163 The Ir-430i complex (445d) was inactive without the addition of DIPEA. Slight variation in the ligand structure, changing the double bond geometry from trans to cis and the amount of DIPEA used all had a strong impact on the reactivity and selectivity of hydrogenation. Alkenes lacking a conjugated cyano group did not react in the present system which rendered the possibility of selectively reducing the cyano-substituted C=C bond of an α,β-unsaturated nitrile, while leaving the less electrophilic C=C bonds intact. The authors proposed the generation of a reactive neutral Ir(I) monohydride via base-mediated deprotonation of an Ir-dihydride complex due to its pronounced Brønsted acidity. The newly generated neutral Ir(I) monohydrate is expected to be less electrophilic which means easy release of nitrile from the Ir-metal center, thus opening the free coordination site required for the hydrogenation reaction. Also, the hydride of neutral Ir (I) monohydrate would be more nucleophilic than hydrides in a cationic dihydride complex, thus aiding hydride transfer to the electrophilic C=C bond of the α,β-unsaturated nitrile.
Scheme 146. Ir-Catalyzed Asymmetric Hydrogenation of α,β-Unsaturated Nitriles.

The application of the ThrePHOX ligand (430j) developed by Pfaltz was demonstrated by Schmalz in a short total synthesis of helioporins C (457) and E (458) (Scheme 147).164 The Ir-catalyzed asymmetric hydrogenation of the exocyclic double bond was established for setting up a homobenzylic stereocenter in a highly diastereoselective manner. The hydrogenated product after subsequent desilylation, oxidative cleavage and vinyl Grignard addition afforded the allylic alcohol 456. This allylic alcohol underwent oxidation to afford helioporins C (457) and acid-catalyzed cyclization to give rise to helioporins E (458).
Scheme 147. Total Synthesis of Helioporins C and E.

Kazmaier described a new efficient P,N-ligand 430k for Ir-catalyzed asymmetric hydrogenations of β-arylated α,β-unsaturated ketones (459) (Scheme 148).165 By using Ir-complex (445f) derived from ligand 430k, various linear as well as cyclic α,β-unsaturated ketone substrates were hydrogenated successfully to the corresponding ketones (460) in excellent yields (>99%) and enantioselectivities (>99% ee).
Scheme 148. Ir-Catalyzed Asymmetric Hydrogenations of β-Arylated α,β-Unsaturated Ketones.

To conclude this section, iridium complexes of PHOX ligands (430a–k) with phosphorus bonded to N- and O- atoms have been mainly employed in asymmetric alkene hydrogenation.
2.2. Mono(oxazoline) N,N-Ligands
Mono(oxazoline) bidentate N,N-ligands are also widely used in asymmetric catalysis. PyOx 461, which contains a pyridine-oxazoline backbone is the most used ligand in this class (Figure 6). These ligand class will not be discussed here since Yang and Zhang recently reviewed the area since Brunner’s original synthesis in 1986.166,167
Figure 6.
PyOx ligand.
2.3. Mono(oxazoline) N,S-Ligands
In 2008 Arai developed a series of novel chiral (sulfinyl)furyl oxazoline-containing ligands 463 and applied them in the Pd-catalyzed allylic alkylation of 1,3-diphenylpropenyl acetate 125-A. After optimization, which included changing the solvent from dichloromethane to THF, it was shown that the product 462 was formed in up to 98% yield and 83% ee. It is thought that the oxazoline nitrogen acts as a π-donor while the sulfur is a π-acceptor leading to the reactive intermediate shown (Scheme 149).168
Scheme 149. Pd-Catalyzed Allylic Alkylation with (Sulfinyl)furyl Oxazoline Ligand.

In 2010 Huang reported the synthesis of axially chiral thiourea-oxazoline ligands in the Pd-catalyzed enantioselective bis(methoxycarbonylation) of styrenes 464. When applied in this catalytic transformation ligand 466 afforded the product 465 in up to 85% yield and 84% ee (Scheme 150). A variety of electronically different styrenes 464 were successfully transformed to the product 465, although when nonstyrenyl alkenes were used, the enantioselectivity dropped dramatically to 21% ee although still in 92% yield.169
Scheme 150. Pd-Catalyzed Esterification of Styrenes.

In 2016 White developed a novel set of aryl sulfoxide-oxazoline ligands 469 and applied them in the Pd-catalyzed synthesis of isochroman motifs 468. A series of oxazoline substituents were screened and the ligand containing the phenyl group gave rise to the desired product in up to 64% yield and 95% ee (Scheme 151). The substrate scope included a range of electronically diverse isochroman motifs with 92% ee being the lowest obtained for all substrates. This methodology was subsequently applied in the synthesis of PNU-109291 a potent 5HT1D agonist avoiding the costly chiral resolution used in its previous synthesis.170 In a subsequent publication White further explored the ligand scaffolds performance in the Pd-catalyzed asymmetric allylic alkylation of pyrazole-5-ones.171
Scheme 151. Pd-Catalyzed Synthesis of Isochroman Motifs.

Han reported the asymmetric intramolecular allylation of aryl ureas 470 to indolines 471 using a novel sulfoxide-oxazoline ligand 472. A variety of dienes 116 were applied in this transformation with all substituents furnishing the products 471 in a range of 80 to 90% ee with yields proving highly variable on the substrate used. Interestingly, this ligand does not possess a chiral oxazoline motif and instead relies on asymmetric induction from the chiral sulfoxide unit. When initial conditions were screened the BOX ligand 600c afforded the product in only trace amounts. In the initial screen, benzoquinone was used as the oxidant furnishing the product in 77% yield and 85% ee, and when 2,5-dimethylquinone 2,5-(DMBQ) was used it improved the results moderately to 79% yield and 88% ee (Scheme 152).172
Scheme 152. Pd-Catalyzed Asymmetric Intramolecular Allylation of Aryl Ureas.

Interestingly, the mono(oxazoline) N,S-ligands mentioned in this section (Figure 7) are all used in Pd-catalyzed asymmetric reactions with unsaturated systems. While the chemistry is synthetically useful it appears this class of ligand is currently limited in their application. Utilizing these ligands in a wider range of chemistry with inexpensive metals could further develop this area greatly.
Figure 7.

Summary of mono(oxazoline) N,S-ligands.
2.4. Mono(oxazoline) N,O-Ligands
N,O-Ferrocenyloxazoline ligands (Rp)- and (Sp)-473a were originally synthesized by Bolm in 1997 (Figure 8).173 Since then, Butenschön has described the synthesis of (Rp)-473b and triferrocenylmethane 474 and applied ligands (Rp)-473a–b and 474 in the diethylzinc addition to aromatic and alkenyl aldehydes, achieving enantioselectivities of up to 97% ee.174
Figure 8.

N,O-Ferrocenyloxazoline ligands 473 and 474.
In almost all of the examples reported, triferrocenylmethane 474 gave the best enantioselectivity. To exemplify, the results of each ligand in the diethylzinc addition to benzaldehyde 475 are summarized in Table 1. The reaction with Bolm’s tBu-473a afforded the chiral alcohol 476 in a higher enantiomeric excess (93% ee) than Butenshön’s iPr-473b (83% ee), but Butenschön’s triferrocenyl iPr-474 gave the best result (97% ee). The increased steric bulk beside the alcohol moiety counteracts the decrease in steric bulk going from tBu to iPr on the oxazoline.
Table 1. Comparison of Bolm and Butenschön’s N,O-Ferrocenyloxazoline Ligands in Asymmetric Diethylzinc Addition to Benzaldehyde 475.

| entry | ligand | yield 478 (%) | % ee |
|---|---|---|---|
| 1 | (Rp)-473a | 83 | 93 |
| 2 | (Rp)-473b | 97 | 83 |
| 3 | (Rp)-474 | 95 | 97 |
More recently, Guiry has studied the gem-dimethyl effect in these N,O-ferrocenyloxazoline ligand systems, describing the synthesis of ligands (Rp)- and (Sp)-479a–b and (Rp)-480, applying them in the diethyl- and diphenylzinc addition to aromatic and aliphatic aldehydes 479, with enantiomeric excesses up to >99% (Scheme 153).175
Scheme 153. Application of Guiry’s gem-Dimethyl N,O-Ferrocenyloxazoline Ligands in Diethylzinc Addition to Various Aldehdyes 477.

The results of the diethylzinc additions to benzaldehyde 475 with each ligand are summarized in Table 2. In the case of benzaldehyde, the triferrocenylmethane ligand (480) performed best, giving the chiral alcohol 476 in 93% ee (entry 5). However, for different aldehydes, different ligands performed better. This study shows that the gem-dimethyl effect can be applied to ligands of this class to reach similar levels of enantioselectivity that can be achieved with less economical tBu-ligands, with (Rp)-479a giving the chiral alcohol 476 in 88% ee (compared to 93% ee) (entry 1). Second, trisubstituted ferrocene derivatives (479b) perform worse than the corresponding disubstituted ferrocene derivatives, with (Rp)-479b giving the chiral alcohol 478 in 82% ee (entry 3). Finally, in this chiral ligand scaffold, planar chirality has a dominant effect over the stereoselectivity induced, with the (Sp)-ligands giving lower levels of enantioselectivity in the diethylzinc additions, while reversing the enantioselectivity in the diphenylzinc additions (entries 2 and 4).
Table 2. Comparison of Guiry’s gem-Dimethyl N,O-Ferrocenyloxazoline Ligands in Asymmetric Diethylzinc Addition to Benzaldehyde 475.

| entry | ligand | yield 478 (%) | % ee |
|---|---|---|---|
| 1 | (Rp)-479a | 93 | 88 |
| 2 | (Sp)-479a | 32 | 33 |
| 3 | (Rp)-479b | 54 | 82 |
| 4 | (Sp)-479b | 11 | –5 |
| 5 | (Rp)-480 | 79 | 93 |
Xiao and Chen reported the synthesis of N,O-ligand 481 bearing a phenyl group at the C-1 position of the oxazoline and an ester at the C-4 stereocenter (Figure 9).
Figure 9.

Xiao and Chen’s N,O-ligand 481 and Ir(481)Cp*Cl 482a and 482b.
They applied this ligand in the asymmetric transfer hydrogenation of aryl ketones 483, having prepared both the N,C- and N,O-chelated Ir complexes 482a and 482b. It was observed that 482a did not induce appreciable levels of enantioselectivity in this reaction, while the reaction with 482b gave the corresponding secondary alcohols 484 in up to 98% yield and with up to >99% ee (Scheme 154).176
Scheme 154. Ir-Complex 482b-Catalyzed Asymmetric Transfer Hydrogenation of Aryl Ketones 483.

A plausible transition state 485 for this process was proposed in which the hydride is delivered to the Re-face of the ketone, with the ammonium cation H-bonding to the N,O-ligand and the ketone (Figure 10).
Figure 10.
Plausible transition state for the asymmetric transfer hydrogenation of ketones 483 with Ir-complex 482b.
In recent years, Wang has significantly contributed to the development of N,O-analogues of PHOX-type ligands, describing the synthesis of Box–OH ligands 486 – 488 and their application in various enantioselective Mg(II)-catalyzed transformations (Figure 11).
Figure 11.

Box–OH ligands 486–488.
Wang first utilized the Box–OH ligands in the Mg-catalyzed asymmetric dearomatization of β-naphthols 489 with meso-aziridines 490. Following optimization, ligand 487c was applied in the synthesis of a range of ring-opened products 491 bearing three contiguous stereocenters in up to 99% yield, with up to >20:1 dr and >99% ee (Scheme 155). Ligands of the type 487, with a methylene spacer between the phenol and the oxazoline, performed better than ligands 486 without the spacer. Changes to the β-naphthols 489 or the meso-aziridines 490 did not have a significant effect on the outcome of the reactions, all of which proceeded with excellent levels of enantioselectivity (all but one example >97% ee).177
Scheme 155. MgBu2/487c-Catalyzed Asymmetric Dearomatization of β-Naphthols 489 with meso-Aziridines 490.

Wang further developed Box–OH/Mg-mediated transformations of naphthols with aziridines, describing two distinct processes for the dearomatization or O-alkylation of naphthols 492. The iPr-487c/MgBu2-catalyzed reaction of 492 with aziridines 490 gives access to the dearomatized products 493 in up to 97% yield, with up to >20:1 dr and >99% ee (Scheme 156). Conducting the reaction of C3-H and C3-halogenated naphthols 492 in the presence of Ph-487a (in place of 487c) gives access to the O-alkylated products 494 in up to 79% yield and with up to 99% ee. So far there is no concrete explanation for the remarkable chemoselectivity observed in these processes. The reaction is certainly substrate dependent as the nature of R1 has the largest effect on chemoselectivity. The ligand plays an important role, and the authors suggest this could be due to π—π or π—anion interactions in the transition state of the reaction with ligand Ph-487a. Finally, the presence of a long alkyl chain at C-1 is essential for O-alkylation to occur, and the nature of this alkyl chain has a slight effect over the C/O ratio (the catenulate ester appears to be optimal).178
Scheme 156. MgBu2/487c-Catalyzed Asymmetric Dearomatization and MgBu2/487a-Catalyzed Asymmetric O-Alkylation of Naphthols 494.

Wang has also reported the 487b/Mg-catalyzed dearomative conjugate addition of naphthols 489 to alkynyl ketones 495 to give the corresponding alkenes 496 in up to 85% yield, with up to 15:1 Z/E selectivity and up to 98% ee, using cyclopentyl methyl ether (CPME) as the solvent (Scheme 157).179 Later, the scope of this reaction was extended to dialkyl acetylenedicarboxylates.180
Scheme 157. MgBu2/487b-Catalyzed Dearomative Conjugate Addition of Naphthols 489 to Alkynyl Ketones 495.

Wang extended the Box–OH/Mg-catalyzed conjugate addition reaction to include C3-pyrrolyl-oxindoles 497 with alkynyl ketones 498. By developing a 499/488c bis-ligand system, a range of oxindoles 500, bearing quaternary stereocenters were accessed via the Mg-catalyzed process, followed by reduction of the alkene with Pd/C and H2 in up to 67% yield and with up to 89% ee (Scheme 158). The reaction with sulfoxide 409 as a coligand only slightly improved the enantioselectivity of the process, compared to the reaction with just 488c, under the exact same reaction conditions (without molecular sieves). The authors did not report the result of the reaction with just ligand 488c under the final optimized conditions (including molecular sieves). The best E/Z selectivity achieved during optimization, without the reduction of the double bond, was 3:1.181
Scheme 158. MgBu2/488b-Catalyzed Dearomative Conjugate Addition of Oxindoles 497 to Alkynyl Ketones 498.

Wang has also applied Box–OH/Mg-catalysts in the asymmetric [3 + 2] cyclization of 3-isothiocyanato oxindoles 501 with alkynyl ketones 502. Utilizing (R)-Ph-487a as the chiral ligand, a range of polycyclic oxindoles 503 were synthesized in up to 99% yield and with up to 94% ee (Scheme 159).182
Scheme 159. MgBu2/487a-Catalyzed Asymmetric [3 + 2] Cyclization of 3-Isothiocyanato Oxindoles 501 with Alkynyl Ketones 502.

Overall, mono(oxazoline) N,O-ligands have been applied in a very limited range of asymmetric transformations in the past decade. In particular, they have been successfully applied in the asymmetric alkyl or aryl zinc addition to aldehydes, and in asymmetric Mg-catalyzed addition reactions to alkynyl ketones.
2.5. Miscellaneous Mono(oxazoline) Ligands
2.5.1. Mono(oxazoline)-Sulfonamide Ligands
Kishi and co-workers reported a library of chiral sulfonamide ligands (Figure 12) and they were successfully employed in the enantioselective Cr-catalyzed allylation, propargylation and vinylation of aldehydes, a reaction famously known as the Nozaki-Hiyama-Kishi (NHK) reaction.
Figure 12.

Chiral sulfonamide ligands.
In 2004, Kishi developed a novel Cr-catalyzed enantioselective allylation of aldehydes using chiral sulfonamide ligands 504–507 (Scheme 160).183 A Cr-504–507 complex generated from a sulfonamide 504–507 and CrCl3/CrBr3 in the presence of Et3N and Mn as a reducing agent of Cr and TMSCl or Zr(Cp)2Cl2 as a Cr alkoxide dissociating reagent. TMSCl and Zr(Cp)2Cl2 reagents upon hydrolysis breaks complex 508d to alcohol 508e. Interestingly, the addition of 2,6-lutidine was found to improve asymmetric inductions significantly by acting as an acid scavenger. Low valent Co phthalocyanine (CoPc) or Fe(TMHD)3 (iron tris(2,2,6,6-tetramethyl-3,5-heptanedione)) were used as an activator since they facilitate radical formation from allyl halides. The proposed mechanism for the Cr-catalyzed allylation of aldehydes is depicted in Scheme 160. The initial reaction between Cr(III) and chiral sulfonamide 504 in the presence of Et3N, 2,6-lutidine, Co/Fe and Mn generates Cr(II)-complex 508a. Metalloallyl species (508b), formed from the allyl bromide and Co/Fe, undergoes transmetalation with Cr(II)-complex (508a) to generate the Cr(III)-allyl complex 508c. This complex would undergo the addition to aldehydes through a six-membered transition state to form complex 508d which further reacts with TMSCl or Zr(Cp)2Cl2 to form the alcohol product 508e.
Scheme 160. Proposed Mechanism for Cr-Catalyzed Allylation of Aldehydes.

The Cr-complex of ligand 504/505 was applied in the synthesis of chiral alcohol 511 (93% ee), a key building block in the halichondrin (a polyether macrolide) synthesis (Scheme 161).184 Ligand 505 exhibited exceptional crystallinity compared to ligand 504 and therefore it was easier to recover when used on a large scale reaction.
Scheme 161. Synthesis of Chiral Alcohol 511.

Furthermore, iterative use of Cr(III)/504-catalyzed asymmetric allylation of aldehyde 512 provided an easy access to stereocontrolled 1,3-polyols, syn/syn- and anti/anti-1,3,5-triols (516a–b) (Scheme 162).185 One iteration cycle is composed of a three-step sequence: (i) oxidative cleavage of the olefin to form an aldehyde; (ii) catalytic asymmetric allylation; and (iii) protection of the resultant alcohol.
Scheme 162. Iterative Asymmetric Allylation.

In 2009, an asymmetric propargylation of aldehydes was developed by using Cr(III) bromide and (R)-sulfonamide 506 (Scheme 163).186 An optically pure homopropargyl alcohol 518 was obtained in 78% yield with 90% ee. Alcohol 518 was further converted to 511, a building block of halichondrins and E7389.
Scheme 163. Asymmetric Propargylation of Aldehyde.

Cr catalysts derived from different chiral sulfonamides were effectively utilized by Kishi to couple various aldehydes with allyl and vinyl halides to construct diverse C–C bonds during their halichondrin synthesis.187−189
In 2009, Kishi designed tethered ligand 507 which then was used to prepare heterobimetallic catalysts with Cr and Ni coordinated to the sulfonamide and phenanthroline moieties, respectively. The Ni/Cr heterobimetallic catalysts performed exceptionally well in the catalytic asymmetric vinylation of aldehydes (519) (Scheme 164).190 The catalyst highlights include low catalytic loading (1 mol%), the formation of a negligible amount of dimer 522, a byproduct formed through the alkenyl Ni species, use of a 1:1 molar ratio of coupling partners aldehyde 519 and vinyl iodide 520, the asymmetric induction was similar to that obtained in the coupling with the Cr/507a and Ni/507b.
Scheme 164. Asymmetric Vinylation of Aldehydes.

The application of this new catalyst was again demonstrated in C–C bond-forming reactions from the synthesis of halichondrin/E7389. The coupling between polyfunctional aldehyde 523 (1.0 equiv) and vinyl iodide 524 (1.2 equiv) in the presence of Cr/Ni catalyst 507 (3 mol%) furnished the desired allylic alcohol in 86% yield with a 19:1 dr whereas the Cr-507a/Ni-507b catalyst (20 mol%) delivered allylic alcohol 525 in 90% yield with a 19:1 dr.
Overall, chiral monooxazoline sulfonamide ligands have found major success in Cr- and Ni-catalyzed allylation, vinylation, and propagylation of aldehydes. The major challenge remains the extension of this methodology to ketones.
2.5.2. Tridentate Mono(oxazoline) Ligands
This section summarizes the progress made in the design and application of tridentate mono(oxazoline) ligands in metal-catalyzed asymmetric catalysis.
A variety of tridentate iminopyridine oxazoline (IPO) ligands (526) (Figure 13) have been applied in asymmetric catalytic transformations.
Figure 13.

Tridentate iminopyridine oxazoline (IPO) ligands.
Huang described the synthesis of novel IPO ligands 526a–c and applied them in the asymmetric Co-catalyzed hydroboration of 1,1-disubstituted aryl alkenes 527. Following optimization, a range of alkenes 527 were successfully subjected to the hydroboration catalyzed by the Co(526a) CH3 complex to give the enantioenriched products 529 in high yields up to 98% and with excellent enantioselectivities up to 99.5% ee (Scheme 165). This methodology was applied to the enantioselective synthesis of Naproxen 529-X in four synthetic steps, in 66% overall yield and with 98% ee.191
Scheme 165. Asymmetric Co-Catalyzed Hydroboration of 1,1-Disubstituted Aryl Alkenes.

Lu independently reported the application of the same IPO ligands in a very similar Co-catalyzed hydroboration of 1,1-disubstituted aryl alkenes 527 (Scheme 166). In this case, the optimized conditions for the hydroboration utilized Co(526b)Cl2 as the catalyst to give the enantioenriched products 529 in up to 96% yield and with up to 99% ee, with very similar results to Huang. Interestingly, this catalyst was tested by Huang to give the model product in 91% ee, compared to Lu’s 98% ee. Both groups used similar reaction conditions during their optimization studies (including the addition of NaBEt3H); however, Lu’s reaction conditions were much more concentrated (5.0 M in toluene, or neat) than Huang’s (0.25 M in THF), leading to different levels of enantioselectivity. Huang excluded NaBEt3H from the reaction mixture and used the Co-CH3 complex (in place of the CoCl2 complex) as the catalyst in their final optimized reaction conditions.192 Lu subsequently described a Bn-IPO Fe(526c)Cl2-catalyzed hydroboration of 1,1,-disubstiuted alkenes with very similar results, giving the enantioenriched products in up to 97% ee.193
Scheme 166. Hydroboration of 1,1-Disubstituted Aryl Alkenes.

Lu has also applied IPO ligands in the Co(526e)Cl2-catalyzed hydroboration of aryl ketones 530 to give the enantioenriched alcohols 531 in moderate to high yields up to 99% and with excellent enantioselectivities up to >99% ee (Scheme 167). The reaction also works with the IPO-FeCl2 complexes, but with lower levels of enantioselectivity.194
Scheme 167. Asymmetric Hydroboration of Aryl Ketones.

Lu has further expanded on the Co-catalyzed asymmetric hydroboration of 1,1-disubstituted aryl alkenes 527, reporting a dual stereocontrolled reaction to give access to both enantiomers of the products 529. Utilizing Bn-IPO ligand 526c, the enantioenriched products 529 were accessed in up to 86% yield and with up to 95% ee (Scheme 168). Under the same reaction conditions, but with novel aminopyridine oxazoline (APO) ligand 532 (8 mol%) and CoCl2 (5 mol%) as the catalytic system, the opposite enantiomers of the products 529 were obtained in up to 81% yield and with up to 95% ee. While the IPO system gave higher yields across the board, both the IPO and APO systems gave better levels of enantioselectivity with different substrates. Preliminary deuterium labeling experiments suggested that the reaction with the IPO ligand operated under a different mechanism than the APO ligand, and this could explain the stereochemical outcomes of the two reactions.195
Scheme 168. Asymmetric Hydroboration of 1,1-Disubstituted Aryl Alkenes.

Co(526b)Cl2 has also been used by Lu in the asymmetric hydrogenation of 1,1-diarylethenes 533. A range of enantioenriched diarylethane derivatives 534 were accessed in up to >99% yield and with up to >99% ee (Scheme 169). In all examples, one of the two aryl groups of the substrates 533 had an ortho-substituent.196
Scheme 169. Asymmetric Hydrogenation of 1,1-Diarylethenes.

Lu has also reported a sequential Ni-catalyzed asymmetric Nazarov cyclization/decarboxylation of divinylketones 535 bearing a tBu-ester group to give enantioenriched cyclopentanones 536 in up to 97% yield and with up to 96% ee (Scheme 170). The reaction tolerated a range of electronically and sterically different aryl groups at R2 and R3, although aliphatic groups at R3 were not tolerated.197
Scheme 170. Ni-Catalyzed Asymmetric Nazarov Cyclization/Decarboxylation.

Huang has reported a highly hindered IPO ligand 538 for use in the Fe-catalyzed hydrosilylation of aryl ketones 530 (Scheme 171). The hindered Fe(538)Br2 catalyst showed high activity for accessing a range of chiral secondary alcohols 531 in up to 99% yield and with moderate to high enantioselectivities up to 93% ee.198
Scheme 171. Fe-Catalyzed Hydrosilylation of Aryl Ketones.

Lu later described derivatives of IPO ligands 538, 539a, and 539b (Figure 14), for the Co- and Fe-catalyzed hydrosilylation of alkenes, respectively.
Figure 14.

IPO ligands 539a–b.
First, Lu developed the Co-catalyzed hydrosilylation of alkenes with phenylsilane 541. During optimization, they found that the sterically hindered ligand 539a performed better than the less hindered ligands 526a and 526d, giving the model product in 98.5% ee under those conditions, compared to 72% ee (526a) and 88% ee (526d). Under optimized conditions with Bn-IPO 539a as the chiral ligand, a range of silylated products 542 were synthesized in up to 97% yield, with >96:4 b/l and with excellent levels of enantioselectivities up to 99.8% ee (Scheme 172). Styrenes were hydrosilylated with excellent levels of enantioselectivity while aliphatic alkenes gave <88% ee.199
Scheme 172. Co-Catalyzed Hydrosilylation of Alkenes with Phenylsilane.

Lu subsequently developed the Fe(539b)Cl2-catalyzed hydrosilylation of aliphatic alkenes 540 under similar reaction conditions to the previous Co-catalyzed report (ligand 539a was not reported). A range of silylated products were isolated in up to 97% yield, with >96:4 b/l and with excellent levels of enantioselectivities up to 99.8% ee (Scheme 173). These reports offer complementary systems, with the same family of chiral ligands, for the enantioselective hydrosilylation of styrenes and aliphatic alkenes in similarly high stereoselectivities.200
Scheme 173. Fe-Catalyzed Hydrosilylation of Aliphatic Alkenes.

Other unsymmetrical tridentate pyridine-oxazoline-containing ligands have been applied in asymmetric catalytic transformations. Yu has described the design of C1-symmetric benzimidazole-pyridyl oxazoline ligands 543a and 543b (Figure 15).
Figure 15.

C1-symmetric benzimidazole-pyridyl oxazoline ligands.
Yu observed that the Ru-complex Ru(543b)(PPh3)Cl2 was a highly active catalyst for the asymmetric transfer hydrogenation of ketones 544, giving the corresponding chiral alcohols in up to >99% yield and with up to 97% ee (Scheme 174).201
Scheme 174. Asymmetric Transfer Hydrogenation of Ketones.

Wang and Shi have reported the synthesis of novel Schiff base oxazoline-containing ligands 546–548 (Figure 16) and their applications in the asymmetric α-chlorination of β-keto esters. The denticity of the ligands was not determined, but the ortho-hydroxy groups were observed to be essential for generating products with appreciable levels of enantioselectivity.
Figure 16.

Novel Schiff base oxazoline-containing ligands.
The results of a screen of ligands 546–548 in the enantioselective Cu-catalyzed α-chlorination of β-keto ester 549 are summarized in Table 3. The reaction with bulkier iPr-oxazoline ligand 546, which bears no substitution on the ortho-phenol, gave the chlorinated product 550 with only 47% ee (entry 1). The reaction with Ph-oxazoline ortho-naphthol 547 gave 550 in 99% yield and with a much improved 78% ee. A screen of other Ph-oxazoline ligands 548a-d showed that ligand 548a, with no substitution on the ortho-phenol, gave 550 in 99% yield and with 78% ee (entries 3–6). A temperature screen was conducted in the reaction with ligand 548a (entries 7–9). The reaction conducted at 0 °C gave 550 with the highest level of enantioselectivity of 83% ee (entry 7).202
Table 3. Enantioselective Cu-Catalyzed α-Chlorination of β-Keto Ester.

| entry | ligand | temp (°C) | yield (%) | % ee |
|---|---|---|---|---|
| 1 | 546 | rt | 99 | 47 |
| 2 | 547 | rt | 99 | 78 |
| 3 | 548a | rt | 99 | 78 |
| 4 | 548b | rt | 95 | 50 |
| 5 | 548c | rt | 99 | 54 |
| 6 | 548d | rt | 99 | 68 |
| 7 | 548a | 0 | 99 | 83 |
| 8 | 548a | –20 | 99 | 72 |
| 9 | 548a | –78 | 99 | 6 |
2.5.3. Mono(oxazoline) Carbene Ligands
In the past decade, limited progress has been made in the design and application of mixed oxazoline-carbene ligands in asymmetric catalysis.
Burgess has applied the Ir-complex 551 (Figure 17) in the asymmetric catalytic hydrogenation of vinyl ethers.
Figure 17.
Mixed oxazoline-carbene ligands.
Under optimized conditions, vinyl ether esters 552 were successfully hydrogenated, under 50 bar of H2, to give the corresponding chiral esters 553 with up to >99% conversion and with mostly moderate to good enantioselectivities of up to 90% ee (Scheme 175). Ir-complex 551 was then applied in the asymmetric hydrogenation of vinyl ether alcohols with better results, under slightly altered reaction conditions. The chiral alcohols 555 were formed with >99% conversion and with very high enantioselectivities of up to 98% ee.203
Scheme 175. Asymmetric Hydrogenation of Vinyl Ether Alcohols.

Ito and Nishiyama reported a set of mixed carbene-oxazoline Rh- and Ru-complexes 556–559(Figure 18), which are analogous to PheBOX complexes (covered in section 2.5.4). The applications of the Rh-complexes in asymmetric conjugate reduction and the Ru-complexes in the asymmetric catalytic hydrogenation and asymmetric transfer hydrogenation of ketones, led to low or moderate enantioselectivities of the isolated products in all cases.
Figure 18.

Mixed carbene-oxazoline Rh- and Ru-complexes.
For example, the asymmetric transfer hydrogenation of 9-acetylanthracene was conducted with 558a–c and 559b as the catalysts (Table 4). Interestingly, the reactions with Ru-catalysts 558a, 558b, and 559b selectively formed secondary alcohols 561a and 561b in which the ketone and part of the anthracene ring are reduced (entries 1–3). The reaction with 559b gave the alcohol 561a with the highest stereoselectivity of 60% ee. Achiral Ru-catalyst 558c gave remarkable selectivity in the formation of 561a. These results suggest that further development of these mixed-carbene-oxazoline systems could be useful for the selective reduction of extended aromatic-ring-systems.204
Table 4. Asymmetric Transfer Hydrogenation of 9-Acetylanthracene.

| entry | cat | yield (%) | 561a/b/c | % ee (561a) | % ee (561b) |
|---|---|---|---|---|---|
| 1 | 558a | 99 | 60:40:0 | 9 (R) | 18 (R) |
| 2 | 558b | 99 | 51:49:0 | 31 (R) | 5 (R) |
| 3 | 559b | 97 | 60:40:0 | 60 (R) | 28 (R) |
| 4 | 558c | 90 | 93:<1:7 |
Wang and Shi have applied Pd-complex 562 (Figure 19), bearing an axially chiral mixed carbene-oxazoline ligand, in an asymmetric allylic arylation for the kinetic resolution of Morita-Baylis-Hillman (MBH) adducts 563.
Figure 19.
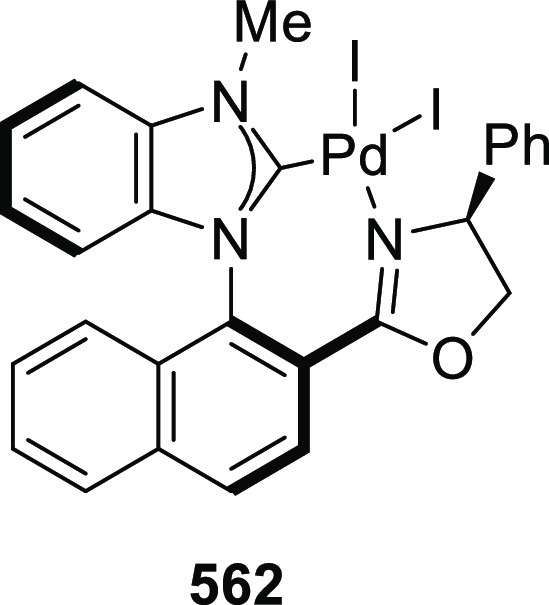
Axially chiral mixed carbene-oxazoline ligand.
Reacting racemic MBH adducts 563 with aryl boronic acids in the presence of 562, the corresponding arylated products 564 were isolated in up to 61% yield, with up to 99:1 E/Z selectivity and >99% ee (Scheme 176). The enantioenriched alcohols 565 were recovered in up to 37% yield and with the best enantioselectivity of 92% ee. This process is clearly a better method for synthesizing the arylated products 564 enantioselectively than it is for resolving the MBH adducts 563.205
Scheme 176. Kinetic Resolution of Morita–Baylis–Hillman (MBH) Adducts.

Ma and Jiang have independently developed novel planar chiral mixed carbene-oxazoline ligands for use in asymmetric catalysis (Figure 20). Jiang has reported the ferrocene-based ligand precursor 566, while Ma has reported the cyclophane-based ligand precursors like 567 and 568. Jiang has applied the RhI(566)(cod) complex in the asymmetric hydrosilylation of ketones, obtaining only low to moderate enantioselectivities of 39–56% ee.206
Figure 20.

Novel planar chiral mixed carbene-oxazoline ligands.
In Ma’s initial report, the cyclophane-containing ligands were applied in the Cu-catalyzed conjugate borylation of α,β-unsaturated ketones, with the products isolated with up to 84% ee when ligand precursor tBu-568b was used.207 The pseudo-ortho-cyclophanes 568 generally perform better than the pseudo-geminal-cyclophanes, like 567 in Ma’s reports. Ma extended the substrates from α,β-unsaturated ketones to α,β-unsaturated esters in the conjugate borylation of 569. Under optimized conditions, a range of esters 569 were borylated with B2pin2, utilizing Cu2O/tBu-568c as the catalytic system, followed by treatment with sodium perborate, to yield the corresponding chiral alcohols 570. The enantioenriched products 570 were generated in up to 95% yield and with high enantioselectivities up to 97% ee (almost all examples >90% ee) (Scheme 177).208
Scheme 177. Cu-Catalyzed Conjugate Borylation of α,β-Unsaturated Esters.

Ma has also developed a Cu2O/iPr-568d-catalyzed 1,2-silylation of N-tosylaldimines 571, generating the silylated amines 573 in up to 98% yield and with excellent enantioselectivities of up to 97% ee (Scheme 178).209
Scheme 178. 1,2-Silylation of N-Tosylaldimines.

2.5.4. Ruthenium(II)/Phenyloxazoline (Ru(II)-Pheox)
Iwasaa designed and synthesized novel chiral Ru(II)/phenyloxazoline (Ru(II)-Pheox) complexes (Figure 21).210 This catalyst system showed excellent reactivity and enantioselectivity in inter- and intramolecular cyclopropanation reactions.
Figure 21.

Chiral Ru(II)/phenyloxazoline (Ru(II)-Pheox) complexes.
Iwasaa employed Ru(II)-Pheox complex (574) for the highly stereoselective cyclopropanation of alkenes (577) with various diazo precursors (578/580/583a) (Scheme 179A–C).211−213 The asymmetric cyclopropanation between diethyl diazomethylphosphonate (578) and a series of alkenes including styrene derivatives, α,β-unsaturated esters, ketones, and amides was carried out to provide the corresponding cyclopropylphosphonate products (579) in high yields and with excellent diastereoselectivity (up to 99:1 dr) and enantioselectivity (up to 97% ee) (Scheme 179A).211 Similarly, diazosulfones (580) reacted with various alkenes including vinyl ethers, vinyl amines, and vinyl carbamates to furnish chiral cyclopropyl sulfones (581) in high yields (up to 80%) with excellent trans-selectivity and enantioselectivity (up to 96% ee) (Scheme 179B).212 An asymmetric synthesis of various trifluoromethyl cyclopropanes (584) was achieved in high yields with excellent diastereoselectivity (up to 98:2 dr) and enantioselectivity (up to 96% ee) by reacting in situ generated CF3CHN2 (583b) with a range of alkenes including vinyl ferrocene, vinyl ethers, vinyl amines, vinyl carbamates, and dienes (Scheme 179 C).213
Scheme 179. Highly Stereoselective Cyclopropanation of Alkenes with Various Diazo Precursors.

In 2012, Iwasaa described the highly enantioselective intramolecular cyclopropanation of trans-allylic diazoacetates (585) and alkenyl diazoketones using a water-soluble Ru(II)/hydroxymethyl (phenyl)oxazoline catalyst, Ru(II)-hm-Pheox (575) (Scheme 180).214 The polar protic functionality of the Ru catalyst was vital to give rise to high yields, due to its ability to form strong hydrogen bonds with water. The catalyst could be reused at least five times without substantial decline in reactivity or enantioselectivity.
Scheme 180. Enantioselective Intramolecular Cyclopropanation.

Later in 2016, chiral Ru(II)–Amm–Pheox complex (576) containing an internal quaternary ammonium unit was first employed in the stereoselective intermolecular cyclopropanation of diazo Weinreb amides (587) with alkenes (577) to furnish the corresponding chiral cyclopropyl Weinreb amides (588) in high yields (up to 98%) with excellent diastereoselectivities (up to 98:2 dr) and enantioselectivities (up to 92% ee) (Scheme 181).215 Additionally, the use of acetoxy-functionalized diazoacetamide (AMD) as the carbene source was found to be crucial for the high trans-selectivity of the cyclopropanation.
Scheme 181. Enantioselective Intermolecular Cyclopropanation of Alkenes with Diazo Weinreb Amides.

In subsequent studies, the intramolecular stereoselective cyclopropanation of a series of trans-allylic diazo Weinreb amide derivatives (589) was developed using the chiral Ru(II)-Amm-Pheox catalyst (576) to afford the corresponding chiral bicyclic cyclopropyl products (590) in excellent yields (up to 99%) with excellent enantioselectivity (up to 97% ee) (Scheme 182).216
Scheme 182. Enantioselective Intramolecular Cyclopropanation of trans-Allylic Diazo Weinreb Amide Derivatives.

Overall, chiral Ru(II)-Pheox complexes have found their applications in inter and intramolecular asymmetric cyclopropanation reactions between various alkenes with diazo derivatives.
2.5.5. Cobalt Oxazoline Palladacycle (COP)
The enantioselective chiral cobalt oxazoline palladacycle (COP) catalyst (591/592) (Figure 22) was first reported by Richards and Overman in 2003 for the enantioselective rearrangement of allylic N-arylimidates.217 A detailed review on Pd(II)-catalyzed enantioselective reactions using the COP catalyst was published by Overman in 2016.218
Figure 22.

COP catalysts.
2.5.6. Pentaphenylferrocene Oxazoline Palladacycle [PPFOP]
Peters reported219,220 the novel Pd(III)-catalyst (595b) for the asymmetric rearrangement of allylic trifluoroacetimidates (593) (Scheme 183). Various spectroscopic and electrochemical methods demonstrated that the Pd-catalyst (595b) unexpectedly had an activated oxidized form of a Pd(III) center bound to a ferrocene core which remained unchanged (Fe(II)) during the oxidative addition. The dimeric paramagnetic ferrocene-derived palladacycle (595b) contains two Pd(III) centers without involving a Pd–Pd bond. The Pd(III)-catalyst was formed by activating the diamagnetic dimeric chloride-bridged pentaphenylferrocene oxazoline palladacycle precatalyst, [PPFOP-Cl]2(595a), by treatment with AgNO3 (4 equiv). Two equivalents of the AgNO3 were required for a Cl-counterion exchange while another 2 equiv oxidized the complex to deliver the dark red-brown paramagnetic species (595b). Pd-complex (595b) was found to be the most efficient enantioselective catalyst for the rearrangement of allylic trifluoroacetimidates in terms of catalyst TON, scope and enantioselectivity. Previously it was assumed that the reaction between [PPFOP-Cl]2 and AgNO3 resulted in the oxidation of Fe(II) to Fe(III) in a ferrocenium core which would decrease the electron density of the Pd(III) center thus generating a more Lewis acidic Pd(III) center.219
Scheme 183. Novel Pd(III)-Catalyst for Asymmetric Rearrangement of Allylic Trifluoroacetimidates.

Peters reported the regio- and enantioselective synthesis of sulfonyl-protected chiral allylic amines (596b) from achiral allylic alcohols (596a) by using a catalytic ferrocene palladacycle, [PPFOP-Cl]2 (595a) and a tertiary amine (proton sponge -1,8-bis(N,N-dimethylamino)naphthalene) (Scheme 184).221,222 This reaction was highly step-economic and proceeded with no inert-gas atmosphere or catalyst activation by a silver salt. The preference for the branched allylic product was due to the Pd(II)-catalyzed cyclization-induced decarboxylative [3,3]-rearrangement of in situ-generated allylic carbamate intermediates. The allylic alcohol with an aliphatic group formed products in good to high yields and with high regio- and enantioselectivity whereas aromatic substituents were not well tolerated. The proposed mechanism of the Pd(II)/base-catalyzed decarboxylative allylic carbamate 597a rearrangement is shown in Scheme 184. The deprotonated carbamate 597b reacts with ferrocene palladacycle 595a to form a chelate complex 597c, the resting state, in which both the olefin and the anionic carbamate moiety are coordinated to the Pd center thus replacing the anionic ligand, Cl. Further dissociation of the carbamate N-atom in complex 597c, which might be triggered by the reversible recoordination of the anionic ligand, Cl, generates complex 597d which subsequently undergoes outer-sphere attack of the nucleophilic deprotonated N-center to the coordinated olefin to form intermediate 597e. The cyclic aminopalladated intermediate 597e featuring a σ-alkyl-Pd bond undergoes a ring-opening and β-hydride elimination to form the deprotonated carbaminic acid derivative 597f. Decomplexation, decarboxylation, and protonation of 597f leads to the formation of N-sulfonylated allylic amine 596b and regenerates the Pd(II) and base catalysts for the next turnovers.
Scheme 184. Regio- and Enantioselective Synthesis of Sulfonyl-Protected Chiral Allylic Amines.

To conclude this section, chiral PPFOP ligands are successfully applied in the synthesis of chiral allyl amines via the asymmetric [3,3] rearrangement of allylic amides.
2.5.7. Olefin-mono(oxazoline) Ligands
In 2010, Glorius developed the η2-binding ligand OlefOx 598 and applied it in the conjugate addition of aryl boronic acids to cyclohexanones 599a (Scheme 185). The ligand 598 had a modular synthesis which allowed for fine-tuning of its structure, furnishing the substituted cyclohexanone product 599b in up to 97% ee and 81% yield. It was thought that the η2-binding mode allowed for alternative coordination geometries to traditional η1-oxazoline ligand motifs.223
Scheme 185. Rhodium Catalyzed Conjugate Addition of Phenylboronic Acids to Cyclohexanones.

3. Bis(oxazoline) Ligands
3.1. Bis(oxazoline) Ligands with One Carbon Separating the Oxazoline Rings
3.1.1. Parent Bis(oxazoline) Ligands with One Carbon Separating the Oxazoline Rings
For C2-symmetric bis(oxazoline) ligands, only examples reported after 2010 will be described in this section. For applications of these ligands prior to 2010, see the 2011 review by Desimoni and Jørgensen.224
Watson has applied 600a (Figure 23) in the alkynylation of benzopyranyl acetals 601 using chiral cuprates (Scheme 186). The reaction proceeded smoothly giving enantiomeric excesses of up to 95% and yields up to 90%. Regarding substrates, the enantioselectivity was enhanced to 95% ee when a methoxy was placed in the 7-position of the chromene acetal. Nonaromatic alkynes were shown to be detrimental to enantioselectivity with results dipping to 70% ee and 49% yield.225
Figure 23.

Parent BOX ligands.
Scheme 186. Cu-Catalyzed Asymmetric Alkynylation of Benzopyranyl Acetals.

Watson developed an asymmetric Cu-catalyzed addition of terminal alkynes 604 to a TMSOTf-generated oxocarbenium ion (Scheme 187). By treating isochroman acetals 603 with TMSOTf, this generates an oxocarbenium ion that can be attacked by the chiral [Cu(MeCN)4]PF6-600a acetylene complex generating the product in up to 94% ee and good yields. It was shown that the p-methoxy electron-donating group on the alkyne lowered the enantioselectivity dramatically to 61% ee.226
Scheme 187. Cu-Catalyzed Asymmetric Addition of Alkynes.

Zeng reported the application of 600c (Figure 23) in the Cu-catalyzed intramolecular cyclization of N-alkenylureas 606 (Scheme 188). It allowed for the generation of chiral vicinal diamino bicyclic heterocycles in good yields and high enantioselectivities of up to 98% ee. Substituted aryl and heteroaryl systems were well tolerated with this catalysis with substrates possessing 2-thienyl substituted sulfonyl groups affording the corresponding product in 90% ee and 86% yield.227
Scheme 188. Cu-Catalyzed Intramolecular Cyclization of N-Alkenylureas.

Gu utilized Cu complexes of ligand 600c in the thiolative ring opening of five-membered cyclic diaryliodonium salts 608 in order to synthesize atropoisomeric products and various axially chiral P,S ligands (Scheme 189). The best results were obtained using Cu(MeCN)4PF6 as the Cu(I) source, which provided high yields and enantioselectivities up to >99% ee. While most substrates were tolerated, bulky aryl thioates 609 appeared to reduce the enantioselectivity to as low as 86% ee.228
Scheme 189. Cu-Catalyzed Ring Opening of Diaryliodonium Salts.

Doyle applied ligand 600d (Figure 23) in the enantioselective carbonyl-ene reactions of 2,3-diketo esters 611 (Scheme 190). The best results were obtained with Cu(SbF6)2 as the Lewis acid and loadings as little as 1 mol% which furnished yields up to 94% and enantioselectivities up to 97% ee. This reaction was also performed on gram-scale while maintaining high yields and excellent enantioselectivities.229
Scheme 190. Enantioselective Carbonyl-Ene Reaction of 2,3-Diketo Esters.

Chemler applied ligand 600c in the Cu-catalyzed aminohalogenation/cyclization reaction of 2-allylaniline 614 (Scheme 191). Isopropyl iodide was used as the halide source providing 2-iodomethylindoline products 615 in 93% ee and high yields. It was also noted that mesyl-protecting groups were not tolerated in this reaction with enantioselectivities lowered to 43% ee.230
Scheme 191. Cu-Catalyzed Asymmetric Aminohalogenation.

Reiser applied ligand 600b (Figure 23) in the Cu-catalyzed cyclopropanation of furans 616 in moderate yields and enantioselectivities of up to 83% ee (Scheme 192). This protocol was subsequently applied in the synthesis of (−)-paeonilide on a 20 g scale but this methodology was limited by the fact that a second cyclopropanation would occur if it was allowed to run to completion.231
Scheme 192. Cu-Catalyzed Asymmetric Cyclopropanation of Furans.

Shi also employed a similar cyclopropanation using a catalyst derived from (CuOTf)2·PhH and ligand 600c achieving 93% yield and a diastereomeric ratio of >10:1 in the synthesis of Cryptotrione (Scheme 193). It was shown that slow addition of ethyl diazoacetate 620 via syringe pump was critical to obtaining the higher yields and enantioselectivities.232
Scheme 193. Cu-Catalyzed Asymmetric Synthesis of Cryptotrione Core.

Minnaard developed a Pd-catalyzed Michael addition of boronic acids to β,β-disubstituted cyclic enones 622 using chiral ligand 600c affording products 624 with up to 99% ee (Scheme 194). The moderate yields were attributed to significant protodeboronation of the arylboronic acid. It was shown that larger ortho-substituents such as aldehydes, nitro groups, trifluoromethyl groups as well as di-ortho substitution were not tolerated on the aryl boronic acid 623. The developed methodology was then applied in the enantioselective synthesis of sesquiterpenes such as herbertenediol.233
Scheme 194. Pd-Catalyzed Michael Addition and Synthesis of Herbertenediol.

Minnaard detailed an efficient Pd-catalyzed conjugate addition of arylbronoic acids to form β-substituted carbocyclic, heterocyclic and acyclic ketones. A PdCl2-600c complex provided the optimal results of up to 91% yield and 99% ee; however, the use of AgBF4 was needed to promote in situ dehalogenation to increase the conversion (Scheme 195). It was shown that the enantioselectivities dramatically dropped for acyclic enones with 60% ee being the highest reported.234
Scheme 195. Pd-Catalyzed Asymmetric Conjugate Addition of Arylboronic Acids.

Pedro applied ligand 600c in the Cu-catalyzed aza-Henry reaction with isatin N-Boc ketimines 627 achieving yields up to 99% and outstanding ees of 99.9% (Scheme 196). The reaction proceeds smoothly with or without protection of the isatin NH. The use of less acidic Cu(II) salts avoided imine hydrolysis but affected the enantioselectivity with CuCl2 affording the product in 97% yield and 27% ee. The best result was obtained with Cu(II) tetrafluoroborate hydrate and ligand 600c. It was also shown that having a nitro group in the 7-position of the isatin significantly reduced the enantioselectivity to 6% ee.235
Scheme 196. Cu-Catalyzed Asymmetric Henry Reaction of Isatin N-Boc Ketimines.

Zhang applied ligand 600a in the Ni-catalyzed cycloaddition of N-tosylaziridines 629 leading to very high yields of up to 99% and enantioselectivities up to 96% ee (Scheme 197). A variety of aziridines were subjected to the optimized reaction conditions, most of which achieved enantioselectivities of greater than 92% ee. Naphthyl-substituted aziridines however led to a reduction in enantioselectivity (89% ee).236
Scheme 197. Ni-Catalyzed Asymmetric Cycloaddition of N-Tosylaziridines.

Chemler utilized a 600c-Cu complex in the synthesis of 6-azabicyclo[3.2.1]octanes 633 via an enantioselective alkene carboamination (Scheme 198). This process allowed for the rapid synthesis of bridged bicyclic rings containing nitrogen in good yields and up to 95% ee. Two new stereocenters are formed in the reaction, and the C–C bond-forming arene addition is a net C–H functionalization. Electron-donating and withdrawing arenes were tolerated in this reaction. However, this reaction was limited in the substitution of the alkene. If the alkene was 1,1 disubstituted the reaction proceeded smoothly but when 1,2-disubstituted the reaction halted completely, not giving any product.237
Scheme 198. Cu-Catalyzed Synthesis of 6-Azabicyclo[3.2.1]octanes.

Tang developed an efficient synthesis of dihydrofuran-3-ones 634 and then applied them as substrates in an asymmetric Claisen rearrangement catalyzed by the Cu complex of ligand 600b. This furnished the corresponding product 635 in quantitative yield and high enantioselectivities up to 91% ee (Scheme 199).238
Scheme 199. Cu-Catalyzed Asymmetric Synthesis of Dihydrofuran-3-ones.

Xiao employed ligand 600d in the Cu-catalyzed inverse electron-demand hetero-Diels–Alder reaction between α-halo-N-acylhydrazones 636 with enol ethers 637. The reaction exhibits high enantioselectivity of up to 90% ee and provides densely substituted chiral pyridazine derivatives 638 in good yields (Scheme 200). It was shown that ester and thienyl-substituted hydrazones led to a drop in enantioselectivity to 34% ee.239
Scheme 200. Cu-Catalyzed Synthesis of Chiral Pyridazine Derivatives.

Gaunt applied chiral Cu-(II)-bisoxazoline complexes derived from ligand 600c in the enantioselective α-arylation of N-acyloxazolidinones 639 by diaryliodonium salts 640 (Scheme 201). Cu(II) triflate was used as the Cu source providing the arylated product in up to 95% ee in excellent yields under mild conditions.240
Scheme 201. Cu-Catalyzed Asymmetric α-Arylation of N-Acyloxazolidinones.

Romo employed a Cu-catalyzed Diels–Alder reaction to synthesize an intermediate of spirocyclic imine marine toxin (−)-Gymnodimine and an unnatural epimer. It provided the spirolactam product 644 in 84% yield and 95% ee (Scheme 202).241
Scheme 202. Cu-Catalyzed Synthesis of Gymnodimine Core.

Ligand 600c was applied successfully in the intramolecular amination/Heck-type coupling of γ-alkenylsulfonamides 645 by Chemler. Using Cu(OTf)2 as the Cu source and 600c as the ligand, enantioselectivities of up to 95% ee and 68% yield were observed for bulky alkenylsulfonamides (Scheme 203).242
Scheme 203. Cu-Catalyzed Asymmetric Amination of γ-Alkenylsulfonamides.

Reisman utilized 600c in the Ni-catalyzed synthesis of α,α-disubstituted ketones 650 from acyl chlorides 648 and racemic benzyl halides 649 (Scheme 204). The best results were obtained by using MnO2 as a stoichiometric reductant and DMBA (7,12-dimethylbenz[a]anthracene) to avoid homocoupling with 79% yield and ees up to 93%. A wide variety of substrates with a range of different substitutions were tolerated, however it was shown that ortho-substituted benzyl chlorides were poor substrates providing products in only 35% yield and 72% ee. It was also noted that an unusual solvent mixture (30% v/v DMA/THF) was needed. It was shown that higher ees but poor yields were obtained in THF while homocoupling issues remained when using pure DMA.243
Scheme 204. Ni-Catalyzed Asymmetric Synthesis of α,α-Disubstituted Ketones.

Fu showed that a catalyst combination of ligand 600c and bench-stable NiCl2·glyme were able to synthesize tertiary stereocenters bearing a CF3 group in a Negishi arylation reaction in up to 92% yield and 99% ee (Scheme 205). It was shown that a variety of functional groups were compatible with the reaction conditions, including silyl ether, primary alkyl chloride, primary alkyl bromide, primary alkyl tosylate, aryl ether, ketone, aryl iodide, carbamate, ester and a furan with all of the above groups maintaining enantioselectivities of greater than 95% ee. It was also shown that a range of electron-withdrawing fluorinated groups could be applied to this methodology.244
Scheme 205. Ni-Catalyzed Asymmetric Cross-Electrophile Coupling.

Tambar developed a novel methodology for the enantioselective allylic amination of terminal olefins 657 by using a Pd(TFA)2-600c catalyst system which promotes the rearrangement of their stable ene-adducts 658 (Scheme 206). This furnished the product 659 in up to 98% ee with good to excellent yields. Also notable is the ability to accommodate a wide variety of functional groups, although carboxylic acids and free alcohols were not compatible in this methodology due to competing addition to the ene-adducts.245
Scheme 206. Enantioselective Allylic Amination of Terminal Olefins.

Hiersemann reported a Cu-catalyzed [1,6]-transannular Gosteli–Claisen rearrangement of cyclic allyl vinyl ethers 660. Throughout their study they showed that enantioselectivity and diastereoselectivity varied significantly depending on the ligand system used (Scheme 207). When diastereoselectivity was high the enantioselectivity dropped dramatically and vice versa when the enantioselectivity was high. For eight-membered ring systems the reaction had outstanding trans-selectivity and reached up to 98% ee and 87:13 dr with ligand 600d. This methodology was extended to larger ring systems and displayed a switch to cis-selectivity affording the 12-membered ring product in up to 98% ee and 95:5 dr once again using chiral ligand 600d.246
Scheme 207. Cu-CAtalyzed [1,6]-Transannular Gosteli–Claisen Rearrangement of Cyclic Allyl Vinyl Ethers.

Hiersemann employed a 600d-Cu(II) complex to prepare a key intermediate in the synthesis of (−)-9,10-dihydroecklonialactone B. This Claisen rearrangement of Gosteli-type allyl vinyl ethers 663 produced the key keto ester 664 in 96% yield, 95:5 dr and 98% ee (Scheme 208). It was shown that a modified catalyst-complex using bis(trifluoroethanol) was more effective for the Claisen rearrangement than the traditionally used bis(aqua) Cu(II) complex. This key intermediate was then converted in eight steps to (−)-9,10-dihydroecklonialactone B.247
Scheme 208. Cu-Catalyzed Claisen Rearrangement of Gosteli-type Allyl Vinyl Ethers.

Walsh reported the asymmetric arylation of α-bromo esters 666 using a CoI2 and 600a catalyst complex with up to 96% yield and 97% ee. The reaction was tolerant of different cyclic/acyclic and aromatic esters. The scope for the alkyl chain showed tolerance for halides/alkenes/aromatic/heteroaromatic/esters while maintaining excellent enantioselectivities (Scheme 209). Interestingly iPr- and cyclopentyl-containing α-bromo esters showed a significant drop in ee. The scope for the aryl-Grignard reagent 665 showed very little variability in ee regardless of sterics or electronics. This methodology was then employed in the synthesis of (S)-fenoprofen and (S)-ar-turmerone without erosion of ee in subsequent steps.248
Scheme 209. Co-Catalyzed Asymmetric Arylation of α-Bromo Ketones.

Maguire highlighted the use of CuCl and 600c as a potent catalyst for intramolecular C–H insertion in the synthesis of a series of thiopyrans 669 (Scheme 210). The optimal catalyst complex in conjunction with NaBARF showed good yields and enantioselectivities of up to 98% ee. Interestingly the electron deficient nitrophenyl substrate did not yield any product. It was also shown that these reactions could be completed in significantly less time using microwave heating, but this came with a cost of reduced enantioselectivity. Further detailed investigations were also carried out by Maguire.249−252
Scheme 210. Cu-Catalyzed Synthesis of Thiopyrans.

Copper triflate and 600e was utilized in the aziridoarylation of aryl cinnamyl ethers 670 for the synthesis of 3-amino-4-arylchromans 671 (Scheme 211). This one-pot, two-step reaction uses the catalyst mixture of 600e and Cu(OTf)2 to effect a stereoselective aziridination, following additional Cu(OTf)2 the arylative ring-opening of the aziridine furnishes substituted chromans in moderate yields and enantioselectivities of up to 95% ee.253
Scheme 211. Cu-Catalyzed Asymmetric Aziridoarylation.

Fu reported the use of NiCl2-glyme and ligand 600c as an effective catalyst for the asymmetric Negishi coupling of sulfonamides and sulfones 672 with organozinc reagents 673 in good yields and reaching up to 96% ee (Scheme 212). A wide range of substrates including aromatic, cyclic and acyclic sulfonamides were investigated, although for sulfones there was a more limited scope which proceeded with high enantioselectivity of 90% ee. The alkenylation of α-bromosulfonamides using zirconium reagents 675 as the nucleophile and 600e as the ligand of choice proceeded in 81% yield and 90% ee.254
Scheme 212. Ni-Catalyzed Asymmetric Negishi Coupling of Sulfonamides.

Gu developed an enantioselective desymmetrization of 1,3-diazoisopropyl diazo(aryl)acetates 677 catalyzed by a Cu complex with ligand 600c to furnish imino esters 678 in very high yields and enantioselectivities of up to 97% ee (Scheme 213). The best results were obtained when using the large noncoordinating counterion, NaBARF, which is often employed in these reactions to increase the enantioselectivity. The substituents on the aromatic ring slightly affected the enantioselectivity. The ortho-groups showed the largest effect on enantioselectivities with electron-donating groups greatly diminishing the enantioselectivity to 58% ee.255
Scheme 213. Cu-Catalyzed Synthesis of Cyclic Iminoesters.

Waser developed the azidation of β-keto esters 679 and silyl enol ethers using a benziodoxole reagent 680 (Scheme 214). This methodology was expanded upon but not fully optimized with an initial test reaction using ligand 600a and a Cu salt showing moderate enantioselectivities of 49% ee.256
Scheme 214. Cu-Catalyzed Asymmetric Azidation of β-Keto Esters.

Nájera disclosed a Cu-catalyzed asymmetric alkylation of β-keto esters 683 using ligand 600d and benzylic alcohols 682 as the alkylating agents (Scheme 215). Xanthydrol and thioxanthydrol were the model alkylating reagents while aryl, alkyl and lactone-based β-keto esters were all shown to be good substrates for this reaction, and good yields with enantioselectivities of up to 90% ee were observed.257
Scheme 215. Cu-Catalyzed Alkylation of β-Keto Esters.

Jia performed the asymmetric Friedel–Crafts alkylation of 3-substituted indoles 685 using Cu(OTf)2 and ligand 600b as the optimal catalyst and ligand system achieving excellent yields and enantioselectivities of up to 93% ee (Scheme 216). This reaction was performed with α,β-unsaturated esters 686 and was sensitive only to bulky substituents on the indole ring.258
Scheme 216. Cu-Catalyzed Friedel–Crafts Alkylation of Indoles.

Pan disclosed an asymmetric cyano-trifluoromethylation of styrenes 688 using Togni’s reagent 689 as the electrophilic source of CF3 and 600e as the chiral ligand (Scheme 217). CuF2 was shown to be the ideal combination with ligand 600e and the products were formed in good yields and enantioselectivites of up to 98% ee. Electron-withdrawing and donating styrenes were tolerated remarkably well with p-methoxystyrene giving the lowest yield of 88% with 91% ee.259
Scheme 217. Cu-Catalyzed Cyano-trifluoromethylation of Styrenes.

Ma disclosed a highly enantioselective decarboxylative Mannich reaction of β-ketoacids 692 and cyclic aldimines 691 (Scheme 218). Using ligand 600c and CuI excellent yields and enantioselectivities of up to 99% ee were achieved. No substrate dipped below 90% ee with a wide variety of electronically different substrates being tolerated by the reaction conditions.260
Scheme 218. Cu-Catalyzed Decarboxylative Mannich Reaction.

Pedro reported the cycloaddition between nitrone 695 and 2-alkenoyl pyridine N-oxides 694 to give the isooxazolidine 696/697 products in high ee and de (Scheme 219). It was shown that N-phenyl nitrones performed much better in catalysis than N-methyl or N-benzyl nitrones which gave a much higher mixture of endo:exo. The nitrone derived from p-nitrobenzaldehyde formed the product in poor yields although the enantioselectivities remained high at 89% ee. The scope for the 2-alkenoyl pyridine N-oxides was remarkably tolerant of varying electronic properties achieving high endo:exo ratios while the enantioselectivities remained high.261
Scheme 219. Cu-Catalyzed Cycloaddition of Nitrones and Pyridine N-Oxides.

Johnston utilized ligand 600d in a three-step sequence for an umpolung amide synthesis (Scheme 220). Aldehydes 477 were reacted with bromonitromethane 698 in an enantioselective Henry addition using Cu(II) ortho-iodo benzoic acid as the catalyst, followed by MOM protection of the corresponding alcohol. This gave the MOM-protected alcohols 699 in good yields and with high enantioselectivities of up to 99% ee. The MOM-protected alcohols 699 were then subjected to the umpolung amide synthesis using enantioenriched (>99% ee) α-methyl benzyl amine 700 and these products 701 were used to provide confirmation that the two diastereomers of 699 are homochiral at the benzylic carbon (differing at the nitro-α-carbon) and remained enantioenriched throughout the three-step sequence (Henry/MOM-protection/umpolung amide synthesis).262
Scheme 220. Cu-Catalyzed Asymmetric Umpolung Amide Synthesis.

Chemler developed a Cu-catalyzed aza-Friedel–Crafts reaction between phenols 702 and aldimines 703 that provides chiral secondary benzylamines 704 with enantioselectivities up to 99% ee using 600c as the chiral ligand (Scheme 221). With regards to the phenol scope most substrates were well tolerated, however some substrates such as 1,3,5-trimethoxyphenol and 5-OTBS phenol gave poor conversions as low as 15% yield and enantioselectivites of <5% ee. When the aryl imine scope was attempted the conditions had to be reoptimized using (R)-600a as the ligand and lower temperatures to provide better enantioselectivity of up to 97% ee.263
Scheme 221. Cu-Catalyzed Asymmetric Aza-Friedel–Crafts Reaction.

Sorensen reported the asymmetric Diels–Alder cycloaddition of 1-hydrazinodienes 708 and N-acryloyl oxazolidinones 709 using ligand 600d and Cu(SbF6)2 as the catalyst complex (Scheme 222). The scope of the reaction was varied and tolerated different substituents on the substituted N-acryloyl oxazolidinones and the lowest enantioselectivities were seen when p-Cl/Br/CF3 substituted aryl rings were used all affording 90% ee. When changing the 1-hydrazinodienes there was little to no drop in enantioselectivity with 97% ee being the lowest recorded.264
Scheme 222. Cu-Catalyzed Asymmetric Diels–Alder of 1-Hydrazinodienes.

An asymmetric addition of nitromethane to 2-acylpyridine N-oxides 711 was reported by Pedro in 2014. Using 600e and Cu(OTf)2 as the catalyst complex and ethanol as the solvent enantioselectivities up to 96% ee were achieved (Scheme 223). Ethanol was chosen as the solvent as it stopped competitive background reactions from occurring. Substrates substituted on the 6-position had a disastrous effect on the enantioselectivity which dropped to as low as 48% ee.265
Scheme 223. Cu-Catalyzed Asymmetric Addition to 2-Acylpyridine N-Oxides.

MacMillan developed an enantioselective cascade arylation-cyclization using Cu(OTf), 600c and aryl iodonium salts 714 to synthesize aryl pyrroloindolines 715 (Scheme 224). In general, the reaction was highly enantioselective with ee’s up to >99% ee. Unprotected indole starting material has the largest negative impact on the catalytic transformation as enantioselectivity was lowered to 90% ee.266
Scheme 224. Copper-Catalyzed Asymmetric Synthesis of Pyrroloindolines.

Waser reported a dynamic kinetic asymmetric annulation of aminocyclopropanes 716 with enol ethers 717 and aldehydes furnishing up to 96% ee and 92% ee, respectively (Scheme 225). The best enantioselectivities were achieved with Cu(ClO4)2-600d as the catalyst system and succinimide as the nitrogen-containing component of the aminocyclopropanes with up to 96% ee being obtained. It was also shown that the counterion had a large influence on the ee, hexafluoroantimonate led to the highest dr’s but when perchlorate was chosen it provided the highest enantioselectivities.267
Scheme 225. Dynamic Kinetic Asymmetric Annulation of Aminocyclopropanes.

MacMillan published the enantioselective α-arylation of carbonyls using aryliodonium salts 722 with ligand 600c and Cu(OTf) as the active catalyst system (Scheme 226). Using silyl enol ethers 721 as the silylated nucleophile they attacked the Cu-oxazoline iodonium salt complex to give good yields and enantioselectivities up to 95% ee. It was shown that using the PF6 counterion and a mixed solvent system of toluene/CH2Cl2 at 0 °C produced the best yields. A diverse range of aryl and heteroaryl rings were tolerated well in catalysis. As well as this, arenes, olefins, ethers, esters, and carbamates were all tolerated for the silyl enol ether scope.268
Scheme 226. Cu-Catalyzed Enantioselective α-Arylation of Carbonyls.

Lu developed a Cu-catalyzed Friedel–Crafts alkylation of indoles 724 with trifluoroethylidene malonates 725 to construct a tertiary stereocenter bearing a trifluoromethyl group (Scheme 227). Using Cu(OTf)2 and 600b as the catalyst system excellent conversions and enantioselectivities of up to 90% ee were achieved. This protocol was then utilized in the synthesis of β-CF3-tryptophan and 4-CF3-β-carboline.269
Scheme 227. Cu-Catalyzed Friedel–Crafts Alkylation of Indoles.

Ohshima reported an enantioselective decarboxylative Mannich-type reaction of unprotected isatin-derived ketimines 727 (Scheme 228). By utilizing a β-keto acid 728 as the nucleophile this led to an irreversible decarboxylation addition process. Using Cu(OTf)2 and 600c as the ligand the desired product was formed in up to 96% ee and very high yields (of up to 90%). With regards to the β-keto acids this reaction was also tolerant of heteroaryl systems such as furan/thiophene and naphthyl with good enantioselectivities (85–92% ee) being obtained.270
Scheme 228. Cu-Catalyzed Enantioselective Decarboxylative Mannich Reaction of Unprotected Isatin-Derived Ketimines.

Wang disclosed a multicomponent cascade inverse electron-demand Aza-Diels–Alder/nucleophilic addition/ring-opening reaction using 2-methoxyfurans 730 as dienophiles (Scheme 229). This reaction was envisaged to synthesize a bicyclic heterocycle, but water does a nucleophilic addition following a ring opening leading instead to a series of tetrahydropyridazine derivatives 732. With this methodology in place a series of substituted tetrahydropyridazines were synthesized. Electron-donating and electron-withdrawing groups were all well tolerated with enantioselectivities in the 92–98% ee range for this Cu-catalyzed process.271
Scheme 229. Cu-Catalyzed Inverse Aza-Diels–Alder Cascade Reaction.

Shibata developed a Zn(OAc)2/Yb(OTf)3 cocatalyst system to induce an intramolecular 5-endo-dig cyclization of β-keto esters 733 and alkynes to produce enantioenriched cyclopentene products of type 734 (Scheme 230). The best results were achieved using 600c as the ligand and 1 equiv of hexafluoroisopropanol (HFIP). It was proposed that the effect of HFIP was to improve catalytic turnover by releasing the products from the catalytic cycle or assist initial enolization. It was believed that the generally high ees were due to the π–π stacking interactions of aromatic ketones with the phenyl groups of the ligand 600c. This hypothesis was backed up by the fact that the reaction with nonaromatic ketones led to poor enantioselectivities as low as 21% ee.272
Scheme 230. Dual-Catalytic Asymmetric Cyclization.

Yamamoto reported an asymmetric synthesis of α-hydroxy-β-keto esters 737 in a Cu-catalyzed O-nitrosocarbonyl aldol reaction (Scheme 231). Using ligand 600c and Cu(OTf)2 as the catalyst complex high levels of enantioselectivities up to 99% ee were observed. MnO2 was chosen as the oxidizing reagent as it was found to be a mild method for the generation of nitrosocarbonyl compounds which avoided overoxidation of the products. This catalysis was shown to be particularly flexible as both oxo-ester and thioesters 735 can be tolerated while retaining high levels of enantioselectivity. This methodology was then applied to the synthesis of antibacterial agent (S)-Kjellmanianone.273
Scheme 231. Cu-Catalyzed Asymmetric Nitrosocarbonyl Aldol Reaction.

Song used ligand 600e and Cu(CH3CN)4PF6 in combination with cinchona alkaloid 74 to promote a [3 + 2] cycloaddition reaction of ethynylethylene carbonates 738 and malononitrile 739 (Scheme 232). This system relied on cinchona alkaloid 741 for activation of the malononitrile and a Cu-mediated decarboxylation of a carbonate followed by a cycloaddition. The ligand 600e was not fully responsible for enantioselectivity as the choice of alkaloid was also shown to have an impact on the enantioselectivity. Good yields and enantioselectivities up to 97% ee were achieved using this copper/organocatalytic system.274
Scheme 232. Bicatalytic [3 + 2] Cycloaddition Reaction of Ethynylethylene Carbonates.

Bar utilized a one-pot- aziridination/Friedel–Crafts cyclization in the synthesis of dopamine D1 agonist A-86929 744 (Scheme 233). Using ligand 600c in combination with Cu(OTf)2 it was shown to be an effective catalyst for the asymmetric aziridination and subsequent Friedel–Crafts cyclization. This provided the key intermediate in good yields, > 99:1 dr and 95% ee.275
Scheme 233. Cu-Catalyzed Asymmetric Synthesis of Dopamine D1 Agonist.

Doyle and Rovis reported a dual Ni- and photoredox-catalyzed enantioselective desymmetrization of cyclic meso-anhydrides 745 (Scheme 234). Using 600c, Ni(cod)2 and photocatalyst 4CzIPN, enantioselectivities up to 94% ee were achieved, with dr values of >20:1. A range of electrophiles were tested such as cyclopentyl, cyclobutyl and cyclopropyl succinic anhydrides, all of which were suitable. However, anhydrides bearing β-substitution provided moderate enantioselectivities of 36% ee. Both electron-deficient and electron-neutral trifluoroborates reacted smoothly. Some ortho-substituted nucleophiles led to a lower diastereoselectivity of 6:1 dr.276
Scheme 234. Ni-Catalyzed Asymmetric Desymmetrisation of Cyclic meso-Anhydrides.

Aziz developed an asymmetric aminolactonization of 1,2 disubstituted alkenoic acid ester 747 via an aziridination-cyclization reaction protocol (Scheme 235). Using Cu(OTf)2 and ligand 600e the aziridination proceeds smoothly and upon treatment with additional Cu(OTf)2 or silica gel the aziridine successfully cyclizes to produce the desired aminolactone 748 in up to 98% ee.277
Scheme 235. Cu-Catalyzed Asymmetric Aminolactonization.

Buchwald developed an efficient Cu-catalyzed synthesis of enantioenriched CF3-containing lactones 751 (Scheme 236). Using 600d as the chiral ligand and[Cu(MeCN)4]PF6 as the Cu source enantioselectivities up to 98% ee after recrystallization were achieved in good yields. Togni’s reagent 750 proved to be a competent radical coupling reagent which promoted an asymmetric cyclization. Mechanistic studies supported a redox radical addition mechanism. This reaction showed a good functional group compatibility with a variety of aryl and heteroaryl systems being tolerated.278
Scheme 236. Cu-Catalyzed Synthesis of Enantioenriched CF3-Containing Lactones.

Fustero and Toste documented a three-component coupling of deactivated alkenes 752, aryl boronic acids 753 and N-fluorobenzenesulfonimide 755 (Scheme 237). This asymmetric Pd-catalyzed functionalization of α,β-unsaturated systems afforded good yields and high enantioselectivities using a Pd(OAc)2 and ligand 600b catalyst system. The reaction goes through a high-valent Pd(IV) intermediate. This methodology is amenable to a wide range of aryl boronic acids without a large drop in enantioselectivity (84–94% ee).279
Scheme 237. Pd-Catalyzed Three-Component Coupling of Deactivated Alkenes.

Fu reported an asymmetric Mukaiyama–Mannich reaction of cyclic N-sulfonyl α-ketiminoesters 756 and silyl enol ethers 757 which synthesized a range of sultams 758 (Scheme 238). The Ni(ClO4)2·6H2O-600c complex promoted the reaction very efficiently, giving the desired product in good yields and up to 99% ee. The reaction appeared to tolerate most substitution on the N-sulfonyl α-ketiminoester substrates in high enantioselectivities. Silyl enol ethers furnished high levels of enantioselectivity regardless of the aromatic/heteroaromatic group attached (97–99% ee). However, the α-difluorinated acetophenone derived silyl enol ether gave the corresponding benzosultam 758 in only 21% yield with a 30% ee value.280
Scheme 238. Ni-Catalyzed Asymmetric Synthesis of Benzosultam.

Onomura developed a catalytic synthesis of chiral oxazoline derivatives 760 via asymmetric desymmetrization of 1,3-diols 759 (Scheme 239). This method employed Cu(OTf)2 and ligand 600c in t-butanol affording good yields and enantioselectivities of up to >99% ee. For the substrate scope of 2-(N-acylamino)-1,3-propanediols, the enantioselectivity remained high regardless of the electronic nature of Ar be it aryl or heteroaryl (87–99% ee). When investigating the R substituents the enantioselectivities remained high (89–97% ee) but the yields were lowered (20–71% yield).281
Scheme 239. Cu-Catalyzed Synthesis of Chiral Oxazoline Derivatives.

Ma reported a novel Cu-catalyzed one-pot cross-coupling of β-ketoacids 692 with in situ generated trifluorodiazoethane 761 to access trifluoromethylated aldol products 762 (Scheme 240).282 Initially the reaction was examined racemically with CuI being the best Lewis acid to promote the formation of the aldol product 762. To move to an asymmetric system the reaction had to be reoptimized with the temperature being lowered and the reaction time extended. Ligand 600c was shown to be optimal giving up to 93% ee in good yields. The proposed reaction mechanism involves nucleophilic attack of the enol form of the ketoacid to activated trifluorodiazoethane and simultaneous termination with water to form intermediate A with the release of diazene which rapidly undergoes a disproportionation reaction to form nitrogen gas and hydrazine (or H2). Finally, decarboxylation leads to trifluoromethylated aldol product 762.
Scheme 240. Cu-Catalyzed Denitrogenative-Decarboxylative Coupling.

Wang described an asymmetric 1,3-dipolar [3 + 4] cycloaddition of azomethine imines 763 and azoalkanes 764 (Scheme 241). Using 600d as the ligand and Cu(OTf)2 as the metal, enantioselectivities up to 98% ee were obtained in good yields. Azomethine imines 763 bearing electron-donating, electron-neutral or electron-withdrawing groups at para-, meta-, or ortho-positions of the aryl ring were well tolerated, giving the corresponding products in up to 90% yield and 98% ee. Naphthyl, heteroaromatic, and alkyl azomethine imines were also tolerated with up to 92% yield and 96% ee. Unfortunately, only racemic product was obtained when an alkyl-substituted hydrazone was employed.283
Scheme 241. Cu-Catalyzed Cycloaddition of Azomethine Imines.

Sen detailed a diversity orientated synthesis (DOS) strategy toward the synthesis of optically active quinolizidinones, piperidinones, and pyrrolidinones. Ligand 600c was applied in the Cu-catalyzed asymmetric Michael reaction of a silyl keteneimide 766 and nitrostyrene 767 to generate valuable intermediates in high diastereomeric ratios and good yields (Scheme 242).284
Scheme 242. Cu-Catalyzed Asymmetric Michael Reaction.

Ohkuma performed a kinetic resolution of sterically hindered racemic α-tert-alkyl-α-hydroxy esters 769via an enantiomer-selective carbamoylation reaction (Scheme 243). Using ligand 600d and Cu(OTf)2 as the catalyst and carrying out the reaction at 0 °C enantioselectivities up to >99.9% ee and selectivity factors of up to 261 were achieved. The racemic α-tert-alkyl-α-hydroxy esters 769 reacted smoothly with 0.5 equiv of isocyanate to give the enriched starting-material 770 and carbamate 771, respectively. Regarding substrates, most were tolerated to give high selectivity and enantioselectivity. However, it was shown that replacing the β-methyl group with a β-methoxy group had a coordinative interaction with the catalyst and this reduced the enantioselectivity to as low as 51%ee.285
Scheme 243. Cu-Catalyzed Kinetic Resolution of α-Tert-alkyl-α-hydroxy Esters.

Beletskaya reported an asymmetric Friedel–Crafts reaction of indoles 724 with coumarin-3-carbonylates 772 in good yields and enantioselectivities (Scheme 244). Using ligand 600d and Cu(OTf)2 up to 82% ee and good yields were achieved. Groups with contrasting electronics in the 5-position of indole did not influence the yield or enantioselectivity, but the introduction of a methoxy group in the 4-position halted the reaction. Electron-withdrawing groups in the 5-position of coumarin greatly increased the enantioselectivities up to 82% ee.286
Scheme 244. Cu-Catalyzed Asymmetric Friedel–Craft Alkylation of Coumarin-3-carbonylates.

Mikami carried out an enantioselective Cu-catalyzed [2 + 2] cycloaddition of silyl enol ethers 774 with trifluoropyruvate 775 in the synthesis of oxetane derivatives 776 (Scheme 245). Using 600d as the ligand and Cu(OTf)2 as the catalyst, excellent cis/trans ratios and enantioselectivites up to >99% ee were achieved.287
Scheme 245. Cu-Catalyzed Asymmetric Synthesis of Oxetane Derivatives.

As shown above parent BOX ligands have been widely applied in asymmetric catalysis to great success. While there are few shortcomings for parent BOX ligands in catalysis, structural modifications are used to modulate the bite angles, electronics and sterics of ligands to gain enhanced activity.
3.1.2. Bis(oxazoline) Ligand with Modified Oxazoline Ring Substitution
Jia reported the construction of cyclic indolyl α-amino esters via a Cu-catalyzed asymmetric Friedel–Crafts alkylation reaction (Scheme 246). Using 777 as the ligand (Figure 24) and Cu(OTf)2 as the Lewis acid excellent yields and up to 99% ee were furnished. A range of N-sulfonyl ketimino ester derivatives 789 and indoles 790 reacted smoothly to give the product in up to 96% yield and >99% ee. This process was then extended to pyrrole and N,N-dimethylaniline while maintaining enantioselectivities up to 98% ee. At the same time indole substrates were very well tolerated with only 5-methoxyindole causing a drop in enantioselectivity to 83% ee.288
Scheme 246. Cu-Catalyzed Friedel–Crafts Alkylation of Indoles and N-Sulfonyl Ketimino Esters.

Figure 24.
Summary of Bis(oxazoline) ligands with modified oxazoline ring substitution.
Jia once again utilized 777 as the ligand but this time in the Ni-catalyzed Friedel–Crafts alkylation of indoles 792 with β-CF3-β-disubstituted nitroalkenes 793 to produce trifluoromethylated all-carbon quaternary stereocenters with enantioselectivities up to 97% ee and good yields (Scheme 247A). Most nitroalkenes furnished the products 784 in 88–97% ee while benzyl substituted nitroalkenes provided the corresponding product in 78% yield and 33% ee. For the indole substrate scope a range of electron-donating and -withdrawing substrates were proven successful (90–97% ee), only methyl substitution in the 1- and 2-positions provided poor results with trace amounts of product being formed.289
Scheme 247. Ni-Catalyzed Asymmetric Friedel–Crafts of Indoles and β-CF3-β-Disubstituted Nitroalkenes.

This methodology was extended to nitroacrylates 795 and indoles 724 garnering access to β-2,2-amino acid derivatives 796 in excellent yields and enantioselectivities up to 97% ee (Scheme 247B). In this case the nitroacrylate scope was very tolerant with different electronic patterns being widely accepted while maintaining high enantioselectivities (88–97% ee). The substitution on the indole ring had a much larger effect on the enantioselectivity. Substrates with 2-Me, 1-Me or 1-allyl all proved to be challenging substrates with enantioselectivities dropping as low as 54% ee.290
Lam reported the use of 778 in a Pd-catalyzed asymmetric addition of alkylazaarenes 798 to N-Boc aldimines 797 to produce Boc-protected α-stereogenic amines 799 (Scheme 248). Addition of the nitro group was integral to activating the alkylazaarenes 797 such as nitrobenzoxazoles and 3-nitropyridines were shown to be appropriate starting materials. A range of electron-donating and withdrawing groups were well tolerated in the optimized conditions achieving good yields, diastereomeric ratios and enantioselectivities (up to 95:5 dr and >99% ee). However, moving the nitro groups to the 4-position of the nitrobenzooxazole was shown to be disastrous for reactivity and enantioselectivity due to co-ordination of Pd and the nitro group furnishing the product in 38% yield, 18% ee and 83:17 dr. Nitroalkenes 245 were shown to be good substrates with similarly impressive enantioselectivities being achieved of up to >99% ee and 95:5 dr.291
Scheme 248. Pd-Catalyzed Asymmetric Addition to N-Boc Aldimines.

In a follow up publication by Lam, the scope of nitroalkenes 245 was investigated extensively, affording good yields and enantioselectivities up to 99% ee (Scheme 249). Regarding the 2-acetylazaarenes 800 scope, substrates containing quinoline, pyrazine, thiazole, benzothiazole, or N-methylimidazole all reacted smoothly to produce the desired product 801 in yields up to 95% and with high levels of enantioselectivity (up to 99%). It was also shown that β-alkyl-substituted nitroalkenes were not optimal substrates even after reoptimization with enantioselectivities reaching a maximum of 82% ee.292
Scheme 249. Ni-Catalyzed Asymmetric Friedel–Crafts Alkylation between Nitrostyrenes and Aryl Ketones.

Zanoni performed an enantioselective Mg-catalyzed Diels–Alder cycloaddition of acetoxyfulvene 802 and 3-acryloyl-1,3-oxazolidin-2-one 803. The resulting cycloadduct 804 was further transformed into a key building block for the formal synthesis of isoprostanoids (Scheme 250). Using Mg(ClO4)2 and 777 the cycloadduct product was isolated in 95% ee and with an endo/exo ratio of >99:1.293
Scheme 250. Mg-Catalyzed Asymmetric Synthesis of 15-F2c-Isoprostane Intermediate.

Doyle reported a catalyst divergent reaction of enoldiazoacetamides 805 with nitrones 695 (Scheme 251). When using the achiral catalyst of Cu(OTf).Tol, the Mannich addition products 808 were observed in up to 98% yield. However, when using Cu(MeCN)4BF4 a [3 + 3] cyclization product was observed. Exploiting this catalyst system using 778 as the ligand enantioselectivities up to 98% ee were obtained with excellent yields. Regarding the substrate scope a range of aryl, heteroaryl and bulky alkyl substrates were tolerated with enantioselectivities and yields remaining high throughout (94–98% ee).294
Scheme 251. Cu-Catalyzed Asymmetric Enoldiazoacetamide Cyclization.

Xu developed an Fe-catalyzed asymmetric intramolecular aminohydroxylation of indoles 809 using Fe(OTf)2 and ligand 778 achieving up to 99% ee and >20:1 dr (Scheme 252). A scope of substituted indoles was attempted, and all were shown to be excellent substrates. Electron donating groups and electron withdrawing groups in the 5-, 6-, and 7-positions were well tolerated (86–99% ee), although 4-bromo-substituted indole caused a substantial drop in enantioselectivity to 74% ee and moderate yields.295
Scheme 252. Fe-Catalyzed Asymmetric Intramolecular Aminohydroxylation.

You reported the asymmetric dearomatization of indole acetamides 811 with 3-indolylphenyliodonium salts 812 catalyzed by a Cu(I) 777 complex (Scheme 253). This process was then applied by carrying out the formal synthesis of folicanthine. Using [Cu(CH3CN)4PF6] and ligand 777 in ethyl acetate enantioselectivities up to 94% ee were achieved with good yields. Electron-withdrawing groups were well tolerated regardless of the position with the 6-fluoro substrate giving 94% ee. It was also shown that placing a methyl group in the 4- and 5-positions led to products formed in 67% ee and 82% ee, respectively.296
Scheme 253. Formal Synthesis of Folicanthine.

Yoon applied ligand 779 in the Fe-catalyzed aminohydroxylation of alkenes 814 using oxaziridines 815 (Scheme 254). Using Fe(NTf)2 and ligand 779 enantioselectivities up to 95% ee and 76% yield were obtained. A variety of aryl alkenes were tested in the reaction providing high enantioselectivities throughout with p-bromostyrene giving 63% yield and 85% ee as the lowest enantioselectivity. It was also shown that aliphatic alkenes and trans-β-methylstyrene were not tolerated in this reaction providing no product whatsoever.297
Scheme 254. Fe-Catalyzed Aminohydroxylation of Alkenes.

Kang reported the dual-functional Cu-catalyzed desymmetrization and subsequent kinetic resolution of serinols 817 to produce enantioenriched oxazolidinones (Scheme 255). Using ligand 780 and a CuCl2 complex enantioselectivities were high across the board tolerating a range of alkyl, aryl and alkenes (94–99% ee).298
Scheme 255. Cu-Catalyzed Synthesis of Chiral Oxazolidinones.

Andrus and Zhou prepared two regioisomers of naphthyl-substituted BOX ligands (781 and 782) and screened them in the allylic oxidation of cyclohexene 819 (Scheme 256). Using ligand 781 and CuPF6 the corresponding product was isolated in 40% yield and 80% ee, while use of ligand 782 gave the product in 80% yield and 85% ee.299
Scheme 256. Cu-Catalyzed Allylic Oxidation of Cyclohexene.

Dixon reported a Pd-catalyzed arylative and vinylative allene carbocyclization cascade. Using silver phosphate and a Pd(OAc)2.783 (s-butyl-substituted BOX ligand), the allene-linked ketoamide 822 pro-nucleophile and aryl iodides 823 were cyclized in good yields and up to 39:1 dr and 89% ee (Scheme 257). While a range of substituted aryl and cis-styrenyl iodides were well tolerated, trans-styrenyl iodides were shown to cause a large drop in enantioselectivity to 61% yield, 21:1 dr, 53% ee.300
Scheme 257. Pd-Catalyzed Asymmetric Carbocyclization Cascade.

Miao developed an asymmetric Cu-catalyzed vinylogous Mukaiyama aldol reaction of α-keto phosphonates 825 and 2-TMSO furan 826 (Scheme 258). Using the ligand 784 with Cu(OTf)2 and 2,2,2-trifluoroethanol as an additive, the aldol products 827 were obtained in enantioselectivities up to 96% ee and diastereoselectivities up to 99:1 dr. It was shown that the size of the phosphonate ester 825 group had little to no effect on the enantioselectivity or diastereoselectivity as both the methyl and ethyl esters gave the same results (95% ee and >99:1 dr). Electron-withdrawing and electron-donating groups were shown to have no effect on the results all giving enantioselectivities in the range of 94–98% ee. However, phosphonates that possessed an ortho-methyl substituent were less reactive giving enantioselectivities as low as 42% ee and diastereoselectivities as low as 58:42 dr.301
Scheme 258. Cu-Catalyzed Asymmetric Mukaiyama Aldol Reaction of α-Keto Phosphonates.

3.1.2.1. Bis(oxazoline) Ligands with Glucose Derived Oxazoline Ring Substitution
Boysen developed a series of glucose-derived ligands (Figure 25) and applied them in Cu-catalyzed cyclopropanations. Initially both ligands 785 and 786 were applied in the cyclopropanation of styrene 577 giving up to 93% ee for the trans-product 828 and 94% ee for the cis-product 829, respectively (Scheme 259A).302 Following on from this initial report, ligand 785 was subsequently applied in the cyclopropanation of 830 affording the product in up to 71% yield and 96% ee (Scheme 259B). The subsequent product was then applied in the synthesis of (−)-desoxyeseroline. A similar strategy was employed using ligand 787 to provide intermediate 833 in 75% yield and 90% ee in the synthesis of (+)-grenadamide (Scheme 259C).303,304
Figure 25.

Glucose-derived BOX ligands.
Scheme 259. Cu-Catalyzed Asymmetric Cyclopropanation.

Reddy reported the use of ligand 785 in the Cu-catalyzed asymmetric Friedel–Crafts reaction of 2-enoylpyridine-N-oxides 835 with indoles 836 (Scheme 260). Using ligand 785 and Cu(OTf)2 enantioselectivities up to 99% ee were obtained in excellent yields. The indole substitution pattern had a large influence on the enantioselectivity, for example, when the electron-donating 4-methoxyindole was used the enantioselectivity dropped to 85% ee although the yield was still high at 94%. For the 2-enoylpyridine-N-oxide substrate scope it was shown the aryl and heteroaryl were suitable substrates with the enantioselectivities remaining high at 90–99% ee. When alkyl substituents were tested the enantioselectivity dropped dramatically with t-butyl affording the product in 65% yield and 33% ee, while the cyclohexyl-containing substrate did not give rise to product.305
Scheme 260. Cu-Catalyzed Asymmetric Friedel–Crafts Reaction of 2-Enoylpyridine-N-oxides.

3.1.2.2. Bis(oxazoline) Ligands with Silyl-Protected Oxazoline Ring Substitution
Desimoni and Faita disclosed the Cu-catalyzed cycloaddition of 2-alkenoylpyridine-N-oxides 835 and dienes 838 (Scheme 261A). Using ligand 788 and Cu(OTf)2 excellent enantioselectivities up to 98% ee and endo:exo ratios up to 99:1 were observed for the products 839/840. Eletron-withdrawing arylidenepyridine-N-oxides were also attempted and showed to have no effect on the enantioselectivity. In a subsequent publication, ligand 788 was applied in the cycloaddition between enol silyl ethers 841 and 2-alkenoylpyridine-N-oxides 835 forming products 842 in poor yield (30%) and with high enantioselectivity (99% ee) (Scheme 261B).306,307
Scheme 261. Cu-Catalyzed Cycloaddition of 2-Alkenoylpyridine-N-oxides and Dienes/Enol Silyl Ethers.

The BOX ligands with modified oxazoline ring substitutions are widely applied in Lewis acid catalysis to great affect as shown above. While many of the modifications rely on costly non-natural amino alcohols more cost-effective ligands have been developed using carbohydrates for their chiral information.
3.1.3. Bis(oxazoline) Ligands with Backbone Modifications
Wang reported the Cu-catalyzed asymmetric cross coupling of α-substituted-β-keto esters 875 with N-substituted glycine esters 876 (Scheme 262). Using a Cu(OTf)2 and ligand 843 complex (Figure 26) gave the product 877 in good yields and up to 96% ee in a 2:1 dr. A variety of α-substituted-β-keto esters 875 were screened affording the products 877 in up to 72% yield and 96% ee. Acyclic α-substituted-β-keto esters however led to a drop in enantioselectivity as low as 81% ee in a 3:2 dr.308
Scheme 262. Cu-Catalyzed Asymmetric Coupling of α-Substituted-β-keto Esters and Glycine Esters.

Figure 26.
BOX ligand with backbone modifications.
Wang developed a Ni-catalyzed asymmetric amination of 3-bromooxindoles 879 with indolines 878 (Scheme 263), a synthetic methodology that was then applied in the formal synthesis of Psychotrimine (Figure 27). Using ligand 843 and Ni(OAc)2 as the optimized catalyst complex enantioselectivities up to 96% ee were obtained. Indolines and oxindoles with a variety of halides in the 4-, 5-, and 6-positions afforded the product in high enantiosleectivities (86–96% ee). However, placing electron-donating substituents (methyl or methoxy) in the 5-position led to a significant drop in the enantioselectivity, to 61% ee and 74% ee, respectively.309
Scheme 263. Ni-Catalyzed Asymmetric Amination of 3-Bromooxindoles.

Figure 27.

Psychotrimine.
Yoon applied ligand 850 in the Fe-catalyzed kinetic resolution of N-sulfonyl oxaziridines 881 achieving up to 99% ee and with selectivity factors up to 30 (Scheme 264). A variety of C-aryl oxaziridines 881 were screened in this reaction with most substrates obtaining >90% ee. A major limitation of this method was that C-alkyl oxaziridines were not compatible with 15% ee being the highest enantioselectivity obtained.310
Scheme 264. Fe-Catalyzed Kinetic Resolution of N-Sulfonyl Oxaziridines.

Sibi developed a Mg-catalyzed asymmetric conjugate addition of malononitrile to pyrazolidone-derived enoates 883 (Scheme 265). Using MgBr2.Et2O and ligand 843 as the catalyst enantioselectivites up to 99% ee and good yields were obtained. It was also shown that when 4 Å MS were taken out of the optimized reaction conditions the enantioselectivity dropped as low as 44% ee. It was shown that the substituents on the enone component of the substrate greatly affected the enantioselectivity with electron donating p-methoxyphenyl groups resulting in enantioselectivities as low as 42% ee.311
Scheme 265. Mg-Catalyzed Asymmetric Conjugate Addition to Pyrazolidone Derivatives.

Singh reported an asymmetric Zn-catalyzed Michael addition of malonates 238 to 2-enoylpyiridine N-oxides 694 (Scheme 266). Using Zn(OTf)2.843 as the catalyst good yields and enantioselectivities of up to 96% ee were observed. It was shown that bulky malonates led to low enantioselectivities, such as di-t-butylmalonate which furnished the product in 60% yield and 16% ee. Interestingly, while many aromatic and heteroaromatic β-substituted 2-enoylpyridine N-oxides were suitable substrates in this reaction, the sterically hindered t-butyl β-substituted 2-enoylpyridine N-oxide did not give rise to any product.312
Scheme 266. Zn-Catalyzed Asymmetric Michael Addition of Malonates to 2-Enoylpyiridine N-Oxides.

Xiao developed a Cu-catalyzed Friedel–Crafts alkylation/N-hemiacetalization cascade reaction of indoles 886 with β,γ-unsaturated α-keto esters 887 (Scheme 267). Using Cu(OTf)2 and the indanol-derived chiral ligand 844 as the catalyst enantioselectivities up to >99% ee and 97:3 dr were obtained. This methodology was then applied in the formal synthesis of flinderole B analogues. A series of β,γ-unsaturated α-keto ester substrates 887, regardless of bulkiness or electronic-nature, resulted in high levels of enantioselectivities of 91 to >99% ee.313
Scheme 267. Cu-Catalyzed Asymmetric Friedel–Crafts Alkylation/N-Hemiacetalization Cascade Reaction of Indoles.

3.1.3.1. Bis(oxazoline) Ligands with Benzyl Substituted Backbone Modifications
Tang developed a Cu-catalyzed [4 + 2] cycloaddition of indoles 889 and cyclobutanes 890 to yield cyclohexyl-fused indolines 891 (Scheme 268). This reaction was initially developed using Cu(SbF6)2 in CH2Cl2 in a racemic manner and was then further developed using the chiral ligand 845 (Figure 28) affording up to 94% ee and 83:17 dr improving upon the parent BOX ligand 600b. This methodology was then applied to the formal total synthesis of (±)-strychnine and the total synthesis of (±)-akuammicine (Figure 29).314
Scheme 268. Cu-Catalyzed Asymmetric Synthesis of Cyclohexyl-Fused Indolines.

Figure 28.

BOX ligands with substituted benzyl backbones.
Figure 29.

(±)-Strychnine and (±)-akuammicine.
Tang applied ligand 846 in the Cu-catalyzed enantioselective synthesis of bicyclic N,O-acetals 892 via a hetero-Diels–Alder reaction (Scheme 269). Catalyzed by a Cu(OTf)2.846 complex, β,γ-unsaturated α-keto esters 893 reacted with cyclic enamines 892 efficiently, affording the desired products 894 in excellent yields with excellent stereoselectivities (up to 99% yields up to >95:5 dr; and 95–99% ee). Aryl and heteroaryl moieties were tolerated in the β,γ-unsaturated α-keto ester 893 substrate scope without any deterioration in the enantioselectivity observed (95–99% ee).315
Scheme 269. Cu-Catalyzed Asymmetric Synthesis of N,O-Acetals.

Tang applied ligand 847 in the asymmetric Cu-catalyzed cyclopropanation of alkenes 895 to yield the 1,1-cyclopropane diesters 897 (Scheme 270). Using phenyliodonium ylides 896 as the carbene transfer reagent, [Cu(CH3CN)4]PF6 as the catalyst and 847 as the chiral ligand, enantioselectivities of up to >99% ee were observed. A variety of terminal alkenes were very well tolerated in the reaction most of which gave enantioselectivities in the range of 87–96% ee. When nonterminal alkenes were subjected to the optimized reaction conditions, 5-, 6- and 7-membered cyclic alkenes yielded the product in up to >99% ee. One limitation of this reaction appeared to be aliphatic-substituted alkenes afforded the product in enantioselectivities as low as 77% ee. In 2018 Tang further developed the asymmetric cyclopropanation of trisubstituted olefins using modified bis(oxazoline) ligands.316,317
Scheme 270. Cu-Catalyzed Enantioselective Cyclopropanation of Alkenes.

Tang also reported the Cu-catalyzed asymmetric construction of cyclobutanes 900 from methylidenemalonate 898 and styrenes 899 (Scheme 271). Using ligand 848 and Cu(ClO4)2·6H2O as the catalyst, enantioselectivities up to >99% ee and >99:1 dr were obtained. A range of styrenes 899 (aryl and heteroaryl) were well tolerated with all enantioselectivities observed ranging between 95 and 99% ee. This methodology was then applied to the synthesis of (+)-piperarborenine B a potential anticancer agent.318
Scheme 271. Cu-Catalyzed Asymmetric Synthesis of Cyclobutanes.

Building on this work, Tang developed a Cu-catalyzed enantioselective [3 + 2] annulation of cyclic enol silyl ethers 901 and cyclopropanes 902 (Scheme 272). Using a catalyst derived from Cu(ClO4)2·6H2O and ligand 849, enantioselectivities of up to 99% ee and >99:1 dr were observed. Bulky adamantyl groups on the cyclopropyl esters were deemed necessary as less bulky ester groups resulting in lower enantioselectivities (94% ee). A range of electronically variable cyclopropanes were tested with optimized conditions showing no significant drop in enantioselectivity (91–99% ee).319
Scheme 272. Cu-Catalyzed [3 + 2] Annulation of Cyclic Enol Silyl Ethers and Cyclopropanes.

Cai utilized ligand 850, a fluorinated derivative of ligand 600c, in the Henry reaction between aldehydes 477 and nitromethane (Scheme 273). Using Cu(OAc)2 and ligand 850 enantioselectivities of up to 99% ee were achieved. While most aldehydes 477 performed well in the catalysis (90–99% ee), electron rich aldehydes such as p-methoxybenzaldehyde provided no product whatsoever. Cai further extended this methodology to the Cu-catalyzed asymmetric addition of acetonitrile 906 to isatins 905 to produce a series of 3-hydroxy-2-oxindoles 907 (Scheme 274). Using Cu(OTf)2 and ligand 850 enantioselectivities up to 92% ee were obtained. Regarding the substrate scope placing the electron-withdrawing chloro-group in the 6-position of the isatin led to lower enantioselectivities of 64% ee.320,321
Scheme 273. Cu-Catalyzed Asymmetric Henry Reaction.

Scheme 274. Cu-Catalyzed Asymmetric Synthesis of 3-Hydroxy-2-oxindoles.

Cai reported the use of ligand 851 in the Cu-catalyzed hydrophosphonylation of aldehydes 477 to give the corresponding products 909 in good yields and with enantioselectivities of up to 98% ee (Scheme 275). Various electron-withdrawing and donating aldehydes 477 were screened in this reaction and these formed the products 909 in moderate to high levels of enantioselectivity (74–98% ee). The highly electron-withdrawing p-trifluoromethylbenzaldehyde gave the lowest enantioselectivity with 74% ee albeit in good yields (91% yield). Heteroaromatic systems largely were not compatible in this reaction with 2-thienyl- and 2-furyl-aldehydes not affording any product; the use of pyridine-2-carboxyladehyde as substrate was more successful, with the product formed in 71% yield albeit with modest enantioselectivity (79% ee).322
Scheme 275. Cu-Catalyzed Asymmetric Hydrophosphonylation of Aldehydes.

Wang reported a Cu-catalyzed Friedel–Crafts reaction of indoles 792 with isatin-derived β,γ-unsaturated α-keto esters 910 (Scheme 276). Using the BINOL-derived ligand 853 (Figure 30), the products 911 were formed in in good yields and enantioselectivities of up to 99% ee while using relatively low catalyst loadings of 0.5 mol %. Regarding the scope of this reaction a wide variety of isatin substrates 910 were well tolerated with most products being formed in 82–99% ee. Interestingly 6-bromoisatin gave rise to the corresponding product in 60% yield and a low enantioselectivity of 75% ee. The scope for indole substrates 792 was similarly impressive with a variety of functional groups being tolerated in good to excellent levels of enantioselectivity (82–99% ee). 7-Methylindole however formed the product in 88% yield and 51% ee.323
Scheme 276. Cu-Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles and Isatin-Derived β,γ-Unsaturated α-Keto Esters.

Figure 30.
BINOL-derived BOX ligand 853.
Wang developed an enantioselective synthesis of trisubstituted allenes 913 from a Cu-catalyzed cross-coupling of diazoalkanes 912 and terminal alkynes 604 (Scheme 277). The optimal catalyst complex was a combination of Cu(MeCN)4PF6 and the naphthyl-containing ligand 854 which gave the product in good yields and up to 98% ee. A series of aryldiazoalkanes 912 bearing electron-deficient or -rich aromatic substituents were smoothly reacted with phenylacetylene to give the corresponding trisubstituted allenes 913 in high yields (78–95% yield) and very high levels of enantioselectivity (84–95% ee). The scope of the terminal alkynes afforded the product in good enantioselectivities (92–98% ee) although the yields were lowered (46–60%)324
Scheme 277. Cu-Catalyzed Asymmetric Synthesis of Trisubstituted Allenes.

3.1.3.2. Bis-Phenyl BOX Ligands with Backbone Modifications
Zhang reported the asymmetric Ni-catalyzed alkenylation of ketimines 914 using alkenylboronic acids 915 (Scheme 278). The optimal catalyst combination of ligand 855 (Figure 31) and Ni(OTf)2 proved to be an extremely effective system providing the product in yields up to >99% and enantioselectivities of up to >99% ee. The initial ketimine 914 scope gave high to excellent enantioselectivities irrespective of the nature of the ketimine (88 to >99% ee). The alkenylboronic acid 915 scope showed tolerance of both alkyl and styrenylboronic acids with enantioselectivities remaining very high (93% to >99% ee). This methodology was then expanded to the π-conjugation-controlled site-selective asymmetric ketimine-alkenylation/ring-expansion (Scheme 279). Once again, this methodology proved robust with routinely high enantioselectivities being obtained (up to >99% ee).325 This methodology was further employed in the allylation of cyclic ketimines.326
Scheme 278. Ni-Catalyzed Asymmetric Alkenylation of Ketimines.

Figure 31.

Bis-phenyl BOX ligands with backbone modifications.
Scheme 279. Ni-Catalyzed Asymmetric Ketimine Alkenylation/Ring-Expansion.

Fu reported the development of an asymmetric Ni-catalyzed Negishi cross-coupling of α,α-dihaloketones 919 to give the corresponding products 920 containing quaternary-fluorinated stereocenters (Scheme 280). Using NiCl2·glyme and ligand 856 enantioselectivities of up to 99% ee were obtained. The structure of the ketone 919 had no major effect on the enantioselectivity with most substrates furnishing the product in high levels of enantioselectivities (92% to 98% ee). In a similar fashion the nature of the nucleophilic zinc reagent 655 had little impact on the enantioselectivity, all giving high enantioselectivities (91 to 99% ee). This methodology was further applied by Futo in the Negishi phenylation of racemic α-bromonitriles and the Kumada cross-coupling of racemic α-bromoketones327−329
Scheme 280. Ni-Catalyzed Asymmetric Negishi Cross-Coupling of α,α-Dihaloketones.

An asymmetric Cu-catalyzed desymmetrization of meso-α,α′-diazido alcohols 921 to produce cyclic α-imino esters 922 was reported by Gu (Scheme 281). The optimized conditions utilized CuPF6(MeCN)4 and ligand 857 as the catalyst complex and NaBARF as the noncoordinating counterion. This combination furnished the corresponding products 922 in up to 97% ee, which was carried out on gram scale. Aryl-substituted meso-α,α′-diazido alcohol substrates 921 afforded the products in very high levels of enantioselectivity (92 to 97% ee). When alkyl-containing substrates were used, the enantioselectivity dropped as low as 77% ee.330
Scheme 281. Cu-Catalyzed Desymmetrisation of meso-α,α′-Diazido Alcohols.

3.1.3.3. Bis(oxazoline) Ligands with Cyclopentyl Backbone Modifications
Liu developed an asymmetric Cu-catalyzed cyanation by C–H activation of benzylic C–H bonds for the synthesis of benzylic nitriles 925 (Scheme 282). This reaction was catalyzed by a Cu(OAc)2·BOX complex using NFSI (N-fluorobenzenesulfonylimide) as the oxidant. Enantioselectivities up to 99% ee were obtained but each substrate had to screened for the optimal ligand, with BOX ligand 843 and the modified BOX ligands 858 and 859 found to be the ligands of choice (Figure 32). Alkyl-naphthylenes, alkyl arenes and alkyl heteroarenes all performed well under the optimized conditions with the lowest enantioselectivities obtained being 80% ee.331
Scheme 282. Cu-Catalyzed Asymmetric Synthesis of Benzylic Nitriles.

Figure 32.

BOX ligands with cyclopentyl backbone modification.
Nishibayashi developed the Cu-catalyzed enantioselective alkylation of β-keto phosphonates 927 using diaryl methanols 926 as the electrophiles (Scheme 283). Cu(OTf)2 and ligand 843 was shown to be the optimal catalyst giving the alkylated product 928 in up to 92% ee. Cyclic phosphonates were shown to be compatible with this catalysis affording the product in up to 92% ee, while acyclic phosphonates furnished the product in as low as 42% ee. Regarding the diarylmethanol the electronic nature of the aryl groups had little effect on the enantioselectivity (84–90% ee), heteroaryl substrates however produced the largest drop in enantioselectivity to 76% ee.332
Scheme 283. Cu-Catalyzed Asymmetric Alkylation of β-Keto Phosphonates.

Stanley reported the Mg-catalyzed asymmetric cycloaddition of nitrile imines 930 with methyleneindolinones 929 to produce enantioenriched spirocyclic products 931 in up to 99% ee employing ligand 843 (Scheme 284). A range of methyleneindolinones were screened in the reaction with electron withdrawing and donating substituents giving rise to the product in up to 99% ee. Interestingly, an ortho-bromo substituent on the methyleneindolinone substrate led to racemic product.333
Scheme 284. Mg-Catalyzed Asymmetric Cycloaddition of Nitrile Imines with Methyleneindolines.

Gong developed an asymmetric Cu-catalyzed cross-coupling of 3-indoylmethyl C–H bonds 932 with diaryl malonates 933 (Scheme 285). With Cu(OTf)2 as the Lewis acid, 843 as the chiral ligand and DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) as the oxidant, the product was formed in up to 99% yield and 96% ee. The substrate scope included a range of 3-arylmethylindole derivatives 932 which proved successful for both electronically rich or poor 3-arylmethyl substituted substrates (up to 99% yield and 94% ee). The electronic properties of the aryl substituent had little effect on the enantioselectivity (86 to 94% ee) The introduction of substituents onto the indole moiety afforded the product in high enantioselectivity (78% yield and 96% ee).334
Scheme 285. Cu-Catalyzed Asymmetric C–H Activation of Indole Derivatives.

Liu reported the trifluoromethylalkynylation of alkenes 464 via Cu-Catalyzed radical relay producing chiral CF3-containing propargylic compounds 936 in up to 97% ee (Scheme 286). Using a complex formed from Cu(CH3CN)4PF6 and ligand 860 as the catalyst complex and Togni’s reagent 750 as the CF3 source, a wide range of products were accessed in good yields (up to 88% yield). The substrate scope for the alkene 464 coupling partner included a series of structurally diverse alkenes with electron-rich and poor styrenes affording the product in enantioselectivities up to 97% ee. Hetereoaryl alkenes also yielded the product in high enantioselectivities (87 to 95% ee).335
Scheme 286. Cu-Catalyzed Asymmetric Trifluoromethylalkynylation of Alkenes.

Pan reported the photoredox cyanoalkylation of akenes 464 using redox active N-hydroxy-phthalimide esters 937. By merging Ir-catalyzed photocatalysis and Cu-catalysis the products were formed in up to 94% ee when using CuBr and ligand 859 (Scheme 287). A range of styrenes 464 were tested using the optimized reaction conditions, most of which gave the product in high enantioselectivities (84 to 94% ee). Large p-t-butylstyrene or 2-naphthylstyrene led to a drop in enantioselectivity (76% ee and 65% ee, respectively). m-Bromostyrene as substrate led to the most substantial drop in enantioselectivity with the product being obtained in 54% ee.336
Scheme 287. Photoredox Cyanoalkylation of Akenes.

Reisman reported the Ni-catalyzed cross-electrophile reductive coupling between vinyl and benzyl electrophiles (Scheme 288). Using NiCl2(dme) as the catalyst and the chiral ligand 859, enantioselectivities up to 97% ee were observed. Both the bromostyrene 939 and benzyl chloride 940 substrate scope had little effect on the enantioselectivity (as low as 85% ee) with pinacol boronate and free phenol functional groups being compatible with the reaction.337
Scheme 288. Ni-Catalyzed Asymmetric Cross-Electrophile Coupling of Vinyl and Benzyl Electrophiles.

3.1.3.4. Bis(oxazoline) Ligands with Unsaturated Backbone Modifications
Fu detailed the synthesis of the novel ligand 861 (Figure 33) and its application in the Cu-catalyzed Friedel–Crafts alkylation of indoles 792 by alkylidene malonates 942 (Scheme 289). The optimal reaction conditions utilized a combination of Cu(OTf)2 and ligand 861, which yielded the product in up to 88% ee. The substrate scope probed electronic properties of the alkylidene malonates with p-substituted substrates leading to a significant drop in enantioselectivities (37% ee). Substitution on the indole ring also played a large effect with 6-chloroindole forming the product in just 10% ee.338
Figure 33.

BOX ligands with unsaturated backbone modifications.
Scheme 289. Cu-Catalyzed Friedel–Crafts Alkylation of Indoles and Alkylidene Malonates.

Fu reported the Cu-catalyzed conjugate addition of indoles 792 to β-substituted unsaturated acyl phosphonates 744 to produce 3-indolyl adducts in good yields (Scheme 290). Heteroarylidene-tethered bis(oxazoline) ligand 862 and Cu(OTf)2 were used as the optimized catalytic conditions forming products in modest to very high yields (73–96%) and achieving very high levels of enantioselectivities of up to 97% ee. Regarding the substrate scope it appears that esters, nitrile, halides and ethers are all well tolerated on the indole ring with enantioselectivities remaining high (90–96% ee). Regarding the scope of the acyl phosphonates 744 a range of aryl and heteroaryl substrates were tested with 2-furyl providing the product in the lowest enantioselectivity (78% ee).339
Scheme 290. Cu-Catalyzed Conjugate Addition of Indoles to β-Substituted Unsaturated Acyl Phosphonates.

Gu applied ligand 863 (Figure 34) in the Cu-catalyzed ring opening of diaryliodonium salts 946 in the synthesis of chiral diarymethanes 948 (Scheme 291). Using thiolates and carboxylates 947 as nucleophiles the asymmetric Cu(OTf)2-catalyzed ring opening gave the products 948 in good yields (up to 99% yield) and enantioselectivities up to 98% ee. Substituted thioacetates appeared to have a small effect on the enantioselectivity with most substrates ranging between 91 to 97% ee. The substrate scope for the carboxylates gave high enantioselectivity once again (93 to 98% ee) but the reaction time was extended from 10 to 48 h.340
Figure 34.

BOX ligand with xylene-derived substituents.
Scheme 291. Cu-Catalzyed Asymmetric Synthesis of Diarylmethanes.

3.1.3.5. Application of Semicorrin Derived Ligands
Toste developed the Re-catalyzed asymmetric reduction of ketones 949 and imines 951 using Semicorrin ligands 864 and 865 developed by Pfaltz.341a,341c When carrying out the asymmetric hydrosilylation of ketones 949 both ligands 864 and 865 (Figure 35) were screened reaching up to 94% ee and 93% ee, respectively, for substituted tetralones (Scheme 292). This methodology appeared to work only with aryl ketone substrates with 4-phenyl-2-butanone furnishing the product in 67% yield and 6% ee. This methodology was then expanded to the reduction of aryl phosphinyl imines 951 which furnished the product in up to >99% ee for some ary and heteroaryl imines (Scheme 293). Similarly, this reaction was limited as alkyl imines furnished the products 952 in lower enantioselectivities (0–32% ee).341d
Figure 35.

Semicorrin derived BOX ligands 864–865.
Scheme 292. Re-Catalyzed Asymmetric Reduction of Ketones.

Scheme 293. Re-Catalyzed Asymmetric Reduction of α-Aryl Phosphinyl Imines.

3.1.3.6. Application of Bi-Functional BOX Ligand with Photosensitizer Backbone Modifications
Xiao developed a bifunctional box derived ligand 866 (Figure 36), which has a built-in diarylketone photosensitizer which allows the ligand to function as a chiral catalyst and a photocatalyst. Using ligand 866 and Ni(acac)2 the oxidation of β-keto esters 953 afforded the desired products 954 in up to 95% ee (Scheme 294). Regarding the substrate scope a range of cyclohexanone-derived β-keto esters 953 formed the products in high enantioselectivity (88–95% ee). Cycloheptanone-derived β-keto esters also formed the products in up to 93% ee.342
Figure 36.
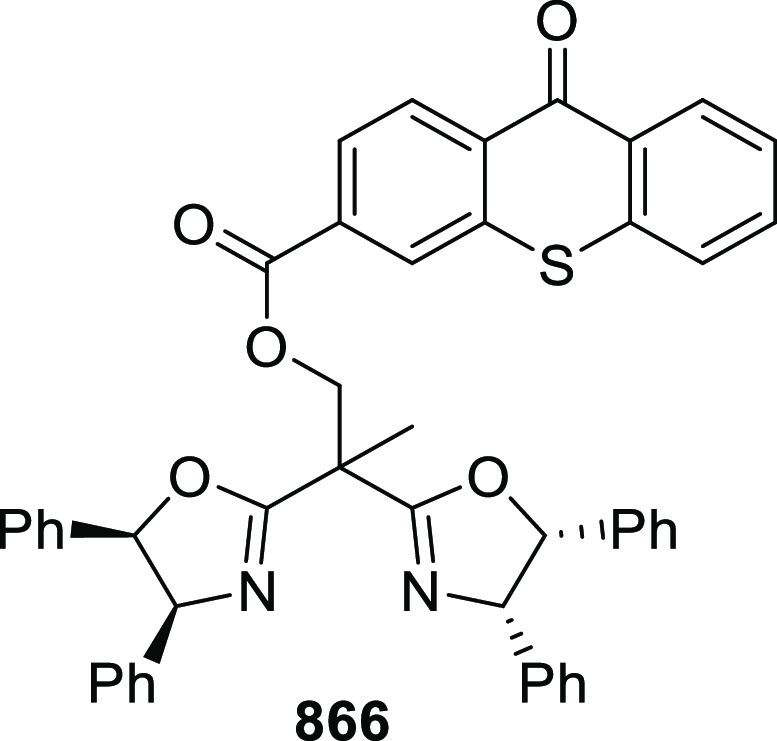
Bifunctional BOX ligand 963 with photosensitizer.
Scheme 294. Ni-Catalyzed Asymmetric Oxidation of β-Keto Esters.

3.1.3.7. Bis(oxazoline) Ligands with Miscellaneous Backbone Modifications
You applied ligand 867 in the Cu-catalyzed dearomative amination of tryptamines 955. Using ligand 867 (Figure 37), CuBr and O-(2,4-dinitrophenyl)hydroxylamine 956, an efficient electrophilic aminating reagent a range of 3-amino-pyrroloindolines 957 were synthesized in up to 95% ee. A range of indole protecting groups were attempted with allyl, methyl and benzyl all affording the product in high enantioselectivities (86–92% ee) (Scheme 295). An indole substrate scope was carried out with a variety of electron withdrawing and donating groups in the 4–7 positions providing the products in high enantioselectivities (82–95% ee). This methodology was then applied to the formal synthesis of tryptophan-derived alkaloids (−)-psychotriasine (Figure 38).343
Figure 37.
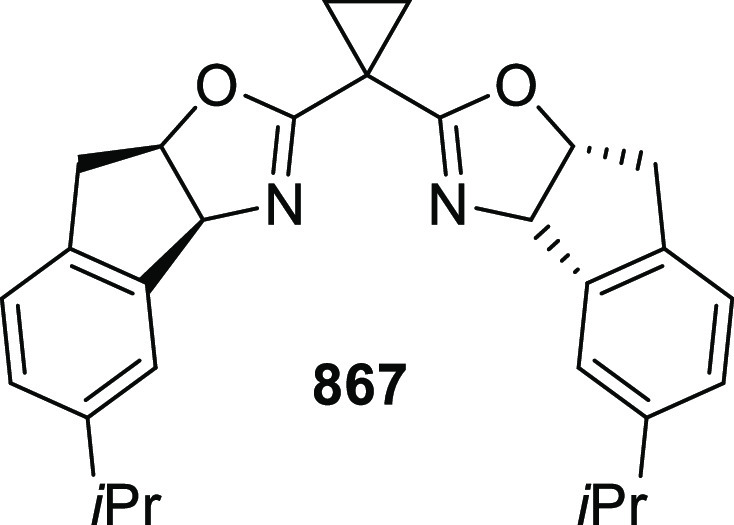
Functionalized indene-derived ligand 867.
Scheme 295. Cu-Catalyzed Enantioselective Synthesis of 3a-Amino-pyrroloinolines.

Figure 38.

(−)-Psychotriasine.
Guo reported the Cu-catalyzed Friedel–Crafts alkylation of β-naphthols 958 and aminocyclopropanes 959 to make γ-aminobutyric acid-like structures 961. Using the chiral ligand 868 and Cu(OTf)2 the desired Friedel–Crafts products were isolated in up to 98% ee (Scheme 296). The undesired O-alkylation product 960 was seen when less sterically demanding β-naphthols were used. Besides the chemoselectivity issue presented by the O-alkylation product a large number or electronically diverse β-naphthols were applied to this catalysis forming the products in high levels of enantioselectivity (90–98% ee).344
Scheme 296. Cu-Catalyzed Asymmetric Friedel–Crafts Alkylation of β-Naphthols and Aminocyclopropanes.

Liu reported the synthesis of axially chiral isoquinolones 963 via a Ni-catalyzed denitrogenative transannulation (Scheme 297). Ni(cod)2 and ligand 869 (Figure 39) proved to be the optimal conditions for the denitrogenative transannualtion of 1,2,3-benzotriazin-4(3H)-ones 962 and bulky internal alkynes 201, to give the product 963 in good yields and enantioselectivities of up to 98% ee. This reaction is amenable to both electron-withdrawing and electron donating groups at the 4-position of the naphthalene ring with 4-bromonaphthalene giving the highest ee of 88%. The stability of these axially chiral products 963 was tested by refluxing them in toluene for 1 day. No erosion of enantioselectivity was observed demonstrating the high barrier to rotation about the naphthyl–isoquinolone bond.345
Scheme 297. Ni-Catalyzed Enantioselective Denitrogenative Transannulation.

Figure 39.

BOX ligand with cyclopropyl-modified backbone.
Nagorny developed a concise Cu-catalyzed synthesis of oxygenated steroids via a sequential Michael-addition/intramolecular aldol cyclization protocol (Scheme 298). Using ligand 870 (Figure 39) and Cu(SbF6)2 to catalyze the Michael addition of β,β′-enones 964 and substituted β,β′-keto esters 965 resulted in Michael adducts which can undergo base-promoted aldol cascade reactions resulting in steroid skeletons 967. Regarding the substrate scope of the Michael addition, five-membered β-keto esters 965 proceeded with high levels of diastereocontrol (up to >20:1 dr and up to 96% ee). Six-membered β-keto esters 965 proceeded in high enantioselectivities (up to 94% ee) but the diastereocontrol dropped to as low as 4:1 dr. Once treated with base the double aldol cyclization occurred to form the steroidal backbone.346
Scheme 298. Cu-Catalyzed Asymmetric Synthesis of Oxygenated Steroid Derivatives.

Kobayashi applied ligand 871 (Figure 40) in the Ca-catalyzed 1,4-addition to form product 969 and a [3 + 2] cycloaddition toward the synthesis of pyrrolidine derivatives 970 (Scheme 299). Using CaCl2 and ligand 871 as the optimized catalyst the tandem reaction sequence between imines 968 and enones 157 proceeded in up to 99% ee and in good yields (up to 98% yield). Imines 968 containing amides appeared to give complete conversion to the pyrrolidine products 970 while ester-containing imines underwent the 1,4-addition with complete selectivity to form 969.347
Figure 40.
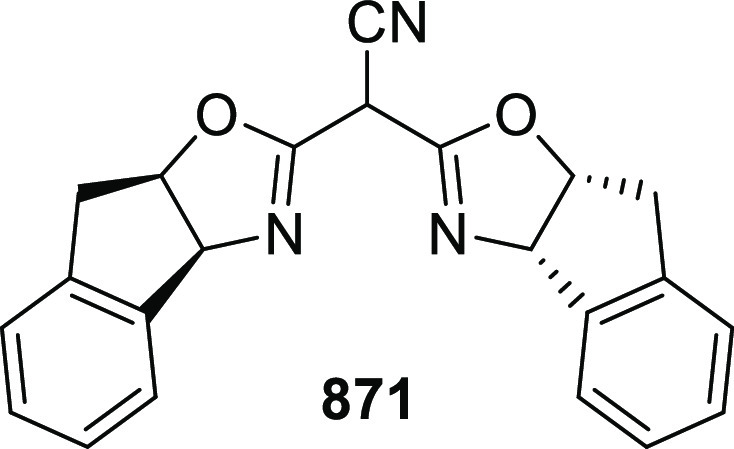
Indene BOX ligand 871 with nitrile backbone modification.
Scheme 299. Calcium-Catalyzed Asymmetric Synthesis of Pyrrolidine Derivatives.

Miñana reported the application of the recyclable ligand 872 (Figure 41) in the Cu-catalyzed Henry reaction of aldehydes 971. Using Cu(OAc)2 and ligand 872 enantioselectivities up to 78% ee were observed with anti/syn ratios of up to 72:28 (Scheme 300). This ligand avails of a release-capture technique which can be recycled after a run of catalysis without the need for a heterogeneous support. After the reaction solvent is removed the ligand is precipitated by washing with hexane/ether removing it from the product solution. The precipitated ligand is dried and reused in another reaction. The viability of 872 as a recyclable ligand was tested by placing it through 14 subsequent runs of catalysis after recovery. This showed a slight decrease in enantioselectivity to 72% ee after 14 cycles in catalysis.348
Figure 41.

Recyclable BOX ligand 872.
Scheme 300. Cu-Catalyzed Enantioselective Henry Reaction with a Non-supported Recyclable Ligand.

Gong reported the Cu-catalyzed asymmetric alkylation of imines 974 driven by light. The light promotes radical formation of the trifluoroborates and acts as a reducing agent for the copper catalyst. Using ligand 873 (Figure 42), a range of N-sulfonylimines 974 were alkylated using alkyl trifluoroborates 975 in high yields and enantioselectivities of up to 94% ee (Scheme 301). The scope of this reaction investigated various electron-withdrawing and donating alkyl trifluoroborates and all were found to maintain high enantioselectivities (81–94% ee). Lower enantioselectivities were observed when using p-methoxyphenyl, heteroaryl and bulky t-butyl trifluoroborates (as low as 24% ee). This methodology was then applied to the benzylation of isatin-derived ketimines with 860 as the ligand. A variety of isatin-derived ketimines were attempted furnishing the product in up to 98% ee (Scheme 302).349
Figure 42.
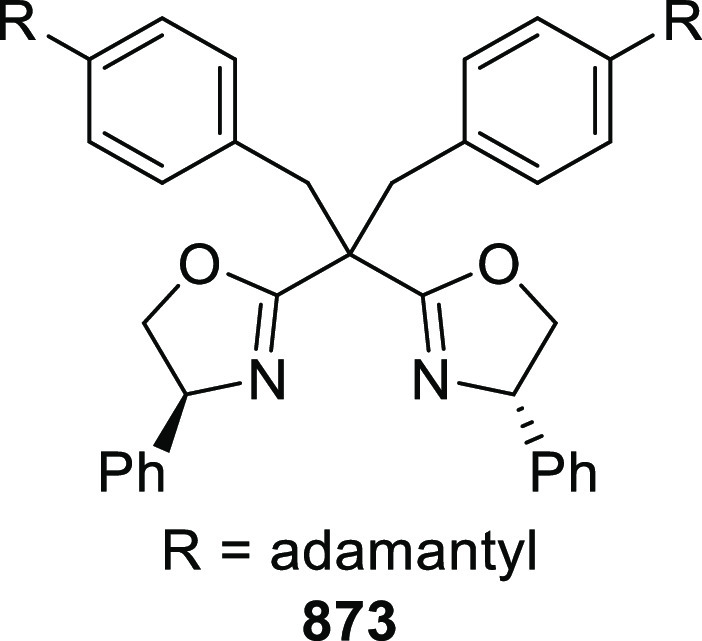
BOX ligand 873 with adamantyl backbone modification.
Scheme 301. Cu-Catalyzed Asymmetric Alkylation of Imines.

Scheme 302. Cu-Catalyzed Enantioselective Alkylation of Isatin-Derived Ketimines.

Nakada reported the Cu-catalyzed catalytic asymmetric intramolecular cyclopropanation of α-diazo ketones 980 in the synthesis of polycyclic polyprenylated acylphloroglucinols (Scheme 303). Using ligand 874 with Cu(CH3CN)4 the cyclopropylated product is formed and undergoes a subsequent rearrangement with water present to furnish the diketone 981 (79% yield, 84% ee), a valuable intermediate in the formal synthesis of (+)-clusianone.350a
Scheme 303. Cu-Catalyzed Asymmetric Intramolecular Cyclopropanation of α-Diazo Ketones.

In 2017, Tang reported the synthesis of Wing-BOX ligand 852 and it is application in the synthesis optically active hexahydrocarbazoles 983 in up to <99% ee and 99% yield (Scheme 304). Ligand 852 exploits the Thorpe–Ingold effect using the cyclopently groups on the backbone to enhance enantioselectivity (the parent bis(oxazoline) ligand 600c furnished the product in 45% ee and 76% yield).350b
Scheme 304. Tandem [2 + 2 + 2] Asymmetric Reaction of Indole with Methylenemalonate.

It has been shown that BOX ligands with backbone modification are used largely within Lewis acid catalysis and cross-coupling to great effect. On multiple occasions they are employed in a scenario where parent BOX ligands achieve moderate enantioselectivity and are used to fine-tune the bite angles, electronics and achieve excellent enantioselectivity. From this section of the review, it is plain to see the far-reaching applications of BOX ligands and modified BOX ligands in asymmetric catalysis. A large proportion of this catalysis entails Lewis acid activation and inherently inexpensive metals such as Zn and Cu. While traditional enantioselective cross-couplings with Pd catalysts are still popular the increased use of inexpensive metals such as Ni or Co has been noted. One key reason for the Nickel’s rise to prominence is its’ use in asymmetric cross-electrophile coulpling with simple coupling partners such as pseudohalides and halides. It is envisaged that the increased research activity in photochemistry and electrochemistry will further enhance and compliment the asymmetric methods developed and discussed above.
3.2. Bis(oxazoline) Ligands with Other Linkers
3.2.1. Bis(oxazoline) Ligands Directly Connected at C1
Bioxazoline (BiOX) ligands 984a–e are bis(oxazoline) ligands with the oxazoline moieties directly attached to each other (Figure 43). They have emerged as a useful class of chiral ligand, especially in asymmetric Ni-catalyzed cross-coupling reactions.
Figure 43.

BiOX ligands.
Molander reported a dual photoredox/Ni-catalyzed cross coupling reaction of benzyltrifluoroborates with aryl bromides. They showed that the application of BiOX ligand 984c in the cross coupling of secondary benzyltrifluoroborate 985 with aryl bromide 986 led to the isolation of the enantioenriched product 987 in 52% yield and with 50% ee (Scheme 305). The stereoconvergent single-electron transmetalation event in this reaction (unlike traditional stereospecific transmetalation) allows for facial discrimination of the prochiral alkyl radical 988 by a chiral ligand.351
Scheme 305. Dual Photoredox/Ni-Catalyzed Cross-Coupling Reaction of Benzyltrifluoroborates with Aryl Bromides.

Doyle and Sigman have applied BiOX ligand 984d in an enantioselective Ni-catalyzed reductive cross-coupling of styrenyl aziridines 989 with aryl iodides 990. A range of racemic aryl aziridines 989 were subjected to the reaction with multiple aryl iodides 990 to give the enantioenriched amines 991 in good yields up to 88% and high enantioselectivities up to 94% ee (Scheme 306). Switching from the initial nonchiral ligand (bpp) for the racemic transformation led to a decrease in the yield of the cross-coupling reaction, however the addition of NaI and catalytic TMSCl was found to increase the yields.352
Scheme 306. Enantioselective Ni-Catalyzed Reductive Cross-Coupling of Styrenyl Aziridines with Aryl Iodides.

Contemporaneously, Reisman reported an enantioselective Ni-catalyzed reductive cross-coupling reaction of aryl iodides 993 with secondary benzyl chlorides 992, utilizing 4-heptyl BiOX ligand 984d. A range of aryl and heteroaryl iodides 993 were well tolerated in the reaction, as were a range of secondary benzyl chlorides 992, giving the diaryl alkanes 994 in moderate to good yields up to 88% and with high enantioselectivities up to 95% ee (Scheme 307). Mn0 was found to be an essential reductant and TMSCl an essential activator for the outcome of the reaction which, unlike the previous example, could be run at ambient temperature.353
Scheme 307. Enantioselective Ni-Catalyzed Reductive Cross-Coupling Reaction of Aryl Iodides with Secondary Benzyl Chlorides.

Yamamoto has reported a regio- and diastereoselective Ni-catalyzed reductive cross-coupling of enantioenriched-3,4-epoxyalcohols 995 with aryl iodides 996, utilizing BiOX ligand 984e (Scheme 308). This general protocol furnishes a new type of enantioenriched 4,4-diaryl alkane 997, which also incorporates an additional 1,3-diol, in up to 61% yield and with up to 99% ee. The diol can be easily transformed to a variety of functional groups.354
Scheme 308. Regio- and Diastereoselective Ni-Catalyzed Reductive Cross-Coupling between Epoxide and Aryl Iodides.

Lu and Xiao have described an enantioselective difluoroalkylation of β-keto esters by dual photoredox/Ni-catalysis. Employing Bn-BiOX ligand 984c, a range of indanone- and tetralone-β-keto esters 998 were successfully difluoroalkylated with iodofluoroacetates 999 to give the corresponding indanones and one tetralone 1000 bearing α-quaternary stereocenters in moderate yields up to 67% and with high enantioselectivities up to 90% ee (Scheme 309). The reaction was found to work with bromofluoroacetates in a slightly lower yield and preliminary studies showed it could also be applied to perfluoroalkylation with iodofluoro compounds under altered reaction conditions.355
Scheme 309. Enantioselective Difluoroalkylation of β-Keto Esters by Dual Photoredox/Ni-Catalysis.

BiOX ligands of the type 984 have also found application in asymmetric Pd-catalyzed transformations. For example, Zeng has reported an asymmetric Pd-catalyzed arylation of α-imino esters 1006 with aryl boronic acids 1007. A range of enantioenriched α-diaryl amino esters 1008 were accessed in low to high yields up to 95% and with excellent enantioselectivities up to 99% ee, utilizing iPr-BiOX ligand 984b as the chiral ligand (Scheme 310). Electron-withdrawing groups on the α-imino ester 1006 were well tolerated while electron-donating groups led to a decrease in reaction yield. Variation of the aryl boronic acid 1007 had a more dramatic effect on the outcome of the reaction, with electron-withdrawing and -donating groups decreasing the reaction yield.356 Yang later reported a related C–H oxidative cross-coupling, in which the same α-imino esters are formed by in situ oxidation with 2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate, which enters the same catalytic cycle as in Zeng’s report.357
Scheme 310. Asymmetric Pd-Catalyzed Arylation of α-Imino Esters with Aryl Boronic Acids.

Manolikakes reported a related enantioselective Pd-catalyzed three-component synthesis of α-substituted amines. An array of aryl or alkyl sulfonamides 1011 and aldehydes 1012 were reacted together with aryl boronic acid 1013 in the presence of Pd(TFA)2 and iPr-BiOX ligand 984b to give enantioenriched α-substituted amines 1014 in up to 99% yield and with excellent enantioselectivities up to 98% ee (almost all >95% ee) (Scheme 311). The reaction was found to be insensitive to air and moisture.358 Later, Manolikakes reported the same reaction with glyoxylic acid derivatives in place of regular aldehydes giving enantioenriched arylglycines with similar results.359
Scheme 311. Enantioselective Pd-Catalyzed Three-Component Synthesis of α-Substituted Amines.

Toste has reported an enantioselective Pd-catalyzed 1,1-fluoroalkylation of amino alkenes 1020 with aryl boronic acids 1021 and Selectfluor. Utilizing Bn-BiOX ligand 984c, a range of fluorinated amines 1022 were synthesized in moderate yields up to 60% and with high enantioselectivities up to 91% ee (Scheme 312). It was found that the exclusion of water from the racemic reaction mixture led to no observable product formation, while the addition of acetonitrile dramatically increased the yield of the product. Interestingly, in the asymmetric reaction, the removal of the nitrile led to trace amounts of product formation, while the use of benzonitrile, in place of acetonitrile, led to the highest levels of enantioselectivity.360
Scheme 312. Enantioselective Pd-Catalyzed 1,1-Fluoroalkylation of Amino Alkenes with Aryl Boronic Acids and Selectfluor.

Correia has developed an enantioselective Heck–Matsuda arylation of (Z)-allyl alcohols with aryl diazonium tetrafluoroborates. When diols 1026 are reacted with diazonium tetrafluoroborates 1027 in the presence of Pd(TFA)2 and Bn-BiOX ligand 984c, followed by a Jones oxidation, the corresponding enantioenriched α-aryl lactones 1028 were isolated in up to 87% yield and with up to 85% ee (Scheme 313). The Heck–Matsuda arylation gives the corresponding lactols, which are then converted to the lactones. The lactol intermediates can be further derivatized by other means to give a range of enantioenriched products. The reaction was found to be stereoconvergent and similar results were obtained when an (E)-allyl diol was used. A small range of allyl alcohols 1029 were subjected to the reaction to give dimethyl acetal intermediates which, following acid hydrolysis, gave the corresponding β-aryl aldehydes 1030 in up to 49% yield and with up to 90% ee. As expected, (Z)- and (E)-allyl alcohols gave opposite enantiomers of the product aldehydes 1030.361
Scheme 313. Enantioselective Heck–Matsuda Arylation of Allyl Alcohols with Aryl Diazonium Tetrafluoroborates.

Fu has developed a Ni-catalyzed Negishi cross-coupling of benzyl mesylates. Employing a disubstituted BiOX ligand 1032 in the reaction between the benzyl alcohols 1031 (which are first converted into the corresponding mesylates) and arylzinc iodides, a wide range of diaryl alkanes 1013 were accessed in good yields up to 98% and with high levels of enantioselectivity up to 95% ee (Scheme 314). This transformation was then applied to the asymmetric synthesis of (S)-sertraline tetralone, a precursor to the antidepressant drug Zoloft.362
Scheme 314. Ni-Catalyzed Negishi Cross-Coupling of Benzyl Mesylates.

Overall, BiOX ligands have been applied in a limited range of Pd- and Ni-catalyzed processes in the past decade. Researchers should seek to continue to expand the use of oxazoline-containing ligands in asymmetric 3d-transition-metal-catalyzed processes, and BiOX ligands have shown great potential for asymmetric induction in new Ni-catalyzed transformations.
3.2.2. Bis(oxazoline) Ligands Directly Connected at C4
As is abundantly clear from the numerous examples shown in this Review, bis(oxazoline) ligands most commonly contain amino alcohol derived stereocenters at the C4-position of the ring. In 1997, Lee reported the synthesis of the novel (L)-tartaric acid-derived bioxazoline ligands 1033, joined at the C4-chiral center, but these have found limited application in asymmetric catalysis as the chiral information is facing away from the coordination sphere (Figure 44). Kesavan has developed novel bioxazoline ligand 1034, which is based on Lee’s design, but contains a second chiral center near the coordination sphere, leading to more effective transfer of chiral information. While variations on this design have also been tested, 1034 has emerged as the best ligand in each application reported thus far.
Figure 44.

Bis(oxazoline) ligands joined at the C4-stereocenter.
Kesevan first applied ligand 1034 in the enantioselective Cu-catalyzed Henry reaction363 and the enantioselective Cu-catalyzed alkynylation of imines,364 achieving enantiomeric excesses of up to 84% ee and 80% ee, respectively (Scheme 315). The stereochemical outcome of the asymmetric Henry reaction is described by the two transition states 1038a and 1038b. The latter is thought to be disfavored due to the steric interaction between the aldehyde and one of the methyl groups of the ligand.
Scheme 315. Asymmetric Transformations Mediated by Chiral Bis(oxazoline) Ligand 1034.

Kesevan then reported the use of ligand 1034 in the asymmetric Pd-catalyzed alkylation of allyl acetates 1042 by malonate 1043. A range of symmetrical allyl acetates 1042 bearing different aryl groups were subjected to the reaction leading to the isolation of enantioenriched products 1044 in up to 99% yield and with up to 95% ee (Scheme 316). An electron-donating group on the allyl acetate 1042 led to a decrease in reaction yield and enantioselectivity, while various electron-withdrawing groups were well tolerated in all positions of the aryl ring. Subjecting nonsymmetrical allyl acetates to the reaction almost always led to a 1:1 regiomeric mixture of products, with mostly high enantioselectivities.365 The scope of this reaction was later extended to include 3-OBoc-oxindoles as the nucleophile with slightly lower levels of enantioselectivity.366
Scheme 316. Asymmetric Pd-Catalyzed Alkylation of Allyl Acetates.

The development of bis(oxazoline) ligands connected at the C4-chiral center remains a significantly underdeveloped area. It is clear from the reports described in this Review that these ligands have the potential to induce high levels of stereoselectivity when a second element of chirality is introduced into the scaffold, and as such, these ligands should be considered by researchers in the future.
3.2.3. Bis(oxazoline) Ligands with Pyrimidine and Pyrazine Linkers
Building on their previous work into the Heck–Matsuda arylation (Scheme 313), Correia and Pfaltz have reported the development of two new classes of bis(oxazoline) ligands, based on pyrimidine- (1045) and pyrazino-oxazoline (1046) motifs, for use in this transformation (Figure 45). With the aim of using the Heck–Matsuda arylation for the enantioselective synthesis of (R)-verapamil, they screened a number of ligands to improve the regioselectivity of the initial arylation reaction. In Correia’s previous report, arylation occurs on a symmetrical allyl diol, giving the lactol intermediate as a single regioisomer, which is then converted to the corresponding lactone by a Jones oxidation. In this report, the diol 1047 is asymmetrical, so an alternative chiral ligand was sought to control the regioselectivity of the process.
Figure 45.

Bis(oxazoline) ligands 1045 and 1046.
Following a screen of ligands, 1045 and 1046 were found to induce high levels of stereo- and regioselectivity, giving the corresponding lactones 1048b in good yields of up to 89% yield, with high enantioselectivities up to >98% ee and with regioselectivities >20:1 γ/β in all cases (Scheme 317). O-Methyl lactol 1048a (the product of the reaction before the Jones oxidation) was synthesized in 89% yield and with 96% ee, utilizing pyrimidine ligand 1045. This was taken forward to synthesize (R)-verapamil in 29% overall yield (six steps). Experimental results obtained for the Heck–Matsuda arylation with the mononuclear Pd(TFA)2(1045) complex and the dinuclear Pd2(TFA)4(1045) complex suggested that the active species in this process is mononuclear.367
Scheme 317. Heck–Matsuda Arylation for Enantioselective Synthesis of (R)-Verapamil.

Correia later developed a Heck–Matsuda arylation of spirocyclic pyrrolidinones 1049 with aryl diazonium tetrafluoroborates 1027 generating the corresponding enantioenriched pyrrolidinones 1050 in good yields up to 93%, with high enantioselectivities up to 92% ee and with high diastereoselectivities up to >98:2 dr, when PyraBOX ligand 1046 was used (Scheme 318). Pyrrolidinone 1050 was then taken forward to the synthesize S1P1 agonist VPC01091 in 40% overall yield (9 steps) and with 94% ee.368
Scheme 318. Heck–Matsuda Arylation of Spirocyclic Pyrrolidinones with Aryl Diazonium Tetrafluoroborates.

The stereochemical outcome of the transformation is explained by the stereoselective migratory insertion of one face of the alkene in 1052a into the Pd–Ar bond to give 1052b (Scheme 319). It is proposed that the alkene coordinates to Pd on the opposite face to the carbamate due to the steric bulk of the −NBoc group, giving the intermediate 1052a with the aryl group on the opposite side to the carbamate. PyraBOX 1046 has also been applied to the Heck–Matsuda reaction of five-membered cyclopentene rings containing S- and P-stereogenic centers with similarly high levels of enantioselectivity.369
Scheme 319. Stereochemical Model for Heck–Matsuda Arylation with Ligand 1046.

Correia has also developed an enantioselective phthalide and isochromanone synthesis via a Heck–Matsuda arylation of dihydrofurans. When 2,3-dihydrofuran 1054 is subjected to the arylation with a range of aryl diazonium tetrafluoroborates 1053, the corresponding lactols 1056 can then be subjected to NaBH4 reduction/cyclization to form enantioenriched phthalides 1055 in moderate yields up to 66% and with high enantioselectivities up to 96% ee (Scheme 320). When 3,4-dihydrofurans 1057 are subjected to the same reaction sequence, the corresponding isochromanones 1059 are isolated in up to 52% yield and with up to 96% ee.
Scheme 320. Heck–Matsuda Arylation of Dihydrofurans.

In recent years, the use of chiral pyrimidine- and pyrazine-linked bis(oxazoline) ligands has been spearheaded by Correia. It is clear that these ligands can be used to induce high levels of enantioselectivity in Pd-catalyzed processes. In future, these unique ligands could be further developed for other transition-metal-catalyzed asymmetric transformations. In particular, the binding mode of these ligands could be suitable for developing chiral nickel catalysts.
3.2.4. Bis(oxazoline) Ligands with Spirocyclic Linkers
The SpiroBOX ligands 1060 have been developed and applied in the asymmetric insertion of carbenoids into O–H bonds by Zhou (Figure 46).
Figure 46.
SpiroBOX ligands 1060.
By utilizing a chiral bis(oxazoline) ligand with a highly rigid backbone, Zhou thought that tighter chelation would give access to successful Fe-catalyzed processes. Subjecting diazo compound 1061 to optimized conditions for Fe-catalyzed O–H insertion, with a range of alcohols 1062, led to the isolation of enantioenriched benzyl ethers 1063 in up to 95% yield and with up to 99% ee, with iPr-SpiroBOX 1060c as the chiral ligand (Scheme 321). Subjecting a range of diazo compounds 1064 to the reaction with water led to the isolation of enantioenriched benzyl alcohols 1065 in up 93% yield and with up to 95% ee, when Ph-SpiroBOX 1060a was used as chiral ligand. Almost all examples are >90% ee, with ortho-substituted diazo compounds giving lower levels of enantioselectivity.370
Scheme 321. Asymmetric Fe-Catalyzed O–H Insertion.

Zhou has applied their Cu-catalyzed O–H insertion chemistry to the enantioselective synthesis of 2-carboxy cyclic ethers 1067. Employing iPr-SpiroBOX ligand 1060c, enantioenriched 2-carboxy cyclic ethers 1067 can be accessed by an intramolecular carbenoid O–H insertion of diazo compounds 1066 in up to 98% yield and with up to 97% ee (Scheme 322).371 Zhou has applied the SpiroBOX ligands 1060 to asymmetric Cu-catalyzed α-diazo phosphonate carbenoid O–H insertions372 and Pd-catalyzed carbenoid insertions into phenol O–H bonds.373
Scheme 322. Asymmetric Cu-Catalyzed Carbenoid O–H Insertion.

Zhou has also extended the application of the SpiroBOX ligands 1060 to include other X-H bonds. Subjecting the dimethylphosphorus-borane adduct 1069 to a Cu-catalyzed carbenoid β-H insertion, utilizing ligand (Ra)-1060a, a range of enantioenriched phosphorus-borane adducts were accessed in high yields up to 96% and with high enantioselectivities up to 94% ee (Scheme 323). The stereochemical outcome of the reaction required very specific 2,6-dichlorophenyl α-diazo esters 1068.374
Scheme 323. Cu-Catalyzed Carbenoid β–H Insertion.

Zhou extended the enantioselective Cu-catalyzed carbenoid insertion reaction to include N-H bonds. A range of α-alkyl-α-diazo esters 1071 were reacted with aniline derivatives 1072 to access a range of enantioenriched secondary amines 1073 in up to 95% yield and with up to 98% ee (Scheme 324). Most of the aniline derivatives performed well in the reaction except p-anisidine which gave the product with only 60% ee. The active catalytic species was found to be a binuclear [Cu2(1060a)2] species.375
Scheme 324. Asymmetric Cu-Catalyzed Carbenoid N–H Insertion.

They proposed a stereochemical model based on this dinuclear Cu-species with a perfect C2-symmetric pocket (Scheme 325).
Scheme 325. Stereochemical Model for Asymmetric Cu-Catalyzed N–H Insertion with Ligand 1060a.

Zhou has also applied an intramolecular Cu/1060a-catalyzed carbenoid N-H insertion reaction to the enantioselective synthesis of 2-carboxytetrahydroquinolines.376
Zhou has described an intramolecular Fe-catalyzed asymmetric cyclopropanation of alkenes with carbenoids. Applying 1060a in this reaction, a range of enantioenriched cyclopropanes 1077 were accessed in high yields up to 96% and with low to high enantioselectivities up to 96% ee (Scheme 326).377
Scheme 326. Intramolecular Fe-Catalyzed Asymmetric Cyclopropanation of Alkenes.

They have also reported an Fe/1060a-catalyzed carbenoid indole C-H insertion, but with only low to moderate enantioselectivities.378 Other bis(oxazoline) ligands with spirocyclic backbones, like HMSI-BOX 1078 and SpanBOX 1079 have been reported (Figure 47).
Figure 47.

HMSI-BOX 1078 and SpanBOX 1079.
Xie and Lin have reported the use of spirocyclic HMSI-BOX ligand 1078 in the Fe-catalyzed carbenoid Si-H insertion. A range of silanes 1081 were reacted with α-aryl-α-diazo esters 1080 to give the corresponding enantioenriched silanes in up to 99% yield and with up to 96% ee (Scheme 327). During optimization, 1078 gave the product of the model reaction with 91% ee, compared to SpiroBOX ligand (Ra)-1060a, which gave the product in 84% ee. As in the other carbenoid insertion reactions, 4-methoxy-substitution on the α-aryl-α-diazo esters 1080 leads to a dramatic decrease in enantioselectivity, in this case to 66% ee.379
Scheme 327. Asymmetric Fe-Catalyzed Carbenoid Si–H Insertion.

Ding has applied SpanBOX 1079 in the Zn-catalyzed α-hydroxylation of β-keto esters 1083 with racemic oxaziridines 1084 to give the enantioenriched α-hydroxy-β-keto esters 1085 in up to >99% yield and with up to 99% ee (Scheme 328). A range of substituted β-keto esters were well tolerated with all reported transformations proceeding with >98% yields and >90% ee. The majority of substrates were successfully hydroxylated using 1 mol % Zn(OTf)2 and 2.2 mol% of ligand 1079.380 Ding also reported the Zn- and Cu-catalyzed α-chlorination of the same β-keto esters, utilizing 1079 with similar results.381
Scheme 328. Asymmetric Zn-Catalyzed α-Hydroxylation of β-Keto Esters.

Overall, bis(oxazoline) ligands with spirocyclic backbones are clearly an optimal choice for asymmetric transformations of carbenoids with both iron and copper catalysts. Researchers developing these types of transformations in the future should consider spirocyclic bis(oxazoline) ligands. More importantly, these ligands could be used to develop new asymmetric Fe-catalyzed processes.
3.2.5. Bis(oxazoline) Ligands with One Boron Separating the Oxazoline Rings
Pfaltz pioneered the use of bis(oxazoline) ligands linked by a boron atom, like the BoraBOX ligands 1086a–d, in asymmetric catalysis (Figure 48).382,383
Figure 48.
BoraBOX ligands 1086.
More recently, Sadow has developed boron-bridged bis(oxazoline) ligands 1087 and 1088 for use in enantioselective transition metal-catalyzed hydroaminations of alkenes (Figure 49). The former ligand exploits the gem-dimethyl effect which forces the iPr-containing ligand to behave like a tBu-substituted ligand. The ligands form complexes like Zr(1087)(NMe2)2, in the case of Zr, in which the Cp ring participates in bonding to the metal center.
Figure 49.

Boron-bridged bis(oxazoline) ligandsand Zr(1087)(NMe2)2.
Sadow has reported detailed studies on the effects of different catalysts, based on Zr, Y, Ti and Hf, which incorporate these BoraBOX ligands.384−386 As an example, a comparison of different BoraBOX precatalysts on the outcome of the hydroamination/cyclization of aminoalkene 1089 to give enantioenriched pyrrolidine 1090 is presented in Table 5. Both the Zr(1087)(NMe2)2 and Hf(1087)(NMe2)2 complexes were found to be efficient catalysts in this transformation, giving the product 1090 in 95% and 98% yield, and with 93% and 97% ee, respectively, with the Hf-catalyst reaction performed at 0 °C (entries 1 and 4). Interestingly, Ti(1087)(NMe2)2 was found to operate at a much lower rate of reaction, and gave the product 1090 in much lower enantioselectivity of 76% ee (entry 5). Therefore, the metal center affects the rate of the reaction, such that Zr > Hf ≫ Ti. It is also apparent that the ancillary ligand 1087 or 1088 has a positive effect on the rate of the reaction, such that Zr(1087)(NMe2)2 (1.25 h) > Zr(1088)(NMe2)2 (18 h), but not the enantioselectivity of the transformation (entry 6). Interestingly, Y(1088)(CH2SiMe3) proved to be an effective catalyst, giving the opposite enantiomer (S) in 100% yield and 94% ee in only 10 min (entry 7).
Table 5. Asymmetric Hydroamination/Cyclization of Aminoalkene 1089 with Ligands 1087 and 1088.

| entry | precatalysta | solvent | temp (°C) | time | yield (%)b | ee (%) |
|---|---|---|---|---|---|---|
| 1 | Zr(S-1087)(NMe2)2 | C6H6 | 25 | 1.25 h | 95 | 93 (R) |
| 2 | Zr(1087)(NMe2)2 | C7H8 | –30 | 5 d | 98 | 98 (R) |
| 3 | Zr(R-1087)(NMe2)2 | C6H6 | 25 | 1.25 h | 95 | 93 (S) |
| 4 | Hf(1087)(NMe2)2 | C7H8 | 0 | 15 h | 98 | 97 (R) |
| 5 | Ti(1087)(NMe2)2 | C6D6 | 25 | 5 d | 93 | 76 (R) |
| 6 | Zr(1088)(NMe2)2 | C6H6 | 25 | 18 h | 95 | 93 (R) |
For Zr, Hf and Ti: 10 mol % precatalyst, for Y: 5 mol % precatalyst.
Determined by 1H NMR spectroscopic analysis of the crude reaction mixture.
Sadow proposed a model to explain the stereochemical outcome of the cyclization catalyzed by the Zr(1087)(NMe2)2 precatalyst (Scheme 329). From their experimental results, they proposed that optimal levels of enantioselectivity could be obtained by using a 2:1 substrate/catatlyst ratio. The coordination of the substrates to the Zr-catalyst occurs, and the cyclization of one Zr-amide is facilitated by the other. Because of the steric interactions in the transition states depicted below, only one of the amides cyclizes to selectively give the (R)-product.
Scheme 329. Stereochemical Model to Explain the Outcome of Cyclization Catalyzed by Zr(1087)(NMe2)2 Precatalyst.

The application of oxazoline-containing ligands in asymmetric Zr-catalyzed transformations is limited. Clearly, the boron-linked bis(oxazoline) ligands described in this Review represent a foundation for the development of more Zr-catalyzed asymmetric transformations. The unique structure of these ligands could also be useful for developing novel chemistry with other metals.
3.2.6. Bis(oxazoline) Ligands with One Nitrogen Separating the Oxazoline Rings
Reiser has developed a related class of bis(oxazoline) ligand, azaBOX, in which there is a central bridging amine (Figure 50).387 The standard azaBOX ligand 1093 has a simple secondary amine bridge, but this amine is an excellent functional handle for further derivatization, and it has been used to create polymer-supported azaBOX ligands, but this is outside the scope of this Review.
Figure 50.

AzaBOX ligands.
The central amine has been used by Hong to develop a base-functionalized azaBOX ligand 1094 for use in bifunctional catalysis. Hong compared the activity of benzyl-substituted azaBOX 1096 and bifunctional piperazine-substituted azaBOX 1094 in the Cu-catalyzed Henry reaction of 2-anisaldehyde 1097 with nitromethane 1036 for the asymmetric synthesis of alcohol 1098. In the reaction with 1096 as the chiral ligand, 1-benzyl-4-ethyl-piperazine 1099 was added as a base. When 2 mol% of 1099 was used with 2 mol% 1096 and 2 mol% CuTC, the alcohol 1098 was furnished in 56% yield and with 95% ee (Table 6, entry 1). Increasing the amount of base was found to increase the yield of the reaction but slightly decrease the enantioselectivity (30 mol % 1099: 85% yield, 90% ee) (entry 2). When 2 mol% of 1094 was used with 2 mol% CuTC, a significant rate acceleration was observed, furnishing the alcohol 1098 90% yield and with 92% ee (entry 3).
Table 6. Asymmetric Cu-Catalyzed Henry Reaction of 2-Anisaldehyde 1097 with Nitromethane 1036.

| entry | CuTC mol % | ligand (mol %) | base (mol %) | yield (%) 1098 | % ee |
|---|---|---|---|---|---|
| 1 | 2 mol % | 1096 (2 mol %) | 1099 (2 mol %) | 56 | 95 |
| 2 | 2 mol % | 1096 (2 mol %) | 1099 (30 mol %) | 85 | 90 |
| 3 | 2 mol % | 1094 (2 mol %) | 90 | 92 |
Applying the 1096/1099 system in the Cu-catalyzed Henry reaction of 2-anisaldehyde 1097 with nitroethane 1100 led to the isolation of alcohol 1101 in 54% yield, 1.3:1 syn/anti, and with 72% ee (syn)/96% ee (anti) (Table 7, entry 1). The same reaction with piperazine-azaBOX 1094 gave alcohol 1101 in 81% yield, 1.3:1 syn/anti, and with 92% ee (syn)/97% ee (anti), an overall significant improvement in the yield and enantioselectivity (entry 3). A di-iso-propylamine-functionalized azaBOX ligand 1095 was also applied in this reaction, giving alcohol 1101 in 99% yield, 1.5:1 syn/anti, and with 92% ee (syn)/96% ee (anti), these results indicated that ligand 1095 is an overall more efficient ligand than 1094 for this transformation. However, neither bifunctional ligand 1094 nor 1095 have a significant advantage over the combination of unfunctionalized azaBOX 1096 with DIPEA in this reaction, which gave alcohol 1101 in 96% yield, 1.6:1 syn/anti, and with 94% ee (syn)/95% ee (anti) (entry 2). While the bifunctional ligand systems do not give largely improved outcomes for the Henry reaction over the unfunctionalized ligand/base systems, they are excellent additions to the library of bis(oxazoline) ligands for the application in new enantioselective catalytic transformations.388
Table 7. Asymmetric Cu-Catalyzed Henry Reaction of 2-Anisaldehyde 1097 with Nitromethane 1036.

| entry | ligand | base | yield (%) 1101 | syn/anti | % ee (syn/anti) |
|---|---|---|---|---|---|
| 1 | 1096 | 1099 | 54 | 1.3:1 | 72/96 |
| 2 | 1096 | DIPEA | 96 | 1.6:1 | 94/95 |
| 3 | 1094 | 81 | 1.3:1 | 92/97 | |
| 4 | 1095 | 99 | 1.5:1 | 92/96 |
Reiser has achieved an enantioselective synthesis of the GABA uptake inhibitor (+)-homo-β-proline 1105 via a key Cu-catalyzed pyrrole cycloproponation. Utilizing tBu-azaBOX 1093a, cyclopropane 1104 was accessed in 44% yield and with 87% ee, which can be recrystallized up to >99% ee (Scheme 330). (+)-Homo-β-proline 1105 was then synthesized in three further synthetic steps, all quantitative with regards to the reaction yield, and with >99% ee.389
Scheme 330. Enantioselective Synthesis of GABA Uptake Inhibitor (+)-Homo-β-proline 1105.

Overall, azaBOX ligands have not received significant attention in the past decade. Clearly, they can be used for inducing high levels of enantioinduction, and the free-amine has been shown to be a useful functional handle for accessing not only novel ligand structures but bifunctionality as well.
3.2.7. C2-Symmetric Bis(oxazoline) Ligands with Diphenylamine Linkers
Since they were first developed by Guiry, diphenylamine-linked bis(oxazoline) ligands of the type 1106a–d have been applied in a diverse range of asymmetric transformations (Figure 51).390 Ligands 1106 can be both bidentate (through the oxazoline moieties) and tridentate (through the oxazoline moieties and the bridging amine), depending on what metal ion the ligand is coordinated to.
Figure 51.

Diphenylamine-linked bis(oxazoline) ligands 1106.
Nishiyama has applied Bn-ligand 1106b in the Fe-catalyzed hydrosilylation of ketones 1107 for the asymmetric synthesis of secondary alcohols 1108. Under two sets of optimized conditions, a range of enantioenriched alcohols 1108 were accessed in up to 99% yield and with up to 95% ee (S) or up to 99% yield and 90% ee (R) (Scheme 331). The stereoselectivity varied depending on whether the preprepared Fe(1106b)Cl2 catalyst and Zn were used in the reaction (method A), or the free ligand 1106b was precomplexed with Fe(OAc)2 directly in the reaction mixture without any Zn (method B). The major difference is the presence of Zn in method A. Both methods gave better levels of enantioselectivity depending on the substrate. For example, in the formation of alcohol 1109, method A gave 1109 in 95% ee (S) as compared to method B, which gave 1109 in 90% ee (R). However, in the case of 1110, method A gave essentially racemic alcohol 1110 (1% ee (S)) while method B gave 1110 in 58% ee (R). Analytical results for the complexes with and without the Zn additive suggested that the Zn was acting to reduce Fe(III) to Fe(II), however no active species has been characterized as of yet.391 Nishiyama has also applied Bn-ligand 1106b and iPr-ligand 1106c in the Fe-catalyzed and Co-catalyzed hydrosilylation of ketones and Co-catalyzed hydrosilylation of enones.392
Scheme 331. Asymmetric Fe-Catalyzed Hydrosilylation of Ketones.

A Cu/1106a-catalyzed solvent-free α-fluorination of β-keto esters/amides 1111 with NSFI, mediated by ball-milling, has been reported by Xu. A range of indanone-, tetralone-, benzosuberone-, benzofuran-3-(2H)-one-, and benzothiophen-3-(2H)-one-based β-keto esters/amides were successfully fluorinated to give the enantioenriched products 1112 in up to 99% yield and with up to 99% ee (Scheme 332). Cyclic aliphatic β-keto esters 1113 were also subjected to the α-fluorination to give the products 1114 in up to 96% yield and 99% ee, while acyclic aliphatic β-keto esters gave the products with lower levels of enantioselectivity up to 75% ee.393
Scheme 332. Asymmetric Cu-Catalyzed Solvent-Free α-Fluorination of β-Keto Esters/Amides.

Du has described the asymmetric Zn-catalyzed Friedel–Crafts alkylation of indoles 1115 with 3-nitro-2H-chromenes 1116, utilizing the Ph-ligand 1106a. The products 1117 were isolated in up to 97% yield, with up to 94% ee and with up to a 92:8 anti/syn ratio (Scheme 333).394
Scheme 333. Asymmetric Zn-Catalyzed Friedel–Crafts Alkylation of Indoles with 3-Nitro-2H-chromenes.

Yuan has reported an asymmetric Zn-catalyzed dearomative [3 + 2] cycloaddition of 2-nitrobenzofurans 1118 and 3-isocyanato oxindoles 1119. Employing Ph-ligand 1106a in the process, followed by treatment with MeI in acetone, a range of spirocyclic products 1120, containing three contiguous stereocenters, were accessed in up to 99% yield, with ≥99% ee in all cases and with up to >99:1 dr. (Scheme 334).395 Yuan later extended the scope of this reaction to include 2-nitroindoles.396
Scheme 334. Asymmetric Zn-Catalyzed Dearomative [3 + 2] Cycloaddition of 2-Nitrobenzofurans and 3-Isocyanato Oxindoles.

Yuan has also described an enantioselective Zn-catalyzed dearomative [3 + 2] cycloaddition of 3-nitrobenzothiophenes 1121 and 3-nitrothiopheno[2,3-b]pyridines 1122 with 3-isocyanato oxindoles 1119. Again, utilizing Ph-ligand 1106a under similar optimized conditions to the previous reactions, a range of spirocyclic products 1123/1124 containing three contiguous stereocenters were accessed in up to 99% yield, with up to >99% ee and with up to >99:1 dr (Scheme 335).397
Scheme 335. Enantioselective Zn-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitrobenzothiophenes/3-Nitrothiopheno[2,3-b]pyridines with 3-Isocyanato Oxindoles.

Xu and Yuan have reported the application of diPh-ligand (R,R)-1125 in the asymmetric Zn-catalyzed dearomative [3 + 2] cycloaddition of 3-nitroindoles 1127 with 3-isocyanato oxindoles 1119 (Figure 52).
Figure 52.

DiPh-Diphenylamine-Linked Bis(oxazoline) Ligands.
The spirocyclic products 1128 were isolated in up to 99% yield, with up to 99% ee and with up to 99:1 dr (Scheme 336).398
Scheme 336. Asymmetric Zn-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitroindoles with 3-Isocyanato Oxindoles.

Chen and Xiao have also applied ligand 1125 in the enantioselective [3 + 2] cycloaddition of 3-nitro-2H-chromenes 1129/1116 with 3-isocyanato oxindoles 1119 to give the spirocyclic products 1130/ 1131 in up to 99% yield, with up to 99% ee and >99:5 dr (Scheme 337).399
Scheme 337. Enantioselective [3 + 2] Cycloaddition of 3-Nitro-2H-chromenes with 3-Isocyanato Oxindoles.

Ding and Xiao have described an asymmetric Zn-catalyzed [4 + 2] cycloaddition of 2-vinyl indoles 1132 with 3-nitro-2H-chromenes, utilizing 1125 as the chiral ligand. The tetracyclic products 1133, containing two contiguous stereocenters were isolated in up to 94% yield, with up to 96% ee and >99:5 dr (Scheme 338).400
Scheme 338. Asymmetric Zn-Catalyzed [4 + 2] Cycloaddition of 2-Vinyl Indoles with 3-Nitro-2H-chromenes.

Ligand 1125 has been applied in the Zn(OTf)2-catalyzed asymmetric Friedel–Crafts alkylation of indoles 1115 with trans-β-nitrostyrene derivatives 1134, for the synthesis of enantioenriched nitroalkanes 1135 in high yields up to >99% and with excellent enantioselectivities up to 97% ee (Scheme 339). Substituients on the trans-β-nitrostyrene derivatives 1134 were well tolerated in terms of steric and electronic changes, except ortho-substitution, for example, in the reaction of indole with 2-chloro-trans-β-nitrostyrene, the product was isolated with only 72% ee, compared to 95% ee for the reaction with 4-chloro-trans-β-nitrostyrene. In the same report, ligand 1106a was applied to the asymmetric Friedel–Crafts alkylation of pyrrole with trans-β-nitrostyrene derivatives, but the products were isolated with low levels of enantioselectivity of 11–91% ee.401
Scheme 339. Zn-Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles with trans-β-Nitrostyrenes.

Du showed that the scope of the reaction of indoles with trans-β-nitrostyrene derivatives, utilizing ligand 1125, could be extended to include 2-propargyloxy-β-nitrostyrenes, giving the products with more inconsistent levels of enantioselectivity of 36–93% ee. In the same report, Du explored the Zn(1125)-catalyzed asymmetric Friedel–Crafts alkylation of indoles 1115 with nitrodienes 1136, giving the enantioenriched products 1137 in up to 91% yield and with lower levels of enantioselectivity compared to trans-β-nitrostyrenes 1134, of up to 87% ee (Scheme 340).402
Scheme 340. Zn-Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles with Nitrodienes.

Guiry has described a one-pot/two-step Zn/1125-catalyzed Friedel–Crafts alkylation/Michael addition sequence of 4-(methylacrylato)indole 1138 with trans-β-nitrostyrene derivatives 1134 for the enantioselective synthesis of tricyclic indoles 1139, representing the C4-substituted core structure of the ergoline derivatives. Subjecting the indole 1138 to the Zn(OTf)2/1125-catalyzed reaction with a range of trans-β-nitrostyrene derivatives 1134, followed by the addition of DBU, the tricyclic products 1139 were isolated as the anti–anti diastereomers in up to 55% yield and with up to 99% ee (Scheme 341). The reaction was very sensitive to changes on the aryl group of the trans-β-nitrostyrene derivative 1134.403
Scheme 341. One-Pot/Two-Step Asymmetric Zn-Catalyzed Friedel–Crafts Alkylation/Michael Addition of 4-(Methylacrylato)indole with trans-β-Nitrostyrenes.

Wang and Xu have applied ligand 1125 in the Cu-catalyzed conjugate addition of 2-substituted benzofuran-3–2H-ones 1140 to α,β-unsaturated ketones 1141 to give the corresponding enantioenriched β-substituted-β-keto esters 1142 in up to 95% yield and with high enantioselectivities up to >99% ee (Scheme 342).404
Scheme 342. Asymmetric Cu-Catalyzed Conjugate Addition of 2-Substituted Benzofuran-3-2H-onesto α,β-Unsaturated Ketones.

In the same report, the substrate scope was extended to include linear β-substituted and cyclic enones, giving the linear products 1143 and 1144 with excellent enantio- and diastereoselectivities, while the cyclic products 1145 and 1146 were isolated in excellent enantioselectivities but low diastereoselectivities (Figure 53).
Figure 53.

Products of the asymmetric Cu-catalyzed conjugate addition of 2-substituted benzofuran-3–2H-onesto β-substituted enones and cyclic enones.
Diphenylamine-linked bis(oxazoline) ligands have been successfully applied in a range of Zn-, Cu- and Fe-catalyzed processes. The future for these ligands probably lies in new asymmetric transformations catalyzed by these metals. In particular, new applications of these ligands in Fe-catalysis should be explored.
3.2.8. C1-Symmetric Bis(oxazoline) Ligands with Diphenylamine Linkers
Most commonly, bis(oxazoline) ligands are designed to incorporate C2-symmetry. This leads to simpler synthetic routes (utilizing a single amino alcohol in one cyclization step) and decreases the amount of transition states possible in the asymmetric transformation, thus increasing the enantioselectivity of the process. However, C1-symmetric bis(oxazoline) ligands have been applied to asymmetric catalytic transformations with excellent results.405
Guiry introduced diphenylamine-linked bis(oxazoline) ligands of the type 1106 by developing a modular synthesis, in which the two oxazoline moieties are linked by a Pd-catalyzed aryl amination. This allowed for the synthesis of both C1-(1147) and C2-symmetric ligands which have been applied in asymmetric catalysis (Figure 54).
Figure 54.

C1-symmetric diphenylamine-linked bis(oxazoline) ligands 1147.
Guiry has reported that C1-symmetric ligands of the type 1147a–d, where R1 ≠ R2, are excellent at controlling the regio- and enantioselectivity in the enantioselective Cr-catalyzed homoallenylation of aldehydes, NHK reaction. In the reaction of allene 1149 with benzaldehyde 1148, in the presence of CrCl3, Mn, TMSCl and the appropriate chiral ligand, C1-symmetric ligand 1147a was found to give the allenyl alcohol 1150, following acid hydrolysis, with perfect regioselectivity (over the diene 1151) and excellent enantioselectivity of 96% ee (Table 8, entry 3). The C2-symmetric ligand 1106c gave the allenyl alcohol 1150 with much lower regioselectivity of 71:29 and almost no enantioselectivity at all (entry 1). The C2-symmetric ligand 1106d gave the allenyl alcohol 1150 with perfect regioselectivity, high enantioselectivity of 84% ee, but with low conversion (entry 2). The methodology was applied to a range of aryl, alkenyl and aliphatic aldehydes, generating allenyl alcohols in moderate to good yields up to 71%, with perfect regioselectivity in all but one case (naphthaldehyde) and with high enantioselectivities of up to 98% ee. This was the first example of a regio- and enantioselective homoallenylation of aldehydes.406
Table 8. Enantioselective Cr-Catalyzed Homoallenylation of Benzaldehyde.

| entry | ligand | conv (%) | yield (%) | 1150/1151 | % ee |
|---|---|---|---|---|---|
| 1 | 1106c | 35 | 16 | 100:0 | 84 (R) |
| 2 | 1106d | 80 | 39 | 71:29 | 8 (S) |
| 3 | 1147a | 87 | 71 | 100:0 | 96 (R) |
| 4 | 1147b | 85 | 40 | 84:16 | 49 (R) |
| 5 | 1147c | 45 | 23 | 100:0 | 91 (R) |
| 6 | 1147d | 93 | 33 | 74:26 | 11 (R) |
A possible transition state, to explain the stereochemical outcome for this transformation with ligand 1147a, is depicted in 1152 (Figure 55). The diene sits in the equatorial position, while the aldehyde coordinates in the apical position in an anti-geometry to avoid a steric interaction between the oxazoline iPr-group (R2). This leads to Re-face attack to yield the (R)-enantiomer. The oxazoline tBu-group (R1) might prevent the aldehyde to coordinate from the opposite apical position.
Figure 55.

Stereochemical model for the enantioselective Cr-catalyzed homoallenylation of benzaldehyde utilizing ligand 1147a.
Guiry later explored the gem-disubstitution effect in the application of C1-symmetric ligands of the type 1153 (Figure 56) in the asymmetric Friedel–Crafts alkylation of indole 1154 and 2-methoxyfuran 1157 with trans-β-nitrostyrene 1155. In a number of transformations, one of the oxazoline moieties must be tBu-substituted to achieve high levels of enantioselectivity. However, l-tert-leucine, the parent amino alcohol of these oxazolines, is very expensive, with d-tert-leucine being prohibitively expensive. They exploited the gem-disubstitution effect with C5-iPr-substituted oxazoline-containing ligands to give the products of the Friedel–Crafts alkylations with enantioselectivities better than the normal iPr-substituted ligands and approaching that of the normal tBu-substituted ligands.
Figure 56.

C1-symmetric gem-dimethyl diphenylamine-linked bis(oxazoline) ligands 1153.
In the Friedel–Crafts reaction of indole 1154, the enantioenriched nitroalkane 1156 was isolated in 95% yield and 68% ee, when the reaction was conducted with C2-symmetric-tBu-ligand 1106d (Scheme 343). gem-Dimethyl-iPr/Ph-ligand 1153a gave the product 1156 in 93% yield and 61% ee, which is a slightly lower enantiomeric excess than 1106d, but actually higher than C1-symmetric-tBu/Ph-ligand 1147b, which gave the product in 89% yield and 45% ee. In the Friedel–Crafts reaction of 2-methoxyfuran 1157, the reaction with C2-symmetric-tBu-ligand 1106d gave the product 1158 in 73% yield and 92% ee, while C1-symmetric-tBu/Bn-ligand 1147c gave the product 1158 in 85% yield and 90% ee. The reaction with gem-dimethyl-iPr/Bn-ligand 1153b gave comparable results, giving product 1158 in 88% yield and 89% ee. In the reactions of both substrates, the gem-diphenyl ligands gave poor levels of enantioselectivity compared to the gem-dimethyl ligands.407
Scheme 343. Asymmetric Friedel–Crafts Alkylation of Indole and 2-Methoxyfuran with trans-β-Nitrostyrene.

A transition state 1159, to explain the stereochemical outcome of the two Friedel–Crafts alkylation reactions, was proposed (Figure 57). The bis(oxazoline) ligand coordinates to the Zn-center in a bidentate fashion (based on X-ray crystallographic structures for Zn(1153)Cl2 complexes), with the nitro-group coordinated to Zn in a bidentate manner through the oxygen atoms. Because of the influence of the oxazoline substituents, the appropriate nucleophile (Nu) preferentially attacks the Si-face of the nitrostyrene, to yield the (R)-enantiomer of the product 1156 in the case of the indole nucleophile and the (S)-enantiomer of the product 1158 in the case of the 2-methoxyfuran nucleophile.
Figure 57.

Stereochemical model for the two Friedel–Crafts alkylation reactions utilizing ligand 1153b.
Guiry has also developed C1-symmetric thiazoline-oxazoline hybrid ligands of the type 1160 (Figure 58). While these ligands are not bis(oxazoline) ligands, they are more comparable to bis(oxazoline) ligands than mono(oxazoline) ligands; therefore, they have been included in this section of the Review.
Figure 58.

C1-symmetric thiazoline-oxazoline hybrid ligands 1160.
The thiazoline-oxazoline ligands 1160 were successfully applied in the Zn-catalyzed asymmetric Friedel–Crafts reaction of indole with trans-β-nitrostyrene derivatives, with the reaction using tBu/Bn-ligand 1160a as the chiral ligand, yielding the corresponding enantioenriched nitroalkanes 1161 in up to >99% and with moderate levels of enantioselectivity up to 76% ee (Scheme 344).408
Scheme 344. Zn-Catalyzed Asymmetric Friedel–Crafts Reaction of Indole with trans-β-Nitrostyrene Derivatives.

Thazoline-oxazoline ligands 1160 were also applied by Guiry in the Cr-catalyzed asymmetric Nozaki-Hiyama-Kishi (NHK) reaction of allyl bromide 1162 with benzaldehyde 1148. A range of these ligands were tested in this reaction, a selection of which are shown in Table 9, which affords, following acidic hydrolysis, the corresponding alcohol 1163. The nature of the substituents on the thiazoline (R1) and oxazoline (R2) moieties of ligands 1160 has a dramatic effect over the enantioselectivity of the reaction, with some ligands giving the (R)-product 1162 (entries 1–2) and some giving the (S)-product 1163 (entries 3–5). As in the Friedel–Crafts reaction, the reaction with tBu/Bn-ligand 1160a gave the best result, yielding alcohol 1163 in 84% yield and with 85% ee (R) (entry 1).409
Table 9. Asymmetric NHK Reaction of Allyl Bromide with Benzaldehyde.

| entry | ligand | conv (%) | yield (%) 1163 | % ee |
|---|---|---|---|---|
| 1 | 1160a | 100 | 84 | 85 (R) |
| 2 | 1160b | 100 | 87 | 10 (R) |
| 3 | 1160c | 100 | 81 | 31 (S) |
| 4 | 1160d | 85 | 67 | 6 (S) |
| 5 | 1160e | 94 | 78 | 39 (S) |
A possible transition state 1164, to explain the stereochemical outcome for this transformation with ligand 1160a, is based on the X-ray crystallographic structure obtained for a tridentate Fe(1160)Cl2 complex (Figure 59). This transition state 1164 is like the transition state 1152, used to explain the stereochemical outcome in the Cr-catalyzed homoallenylation with ligand 1147a. In this case, the allyl moiety binds to chromium in the equatorial position, while the aldehyde binds in the apical position in the anti-geometry to avoid a steric interaction between the aldehyde Ph-group and the oxazoline-benzyl (R2) Ph-group. The allyl moiety then attacks the aldehyde from the Re-face to give the (R)-enantiomer. As in the homoallylation transition state 1152, the thiazoline tBu group (R1) might block the aldehyde from coordinating from the opposite apical position.
Figure 59.

Stereochemical model for the asymmetric NHK reaction utilizing ligand 1160a.
While conventional wisdom suggests that C2-symmetry is important for applying chiral bis(oxazoline) ligands in highly enantioselective transformations, the reports described in this Review show that there is a place for C1-symmetric diphenylamine-linked bis(oxazoline) ligands. Significant progress has been made in the Cr-catalyzed asymmetric NHK reaction and in particular, the asymmetric and regioselective homoallenylation of aldehydes. This chemistry requires the use of chemical reductants, and a potential future direction could be the application of electrochemistry to develop more environmentally sustainable processes.
3.2.9. C1-Symmetric Bis(oxazoline) Ligands with Modified Diphenylamine Linkers
Guiry has also developed related C1-symmetric bis(oxazoline) ligands 1165 based on a phenyl-thiophene-amine backbone (Figure 60).
Figure 60.

C1-Symmetric phenyl-thiophene-amine-linked bis(oxazoline) ligands 1165.
These ligands 1165 were applied in the same Cr-catalyzed NHK reaction of allyl bromide 1162 with benzaldehyde 1148 to yield the enantioenriched alcohol 1163. As with the previous report with C1-symmetric ligands 1160 (Table 9), the reaction with the tBu/Bn-substituted ligand (1165a) gave the best result, yielding the alcohol 1163 in 83% and with 73% ee (R) (Table 10, entry 1). As before, the same trend in the formation of the (R)- or (S)-enantiomer of the product 1163 was observed for the different substituents on the oxazoline moieties.410
Table 10. Asymmetric NHK Reaction of Allyl Bromide with Benzaldehyde.

| entry | ligand | conv (%) | yield (%) 1163 | % ee |
|---|---|---|---|---|
| 1 | 1165a | 88 | 83 | 73 (R) |
| 2 | 1165b | 83 | 79 | 22 (R) |
| 3 | 1165c | 80 | 72 | 38 (S) |
| 4 | 1165d | 80 | 78 | 8 (S) |
| 5 | 1165e | 82 | 78 | 63 (S) |
3.2.10. Bis(oxazoline) Ligands with Carbazole Linkers
Developed by Nakada, carbazole-linked bis(oxazoline) ligands 1166 and 1167 are tridentate ligands with a more rigid backbone than their diphenylamine-linked counterparts (Figure 61).411 They have mostly been applied to Cr-catalyzed NHK-type transformations, with some exceptions.
Figure 61.

Carbazole-linked bis(oxazoline) ligands.
Connell has reported the enantioselective Cr-catalyzed addition of (4-bromobut-2-ynyl)-trimethylsilane 1168 to aldehydes 1021 for the asymmetric synthesis of (1,3-butadien-2-yl)methanols 1170 via [1-(silylmethyl)allenyl]methanols 1169. Using iPr-ligand 1166a as the chiral ligand, a small range of [1-(silylmethyl)allenyl]methanols 1169 were accessed in up to 88% yield and with up to 78% ee (Scheme 345). These were converted to (1,3-butadien-2-yl)methanols 1170 by treating with TBAF in up to 86% yield and with up to 77% ee, with a slight decrease in the enantiomeric excess (for R = Ph, 78% ee to 70% ee).412
Scheme 345. Enantioselective Cr-Catalyzed Addition of (4-Bromobut-2-ynyl)-trimethylsilane to Aldehydes.

Nakada has reported that the Fe(III) complex of Ph-ligand 1166c Fe(1166c)Cl2 catalyzes the asymmetric epoxidation of trans-alkenes 1171 in up to 93% yield and with up to 97% ee (Scheme 346). Trans-stilbene derivatives and cinnamyl alcohol derivatives performed well in the reaction, although substrates bearing an electron-rich arene gave the epoxides with only moderate levels of enantioselectivity. The only cyclic alkene tested (1,2-dihydronaphthalene) gave the epoxide product with only 48% ee. The diphenylamine-linked ligand 1106a did not catalyze the epoxidation, indicating that the π–π conjugation system of the carbazole ligand 1166c is essential to the catalytic activity of the Fe(III) complex in this transformation.413
Scheme 346. Asymmetric Fe-Catalyzed Epoxidation of trans-Alkenes.

Zhang has reported the use of ligands 1167 in a variety of Cr-catalyzed processes, like the enantioselective synthesis of α-exo-γ-butyrolactones 1174 via a sequential 2-(alkoxycarbonyl)allylation/lactonization process. Ethyl 2-(bromomethyl) acrylate 1173 was reacted with a variety of aldehydes 1021 in the presence of 10 mol % CrCl2 and iPr-ligand 1167a, followed by acidic workup or treatment with K2CO3 overnight to give the enantioenriched lactones 1174 in up to 93% yield and with up to 99% ee (Scheme 347). The diphenylamine-linked bis(oxazoline) ligand 1106c also catalyzed the model reaction but with a lower enantiomeric excess (79% ee compared to 93% ee).414
Scheme 347. Cr-Catalyzed Enantioselective Synthesis of α-exo-γ-Butyrolactones.

A possible transition state 1175, similar to the other Cr-catalyzed processes utilizing these ligands and diphenyl-amine-linked bis(oxazoline) ligands, but invoking tridentate chelation, was proposed to explain the formation of the (S)-enantiomer of the product with the iPr-ligand 1167a (Figure 62).
Figure 62.

Stereochemical model for the sequential 2-(alkoxycarbonyl)allylation/lactonization process utilizing ligand 1167a.
The asymmetric Cr-catalyzed dearomative addition of halomethylarenes 1176 to aldehydes 1021 for the asymmetric synthesis of secondary benzylic alcohols 1177 was also reported by Zhang, employing Et-ligand 1167d. 2-(Chloromethyl)benzofurans, -benzothiophenes, and -indenes 1176 were successfully subjected to the reaction conditions with a range of aryl, alkenyl and aliphatic aldehydes 1021 to give the corresponding benzylic alcohols in up to 92% yield and with enantiomeric excesses up to 99% ee (Scheme 348). 3-(Chloromethyl)benzofuran 1178 was also subjected to the reaction to yield the corresponding secondary benzylic alcohols 1179 in up to 88% yield and with up to 96% ee.415 The scope of this process was extended to include bromomethylnaphthalenes,416 bromomethyl- and chloromethyloxazoles, and bromomethyl- and chloromethylindoles417 with similar results.
Scheme 348. Asymmetric Cr-Catalyzed Dearomative Addition of Chloro(methyl) Heteroarenes to Aldehydes.

An enantioselective 1,2-difunctionalization of 1,3-butadienes 1181 via an asymmetric Cr/Co-bimetallic alkylation/carbonyl allylation sequence was achieved by Zhang, with the iPr-ligand 1167a. A range of 1,3-butadienes 1181 were difunctionalized with various alkyl bromides and iodides 1180, and aliphatic and aryl aldehydes 1021. The enantioenriched alcohols 1182 were isolated in good yields up to 88%, with high levels of enantioselectivity up to 98% ee and with high diastereoselectivities up to >15:1 dr (Scheme 349). A range of fluorinated and nonfluorinated alkyl iodides/bromides were successfully applied in this transformation.418
Scheme 349. Asymmetric Cr/Co-Bimetallic Alkylation/Carbonyl Allylation.

Like the diphenylamine-linked bis(oxazoline) ligands, carbazole-linked bis(oxazoline) ligands have been successfully applied in enantioselective Cr-catalyzed processes. The carbazole backbone is rigid compared to the flexible diphenylamine backbone; as a result, both ligand families have their own unique properties.
3.2.11. Miscellaneous Bis(oxazoline) Ligands with Free-Amine-Based Linkers
Zhang has reported the asymmetric Ru-catalyzed hydrogenation of ketones 1107 with a tridentate, bifunctional indaBOX ligand 1183 for the synthesis of enantioenriched alcohols 1108 with up to >99% conversion and up to 95% ee (Scheme 350). Monoaryl and cyclohexyl ketones performed well in the reaction, yielding the secondary alcohols with enantiomeric excesses >80%, but linear aliphatic ketones gave the corresponding alcohols in moderate enantioselectivities 42–65% ee.419
Scheme 350. Asymmetric Ru-Catalyzed Hydrogenation of Ketones.

A possible transition state 1184 was proposed to explain the stereochemical outcome of the transformation, highlighting the suggested ligand-assisted mechanistic pathway (Figure 63).
Figure 63.

Stereochemical model for the asymmetric Ru-catalyzed hydrogenation utilizing ligand 1183.
Gade has developed a new class of chiral, planar, rigid pincer-type ligand, bis(oxazolinylmethylidene)isoindolines 1185– 1187 (boxmi), which contain a phthalimide-based linker in the backbone (Figure 64).
Figure 64.

Boxmi ligands.
Gade first applied these ligands in the Ni-catalyzed asymmetric α-fluorination of oxindoles 1188 with NFSI. A range of α-aryl-α-fluorooxindoles 1189 (and one α-methyl example) were accessed in up to 95% yield and with up to >99% ee, using opitimized reaction conditions with Ph-boxmi 1185c as the chiral ligand (Scheme 351). They also extended the scope to include indanone-based β-keto esters with enantioselectivities up to 97% ee.420
Scheme 351. Ni-Catalyzed Asymmetric α-Fluorination of Oxindoles.

In same report, Gade applied the ligands in the NHK-reaction of allyl bromide 1062 with benzaldehyde 1021 and Table 11 summarizes the results of this study. The reaction with iPr-ligand 1186a gave the best outcome, giving alcohol 1063 in 92% yield and with 86% ee (S) (entry 4). Unlike the diphenylamine- and carbazole-linked bis(oxazoline) ligands, all ligands tested in the boxmi series led to the formation of the (S)-alcohol.
Table 11. Asymmetric NHK Reaction of Allyl Bromide with Benzaldehyde.

| entry | ligand | time (h) | yield (%) 1063 | % ee |
|---|---|---|---|---|
| 1 | 1185a | 12 | 92 | 83 (S) |
| 2 | 1185b | 10 | 91 | 73 (S) |
| 3 | 1185c | 12 | 91 | 84 (S) |
| 4 | 1186a | 10 | 92 | 86 (S) |
| 5 | 1186b | 10 | 93 | 68 (S) |
| 6 | 1186c | 10 | 89 | 54 (S) |
| 7 | 1186d | 12 | 92 | 73 (S) |
| 8 | 1187a | 12 | 91 | 83 (S) |
| 9 | 1187b | 12 | 92 | 63 (S) |
| 10 | 1187c | 12 | 93 | 79 (S) |
Gade has developed an asymmetric Cu/1186c-catalyzed α-alkylation of β-keto esters 1190 with benzyl alcohols 1191, for the enantioselective synthesis of enantioenriched β-keto esters 1192 in up to 94% yield and with 98% ee (Scheme 352). A variety of indanone-, cyclopentanone- and cyclopentenone-based β-keto esters 1192 were tolerated in the reaction. The initial step in the reaction converts the alcohols 1191 to the corresponding iodides with CsI, and the iodides then participate in the alkylation reaction in the second step. Benzyl alcohols 1191 bearing different substitutions were well tolerated; however, no heteroaromatic substrates were reported. The reaction was then extended to include allyl alcohols 1194 as the alkylating agent. Both di- and trisubstituted alkenes were tolerated in the reaction, giving the enantioenriched β-keto esters 1195 in up to 94% yield and with up to 99% ee.421
Scheme 352. Asymmetric Cu-Catalyzed α-Alkylation/Allylation of β-Keto Esters.

In the same report, Gade reported that treating the allylated-β-keto ester products from the reaction with cycloalkenes 1196 with BF3·OEt2, the allylation could be further extended to the synthesis of bispirolactones 1197 in up to 86% yield and with >99% ee (Scheme 353). When 3-iodo-2-methylpropene 1198 (which cannot be prepared from the corresponding alcohol by the CsI method) was used directly in the allylation/spirolactonization sequence, spirolactones 1199 were isolated in up to 89% yield and with 99% ee.
Scheme 353. Asymmetric Synthesis of Spirolactones.

Gade later reported an asymmetric Cu/1186c-catalyzed trifluoromethylation of β-keto esters 1200 and 1203. The 5-membered-ring-based β-keto esters 1200 were trifluoromethylated under optimized conditions with Togni’s reagent 1201 to give the α-trifluoromethyl-β-keto esters 1202 in up to 99% yield and with up to 94% ee (Scheme 354). The 6-membered-ring-based β-keto esters 1203, with a more enolizable ketone were found to undergo the trifluoromethylation with higher levels of enantioselectivity with Umemoto’s reagent 1204 and DIPEA in place of Togni’s reagent 1201. The 6-membered-ring-based products 1205 were isolated in up to 96% yield and with up to 93% ee.422 Gade subsequently developed a very similar methodology for a highly enantioselective Cu/1185c-catalyzed trifluoromethylthiolation of β-keto esters with the -SCF3 analogue of Togni’s reagent.423
Scheme 354. Asymmetric Cu-Catalyzed Trifluoromethylation of β-Keto Esters.

Further expanding on their use of boxmi-ligands in the α-functionalization of oxindoles and β-keto esters, Gade described an asymmetric Fe-catalyzed azidation of β-keto esters 1206 and oxindoles 1188 with an azide-analogue of Togni’s reagent 1207 (Scheme 355). During optimization they found that iron carboxylates gave the highest levels of enantioselectivity in the reaction. A screen of silver carboxylates in combination with the [Fe(1185c)Cl] complex led to the best result being obtained with silver p-nitrobenzoate. Using the optimized conditions, a range of 5- and 6-membered-ring-based β-keto esters 1206 were azidated to give the enantioenriched products 1208 in up to 93% yield and with up to 90% ee. A range of α-aryl-oxindoles 1188 were azidated to give the products in up to 94% yield and with up to 90% ee, utilizing Fe(OOCOEt)2/1185c and no silver salts.424
Scheme 355. Asymmetric Fe-Catalyzed Azidation of β-Keto Esters and Oxindoles.

Gade has furthered the application of boxmi ligands in Fe-catalysis by developing a highly reactive Fe/1185c system for the hydrosilylation of ketones 1210. Under optimized conditions, Fe-catalyst 1212 successfully catalyzed the hydrosilylation of a range of ketones 1210, the first Fe-catalyst to achieve >95% ee in this reaction and the first Fe-catalyst to operate at such relatively low temperatures. Monoarylated alcohols 1211 were isolated in up to >95% yield and with up to 99% ee, whereas diaryl ketones gave lower levels of enantioselectivity due to less discrimination between the two groups (Scheme 356).425
Scheme 356. Asymmetric Fe-Catalyzed Hydrosilylation of Ketones.

Tridentate bis(oxazoline) ligands bearing free-amine linkers have been shown to be useful in a range of transformations. In particular, Gade has pioneered the use of Boxmi ligands in Fe-catalysis. These ligands can be used to form very active Fe-catalysts, and future developments in this area are expected.
3.2.12. Bis(oxazoline) Ligands with Pyridine Linkers and Monosubstituted Oxazoline Rings
PyBOX ligands 1213a–h are an extensively used class of pyridine-based tridentate bis(oxazoline) ligand (Figure 65).
Figure 65.

PyBOX ligands.
Bolm has applied ligand 1213a in the Fe-catalyzed enantioselective transfer of nitrenes to sulfides to afford chiral sulfimides (nitrogen analogues of sulfoxides) in excellent yields and good enantiomeric excesses of up to 90% ee. Only Ph-PyBOX 1213a was found to induce good levels of stereoinduction, while the other PyBOX ligands tested 1213b-c gave almost racemic products. The Fe(III) source was found to have a dramatic effect on the stereoselectivity of the reaction, with only Fe(III) acetylacetonate derivatives yielding the product in good enantioselectivity (Table 12). Ultimately, the catalyst derived from [Fe(dmhdCl)3] was found to be optimal giving sulfimide 1216 in 98% yield and 86% ee (entry 6). Only sulfides 1214 bearing at least one nonaliphatic group facilitated nitrene transfer with good stereoselectivity. For example, when the benzyl analogue of sulfide 1214 was subjected to the reaction, the product was isolated in 55% yield and 10% ee, the only example with an enantiomeric excess of less than 60% ee.426
Table 12. Fe(III) Source Effect on Stereoselectivity of the Reaction.

| entry | Fe source | ligand | time | yield | % ee |
|---|---|---|---|---|---|
| 1 | [Fe(acac)3] | (S,S)-1213a | 14 h | 75% | 50% |
| 2 | [Fe(acacCl)3] | (S,S)-1213a | 1 h | 97% | 64% |
| 3 | [Fe(dmhd)3] | (S,S)-1213a | 6 h | 99% | 68% |
| 5a | [Fe(dmhdCl)3] | (S,S)-1213a | 2 h | 99% | 82% |
| 6a | [Fe(dmhdCl)3] | (R,R)-1213a | 16 h | 98% | –86% |
Acetone instead of MeCN.
Bolm applied the same ligand 1213a in the Fe(III)-catalyzed imidative kinetic resolution of racemic sulfoxides, accessing the sulfamidates in moderate to good yields and enantioselectivities. When racemic sulfoxides 1217 are subjected to the reaction conditions, sulfamidates 1218 were obtained in up to 37% yield and 88% ee, with an S factor of up to 26.2 (Scheme 357). The reaction outcome, especially with regard to the yield of the reaction, was sensitive to steric and electronic modifications on the aryl ring of the sulfoxide 1217. For example, the 2-methyl and 4-nitro-substituted examples gave the corresponding products in 4% and 6% yield with enantiomeric excesses of 58% and 80% ee, respectively.427
Scheme 357. Fe(III)-Catalyzed Imidative Kinetic Resolution of Racemic Sulfoxides.

Ligand 1213a was applied by Kobayashi in the asymmetric protonation of chiral calcium enolates formed from the 1,4-addition of malonates to oxazolidinone-based Michael acceptors. Following addition of the malonate 1220 to the Michael acceptor 1219, the chiral Ca(II)-PyBOX complex coordinates to and rigidifies the enolate, controlling its geometry, yielding enantiomerically enriched 1221 after protonation (Scheme 358). Only Ph-PyBOX 1213a was found to induce good stereoselectivity and interestingly, best results were obtained when 10 mol% phenol 1222 and 200 mol% EtOH were used as additives in cyclopentyl methyl ether (CPME) at −20 °C. The products 1221 were formed in excellent yields and enantioselectivities of up to 91% and 96% ee, respectively, with a variety of alkyl groups. In the case of an aryl group (R = Ph) a diminished enantiomeric excess of 42% was achieved.428
Scheme 358. Asymmetric Protonation of Chiral Calcium Enolates.

Ligand 1213a was employed by Blay and Pedro in the La(III)-catalyzed asymmetric conjugate addition of malonate esters to α,β-unsaturated N-sulfonyl imines 1223. Of the ligands tested, only the Ph-PyBOX 1213a gave appreciable levels of enantiomeric excess, with a range of (E)-enamine products 1224 isolated in moderate to excellent yields, moderate to high E/Z selectivities (up to 95:5 E/Z) and good enantioselectivities up to 94% ee (Scheme 359). The enamine products can be used to synthesize chiral δ-aminoesters and lactams with good efficiency, for example, optically pure lactam (R,S)-1227 was accessed in 2 steps and 52% yield from (R,E)-enamine 1226.429
Scheme 359. La(III)-Catalyzed Asymmetric Conjugate Addition of Malonate Esters to α,β-Unsaturated N-Sulfonyl Imines.

Pedro and Blay described a new naphthyl-PyBOX ligand 1213e in the related La(III)-catalyzed conjugate addition of nitroalkanes to (E)-2-azachalcones 1228, providing the nitro-Michael product 1230 in up to 74% yield and moderate to good enantioselectivities of up to 87% ee for a range of substrates (Scheme 360). The reaction proceeded well when R1 was either aryl or alkyl, and R2 was methyl. Where R2 = ethyl or propyl, the dr of the corresponding products was often low.430
Scheme 360. La(III)-Catalyzed Asymmetric Conjugate Addition of Nitroalkanes to (E)-2-Azachalcones.

Maarseveen prepared a range of chiral propargylic amines via a Cu-1213g-catalyzed propargylation of both aromatic and nonaromatic amines with propargylic esters 1231. Using a range of primary (1232) and secondary amines (1233), several propargylic amines 1234 were synthesized in up to 97% yield and 90% ee (Scheme 361). Aniline derivatives performed best as nucleophiles, giving the corresponding products in high yields and enantioselectivities, while other amines generally performed poorly. A number of C-nucleophiles were also tested, with indole derivatives providing the products in up to 91% yield and 98% ee. Some of the propargylic amines synthesized by this protocol were converted into α-amino acid derivatives and further elaborated to provide formal total syntheses of the biologically active compounds (+)-anisomycin and (−)-cytoxazone.431
Scheme 361. Asymmetric Cu-Catalyzed Propargylation of Amines.

Nishibayashi employed a Cu-1213g complex in the enantioselective propargylic etherification of propargylic esters 1235 with alcohols (Scheme 362). Initially, a range of PyBOX ligands were tested in the reaction, giving low enantioselectivities when the reaction was performed at rt. Performing the reaction at a lower temperature of −10 °C for a longer reaction time (72 h) gave the propargyl ether products in generally high yields (57–91%) and enantioselectivities (79–99% ee) when using Me-PyBOX 1213g as the chiral ligand. For reactions with MeOH and EtOH, the alcohol can be used as the reaction solvent. For reactions with phenol derivatives, 2 equiv of the corresponding phenol can be used in MeOH as the reaction solvent. A dimeric [Cu2(PyBOX)2][OTf] species was proposed as the active catalytic agent in the reaction based on experimental evidence.432
Scheme 362. Asymmetric Cu-Catalyzed Propargylation of Alcohols.

Nishibayashi employed Ph- and Me-PyBOX ligands 1213a and 1213g in a related Cu-catalyzed intramolecular amination of propargylic acetates 1237 to yield nitrogen-containing heterocycles 1238 bearing ethynyl groups in the α-position. A range of PyBOX ligands bearing both mono- and disubstitution on the oxazoline ring were tested, with both 1213a and 1213g performing well. A range of nitrogen heterocycles were successfully synthesized in good yields and high enantioselectivities of up to 98% ee (Scheme 363).433
Scheme 363. Asymmetric Cu-Catalyzed Intramolecular Amination of Propargylic Acetates.

A Cu/boronic acid dual catalyzed enantioselective propargylation of polyols was reported by Niu and applied to the desymmetrization of meso 1,2-diols 1240 to furnish products with up to three stereocenters in one operation. Generally, aliphatic alcohols are not nucleophilic enough to engage in Cu-catalyzed propargylation reactions unless they are used as the reaction solvent, thus limiting the scope of the reaction to simple alcohols like MeOH and EtOH. Niu addressed this issue by taking advantage of the increased nucleophilicity of boronate complexes formed between diols and boronic acids. Subjecting propargylic esters 1239 to the reaction with various diols 1240 in the presence of [Cu(MeCN)4]PF6 and chiral Me-PyBOX ligand 1213g the desymmetrized propargylic ethers 1241 were obtained in yields up to 99% and enantioselectivities up to 99% ee (Scheme 364). The authors suggested the presence of a dinuclear Cu species as the active catalyst in the reaction, based on Nishibayashi’s reports.434
Scheme 364. Asymmetric Cu-Catalyzed Desymmetrization of Diols with Propargylic Acetates.

Maruoka has applied 1213a in the catalytic asymmetric alkynylation of C1-substituted C,N-cyclic azomethine imines 1243 with a Cu(I)/chiral Bronsted acid cocatalyst system (Scheme 365). This is the first example of an asymmetric direct alkyne addition to a CN double bond to give tetrasubstituted carbon centers in high stereoselectivity. A range of nitrogen heterocyclic products 1244 with tertiary and quaternary alkyl-substituted stereocenters were generated in excellent yields up to >99% and excellent enantioselectivities up to 96% ee. The tertiary stereocenters were formed in excellent yields and enantioselectivities without the need for a chiral Bronsted acid additive 1245, however in the case of products containing a quaternary stereocenter, the additive dramatically improved the stereoselectivity of the process.435
Scheme 365. Cu/chiral Bronsted Acid-Catalyzed Asymmetric Alkynylation of C1-Substituted C,N-Cyclic Azomethine Imines.

Watson later reported a similar Cu-catalyzed asymmetric alkynylation of diaryl ketimines 1246 (Scheme 366). Utilizing Ph-PyBOX ligand 1213a, diaryl ketimines 1246 were first reacted with ClCO2Me to form an iminium ion, followed by the CuI/1213a-catalyzed addition of an alkyne to form a range of C1-tetrasubstituted nitrogen heterocyclic products 1247 in generally excellent yields up to 93% and enantioselectivities up to 98% ee. The reactions were run under basic conditions without the need for a chiral Bronsted acid additive. A wide range of aryl groups were tolerated on the ketimine 1246; however, no heteroaryl groups were reported. The alkyne was found to tolerate a range of substituted aryl groups, however alkyl and silyl groups were detrimental to the stereoselectivity of the process.436
Scheme 366. Cu-Catalyzed Asymmetric Alkynylation of Diaryl Ketimines.

Zhou has applied ligand 1213a in the first highly enantioselective Cu-catalyzed azide–alkyne cycloaddition (CuAAC) via desymmetrization of oxindole-based 1,5-heptadiynes 1248 to furnish chiral quaternary oxindoles 1250 bearing a 1,2,3-triazole moiety (Scheme 367). A range of azides and oxindoles were demonstrated to perform well in the reaction providing the products in good yields of up to 82% and excellent enantioselectivities of up to 98% ee, with the construction of all-carbon quaternary stereogenic centers.437
Scheme 367. Enantioselective Cu-Catalyzed Azide–Alkyne Cycloaddition via Desymmetrization of Oxindole-Based 1,5-Heptadiynes.

Li and Xiao reported the first decarboxylative [4 + 1] cycloaddition of propargylic carbamates 1251 with sulfur ylides (Scheme 368). A range of ethynyl benzoxazinanones 1251 with varying aryl substitution patterns were subjected to the reaction with sulfonium salts 1252, bearing different ketonic moieties, in the presence of Cu(OTf)2 and chiral Ph-PyBOX ligand 1213a to yield chiral indolines 1253 in high yields of up to 99% and with excellent enantioselectivities of up to 98% ee. The proposed mechanism for the reaction involves a key Cu-allenylidene intermediate 1254, which undergoes a formal [4 + 1] cycloaddition with the in situ formed sulfur ylide.438
Scheme 368. Asymmetric Decarboxylative [4 + 1] Cycloaddition of Propargylic Carbamates with Sulfur Ylides.

Gong439 and Cao and Wu440 independently reported the use of ethynyl benzoxazinanones 1251 in a similar Cu-catalyzed [4 + 2] annulation of the same copper allenylidene intermediate 1254 with in situ generated carboxylic acid/nucleophilic Lewis base derived enolates. Gong utilized iPr-PyBOX ligand 1213b to achieve high levels of stereoinduction of up to 99% ee, while Cao and Wu utilized Ph-PyBOX 1213a, also achieving enantioselectivities of up to 99% ee (Scheme 369). Interestingly, Cao and Wu reported the opposite absolute stereochemistry of the quinolinone products 1256 to that reported by Gong (both reports determined the absolute stereochemistry by X-ray crystallographic analysis of the product). The (R,R)-PyBOX ligands (Ph 1213a or iPr 1213b) were applied in both cases, while the nucleophilic Lewis base catalysts 1257 and 1258, with opposite senses of stereochemistry were used. In both reports, a switch in the stereochemistry of the nucleophilic Lewis base catalyst led to the formation of the opposite enantiomer, so the nucleophilic Lewis base catalyst controls the absolute stereochemistry of the product of this cascade process. In fact, Gong performed the reaction in the presence of an achiral PyBOX ligand, isolating the product quinolinone in a slightly lower 92% ee, but with a much lower diastereoselectivity. Multiple Cu(PyBOX) catalytic systems have been shown to be proficient in the enantioselective [4 + 2] cycloaddition of Cu-allenylidenes (derived from ethynyl benzoxazinanones) with multiple synthetic partners.441−443
Scheme 369. Asymmetric Cu-Catalyzed [4 + 2] Annulation of Ethynyl Benzoxazinanones with In Situ Generated Carboxylic Acid/Nucleophilic Lewis Base Derived Enolates.

Kleij has developed a somewhat related Cu-catalyzed asymmetric synthesis of γ-amino acids bearing quaternary stereocenters. Utilizing chiral naphthyl-PyBOX ligand 1213e, propargylic lactones 1259 undergo an enantioselective Cu-catalyzed amination with a variety of primary amines and one secondary amine 1260 to give the corresponding γ-amino acids 1261 in up to 98% yield and with up to 96% ee (Scheme 370). The proposed mechanism for this transformation involves a similar Cu-allenylidene intermediate 1262 which is nucleophilically attacked by the amine.444 Zhang has described a very similar Cu(1213d)-catalyzed enantioselective synthesis of β-amino alcohols, utilizing cyclic carbonates in place of lactones.445
Scheme 370. Cu-Catalyzed Asymmetric Synthesis of γ-Amino Acids.

Carreria has utilized the electron rich 3,4,5-trimethoxyphenyl PyBOX ligand (R,R)-1265 in a Cu-catalyzed propargylation of an internal C-nucelophile as the key step in the enantioselective total synthesis of three natural products. Propargyl acetate 1263 was subjected to the reaction, which proceeds through a Cu-allenylidene intermediate, to give pyrrole 1264 containing the core stereogenic center of (−)-rhazinilam, in 90% yield and 78% ee (Scheme 371). (−)-Rhazinilam was synthesized in six additional steps. Pyrrole 1264 was also taken forward to synthesize (+)-eburenine in five additional steps. (+)-Aspidospermidine was synthesized in one step from (+)-eburenine.446
Scheme 371. Asymmetric Cu-Catalyzed Propargylation for the Total Synthesis of (−)-Rhazinilam, Aspidospermidine, and (+)-Eburenine.

Uozumi has developed an enantioposition-selective CuAAC to construct axially chiral biaryl derivatives by employing the l-serine-derived, di-OTBS protected PyBOX ligand 1213h. The reaction of benzyl azide 1267 with prochiral biaryl dialkynes 1266 led to the formation of 1,2,3-triazoles 1268 bearing axially chiral biaryl groups in up to 76% yield and 99% ee (Scheme 372). The reaction was limited to the use of benzyl azide for good enantioposition-selectivity; however, changes to the top aryl ring were well tolerated, as was the use of both naphthalene and ortho-substituted benzene rings as the bottom aryl moiety.447
Scheme 372. Enantioposition-Selective CuAAC to Construct Axially Chiral Biaryl Derivatives.

Mlynarski applied the highly hindered TBDPS O-protected chiral PyBOX ligand 1271 in an unprecedented asymmetric Zn-catalyzed Mukaiyama aldol reaction of 2-(trimethoxylsiloxy)-furans 1269 with various aldehydes (Scheme 373). Some commercially available PyBOX ligands were tested in the reaction with little success, leading the authors to employ a more hindered PyBOX ligand 1271 which proved essential in obtaining decent levels of asymmetric induction. Several chiral α-butenolides 1270 were obtained in good yields of up to 82% and moderate to good enantioselectivies of up to 70% ee. A benzoic acid additive (10 mol %) was found to promote the reaction in the case of aryl aldehydes, but was not welcome in the reaction of aliphatic aldehydes.448
Scheme 373. Asymmetric Zn-Catalyzed Mukaiyama Aldol Reaction of 2-(Trimethoxylsiloxy)-furans.

Mlynarski has applied the same chiral PyBOX ligand 1271 in an Fe-catalyzed nitro-Mannich reaction for the enantioselective synthesis of β-nitroamines 1273 (Scheme 374). A wide range of chiral PyBOX ligands were found to induce good enantioselectivities with the very hindered PyBOX 1271 giving the best results. A range of β-nitroamines 1273 were prepared with good yields of up to 91% and consistently high enantioselectivies of up to 98% ee.449
Scheme 374. Fe-Catalyzed Nitro-Mannich Reaction for Enantioselective Synthesis of β-Nitroamines.

Utilizing the same hindered PyBOX ligand 1271, Mlynarski has developed an enantioselective Zn-catalyzed hydrosilylation of aromatic ketones 1274 with (EtO)2MeSiH (Scheme 375). A range of chiral secondary alcohols 1275 were obtained in good yields up to 96% (generally >99% conv.) and moderate to good enantiomeric excesses of up to 85% ee.450
Scheme 375. Enantioselective Zn-Catalyzed Hydrosilylation of Aromatic Ketones.

Huang utilized 1271 in an asymmetric Co-catalyzed regioselective alkyne hydrosilylation with dihydrosilane 1277. A range of aryl, alkyl, internal and terminal alkynes 1276 were applied in the hydrosilylation to yield alkenes 1278 bearing silicon stereogenic centers in up to 99% yield and with up to 91% ee (Scheme 376). Reactions with terminal alkynes proceeded with high Markovnikov regioselectivity.451
Scheme 376. Asymmetric Co-Catalyzed Regioselective Alkyne Hydrosilylation.

Watson has reported a Cu-catalyzed alkynylation of oxocarbenium ions 1281 derived from isochroman acetals 1279, utilizing chiral PyBOX ligand 1213a to access tetrasubstituted biaryl stereocenters in high enantioselectivities. A range of alkynes 1040 can be added to isochroman oxocarbenium species 1281 bearing a variety of aryl groups to access isochroman ketals 1280 in high yields of up to 97% and enantioselectivities of up to 97% ee (Scheme 377). Changes to the substitution on the aryl group of the isochroman acetals 1279 affected the stereoselectivity of the reaction, for example when the 2-methyl substituted substrate was reacted with phenylacetylene, the product was isolated in 36% ee. Generally, aryl- and silane-based acetylenes performed well, with alkylacetylenes giving the product with lower enantioselectivities. For example, subjecting n-hexaneacetylene to the reaction with the 3-MeO-substituted isochroman acetal gave the product in only 66% ee, as compared to 81% ee for -SiPhMe2 acetylene.452
Scheme 377. Asymmetric Cu-Catalyzed Alkynylation of Oxocarbenium Ions.

Yoda has described an asymmetric In-catalyzed amide allylation of N-methyl isatin 1282 with N-substituted-β-amido allyltributylstannanes 1283 to synthesize the corresponding chiral allylated products 1284 in excellent yields of up to >99% and enantioselectivities of up to 99% ee (Scheme 378). Further derivatization of the R = NH(4-MeC6H4) product, by means of an acid promoted cyclization followed by C5-iodination, led to the formation of an antineoplastic spiro-fused 2-oxindole/R-methylene-γ-butyrolactone 1285, retaining the high optical purity of 99% ee.453 A follow-up report by Yoda expanded the substrate scope to isatins bearing different N-protecting groups and C5-substitution, retaining the excellent yields and enantioselectivities from the previous report.454
Scheme 378. Asymmetric In-Catalyzed Amide Allylation of N-Methyl Isatin.

Yoda further explored the In-catalyzed amide allylation with the allylation/lactonization of α-keto esters 1286 for the enantioselective synthesis of ester-functionalized α-methylene-γ-butyrolactones 1288 in excellent yields of up to 99% and enantiomeric excesses of up to 99% (Scheme 379). Following the [In(1213a)(OTf)3]-catalyzed allylation of α-keto esters 1286 with allylstannanes 1283, an acid promoted lactonization of the corresponding amides 1287 gives access to the α-methylene-γ-butyrolactones 1288.455
Scheme 379. In-Catalyzed Amide Allylation with Allylation/Lactonization of α-Keto Esters.

Watanabe and Shibasaki reported the use of chiral PyBOX ligand 1213a in an asymmetric Cu-catalyzed A3-coupling reaction for the synthesis of an oseltamivir phosphate precursor 1293 in 84% yield and 76% ee (Scheme 380). This propargylic amine 1292 was taken forward to synthesize a direct precursor of oseltamivir phosphate consisting of 5 purification steps, 25.7% overall yield and an optical purity of 76% ee. This sequence represents a formal total synthesis of the antiviral drug Tamiflu.456
Scheme 380. Asymmetric Synthesis of Tamiflu.

Porco and Schaus have reported the asymmetric Sc-catalyzed rearrangement of 3-allyloxyflavones 1294 for the preparation of chiral 3,4-chromanediones 1295. For purification purposes, the authors condensed the dicarbonyl products with various diamines to yield the corresponding dihydopyrazines 1296. Employing chiral PyBOX ligand 1213a, the dihydropyrazines 1296 could be accessed in high yields of up to 98% and enantioselectivities of up to 96% ee (Scheme 381). Mechanistic studies support the intramolecular rearrangement pathway proceeding through a benzopyrylium intermediate.457
Scheme 381. Asymmetric Sc-Catalyzed Rearrangement of 3-Allyloxyflavones for the Preparation of Chiral 3,4-Chromanediones.

Desimoni studied the enantioselective formal hetero-Diels–Alder (HDA) reaction of (E)-4-aryl-2-oxo-3-butenoates 1297 utilizing 1213-Sc(III) catalysts (Scheme 382). The authors found that these compounds, behaving as α-dicarbonyl derivatives, operate through a bidentate coordination to form rigid complexes that are characterized by a 5-membered structure. The reaction intermediate (as shown in 1300) gives excellent facial discrimination, determined by the configuration of the PyBOX C4-substituent in a tandem Mukaiyama–Michael addition/intramolecular ring closure, which is a formal HDA reaction. This process yields the products 1299 with trans–trans-fused junctions in bicyclic six to six-membered ring systems in up to 73% yield and >99% ee.458
Scheme 382. Enantioselective Formal Hetero-Diels–Alder Reaction of (E)-4-Aryl-2-oxo-3-butenoates.

Karimi and Enders have described a highly efficient Yb(1213b)-catalyzed Mannich reaction of malonates with N-tert-butoxycarbonyl (Boc) imines 1301 to access the corresponding chiral amines 1302 in excellent yields and enantioselectivities (Scheme 383). Using MeOH as an additive, the authors could synthesize a range of chiral amines in up to 95% yield and 99% ee. They found the substituent on the N-atom of the imine played an important role in the formation of the product, with only N-Boc imines allowing the reaction to proceed.459
Scheme 383. Asymmetric Yb-Catalyzed Mannich Reaction of Malonates with N-tert-Butoxycarbonyl (Boc) Imines.

Kawatsura and Itoh have demonstrated a RuCl3/1213b-catalyzed allylic amination of racemic 1-aryl allyl esters 1303 with amines 1233. Using mainly cyclic secondary amines, they could access enantiomerically enriched allylic amine products 1304 in good yields up to 93% and high enantioselectivities of up to 94% ee (Scheme 384). The reaction was shown to proceed with perfect regioselectivity for several allyl esters.460
Scheme 384. Ru-Catalyzed Asymmetric Allylic Amination of Racemic 1-Aryl Allyl Esters with Amines.

Kawatsura has reported a related kinetic resolution of allyl acetates via a Ru-catalyzed asymmetric etherification. A variety of aryl allyl acetates 1303 were subjected to the reaction with alkyl and benzyl alcohols 1062 in the presence of [RuCl2(p-cymene)]2 and iPr-PyBOX 1213b to give the corresponding ethers 1305 in up to 48% yield, 96% ee and with selectivity factors (S) of up 103 (Scheme 385). The enantiomerically enriched allyl acetate (R)-1303 could also be isolated in up to 98% ee. The reaction proceeded with perfect regioselectivity in all cases.461
Scheme 385. Kinetic Resolution of Allyl Acetates via a Ru-Catalyzed Asymmetric Etherification.

Gamasa and Pizzano have described a Ru-1213a-catalyzed hydrogenation and transfer hydrogenation of imines for the asymmetric synthesis of benzylamines. Subjecting diaryl imines 1306 to 1 mol% of the Ru catalyst in iPrOH at 60 °C under 20 bar H2 led to the isolation of enantioenriched benzyl amines 1307 in up to 93% yield and up to 99% ee (Scheme 386). Conducting the transfer hydrogenation under N2 in place of H2 led to the isolation of the enantioenriched benzyl amines 1307 in up to 99% yield and up to 99% ee. Overall, the two processes gave similar results. Substrates bearing Ar1 = 4-ClC6H4 or 4-BrC6H4 did not perform well under catalytic hydrogenation, while they were found to be completely incompatible with transfer hydrogenation.462
Scheme 386. Ru-Catalyzed Asymmetric Hydrogenation and Asymmetric Transfer Hydrogenation of Imines.

Van Vranken has reported an enantioselective Pd-catalyzed carbene insertion into carbazole derivatives 1308 (Scheme 387). While a range of oxazoline-containing chiral ligands were tested, the iPr-PyBOX ligand 1213b was found to induce the highest levels of enantioselectivity, giving the chiral amine products 1309 in up to 99% yield and 99% ee. Carbenes bearing a strongly electron-withdrawing group were found to perform poorly in the reaction. For example, the reaction of a 4-nitro-substituted carbene with carbazole led to the formation of the product as a racemate.463
Scheme 387. Enantioselective Pd-Catalyzed Carbene Insertion into Carbazole Derivatives.

Zhou has reported a Ni-catalyzed asymmetric cross-coupling reaction of secondary benzylic bromides 1310 with aryl alkynyl aluminum reagents 1311 (Scheme 388). iPr-PyBOX ligand 1213b was found to effectively induce good to high levels of enantioselectivity, leading to the isolation of a range of internal alkyne products 1312 in up to 94% yield and with up to 93% ee. Other PyBOX and BOX ligands were tested in the reaction, but these did not give comparable levels of stereoinduction. A range of aryl groups were well tolerated on the alkyl bromide coupling partner, while aryl and heteroaryl groups performed well on the alkynyl aluminum partner.464
Scheme 388. Ni-Catalyzed Asymmetric Cross-Coupling Reaction of Secondary Benzylic Bromides with Aryl Alkynyl Aluminum Reagents.

Morken has applied 3,5-EtC6H3–PyBOX ligand 1316 in the development of an asymmetric Ni-catalyzed Kumada cross-coupling of symmetric cyclic sulfates 1313. A range of aryl Grignard reagents 1314 were successfully coupled with the cyclic sulfates 1313 to give, following acid hydrolysis, chiral alcohols 1315 in up to 98% yield and with high enantioselectivities up to 92% ee (Scheme 389).465
Scheme 389. Asymmetric Ni-Catalyzed Kumada Cross-Coupling of Symmetric Cyclic Sulfates.

Luan achieved an asymmetric dearomatization of naphthols 1317via a Sc-catalyzed electrophilic amination reaction with DEAD 1318. A series of chiral PyBOX ligands were screened, but only ligand 1213d gave high levels of asymmetric induction. Both monosubstituted 2-naphthols and disubstituted 1,3-naphthols performed well in the reaction, allowing access to the products 1319 in up to 98% yield and 98% ee with a wide substrate scope and at gram scale (Scheme 390).466
Scheme 390. Asymmetric Dearomatization of Naphthols via a Sc-Catalyzed Electrophilic Amination Reaction with DEAD.

Fu and Gu have also utilized Bn-PyBOX ligand 1213d in a Cu-catalyzed asymmetric ring-opening reaction of diaryliodonium salts 1320 with amines 1232 (Scheme 391). Diaryliodonium salts 1320 exist in two rapidly interconverting conformers. The interaction of the two conformers with the Cu(1213d) species leads to the formation of 1322a and 1322b. 1322b is sterically disfavored due to the steric interaction between the Bn-group of the ligand and the methyl group of the diaryliodonium salt. Thus, 1322b rapidly converts to 1322a and undergoes the ring-opening reaction with the Cu(1213d) species, selectively establishing axial chirality. Base promoted coordination of the amine with the Cu-species, followed by reductive elimination leads to the formation of the products 1321. A range of di-o-substituted diaryliodonium salts 1320 were successfully reacted with a range of aryl and benzylic amine 1232 giving the axially chiral products 1321 in up to 99% yield and >99% ee. It should be noted that the reaction works equally well when indanyl PyBOX ligand (R,S)-1337 (below) is utilized.467 Further mechanistic insights, and an improved experimental procedure, in which the amine is added slowly via a syringe pump, leading to an expanded substrate scope (benzylic and aliphatic amines) were subsequently reported by Gu.468
Scheme 391. Cu-Catalyzed Asymmetric Ring-Opening Reaction of Diaryliodonium Salts.

You has described a MgI2/1213c-catalyzed asymmetric dearomative [3 + 2] cycloaddition of benzotriazoles 1324 with cyclopropane-1,1-dicarboxylates 1323 (Scheme 392). By subjecting a range of aryl- and alkene-substituted cyclopropanes 1323 to the reaction with a range of benzotriazoles 1324 in the presence of 4 Å MS at 0 °C for 5 days, the polycyclic products 1325 containing two stereocenters were isolated in excellent yields of up to 97%, with excellent enantioselectivities of up to 97% ee (only one example <90% ee) and all >20:1 dr. Conducting the reaction with both the enantiomerically pure (S)- and (R)-enantiomers of the cyclopropane 1323 showed that the catalyst preferentially reacts with the (R)-enantiomer. The results also suggested that this transformation is a simple kinetic resolution and not a dynamic kinetic resolution, with >2.0 equiv of the racemic cyclopropane required to achieve a high yield.469
Scheme 392. Mg-Catalyzed Asymmetric Dearomative [3 + 2] Cycloaddition of Benzotriazoles with Cyclopropane-1,1-dicarboxylates.

Yoon has reported an asymmetric α-amino radical conjugate addition by the merging of photoredox catalysis and Lewis acid catalysis. Following a screen of isomers of chiral PyBOX ligand 1213c they found that the iBu-containing PyBOX ligand 1213i in combination with Sc(OTf)3 and Ru(bpy)3Cl2, provided a system that was optimal for catalyzing the reaction and achieving high levels of stereoinduction. A range of chiral amines 1328 were synthesized in good yields of up to 96% and enantioselectivities of up to 96% ee (Scheme 393). Aryl amines appeared to perform best in the reaction, while the Michael acceptor bearing an auxiliary group X was required.470
Scheme 393. Asymmetric α-Amino Radical Conjugate Addition.

Yoon subsequently described a chiral Lewis acid-catalyzed triplet energy transfer strategy for the asymmetric [2 + 2] cycloaddition of 2′-hydroxychalchones 1329 with dienes 1330. They discovered that the coordination of a Sc(OTf)3-1213c complex to the 2′-hydroxychalchone substrates 1329 significantly decreased their triplet state energy, giving access to enantioselective reactions of the electronically excited states. A range of 2′-hydroxychalcones 1329 were subjected to the [2 + 2] cycloaddition with a small range of dienes 1330, in the presence of Ru(bpy)(PF6) as the photosensitizer, to yield chiral cyclobutanes 1331 in high yields of up to 86%, excellent enantioselectivities of up to 98% ee and low diastereoselectivities of up to 4:1 dr (Scheme 394).471
Scheme 394. Chiral Lewis Acid-Catalyzed Triplet Energy Transfer Strategy for the Asymmetric [2 + 2] Cycloaddition of 2′-Hydroxychalchones with Dienes.

A later report by Yoon expanded the scope of the [2 + 2] cycloaddition to include styrene substrates in place of the dienes 1330, maintaining the high levels of enantioselectivity. This strategy was applied in the total synthesis of norlignan, a diaryl cyclobutane natural product (Scheme 395).472
Scheme 395. Asymmetric Total Synthesis of Norlignan.

Overall, PyBOX ligands are some of the most used bis(oxazoline) ligands in asymmetric catalysis. They have been combined with a range of metal catalysts to induce excellent levels of stereoselectivity. The tridentate and planar PyBOX ligands appear to be particularly useful in Cu-catalyzed asymmetric transformations, such as progargylation and cycloaddition reactions. However, they have also been applied inter alia in the chemistries of Pd, Ni, Ru, Fe, Sc, and In. Notably, the application of these ligands in asymmetric photoredox catalysis is an important step forward. We predict there will be further developments in this area.
3.2.13. Bis(oxazoline) Ligands with Pyridine Linkers and Disubstituted Oxazoline Rings
PyBOX ligands derived from disubstituted amino alcohols are commonly applied in a wide range of transformations in asymmetric catalysis (Figure 66).
Figure 66.

PyBOX ligands derived from disubstituted amino alcohols.
Vallribera has reported an enantioselective synthesis of l-carbidopa in 7 steps from β-keto ester 1338b. The synthesis involves a key Eu-catalyzed α-amination of acyclic β-keto esters, utilizing diphenyl-PyBOX ligand (R,R)-1335. Monosubstituted PyBOX ligands 1213a–b were found to give the product 1340a with good enantioselectivities, although lower than (R,R)-1335. β-Ketoesters 1338a–b were reacted with di-tert-butyl azodicarboxylate 1339 in the presence of Eu(OTf)3 and 1335 to yield the chiral amines 1340a and 1340b in 83% and 95% yields, respectively, and >99.9% and 98% ee (Scheme 396). Chiral amine 1340b was brought forward to synthesize l-carbidopa in a 50% overall yield.473 Sodupe and Vallribera later applied this methodology to the asymmetric synthesis of fluorous l-carbidopa precursors with similarly high enantioselectivities of up to 99% ee.474
Scheme 396. Asymmetric Synthesis of l-Carbidopa via an Enantioselective Eu-Catalyzed α-Amination of Acyclic β-Keto Esters.

Suga has described the chiral Lewis acid-catalyzed asymmetric cycloaddition of carbonyl ylides, employing chiral PyBOX ligand 1335, for the synthesis of indolizidine alkaloids. The carbonyl ylides 1344 were first derived from various sized (n = 5, 6, 7) N-diazoacetyl lactams 1341 in the presence of Rh2(OAc)4, followed by the chiral Lewis acid-catalyzed cycloaddition with the appropriate dienophile 1342, to yield the corresponding heterocycle 1343 (n = 5, 6, 7) in up to >99% yield and 95% ee (Scheme 397). This methodology was applied to the asymmetric total synthesis of the indolizidine alkaloid (+)-tashiromine in 27% overall yield.475
Scheme 397. Chiral Lewis Acid-Catalyzed Asymmetric Cycloaddition of Carbonyl Ylides.

Gong has reported a Pd-catalyzed regioselective asymmetric aminohydroxylation of 1,3-dienes 1345 with N-tosyl-2-aminophenols 1344. Employing chiral PyBOX ligand 1336, the corresponding 3,4-dihydro-2H-1,4-benzoxazine 1346 was formed with perfect regioselectivity for a range of substrates in up to 84% yield and 92% ee (Scheme 398). The reaction proceeds via a cascade aminopalladation (aza-Wacker)/asymmetric allylation sequence, with the ligand 1336 chelating in a bidentate-mode throughout.476
Scheme 398. Pd-Catalyzed Regioselective Asymmetric Aminohydroxylation of 1,3-Dienes with N-Tosyl-2-aminophenols.

Nishibayashi has described an enantioselective Cu-catalyzed propargylation of indoles 1348 with CF3-substituted propargylic esters 1347 for the construction of all-carbon quaternary stereocenters. Employing chiral PyBOX ligand (S,R)-1336 in the Cu(OTf)·0.5C6H6-catalyzed process, a range of propargylic esters 1349 bearing different aryl groups were reacted with a range of indoles 1348 for the enantioselective synthesis of propargylic indoles 1349 in excellent yields of up to 90% yield and with up to 97% ee (Scheme 399).477
Scheme 399. Enantioselective Cu-Catalyzed Propargylation of Indoles.

Liu has described a Cu-catalyzed asymmetric dehydrogenative C(sp3)H–C(sp)H cross-coupling employing O2 as the terminal oxidant. Following a screen of multiple oxazoline-based chiral ligands, indanyl PyBOX ligand (R,S)-1337 was found to induce moderate to high levels of enantioselectivity in the Cu-catalyzed cross-coupling of N-aryl glycine esters 1350 (C(sp3)H) with terminal alkynes 1040 (C(sp)H) to give the enantioenriched α-amino esters 1351 in up to 80% yield and with up to 87% ee (Scheme 400). A range of aryl and alkyl alkynes 1040 were well tolerated in the reaction giving access to enantioenriched non-natural α-amino acids.478
Scheme 400. Cu-Catalyzed Asymmetric Dehydrogenative C(sp3)H–C(sp)H Cross-Coupling.

Loh has reported a chiral In(II)-(R,S)-1337 complex-catalyzed asymmetric ketone-ene reaction of trifluoropyruvate 1352 conducted in the ionic liquid [hmim]PF6. Subjecting a range of alkenes 1353 to the reaction led to the isolation of the corresponding tertiary allylic alcohols 1354 in high yields of up to 98% and excellent enantioselectivities of up to 98% ee (Scheme 401). The chiral In(II)-(R,S)-1337 complex in the ionic liquid was recycled up to seven times with retention of high yields and enantioselectivities.479
Scheme 401. In-Catalyzed Asymmetric Ketone-ene Reaction of Trifluoropyruvate.

Loh has described a study on the asymmetric carbonyl-ene and cationic polyene cyclization reactions of 1,5-keto-olefins 1355 with Sc(III)-(R,S)-1337 complexes. First, promoting the cyclization of 1,5-keto-olefins 1355 with a Sc(III)-(R,S)-1337 complex led to the isolation of carbonyl-ene product 1356 in high yields up to 87% and enantioselectivities up to 95% ee (Scheme 402). Treatment of the ene-product 1356 with TiCl4 led to formation of the polyene-products 1357 without much loss in enantioselectivity. Subjecting substrates with terminator groups which are more nucleophilic, like furan, indole or tetrasubstituted alkenes, to the reaction conditions led to formation of the polyene-products 1359 in high yields and enantioselectivities without the need for TiCl4.480
Scheme 402. Asymmetric Carbonyl-ene and Cationic Polyene Cyclization Reactions.

Zhu has reported a highly enantioselective intramolecular allylic C–H insertion reaction. Using indanyl PyBOX ligand (R,S)-1337 in this Ru-catalyzed process, a variety of N-allylic enynones 1360 were transformed into di-syn-substituted indolines 1361 in just one step, giving the products in up to 87% yield, with excellent enantioselectivities up to >99% ee, and in all cases with diastereoselectivities of >99:1 dr (Scheme 403).481
Scheme 403. Enantioselective Intramolecular Allylic C–H Insertion Reaction.

Loh has applied (R,S)-1337 in an In-catalyzed HDA reaction of Danishefsky-type dienes 1362 with α-carbonyl esters 1286. A variety of substrates were successfully applied in the reaction to give the corresponding chiral 2,3-dihydro-4-pyranones 1363 in high yields up to 84% and enantioselectivities up to 95% ee (Scheme 404). Products containing both tertiary and quaternary stereocenters could be isolated with similarly good results.482
Scheme 404. In-Catalyzed HDA Reaction of Danishefsky-type Dienes with α-Carbonyl Esters.

Franz has reported an asymmetric synthesis of 3-hydroxy-2-oxindoles 1366 via the Friedel–Crafts alkylation of indoles 1364 and electron-rich arenes with isatins catalyzed by Sc(III)- and In(III)-(S,R)-1337 complexes. Other PyBOX ligands tested were found to induce only moderate to good enantioselectivities. The product oxindoles 1366 were isolated in up to 99% yield and 99% ee for a range of substrates (Scheme 405). The catalysts were also found to promote asymmetric allylation and aldol reactions of isatins in up to 97% yield and 99% ee. An octahedral model 1369 for this process is invoked to explain the sense of stereoinduction. If the isatin amide carbonyl group is bound to the apical position of the complex, the nucleophile is forced to attack the Si-face. This study into the activity of both Sc(III) and In(III) complexes provides a useful guide for further research in this area.483 Franz has extended this work to include N-methyl-pyrrole484 and allylsilanes485 as nucleophiles.
Scheme 405. Asymmetric Synthesis of 3-Hydroxy-2-oxindoles.

Franz has described another extension of this work wherein allyl silanes based on a bulky −Si(iPr)3 moiety is used as nucleophiles in an asymmetric [3 + 2] annulation with isatins. Various isatin derivatives 1365 react with allylsilanes 1370, which bear a bulky silyl-group, in an ScCl2(SbF6)-(S,R)-1337-catalyzed nucleophilic addition/1,2-silyl migration/annulation to give a range of spirooxindoles 1371 in up to 82% yield and 99% ee (Scheme 406). Subjecting allyl silane 1372, bearing an −SiMe2(CHPh2) group, to the annulation reaction conditions followed by C–Si oxidation affords the corresponding secondary alcohols 1373 in excellent yields and enantioselectivities.486
Scheme 406. Asymmetric [3 + 2] Annulation of Isatins with Allyl Silanes.

Franz has extended this carboannulation methodology to include alkylidene oxindoles 1374 as electrophiles, utilizing a ScCl3/(R,S)-1337/NaBARF catalytic system, to give the corresponding spirooxindoles 1375 in up to 99% yield and 99% ee (Scheme 407).487
Scheme 407. Asymmetric [3 + 2] Annulation of Alkylidene Oxindoles with Allyl Silanes.

Wang has described a related Yb-catalyzed decarboxylative asymmetric addition of β-ketoacids 1376 to isatins 1365, employing chiral PyBOX ligand (R,S)-1337. A range of 3-hydroxy-oxindoles 1377 were synthesized in up to 98% yield and 99% ee (Scheme 408). The same stereochemical model is used to explain the outcome of this reaction as in Franz’s previous example, with the opposite enantiomer of the ligand giving Re-face attack in this instance.488
Scheme 408. Yb-Catalyzed Decarboxylative Asymmetric Addition of β-Ketoacidsto Isatins.

Kesavan has reported a highly enantioselective Sc(OTf)3-(R,S)-1337-catalyzed intramolecular amidation of imines for the synthesis of a range of 2,3-dihydroquinazolinones 1379 in high yields of up to 97% and excellent enantiomeric excesses of up to 98% (Scheme 409). Aryl aldehydes 1021 were reacted with ortho-amide-substituted aniline derivatives 1378 with high to excellent enantioselectivities in all reported cases.489
Scheme 409. Enantioselective Sc-Catalyzed Intramolecular Amidation of Imines.

Singh has reported a Sc(III)- and In(III)-catalyzed Mukaiyama–Michael addition of silyl enol ethers 1380 to α,β-unsaturated 2-acyl imidazoles 1381. Utilizing indanyl PyBOX ligand (S,R)-1337, the enantioselectivity of the process could be switched by using either Sc(OTf)3-(S,R)-1337 or In(OTf)3-(S,R)-1337 as the catalyst. In the reaction catalyzed by Sc(OTf)3-(S,R)-1337, a range of aryl-substituted silyl enol ethers 1380 were tolerated, as were a range of aryl groups on the Michael acceptors 1381, to yield the enantioenriched 1,5-carbonyl products 1382 in up to 93% yield and with up to 84% ee (Scheme 410). The reaction catalyzed by In(OTf)3-(S,R)-1337 gave similar, but opposite results, leading to the formation of the products ent-1382 in up to 92% yield and with higher levels of enantioselectivity of up to 94% ee. The larger ionic radius of In(III), which leads to the formation of a bipyramidal trigonal complex in the key intermediate, as compared to Sc(III), which leads to the formation of an octahedral complex intermediate, was presented as an explanation for the observed switch in enantioselectivity.490 Singh has also employed (S,R)-1337 in the Sc(III)- and Er(III)-catalyzed Mukaiyama–Michael addition of siloxyfurans to the same α,β-unsaturated 2-acyl imidazoles.491
Scheme 410. Enantiodivergent Sc(III)- and In(III)-Catalyzed Mukaiyama–Michael Addition of Silyl Enol Ethers.

Kobayashi has described a Ca-catalyzed reaction of oxindoles 1383 with N-Boc imines 1301 for the asymmetric synthesis of 3-tetrasubstituted oxindoles 1384. Utilizing chiral PyBOX ligand 1385, a range of oxindoles 1383 and imines 1301 were successfully subjected to the reaction to give the corresponding chiral amine products 1384 in up to 99% yield, >99% ee, and 98:2 anti/syn dr (Scheme 411). A DFT study of the CaCl2-1385 complex and the corresponding oxindole enolate complex suggested the formation of a C2-symmetric complex with all three nitrogen atoms coordinated to the metal center. The distance between the Ca and O (of the methoxy groups) suggested a weak coordination of O to Ca, and a stereochemical model 1386 in which the −CH2OMe moiety of the ligand shields the Re-face of the enolate was proposed (Figure 67).492
Scheme 411. Asymmetric Ca-Catalyzed Reaction of Oxindoles with N-Boc Imines.

Figure 67.
Stereochemical model for the asymmetric Ca-catalyzed reaction of oxindoles with N-Boc imines.
Singh has reported the use of gem-diPh-iPr PyBOX ligand 1387 in several enantioselective catalytic transformations (Figure 68).
Figure 68.
PyBOX ligand 1387.
They reported an asymmetric synthesis of coumarin derivatives 1390via a Zn(II)-1387-catalyzed Michael addition of 4-hydroxycoumarin 1388 with 2-enolpyridine N-oxides 1389 in up to 99% yield and 97% ee (Scheme 412). PyBOX ligands without gem-disubstitution did not perform well in the transformation. The coumarin products 1390 were found to be in equilibrium with both diastereomeric forms of cyclized 1391. Washing the products of the reaction with ethyl acetate significantly increased their enantiomeric purity up to >99.9% ee.493 The scope of this reaction was further extended to include aliphatic cyclic 1,3-diketones494 and silyl enol ethers495 as the nucleophiles with similar results.
Scheme 412. Asymmetric Synthesis of Coumarin Derivatives.

Singh also reported a Cu(I)-1387-catalyzed asymmetric alkynylation/lactamization cascade for the synthesis of chiral isoindolinones 1393. The three component reaction of various methyl-2-formylbenzoate derivatives 1392 with aryl amines 1072 and alkynes 1040 proceeds through an A3-coupling to give, following intramolecular lactamization of the intermediate chiral amine, the corresponding isoindolinones 1393 in high yields of up to 98% and enantiomeric excesses of up to >99% (Scheme 413).496 Singh further employed this methodology in the asymmetric synthesis of medicinally relevant target compounds.497
Scheme 413. Cu(I)-Catalyzed Asymmetric Alkynylation/Lactamization.

Kang has reported a desymmetrization reaction of 2,2-disubstituted 1,3-propanediols for the synthesis of all-carbon quaternary stereocenters. Utilizing gem-dibutyl CuCl2-1396, an asymmetric benzoylation of one of the two alcohols was achieved by reacting various 1,3-propanediols 1394 with benzoyl chloride under basic conditions to give the desymmetrized products 1395 in up to 99% yield and 99% ee (Scheme 414). A stereochemical model 1397 was proposed to explain the asymmetric induction in the reaction (Figure 69). If the tridentate chiral PyBOX ligand sits equatorial in the octahedral complex, the benzoyl chloride axial and the diol axial and equatorial, the smaller R-group on the diol is most likely to occupy the space near the 4-Ph group of the oxazoline ring. The benzoyl cation is then attacked by the equatorial alcohol group to give the desymmetrized products. As a result, substrates with lower levels of steric differentiation between the two R-groups gave lower enantioselectivities, for example in the case of R1 = Me, R2 = −CH=CH2, the product was obtained in only 54% ee.498
Scheme 414. Desymmetrization of 2,2-Disubstituted 1,3-Propanediols.

Figure 69.

Stereochemical model for the desymmetrization of 2,2-disubstituted 1,3-propanediols.
Overall, PyBOX ligands with disubstituted oxazoline rings appear to be more useful in asymmetric lanthanide-metal-catalyzed processes than their monosubstituted counterparts. They have also been applied in a range of Sc- and In-catalyzed processes.
3.2.14. Bis(oxazoline) Ligands with Modified Pyridine Linkers
Less common modifications to the chiral PyBOX ligand structure are pyridine ring-based modifications. Johnson has applied 4-halogenated chiral tBu-PyBOX ligands 1398a–b in dynamic kinetic asymmetric reactions of strained cyclopropane rings (Figure 70).
Figure 70.

4-Halogenated PyBOX ligands.
For example, they applied chlorinated chiral PyBOX ligand 1398a in a dynamic kinetic asymmetric [3 + 2] cycloaddition of racemic cyclopropanes 1399 bearing electron-rich donor groups, such as p-methoxybenzene, and aldehydes 1021. A range of chiral tetrahydrofurans 1400 were synthesized in the MgI2-catalyzed process in up to 92% yield and 94% ee (Scheme 415). A range of alkyl and aryl aldehydes 1021 performed well in the reaction, with the lowest enantiomeric excess obtained for the iso-propyl aldehyde of 82%. A range of ligands with pyridine ring substitution were tested, with the halogenated substrates giving the best results. For comparison, the halogenated tBu-PyBOX ligand 1213c gave the product in 62% yield and 91% ee, compared to 74% yield and 92% ee with ligand 1398a for the same substrate.499
Scheme 415. Dynamic Kinetic Asymmetric [3 + 2] Cycloaddition of Racemic Cyclopropanes.

Johnson subsequently extended the dynamic kinetic [3 + 2] annulation to N-benzyl-(E)-aldimines 1401 utilizing brominated chiral PyBOX ligand 1398b under similar conditions to the previous reaction. Chiral 2,5-cis-pyrrolidines 1402 were synthesized from cyclopropanes 1399 bearing electron-rich donor groups and aryl aldimines 1401 in up to 86% yield and 96% ee (Scheme 416). Mechanistic studies suggest that the aldimine reacts through the (E)-isomer, and that isomerization to the (Z)-isomer is not a pathway that furnishes the major 2,5-cis-disubstituted products.500
Scheme 416. Dynamic Kinetic [3 + 2] Annulation to N-Benzyl-(E)-aldimine with Cyclopropanes.

Johnson has further described a MgI2-catalyzed dynamic kinetic Friedel–Crafts alkylation of indoles with cyclopropanes for the asymmetric synthesis of 3-substituted indoles. A range of N-TBS protected indoles 1403 were reacted with racemic substituted cyclopropanes 1399 in the presence of Br-modified chiral PyBOX ligand 1398b to give the chiral 3-substituted indoles 1404 in up to 96% yield and 94% ee (Scheme 417).501
Scheme 417. Mg-Catalyzed Dynamic Kinetic Friedel–Crafts Alkylation of Indoles with Cyclopropanes.

Yoon has applied dimethylamino-substituted s-Bu-PyBOX ligand 1408 in the asymmetric photocatalytic [3 + 2] cycloaddition of aryl cyclopropyl ketones 1405 with alkenes 1406 for the synthesis of enantioenriched cylopentanes 1407. Employing Gd(OTf)3 as the Lewis acid in this process, a range of substituted and spirocyclic cyclopentanes 1407 were accessed in up to 95% yield and with up to >99% ee (Scheme 418). Generally, the diastereoselectivity of the reaction was relatively low, giving the products in 2:1 to 5:1 dr, with two examples >20:1 dr502
Scheme 418. Asymmetric Photocatalytic [3 + 2] Cycloaddition of Aryl Cyclopropyl Ketones with Alkenes.

Zhou has reported a one-pot/sequential tandem asymmetric A3-coupling/carboxylation/cyclization for the enantioselective synthesis of 2-oxazolidinones 1409. Employing the novel −OBn substituted Ph-PyBOX 1411, aryl aldehydes 1035, mostly aryl alkynes 1040 and aniline derivatives 1072 were successfully reacted in the Cu-catalyzed A3-coupling/Ag-catalyzed carboxylation sequence to access a range of chiral 2-oxazolidinones 1409 in up to 99% yield and with excellent enantioselectivities up to 96% ee (all examples reported were >90% ee) (Scheme 419). An interesting ligand-accelerating effect was found for the cyclization step. The cyclization occurs without the presence of the ligand 1411 or Cu(OTf)2, but the rate of the reaction and subsequent yield of the isolated product was found to increase dramatically when the ligand was present, and more so when Cu(OTf)2 was also present. More interesting still, the presence of excess aniline was found to increase the rate of the cyclization step. Thus, the leftover reagents from the first step were found to promote the upstream cyclization.503
Scheme 419. One-Pot/Sequential Tandem Asymmetric A3-Coupling/Carboxylation/Cyclization.

Loh and Xu have utilized the Ph-derivative of the s-Bu-PyBOX ligand 1415 in a Cu-catalyzed enantioselective 1,4-protosilylation of α,β-unsaturated ketimines 1412. A range of aryl-substituted ketimines 1412, bearing a variety of protecting groups on N, were successfully employed in the reaction for the synthesis of (E)-allyl silanes 1414 in up to 95% yield, with up to 92% ee and excellent E/Z selectivity of up to >99:1 (Scheme 420). The choice of chiral ligand, bearing Ph-substitution on the pyridine ring, was based on the formation of a favorable π—π interaction between the Si-Ph moiety and the pyridine-Ph moiety in the transition state. The enantioselectivity is rationalized by the disfavored steric interaction between the N-PG of the substrate with one of the s-Bu groups of the ligand in 1416b, forcing the ketimine to approach as in 1416a with the N-PG pointing away, for selective delivery of the silyl group.504
Scheme 420. Cu-Catalyzed Enantioselective 1,4-Protosilylation of α,β-Unsaturated Ketimines.

Jayashankaran has also employed the novel 4-substituted pyridine ring-modified chiral PyBOX ligands in asymmetric catalysis. A range of modified iPr-PyBOX ligands of the type 1419 bearing a substituted aryl ring in the 4-position of the pyridine ring were screened in the Rh-catalyzed hydrosilylation of aromatic ketones with Ph2SiH. Acetophenone 1417 was chosen as a model substrate and subjected to the reaction with various preprepared Rh-catalysts [RhCl3(1419a–f)] (Table 13). While all the ligands induced good levels of enantiocontrol, ligand 1419d bearing a 4-EtC6H4 group at the 4-position of the pyridine ring gave the best result, yielding the alcohol 1418 in 94% yield and 98% ee (entry 4). A variety of aromatic ketones were subjected to the hydrosilylation using [RhCl3(1419d)] as the catalyst to afford the corresponding alcohols in up to 94% yield and 99% ee.505
Table 13. Rh-Catalyzed Hydrosilylation of Aromatic Ketones with Ph2SiH.

| entry | 1419 | R | yield (%) | % ee |
|---|---|---|---|---|
| 1 | a | H | 88 | 90 |
| 2 | b | OMe | 90 | 78 |
| 3 | c | CN | 96 | 75 |
| 4 | d | Et | 94 | 98 |
| 5 | e | Cl | 89 | 84 |
| 6 | f | CO2Et | 78 | 73 |
The modification of the pyridine ring of the PyBOX scaffold offers an opportunity to alter the electronic properties of these ligands. It is clear from the reports described in this Review that this can be a successful strategy for increasing the levels of stereoinduction in a particular transformation, and this should be considered by researchers in the future.
3.2.15. Bis(oxazoline) Ligands with Phenyl Linkers
Nishiyama has pioneered the development of N,C,N-tridentate bis(oxazoline) ligands linked by a phenyl-anion unit (PheBOX).506 These ligands form strong C–M (M = metal) covalent bonds that stabilize the resulting chiral ligand–metal complexes. The most widely applied PheBOX-complexes in asymmetric catalysis are the Rh-complexes like 1420 and 1421, although other metals have also been used like Ru, Pt, Pd, and Ni (Figure 71).
Figure 71.

Rh(PheBOX) complexes.
While a range of PheBOX analogues with different phenyl- and oxazoline-ring substitution patterns have been developed, the standard PheBOX metal complexes are more abundantly reported. For example, Nishiyama has developed a diastereo- and enantioselective 1420a-catalyzed reductive coupling of cyclopentenone 1422 with aromatic aldehydes 1035 to yield a range of anti-alcohols 1423 in up to 90% yield, with up to 95:5 dr and 93% ee. The outcome of the reaction was sensitive to substitution on the aryl ring of the aldehyde 1035, for example, p-anisaldehyde gave the alcohol in only 65% ee (Scheme 421).507
Scheme 421. Diastereo- and Enantioselective Rh-Catalyzed Reductive Coupling of Cyclopentenone with Aromatic Aldehydes.

Some other enones were tested, with cyclic enones giving the corresponding alcohols 1424–1427 with better enantioselectivities than the only acyclic example, methylvinyl ketone, which gave the anti-alcohol 1426 with 80:20 dr and 57% ee (Figure 72).
Figure 72.

Products of the Rh-catalyzed reductive coupling of various enones with naphthaldehyde.
The possible transition state 1428 was proposed to explain the stereochemical outcome of the reaction, in which the Re-face of the enolate, which forms following hydride addition to the enone, attacks the Re-face of the aldehyde to avoid a steric interaction with one of the oxazoline-Ph groups (Figure 73).
Figure 73.
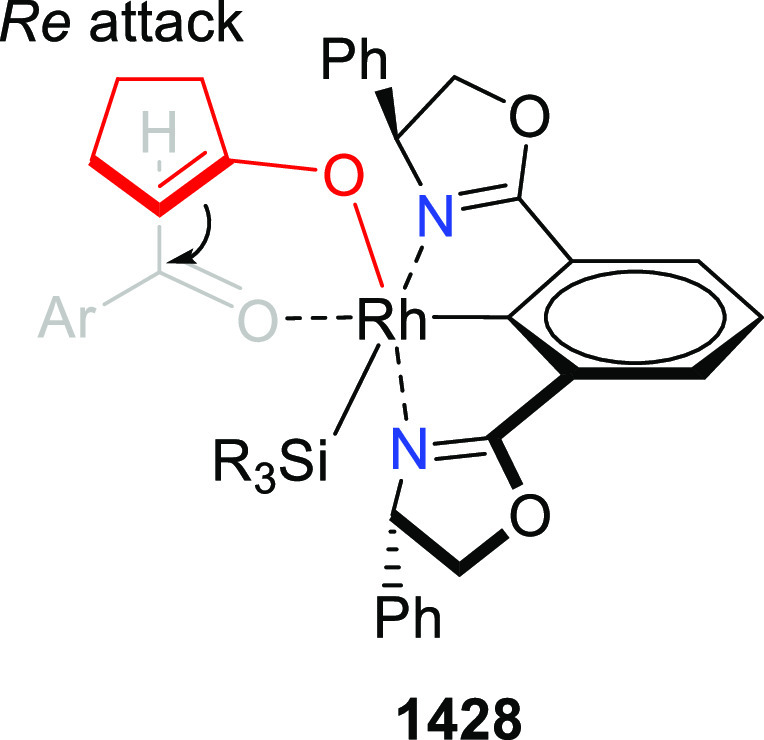
Stereochemical model for the Rh-catalyzed reductive coupling of cyclopentenone with aromatic aldehydes.
Nishiyama also developed the asymmetric hydrosilylation of α,β-unsaturated esters 1429 catalyzed by iPr-PheBOX-Rh 1420c. Under optimized conditions, a range of enantioenriched, diaryl propanoates 1430 were accessed in up to 99% yield and with up to 99% ee with all reported examples isolated with >95% ee (Scheme 422). The reaction of heteroaromatic substrates did not proceed smoothly under the optimized conditions.508
Scheme 422. Rh-Catalyzed Asymmetric Hydrosilylation of α,β-Unsaturated Esters.

Nishiyama has applied sBu-PheBOX-Rh 1420d in the asymmetric hydrosilylation of cyclohexadienones 1431 with asymmetric induction at remote quaternary centers. A small range of nonspirocyclic products 1432 were accessed in up to 99% yield and with up to 81% ee, generally only giving moderate levels of enantioselectivity (<80% ee) (Scheme 423). Spirocyclic cyclohexadienones were found to perform better in this process giving the enantioenriched products in up to 99% yield and with 93% ee with most examples giving high levels of enantioselectivity >80% ee.509
Scheme 423. Rh-Catalyzed Asymmetric Hydrosilylation of Cyclohexadienones.

A possible transition state 1433a was proposed to explain the stereochemical outcome of this hydrosilylation, with stereoinduction at a nonreacting quaternary center (Scheme 424). The Rh–H bond sits in the equatorial position with bond formation occurring on the Re-face of the enone, due to the steric repulsion of the oxazoline-sBu-group and the cyclohexadienone, as shown in 1433b. The steric environment of the γ-position (Ph and Me) is differentiated due to steric repulsion of the bulky trimethoxysilyl group with the Ph-group of the cyclohexadienone, as shown in 1433c.
Scheme 424. Stereochemical Model for Rh-Catalyzed Asymmetric Hydrosilylation of Cyclohexadienones.

The asymmetric β-borylation of α,β-unsaturated esters, amides and ketones 1341 with iPr- and sBu-PheBOX-Rh catalysts 1420c and 1420d was reported by Nishiyama. Enantioenriched esters 1434 were formed in this transformation with a number of examples isolated in up to 91% yield and with up to 97% ee (Scheme 425). Methyl ketones did not perform well, with two examples isolated in up to 89% yield and with up to 70% ee. Finally, a single dimethylamide example was isolated in 70% yield and with 97% ee (1420c) and 74% yield and with 93% ee (1420d). When the reaction was run with (Z)-ethyl cinnamate as the substrate and 1420c as the catalyst, the borylated product was isolated as the same (S)-enantiomer, as in the reaction with (E)-ethyl cinnamate (84% yield, 95% ee), in 70% yield and with 93% ee. While a stereochemical model for this transformation was proposed, it did not take the outcome of the reaction with the (Z)-alkene into account.510
Scheme 425. Asymmetric β-Borylation of α,β-Unsaturated Esters, Amides, and Ketones.

Nishiyama continued to explore asymmetric borylation reactions of alkenes and reported the iPr-PheBOX-Rh 1420c-catalyzed diboration/oxidation of unactivated terminal alkenes 1406 for the asymmetric synthesis of 1,2-diols 1435. A wide range of aryl, alkenyl and aliphatic substituted-alkenes 1406 were successfully transformed into enantioenriched 1,2-diols 1435 in up to 96% yield and with up to >99% ee (Scheme 426).511
Scheme 426. Asymmetric Synthesis of 1,2-Diols.

A stereochemical model was proposed to explain the formation of the major (R)-enantiomer in this transformation. In transition states 1436a and 1436b, the Bpin (B) sits in one of the apical positions (Figure 74). In 1436a, C–B bond formation on the Si-face of the alkene gives the major (R)-enantiomer of the product, while transition state 1436b, with C–B bond formation on the Re-face of the alkene, is disfavored due to the steric interaction between the Ph-group of the alkene and one of the oxazoline R-groups. Transition states 1436c and 1436d show the Bpin in one of the equatorial positions. Transition state 1436c shows a disfavored steric interaction as in 1436b. 1436d has no disfavored interactions, but C–B bond formation on the Re-face of the alkene gives rise to the minor (S)-enantiomer of product.
Figure 74.

Stereochemical mode for the Rh-catalyzed borylation of alkenes.
Aggarwal built on the work by Nishiyama in the diboration of terminal alkenes and has reported an iPr-PheBOX-Rh 1420c-catalyzed Markovnikov hydroboration of unactivated terminal alkenes 1406 with a novel borylating reagent 1438 and H2O as a proton source. Following optimized conditions, the enantioenriched products 1437 were isolated in moderate to good yields up to 86%, with regioselectivities up to 99:1 rr and enantioselectivities up to 96% ee (Scheme 427). Aliphatic and aryl substrates were reported with the process showing good functional group tolerance.512
Scheme 427. Rh-Catalyzed Markovnikov Hydroboration of Unactivated Terminal Alkenes.

Analogues of the PheBOX ligands have been developed for use in Ru-catalysis. For example, Nishiyama has developed complexes of the type 1439–1442, bearing PheBOX ligands with dimethyl-substituted bridging-phenyl rings to prevent Ru C-H insertion at these positions (Figure 75).
Figure 75.

Ru(PheBOX) complexes.
Nishiyama first applied Ru-complexes 1440–1442 in the asymmetric hydrogenation of ketones. Reactions with Ru-complexes 1440a and 1442a were found to give access to enantioenriched alcohols in high yields up to 99% and with up to 98% ee (S). For example, ketone 1443 was subjected to the reaction with both complexes to give alcohol 1444 in 94% yield and with 97% ee (1440a) and in 99% yield and with 98% ee (1442a) (Scheme 428). These complexes were also applied in enantioselective transfer hydrogenation to give the alcohols with overall lower levels of enantioselectivity. For example, the same ketone 1443 was subjected to the transfer hydrogenation in the presence of both complexes to give the alcohol 1444 in 88% yield and with 97% ee (1440a) and in 95% yield and with 92% ee (1442a).513 In a report earlier that year, complex 1441a was applied in the same reactions, giving overall lower levels of enantioselectivity to 1440a and 1442a.514
Scheme 428. Ru-Catalyzed Asymmetric Hydrogenation and Asymmetric Transfer Hydrogenation of Ketone 1443.

Nishiyama later reported an enhancement of the enantioselectivity in the asymmetric hydrogenation of ketones by including alcohol (S)-1444 as an additive. For example, for the asymmetric hydrogenation of ketone 1445, catalyzed by complex 1442a, the enantioselectivity increased from 69% ee without the additive to 88% ee with the additive. In all but one reported case, an increase in the enantioselectivity was observed (Scheme 429).515
Scheme 429. Ru-Catalyzed Asymmetric Hydrogenation of Ketone 1445.

Nishiyama applied the Ph-PheBOX-Ru complex 1442a in the asymmetric Ru-catalyzed alkynylation of aldehydes 1021. A range of enantioenriched alcohols 1447 were isolated in up to 98% yield and with up to 95% ee (Scheme 430). Aryl alkynes were well tolerated in the reaction, while cyclohexyl and trimethylsilyl alkynes gave the corresponding alcohols in low yields but with high levels of enantioselectivity.516
Scheme 430. Asymmetric Ru-Catalyzed Alkynylation of Aldehydes.

Nishiyama later applied Ph-PheBOX-Ru complex 1441a in the racemic conjugate alkyne addition to α,β-unsaturated carbonyls. A single asymmetric example of an alkyne addition to a β-substituted enone was reported, giving the product in 82% ee.517 This led to the development of the 1441a-catalyzed enantioselective 3-component coupling reaction of alkynes 1040, enones 1141, and aldehydes 1021. The levels of enantioselectivity for the formation of the isolated products were mostly low to moderate, up to 78% ee (Scheme 431). The diastereoselectivity was also low, up to 3:1 dr, and in some cases there were regioselectivity issues.518
Scheme 431. Ru-Catalyzed Enantioselective 3-Component Coupling Reaction of Alkynes, Enones, and Aldehydes.

Ohshima has reported the asymmetric Rh-catalyzed alkynylation of α-keto esters519 and α-ketiminoesters520 with C1-symmetric indanyl-PheBOX-Rh complexes 1449a and 1449b and C2-symmetric indanyl-PheBOX-Rh complexes 1450a and 1450b (Figure 76).
Figure 76.

Rh(indanyl-PheBOX) complexes.
Under optimized conditions, enantioenriched tertiary alcohols 1452 could be accessed from α-keto ester 1451 in up to 99% yield and with up to >99% ee (Scheme 432). Initially, complexes 1449a and 1450a were applied in the reaction with aryl-substituted alkynes. C1-Symmetric 1449a was found to outperform C2-symmetric 1450a giving the products in generally higher yields and in most cases, slightly higher enantioselectivities. Nitro-substituted C1-symmetric ligand 1449b was applied in the reactions of less reactive substrates and was found to drastically improve the isolated yields and enantiomeric excesses of the tertiary alcohol products 1452 when compared to 1449a. Interestingly, in a competition experiment between benzaldehyde and α-keto ester 1451, the reaction catalyzed by 1449a gave no observable alkynylation of the aldehyde.
Scheme 432. Asymmetric Rh-Catalyzed Alkynylation of α-Keto Esters.

Later, Ohshima found that the C2-symmetric indanyl-PheBOX-Rh complexes 1450a and 1450b outperformed their C1-symmetric counterparts in the asymmetric alkynylation of α-ketiminoester 1453. Under optimized conditions with nitro-substituted 1450b, a range of aryl, alkenyl, alkyl and silyl-substituted alkynes 1040 were subjected to this reaction with α-ketiminoester 1453 to give a range of tertiary amines 1454 in up to >99% yield and with up to 96% ee (Scheme 433).
Scheme 433. Asymmetric Rh-Catalyzed Alkynylation of α-Ketiminoesters.

A follow-up mechanistic study found that the application of (trimethylsilylethynyl)(PheBOX)Rh complexes 1455a–b in the alkynylation of α-ketoiminoesters allowed for the alkynylation of less reactive α-ketiminoesters, for example cyclic N-sulfonyl ketiminoester- and α-ketiminophosphonate- derived amines 1456 and 1457 were accessed in high yields of 97% and 98%, respectively, and enantioselectivities of 93% ee and 82% ee, respectively (Figure 77).521
Figure 77.

Rh(PheBOX) complex 1455 and cyclic N-sulfonyl ketiminoester- and α-ketiminophosphonate- derived amines 1456 and 1457.
Nishiyama has reported the asymmetric Ru-catalyzed cyclopropanation of terminal alkenes 1458 utilizing diPh-PheBOX-Ru 1461. A range of trans-cyclopropanes 1460 were isolated from the reaction with tert-butyl-α-diazoacetate 1459 in up to 91% yield, with up to 96:4 dr and 99% ee (Scheme 434). Styrene derivatives performed well in the reaction, giving the trans-cyclopropanes with high dr, while the only reported disubstituted alkene example gave the opposite diastereoselectivity (35:65 anti/syn).522
Scheme 434. Asymmetric Ru-Catalyzed Cyclopropanation of Terminal Alkenes.

The vast majority of PheBOX applications over the past decade have been in Rh- and Ru-catalyzed transformations. However, Davies and Blakey have reported a PheBOX-Ir-catalyzed asymmetric C–H activation of 1,4-cyclohexadiene 1462 with carbenoids. A range of enantioenriched products were isolated from the reaction catalyzed by tBu-substituted-Bn-PheBOX-Ir complex 1465 in up to 99% yield and with up to 99% ee (Scheme 435). The tBu-group in the backbone of the ligand was found to increase the isolated yield of the product without affecting the high levels of enantioselectivity.523
Scheme 435. Ir-Catalyzed Asymmetric C–H Activation of 1,4-Cyclohexadiene.

In the same report, a small range of substituted cyclohexadienes 1466 were also subjected to the site selective C–H activation with subsequent oxidation by DDQ to give a range of enantioenriched α-diarylesters 1468 in up to 98% yield and with up to 99% ee (Scheme 436).
Scheme 436. Ir-Catalyzed Asymmetric C–H Activation of Substituted 1,4-Cyclohexadienes.

PheBOX ligands have been successfully applied in a whole range of asymmetric transformations of Rh, Ru and Ir. The main disadvantage of these ligands is that the catalysts must be synthesized prior to use. While this will most likely prevent the widespread use of these ligands in asymmetric catalysis, we expect to see future developments, especially in asymmetric Ir-catalysis, which has not been extensively explored.
3.2.16. Bis(oxazoline) Ligands with Dibenzofuran Linkers
Dibenzofuran-4,6-bis(oxazoline) (DBFOX) ligands of the type 1469 and 1470, which bear a coordinating O atom in the bridging dibenzofuran moiety, have been developed for a range of applications in asymmetric metal catalysis (Figure 78). In fact, this area was comprehensively reviewed in 2018 by Itoh and Sibi.524 As a result, we will only discuss some recent examples in detail.
Figure 78.

Dibenzofuran-4,6-bis(oxazoline) (DBFOX) ligands 1469–1470.
Since 2008, DBFOX ligands have been employed in, for example, enantioselective Friedel–Crafts of pyrroles,525,526 hydride shift/ring closure cascade,527 α-cyanation,528 α-fluorination,529−531 and α-hydroxylation of carbonyls,532 enolate protonation533 and radical conjugate addition534 chemistry, utilizing Zn-, Cu-, Ni- and Mg-catalysis.
Recently, Huang and Shibata reported the catalytic asymmetric 1,3-dipolar cycloaddition of β-fluoroalkylated α,β-unsaturated 2-pyridylsulfones 1471 with nitrones 1472 for the enantioselective synthesis of chiral fluoroalkylated isoxalidinones 1473, which can be converted into γ-amino alcohols. Under optimized conditions with Ni(ClO4)2·6H2O and Ph-DBFOX 1469a as the chiral ligand, a range of isoxalidinones 1473 were accessed in up to 97% yield, with up to 99:1 dr and 99% ee (Scheme 437). A range of fluoroalkylated groups (Rf) were tolerated in the reaction.535
Scheme 437. Asymmetric 1,3-Dipolar Cycloaddition of β-Fluoroalkylated α,β-Unsaturated 2-Pyridylsulfones with Nitrones.

Gong has developed a visible-light-promoted Ni-1469a-catalyzed asymmetric radical conjugate addition reaction. The bifunctional Ni-catalyst initiates single electron transfer and provides a chiral environment for effective asymmetric induction. A range of tertiary silylamines 1475 were reacted with α,β-unsaturated N-acyl pyrazoles 1474 to give γ-amino acids 1476 in up to 89% yield and with up to 99% ee (Scheme 438). When secondary amines were used in the reaction under the optimized conditions, subsequent lactamization occurred to give enantioenriched γ-lactams 1477 in up to 80% yield and with up to 93% ee.536
Scheme 438. Visible-Light-Promoted Ni-Catalyzed Asymmetric Radical Conjugate Addition.

We chose to highlight the recent Ni-catalyzed transformations that the DBFOX ligands have been applied in because the development of asymmetric 3d-transition-metal-catalyzed reactions is an important area in modern organic chemistry. We hope that these ligands can be further applied in this area.
4. Tetradentate Bis(oxazoline) Ligands
In 2013, Li reported the use of chiral biphosphinobioxazoline ligand 1478 in the asymmetric reduction of α,β-unsaturated ketones 1486 furnishing the product 1487 in up to 89% yield and 97% ee (Figure 79, Scheme 439). Interestingly when tBuPHOX ligand was used instead in this catalytic transformation only <5% product 1489 was observed. This Ru-catalyst system was also noted to be tolerant of water and air, providing very mild conditions for access to enantioenriched allylic alcohols.537
Figure 79.

Summary of tetradentate bis(oxazolines) ligands.
Scheme 439. Ru-Catalyzed Asymmetric Reduction of Ketones.

Gao reported the use of the tetradentate porphyrin inspired ligand 1479 in the Fe-catalyzed asymmetric epoxidation of electron deficient alkenes 1488 (Scheme 440). A range of electronically varied alkenes 1488 were screened using the optimized reaction conditions furnishing the epoxide 1489 in up to 80% yield and 99% ee. The utility of this reaction was further tested by carrying out gram-scale epoxidations with no erosion of enantioselectivity observed.538
Scheme 440. Fe-Catalyzed Asymmetric Epoxidation.

Ishihara developed a novel sulfonamide-based bis(oxazoline) ligand 1480 and applied it in the inverse-electron-demand hetero Diels–Alder reaction of β,γ-unsaturated α-keto esters 1491 with allyl silanes 1490 (Scheme 441). This methodology gives access to chiral oxanes 1492 rather than cyclic acetals which are the product of the conventional inverse-electron-demand hetero Diels–Alder reaction. A broad substrate scope was carried out furnishing the product 1492 in up to 99% yield and 99% ee in a 92:8 cis/trans ratio.539 It was also applied in the asymmetric Cu-catalyzed Diels–Alder reaction to great effect.540
Scheme 441. Cu-Catalyzed Asymmetric Synthesis of Oxanes.

Waser reported the Cu-catalyzed desymmetrization of meso-diaminocyclopropanes 1493 in 2018. This modified Friedel–Crafts reaction was originally attempted using BOX ligand 600c which only furnished the product 1495 in 74% yield, 64% ee, and >20:1 dr. To increase the enantioselectivity the native BOX ligand 600c was modified in the α-position to furnish a large range of ligands containing bulky alcohols. After screening the structure of the ligand further by fine-tuning the sterics, the Cu complex of ligand 1481 afforded the product 1495 in 80% yield, 86% ee, and >20:1 dr. With the optimized conditions in hand a range of electronically and sterically diverse N-TBS indoles 1494 were screened in this catalytic transformation, furnishing the product 1495 in up to 82% yield, 92% ee, and all in >20:1 dr (Scheme 442).541
Scheme 442. Cu-Catalyzed Desymmetrization of meso-Diaminocyclopropanes.

Zhang reported the Cu-catalyzed asymmetric Mannich reaction of cyclic ketimines 1496 and glycine Schiff bases 1497. Planar chiral ferrocene ligands have been widely used in asymmetric catalysis in recent times, however in contrast, ruthenocene based ligands have not been as widely applied in asymmetric catalysis. In this reaction the tetradentate planar-chiral ruthenocene ligand 1482 showed excellent selectivity giving the product in up to 83% yield, 99% ee, and 7:1 dr. The substrate scope highlighted the trend that the ester functionality in the cyclic ketimine 1496 had a large effect on the enantioselectivity. Sterically bulky esters such as an isopropropyl ester led to an increase in enantioselectivity and diastereoselectivity (96% ee, >20:1 dr, and 90% yield) compared to methyl esters (99% yield, 93% ee, 8:1 dr). If the cyclic ketimine ester was replaced by a methyl group this caused a pronounced decrease in all selectivity to 26% yield, 8% ee, and 1:1 dr (Scheme 443).542 In other publications by Zhang’s group, this ligand was applied in the Pd-catalyzed allylation of amino acid derivatives, N-sulfonylimines and in the asymmetric synthesis of chromanols.127,543−546
Scheme 443. Cu-Catalyzed Enantioselective Mannich Reaction of Cyclic Ketimines.

In 2013 Gao reported the synthesis of ligand 1479 as a porphyrin mimic and subsequently utilized it in the Mn-catalyzed asymmetric epoxidation of alkenes 1499 (Scheme 444). This methodology was applied to a range of alkenes 1499 forming the product 1500 in up to 95% yield and >99% ee.547 In a follow up publication this methodology was further developed to include a wider substrate scope of cyclic and acyclic alkenes 1499.548
Scheme 444. Mn-Catalyzed Enantioselective Epoxidation of Alkenes.

Ligand 1479 was further used by Gao in the Mn-catalyzed asymmetric oxidation of aryl sulfides 1501. This methodology encompassed a large substrate scope with all aryl methyl sulfides 1501 affording the product 1502 in up to 84% yield and >99% ee (Scheme 445). This methodology was expanded in a subsequent publication and transferred from a batch reaction to a flow set up. This allowed for quicker reactions, the lowering of catalyst loading from 1.0% to 0.5% and for a gram scale synthesis of chiral sulfoxides.549,550
Scheme 445. Mn-Catalyzed Enantioselective Epoxidation of Sulfides.

Meyer reported the synthesis of a pyrazole-bridged bis(oxazoline) ligands 1483/1484 and their application in the Pd-catalyzed allylic alkylation of 1,3-diphenylallyl acetate 125A (Scheme 446). Interestingly, the active Pd-complex was shown to be a dinuclear palladium species with ligands 1483/1484. While all Pd complexes of ligands 1483/1484 yielded the desired product 1503 there was a large variation in the levels of enantioselectivities observed, the phenyl-substituted ligand 1483 gave the product in 40% yield and 68% ee while 1484 did so in 67% yield and 44% ee.551
Scheme 446. Pd-Catalyzed Enantioselective Allylic Alkylation.

In 2017 Ollevier reported the asymmetric oxidation and tandem kinetic resolution of aryl sulfides 1501 to sulfoxides 1502 (Scheme 447). Ligand 1485 was chosen as a porphyrin mimick creating a bioinspired nonhaem FeII catalyst with FeCl2. This system was employed and furnished the products in up to 96% ee and 21% yield. This catalysis was shown to be highly sensitive to the nature of the aryl substituents on the aryl sulfides. Placing a methyl group in the ortho-position drastically deminished the enantioselectivity to 44% ee and 50% yield.552
Scheme 447. Fe-Catalyzed Asymmetric Oxidation of Aromatic Sulfides.

5. Tris(oxazoline) and Tetra(oxazoline) Ligands
5.1. Tris(oxazoline) Ligands
Tang, one of the key pioneers in the design, synthesis and applications of metal complexes of trisoxazolines, developed a library of pseudo-C3-symmetric trisoxazolines 1505–1507 (TOX; Figure 80) by a “side arm” approach. The metal complexes of TOX ligands 1505–1507 were found to be efficient catalysts for the asymmetric Friedel–Crafts reaction of indole with alkylidene malonate, the asymmetric intramolecular Cannizzaro reaction of aryl and alkyl glyoxals, the asymmetric [3 + 3] cycloaddition of donor–acceptor (D-A)-substituted cyclopropane diesters with aromatic azomethine imines and the asymmetric oxa-[3 + 3]-annulation of oxygenated phenols and 3-aminophenols with β,γ-unsaturated α-keto esters. Compared with the corresponding bisoxazoline ligands, these metal-TOX complexes showed some promising properties, such as better enantioselectivity and stronger tolerance toward water and air.
Figure 80.

Trisoxazoline ligands (TOX).
In 2011, Tang reported a new pseudo-C3-symmetric heterotrisoxazoline (1505), which in combination with Cu(OTf)2 displayed excellent enantioselectivity in the asymmetric Friedel–Crafts alkylation between indoles (1508) and pyrroles and alkylidene malonates (1509) (Scheme 448).553 Enantioselectivities of up to 94% ee were achieved with only 0.5 mol% of catalyst loading.
Scheme 448. Asymmetric Friedel–Crafts Alkylation between Indoles and Pyrroles and Alkylidene Malonates.

Later in 2013, the first asymmetric intramolecular Cannizzaro reaction of aryl and alkyl glyoxals (1511) with alcohols (1512) was reported with excellent levels of enantioselectivity by using a newly developed congested TOX ligand (1506) and Cu(OTf)2 with a gradual liberation protocol of active glyoxals from glyoxal monohydrates (1511) (Scheme 449).554 This method allowed a facile entry to the preparation of a variety of α-hydroxy carboxylic acid derivatives (1513) with high optical purity (up to 96% ee). The proposed reaction pathway suggested that the chiral induction occurred at the catalyst-controlled face selective addition of alcohols to coordinated glyoxals. The improved stereocontrol with the TOX ligand 1506 over the addition of the alcohol to 1515-Cu could be described in terms of a more congested environment created around the reactive site with the aid of the extra t-butyl oxazoline, while the more typical bisoxazoline (BOX) ligands lack such a side arm regulator.
Scheme 449. Asymmetric Intramolecular Cannizzaro Reaction of Aryl and Alkyl Glyoxals with Alcohols.

In 2013, Tang reported the Ni/In-TOX (1507-A)-catalyzed highly enantioselective asymmetric [3 + 3] cycloaddition of donor–acceptor (D–A)-substituted cyclopropane diesters (1517) with aromatic azomethine imines (1516) (Scheme 450).555 A variety of 6,6,6-tricyclic dihydroisoquinoline derivatives (1518) were synthesized in up to 98% yields with excellent diastereo- and enantioselectivities (>20:1 dr and up to 94% ee). Preliminary results and DFT studies suggested that the π–π interaction between the indane group of the ligated side arm and the phenyl group of the cyclopropane plays a vital role in the control of enantioselectivity. On the basis of computational studies, the optimized model of Ni(II)/TOX (1507-A) was proposed to be a six-coordinate Ni(II) complex with one molecule of isoquinoline azomethine imine coordinating to the Ni center.
Scheme 450. Ni/In-TOX-Catalyzed Highly Enantioselective Asymmetric [3 + 3] Cycloaddition.

In 2018, the first Ni(II)/TOX-catalyzed asymmetric oxa-[3 + 3]-annulation of oxygenated phenols and 3-aminophenols (1519) with β,γ-unsaturated α-keto esters (1520) was disclosed (Scheme 451).556 This method allowed a rapid access to a variety of oxygenated and 7-aminated chromans (1521) in excellent yields (up to 95%) with excellent diastereoselectivities (90:1 dr) and enantioselectivities (up to 90% ee). The improved functionality tolerance was attributed to the probable interference of the oxazoline side arm with the coordination of the amino group to the catalyst. Also of note Tang applied the aforementioned TOX ligands in other chemistries such as the asymmetric Nazarov reaction, the enantioselective ring-opening of cyclopropanes and the asymmetric [4 + 3] annulation reaction.557−560 Greater detail of TOX ligands and side arm modification strategies for bis(oxazoline) ligand design has been discussed by Tang in the 2014 account.561
Scheme 451. Ni(II)/TOX-Catalyzed Asymmetric Oxa-[3 + 3]-annulation.

5.2. Tetra(oxazoline) Ligands
In 2010, Li reported a novel D2-symmetrical chiral tetraoxazoline ligand (1524) and applied it in a Cu-catalyzed asymmetric hydrosilylation of aromatic ketones (1522) with diphenylsilane to give optically active secondary alcohols (1523) (Scheme 452).562 The chiral catalyst showed excellent activities and enantioselectivities in the hydrosilylation of aryl ketones (1522) with up to 89% ee. A transition state model was proposed wherein diphenylsilane could only approach the Re-face of the aromatic ketones due to steric hindrance leading to the (S)-absolute configuration observed for all secondary alcohol products.
Scheme 452. D2-Symmetrical Chiral Tetraoxazoline Ligand for Asymmetric Hydrosilylation.

Bellemin-Laponnaz reported the synthesis and application of self-supported recyclable oxazoline catalysts 1525–1527 (Figure 81) in the α-hydrazination of β-keto esters 1535.
Figure 81.

Recyclable oxazoline catalysts.
Di-, tri- and tetratopic ligands bearing isopropyl substituents 1525-A, 1526, and 1527 were synthesized and screened in the α-hydrazination of β-keto esters 1528, while enantioselectivities were similar (78–82% ee), it was shown that the recyclability of the ligands varied greatly. After five catalytic runs ditopic tetraoxazoline ligand 1525-A gave the product 1530 in 83% ee and 87% yield, tritopic hexaoxazoline ligand 1526 afforded the product in 38% ee and <5% yield and tetratopic octaoxazoline ligand 1527 did not give rise to the product 1530 at all. Catalysts were recycled by a solvent swap which allowed them to be decanted from the reaction mixture. It was thought that during the solvent swap Cu was leading to the reduced results for the tri- and tetratopic ligands. When a substrate scope was carried out with 1525-B enantioselectivities of up to 99% ee and 99% yield were achieved with up to 10 catalytic runs possible with recycled catalysts (Scheme 453).563
Scheme 453. Asymmetric α-Hydrazination of β-Keto Esters.

6. Conclusion
This Review reports on further developments in the design and application of oxazoline-derived ligands in asymmetric catalysis over a 10-year period since 2009, when the area was previously reviewed by us.
The exceptionally wide utility of this ligand class in catalytic asymmetric synthesis has continued to be illustrated by the high levels of asymmetric induction in a variety of metal-catalyzed transformations in the review period. Many of the tried and trusted oxazoline-containing ligands like PHOX, BOX, and PyBOX continue to be applied with success to new transformations, and there are few examples where the levels of enantioselectivies obtained do not reach 99% ee. Variation in the ligand structures mainly relies upon small changes to well-established motifs with additional steric or electronic features being modified. Such structural changes to the parent ligands have been aided by mechanistic, spectroscopic, X-ray crystallographic, and computational studies. While there has been a considerable renaissance in metalla-photoredox and metalla-electrocatalysis in recent times, the combination of these methods with metal-catalyzed asymmetric synthesis has remained limited. As society moves toward increased sustainability, organic chemists must keep up, and there is considerable scope for adapting the privileged oxazoline motifs described in this Review to photoredox- and electrocatalytic methodologies. In a similar vein, we have seen a shift toward the development of catalysts based around the earth-abundant 3d transition metals such as cobalt and nickel. 3d Transition metals have been shown to have similar and, in some cases, superior reactivities compared to their more unsustainable, expensive and toxic late-transition-metal counterparts like palladium and rhodium. We hope to see considerable progress in 3d-transition-metal-catalyzed asymmetric synthesis in the coming years, and the use of oxazoline-based ligands in these transformations is a challenge that is just beginning to be addressed.
Although this Review highlights extensive research in the design, synthesis, and application of oxazoline-derived ligands, this topic is far from being exhausted and it is hoped that this Review will again stimulate both the development/design of new ligands and their applications in novel metal-catalyzed asymmetric transformations.
Chart 1. Master List of Ligands.
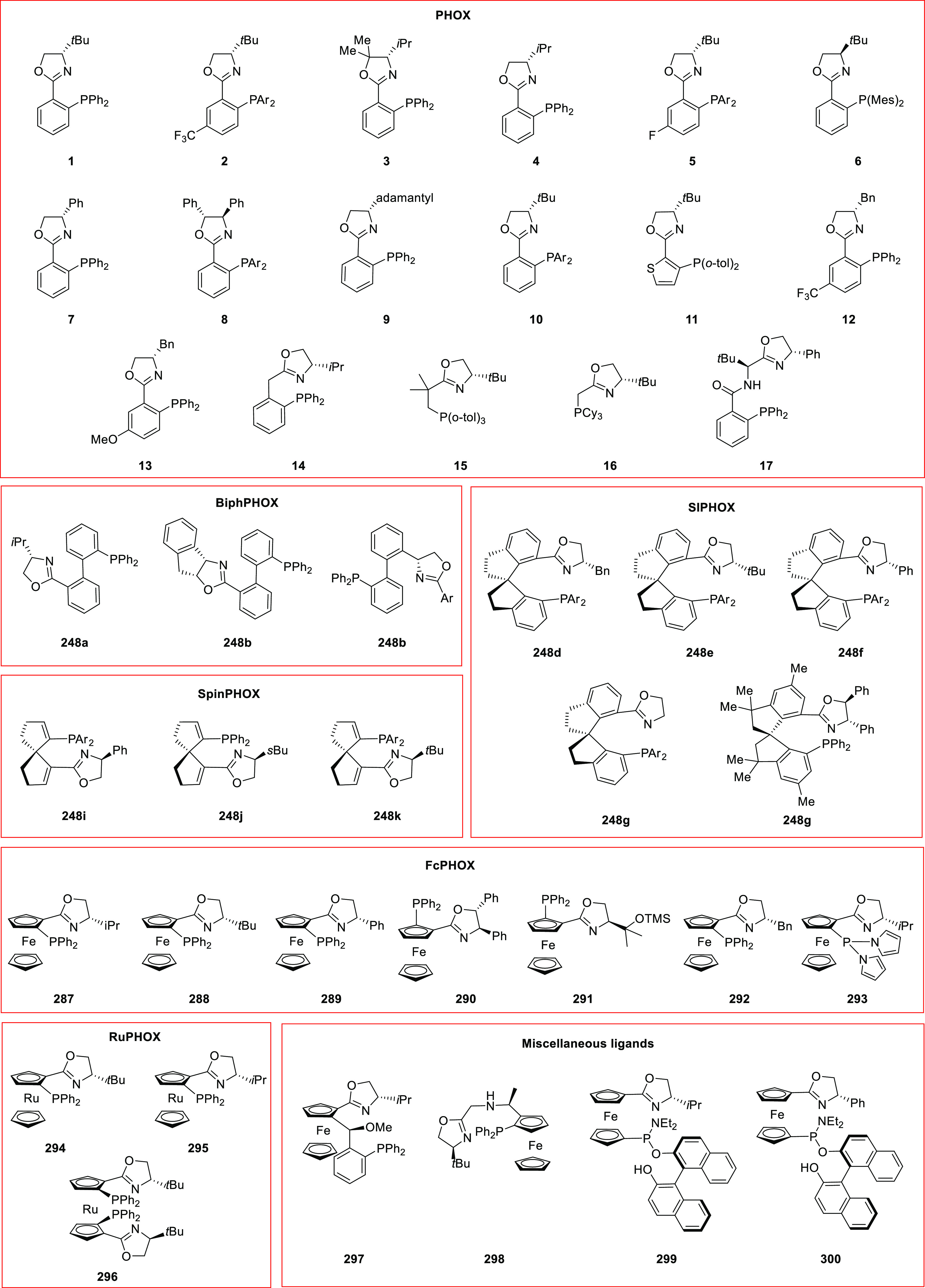
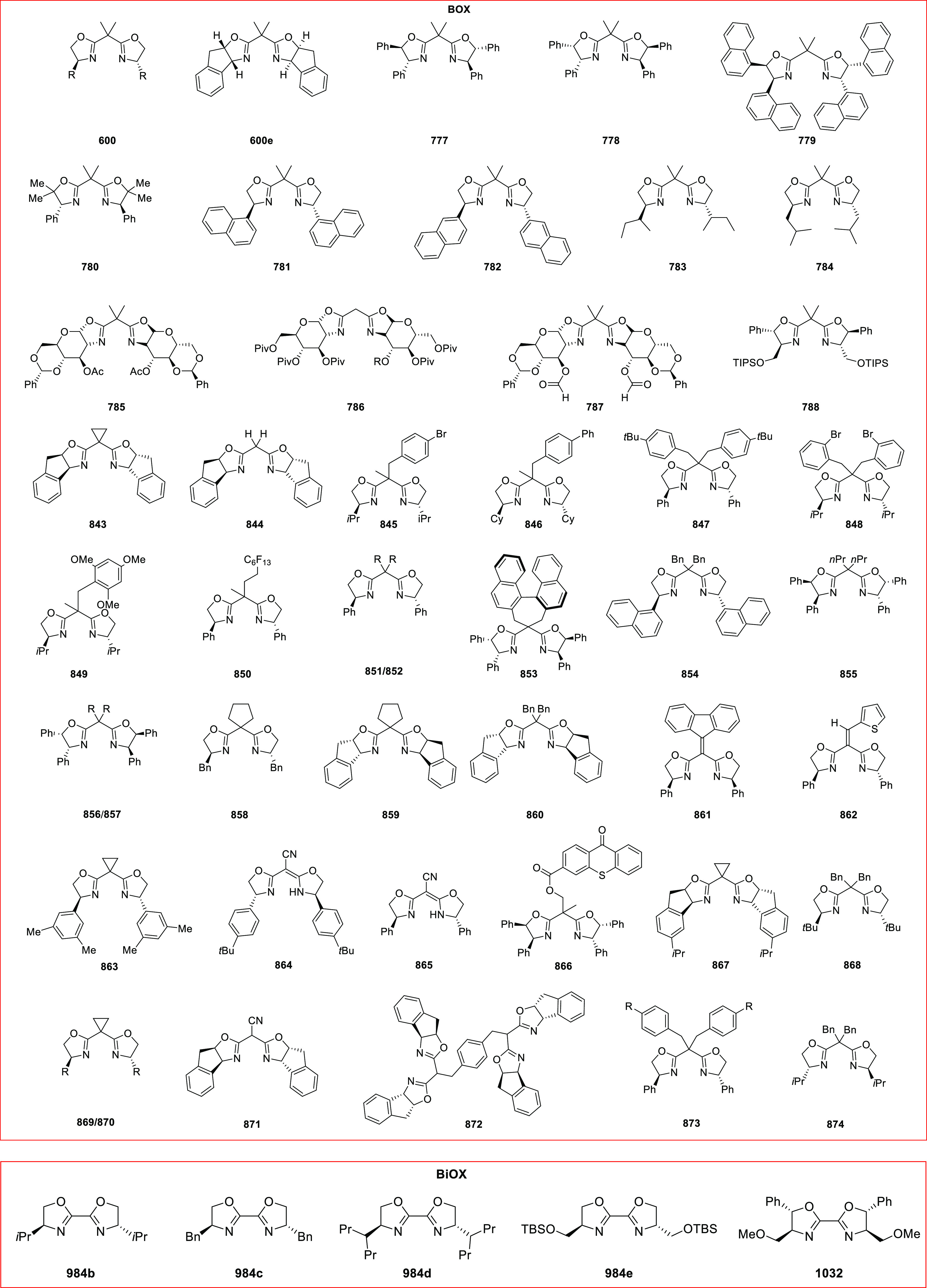
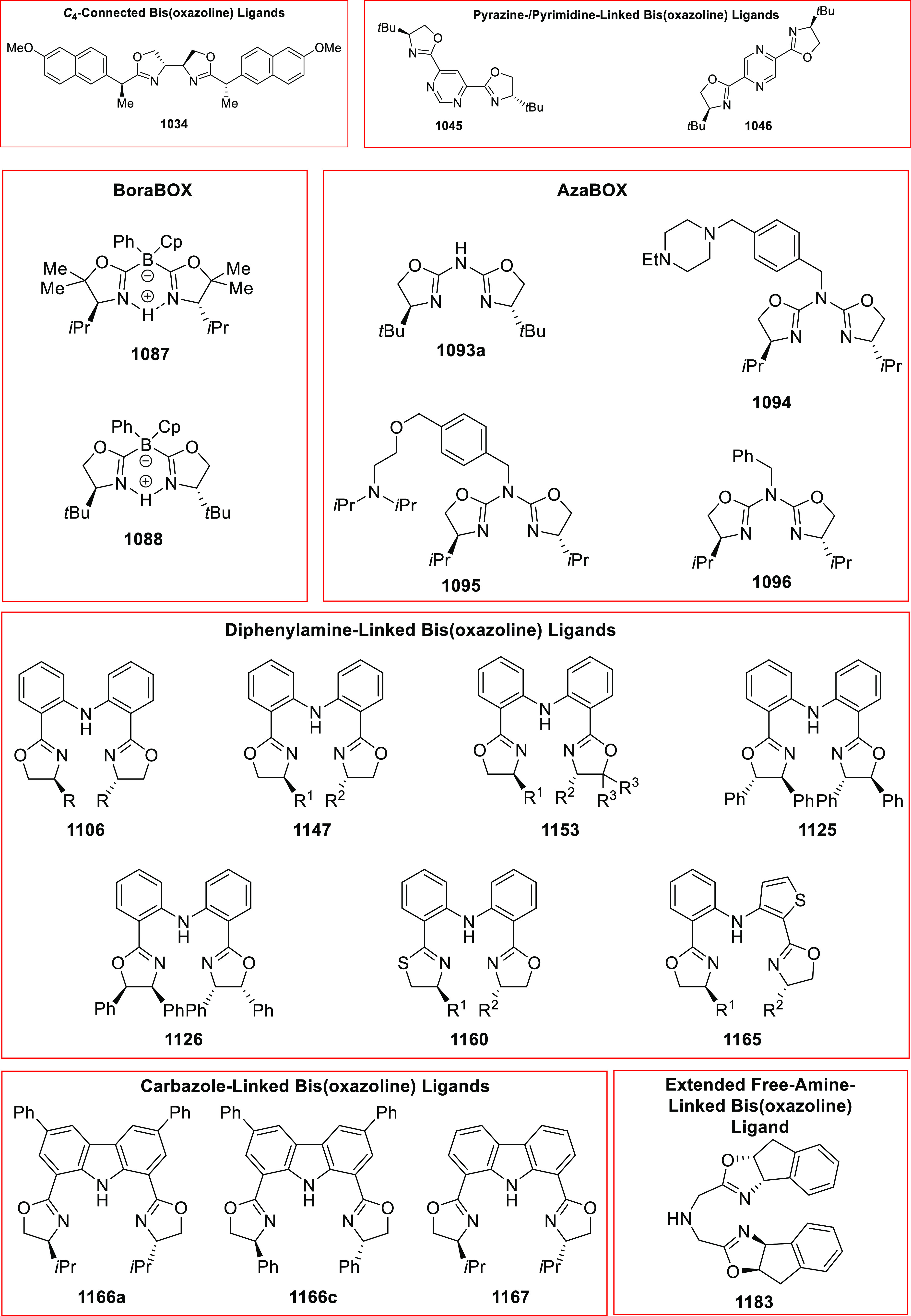
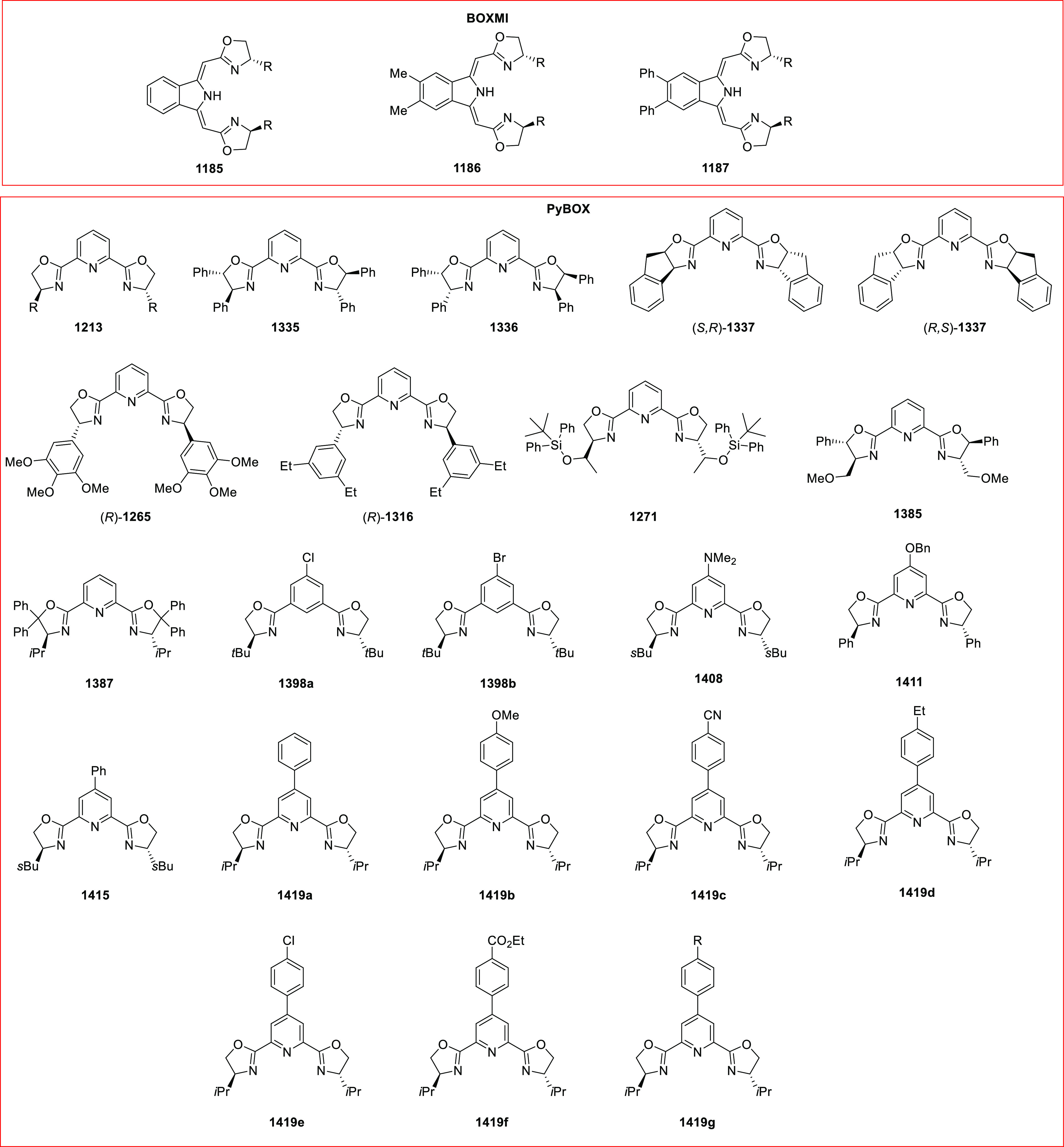
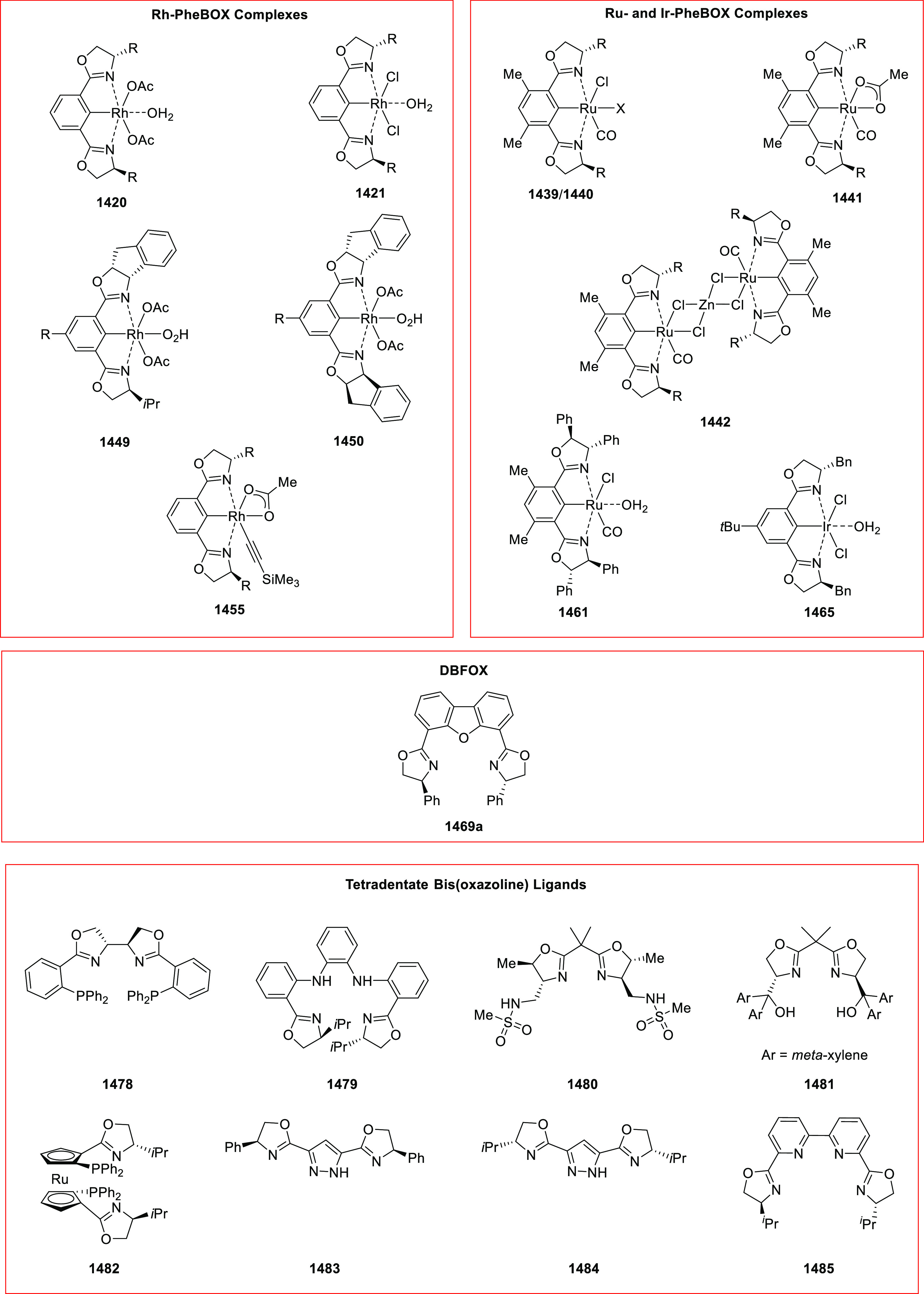
Acknowledgments
This publication has emanated from research conducted with the financial support of the Synthesis and Solid State Pharmaceutical Centre (SSPC), funded by Science Foundation Ireland (SFI) under Grant No. 12/RC/2275. B.V.R. is grateful for the award of a BiOrbic (Bioeconomy Research Centre) postdoctoral fellowship (16/RC/23889). B.V.R. also thanks the Irish Research Council (IRC) for the award of a postdoctoral fellowship (GOIPD/2015/453).
Biographies
Robert Connon received his B.Sc. in Chemistry from University College Dublin in 2015. He was then awarded his Ph.D. degree in 2020, also from University College Dublin, under the supervision of Prof. Pat Guiry. He is currently working as a postdoctoral researcher with Prof. Dr. Lutz Ackermann in the Georg-August-Universität Göttingen, Germany. His current research is focussed on the development of sustainable and asymmetric C–H activation methodologies.
Brendan Roche received his B.Sc. from the University College Dublin in 2015 in Chemistry. He was awarded his Ph.D degree in 2020 from University College Dublin under the supervision of Prof. Pat Guiry. His research interests include carboxylation chemistry and the development and design of oxazoline ligands for asymmetric catalysis.
Balaji Vasantrao Rokade received B.Sc. and M.Sc. degrees from the Swami Ramanand Teerth Marathwada University, Nanded, India. He was awarded a Ph.D. degree in 2014 from the Indian Institute of Science, Bangalore, under the supervision of Prof. K. R. Prabhu. He then moved to University College Dublin, Ireland, where he is presently working as a postdoctoral researcher with Prof. Pat Guiry. His current research is focused on the design and development of chiral P,N-ligands for asymmetric catalysis.
Patrick J. Guiry was born in County Tipperary and studied at University College Dublin (UCD), B.Sc. 1986 and Ph.D. 1990, with Professor Dervilla Donnelly and Professor Sir Derek Barton as Ph.D. supervisors. He carried out postdoctoral research in asymmetric catalysis with John M. Brown FRS (Oxford University). He joined UCD in 1993 and is currently the Director of the CSCB and Full Professor of Synthetic Organic Chemistry since 2006. His research interests include the design, synthesis, and application of novel ligands in asymmetric catalysis, natural product synthesis, and medicinal chemistry. He has supervised 56 Ph.D. students to graduation to date. He was elected a Member of the Royal Irish Academy in 2013 and is an elected member of the UCD Governing Authority and of the Senate of the National University of Ireland. He was the Science Secretary of the Royal Irish Academy from 2016–2020 and is the Vice President of the Institute of Chemistry in Ireland from 2019–present. He was recipient of the Institute of Chemistry of Ireland’s Boyle-Higgins Medal in 2014 and the Science Foundation Ireland Mentorship Award in 2020. A keen tennis player, with 18 Irish national titles, he was selected to represent Ireland in 2020 in the Austria Cup (ITF World Team Competition) in Florida, which was unfortunately canceled due to COVID-19.
Author Contributions
§ R.C., B.R., and B.V.R. contributed equally.
The authors declare no competing financial interest.
Dedication
Dedicated with respect and admiration to Dr. John Brown FRS, on the occasion of his 80th birthday, and with whom P.J.G. made his first bis(oxazoline) ligand in 1991.
References
- Hargaden G. C.; Guiry P. J. Recent Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2009, 109, 2505–2550. 10.1021/cr800400z. [DOI] [PubMed] [Google Scholar]
- McManus H. A.; Guiry P. J. Recent Developments in the Application of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2004, 104, 4151–4202. 10.1021/cr040642v. [DOI] [PubMed] [Google Scholar]
- Koch G.; Lloyd-Jones G. C.; Loiseleur O.; Pfaltz A.; Prétôt R.; Schaffner S.; Schnider P.; von Matt P. Synthesis of Chiral (Phosphinoaryl)Oxazolines, a Versatile Class of Ligands for Asymmetric Catalysis. Recl. des Trav. Chim. des Pays-Bas 1995, 114, 206–210. 10.1002/recl.19951140413. [DOI] [Google Scholar]
- Helmchen G.; Pfaltz A. Phosphinooxazolines - A New Class of Versatile, Modular P,N-Ligands for Asymmetric Catalysis. Acc. Chem. Res. 2000, 33, 336–345. 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- Tani K.; Behenna D. C.; McFadden R. M.; Stoltz B. M. A Facile and Modular Synthesis of Phosphinooxazoline Ligands. Org. Lett. 2007, 9, 2529–2531. 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- Carroll M. P.; Guiry P. J. P,N Ligands in Asymmetric Catalysis. Chem. Soc. Rev. 2014, 43, 819–833. 10.1039/C3CS60302D. [DOI] [PubMed] [Google Scholar]
- Behenna D. C.; Stoltz B. M. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Xu J. Regio- and Enantioselective Pd-Catalyzed Allylic Alkylation of Ketones through Allyl Enol Carbonates. J. Am. Chem. Soc. 2005, 127, 2846–2847. 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]
- James J.; Jackson M.; Guiry P. J. Palladium-Catalyzed Decarboxylative Asymmetric Allylic Alkylation: Development, Mechanistic Understanding and Recent Advances. Adv. Synth. Catal. 2019, 361, 3016–3049. 10.1002/adsc.201801575. [DOI] [Google Scholar]
- Enquist J. A.; Stoltz B. M. The Total Synthesis of (−)-Cyanthiwigin F by Means of Double Catalytic Enantioselective Alkylation. Nature 2008, 453, 1228–1231. 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist J. A.; Virgil S. C.; Stoltz B. M. Total Syntheses of Cyanthiwigins B, F, and G. Chem. - Eur. J. 2011, 17, 9957–9969. 10.1002/chem.201100425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. E.; Stoltz B. M. A Second-Generation Synthesis of the Cyanthiwigin Natural Product Core. Org. Lett. 2016, 18, 5720–5723. 10.1021/acs.orglett.6b02962. [DOI] [PubMed] [Google Scholar]
- Hong A. Y.; Krout M. R.; Jensen T.; Bennett N. B.; Harned A. M.; Stoltz B. M. Ring-Contraction Strategy for the Practical, Scalable, Catalytic Asymmetric Synthesis of Versatile γ-Quaternary Acylcyclopentenes. Angew. Chem., Int. Ed. 2011, 50, 2756–2760. 10.1002/anie.201007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves C. M.; Eidamshaus C.; Kim J.; Stoltz B. M. Enantioselective Construction of α-Quaternary Cyclobutanones by Catalytic Asymmetric Allylic Alkylation. Angew. Chem., Int. Ed. 2013, 52, 6718–6721. 10.1002/anie.201301815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartshore C. J.; Lupton D. W. Enantioselective Palladium-Catalyzed Decarboxylative Allylation of Carbazolones and Indolones: Formal Synthesis of (+)-KopsihainanineA. Angew. Chem., Int. Ed. 2013, 52, 4113–4116. 10.1002/anie.201209069. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang S.; Wu S.; Shen X.; Zou L.; Wang F.; Li X.; Peng F.; Zhang H.; Shao Z. Enantioselective Palladium-Catalyzed Decarboxylative Allylation of Carbazolones: Total Synthesis of (−)-Aspidospermidine and (+)-KopsihainanineA. Angew. Chem., Int. Ed. 2013, 52, 4117–4121. 10.1002/anie.201209878. [DOI] [PubMed] [Google Scholar]
- Bennett N. B.; Duquette D. C.; Kim J.; Liu W. B.; Marziale A. N.; Behenna D. C.; Virgil S. C.; Stoltz B. M. Expanding Insight into Asymmetric Palladium-Catalyzed Allylic Alkylation of N-Heterocyclic Molecules and Cyclic Ketones. Chem. - Eur. J. 2013, 19, 4414–4418. 10.1002/chem.201300030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behenna D. C.; Liu Y.; Yurino T.; Kim J.; White D. E.; Virgil S. C.; Stoltz B. M. Enantioselective Construction of Quaternary N-Heterocycles by Palladium-Catalysed Decarboxylative Allylic Alkylation of Lactams. Nat. Chem. 2012, 4, 130–133. 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korch K. M.; Eidamshaus C.; Behenna D. C.; Nam S.; Horne D.; Stoltz B. M. Enantioselective Synthesis of α-Secondary and α-Tertiary Piperazin-2- Ones and Piperazines by Catalytic Asymmetric Allylic Alkylation. Angew. Chem., Int. Ed. 2015, 54, 179–183. 10.1002/anie.201408609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numajiri Y.; Jiménez-Osés G.; Wang B.; Houk K. N.; Stoltz B. M. Enantioselective Synthesis of Dialkylated N-Heterocycles by Palladium-Catalyzed Allylic Alkylation. Org. Lett. 2015, 17, 1082–1085. 10.1021/ol503425t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambla M.; Duroure L.; Chabaud L.; Guillou C. Enantioselective Synthesis of Spiroimines by Asymmetric Decarboxylative Alkylation/Isomerization/[3 + 2]-Cycloaddition Reaction of Azidoalkenes. Eur. J. Org. Chem. 2014, 2014, 7716–7720. 10.1002/ejoc.201403161. [DOI] [Google Scholar]
- Pritchett B. P.; Kikuchi J.; Numajiri Y.; Stoltz B. M. Enantioselective Pd-Catalyzed Allylic Alkylation Reactions of Dihydropyrido[1,2-a]Indolone Substrates: Efficient Syntheses of (−)-Goniomitine, (+)-Aspidospermidine, and (−)-Quebrachamine. Angew. Chem., Int. Ed. 2016, 55, 13529–13532. 10.1002/anie.201608138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Goldstein E. L.; Stoltz B. M. Palladium-Catalyzed Enantioselective Csp3-Csp3 Cross-Coupling for the Synthesis of (Poly)Fluorinated Chiral Building Blocks. Org. Lett. 2018, 20, 5657–5660. 10.1021/acs.orglett.8b02369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuff J.; White D. E.; Virgil S. C.; Stoltz B. M. A Palladium-Catalysed Enolate Alkylation Cascade for the Formation of Adjacent Quaternary and Tertiary Stereocenters. Nat. Chem. 2010, 2, 192–196. 10.1038/nchem.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger É.; Houzé C.; Guimond N.; Cantin K.; Paquin J. F. Unexpected Effect of the Fluorine Atom on the Optimal Ligand-to-Palladium Ratio in the Enantioselective Pd-Catalyzed Allylation Reaction of Fluorinated Enol Carbonates. Chem. Commun. 2008, 1, 3251–3253. 10.1039/b803097a. [DOI] [PubMed] [Google Scholar]
- White D. E.; Stewart I. C.; Grubbs R. H.; Stoltz B. M. The Catalytic Asymmetric Total Synthesis of Elatol. J. Am. Chem. Soc. 2008, 130, 810–811. 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J. J.; McFadden R. M.; Virgil S. C.; Kolding H.; Alleva J. L.; Stoltz B. M. The Catalytic Enantioselective Total Synthesis of (+)-Liphagal. Angew. Chem., Int. Ed. 2011, 50, 6814–6818. 10.1002/anie.201101842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee H.; McDougal N. T.; Virgil S. C.; Stoltz B. M. A Catalytic, Asymmetric Formal Synthesis of (+)-Hamigeran B. Org. Lett. 2011, 13, 825–827. 10.1021/ol102669z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. W.; Wang C. C.; Xue H.; Dong Y.; Yang J. H.; Liu S.; Liu W. Q.; Li W. D. Z. Asymmetric Formal Synthesis of (−)-Cephalotaxine via Palladium-Catalyzed Enantioselective Tsuji Allylation. Org. Lett. 2018, 20, 1050–1053. 10.1021/acs.orglett.7b04008. [DOI] [PubMed] [Google Scholar]
- Hanessian S.; Chénard E. A New Approach to the Synthesis of Peptidomimetic Renin Inhibitors: Palladium-Catalyzed Asymmetric Allylation of Acyclic Alkyl Aryl Ketones. Org. Lett. 2012, 14, 3222–3225. 10.1021/ol301332f. [DOI] [PubMed] [Google Scholar]
- Alexy E. J.; Zhang H.; Stoltz B. M. Catalytic Enantioselective Synthesis of Acyclic Quaternary Centers: Palladium-Catalyzed Decarboxylative Allylic Alkylation of Fully Substituted Acyclic Enol Carbonates. J. Am. Chem. Soc. 2018, 140, 10109–10112. 10.1021/jacs.8b05560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto M.; Roizen J. L.; Stoltz B. M. Catalytic Enantioselective Alkylation of Substituted Dioxanone Enol Ethers: Ready Access to C(α)-Tetrasubstituted Hydroxyketones, Acids, and Esters. Angew. Chem., Int. Ed. 2008, 47, 6873–6876. 10.1002/anie.200801424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger É.; Pouliot M. F.; Paquin J. F. Use of 5,5-(Dimethyl)-i-Pr-PHOX as a Practical Equivalent to t-Bu-PHOX in Asymmetric Catalysis. Org. Lett. 2009, 11, 2201–2204. 10.1021/ol9005618. [DOI] [PubMed] [Google Scholar]
- Nahra F.; Macé Y.; Lambin D.; Riant O. Copper/Palladium-Catalyzed 1,4 Reduction and Asymmetric Allylic Alkylation of α,β-Unsaturated Ketones: Enantioselective Dual Catalysis. Angew. Chem., Int. Ed. 2013, 52, 3208–3212. 10.1002/anie.201208612. [DOI] [PubMed] [Google Scholar]
- Grenning A. J.; Van Allen C. K.; Maji T.; Lang S. B.; Tunge J. A. Development of Asymmetric Deacylative Allylation. J. Org. Chem. 2013, 78, 7281–7287. 10.1021/jo400793a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith J. A.; Behenna D. C.; Sherden N.; Mohr J. T.; Ma S.; Marinescu S. C.; Nielsen R. J.; Oxgaard J.; Stoltz B. M.; Goddard W. A. The Reaction Mechanism of the Enantioselective Tsuji Allylation: Inner-Sphere and Outer-Sphere Pathways, Internal Rearrangements, and Asymmetric C-C Bond Formation. J. Am. Chem. Soc. 2012, 134, 19050–19060. 10.1021/ja306860n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Mishra S.; Aponick A. Enol Acetates: Versatile Substrates for the Enantioselective Intermolecular Tsuji Allylation. J. Am. Chem. Soc. 2018, 140, 16152–16158. 10.1021/jacs.8b08746. [DOI] [PubMed] [Google Scholar]
- Craig R. A. II; Roizen J. L.; Smith R. C.; Jones A. C.; Virgil S. C.; Stoltz B. M. Enantioselective, Convergent Synthesis of the Ineleganolide Core by a Tandem Annulation Cascade. Chem. Sci. 2017, 8, 507–514. 10.1039/C6SC03347D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Shen H.; Wan X. L.; Chen Q. Y.; Guo Y. Enantioselective Pd-Catalyzed Allylation of Acyclic α-Fluorinated Ketones. J. Org. Chem. 2014, 79, 6347–6353. 10.1021/jo500923u. [DOI] [PubMed] [Google Scholar]
- Adamson N. J.; Hull E.; Malcolmson S. J. Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd-PHOX Catalyst. J. Am. Chem. Soc. 2017, 139, 7180–7183. 10.1021/jacs.7b03480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Malcolmson S. J. Development and Mechanistic Investigations of Enantioselective Pd-Catalyzed Intermolecular Hydroaminations of Internal Dienes. ACS Catal. 2018, 8, 8468–8476. 10.1021/acscatal.8b01914. [DOI] [Google Scholar]
- Adamson N. J.; Wilbur K. C. E.; Malcolmson S. J. Enantioselective Intermolecular Pd-Catalyzed Hydroalkylation of Acyclic 1,3-Dienes with Activated Pronucleophiles. J. Am. Chem. Soc. 2018, 140, 2761–2764. 10.1021/jacs.7b13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman K.; Wolf C. Catalytic Enantioselective and Diastereoselective Allylic Alkylation with Fluoroenolates: Efficient Access to C3-Fluorinated and All-Carbon Quaternary Oxindoles. Angew. Chem., Int. Ed. 2017, 56, 1390–1395. 10.1002/anie.201608752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Zhang H. J.; Yue W. J.; You S. L. Palladium-Catalyzed Highly Stereoselective Dearomative [3 + 2] Cycloaddition of Nitrobenzofurans. Chem. 2017, 3, 428–436. 10.1016/j.chempr.2017.06.015. [DOI] [Google Scholar]
- Cheng Q.; Zhang F.; Cai Y.; Guo Y. L.; You S. L. Stereodivergent Synthesis of Tetrahydrofuroindoles through Pd-Catalyzed Asymmetric Dearomative Formal [3 + 2] Cycloaddition. Angew. Chem., Int. Ed. 2018, 57, 2134–2138. 10.1002/anie.201711873. [DOI] [PubMed] [Google Scholar]
- Suetsugu S.; Nishiguchi H.; Tsukano C.; Takemoto Y. Asymmetric Synthesis of (−)-Aurantioclavine via Palladium-Catalyzed Intramolecular Allylic Amination. Org. Lett. 2014, 16, 996–999. 10.1021/ol4037314. [DOI] [PubMed] [Google Scholar]
- Linton E. C.; Kozlowski M. C. Catalytic Enantioselective Meerwein-Eschenmoser Claisen Rearrangement: Asymmetric Synthesis of Allyl Oxindoles. J. Am. Chem. Soc. 2008, 130, 16162–16163. 10.1021/ja807026z. [DOI] [PubMed] [Google Scholar]
- Cao T.; Deitch J.; Linton E. C.; Kozlowski M. C. Asymmetric Synthesis of Allenyl Oxindoles and Spirooxindoles by a Catalytic Enantioselective Saucy-Marbet Claisen Rearrangement. Angew. Chem., Int. Ed. 2012, 51, 2448–2451. 10.1002/anie.201107417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston C.; James J.; Guiry P. J. Development of and Recent Advances in Pd-Catalyzed Decarboxylative Asymmetric Protonation. J. Org. Chem. 2019, 84, 473–485. 10.1021/acs.joc.8b02478. [DOI] [PubMed] [Google Scholar]
- Marinescu S. C.; Nishimata T.; Mohr J. T.; Stoltz B. M. Homogeneous Pd-Catalyzed Enantioselective Decarboxylative Protonation. Org. Lett. 2008, 10, 1039–1042. 10.1021/ol702821j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Sun Z.; Mo M.; Peng F.; Shao Z. Catalytic Asymmetric Assembly of C3-Monosubstituted Chiral Carbazolones and Concise Formal Synthesis of (−)-Aspidofractinine: Application of Enantioselective Pd-Catalyzed Decarboxylative Protonation of Carbazolones. Org. Lett. 2014, 16, 4178–4181. 10.1021/ol501877x. [DOI] [PubMed] [Google Scholar]
- Carroll M. P.; Müller-Bunz H.; Guiry P. J. Enantioselective Construction of Sterically Hindered Tertiary α-Aryl Ketones: A Catalytic Asymmetric Synthesis of Isoflavanones. Chem. Commun. 2012, 48, 11142–11144. 10.1039/c2cc36452b. [DOI] [PubMed] [Google Scholar]
- Doran R.; Carroll M. P.; Akula R.; Hogan B. F.; Martins M.; Fanning S.; Guiry P. J. A Stereoselective Switch: Enantiodivergent Approach to the Synthesis of Isoflavanones. Chem. - Eur. J. 2014, 20, 15354–15359. 10.1002/chem.201405246. [DOI] [PubMed] [Google Scholar]
- Doran R.; Guiry P. J. Catalytic Asymmetric Synthesis of Sterically Hindered Tertiary α-Aryl Ketones. J. Org. Chem. 2014, 79, 9112–9124. 10.1021/jo5014806. [DOI] [PubMed] [Google Scholar]
- Kingston C.; Guiry P. J. Enantiodivergent Synthesis of Tertiary α-Aryl 1-Indanones: Evidence Toward Disparate Mechanisms in the Palladium-Catalyzed Decarboxylative Asymmetric Protonation. J. Org. Chem. 2017, 82, 3806–3819. 10.1021/acs.joc.7b00303. [DOI] [PubMed] [Google Scholar]
- Lu S. M.; Bolm C. Highly Enantioselective Synthesis of Optically Active Ketones by Iridium-Catalyzed Asymmetric Hydrogenation. Angew. Chem., Int. Ed. 2008, 47, 8920–8923. 10.1002/anie.200803709. [DOI] [PubMed] [Google Scholar]
- Lu W. J.; Chen Y. W.; Hou X. L. Iridium-Catalyzed Highly Enantioselective Hydrogenation of the C=C Bond of α, β-Unsaturated Ketones. Angew. Chem., Int. Ed. 2008, 47, 10133–10136. 10.1002/anie.200803872. [DOI] [PubMed] [Google Scholar]
- Zheng Z.; Cao Y.; Chong Q.; Han Z.; Ding J.; Luo C.; Wang Z.; Zhu D.; Zhou Q. L.; Ding K. Chiral Cyclohexyl-Fused Spirobiindanes: Practical Synthesis, Ligand Development, and Asymmetric Catalysis. J. Am. Chem. Soc. 2018, 140, 10374–10381. 10.1021/jacs.8b07125. [DOI] [PubMed] [Google Scholar]
- Baeza A.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of Unfunctionalized Enamines. Chem. - Eur. J. 2009, 15, 2266–2269. 10.1002/chem.200802576. [DOI] [PubMed] [Google Scholar]
- Weise C. F.; Pischl M.; Pfaltz A.; Schneider C. A Non-Iterative, Flexible, and Highly Stereoselective Synthesis of Polydeoxypropionates—Synthesis of (+)-Vittatalactone. Chem. Commun. 2011, 47, 3248–3250. 10.1039/c0cc05215a. [DOI] [PubMed] [Google Scholar]
- Pischl M. C.; Weise C. F.; Haseloff S.; Müller M. A.; Pfaltz A.; Schneider C. A Highly Stereoselective and Flexible Strategy for the Convergent Synthesis of Long-Chain Polydeoxypropionates: Application towards the Synthesis of the Glycolipid Membrane Components Hydroxyphthioceranic and Phthioceranic Acid. Chem. - Eur. J. 2014, 20, 17360–17374. 10.1002/chem.201404034. [DOI] [PubMed] [Google Scholar]
- Lu W. J.; Chen Y. W.; Hou X. L. Highly Enantioselective Iridium-Catalyzed Hydrogenation of Trisubstituted Olefins, α,β-Unsaturated Ketones and Imines with Chiral Benzylic Substituted P,N Ligands. Adv. Synth. Catal. 2010, 352, 103–107. 10.1002/adsc.200900618. [DOI] [Google Scholar]
- Schrems M. G.; Pfaltz A. NeoPHOX - An Easily Accessible P,N-Ligand for Iridium-Catalyzed Asymmetric Hydrogenation: Preparation, Scope and Application in the Synthesis of Demethyl Methoxycalamenene. Chem. Commun. 2009, (41), 6210–6212. 10.1039/b912680e. [DOI] [PubMed] [Google Scholar]
- Baeza A.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of N-Protected Indoles. Chem. - Eur. J. 2010, 16, 2036–2039. 10.1002/chem.200903105. [DOI] [PubMed] [Google Scholar]
- Ikeda R.; Kuwano R. Asymmetric Hydrogenation of Isoxazolium Triflates with a Chiral Iridium Catalyst. Chem. - Eur. J. 2016, 22, 8610–8618. 10.1002/chem.201600732. [DOI] [PubMed] [Google Scholar]
- Clarke C.; Incerti-Pradillos C. A.; Lam H. W. Enantioselective Nickel-Catalyzed Anti-Carbometallative Cyclizations of Alkynyl Electrophiles Enabled by Reversible Alkenylnickel E/Z Isomerization. J. Am. Chem. Soc. 2016, 138, 8068–8071. 10.1021/jacs.6b04206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karad S. N.; Panchal H.; Clarke C.; Lewis W.; Lam H. W. Enantioselective Synthesis of Chiral Cyclopent-2-Enones by Nickel-Catalyzed Desymmetrization of Malonate Esters. Angew. Chem., Int. Ed. 2018, 57, 9122–9125. 10.1002/anie.201805578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchal H.; Clarke C.; Bell C.; Karad S. N.; Lewis W.; Lam H. W. Nickel-Catalyzed, Ligand-Free, Diastereoselective Synthesis of 3-Methyleneindan-1-Ols. Chem. Commun. 2018, 54, 12389–12392. 10.1039/C8CC06388E. [DOI] [PubMed] [Google Scholar]
- Chen M. H.; Hsieh J. C.; Lee Y. H.; Cheng C. H. Controlled Synthesis of Enantioselective 1-Aminoindenes via Cobalt-Catalyzed [3 + 2] Annulation Reaction. ACS Catal. 2018, 8, 9364–9369. 10.1021/acscatal.8b02566. [DOI] [Google Scholar]
- Nguyen T. L. N.; Incerti-Pradillos C. A.; Lewis W.; Lam H. W. Enantioselective Nickel-Catalyzed Arylative Intramolecular 1,4-Allylations. Chem. Commun. 2018, 54, 5622–5625. 10.1039/C8CC03204A. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Jiang C.; Gandi V. R.; Lu Y.; Hayashi T. Palladium-Catalyzed Asymmetric Arylation of Trifluoromethylated/Perfluoroalkylated 2-Quinazolinones with High Enantioselectivity. Chem. - Eur. J. 2016, 22, 13068–13071. 10.1002/chem.201603105. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Wu B.; Gao X.; Zhou Y. G. Enantioselective Synthesis of Quaternary α-Aminophosphonates by Pd-Catalyzed Arylation of Cyclic α-Ketiminophosphonates with Arylboronic Acids. Chem. Commun. 2016, 52, 10882–10885. 10.1039/C6CC04096A. [DOI] [PubMed] [Google Scholar]
- Chen W.; Meng D.; N’Zemba B.; Morris W. J. Palladium-Catalyzed Enantioselective Synthesis of Cyclic Sulfamidates and Application to a Synthesis of Verubecestat. Org. Lett. 2018, 20, 1265–1268. 10.1021/acs.orglett.7b03639. [DOI] [PubMed] [Google Scholar]
- Mazet C.; Gérard D. Highly Regio- and Enantioselective Catalytic Asymmetric Hydroboration of α-Substituted Styrenyl Derivatives. Chem. Commun. 2011, 47, 298–300. 10.1039/C0CC01547D. [DOI] [PubMed] [Google Scholar]
- García-Fortanet J.; Buchwald S. L. Asymmetric Palladium-Catalyzed Intramolecular α-Arylation of Aldehydes. Angew. Chem., Int. Ed. 2008, 47, 8108–8111. 10.1002/anie.200803809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagar V. V.; RajanBabu T. V. Tandem Catalysis for Asymmetric Coupling of Ethylene and Enynes to Functionalized Cyclobutanes. Science 2018, 361, 68–72. 10.1126/science.aat6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dounay A. B.; Humphreys P. G.; Overman L. E.; Wrobleski A. D. Total Synthesis of the Strychnos Alkaloid (+)-Minfiensine: Tandem Enantioselective Intramolecular Heck-Iminium Ion Cyclization. J. Am. Chem. Soc. 2008, 130, 5368–5377. 10.1021/ja800163v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick M. O.; Muller-Bunz H.; Guiry P. J. The Synthesis of New HetPHOX Ligands and Their Application to the Intermolecular Asymmetric Heck Reaction. Eur. J. Org. Chem. 2009, 2009, 1889–1895. 10.1002/ejoc.200800761. [DOI] [Google Scholar]
- Kong W.; Wang Q.; Zhu J. Water as a Hydride Source in Palladium-Catalyzed Enantioselective Reductive Heck Reactions. Angew. Chem., Int. Ed. 2017, 56, 3987–3991. 10.1002/anie.201700195. [DOI] [PubMed] [Google Scholar]
- Jiang Z.; Hou L.; Ni C.; Chen J.; Wang D.; Tong X. Enantioselective Construction of Quaternary Tetrahydropyridines by Palladium-Catalyzed Vinylborylation of Alkenes. Chem. Commun. 2017, 53, 4270–4273. 10.1039/C7CC01488K. [DOI] [PubMed] [Google Scholar]
- Hou L.; Yuan Y.; Tong X. Pd(0)-Catalysed Asymmetric Reductive Heck-Type Cyclization of (: Z)-1-Iodo-1,6-Dienes and Enantioselective Synthesis of Quaternary Tetrahydropyridines. Org. Biomol. Chem. 2017, 15, 4803–4806. 10.1039/C7OB00762K. [DOI] [PubMed] [Google Scholar]
- Babu K. N.; Kinthada L. K.; Pratim Das P.; Bisai A. Cu(II)-TBu-PHOX Catalyzed Enantioselective Malonate Addition onto 3-Hydroxy 2-Oxindoles: Application in the Synthesis of Dimeric Pyrroloindoline Alkaloids. Chem. Commun. 2018, 54, 7963–7966. 10.1039/C8CC04338H. [DOI] [PubMed] [Google Scholar]
- Cook M. J.; Rovis T. Enantioselective Rhodium-Catalyzed Alkylative Desymmetrization of 3,5-Dimethylglutaric Anhydride. Synthesis 2009, 2009 (2), 335–338. 10.1055/s-0028-1083275. [DOI] [Google Scholar]
- Shin M.; Gu M.; Lim S. S.; Kim M.-J.; Lee J.; Jin H.; Jang Y. H.; Jung B. Cu I -Catalysed Enantioselective Alkyl 1,4-Additions to (E)-Nitroalkenes and Cyclic Enones with Phosphino-Oxazoline Ligands. Eur. J. Org. Chem. 2018, 2018, 3122–3130. 10.1002/ejoc.201800476. [DOI] [Google Scholar]
- Liu Y.; Zhang W. Iridium-Catalyzed Asymmetric Hydrogenation of α-Alkylidene Succinimides. Angew. Chem., Int. Ed. 2013, 52, 2203–2206. 10.1002/anie.201209126. [DOI] [PubMed] [Google Scholar]
- Xia J.; Yang G.; Zhuge R.; Liu Y.; Zhang W. Iridium-Catalyzed Asymmetric Hydrogenation of Unfunctionalized Exocyclic C=C Bonds. Chem. - Eur. J. 2016, 22, 18354–18357. 10.1002/chem.201604298. [DOI] [PubMed] [Google Scholar]
- Xia J.; Nie Y.; Yang G.; Liu Y.; Zhang W. Iridium-Catalyzed Asymmetric Hydrogenation of 2H-Chromenes: A Highly Enantioselective Approach to Isoflavan Derivatives. Org. Lett. 2017, 19, 4884–4887. 10.1021/acs.orglett.7b02341. [DOI] [PubMed] [Google Scholar]
- Meng K.; Xia J.; Wang Y.; Zhang X.; Yang G.; Zhang W. Ir/BiphPHOX-Catalyzed Asymmetric Hydrogenation of 3-Substituted 2,5-Dihydropyrroles and 2,5-Dihydrothiophene 1,1-Dioxides. Org. Chem. Front. 2017, 4, 1601–1605. 10.1039/C7QO00248C. [DOI] [Google Scholar]
- Quan M.; Tang L.; Shen J.; Yang G.; Zhang W. Ni(Ii)-Catalyzed Asymmetric Addition of Arylboronic Acids to Cyclic Imines. Chem. Commun. 2017, 53, 609–612. 10.1039/C6CC08759K. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Zhang Z.; Chen C.; Yang H.; Han Z.; Dong X. Q.; Zhang X. Iridium Catalysts with Modular Axial-Unfixed Biphenyl Phosphine-Oxazoline Ligands: Asymmetric Hydrogenation of α,β-Unsaturated Carboxylic Acids. Org. Chem. Front. 2017, 4, 627–630. 10.1039/C6QO00677A. [DOI] [Google Scholar]
- Song S.; Zhu S. F.; Li Y.; Zhou Q. L. Iridium-Catalyzed Enantioselective Hydrogenation of α,β- Unsaturated Carboxylic Acids with Tetrasubstituted Olefins. Org. Lett. 2013, 15, 3722–3725. 10.1021/ol401593a. [DOI] [PubMed] [Google Scholar]
- Song S.; Zhu S. F.; Yu Y. B.; Zhou Q. L. Carboxy-Directed Asymmetric Hydrogenation of 1,1-Diarylethenes and 1,1-Dialkylethenes. Angew. Chem., Int. Ed. 2013, 52, 1556–1559. 10.1002/anie.201208606. [DOI] [PubMed] [Google Scholar]
- Race N. J.; Faulkner A.; Fumagalli G.; Yamauchi T.; Scott J. S.; Rydén-Landergren M.; Sparkes H. A.; Bower J. F. Enantioselective Narasaka-Heck Cyclizations: Synthesis of Tetrasubstituted Nitrogen-Bearing Stereocenters. Chem. Sci. 2017, 8, 1981–1985. 10.1039/C6SC04466B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. N.; Yang L. C.; Rong Z. Q.; Liu T. L.; Liu R.; Zhao Y. Pd-Catalyzed Enantioselective [6 + 4] Cycloaddition of Vinyl Oxetanes with Azadienes to Access Ten-Membered Heterocycles. Angew. Chem., Int. Ed. 2018, 57, 1596–1600. 10.1002/anie.201711648. [DOI] [PubMed] [Google Scholar]
- Sun W.; Gu H.; Lin X. Synthesis and Application of Hexamethyl-1,1′-Spirobiindane-Based Phosphine-Oxazoline Ligands in Ni-Catalyzed Asymmetric Arylation of Cyclic Aldimines. J. Org. Chem. 2018, 83, 4034–4043. 10.1021/acs.joc.8b00422. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Han Z.; Li F.; Ding K.; Zhang A. Highly Enantioselective Hydrogenation of α-Aryl-β-Substituted Acrylic Acids Catalyzed by Ir-SpinPHOX. Chem. Commun. 2010, 46, 156–158. 10.1039/B919902K. [DOI] [PubMed] [Google Scholar]
- Liu X.; Han Z.; Wang Z.; Ding K. SpinPhox/Iridium(I)-Catalyzed Asymmetric Hydrogenation of Cyclic α-Alkylidene Carbonyl Compounds. Angew. Chem., Int. Ed. 2014, 53, 1978–1982. 10.1002/anie.201309521. [DOI] [PubMed] [Google Scholar]
- Ge Y.; Han Z.; Wang Z.; Feng C. G.; Zhao Q.; Lin G. Q.; Ding K. Ir-SpinPHOX Catalyzed Enantioselective Hydrogenation of 3-Ylidenephthalides. Angew. Chem., Int. Ed. 2018, 57, 13140–13144. 10.1002/anie.201807639. [DOI] [PubMed] [Google Scholar]
- Qiu Z.; Sun R.; Teng D. Synthesis of Highly Rigid Phosphine-Oxazoline Ligands for Palladium-Catalyzed Asymmetric Allylic Alkylation. Org. Biomol. Chem. 2018, 16, 7717–7724. 10.1039/C8OB02265H. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Qiu Z.; Sun R.; Gao N.; Cao G.; Teng D. New Spiro Phosphinooxazolines for Palladium-Catalyzed Asymmetric Allylic Amination. Tetrahedron Lett. 2018, 59, 3938–3941. 10.1016/j.tetlet.2018.09.044. [DOI] [Google Scholar]
- Nishibayashi Y.; Uemura S. Asymmetric Synthesis and Highly Diastereoselective Ortho-Lithiation of Oxazolinylferrocenes. Synlett 1995, 1995, 79–81. 10.1055/s-1995-4881. [DOI] [Google Scholar]
- Tong M. C.; Chen X.; Li J.; Huang R.; Tao H.; Wang C. J. Catalytic Asymmetric Synthesis of [2,3]-Fused Indoline Heterocycles through Inverse-Electron-Demand Aza-Diels-Alder Reaction of Indoles with Azoalkenes. Angew. Chem., Int. Ed. 2014, 53, 4680–4684. 10.1002/anie.201400109. [DOI] [PubMed] [Google Scholar]
- Wei L.; Yao L.; Wang Z. F.; Li H.; Tao H. Y.; Wang C. J. Copper(I)-Catalyzed Asymmetric 1,3-Dipolar [3 + 4] Cycloaddition of Nitrones with Azoalkenes. Adv. Synth. Catal. 2016, 358, 3748–3752. 10.1002/adsc.201600457. [DOI] [Google Scholar]
- Yang W. L.; Li C. Y.; Qin W. J.; Tang F. F.; Yu X.; Deng W. P. Cu(I)-Catalyzed Chemoselective and Stereoselective [3 + 3] Cycloaddition of Azomethine Ylides with 2-Indolylnitroethylenes: Facile Access to Highly Substituted Tetrahydro-γ-Carbolines. ACS Catal. 2016, 6, 5685–5690. 10.1021/acscatal.6b01596. [DOI] [Google Scholar]
- Liu Y. Z.; Shang S. J.; Zhu J. Y.; Yang W. L.; Deng W. P. Regioselective and Stereoselective [3 + 3] Annulation of Ketones Derived Azomethine Ylides with 2-Indolylethylenes: Direct Access to Highly Substituted Tetrahydro-γ-Carbolines. Adv. Synth. Catal. 2018, 360, 2191–2203. 10.1002/adsc.201800274. [DOI] [Google Scholar]
- Yang Q. L.; Xie M. S.; Xia C.; Sun H. L.; Zhang D. J.; Huang K. X.; Guo Z.; Qu G. R.; Guo H. M. A Rapid and Divergent Access to Chiral Azacyclic Nucleoside Analogues via Highly Enantioselective 1,3-Dipolar Cycloaddition of β-Nucleobase Substituted Acrylates. Chem. Commun. 2014, 50, 14809–14812. 10.1039/C4CC06632D. [DOI] [PubMed] [Google Scholar]
- Tang L. W.; Zhao B. J.; Dai L.; Zhang M.; Zhou Z. M. Asymmetric Construction of Pyrrolidines Bearing a Trifluoromethylated Quaternary Stereogenic Center via CuI-Catalyzed 1,3-Dipolar Cycloaddition of Azomethine Ylides with β-CF3-β,β-Disubstituted Nitroalkenes. Chem. - Asian J. 2016, 11, 2470–2477. 10.1002/asia.201600941. [DOI] [PubMed] [Google Scholar]
- Liu B.; Zhang Z. M.; Xu B.; Xu S.; Wu H. H.; Liu Y.; Zhang J. Cu(i)-Catalyzed Michael Addition of Ketiminoesters to β-Trifluoromethyl β,β-Disubstituted Enones: Rapid Access to 1-Pyrrolines Bearing a Quaternary All-Carbon Stereocenter. Org. Chem. Front. 2017, 4, 1772–1776. 10.1039/C7QO00291B. [DOI] [Google Scholar]
- He F. S.; Li C. S.; Deng H.; Zheng X.; Yang Z. T.; Deng W. P. The Facile and Stereoselective Synthesis of Pyrrolidine β-Amino Acids: Via Copper(i)-Catalyzed Asymmetric 1,3-Dipolar Cycloaddition. Org. Chem. Front. 2017, 4, 52–56. 10.1039/C6QO00512H. [DOI] [Google Scholar]
- Deng H.; Yang W. L.; Tian F.; Tang W.; Deng W. P. Asymmetric Construction of 3-Azabicyclo[3.1.0]Hexane Skeleton with Five Contiguous Stereogenic Centers by Cu-Catalyzed 1,3-Dipolar Cycloaddition of Trisubstituted Cyclopropenes. Org. Lett. 2018, 20, 4121–4125. 10.1021/acs.orglett.8b01686. [DOI] [PubMed] [Google Scholar]
- Xu S.; Zhang Z. M.; Xu B.; Liu B.; Liu Y.; Zhang J. Enantioselective Regiodivergent Synthesis of Chiral Pyrrolidines with Two Quaternary Stereocenters via Ligand-Controlled Copper(I)-Catalyzed Asymmetric 1,3-Dipolar Cycloadditions. J. Am. Chem. Soc. 2018, 140, 2272–2283. 10.1021/jacs.7b12137. [DOI] [PubMed] [Google Scholar]
- Ma C.; Huang Y.; Zhao Y. Stereoselective 1,6-Conjugate Addition/Annulation of Para-Quinone Methides with Vinyl Epoxides/Cyclopropanes. ACS Catal. 2016, 6, 6408–6412. 10.1021/acscatal.6b01845. [DOI] [Google Scholar]
- Yamauchi M.; Morimoto M.; Miura T.; Murakami M. Enantioselective Synthesis of 3,4-Dihydroisoquinolin-1(2H)-Ones by Nickel-Catalyzed Denitrogenative Annulation of 1,2,3-Benzotriazin-4(3H)-Ones with Allenes. J. Am. Chem. Soc. 2010, 132, 54–55. 10.1021/ja909603j. [DOI] [PubMed] [Google Scholar]
- Miura T.; Morimoto M.; Murakami M. Enantioselective [2 + 2 + 2] Cycloaddition Reaction of Isocyanates and Allenes Catalyzed by Nickel. J. Am. Chem. Soc. 2010, 132, 15836–15838. 10.1021/ja105541r. [DOI] [PubMed] [Google Scholar]
- Ochi Y.; Kurahashi T.; Matsubara S. Decarbonylative Cycloaddition of Phthalic Anhydrides with Allenes. Org. Lett. 2011, 13, 1374–1377. 10.1021/ol200044y. [DOI] [PubMed] [Google Scholar]
- Hernandez L. W.; Pospech J.; Klöckner U.; Bingham T. W.; Sarlah D. Synthesis of (+)-Pancratistatins via Catalytic Desymmetrization of Benzene. J. Am. Chem. Soc. 2017, 139, 15656–15659. 10.1021/jacs.7b10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L. W.; Klöckner U.; Pospech J.; Hauss L.; Sarlah D. Nickel-Catalyzed Dearomative Trans −1,2-Carboamination. J. Am. Chem. Soc. 2018, 140, 4503–4507. 10.1021/jacs.8b01726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura M.; Shved A. S.; Sarlah D. Palladium-Catalyzed Dearomative Syn-1,4-Carboamination. J. Am. Chem. Soc. 2017, 139, 17787–17790. 10.1021/jacs.7b11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeier M.; Leach K.; Zajac M. A. Asymmetric Conjugate Addition of Glycine Derivatives under Copper Catalysis. Angew. Chem., Int. Ed. 2011, 50, 12335–12338. 10.1002/anie.201105258. [DOI] [PubMed] [Google Scholar]
- Li Q.; Ding C. H.; Hou X. L.; Dai L. X. Diastereo- And Enantioselective Synthesis of α,γ-Diaminobutyric Acid Derivatives via Cu-Catalyzed Asymmetric Michael Reaction. Org. Lett. 2010, 12, 1080–1083. 10.1021/ol100060t. [DOI] [PubMed] [Google Scholar]
- Chen C. G.; Hou X. L.; Pu L. Highly Enantioselective Cu-Catalyzed Conjugate Addition-Elimination of Activated Ally Lie Acetates with Glycine Derivatives. Org. Lett. 2009, 11, 2073–2075. 10.1021/ol900439h. [DOI] [PubMed] [Google Scholar]
- He Z. T.; Zhao Y. S.; Tian P.; Wang C. C.; Dong H. Q.; Lin G. Q. Copper-Catalyzed Asymmetric Hydroboration of α-Dehydroamino Acid Derivatives: Facile Synthesis of Chiral β-Hydroxy-α-Amino Acids. Org. Lett. 2014, 16, 1426–1429. 10.1021/ol500219e. [DOI] [PubMed] [Google Scholar]
- He F. S.; Jin J. H.; Yang Z. T.; Yu X.; Fossey J. S.; Deng W. P. Direct Asymmetric Synthesis of β-Bis-Aryl-α-Amino Acid Esters via Enantioselective Copper-Catalyzed Addition of p-Quinone Methides. ACS Catal. 2016, 6, 652–656. 10.1021/acscatal.5b02619. [DOI] [Google Scholar]
- Ma Z.; Xie F.; Yu H.; Zhang Y.; Wu X.; Zhang W. Copper-Catalyzed Asymmetric 1,4-Conjugate Addition of Grignard Reagents to Linear α,β,γ,δ-Unsaturated Ketones. Chem. Commun. 2013, 49, 5292–5294. 10.1039/c3cc42088d. [DOI] [PubMed] [Google Scholar]
- Zhu J. Y.; Yang W. L.; Liu Y. Z.; Shang S. J.; Deng W. P. A Copper(i)-Catalyzed Asymmetric Mannich Reaction of Glycine Schiff Bases with Isatin-Derived Ketimines: Enantioselective Synthesis of 3-Substituted 3-Aminooxindoles. Org. Chem. Front. 2018, 5, 70–74. 10.1039/C7QO00691H. [DOI] [Google Scholar]
- Quan M.; Butt N.; Shen J.; Shen K.; Liu D.; Zhang W. The Synthesis of Chiral β-Aryl-α,β-Unsaturated Amino Alcohols via a Pd-Catalyzed Asymmetric Allylic Amination. Org. Biomol. Chem. 2013, 11, 7412–7419. 10.1039/c3ob41642a. [DOI] [PubMed] [Google Scholar]
- Huo X.; He R.; Fu J.; Zhang J.; Yang G.; Zhang W. Stereoselective and Site-Specific Allylic Alkylation of Amino Acids and Small Peptides via a Pd/Cu Dual Catalysis. J. Am. Chem. Soc. 2017, 139, 9819–9822. 10.1021/jacs.7b05460. [DOI] [PubMed] [Google Scholar]
- Huo X.; Fu J.; He X.; Chen J.; Xie F.; Zhang W. Pd/Cu Dual Catalysis: Highly Enantioselective Access to α-Substituted α-Amino Acids and α-Amino Amides. Chem. Commun. 2018, 54, 599–602. 10.1039/C7CC08732B. [DOI] [PubMed] [Google Scholar]
- Liu P.; Huo X.; Li B.; He R.; Zhang J.; Wang T.; Xie F.; Zhang W. Stereoselective Allylic Alkylation of 1-Pyrroline-5-Carboxylic Esters via a Pd/Cu Dual Catalysis. Org. Lett. 2018, 20, 6564–6568. 10.1021/acs.orglett.8b02902. [DOI] [PubMed] [Google Scholar]
- Huo X.; Zhang J.; Fu J.; He R.; Zhang W. Ir/Cu Dual Catalysis: Enantio- and Diastereodivergent Access to α,α-Disubstituted α-Amino Acids Bearing Vicinal Stereocenters. J. Am. Chem. Soc. 2018, 140, 2080–2084. 10.1021/jacs.8b00187. [DOI] [PubMed] [Google Scholar]
- Wei L.; Xu S. M.; Zhu Q.; Che C.; Wang C. J. Synergistic Cu/Pd Catalysis for Enantioselective Allylic Alkylation of Aldimine Esters: Access to α,α-Disubstituted α-Amino Acids. Angew. Chem., Int. Ed. 2017, 56, 12312–12316. 10.1002/anie.201707019. [DOI] [PubMed] [Google Scholar]
- Wei L.; Xiao L.; Wang C. J. Synergistic Cu/Pd Catalysis for Enantioselective Allylation of Ketimine Esters: The Direct Synthesis of α-Substituted α-Amino Acids and 2H-Pyrrols. Adv. Synth. Catal. 2018, 360, 4715–4719. 10.1002/adsc.201800986. [DOI] [Google Scholar]
- Wei L.; Zhu Q.; Xu S. M.; Chang X.; Wang C. J. Stereodivergent Synthesis of α,α-Disubstituted α-Amino Acids via Synergistic Cu/Ir Catalysis. J. Am. Chem. Soc. 2018, 140, 1508–1513. 10.1021/jacs.7b12174. [DOI] [PubMed] [Google Scholar]
- Nakao Y.; Ebata S.; Yada A.; Hiyama T.; Ikawa M.; Ogoshi S. Intramolecular Arylcyanation of Alkenes Catalyzed by Nickel/AlMe 2Cl. J. Am. Chem. Soc. 2008, 130, 12874–12875. 10.1021/ja805088r. [DOI] [PubMed] [Google Scholar]
- Hsieh J. C.; Ebata S.; Nakao Y.; Hiyama T. Asymmetric Synthesis of Indolines Bearing a Benzylic Quaternary Stereocenter through Intramolecular Arylcyanation of Alkenes. Synlett 2010, 2010, 1709–1711. 10.1055/s-0029-1219964. [DOI] [Google Scholar]
- Wang K.; Ding Z.; Zhou Z.; Kong W. Ni-Catalyzed Enantioselective Reductive Diarylation of Activated Alkenes by Domino Cyclization/Cross-Coupling. J. Am. Chem. Soc. 2018, 140, 12364–12368. 10.1021/jacs.8b08190. [DOI] [PubMed] [Google Scholar]
- Lu W. J.; Hou X. L. Highly Enantioselective Construction of the α-Chiral Center of Amides via Iridium-Catalyzed Hydrogenation of α,β-Unsaturated Amides. Adv. Synth. Catal. 2009, 351, 1224–1228. 10.1002/adsc.200900080. [DOI] [Google Scholar]
- Wang Y.; Liu Y.; Li K.; Yang G.; Zhang W. Iridium-Catalyzed Asymmetric Hydrogenation of Unsaturated Piperazin-2-Ones. Adv. Synth. Catal. 2017, 359, 1933–1941. 10.1002/adsc.201700175. [DOI] [Google Scholar]
- Wang Y.; Yang G.; Xie F.; Zhang W. A Ferrocene-Based NH-Free Phosphine-Oxazoline Ligand for Iridium-Catalyzed Asymmetric Hydrogenation of Ketones. Org. Lett. 2018, 20, 6135–6139. 10.1021/acs.orglett.8b02591. [DOI] [PubMed] [Google Scholar]
- Wu W.; Liu S.; Duan M.; Tan X.; Chen C.; Xie Y.; Lan Y.; Dong X. Q.; Zhang X. Iridium Catalysts with F-Amphox Ligands: Asymmetric Hydrogenation of Simple Ketones. Org. Lett. 2016, 18, 2938–2941. 10.1021/acs.orglett.6b01290. [DOI] [PubMed] [Google Scholar]
- Wu W.; Xie Y.; Li P.; Li X.; Liu Y.; Dong X. Q.; Zhang X. Asymmetric Hydrogenation of α-Hydroxy Ketones with an Iridium/f-Amphox Catalyst: Efficient Access to Chiral 1,2-Diols. Org. Chem. Front. 2017, 4, 555–559. 10.1039/C6QO00810K. [DOI] [Google Scholar]
- Hu Y.; Wu W.; Dong X. Q.; Zhang X. Efficient Access to Chiral 1,2-Amino Alcohols: Via Ir/f-Amphox-Catalyzed Asymmetric Hydrogenation of α-Amino Ketones. Org. Chem. Front. 2017, 4, 1499–1502. 10.1039/C7QO00237H. [DOI] [Google Scholar]
- Wu W.; You C.; Yin C.; Liu Y.; Dong X. Q.; Zhang X. Enantioselective and Diastereoselective Construction of Chiral Amino Alcohols by Iridium-f-Amphox-Catalyzed Asymmetric Hydrogenation via Dynamic Kinetic Resolution. Org. Lett. 2017, 19, 2548–2551. 10.1021/acs.orglett.7b00844. [DOI] [PubMed] [Google Scholar]
- Gu G.; Yang T.; Yu O.; Qian H.; Wang J.; Wen J.; Dang L.; Zhang X. Enantioselective Iridium-Catalyzed Hydrogenation of α-Keto Amides to α-Hydroxy Amides. Org. Lett. 2017, 19, 5920–5923. 10.1021/acs.orglett.7b02912. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Yin X.; Chen Z.; Dong X. Q.; Zhang X. Highly Enantioselective Ir/f-Amphox-Catalyzed Hydrogenation of Ketoamides: Efficient Access to Chiral Hydroxy Amides. Org. Chem. Front. 2018, 5, 2000–2003. 10.1039/C8QO00307F. [DOI] [Google Scholar]
- Yin C.; Wu W.; Hu Y.; Tan X.; You C.; Liu Y.; Chen Z.; Dong X. Q.; Zhang X. Iridium-Catalyzed Asymmetric Hydrogenation of Halogenated Ketones for the Efficient Construction of Chiral Halohydrins. Adv. Synth. Catal. 2018, 360, 2119–2124. 10.1002/adsc.201800267. [DOI] [Google Scholar]
- Zhang K.; Peng Q.; Hou X. L.; Wu Y. D. Highly Enantioselective Palladium-Catalyzed Alkylation of Acyclic Amides. Angew. Chem., Int. Ed. 2008, 47, 1741–1744. 10.1002/anie.200704629. [DOI] [PubMed] [Google Scholar]
- Lei B. L.; Ding C. H.; Yang X. F.; Wan X. L.; Hou X. L. Kinetic Resolution of 2,3-Dihydro-2-Substituted 4-Quinolones by Palladium-Catalyzed Asymmetric Allylic Alkylation. J. Am. Chem. Soc. 2009, 131, 18250–18251. 10.1021/ja9082717. [DOI] [PubMed] [Google Scholar]
- Yang X. F.; Ding C. H.; Li X. H.; Huang J. Q.; Hou X. L.; Dai L. X.; Wang P. J. Regio- and Enantioselective Palladium-Catalyzed Allylic Alkylation of Nitromethane with Monosubstituted Allyl Substrates: Synthesis of (R)-Rolipram and (R)-Baclofen. J. Org. Chem. 2012, 77, 8980–8985. 10.1021/jo301506p. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Zhang Q. S.; Fang P.; Chen T. G.; Zhu J.; Hou X. L. Pd-Catalyzed Highly Regio-, Diastereo-, and Enantioselective Allylic Alkylation of α-Fluorophosphonates. Chem. Commun. 2014, 50, 6751–6753. 10.1039/C4CC02158D. [DOI] [PubMed] [Google Scholar]
- Cheruku P.; Gohil S.; Andersson P. G. Asymmetric Hydrogenation of Enol Phosphinates by Iridium Catalysts Having N,P Ligands. Org. Lett. 2007, 9, 1659–1661. 10.1021/ol070325l. [DOI] [PubMed] [Google Scholar]
- Cheruku P.; Diesen J.; Andersson P. G. Asymmetric Hydrogenation of Di and Trisubstituted Enol Phosphinates with N,P-Ligated Iridium Complexes. J. Am. Chem. Soc. 2008, 130, 5595–5599. 10.1021/ja711372c. [DOI] [PubMed] [Google Scholar]
- Paptchikhine A.; Cheruku P.; Engman M.; Andersson P. G. Iridium-Catalyzed Enantioselective Hydrogenation of Vinyl Boronates. Chem. Commun. 2009, (40), 5996–5998. 10.1039/b912590f. [DOI] [PubMed] [Google Scholar]
- Verendel J. J.; Zhou T.; Li J. Q.; Paptchikhine A.; Lebedev O.; Andersson P. G. Highly Flexible Synthesis of Chiral Azacycles via Iridium-Catalyzed Hydrogenation. J. Am. Chem. Soc. 2010, 132, 8880–8881. 10.1021/ja103901e. [DOI] [PubMed] [Google Scholar]
- Li J. Q.; Peters B.; Andersson P. G. Highly Enantioselective Asymmetric Isomerization of Primary Allylic Alcohols with an Iridium-N,P Complex. Chem. - Eur. J. 2011, 17, 11143–11145. 10.1002/chem.201101524. [DOI] [PubMed] [Google Scholar]
- Li J. Q.; Quan X.; Andersson P. G. Highly Enantioselective Iridium-Catalyzed Hydrogenation of α,β-Unsaturated Esters. Chem. - Eur. J. 2012, 18, 10609–10616. 10.1002/chem.201200907. [DOI] [PubMed] [Google Scholar]
- Ponra S.; Rabten W.; Yang J.; Wu H.; Kerdphon S.; Andersson P. G. Diastereo- and Enantioselective Synthesis of Fluorine Motifs with Two Contiguous Stereogenic Centers. J. Am. Chem. Soc. 2018, 140, 13878–13883. 10.1021/jacs.8b08778. [DOI] [PubMed] [Google Scholar]
- Orgué S.; Flores-Gaspar A.; Biosca M.; Pàmies O.; Diéguez M.; Riera A.; Verdaguer X. Stereospecific SN2@P Reactions: Novel Access to Bulky P-Stereogenic Ligands. Chem. Commun. 2015, 51, 17548–17551. 10.1039/C5CC07504A. [DOI] [PubMed] [Google Scholar]
- Salomó E.; Orgué S.; Riera A.; Verdaguer X. Highly Enantioselective Iridium-Catalyzed Hydrogenation of Cyclic Enamides. Angew. Chem., Int. Ed. 2016, 55, 7988–7992. 10.1002/anie.201602219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomó E.; Rojo P.; Hernández-Lladó P.; Riera A.; Verdaguer X. P-Stereogenic and Non-P-Stereogenic Ir-MaxPHOX in the Asymmetric Hydrogenation of N -Aryl Imines. Isolation and X-Ray Analysis of Imine Iridacycles. J. Org. Chem. 2018, 83, 4618–4627. 10.1021/acs.joc.8b00361. [DOI] [PubMed] [Google Scholar]
- Cabré A.; Khaizourane H.; Garçon M.; Verdaguer X.; Riera A. Total Synthesis of (R)-Sarkomycin Methyl Ester via Regioselective Intermolecular Pauson-Khand Reaction and Iridium-Catalyzed Asymmetric Isomerization. Org. Lett. 2018, 20, 3953–3957. 10.1021/acs.orglett.8b01525. [DOI] [PubMed] [Google Scholar]
- Bess E. N.; Sigman M. S. Distinctive Meta-Directing Group Effect for Iridium-Catalyzed 1,1-Diarylalkene Enantioselective Hydrogenation. Org. Lett. 2013, 15, 646–649. 10.1021/ol303465c. [DOI] [PubMed] [Google Scholar]
- Müller M. A.; Pfaltz A. Asymmetric Hydrogenation of α,β-Unsaturated Nitriles with Base-Activated Iridium N,P Ligand Complexes. Angew. Chem., Int. Ed. 2014, 53, 8668–8671. 10.1002/anie.201402053. [DOI] [PubMed] [Google Scholar]
- Lölsberg W.; Werle S.; Neudörfl J. M.; Schmalz H. G. An Enantioselective Total Synthesis of Helioporins C and E. Org. Lett. 2012, 14, 5996–5999. 10.1021/ol302898h. [DOI] [PubMed] [Google Scholar]
- Maurer F.; Huch V.; Ullrich A.; Kazmaier U. Development of Catalysts for the Stereoselective Hydrogenation of α,β-Unsaturated Ketones. J. Org. Chem. 2012, 77, 5139–5143. 10.1021/jo300246c. [DOI] [PubMed] [Google Scholar]
- Yang G.; Zhang W. Renaissance of Pyridine-Oxazolines as Chiral Ligands for Asymmetric Catalysis. Chem. Soc. Rev. 2018, 47, 1783–1810. 10.1039/C7CS00615B. [DOI] [PubMed] [Google Scholar]
- Brunner H.; Obermann U.; Wimmer P. Asymmetrische Katalysen. XXXII. Enantioselektive Phenylierung von Cis-Cyclohexan-1,2-Diol Und Meso-Butan-2,3-Diol. J. Organomet. Chem. 1986, 316, 6–8. [Google Scholar]
- Arai Y.; Bunya Y.; Sengoku T.; Imamura Y. Synthesis of Chiral (Sulfinyl)Furyloxazoline Ligands and Its Application to Enantioselective Palladium-Catalyzed Allylic Alkylation. Heterocycles 2008, 76, 833–843. 10.3987/COM-08-S(N)18. [DOI] [Google Scholar]
- Gao Y. X.; Chang L.; Shi H.; Liang B.; Wongkhan K.; Chaiyaveij D.; Batsanov A. S.; Marder T. B.; Li C. C.; Yang Z.; et al. A Thiourea-Oxazoline Library with Axial Chirality: Ligand Synthesis and Studies of the Palladium-Catalyzed Enantioselective Bis(Methoxycarbonylation) of Terminal Olefins. Adv. Synth. Catal. 2010, 352, 1955–1966. 10.1002/adsc.201000070. [DOI] [Google Scholar]
- Ammann S. E.; Liu W.; White M. C. Enantioselective Allylic C-H Oxidation of Terminal Olefins to Isochromans by Palladium(II)/Chiral Sulfoxide Catalysis. Angew. Chem., Int. Ed. 2016, 55, 9571–9575. 10.1002/anie.201603576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Ali S. Z.; Ammann S. E.; White M. C. Asymmetric Allylic C-H Alkylation via Palladium(II)/ Cis-ArSOX Catalysis. J. Am. Chem. Soc. 2018, 140, 10658–10662. 10.1021/jacs.8b05668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.-S.; Wu M.-S.; Han Z.-Y. Palladium-Catalyzed Cascade Sp2 C-H Functionalization/Intramolecular Asymmetric Allylation: From Aryl Ureas and 1,3-Dienes to Chiral Indolines. Angew. Chem., Int. Ed. 2017, 56, 6641–6645. 10.1002/anie.201702745. [DOI] [PubMed] [Google Scholar]
- Bolm C.; Muñiz Fernández K.; Seger A.; Raabe G. A New Ferrocene in the Asymmetric Addition of Diethylzinc to Aldehydes. Synlett 1997, 1997, 1051–1052. 10.1055/s-1997-1528. [DOI] [Google Scholar]
- Garabatos-Perera J. R.; Butenschön H. New Chiral Ferrocenyloxazolines: The First Planar Chiral Triferrocenylmethane Derivative and Its Use in Asymmetric Catalysis. J. Organomet. Chem. 2009, 694, 2047–2052. 10.1016/j.jorganchem.2009.01.057. [DOI] [Google Scholar]
- Nottingham C.; Benson R.; Müller-Bunz H.; Guiry P. J. Synthesis of Ferrocene Oxazoline N,O Ligands and Their Application in Asymmetric Ethyl- and Phenylzinc Additions to Aldehydes. J. Org. Chem. 2015, 80, 10163–10176. 10.1021/acs.joc.5b01766. [DOI] [PubMed] [Google Scholar]
- Zhou G.; Aboo A. H.; Robertson C. M.; Liu R.; Li Z.; Luzyanin K.; Berry N. G.; Chen W.; Xiao J. N,O- vs N,C-Chelation in Half-Sandwich Iridium Complexes: A Dramatic Effect on Enantioselectivity in Asymmetric Transfer Hydrogenation of Ketones. ACS Catal. 2018, 8, 8020–8026. 10.1021/acscatal.8b02068. [DOI] [Google Scholar]
- Yang D.; Wang L.; Han F.; Li D.; Zhao D.; Wang R. Intermolecular Enantioselective Dearomatization Reaction of β-Naphthol Using Meso-Aziridine: A Bifunctional in Situ Generated Magnesium Catalyst. Angew. Chem., Int. Ed. 2015, 54, 2185–2189. 10.1002/anie.201410257. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang D.; Li D.; Zhu H.; Wang P.; Liu X.; Bai L.; Wang R. Diversiform Reactivity of Naphthols in Asymmetric Dearomatization or O-Alkylation Reactions with Aziridines. Adv. Synth. Catal. 2018, 360, 4491–4496. 10.1002/adsc.201801041. [DOI] [Google Scholar]
- Yang D.; Wang L.; Kai M.; Li D.; Yao X.; Wang R. Application of a C-C Bond-Forming Conjugate Addition Reaction in Asymmetric Dearomatization of β-Naphthols. Angew. Chem., Int. Ed. 2015, 54, 9523–9527. 10.1002/anie.201503056. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang D.; Li D.; Wang P.; Wang K.; Wang J.; Jiang X.; Wang R. Mg(II)-Mediated Catalytic Asymmetric Dearomatization (CADA) Reaction of β-Naphthols with Dialkyl Acetylenedicarboxylates. Chem. - Eur. J. 2016, 22, 8483–8487. 10.1002/chem.201601399. [DOI] [PubMed] [Google Scholar]
- Wang K.; Wang L.; Liu X.; Li D.; Zhu H.; Wang P.; Liu Y.; Yang D.; Wang R. Development of Biligands Magnesium Catalysis in Asymmetric Conjugate Reactions of C3-Pyrrolyl-Oxindoles. Org. Lett. 2017, 19, 4351–4354. 10.1021/acs.orglett.7b02044. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang D.; Li D.; Liu X.; Zhao Q.; Zhu R.; Zhang B.; Wang R. Catalytic Asymmetric [3 + 2] Cyclization Reactions of 3-Isothiocyanato Oxindoles and Alkynyl Ketones Via an in Situ Generated Magnesium Catalyst. Org. Lett. 2015, 17, 4260–4263. 10.1021/acs.orglett.5b02052. [DOI] [PubMed] [Google Scholar]
- Kurosu M.; Lin M. H.; Kishi Y. Fe/Cr- and Co/Cr-Mediated Catalytic Asymmetric 2-Haloallylations of Aldehydes. J. Am. Chem. Soc. 2004, 126, 12248–12249. 10.1021/ja045557j. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Huang J.; Ma B.; Kishi Y. Further Improvement on Sulfonamide-Based Ligand for Catalytic Asymmetric 2-Haloallylation and Allylation. Org. Lett. 2008, 10, 3073–3076. 10.1021/ol801093p. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Aubry S.; Kishi Y. Iterative Cr-Mediated Catalytic Asymmetric Allylation to Synthesize Syn/Syn- and Anti/Anti-1,3,5-Triols. Org. Lett. 2008, 10, 3077–3080. 10.1021/ol801094e. [DOI] [PubMed] [Google Scholar]
- Liu S.; Kim J. T.; Dong C.-G.; Kishi Y. Catalytic Enantioselective Cr-Mediated Propargylation: Application to Halichondrin Synthesis. Org. Lett. 2009, 11, 4520–4523. 10.1021/ol9016595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Dong C. G.; Kim D. S.; Urabe D.; Wang J.; Kim J. T.; Liu X.; Sasaki T.; Kishi Y. Toolbox Approach to the Search for Effective Ligands for Catalytic Asymmetric Cr-Mediated Coupling Reactions. J. Am. Chem. Soc. 2009, 131, 15387–15393. 10.1021/ja905843e. [DOI] [PubMed] [Google Scholar]
- Kim D. S.; Dong C. G.; Kim J. T.; Guo H.; Huang J.; Tiseni P. S.; Kishi Y. New Syntheses of E7389 C14-C35 and Halichondrin C14-C38 Building Blocks: Double-Inversion Approach. J. Am. Chem. Soc. 2009, 131, 15636–15641. 10.1021/ja9058475. [DOI] [PubMed] [Google Scholar]
- Dong C. G.; Henderson J. A.; Kaburagi Y.; Sasaki T.; Kim D. S.; Kim J. T.; Urabe D.; Guo H.; Kishi Y. New Syntheses of E7389 C14-C35 and Halichondrin C14-C38 Building Blocks: Reductive Cyclization and Oxy-Michael Cyclization Approaches. J. Am. Chem. Soc. 2009, 131, 15642–15646. 10.1021/ja9058487. [DOI] [PubMed] [Google Scholar]
- Liu X.; Henderson J. A.; Sasaki T.; Kishi Y. Dramatic Improvement in Catalyst Loadings and Molar Ratios of Coupling Partners for Ni/Cr-Mediated Coupling Reactions: Heterobimetallic Catalysts. J. Am. Chem. Soc. 2009, 131, 16678–16680. 10.1021/ja9079308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zuo Z.; Wan X.; Huang Z. Cobalt-Catalyzed Enantioselective Hydroboration of 1,1-Disubstituted Aryl Alkenes. J. Am. Chem. Soc. 2014, 136, 15501–15504. 10.1021/ja5093908. [DOI] [PubMed] [Google Scholar]
- Chen J.; Xi T.; Ren X.; Cheng B.; Guo J.; Lu Z. Asymmetric Cobalt Catalysts for Hydroboration of 1,1-Disubstituted Alkenes. Org. Chem. Front. 2014, 1, 1306–1309. 10.1039/C4QO00295D. [DOI] [Google Scholar]
- Chen J.; Xi T.; Lu Z. Iminopyridine Oxazoline Iron Catalyst for Asymmetric Hydroboration of 1,1-Disubstituted Aryl Alkenes. Org. Lett. 2014, 16, 6452–6455. 10.1021/ol503282r. [DOI] [PubMed] [Google Scholar]
- Guo J.; Chen J.; Lu Z. Cobalt-Catalyzed Asymmetric Hydroboration of Aryl Ketones with Pinacolborane. Chem. Commun. 2015, 51, 5725–5727. 10.1039/C5CC01084E. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Lu Z. Dual-Stereocontrol Asymmetric Cobalt-Catalyzed Hydroboration of Sterically Hindered Styrenes. ACS Catal. 2016, 6, 6596–6600. 10.1021/acscatal.6b02278. [DOI] [Google Scholar]
- Chen J.; Chen C.; Ji C.; Lu Z. Cobalt-Catalyzed Asymmetric Hydrogenation of 1,1-Diarylethenes. Org. Lett. 2016, 18, 1594–1597. 10.1021/acs.orglett.6b00453. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Lu Z. Nickel-Catalyzed Enantioselective Sequential Nazarov Cyclization/Decarboxylation. Org. Chem. Front. 2018, 5, 1763–1767. 10.1039/C8QO00279G. [DOI] [Google Scholar]
- Zuo Z.; Zhang L.; Leng X.; Huang Z. Iron-Catalyzed Asymmetric Hydrosilylation of Ketones. Chem. Commun. 2015, 51, 5073–5076. 10.1039/C5CC00612K. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Lu P.; Zhang H.; Cheng X.; Lu Z. Highly Enantioselective Cobalt-Catalyzed Hydrosilylation of Alkenes. J. Am. Chem. Soc. 2017, 139, 9439–9442. 10.1021/jacs.7b04137. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Liu W.; Lu Z. Iron-Catalyzed Highly Enantioselective Hydrosilylation of Unactivated Terminal Alkenes. J. Am. Chem. Soc. 2018, 140, 5014–5017. 10.1021/jacs.8b01638. [DOI] [PubMed] [Google Scholar]
- Ye W.; Zhao M.; Du W.; Jiang Q.; Wu K.; Wu P.; Yu Z. Highly Active Ruthenium(II) Complex Catalysts Bearing an Unsymmetrical NNN Ligand in the (Asymmetric) Transfer Hydrogenation of Ketones. Chem. - Eur. J. 2011, 17, 4737–4741. 10.1002/chem.201002039. [DOI] [PubMed] [Google Scholar]
- Qi M. H.; Wang F. J.; Shi M. Synthesis of Novel Chiral Oxazoline-Schiff Base Ligands for the Catalytic Asymmetric Chlorination of β-Keto Esters. Tetrahedron: Asymmetry 2010, 21, 247–253. 10.1016/j.tetasy.2010.02.003. [DOI] [Google Scholar]
- Zhu Y.; Burgess K. Iridium-Catalyzed Asymmetric Hydrogenation of Vinyl Ethers. Adv. Synth. Catal. 2008, 350, 979–983. 10.1002/adsc.200700546. [DOI] [Google Scholar]
- Ito J. I.; Sugino K.; Matsushima S.; Sakaguchi H.; Iwata H.; Ishihara T.; Nishiyama H. Synthesis of NHC-Oxazoline Pincer Complexes of Rh and Ru and Their Catalytic Activity for Hydrogenation and Conjugate Reduction. Organometallics 2016, 35, 1885–1894. 10.1021/acs.organomet.6b00239. [DOI] [Google Scholar]
- Wang F.; Li S.; Qu M.; Zhao M. X.; Liu L. J.; Shi M. A Highly Efficient Kinetic Resolution of Morita-Baylis-Hillman Adducts Achieved by N-Ar Axially Chiral Pd-Complexes Catalyzed Asymmetric Allylation. Chem. Commun. 2011, 47, 12813–12815. 10.1039/c1cc15543a. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Sun X.; Chen H.; Liu P.; Jiang R. A Novel Planar Chiral N-Heterocyclic Carbene-Oxazoline Ligand for the Asymmetric Hydrosilylation of Ketones. Catal. Commun. 2009, 10, 1493–1496. 10.1016/j.catcom.2009.03.027. [DOI] [Google Scholar]
- Hong B.; Ma Y.; Zhao L.; Duan W.; He F.; Song C. Synthesis of Planar Chiral Imidazo[1,5-a]Pyridinium Salts Based on [2.2]Paracyclophane for Asymmetric β-Borylation of Enones. Tetrahedron: Asymmetry 2011, 22, 1055–1062. 10.1016/j.tetasy.2011.06.023. [DOI] [Google Scholar]
- Niu Z.; Chen J.; Chen Z.; Ma M.; Song C.; Ma Y. Application of Bidentate Oxazoline-Carbene Ligands with Planar and Central Chirality in Asymmetric β-Boration of α,β-Unsaturated Esters. J. Org. Chem. 2015, 80, 602–608. 10.1021/jo5021135. [DOI] [PubMed] [Google Scholar]
- Wang X.; Chen Z.; Duan W.; Song C.; Ma Y. Synthesis of [2.2]Paracyclophane-Based Bidentate Oxazoline-Carbene Ligands for the Asymmetric 1,2-Silylation of N-Tosylaldimines. Tetrahedron: Asymmetry 2017, 28, 783–790. 10.1016/j.tetasy.2017.05.001. [DOI] [Google Scholar]
- Abu-Elfotoh A. M.; Phomkeona K.; Shibatomi K.; Iwasa S. Asymmetric Inter- and Intramolecular Cyclopropanation Reactions Catalyzed by a Reusable Macroporous-Polymer-Supported Chiral Ruthenium(II)/Phenyloxazoline Complex. Angew. Chem., Int. Ed. 2010, 49, 8439–8443. 10.1002/anie.201002240. [DOI] [PubMed] [Google Scholar]
- Chanthamath S.; Ozaki S.; Shibatomi K.; Iwasa S. Highly Stereoselective Synthesis of Cyclopropylphosphonates Catalyzed by Chiral Ru(II)-Pheox Complex. Org. Lett. 2014, 16, 3012–3015. 10.1021/ol501135p. [DOI] [PubMed] [Google Scholar]
- Kotozaki M.; Chanthamath S.; Fujisawa I.; Shibatomi K.; Iwasa S. Highly Stereoselective Cyclopropanation of Various Olefins with Diazosulfones Catalyzed by Ru(II)-Pheox Complexes. Chem. Commun. 2017, 53, 12193–12196. 10.1039/C7CC05951E. [DOI] [PubMed] [Google Scholar]
- Kotozaki M.; Chanthamath S.; Fujii T.; Shibatomi K.; Iwasa S. Highly Enantioselective Synthesis of Trifluoromethyl Cyclopropanes by Using Ru(II)-Pheox Catalysts. Chem. Commun. 2018, 54, 5110–5113. 10.1039/C8CC02286K. [DOI] [PubMed] [Google Scholar]
- Abu-Elfotoh A. M.; Nguyen D. P. T.; Chanthamath S.; Phomkeona K.; Shibatomi K.; Iwasa S. Water-Soluble Chiral Ruthenium(II) Phenyloxazoline Complex: Reusable and Highly Enantioselective Catalyst for Intramolecular Cyclopropanation Reactions. Adv. Synth. Catal. 2012, 354, 3435–3439. 10.1002/adsc.201200508. [DOI] [Google Scholar]
- Chanthamath S.; Mandour H. S. A.; Tong T. M. T.; Shibatomi K.; Iwasa S. Highly Stereoselective Cyclopropanation of Diazo Weinreb Amides Catalyzed by Chiral Ru(II)-: Amm -Pheox Complexes. Chem. Commun. 2016, 52, 7814–7817. 10.1039/C6CC02498J. [DOI] [PubMed] [Google Scholar]
- Mandour H. S. A.; Chanthamath S.; Shibatomi K.; Iwasa S. Inter- and Intramolecular Cyclopropanations of Diazo Weinreb Amides Catalyzed by Ruthenium(II)-Amm-Pheox. Adv. Synth. Catal. 2017, 359, 1742–1746. 10.1002/adsc.201601345. [DOI] [Google Scholar]
- Overman L. E.; Owen C. E.; Pavan M. M.; Richards C. J. Catalytic Asymmetric Rearrangement of Allylic N-Aryl Trifluoroacetimidates. A Useful Method for Transforming Prochiral Allylic Alcohols to Chiral Allylic Amines. Org. Lett. 2003, 5, 1809–1812. 10.1021/ol0271786. [DOI] [PubMed] [Google Scholar]
- Cannon J. S.; Overman L. E. Palladium(II)-Catalyzed Enantioselective Reactions Using COP Catalysts. Acc. Chem. Res. 2016, 49, 2220–2231. 10.1021/acs.accounts.6b00398. [DOI] [PubMed] [Google Scholar]
- Fischer D. F.; Barakat A.; Xin Z.; Weiss M. E.; Peters R. The Asymmetric Aza-Claisen Rearrangement: Development of Widely Applicable Pentaphenylferrocenyl Palladacycle Catalysts. Chem. - Eur. J. 2009, 15, 8722–8741. 10.1002/chem.200900712. [DOI] [PubMed] [Google Scholar]
- Eitel S. H.; Bauer M.; Schweinfurth D.; Deibel N.; Sarkar B.; Kelm H.; Krüger H. J.; Frey W.; Peters R. Paramagnetic Palladacycles with Pd III Centers Are Highly Active Catalysts for Asymmetric Aza-Claisen Rearrangements. J. Am. Chem. Soc. 2012, 134, 4683–4693. 10.1021/ja2098222. [DOI] [PubMed] [Google Scholar]
- Bauer J. M.; Frey W.; Peters R. Asymmetric Cascade Reaction to Allylic Sulfonamides from Allylic Alcohols by Palladium(II)/Base-Catalyzed Rearrangement of Allylic Carbamates. Angew. Chem., Int. Ed. 2014, 53, 7634–7638. 10.1002/anie.201403090. [DOI] [PubMed] [Google Scholar]
- Bauer J. M.; Frey W.; Peters R. Dual Palladium(II)/Tertiary Amine Catalysis for Asymmetric Regioselective Rearrangements of Allylic Carbamates. Chem. - Eur. J. 2016, 22, 5767–5777. 10.1002/chem.201600138. [DOI] [PubMed] [Google Scholar]
- Hahn B. T.; Tewes F.; Fröhlich R.; Glorius F. Olefin-Oxazolines (OlefOx): Highly Modular, Easily Tunable Ligands for Asymmetric Catalysis. Angew. Chem., Int. Ed. 2010, 49, 1143–1146. 10.1002/anie.200905712. [DOI] [PubMed] [Google Scholar]
- Desimoni G.; Faita G.; Jørgensen K. A. Update 1 of: C 2 -Symmetric Chiral Bis(Oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev. 2011, 111, 284–437. 10.1021/cr100339a. [DOI] [PubMed] [Google Scholar]
- Srinivas H. D.; Maity P.; Yap G. P. A.; Watson M. P. Enantioselective Copper-Catalyzed Alkynylation of Benzopyranyl Oxocarbenium Ions. J. Org. Chem. 2015, 80, 4003–4016. 10.1021/acs.joc.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity P.; Srinivas H. D.; Watson M. P. Copper-Catalyzed Enantioselective Additions to Oxocarbenium Ions: Alkynylation of Isochroman Acetals. J. Am. Chem. Soc. 2011, 133, 17142–17145. 10.1021/ja207585p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S.; Yang H.; Li G.; Deng Y.; Jiang H.; Zeng W. Copper(II)-Catalyzed Enantioselective Intramolecular Cyclization of N-Alkenylureas. Org. Lett. 2015, 17, 1018–1021. 10.1021/acs.orglett.5b00131. [DOI] [PubMed] [Google Scholar]
- Hou M.; Deng R.; Gu Z. Cu-Catalyzed Enantioselective Atropisomer Synthesis via Thiolative Ring Opening of Five-Membered Cyclic Diaryliodoniums. Org. Lett. 2018, 20, 5779–5783. 10.1021/acs.orglett.8b02477. [DOI] [PubMed] [Google Scholar]
- Truong P. M.; Zavalij P. Y.; Doyle M. P. Highly Enantioselective Carbonyl-Ene Reactions of 2,3-Diketoesters: Efficient and Atom-Economical Process to Functionalized Chiral α-Hydroxy-β-Ketoesters. Angew. Chem., Int. Ed. 2014, 53, 6468–6472. 10.1002/anie.201402233. [DOI] [PubMed] [Google Scholar]
- Bovino M. T.; Chemler S. R. Catalytic Enantioselective Alkene Aminohalogenation/Cyclization Involving Atom Transfer. Angew. Chem., Int. Ed. 2012, 51, 3923–3927. 10.1002/anie.201109044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrar K.; Reiser O. Enantioselective Synthesis of (−)-Paeonilide. Chem. Commun. 2012, 48, 3457–3459. 10.1039/c2cc18172j. [DOI] [PubMed] [Google Scholar]
- Chen S.; Rong C.; Feng P.; Li S.; Shi Y. Stereoselective Construction of the Tetracyclic Core of Cryptotrione. Org. Biomol. Chem. 2012, 10, 5518–5520. 10.1039/c2ob25923k. [DOI] [PubMed] [Google Scholar]
- Buter J.; Moezelaar R.; Minnaard A. J. Enantioselective Palladium Catalyzed Conjugate Additions of Ortho-Substituted Arylboronic Acids to β,β-Disubstituted Cyclic Enones: Total Synthesis of Herbertenediol, Enokipodin A and Enokipodin B. Org. Biomol. Chem. 2014, 12, 5883–5890. 10.1039/C4OB01085J. [DOI] [PubMed] [Google Scholar]
- Gottumukkala A. L.; Matcha K.; Lutz M.; De Vries J. G.; Minnaard A. J. Palladium-Catalyzed Asymmetric Quaternary Stereocenter Formation. Chem. - Eur. J. 2012, 18, 6907–6914. 10.1002/chem.201200694. [DOI] [PubMed] [Google Scholar]
- Holmquist M.; Blay G.; Pedro J. R. Highly Enantioselective Aza-Henry Reaction with Isatin N-Boc Ketimines. Chem. Commun. 2014, 50, 9309–9312. 10.1039/C4CC04051A. [DOI] [PubMed] [Google Scholar]
- Wu X.; Zhou W.; Wu H. H.; Zhang J. Enantioselective [3 + 2] Cycloaddition of Azomethine Ylides and Aldehydes: Via Ni/Bis(Oxazoline)-Catalyzed Ring Opening of N-Tosylaziridines through a Chirality Transfer Approach. Chem. Commun. 2017, 53, 5661–5664. 10.1039/C7CC02906C. [DOI] [PubMed] [Google Scholar]
- Casavant B. J.; Hosseini A. S.; Chemler S. R. 6-Azabicyclo[3.2.1]Octanes Via Copper-Catalyzed Enantioselective Alkene Carboamination. Adv. Synth. Catal. 2014, 356, 2697–2702. 10.1002/adsc.201400317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Shang H.; Wang Z.; Chang L.; Shao W.; Yang Z.; Tang Y. Gold-Catalyzed Rearrangement of Allylic Oxonium Ylides: Efficient Synthesis of Highly Functionalized Dihydrofuran-3-Ones. Angew. Chem., Int. Ed. 2013, 52, 4198–4202. 10.1002/anie.201208305. [DOI] [PubMed] [Google Scholar]
- Gao S.; Chen J. R.; Hu X. Q.; Cheng H. G.; Lu L. Q.; Xiao W. J. Copper-Catalyzed Enantioselective Inverse Electron-Demand Hetero-Diels-Alder Reactions of Diazadienes with Enol Ethers: Efficient Synthesis of Chiral Pyridazines. Adv. Synth. Catal. 2013, 355, 3539–3544. 10.1002/adsc.201300723. [DOI] [Google Scholar]
- Bigot A.; Williamson A. E.; Gaunt M. J. Enantioselective α-Arylation of N-Acyloxazolidinones with Copper(II)-Bisoxazoline Catalysts and Diaryliodonium Salts. J. Am. Chem. Soc. 2011, 133, 13778–13781. 10.1021/ja206047h. [DOI] [PubMed] [Google Scholar]
- Kong K.; Moussa Z.; Lee C.; Romo D. Total Synthesis of the Spirocyclic Imine Marine Toxin (−)-Gymnodimine and an Unnatural C4-Epimer. J. Am. Chem. Soc. 2011, 133, 19844–19856. 10.1021/ja207385y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liwosz T. W.; Chemler S. R. Copper-Catalyzed Enantioselective Intramolecular Alkene Amination/Intermolecular Heck-Type Coupling Cascade. J. Am. Chem. Soc. 2012, 134, 2020–2023. 10.1021/ja211272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney A. H.; Kadunce N. T.; Reisman S. E. Catalytic Asymmetric Reductive Acyl Cross-Coupling: Synthesis of Enantioenriched Acyclic α,α-Disubstituted Ketones. J. Am. Chem. Soc. 2013, 135, 7442–7445. 10.1021/ja402922w. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Fu G. C. Stereoconvergent Negishi Arylations of Racemic Secondary Alkyl Electrophiles: Differentiating between a CF3 and an Alkyl Group. J. Am. Chem. Soc. 2015, 137, 9523–9526. 10.1021/jacs.5b04725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H.; Tambar U. K. Catalytic Enantioselective Allylic Amination of Unactivated Terminal Olefins via an Ene Reaction/[2,3]-Rearrangement. J. Am. Chem. Soc. 2012, 134, 18495–18498. 10.1021/ja307851b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaschinski T.; Hiersemann M. {1,6}-Transannular Catalytic Asymmetric Gosteli-Claisen Rearrangement. Org. Lett. 2012, 14, 4114–4117. 10.1021/ol3017676. [DOI] [PubMed] [Google Scholar]
- Becker J.; Butt L.; Von Kiedrowski V.; Mischler E.; Quentin F.; Hiersemann M. Catalytic Asymmetric Claisen Rearrangement of Gosteli-Type Allyl Vinyl Ethers: Total Synthesis of (−)-9,10-Dihydroecklonialactone B. J. Org. Chem. 2014, 79, 3040–3051. 10.1021/jo5001466. [DOI] [PubMed] [Google Scholar]
- Mao J.; Liu F.; Wang M.; Wu L.; Zheng B.; Liu S.; Zhong J.; Bian Q.; Walsh P. J. Cobalt-Bisoxazoline-Catalyzed Asymmetric Kumada Cross-Coupling of Racemic α-Bromo Esters with Aryl Grignard Reagents. J. Am. Chem. Soc. 2014, 136, 17662–17668. 10.1021/ja5109084. [DOI] [PubMed] [Google Scholar]
- Flynn C. J.; Elcoate C. J.; Lawrence S. E.; Maguire A. R. Highly Enantioselective Intramolecular Copper Catalysed C-H Insertion Reactions of α -Diazosulfones Supporting Information Table of Contents:. J. Am. Chem. Soc. 2010, 132, 1184–1185. 10.1021/ja909713a. [DOI] [PubMed] [Google Scholar]
- Slattery C. N.; Maguire A. R. Asymmetric Copper-Catalysed Intramolecular C-H Insertion Reactions of α-Diazo-β-Keto Sulfones. Org. Biomol. Chem. 2011, 9, 667–669. 10.1039/C0OB00914H. [DOI] [PubMed] [Google Scholar]
- Shiely A. E.; Slattery C. N.; Ford A.; Eccles K. S.; Lawrence S. E.; Maguire A. R. Enantioselective Copper Catalysed Intramolecular C-H Insertion Reactions of α-Diazo-β-Keto Sulfones, α-Diazo-β-Keto Phosphine Oxides and 2-Diazo-1,3-Diketones; the Influence of the Carbene Substituent. Org. Biomol. Chem. 2017, 15, 2609–2628. 10.1039/C7OB00214A. [DOI] [PubMed] [Google Scholar]
- Shiely A. E.; Clarke L.-A.; Flynn C. J.; Buckley A. M.; Ford A.; Lawrence S. E.; Maguire A. R. Substrate and Catalyst Effects in the Enantioselective Copper-Catalysed C-H Insertion Reactions of α-Diazo-β-Oxo Sulfones. Eur. J. Org. Chem. 2018, 2018, 2277–2289. 10.1002/ejoc.201800077. [DOI] [Google Scholar]
- Hajra S.; Sinha D. Catalytic Enantioselective Aziridoarylation of Aryl Cinnamyl Ethers toward Synthesis of Trans-3-Amino-4-Arylchromans. J. Org. Chem. 2011, 76, 7334–7340. 10.1021/jo200711s. [DOI] [PubMed] [Google Scholar]
- Choi J.; Martín-Gago P.; Fu G. C. Stereoconvergent Arylations and Alkenylations of Unactivated Alkyl Electrophiles: Catalytic Enantioselective Synthesis of Secondary Sulfonamides and Sulfones. J. Am. Chem. Soc. 2014, 136, 12161–12165. 10.1021/ja506885s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X. P.; Su Y.; Gu P. Catalytic Enantioselective Desymmetrization of 1,3-Diazido-2-Propanol via Intramolecular Interception of Alkyl Azides with Diazo(Aryl)Acetates. Org. Lett. 2014, 16, 5339–5341. 10.1021/ol502608d. [DOI] [PubMed] [Google Scholar]
- Vita M. V.; Waser J. Azidation of β-Keto Esters and Silyl Enol Ethers with a Benziodoxole Reagent. Org. Lett. 2013, 15, 3246–3249. 10.1021/ol401229v. [DOI] [PubMed] [Google Scholar]
- Trillo P.; Baeza A.; Nájera C. Copper-Catalyzed Asymmetric Alkylation of β-Keto Esters with Xanthydrols. Adv. Synth. Catal. 2013, 355, 2815–2821. 10.1002/adsc.201300627. [DOI] [Google Scholar]
- Weng J. Q.; Fan R. J.; Deng Q. M.; Liu R. R.; Gao J. R.; Jia Y. X. Enantioselective Friedel-Crafts Alkylation Reactions of 3-Substituted Indoles with Electron-Deficient Alkenes. J. Org. Chem. 2016, 81, 3023–3030. 10.1021/acs.joc.6b00123. [DOI] [PubMed] [Google Scholar]
- Sha W.; Zhu Y.; Mei H.; Han J.; Soloshonok V. A.; Pan Y. Catalytic Enantioselective Cyano-Trifluoromethylation of Styrenes. Chem. Sel. 2017, 2, 1129–1132. 10.1002/slct.201601893. [DOI] [Google Scholar]
- Zhang H. X.; Nie J.; Cai H.; Ma J. A. Cyclic Aldimines as Superior Electrophiles for Cu-Catalyzed Decarboxylative Mannich Reaction of β-Ketoacids with a Broad Scope and High Enantioselectivity. Org. Lett. 2014, 16, 2542–2545. 10.1021/ol500929d. [DOI] [PubMed] [Google Scholar]
- Barroso S.; Blay G.; Munoz M. C.; Pedro J. R. Highly Enantioselective Nitrone Cycloadditions with 2-Alkenoyl Pyridine N-Oxides Catalyzed by Cu(II)-BOX Complexes. Org. Lett. 2011, 13, 402 10.1021/ol102716e. [DOI] [PubMed] [Google Scholar]
- Leighty M. W.; Shen B.; Johnston J. N. Enantioselective Synthesis of α-Oxy Amides via Umpolung Amide Synthesis. J. Am. Chem. Soc. 2012, 134, 15233–15236. 10.1021/ja306225u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikora J. M.; Chemler S. R. Synthesis of Benzyl Amines via Copper-Catalyzed Enantioselective Aza-Friedel-Crafts Addition of Phenols to N -Sulfonyl Aldimines. Org. Lett. 2018, 20, 2133–2137. 10.1021/acs.orglett.8b00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Sammis G. M.; Flamme E. M.; Kraml C. M.; Sorensen E. J. The Catalytic Asymmetric Diels-Alder Reactions and Post-Cycloaddition Reductive Transpositions of 1-Hydrazinodienes. Chem. - Eur. J. 2011, 17, 11131–11134. 10.1002/chem.201102394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist M.; Blay G.; Muñoz M. C.; Pedro J. R. Enantioselective Addition of Nitromethane to 2-Acylpyridine N -Oxides. Expanding the Generation of Quaternary Stereocenters with the Henry Reaction. Org. Lett. 2014, 16, 1204–1207. 10.1021/ol500082d. [DOI] [PubMed] [Google Scholar]
- Zhu S.; MacMillan D. W. C. Enantioselective Copper-Catalyzed Construction of Aryl Pyrroloindolines via an Arylation-Cyclization Cascade. J. Am. Chem. Soc. 2012, 134, 10815–10818. 10.1021/ja305100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nanteuil F.; Serrano E.; Perrotta D.; Waser J. Dynamic Kinetic Asymmetric [3 + 2] Annulation Reactions of Aminocyclopropanes. J. Am. Chem. Soc. 2014, 136, 6239–6242. 10.1021/ja5024578. [DOI] [PubMed] [Google Scholar]
- Harvey J. S.; Simonovich S. P.; Jamison C. R.; MacMillan D. W. C. Enantioselective α-Arylation of Carbonyls via Cu(I)-Bisoxazoline Catalysis. J. Am. Chem. Soc. 2011, 133, 13782–13785. 10.1021/ja206050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L.; Shen Q.; Wan X.; Lu L. Enantioselective Friedel-Crafts Alkylation of Indoles with Trifluoroethylidene Malonates by Copper-Bis(Oxazoline) Complexes: Construction of Trifluoromethyl-Substituted Stereogenic Tertiary Carbon Center. J. Org. Chem. 2011, 76, 2282–2285. 10.1021/jo1024333. [DOI] [PubMed] [Google Scholar]
- Sawa M.; Miyazaki S.; Yonesaki R.; Morimoto H.; Ohshima T. Catalytic Enantioselective Decarboxylative Mannich-Type Reaction of N-Unprotected Isatin-Derived Ketimines. Org. Lett. 2018, 20, 5393–5397. 10.1021/acs.orglett.8b02306. [DOI] [PubMed] [Google Scholar]
- Huang R.; Chang X.; Li J.; Wang C. J. Cu(I)-Catalyzed Asymmetric Multicomponent Cascade Inverse Electron-Demand Aza-Diels-Alder/Nucleophilic Addition/Ring-Opening Reaction Involving 2-Methoxyfurans as Efficient Dienophiles. J. Am. Chem. Soc. 2016, 138, 3998–4001. 10.1021/jacs.6b01008. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Tokunaga E.; Reddy D. S.; Matsumoto T.; Shiro M.; Shibata N. Enantioselective 5-Endo-Dig Carbocyclization of β-Ketoesters with Internal Alkynes Employing a Four-Component Catalyst System. Angew. Chem., Int. Ed. 2012, 51, 4131–4135. 10.1002/anie.201201060. [DOI] [PubMed] [Google Scholar]
- Baidya M.; Griffin K. A.; Yamamoto H. Catalytic Enantioselective O -Nitrosocarbonyl Aldol Reaction of β-Dicarbonyl Compounds. J. Am. Chem. Soc. 2012, 134, 18566–18569. 10.1021/ja309311z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. C.; Zhang B. W.; Geng R. L.; Song J. Enantioselective [3 + 2] Cycloaddition Reaction of Ethynylethylene Carbonates with Malononitrile Enabled by Organo/Metal Cooperative Catalysis. Org. Lett. 2018, 20, 7907–7911. 10.1021/acs.orglett.8b03454. [DOI] [PubMed] [Google Scholar]
- Hajra S.; Bar S. Catalytic Enantioselective Synthesis of A-86929, a Dopamine D1 Agonist. Chem. Commun. 2011, 47, 3981–3982. 10.1039/c1cc10263j. [DOI] [PubMed] [Google Scholar]
- Stache E. E.; Rovis T.; Doyle A. G. Dual Nickel- and Photoredox-Catalyzed Enantioselective Desymmetrization of Cyclic Meso-Anhydrides. Angew. Chem., Int. Ed. 2017, 56, 3679–3683. 10.1002/anie.201700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajra S.; Akhtar S. M. S.; Aziz S. M. Catalytic Asymmetric Aminolactonization of 1,2-Disubstituted Alkenoic Acid Esters: Efficient Construction of Aminolactones with an All-Carbon Quaternary Stereo-Centre. Chem. Commun. 2014, 50, 6913–6916. 10.1039/C4CC01944J. [DOI] [PubMed] [Google Scholar]
- Zhu R.; Buchwald S. L. Enantioselective Functionalization of Radical Intermediates in Redox Catalysis: Copper-Catalyzed Asymmetric Oxytrifluoromethylation of Alkenes. Angew. Chem., Int. Ed. 2013, 52, 12655–12658. 10.1002/anie.201307790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miró J.; del Pozo C.; Toste F. D.; Fustero S. Enantioselective Palladium-Catalyzed Oxidative β,β-Fluoroarylation of α,β-Unsaturated Carbonyl Derivatives. Angew. Chem., Int. Ed. 2016, 55, 9045–9049. 10.1002/anie.201603046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.; Ma H.; Li J.; Yu Y.; Qin Z.; Fu B. Ni(II)-Catalyzed Enantioselective Mukaiyama-Mannich Reaction between Silyl Enol Ethers and Cyclic: N -Sulfonyl α-Ketiminoesters. Org. Chem. Front. 2017, 4, 1858–1862. 10.1039/C7QO00370F. [DOI] [Google Scholar]
- Tsuda Y.; Kuriyama M.; Onomura O. Synthesis of Optically Active Oxazoline Derivatives via Catalytic Asymmetric Desymmetrization of 1,3-Diols. Chem. - Eur. J. 2012, 18, 2481–2483. 10.1002/chem.201103800. [DOI] [PubMed] [Google Scholar]
- Xiong H. Y.; Yang Z. Y.; Chen Z.; Zeng J. L.; Nie J.; Ma J. A. Copper-Catalyzed One-Pot Denitrogenative-Dehydrogenative-Decarboxylative Coupling of β-Ketoacids with Trifluorodiazoethane: Facile Access to Trifluoromethylated Aldol Products. Chem. - Eur. J. 2014, 20, 8325–8329. 10.1002/chem.201403073. [DOI] [PubMed] [Google Scholar]
- Wei L.; Wang Z. F.; Yao L.; Qiu G.; Tao H.; Li H.; Wang C. J. Copper(II)-Catalyzed Asymmetric 1,3-Dipolar [3 + 4] Cycloaddition and Kinetic Resolution of Azomethine Imines with Azoalkenes. Adv. Synth. Catal. 2016, 358, 3955–3959. 10.1002/adsc.201600961. [DOI] [Google Scholar]
- Sen S.; Kamma S. R.; Gundla R.; Adepally U.; Kuncha S.; Thirnathi S.; Prasad U. V. A Reagent Based DOS Strategy via Evans Chiral Auxiliary: Highly Stereoselective Michael Reaction towards Optically Active Quinolizidinones, Piperidinones and Pyrrolidinones. RSC Adv. 2013, 3, 2404–2411. 10.1039/c2ra22115b. [DOI] [Google Scholar]
- Kurono N.; Ohtsuga K.; Wakabayashi M.; Kondo T.; Ooka H.; Ohkuma T. Kinetic Resolution of Racemic α-Tert-Alkyl-α-Hydroxy Esters by Enantiomer-Selective Carbamoylation. J. Org. Chem. 2011, 76, 10312–10318. 10.1021/jo201940w. [DOI] [PubMed] [Google Scholar]
- Desyatkin V. G.; Beletskaya I. P. Asymmetric Friedel-Crafts/Michael Reaction of Indoles and Pyrroles with Coumarin-3-Carbonylates. Synthesis 2017, 49, 4327–4334. 10.1055/s-0036-1589024. [DOI] [Google Scholar]
- Mikami K.; Aikawa K.; Aida J. Fragment-Based Reaction Discovery of Non-Ene-Type Carbon-Carbon Bond-Forming Reactions: Catalytic Asymmetric Oxetane Synthesis by Screening Olefinic Reactants without Allylic Hydrogen. Synlett 2011, 18, 2719–2724. 10.1055/s-0031-1289540. [DOI] [Google Scholar]
- Wu L.; Liu R. R.; Zhang G.; Wang D. J.; Wu H.; Gao J.; Jia Y. X. Enantioselective Construction of Cyclic Indolyl α-Amino Esters via a Friedel-Crafts Alkylation Reaction. Adv. Synth. Catal. 2015, 357, 709–713. 10.1002/adsc.201400987. [DOI] [Google Scholar]
- Gao J.-R.; Wu H.; Xiang B.; Yu W.-B.; Han L.; Jia Y.-X. Highly Enantioselective Construction of Trifluoromethylated All-Carbon Quaternary Stereocenters via Nickel-Catalyzed Friedel-Crafts Alkylation Reaction. J. Am. Chem. Soc. 2013, 135, 2983–2986. 10.1021/ja400650m. [DOI] [PubMed] [Google Scholar]
- Weng J. Q.; Deng Q. M.; Wu L.; Xu K.; Wu H.; Liu R. R.; Gao J. R.; Jia Y. X. Asymmetric Friedel-Crafts Alkylation of α-Substituted β-Nitroacrylates: Access to B2,2-Amino Acids Bearing Indolic All-Carbon Quaternary Stereocenters. Org. Lett. 2014, 16, 776–779. 10.1021/ol403480v. [DOI] [PubMed] [Google Scholar]
- Best D.; Kujawa S.; Lam H. W. Diastereo- and Enantioselective Pd(II)-Catalyzed Additions of 2-Alkylazaarenes to N -Boc Imines and Nitroalkenes. J. Am. Chem. Soc. 2012, 134, 18193–18196. 10.1021/ja3083494. [DOI] [PubMed] [Google Scholar]
- Simpson A. J.; Lam H. W. Enantioselective Nickel-Catalyzed Michael Additions of 2-Acetylazaarenes to Nitroalkenes. Org. Lett. 2013, 15, 2586–2589. 10.1021/ol400578c. [DOI] [PubMed] [Google Scholar]
- Valli M.; Chiesa F.; Gandini A.; Porta A.; Vidari G.; Zanoni G. A Unified Stereodivergent Strategy for Prostaglandin and Isoprostanoid Synthesis. J. Org. Chem. 2014, 79, 2632–2639. 10.1021/jo500093k. [DOI] [PubMed] [Google Scholar]
- Cheng Q. Q.; Yedoyan J.; Arman H.; Doyle M. P. Copper-Catalyzed Divergent Addition Reactions of Enoldiazoacetamides with Nitrones. J. Am. Chem. Soc. 2016, 138, 44–47. 10.1021/jacs.5b10860. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Q.; Yuan Y. A.; Liu G. S.; Xu H. Iron(II)-Catalyzed Asymmetric Intramolecular Aminohydroxylation of Indoles. Org. Lett. 2013, 15, 3910–3913. 10.1021/ol401666e. [DOI] [PubMed] [Google Scholar]
- Liu C.; Yi J. C.; Liang X. W.; Xu R. Q.; Dai L. X.; You S. L. Copper(I)-Catalyzed Asymmetric Dearomatization of Indole Acetamides with 3-Indolylphenyliodonium Salts. Chem. - Eur. J. 2016, 22, 10813–10816. 10.1002/chem.201602229. [DOI] [PubMed] [Google Scholar]
- Williamson K. S.; Yoon T. P. Iron Catalyzed Asymmetric Oxyamination of Olefins. J. Am. Chem. Soc. 2012, 134, 12370–12373. 10.1021/ja3046684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y. S.; Kim T. W.; Kang S. H. Asymmetric Formation of Tert-Alkylamines from Serinols by a Dual Function Catalyst. Chem. Commun. 2013, 49, 9669–9671. 10.1039/c3cc45099f. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Andrus M. B. Naphthyl-Substituted Bisoxazoline and Pyridylbisoxazoline-Copper(I) Catalysts for Asymmetric Allylic Oxidation. Tetrahedron Lett. 2012, 53, 4518–4521. 10.1016/j.tetlet.2012.06.010. [DOI] [Google Scholar]
- Li M.; Hawkins A.; Barber D. M.; Bultinck P.; Herrebout W.; Dixon D. J. Enantio- and Diastereoselective Palladium Catalysed Arylative and Vinylative Allene Carbocyclisation Cascades. Chem. Commun. 2013, 49, 5265–5267. 10.1039/c3cc42079e. [DOI] [PubMed] [Google Scholar]
- Hou G.; Yu J.; Yu C.; Wu G.; Miao Z. Enantio- and Diastereoselective Vinylogous Mukaiyama Aldol Reactions of α-Keto Phosphonates with 2-(Trimethylsilyloxy)-Furan Catalyzed by Bis(Oxazoline)-Copper Complexes. Adv. Synth. Catal. 2013, 355, 589–593. 10.1002/adsc.201200810. [DOI] [Google Scholar]
- Minuth T.; Boysen M. M. K. Bis(Oxazolines) Based on Glycopyranosides - Steric, Configurational and Conformational Influences on Stereoselectivity. Beilstein J. Org. Chem. 2010, 6, 1–7. 10.3762/bjoc.6.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özüduru G.; Schubach T.; Boysen M. M. K. Enantioselective Cyclopropanation of Indoles: Construction of All-Carbon Quaternary Stereocenters. Org. Lett. 2012, 14, 4990–4993. 10.1021/ol302388t. [DOI] [PubMed] [Google Scholar]
- Minuth T.; Boysen M. M. K. Carbohydrate-Derived Bis(Oxazoline) Ligand in the Total Synthesis of Grenadamide. Synthesis 2010, 2010 (16), 2799–2803. 10.1055/s-0030-1258143. [DOI] [Google Scholar]
- George J.; Reddy B. V. S. Enantioselective Friedel-Crafts Alkylation of Indoles with 2-Enoylpyridine-N-Oxides Catalyzed by GlucoBOX-Cu(II) Complex. Org. Biomol. Chem. 2012, 10, 4731–4738. 10.1039/c2ob25315a. [DOI] [PubMed] [Google Scholar]
- Livieri A.; Boiocchi M.; Desimoni G.; Faita G. Enantioselective Cycloadditions of 2-Alkenoylpyridine-N-Oxides Catalysed by a Bis(Oxazoline)/Cu(II) Complex: Structure of the Reactive Intermediate. Chem. - Eur. J. 2011, 17, 516–520. 10.1002/chem.201002017. [DOI] [PubMed] [Google Scholar]
- Livieri A.; Boiocchi M.; Desimoni G.; Faita G. Enantioselective Addition of Cyclic Enol Silyl Ethers to 2-Alkenoyl-Pyridine-N-Oxides Catalysed by Cu(II)-Bis(Oxazoline) Complexes. Chem. - Eur. J. 2012, 18, 11662–11668. 10.1002/chem.201201214. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Zhang Y.; Wang R. Catalytic Asymmetric Activation of a Csp3-H Bond Adjacent to a Nitrogen Atom: A Versatile Approach to Optically Active α-Alkyl α-Amino Acids and C1-Alkylated Tetrahydroisoquinoline Derivatives. Angew. Chem., Int. Ed. 2011, 50, 10429–10432. 10.1002/anie.201105123. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Kang H.; Hong L.; Dong W.; Li G.; Zheng X.; Wang R. Construction of the N1-C3 Linkage Stereogenic Centers by Catalytic Asymmetric Amination Reaction of 3-Bromooxindoles with Indolines. Org. Lett. 2014, 16, 2394–2397. 10.1021/ol5007423. [DOI] [PubMed] [Google Scholar]
- Williamson K. S.; Sawicki J. W.; Yoon T. P. Iron-Catalyzed Kinetic Resolution of N-Sulfonyl Oxaziridines. Chem. Sci. 2014, 5, 3524–3527. 10.1039/C4SC01371A. [DOI] [Google Scholar]
- Adachi S.; Takeda N.; Sibi M. P. Evaluation of Achiral Templates with Fluxional Brønsted Basic Substituents in Enantioselective Conjugate Additions. Org. Lett. 2014, 16, 6440–6443. 10.1021/ol503275w. [DOI] [PubMed] [Google Scholar]
- Ray S. K.; Singh P. K.; Singh V. K. Enantioselective Michael Addition of Malonates to 2-Enoylpyridine N-Oxides Catalyzed by Chiral Bisoxazoline-Zn(II) Complex. Org. Lett. 2011, 13, 5812–5815. 10.1021/ol202405v. [DOI] [PubMed] [Google Scholar]
- Cheng H. G.; Lu L. Q.; Wang T.; Yang Q. Q.; Liu X. P.; Li Y.; Deng Q. H.; Chen J. R.; Xiao W. J. Highly Enantioselective Friedel-Crafts Alkylation/N-Hemiacetalization Cascade Reaction with Indoles. Angew. Chem., Int. Ed. 2013, 52, 3250–3254. 10.1002/anie.201209998. [DOI] [PubMed] [Google Scholar]
- Feng L. W.; Ren H.; Xiong H.; Wang P.; Wang L.; Tang Y. Reaction of Donor-Acceptor Cyclobutanes with Indoles: A General Protocol for the Formal Total Synthesis of (±)-Strychnine and the Total Synthesis of (±)-Akuammicine. Angew. Chem., Int. Ed. 2017, 56, 3055–3058. 10.1002/anie.201611734. [DOI] [PubMed] [Google Scholar]
- Liu Q. J.; Wang L.; Kang Q. K.; Zhang X. P.; Tang Y. Cy-SaBOX/Copper(II)-Catalyzed Highly Diastereo- and Enantioselective Synthesis of Bicyclic N,O Acetals. Angew. Chem., Int. Ed. 2016, 55, 9220–9223. 10.1002/anie.201603911. [DOI] [PubMed] [Google Scholar]
- Deng C.; Wang L. J.; Zhu J.; Tang Y. A Chiral Cagelike Copper(I) Catalyst for the Highly Enantioselective Synthesis of 1,1-Cyclopropane Diesters. Angew. Chem., Int. Ed. 2012, 51, 11620–11623. 10.1002/anie.201206376. [DOI] [PubMed] [Google Scholar]
- Li J.; Zheng L.; Chen H.; Wang L.; Sun X. L.; Zhu J.; Tang Y. Highly Enantioselective Cyclopropanation of Trisubstituted Olefins. Sci. China: Chem. 2018, 61, 526–530. 10.1007/s11426-017-9200-9. [DOI] [Google Scholar]
- Hu J. L.; Feng L. W.; Wang L.; Xie Z.; Tang Y.; Li X. Enantioselective Construction of Cyclobutanes: A New and Concise Approach to the Total Synthesis of (+)-Piperarborenine B. J. Am. Chem. Soc. 2016, 138, 13151–13154. 10.1021/jacs.6b08279. [DOI] [PubMed] [Google Scholar]
- Xu H.; Qu J. P.; Liao S.; Xiong H.; Tang Y. Highly Enantioselective [3 + 2] Annulation of Cyclic Enol Silyl Ethers with Donor-Acceptor Cyclopropanes: Accessing 3a-Hydroxy [n.3.0]Carbobicycles. Angew. Chem., Int. Ed. 2013, 52, 4004–4007. 10.1002/anie.201300032. [DOI] [PubMed] [Google Scholar]
- Deng T.; Cai C. Fluorous Chiral Bis(Oxazolines): Synthesis and Application in Asymmetric Henry Reaction. J. Fluorine Chem. 2013, 156, 183–186. 10.1016/j.jfluchem.2013.09.014. [DOI] [Google Scholar]
- Deng T.; Wang H.; Cai C. Copper-Catalyzed Asymmetric Addition to Isatins to Give 3-Hydroxy-2-Oxindoles by C-H Activation. Eur. J. Org. Chem. 2014, 2014, 7259–7264. 10.1002/ejoc.201402852. [DOI] [Google Scholar]
- Deng T.; Cai C. Bis(Oxazoline)-Copper Catalyzed Enantioselective Hydrophosphonylation of Aldehydes. RSC Adv. 2014, 4, 27853–27856. 10.1039/C4RA03269A. [DOI] [Google Scholar]
- Li N. K.; Kong L. P.; Qi Z. H.; Yin S. J.; Zhang J. Q.; Wu B.; Wang X. W. Friedel-Crafts Reaction of Indoles with Isatin-Derived β,γ-Unsaturated α-Keto Esters Using a BINOL-Derived Bisoxazoline (BOX)/Copper(II) Complex as Catalyst. Adv. Synth. Catal. 2016, 358, 3100–3112. 10.1002/adsc.201600272. [DOI] [Google Scholar]
- Chu W. D.; Zhang L.; Zhang Z.; Zhou Q.; Mo F.; Zhang Y.; Wang J. Enantioselective Synthesis of Trisubstituted Allenes via Cu(I)-Catalyzed Coupling of Diazoalkanes with Terminal Alkynes. J. Am. Chem. Soc. 2016, 138, 14558–14561. 10.1021/jacs.6b09674. [DOI] [PubMed] [Google Scholar]
- Quan M.; Wang X.; Wu L.; Gridnev I. D.; Yang G.; Zhang W. Ni(II)-Catalyzed Asymmetric Alkenylations of Ketimines. Nat. Commun. 2018, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Shao Q.; Yang G.; Zhang W. Cobalt-Catalyzed Asymmetric Allylation of Cyclic Ketimines. Chem. - Eur. J. 2018, 24, 1241–1245. 10.1002/chem.201704760. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Fu G. C. Catalytic Asymmetric Synthesis of Tertiary Alkyl Fluorides: Negishi Cross-Couplings of Racemic α,α-Dihaloketones. J. Am. Chem. Soc. 2014, 136, 5520–5524. 10.1021/ja501815p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.; Fu G. C. Catalytic Asymmetric Synthesis of Secondary Nitriles via Stereoconvergent Negishi Arylations and Alkenylations of Racemic α-Bromonitriles. J. Am. Chem. Soc. 2012, 134, 9102–9105. 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou S.; Fu G. C. Nickel/Bis(Oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones. J. Am. Chem. Soc. 2010, 132, 1264–1266. 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J.-B.; Zhao Y.-M.; Gu P. Asymmetric Intramolecular Desymmetrization of Meso -α,α ′-Diazido Alcohols with Aryldiazoacetates: Assembly of Chiral C3 Fragments with Three Continuous Stereocenters. Org. Lett. 2016, 18, 1984–1987. 10.1021/acs.orglett.6b00570. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Wang F.; McCann S. D.; Wang D.; Chen P.; Stahl S. S.; Liu G. Enantioselective Cyanation of Benzylic C-H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. 10.1126/science.aaf7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M.; Ikeda M.; Motoyama K.; Miyake Y.; Nishibayashi Y. Enantioselective Alkylation of β-Keto Phosphonates by Direct Use of Diaryl Methanols as Electrophiles. Chem. Commun. 2012, 48, 9528–9530. 10.1039/c2cc35262a. [DOI] [PubMed] [Google Scholar]
- Gerten A. L.; Slade M. C.; Pugh K. M.; Stanley L. M. Catalytic, Enantioselective 1,3-Dipolar Cycloadditions of Nitrile Imines with Methyleneindolinones. Org. Biomol. Chem. 2013, 11, 7834–7837. 10.1039/c3ob41815d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Song J.; Luo S. W.; Gong L. Z. Enantioselective Oxidative Cross-Coupling Reaction of 3indolylmethyl c-h Bonds with 1, 3-Dicarbonyls Using a Chiral Lewis Acid-Bonded Nucleophile to Control Stereochemistry. Angew. Chem., Int. Ed. 2010, 49, 5558–5562. 10.1002/anie.201002108. [DOI] [PubMed] [Google Scholar]
- Fu L.; Zhou S.; Wan X.; Chen P.; Liu G. Enantioselective Trifluoromethylalkynylation of Alkenes via Copper-Catalyzed Radical Relay. J. Am. Chem. Soc. 2018, 140, 10965–10969. 10.1021/jacs.8b07436. [DOI] [PubMed] [Google Scholar]
- Sha W.; Deng L.; Ni S.; Mei H.; Han J.; Pan Y. Merging Photoredox and Copper Catalysis: Enantioselective Radical Cyanoalkylation of Styrenes. ACS Catal. 2018, 8, 7489–7494. 10.1021/acscatal.8b01863. [DOI] [Google Scholar]
- Cherney A. H.; Reisman S. E. Nickel-Catalyzed Asymmetric Reductive Cross-Coupling between Vinyl and Benzyl Electrophiles. J. Am. Chem. Soc. 2014, 136, 14365–14368. 10.1021/ja508067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Chen H. L.; Liu L.; Fu B. Synthesis of New C2- Symmetric Fluoren-9-Ylidene Malonate Derived Bis(Oxazoline) Ligands and Their Application in Friedel-Crafts Reactions. Molecules 2010, 15, 8582–8592. 10.3390/molecules15128582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H. L.; Xie L.; Zhang Z. H.; Li J. Q.; Qin Z. H.; Fu B. Enantioselective Copper(II)-Catalyzed Conjugate Addition of Indoles to β-Substituted Unsaturated Acyl Phosphonates. Adv. Synth. Catal. 2016, 358, 1011–1016. 10.1002/adsc.201500923. [DOI] [Google Scholar]
- Li B.; Chao Z.; Li C.; Gu Z. Cu-Catalyzed Enantioselective Ring Opening of Cyclic Diaryliodoniums toward the Synthesis of Chiral Diarylmethanes. J. Am. Chem. Soc. 2018, 140, 9400–9403. 10.1021/jacs.8b05743. [DOI] [PubMed] [Google Scholar]
- a Fritschi H.; Leutenegger U.; Siegmann K.; Pfaltz A.; Keller W.; Kratky C. Synthesis of Chiral Semicorrin Ligands and General Concept. Helv. Chim. Acta 1988, 71, 1541–1552. 10.1002/hlca.19880710620. [DOI] [Google Scholar]; b Fritschi H.; Leutenegger U.; Pfaltz A. Enantioselective Cyclopropane Formation from Olefins with Diazo Compounds Catalyzed by Chiral (Semicorrinato)copper Complexes. Helv. Chim. Acta 1988, 71, 1552–1565. [Google Scholar]; c von Matt P.; Pfaltz A. Enantioselective Conjugate Reduction of α,β-Unsaturated Carboxamides with Semicorrin Cobalt Catalysts. Tetrahedron: Asymmetry 1991, 7, 691–700. 10.1016/S0957-4166(00)86123-6. [DOI] [Google Scholar]; d Nolin K. A.; Ahn R. W.; Kobayashi Y.; Kennedy-Smith J. J.; Toste F. D. Enantioselective Reduction of Ketones and Imines Catalyzed by (CN-Box)ReV-Oxo Complexes. Chem. - Eur. J. 2010, 16, 9555–9562. 10.1002/chem.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W.; Lu L. Q.; Zhou Q. Q.; Wei Y.; Chen J. R.; Xiao W. J. Bifunctional Photocatalysts for Enantioselective Aerobic Oxidation of β-Ketoesters. J. Am. Chem. Soc. 2017, 139, 63–66. 10.1021/jacs.6b11418. [DOI] [PubMed] [Google Scholar]
- Liu C.; Yi J. C.; Zheng Z. B.; Tang Y.; Dai L. X.; You S. L. Enantioselective Synthesis of 3a-Amino-Pyrroloindolines by Copper-Catalyzed Direct Asymmetric Dearomative Amination of Tryptamines. Angew. Chem., Int. Ed. 2016, 55, 751–754. 10.1002/anie.201508570. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Wang D. C.; Xie M. S.; Qu G. R.; Guo H. M. Enantioselective Friedel-Crafts Alkylation Reactions of β-Naphthols with Donor-Acceptor Aminocyclopropanes. Chem. - Eur. J. 2018, 24, 15512–15516. 10.1002/chem.201804032. [DOI] [PubMed] [Google Scholar]
- Fang Z. J.; Zheng S. C.; Guo Z.; Guo J. Y.; Tan B.; Liu X. Y. Asymmetric Synthesis of Axially Chiral Isoquinolones: Nickel-Catalyzed Denitrogenative Transannulation. Angew. Chem., Int. Ed. 2015, 54, 9528–9532. 10.1002/anie.201503207. [DOI] [PubMed] [Google Scholar]
- Cichowicz N. R.; Kaplan W.; Khomutnyk Y.; Bhattarai B.; Sun Z.; Nagorny P. Concise Enantioselective Synthesis of Oxygenated Steroids via Sequential Copper(II)-Catalyzed Michael Addition/Intramolecular Aldol Cyclization Reactions. J. Am. Chem. Soc. 2015, 137, 14341–14348. 10.1021/jacs.5b08528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hut’Ka M.; Tsubogo T.; Kobayashi S. Synthesis of Glutamic Acid and Highly Functionalized Pyrrolidine Derivatives by Utilizing Tunable Calcium Catalysts for Chemoselective Asymmetric 1,4-Addition and [3 + 2] Cycloaddition Reactions. Adv. Synth. Catal. 2013, 355, 1561–1569. 10.1002/adsc.201300171. [DOI] [Google Scholar]
- Angulo B.; García J. I.; Herrerías C. I.; Mayoral J. A.; Miñana A. C. Polytopic Bis(Oxazoline)-Based Ligands for Recoverable Catalytic Systems Applied to the Enantioselective Henry Reaction. Org. Biomol. Chem. 2015, 13, 9314–9322. 10.1039/C5OB01033K. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhou K.; Wen Z.; Cao S.; Shen X.; Lei M.; Gong L. Copper(II)-Catalyzed Asymmetric Photoredox Reactions: Enantioselective Alkylation of Imines Driven by Visible Light. J. Am. Chem. Soc. 2018, 140, 15850–15858. 10.1021/jacs.8b09251. [DOI] [PubMed] [Google Scholar]
- a Uetake Y.; Uwamori M.; Nakada M. Enantioselective Approach to Polycyclic Polyprenylated Acylphloroglucinols via Catalytic Asymmetric Intramolecular Cyclopropanation. J. Org. Chem. 2015, 80, 1735–1745. 10.1021/jo5026699. [DOI] [PubMed] [Google Scholar]; b Chen H.; Wang L.; Wang F.; Zhao L.-P.; Wang P.; Tang Y. Access to Hexahydrocarbazoles: The Thorpe-Ingold Effects of the Ligand on Enantioselectivity. Angew. Chem., Int. Ed. 2017, 56, 6942–6945. 10.1002/anie.201700042. [DOI] [PubMed] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods B. P.; Orlandi M.; Huang C. Y.; Sigman M. S.; Doyle A. G. Nickel-Catalyzed Enantioselective Reductive Cross-Coupling of Styrenyl Aziridines. J. Am. Chem. Soc. 2017, 139, 5688–5691. 10.1021/jacs.7b03448. [DOI] [PubMed] [Google Scholar]
- Poremba K. E.; Kadunce N. T.; Suzuki N.; Cherney A. H.; Reisman S. E. Nickel-Catalyzed Asymmetric Reductive Cross-Coupling To Access 1,1-Diarylalkanes. J. Am. Chem. Soc. 2017, 139, 5684–5687. 10.1021/jacs.7b01705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.; Yamamoto H. Nickel Catalyzed Regio-, Diastereo-, and Enantioselective Cross-Coupling of 3,4-Epoxyalcohol with Aryl Iodides. Org. Lett. 2017, 19, 4363–4366. 10.1021/acs.orglett.7b02076. [DOI] [PubMed] [Google Scholar]
- Liu J.; Ding W.; Zhou Q. Q.; Liu D.; Lu L. Q.; Xiao W. J. Enantioselective Di-/Perfluoroalkylation of β-Ketoesters Enabled by Cooperative Photoredox/Nickel Catalysis. Org. Lett. 2018, 20, 461–464. 10.1021/acs.orglett.7b03826. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lu X.; Lou W.; Ye Y.; Jiang H.; Zeng W. Palladium(II)-Catalyzed Enantioselective Arylation of α-Imino Esters. J. Org. Chem. 2012, 77, 8541–8548. 10.1021/jo301423e. [DOI] [PubMed] [Google Scholar]
- Wei X. H.; Wang G. W.; Yang S. D. Enantioselective Synthesis of Arylglycine Derivatives by Direct C-H Oxidative Cross-Coupling. Chem. Commun. 2015, 51, 832–835. 10.1039/C4CC07361D. [DOI] [PubMed] [Google Scholar]
- Beisel T.; Manolikakes G. Palladium-Catalyzed Enantioselective Three-Component Synthesis of α-Substituted Amines. Org. Lett. 2015, 17, 3162–3165. 10.1021/acs.orglett.5b01502. [DOI] [PubMed] [Google Scholar]
- Beisel T.; Diehl A. M.; Manolikakes G. Palladium-Catalyzed Enantioselective Three-Component Synthesis of α-Arylglycines. Org. Lett. 2016, 18, 4116–4119. 10.1021/acs.orglett.6b02045. [DOI] [PubMed] [Google Scholar]
- He Y.; Yang Z.; Thornbury R. T.; Toste F. D. Palladium-Catalyzed Enantioselective 1,1-Fluoroarylation of Aminoalkenes. J. Am. Chem. Soc. 2015, 137, 12207–12210. 10.1021/jacs.5b07795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira C. C.; Angnes R. A.; Correia C. R. D. Intermolecular Enantioselective Heck-Matsuda Arylations of Acyclic Olefins: Application to the Synthesis of β-Aryl-γ-Lactones and β-Aryl Aldehydes. J. Org. Chem. 2013, 78, 4373–4385. 10.1021/jo400378g. [DOI] [PubMed] [Google Scholar]
- Do H.-Q.; Chandrashekar E. R. R.; Fu G. C. Nickel/Bis(Oxazoline)-Catalyzed Asymmetric Negishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J. Am. Chem. Soc. 2013, 135, 16288–16291. 10.1021/ja408561b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman K.; Vasanthan R.; Kesavan V. Asymmetric Henry Reaction Catalyzed by Novel Chiral Bioxazolines from Tartaric Acid. Synthesis 2012, 44, 2455–2462. 10.1055/s-0031-1289811. [DOI] [Google Scholar]
- Balaraman K.; Vasanthan R.; Kesavan V. Application of Tartarate Derived Bidentate Bioxazolines in Enantioselective Addition of Terminal Alkynes to Imines. Tetrahedron Lett. 2013, 54, 3613–3616. 10.1016/j.tetlet.2013.04.108. [DOI] [Google Scholar]
- Jayakumar S.; Prakash M.; Balaraman K.; Kesavan V. Highly Enantioselective Alkylation of Allyl Acetates Using Tartrate-Derived Bioxazoline Ligands. Eur. J. Org. Chem. 2014, 2014, 606–615. 10.1002/ejoc.201301208. [DOI] [Google Scholar]
- Jayakumar S.; Kumarswamyreddy N.; Prakash M.; Kesavan V. Palladium Catalyzed Asymmetric Allylation of 3-OBoc-Oxindoles: An Efficient Synthesis of 3-Allyl-3-Hydroxyoxindoles. Org. Lett. 2015, 17, 1066–1069. 10.1021/acs.orglett.5b00034. [DOI] [PubMed] [Google Scholar]
- Oliveira C. C.; Pfaltz A.; Correia C. R. D. Quaternary Stereogenic Centers through Enantioselective Heck Arylation of Acyclic Olefins with Aryldiazonium Salts: Application in a Concise Synthesis of (R)-Verapamil. Angew. Chem., Int. Ed. 2015, 54, 14036–14039. 10.1002/anie.201507927. [DOI] [PubMed] [Google Scholar]
- Khan I. U.; Kattela S.; Hassan A.; Correia C. R. D. Enantioselective Total Synthesis of the Highly Selective Sphingosine-1-Receptor VPC01091 by the Heck Desymmetrization of a Non-Activated Cyclopentene-Fused Spiro-Pyrrolidinone. Org. Biomol. Chem. 2016, 14, 9476–9480. 10.1039/C6OB01892K. [DOI] [PubMed] [Google Scholar]
- de Azambuja F.; Carmona R. C.; Chorro T. H. D.; Heerdt G.; Correia C. R. D. Noncovalent Substrate-Directed Enantioselective Heck Reactions: Synthesis of S- and P-Stereogenic Heterocycles. Chem. - Eur. J. 2016, 22, 11205–11209. 10.1002/chem.201602572. [DOI] [PubMed] [Google Scholar]
- Zhu S. F.; Cai Y.; Mao H. X.; Xie J. H.; Zhou Q. L. Enantioselective Iron-Catalysed O-H Bond Insertions. Nat. Chem. 2010, 2, 546–551. 10.1038/nchem.651. [DOI] [PubMed] [Google Scholar]
- Zhu S. F.; Song X. G.; Li Y.; Cai Y.; Zhou Q. L. Enantioselective Copper-Catalyzed Intramolecular O-H Insertion: An Efficient Approach to Chiral 2-Carboxy Cyclic Ethers. J. Am. Chem. Soc. 2010, 132, 16374–16376. 10.1021/ja1078464. [DOI] [PubMed] [Google Scholar]
- Zhu S. F.; Chen W. Q.; Zhang Q. Q.; Mao H. X.; Zhou Q. L. Enantioselective Copper-Catalyzed O-H Insertion of α-Diazo Phosphonates. Synlett 2011, 2011, 919–922. 10.1055/s-0030-1259710. [DOI] [Google Scholar]
- Xie X. L.; Zhu S. F.; Guo J. X.; Cai Y.; Zhou Q. L. Enantioselective Palladium-Catalyzed Insertion of α-Aryl-α- Diazoacetates into the O-H Bonds of Phenols. Angew. Chem., Int. Ed. 2014, 53, 2978–2981. 10.1002/anie.201309820. [DOI] [PubMed] [Google Scholar]
- Cheng Q.-Q.; Zhu S.-F.; Zhang Y.-Z.; Xie X.-L.; Zhou Q.-L. Copper-Catalyzed B-H Bond Insertion Reaction: A Highly Efficient and Enantioselective C-B Bond-Forming Reaction with Amine-Borane and Phosphine-Borane Adducts. J. Am. Chem. Soc. 2013, 135, 14094–14097. 10.1021/ja408306a. [DOI] [PubMed] [Google Scholar]
- Zhu S. F.; Xu B.; Wang G. P.; Zhou Q. L. Well-Defined Binuclear Chiral Spiro Copper Catalysts for Enantioselective N-H Insertion. J. Am. Chem. Soc. 2012, 134, 436–442. 10.1021/ja2084493. [DOI] [PubMed] [Google Scholar]
- Song X. G.; Ren Y. Y.; Zhu S. F.; Zhou Q. L. Enantioselective Copper-Catalyzed Intramolecular N-H Bond Insertion: Synthesis of Chiral 2-Carboxytetrahydroquinolines. Adv. Synth. Catal. 2016, 358, 2366–2370. 10.1002/adsc.201600390. [DOI] [Google Scholar]
- Shen J. J.; Zhu S. F.; Cai Y.; Xu H.; Xie X. L.; Zhou Q. L. Enantioselective Iron-Catalyzed Intramolecular Cyclopropanation Reactions. Angew. Chem., Int. Ed. 2014, 53, 13188–13191. 10.1002/anie.201406853. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Zhu S. F.; Wang G. P.; Zhou Q. L. Iron-Catalyzed C-H Fuctionalization of Indoles. Adv. Synth. Catal. 2011, 353, 2939–2944. 10.1002/adsc.201100334. [DOI] [Google Scholar]
- Gu H.; Han Z.; Xie H.; Lin X. Iron-Catalyzed Enantioselective Si-H Bond Insertions. Org. Lett. 2018, 20, 6544–6549. 10.1021/acs.orglett.8b02868. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen G.; Wang Z.; Zhang R.; Zhang X.; Ding K. Spiro-2,2′-Bichroman-Based Bisoxazoline (SPANbox) Ligands for ZnII Catalyzed Enantioselective Hydroxylation of β-Keto Esters and 1,3-Diester. Chem. Sci. 2011, 2, 1141–1144. 10.1039/c0sc00607f. [DOI] [Google Scholar]
- Li J.; Pan W.; Wang Z.; Zhang X.; Ding K. Access to Both Enantiomers of α-Chloro-β-Keto Esters with a Single Chiral Ligand: Highly Efficient Enantioselective Chlorination of Cyclic β-Keto Esters Catalyzed by Chiral Copper(II) and Zinc(II) Complexes of a Spiro-2,2′-Bischroman-Based Bisoxazoline Li. Adv. Synth. Catal. 2012, 354, 1980–1986. 10.1002/adsc.201200088. [DOI] [Google Scholar]
- Toussaint A.; Pfaltz A. Asymmetric Henry Reactions Catalyzed by Metal Complexes of Chiral Boron-Bridged Bisoxazoline (Borabox) Ligands. Eur. J. Org. Chem. 2008, 2008, 4591–4597. 10.1002/ejoc.200800570. [DOI] [PubMed] [Google Scholar]
- Köhler V.; Mazet C.; Toussaint A.; Kulicke K.; Häussinger D.; Neuburger M.; Schaffner S.; Kaiser S.; Pfaltz A. Chiral Boron-Bridged Bisoxazoline (Borabox) Ligands: Structures and Reactivities of Pd and Cu Complexes. Chem. - Eur. J. 2008, 14, 8530–8539. 10.1002/chem.200800822. [DOI] [PubMed] [Google Scholar]
- Manna K.; Xu S.; Sadow A. D. A Highly Enantioselective Zirconium Catalyst for Intramolecular Alkene Hydroamination: Significant Isotope Effects on Rate and Stereoselectivity. Angew. Chem., Int. Ed. 2011, 50, 1865–1868. 10.1002/anie.201006163. [DOI] [PubMed] [Google Scholar]
- Manna K.; Everett W. C.; Schoendorff G.; Ellern A.; Windus T. L.; Sadow A. D. Highly Enantioselective Zirconium-Catalyzed Cyclization of Aminoalkenes. J. Am. Chem. Soc. 2013, 135, 7235–7250. 10.1021/ja4000189. [DOI] [PubMed] [Google Scholar]
- Manna K.; Kruse M. L.; Sadow A. D. Concerted C-N/C-H Bond Formation in Highly Enantioselective Yttrium(III)-Catalyzed Hydroamination. ACS Catal. 2011, 1, 1637–1642. 10.1021/cs200511z. [DOI] [Google Scholar]
- Glos M.; Reiser O. Aza-Bis(Oxazolines): New Chiral Ligands for Asymmetric Catalysis. Org. Lett. 2000, 2, 2045–2048. 10.1021/ol005947k. [DOI] [PubMed] [Google Scholar]
- Lang K.; Park J.; Hong S. Development of Bifunctional Aza-Bis(Oxazoline) Copper Catalysts for Enantioselective Henry Reaction. J. Org. Chem. 2010, 75, 6424–6435. 10.1021/jo1009867. [DOI] [PubMed] [Google Scholar]
- Pilsl L. K. A.; Ertl T.; Reiser O. Enantioselective Three-Step Synthesis of Homo-β-Proline: A Donor-Acceptor Cyclopropane as Key Intermediate. Org. Lett. 2017, 19, 2754–2757. 10.1021/acs.orglett.7b01111. [DOI] [PubMed] [Google Scholar]
- McManus H. A.; Guiry P. J. Coupling of Bulky, Electron-Deficient Partners in Aryl Amination in the Preparation of Tridentate Bis(Oxazoline) Ligands for Asymmetric Catalysis. J. Org. Chem. 2002, 67, 8566–8573. 10.1021/jo0262558. [DOI] [PubMed] [Google Scholar]
- Inagaki T.; Ito A.; Ito J. I.; Nishiyama H. Asymmetric Iron-Catalyzed Hydrosilane Reduction of Ketones: Effect of Zinc Metal upon the Absolute Configuration. Angew. Chem., Int. Ed. 2010, 49, 9384–9387. 10.1002/anie.201005363. [DOI] [PubMed] [Google Scholar]
- Inagaki T.; Phong L. T.; Furuta A.; Ito J. I.; Nishiyama H. Iron- and Cobalt-Catalyzed Asymmetric Hydrosilylation of Ketones and Enones with Bis(Oxazolinylphenyl)Amine Ligands. Chem. - Eur. J. 2010, 16, 3090–3096. 10.1002/chem.200903118. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang H.; Jiang Y.; Zhang C.; Shao J.; Xu D. Fast, Solvent-Free and Highly Enantioselective Fluorination of β-Keto Esters Catalyzed by Chiral Copper Complexes in a Ball Mill. Green Chem. 2017, 19, 1674–1677. 10.1039/C6GC03306G. [DOI] [Google Scholar]
- Jia Y.; Yang W.; Du D. M. Asymmetric Friedel-Crafts Alkylation of Indoles with 3-Nitro-2H-Chromenes Catalyzed by Diphenylamine-Linked Bis(Oxazoline) and Bis(Thiazoline) Zn(II) Complexes. Org. Biomol. Chem. 2012, 10, 4739–4746. 10.1039/c2ob25360g. [DOI] [PubMed] [Google Scholar]
- Zhao J. Q.; Zhou X. J.; Zhou Y.; Xu X. Y.; Zhang X. M.; Yuan W. C. Diastereo- and Enantioselective Dearomative [3 + 2] Cycloaddition Reaction of 2-Nitrobenzofurans with 3-Isothiocyanato Oxindoles. Org. Lett. 2018, 20, 909–912. 10.1021/acs.orglett.7b03667. [DOI] [PubMed] [Google Scholar]
- Zhao J. Q.; Zhou X. J.; Chen Y. Z.; Xu X. Y.; Zhang X. M.; Yuan W. C. Zn-Catalyzed Diastereo- and Enantioselective Dearomative [3 + 2] Cycloaddition Reaction of 2-Nitroindoles and 2-Nitrobenzothiophenes. Adv. Synth. Catal. 2018, 360, 2482–2487. 10.1002/adsc.201800266. [DOI] [Google Scholar]
- Yue D.-F.; Zhao J.-Q.; Chen Y.-Z.; Zhang X.-M.; Xu X.-Y.; Yuan W.-C. Zinc-Catalyzed Enantioselective Dearomative [3 + 2] Cycloaddition Reaction of 3-Nitrobenzothiophenes and 3-Nitrothieno[2,3-b]yridine with 3-Isothiocyanato Oxindoles. Adv. Synth. Catal. 2018, 360, 1420–1425. 10.1002/adsc.201701557. [DOI] [Google Scholar]
- Zhao J. Q.; Wu Z. J.; Zhou M. Q.; Xu X. Y.; Zhang X. M.; Yuan W. C. Zn-Catalyzed Diastereo- and Enantioselective Cascade Reaction of 3-Isothiocyanato Oxindoles and 3-Nitroindoles: Stereocontrolled Syntheses of Polycyclic Spirooxindoles. Org. Lett. 2015, 17, 5020–5023. 10.1021/acs.orglett.5b02489. [DOI] [PubMed] [Google Scholar]
- Tan F.; Lu L. Q.; Yang Q. Q.; Guo W.; Bian Q.; Chen J. R.; Xiao W. J. Enantioselective Cascade Michael Addition/Cyclization Reactions of 3-Nitro-2H-Chromenes with 3-Isothiocyanato Oxindoles: Efficient Synthesis of Functionalized Polycyclic Spirooxindoles. Chem. - Eur. J. 2014, 20, 3415–3420. 10.1002/chem.201303583. [DOI] [PubMed] [Google Scholar]
- Tan F.; Xiao C.; Cheng H.-G.; Wu W.; Ding K.-R.; Xiao W.-J. Enantioselective [4 + 2] Cycloadditions of 2-Vinyl-1 H-Indoles with 3-Nitro-2 H-Chromenes Catalyzed by a Zn(OTf)2/Bis(Oxazoline) Complex: An Efficient Approach to Fused Heterocycles with a Quaternary Stereocenter. Chem. - Asian J. 2012, 7, 493–497. 10.1002/asia.201100820. [DOI] [PubMed] [Google Scholar]
- Liu H.; Lu S.; Xu J.; Du D. Asymmetric Friedel-Crafts Alkylation of Electron-Rich N-Heterocycles with Nitroalkenes Catalyzed by Diphenylamine-Tethered Bis(Oxazoline) and Bis(Thiazoline) ZnII Complexes. Chem. - Asian J. 2008, 3, 1111–1121. 10.1002/asia.200800071. [DOI] [PubMed] [Google Scholar]
- Peng J.; Du D.-M. Asymmetric Friedel-Crafts Alkylation of Indoles with Nitrodienes and 2-Propargyloxy-β-Nitrostyrenes Catalyzed by Diphenylamine-Linked Bis(Oxazoline)-Zn-(OTf)2 Complexes. Eur. J. Org. Chem. 2012, 2012, 4042–4051. 10.1002/ejoc.201200382. [DOI] [Google Scholar]
- Despotopoulou C.; McKeon S. C.; Connon R.; Coeffard V.; Müller-Bunz H.; Guiry P. J. Application of a One-Pot Friedel-Crafts Alkylation/Michael Addition Methodology to the Asymmetric Synthesis of Ergoline Derivatives. Eur. J. Org. Chem. 2017, 2017 (45), 6734–6738. 10.1002/ejoc.201701480. [DOI] [Google Scholar]
- Wang H.; Wang Y.; Zhang C.; Jiang Y.; Chu M.; Li Z.; Du X.; Xu D. Asymmetric Conjugate Additions of 2-Substituted Benzofuran-3(2H)-Ones to α,β-Unsaturated Ketones Catalyzed by Chiral Copper Complexes. Org. Biomol. Chem. 2017, 15, 4191–4198. 10.1039/C7OB00677B. [DOI] [PubMed] [Google Scholar]
- O’Reilly S.; Guiry P. Recent Applications of C1-Symmetric Bis(Oxazoline)-Containing Ligands in Asymmetric Catalysis. Synthesis 2014, 46, 722–739. 10.1055/s-0033-1340829. [DOI] [Google Scholar]
- Coeffard V.; Aylward M.; Guiry P. J. First Regio- and Enantioselective Chromium-Catalyzed Homoallenylation of Aldehydes. Angew. Chem., Int. Ed. 2009, 48, 9152–9155. 10.1002/anie.200903647. [DOI] [PubMed] [Google Scholar]
- O’Reilly S.; Aylward M.; Keogh-Hansen C.; Fitzpatrick B.; McManus H. A.; Müller-Bunz H.; Guiry P. J. Synthesis of Bis(Oxazoline) Ligands Possessing C-5 Gem-Disubstitution and Their Application in Asymmetric Friedel-Crafts Alkylations. J. Org. Chem. 2015, 80, 10177–10186. 10.1021/acs.joc.5b01767. [DOI] [PubMed] [Google Scholar]
- McKeon S. C.; Müller-Bunz H.; Guiry P. J. New Thiazoline-Oxazoline Ligands and Their Application in the Asymmetric Friedel-Crafts Reaction. Eur. J. Org. Chem. 2009, 2009, 4833–4841. 10.1002/ejoc.200900683. [DOI] [Google Scholar]
- McKeon S. C.; Müller-Bunz H.; Guiry P. J. Synthesis of Thiazoline-Oxazoline Ligands and Their Application in Asymmetric Catalysis. Eur. J. Org. Chem. 2011, 2011, 7107–7115. 10.1002/ejoc.201101335. [DOI] [Google Scholar]
- Hargaden G. C.; O’Sullivan T. P.; Guiry P. J. Synthesis of Non-Symmetric Bis(Oxazoline)-Containing Ligands and Their Application in the Catalytic Enantioselective Nozaki-Hiyama-Kishi Allylation of Benzaldehyde. Org. Biomol. Chem. 2008, 6, 562–566. 10.1039/B715834C. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Kinoshita A.; Kawada H.; Nakada M. A New Asymmetric Tridentate Carbazole Ligand: Its Preparation and Application to Nozaki-Hiyama Allylation. Synlett 2003, 0570–0572. 10.1055/s-2003-37535. [DOI] [Google Scholar]
- Durán-Galván M.; Connell B. T. Asymmetric Synthesis of (1,3-Butadien-2-Yl)Methanols from Aldehydes via [1-(Silylmethyl)Allenyl]Methanols. Eur. J. Org. Chem. 2010, 2010, 2445–2448. 10.1002/ejoc.201000199. [DOI] [Google Scholar]
- Niwa T.; Nakada M. A Non-Heme Iron(III) Complex with Porphyrin-like Properties That Catalyzes Asymmetric Epoxidation. J. Am. Chem. Soc. 2012, 134, 13538–13541. 10.1021/ja304219s. [DOI] [PubMed] [Google Scholar]
- Chen W.; Yang Q.; Zhou T.; Tian Q.; Zhang G. Enantioselective Synthesis of α-Exo-Methylene γ-Butyrolactones via Chromium Catalysis. Org. Lett. 2015, 17, 5236–5239. 10.1021/acs.orglett.5b02597. [DOI] [PubMed] [Google Scholar]
- Tian Q.; Bai J.; Chen B.; Zhang G. Chromium-Catalyzed Asymmetric Dearomatization Addition Reactions of Halomethyl Heteroarenes. Org. Lett. 2016, 18, 1828–1831. 10.1021/acs.orglett.6b00559. [DOI] [PubMed] [Google Scholar]
- Chen W.; Bai J.; Zhang G. Chromium-Catalysed Asymmetric Dearomatization Addition Reactions of Bromomethylnaphthalenes. Adv. Synth. Catal. 2017, 359, 1227–1231. 10.1002/adsc.201600962. [DOI] [Google Scholar]
- Wang Z.; Ji H.; He W. M.; Xiong Y.; Zhang G. Chromium-Catalyzed Asymmetric Dearomatization-Addition Reactions of Halomethyloxazoles and Indoles. Synthesis 2018, 50, 4915–4921. 10.1055/s-0037-1609753. [DOI] [Google Scholar]
- Xiong Y.; Zhang G. Enantioselective 1,2-Difunctionalization of 1,3-Butadiene by Sequential Alkylation and Carbonyl Allylation. J. Am. Chem. Soc. 2018, 140, 2735–2738. 10.1021/jacs.7b12760. [DOI] [PubMed] [Google Scholar]
- Li W.; Hou G.; Wang C.; Jiang Y.; Zhang X. Asymmetric Hydrogenation of Ketones Catalyzed by a Ruthenium(II)-Indan-Ambox Complex. Chem. Commun. 2010, 46, 3979. 10.1039/b927028k. [DOI] [PubMed] [Google Scholar]
- Deng Q. H.; Wadepohl H.; Gade L. H. The Synthesis of a New Class of Chiral Pincer Ligands and Their Applications in Enantioselective Catalytic Fluorinations and the Nozaki-Hiyama-Kishi Reaction. Chem. - Eur. J. 2011, 17, 14922–14928. 10.1002/chem.201102375. [DOI] [PubMed] [Google Scholar]
- Deng Q. H.; Wadepohl H.; Gade L. H. Highly Enantioselective Copper-Catalyzed Alkylation of β-Ketoesters and Subsequent Cyclization to Spirolactones/Bi-Spirolactones. J. Am. Chem. Soc. 2012, 134, 2946–2949. 10.1021/ja211859w. [DOI] [PubMed] [Google Scholar]
- Deng Q.-H.; Wadepohl H.; Gade L. H. Highly Enantioselective Copper-Catalyzed Electrophilic Trifluoromethylation of β-Ketoesters. J. Am. Chem. Soc. 2012, 134, 10769–10772. 10.1021/ja3039773. [DOI] [PubMed] [Google Scholar]
- Deng Q. H.; Rettenmeier C.; Wadepohl H.; Gade L. H. Copper-Boxmi Complexes as Highly Enantioselective Catalysts for Electrophilic Trifluoromethylthiolations. Chem. - Eur. J. 2014, 20, 93–97. 10.1002/chem.201303641. [DOI] [PubMed] [Google Scholar]
- Deng Q. H.; Bleith T.; Wadepohl H.; Gade L. H. Enantioselective Iron-Catalyzed Azidation of β-Keto Esters and Oxindoles. J. Am. Chem. Soc. 2013, 135, 5356–5359. 10.1021/ja402082p. [DOI] [PubMed] [Google Scholar]
- Bleith T.; Wadepohl H.; Gade L. H. Iron Achieves Noble Metal Reactivity and Selectivity: Highly Reactive and Enantioselective Iron Complexes as Catalysts in the Hydrosilylation of Ketones. J. Am. Chem. Soc. 2015, 137, 2456–2459. 10.1021/ja512986m. [DOI] [PubMed] [Google Scholar]
- Wang J.; Frings M.; Bolm C. Enantioselective Nitrene Transfer to Sulfides Catalyzed by a Chiral Iron Complex. Angew. Chem., Int. Ed. 2013, 52, 8661–8665. 10.1002/anie.201304451. [DOI] [PubMed] [Google Scholar]
- Wang J.; Frings M.; Bolm C. Iron-Catalyzed Imidative Kinetic Resolution of Racemic Sulfoxides. Chem. - Eur. J. 2014, 20, 966–969. 10.1002/chem.201303850. [DOI] [PubMed] [Google Scholar]
- Poisson T.; Yamashita Y.; Kobayashi S. Catalytic Asymmetric Protonation of Chiral Calcium Enolates. J. Am. Chem. Soc. 2010, 2010, 7890–7892. 10.1021/ja102555a. [DOI] [PubMed] [Google Scholar]
- Espinosa M.; Blay G.; Cardona L.; Pedro J. R. Asymmetric Conjugate Addition of Malonate Esters to α,β-Unsaturated N -Sulfonyl Imines: An Expeditious Route to Chiral δ-Aminoesters and Piperidones. Chem. - Eur. J. 2013, 19, 14861–14866. 10.1002/chem.201302687. [DOI] [PubMed] [Google Scholar]
- Blay G.; Incerti C.; Muñoz M. C.; Pedro J. R. Enantioselective LaIII-PyBOX-Catalyzed Nitro-Michael Addition to (E)-2-Azachalcones. Eur. J. Org. Chem. 2013, 2013, 1696–1705. 10.1002/ejoc.201201579. [DOI] [Google Scholar]
- Detz R. J.; Abiri Z.; Le Griel R.; Hiemstra H.; Van Maarseveen J. H. Enantioselective Copper-Catalysed Propargylic Substitution: Synthetic Scope Study and Application in Formal Total Syntheses of (+)-Anisomycin and (−)-Cytoxazone. Chem. - Eur. J. 2011, 17, 5921–5930. 10.1002/chem.201003727. [DOI] [PubMed] [Google Scholar]
- Nakajima K.; Shibata M.; Nishibayashi Y. Copper-Catalyzed Enantioselective Propargylic Etherification of Propargylic Esters with Alcohols. J. Am. Chem. Soc. 2015, 137, 2472–2475. 10.1021/jacs.5b00004. [DOI] [PubMed] [Google Scholar]
- Shibata M.; Nakajima K.; Nishibayashi Y. Enantioselective Intramolecular Propargylic Amination Using Chiral Copper-Pybox Complexes as Catalysts. Chem. Commun. 2014, 50, 7874–7877. 10.1039/C4CC01676A. [DOI] [PubMed] [Google Scholar]
- Li R. Z.; Tang H.; Yang K. R.; Wan L. Q.; Zhang X.; Liu J.; Fu Z.; Niu D. Enantioselective Propargylation of Polyols and Desymmetrization of Meso 1,2-Diols by Copper/Borinic Acid Dual Catalysis. Angew. Chem., Int. Ed. 2017, 56, 7213–7217. 10.1002/anie.201703029. [DOI] [PubMed] [Google Scholar]
- Hashimoto T.; Omote M.; Maruoka K. Catalytic Asymmetric Alkynylation of C1-Substituted C,N-Cyclic Azomethine Imines by CuI/Chiral Brønsted Acid Co-Catalyst. Angew. Chem., Int. Ed. 2011, 50, 8952–8955. 10.1002/anie.201104017. [DOI] [PubMed] [Google Scholar]
- Dasgupta S.; Liu J.; Shoffler C. A.; Yap G. P. A.; Watson M. P. Enantioselective, Copper-Catalyzed Alkynylation of Ketimines to Deliver Isoquinolines with α-Diaryl Tetrasubstituted Stereocenters. Org. Lett. 2016, 18, 6006–6009. 10.1021/acs.orglett.6b02787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F.; Tan C.; Tang J.; Zhang Y. Y.; Gao W. M.; Wu H. H.; Yu Y. H.; Zhou J. Asymmetric Copper(I)-Catalyzed Azide-Alkyne Cycloaddition to Quaternary Oxindoles. J. Am. Chem. Soc. 2013, 135, 10994–10997. 10.1021/ja4066656. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Li T.-R.; Lu L.-Q.; Li M.-M.; Zhang K.; Xiao W.-J. Catalytic Asymmetric [4 + 1] Annulation of Sulfur Ylides with Copper-Allenylidene Intermediates. J. Am. Chem. Soc. 2016, 138, 8360–8363. 10.1021/jacs.6b04414. [DOI] [PubMed] [Google Scholar]
- Song J.; Zhang Z.-J.; Gong L.-Z. Asymmetric [4 + 2] Annulation of C1 Ammonium Enolates with Copper-Allenylidenes. Angew. Chem., Int. Ed. 2017, 56, 5212–5216. 10.1002/anie.201700105. [DOI] [PubMed] [Google Scholar]
- Lu X.; Ge L.; Cheng C.; Chen J.; Cao W.; Wu X. Enantioselective Cascade Reaction for Synthesis of Quinolinones through Synergistic Catalysis Using Cu-Pybox and Chiral Benzotetramisole as Catalysts. Chem. - Eur. J. 2017, 23, 7689–7693. 10.1002/chem.201701741. [DOI] [PubMed] [Google Scholar]
- Ji D.; Wang C.; Sun J. Asymmetric [4 + 2]-Cycloaddition of Copper-Allenylidenes with Hexahydro-1,3,5-Triazines: Access to Chiral Tetrahydroquinazolines. Org. Lett. 2018, 20, 3710–3713. 10.1021/acs.orglett.8b01584. [DOI] [PubMed] [Google Scholar]
- Chen H.; Lu X.; Xia X.; Zhu Q.; Song Y.; Chen J.; Cao W.; Wu X. Asymmetric Catalytic [4 + 2] Cycloaddition via Cu-Allenylidene Intermediate: Stereoselective Synthesis of Tetrahydroquinolines Fused with a γ-Lactone Moiety. Org. Lett. 2018, 20, 1760–1763. 10.1021/acs.orglett.8b00253. [DOI] [PubMed] [Google Scholar]
- Shao W.; You S. L. Highly Diastereo- and Enantioselective Synthesis of Tetrahydro-5H-Indolo[2,3-b]Quinolines through Copper-Catalyzed Propargylic Dearomatization of Indoles. Chem. - Eur. J. 2017, 23, 12489–12493. 10.1002/chem.201703443. [DOI] [PubMed] [Google Scholar]
- Gómez J. E.; Guo W.; Gaspa S.; Kleij A. W. Copper-Catalyzed Synthesis of γ-Amino Acids Featuring Quaternary Stereocenters. Angew. Chem., Int. Ed. 2017, 56, 15035–15038. 10.1002/anie.201709511. [DOI] [PubMed] [Google Scholar]
- Tian L.; Gong L.; Zhang X. Copper-Catalyzed Enantioselective Synthesis of β-Amino Alcohols Featuring Tetrasubstituted Tertiary Carbons. Adv. Synth. Catal. 2018, 360, 2055–2059. 10.1002/adsc.201701613. [DOI] [Google Scholar]
- Shemet A.; Carreira E. M. Total Synthesis of (−)-Rhazinilam and Formal Synthesis of (+)-Eburenine and (+)-Aspidospermidine: Asymmetric Cu-Catalyzed Propargylic Substitution. Org. Lett. 2017, 19, 5529–5532. 10.1021/acs.orglett.7b02619. [DOI] [PubMed] [Google Scholar]
- Osako T.; Uozumi Y. Enantioposition-Selective Copper-Catalyzed Azide-Alkyne Cycloaddition for Construction of Chiral Biaryl Derivatives. Org. Lett. 2014, 16, 5866–5869. 10.1021/ol502778j. [DOI] [PubMed] [Google Scholar]
- Woyciechowska M.; Forcher G.; Buda S.; Mlynarski J. General Switch in Regioselectivity in the Mukaiyama Aldol Reaction of Silyloxyfuran with Aldehydes in Aqueous Solvents. Chem. Commun. 2012, 48, 11029–11031. 10.1039/c2cc36656h. [DOI] [PubMed] [Google Scholar]
- Dudek A.; Mlynarski J. Iron-Catalyzed Asymmetric Nitro-Mannich Reaction. J. Org. Chem. 2017, 82, 11218–11224. 10.1021/acs.joc.7b01786. [DOI] [PubMed] [Google Scholar]
- Lowicki D.; Bezlada A.; Mlynarski J. Asymmetric Hydrosilylation of Ketones Catalyzed by Zinc Acetate with Hindered Pybox Ligands. Adv. Synth. Catal. 2014, 356, 591–595. 10.1002/adsc.201300682. [DOI] [Google Scholar]
- Wen H.; Wan X.; Huang Z. Asymmetric Synthesis of Silicon-Stereogenic Vinylhydrosilanes by Cobalt-Catalyzed Regio- and Enantioselective Alkyne Hydrosilylation with Dihydrosilanes. Angew. Chem., Int. Ed. 2018, 57, 6319–6323. 10.1002/anie.201802806. [DOI] [PubMed] [Google Scholar]
- Dasgupta S.; Rivas T.; Watson M. P. Enantioselective Copper(I)-Catalyzed Alkynylation of Oxocarbenium Ions to Set Diaryl Tetrasubstituted Stereocenters. Angew. Chem., Int. Ed. 2015, 54, 14154–14158. 10.1002/anie.201507373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y.; Takahashi M.; Yagishita F.; Sakamoto M.; Sengoku T.; Yoda H. Construction of Spiro-Fused 2-Oxindole/α-Methylene- γ-Butyrolactone Systems with Extremely High Enantioselectivity via Indium-Catalyzed Amide Allylation of N-Methyl Isatin. Org. Lett. 2013, 15, 6182–6185. 10.1021/ol403014u. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Murata Y.; Yagishita F.; Sakamoto M.; Sengoku T.; Yoda H. Catalytic Enantioselective Amide Allylation of Isatins and Its Application in the Synthesis of 2-Oxindole Derivatives Spiro-Fused to the α-Methylene-γ-Butyrolactone Functionality. Chem. - Eur. J. 2014, 20, 11091–11100. 10.1002/chem.201403357. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Murata Y.; Ishida M.; Yagishita F.; Sakamoto M.; Sengoku T.; Yoda H. Catalytic Amide Allylation of α-Ketoesters: Extremely High Enantioselective Synthesis of Ester Functionalised α-Methylene-γ-Butyrolactones. Org. Biomol. Chem. 2014, 12, 7686–7689. 10.1039/C4OB01508H. [DOI] [PubMed] [Google Scholar]
- Alagiri K.; Furutachi M.; Yamatsugu K.; Kumagai N.; Watanabe T.; Shibasaki M. Two Approaches toward the Formal Total Synthesis of Oseltamivir Phosphate (Tamiflu): Catalytic Enantioselective Three-Component Reaction Strategy and l-Glutamic Acid Strategy. J. Org. Chem. 2013, 78, 4019–4026. 10.1021/jo400360j. [DOI] [PubMed] [Google Scholar]
- Marié J. C.; Xiong Y.; Min G. K.; Yeager A. R.; Taniguchi T.; Berova N.; Schaus S. E.; Porco J. A. Enantioselective Synthesis of 3,4-Chromanediones via Asymmetric Rearrangement of 3-Allyloxyflavones. J. Org. Chem. 2010, 75, 4584–4590. 10.1021/jo100889c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desimoni G.; Faita G.; Livieri A.; Mella M.; Ponta L.; Boiocchi M. The Asymmetric Formal Hetero-Diels-Alder Reaction of Methyl (E)-4-Aryl-2-Oxo-3-Butenoates Catalyzed by [Sc(OTf)3/Pybox] Complexes. Eur. J. Org. Chem. 2012, 2012, 2916–2928. 10.1002/ejoc.201200056. [DOI] [Google Scholar]
- Karimi B.; Jafari E.; Enders D. Highly Efficient Catalytic Enantioselective Mannich Reaction of Malonates with N-Tert-Butoxycarbonyl Imines by Using Yb(OTf)3/Pybox Catalysts at Room Temperature. Chem. - Eur. J. 2013, 19, 10142–10145. 10.1002/chem.201300241. [DOI] [PubMed] [Google Scholar]
- Kawatsura M.; Uchida K.; Terasaki S.; Tsuji H.; Minakawa M.; Itoh T. Ruthenium-Catalyzed Regio- and Enantioselective Allylic Amination of Racemic 1-Arylallyl Esters. Org. Lett. 2014, 16, 1470–1473. 10.1021/ol5002768. [DOI] [PubMed] [Google Scholar]
- Shinozawa T.; Terasaki S.; Mizuno S.; Kawatsura M. Kinetic Resolution of Racemic and Branched Monosubstituted Allylic Acetates by a Ruthenium-Catalyzed Regioselective Allylic Etherification. J. Org. Chem. 2016, 81, 5766–5774. 10.1021/acs.joc.6b00939. [DOI] [PubMed] [Google Scholar]
- de Julián E.; Menéndez-Pedregal E.; Claros M.; Vaquero M.; Díez J.; Lastra E.; Gamasa P.; Pizzano A. Practical Synthesis of Enantiopure Benzylamines by Catalytic Hydrogenation or Transfer Hydrogenation Reactions in Isopropanol Using a Ru-Pybox Catalyst. Org. Chem. Front. 2018, 5, 841–849. 10.1039/C7QO00908A. [DOI] [Google Scholar]
- Arredondo V.; Hiew S. C.; Gutman E. S.; Premachandra I. D. U. A.; Van Vranken D. L. Enantioselective Palladium-Catalyzed Carbene Insertion into the N-H Bonds of Aromatic Heterocycles. Angew. Chem., Int. Ed. 2017, 56, 4156–4159. 10.1002/anie.201611845. [DOI] [PubMed] [Google Scholar]
- Fang H.; Yang Z.; Zhang L.; Wang W.; Li Y.; Xu X.; Zhou S. Transmetal-Catalyzed Enantioselective Cross-Coupling Reaction of Racemic Secondary Benzylic Bromides with Organoaluminum Reagents. Org. Lett. 2016, 18, 6022–6025. 10.1021/acs.orglett.6b02933. [DOI] [PubMed] [Google Scholar]
- Eno M. S.; Lu A.; Morken J. P. Nickel-Catalyzed Asymmetric Kumada Cross-Coupling of Symmetric Cyclic Sulfates. J. Am. Chem. Soc. 2016, 138, 7824–7827. 10.1021/jacs.6b03384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan J.; Liu J.; Zheng H.; Zuo Z.; Hou L.; Hu H.; Wang Y.; Luan X. Direct Asymmetric Dearomatization of 2-Naphthols by Scandium-Catalyzed Electrophilic Amination. Angew. Chem., Int. Ed. 2015, 54, 2356–2360. 10.1002/anie.201409565. [DOI] [PubMed] [Google Scholar]
- Zhao K.; Duan L.; Xu S.; Jiang J.; Fu Y.; Gu Z. Enhanced Reactivity by Torsional Strain of Cyclic Diaryliodonium in Cu-Catalyzed Enantioselective Ring-Opening Reaction. Chem. 2018, 4, 599–612. 10.1016/j.chempr.2018.01.017. [DOI] [Google Scholar]
- Xu S.; Zhao K.; Gu Z. Copper-Catalyzed Asymmetric Ring-Opening of Cyclic Diaryliodonium with Benzylic and Aliphatic Amines. Adv. Synth. Catal. 2018, 360, 3877–3883. 10.1002/adsc.201800637. [DOI] [Google Scholar]
- Wang D.-C.; Xie M.-S.; Guo H.-M.; Qu G.-R.; Zhang M.-C.; You S.-L. Enantioselective Dearomative [3 + 2] Cycloaddition Reactions of Benzothiazoles. Angew. Chem., Int. Ed. 2016, 55, 14111–14115. 10.1002/anie.201607852. [DOI] [PubMed] [Google Scholar]
- Ruiz Espelt L.; McPherson I. S.; Wiensch E. M.; Yoon T. P. Enantioselective Conjugate Additions of α-Amino Radicals via Cooperative Photoredox and Lewis Acid Catalysis. J. Am. Chem. Soc. 2015, 137, 2452–2455. 10.1021/ja512746q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T. R.; Miller Z. D.; Bates D. M.; Guzei I. A.; Yoon T. P. Enantioselective Photochemistry through Lewis Acid-Catalyzed Triplet Energy Transfer. Science 2016, 354, 1391–1395. 10.1126/science.aai8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Z. D.; Lee B. J.; Yoon T. P. Enantioselective Crossed Photocycloadditions of Styrenic Olefins by Lewis Acid Catalyzed Triplet Sensitization. Angew. Chem., Int. Ed. 2017, 56, 11891–11895. 10.1002/anie.201706975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pericas L.; Shafir A.; Vallribera A. Asymmetric Synthesis of L-Carbidopa Based on a Highly Enantioselective α-Amination. Org. Lett. 2013, 15, 1448–1451. 10.1021/ol400136y. [DOI] [PubMed] [Google Scholar]
- Granados A.; del Olmo A.; Peccati F.; Billard T.; Sodupe M.; Vallribera A. Fluorous L-Carbidopa Precursors: Highly Enantioselective Synthesis and Computational Prediction of Bioactivity. J. Org. Chem. 2018, 83, 303–313. 10.1021/acs.joc.7b02685. [DOI] [PubMed] [Google Scholar]
- Suga H.; Hashimoto Y.; Yasumura S.; Takezawa R.; Itoh K.; Kakehi A. Chiral Lewis Acid Catalyzed Asymmetric Cycloadditions of Carbonyl Ylides Generated from Diazoimide Derivatives and Their Synthetic Applications to Indolizidine Alkaloids. J. Org. Chem. 2013, 78, 10840–10852. 10.1021/jo401837d. [DOI] [PubMed] [Google Scholar]
- Shen H. C.; Wu Y. F.; Zhang Y.; Fan L. F.; Han Z. Y.; Gong L. Z. Palladium-Catalyzed Asymmetric Aminohydroxylation of 1,3-Dienes. Angew. Chem., Int. Ed. 2018, 57, 2372–2376. 10.1002/anie.201712350. [DOI] [PubMed] [Google Scholar]
- Tsuchida K.; Senda Y.; Nakajima K.; Nishibayashi Y. Construction of Chiral Tri- and Tetra-Arylmethanes Bearing Quaternary Carbon Centers: Copper-Catalyzed Enantioselective Propargylation of Indoles with Propargylic Esters. Angew. Chem., Int. Ed. 2016, 55, 9728–9732. 10.1002/anie.201604182. [DOI] [PubMed] [Google Scholar]
- Xie Z.; Liu X.; Liu L. Copper-Catalyzed Aerobic Enantioselective Cross-Dehydrogenative Coupling of N-Aryl Glycine Esters with Terminal Alkynes. Org. Lett. 2016, 18, 2982–2985. 10.1021/acs.orglett.6b01328. [DOI] [PubMed] [Google Scholar]
- Zhao J. F.; Tan B. H.; Zhu M. K.; Tjan T. B. W.; Loh T. P. Enantioselective Carbonyl-Ene Reactions of Trifluoropyruvate in Ionic Liquid via a Recyclable Indium(III)-Pybox Complex. Adv. Synth. Catal. 2010, 352, 2085–2088. 10.1002/adsc.201000170. [DOI] [Google Scholar]
- Zhao Y. J.; Li B.; Tan L. J. S.; Shen Z. L.; Loh T. P. Enantioselective Cationic Polyene Cyclization vs Enantioselective Intramolecular Carbonyl-Ene Reaction. J. Am. Chem. Soc. 2010, 132, 10242–10244. 10.1021/ja104119j. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Chen L.; Zhang H.; Ma Z.; Jiang H.; Zhu S. Highly Chemo- and Stereoselective Catalyst-Controlled Allylic C-H Insertion and Cyclopropanation Using Donor/Donor Carbenes. Angew. Chem., Int. Ed. 2018, 57, 12405–12409. 10.1002/anie.201805676. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Loh T. P. Asymmetric Hetero-Diels-Alder Reaction of Danishefsky’s Dienes with α-Carbonyl Esters Catalyzed by an Indium(III)-PyBox Complex. Org. Lett. 2013, 15, 2914–2917. 10.1021/ol400841s. [DOI] [PubMed] [Google Scholar]
- Hanhan N. V.; Sahin A. H.; Chang T. W.; Fettinger J. C.; Franz A. K. Catalytic Asymmetric Synthesis of Substituted 3-Hydroxy-2oxindoles. Angew. Chem., Int. Ed. 2010, 49, 744–747. 10.1002/anie.200904393. [DOI] [PubMed] [Google Scholar]
- Gutierrez E. G.; Wong C. J.; Sahin A. H.; Franz A. K. Enantioselective and Regioselective Indium(III)-Catalyzed Addition of Pyrroles to Isatins. Org. Lett. 2011, 13, 5754–5757. 10.1021/ol202329s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanhan N. V.; Tang Y. C.; Tran N. T.; Franz A. K. Scandium(III)-Catalyzed Enantioselective Allylation of Isatins Using Allylsilanes. Org. Lett. 2012, 14, 2218–2221. 10.1021/ol300496v. [DOI] [PubMed] [Google Scholar]
- Hanhan N. V.; Ball-Jones N. R.; Tran N. T.; Franz A. K. Catalytic Asymmetric [3 + 2] Annulation of Allylsilanes with Isatins: Synthesis of Spirooxindoles. Angew. Chem., Int. Ed. 2012, 51, 989–992. 10.1002/anie.201105739. [DOI] [PubMed] [Google Scholar]
- Ball-Jones N. R.; Badillo J. J.; Tran N. T.; Franz A. K. Catalytic Enantioselective Carboannulation with Allylsilanes. Angew. Chem., Int. Ed. 2014, 53, 9462–9465. 10.1002/anie.201403607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z.; Han J.; Qian P.; Zhang Z.; Wang Y.; Pan Y. Enantioselective Synthesis of 3-Hydroxy Oxindoles by Ytterbium-Catalysed Decarboxylative Addition of β-Ketoacids to Isatins. Org. Biomol. Chem. 2013, 11, 6456–6459. 10.1039/c3ob41460d. [DOI] [PubMed] [Google Scholar]
- Prakash M.; Kesavan V. Highly Enantioselective Synthesis of 2,3-Dihydroquinazolinones through Intramolecular Amidation of Imines. Org. Lett. 2012, 14, 1896–1899. 10.1021/ol300518m. [DOI] [PubMed] [Google Scholar]
- Rout S.; Das A.; Singh V. K. Metal-Controlled Switching of Enantioselectivity in the Mukaiyama-Michael Reaction of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Chiral Metal-Pybox Complexes. J. Org. Chem. 2018, 83, 5058–5071. 10.1021/acs.joc.8b00399. [DOI] [PubMed] [Google Scholar]
- Rout S.; Das A.; Singh V. K. An Asymmetric Vinylogous Mukaiyama-Michael Reaction of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Chiral Sc(III)- or Er(III)-Pybox Complexes. Chem. Commun. 2017, 53, 5143–5146. 10.1039/C7CC01763D. [DOI] [PubMed] [Google Scholar]
- Shimizu S.; Tsubogo T.; Xu P.; Kobayashi S. Calcium-Catalyzed Asymmetric Synthesis of 3-Tetrasubstituted Oxindoles: Efficient Construction of Adjacent Quaternary and Tertiary Chiral Centers. Org. Lett. 2015, 17, 2006–2009. 10.1021/acs.orglett.5b00749. [DOI] [PubMed] [Google Scholar]
- Ray S. K.; Singh P. K.; Molleti N.; Singh V. K. Enantioselective Synthesis of Coumarin Derivatives by PYBOX-DIPH-Zn(II) Complex Catalyzed Michael Reaction. J. Org. Chem. 2012, 77, 8802–8808. 10.1021/jo301513x. [DOI] [PubMed] [Google Scholar]
- Ray S. K.; Rout S.; Singh V. K. Enantioselective Synthesis of 3,4-Dihydropyran Derivatives via a Michael Addition Reaction Catalysed by Chiral Pybox-Diph-Zn(II) Complex. Org. Biomol. Chem. 2013, 11, 2412. 10.1039/c3ob40246k. [DOI] [PubMed] [Google Scholar]
- Rout S.; Ray S. K.; Singh V. K. Enantioselective Mukaiyama-Michael with 2-Enoyl Pyridine N-Oxides Catalyzed by PYBOX-DIPH-Zn(II)-Complexes at Ambient Temperature. Org. Biomol. Chem. 2013, 11, 4537–4545. 10.1039/c3ob40445e. [DOI] [PubMed] [Google Scholar]
- Bisai V.; Suneja A.; Singh V. K. Asymmetric Alkynylation/Lactamization Cascade: An Expeditious Entry to Enantiomerically Enriched Isoindolinones. Angew. Chem., Int. Ed. 2014, 53, 10737–10741. 10.1002/anie.201405074. [DOI] [PubMed] [Google Scholar]
- Suneja A.; Bisai V.; Singh V. K. Asymmetric Syntheses of Medicinally Important Isoindolinones (S)-PD 172938, (R)-JM 1232, and Related Structures. J. Org. Chem. 2016, 81, 4779–4788. 10.1021/acs.joc.6b00770. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; You Y. S.; Kang S. H. Asymmetric Synthesis of All-Carbon Quaternary Stereocenters via Desymmetrization of 2,2-Disubstituted 1,3-Propanediols. J. Am. Chem. Soc. 2011, 133, 1772–1774. 10.1021/ja1103102. [DOI] [PubMed] [Google Scholar]
- Parsons A. T.; Johnson J. S. Catalytic Enantioselective Synthesis of Tetrahydrofurans: A Dynamic Kinetic Asymmetric [3 + 2] Cycloaddition of Racemic Cyclopropanes and Aldehydes. J. Am. Chem. Soc. 2009, 131, 3122–3123. 10.1021/ja809873u. [DOI] [PubMed] [Google Scholar]
- Parsons A. T.; Smith A. G.; Neel A. J.; Johnson J. S. Dynamic Kinetic Asymmetric Synthesis of Substituted Pyrrolidines from Racemic Cyclopropanes and Aldimines: Reaction Development and Mechanistic Insights. J. Am. Chem. Soc. 2010, 132, 9688–9692. 10.1021/ja1032277. [DOI] [PubMed] [Google Scholar]
- Wales S. M.; Walker M. M.; Johnson J. S. Asymmetric Synthesis of Indole Homo-Michael Adducts via Dynamic Kinetic Friedel-Crafts Alkylation with Cyclopropanes. Org. Lett. 2013, 15, 2558–2561. 10.1021/ol4010646. [DOI] [PubMed] [Google Scholar]
- Amador A. G.; Sherbrook E. M.; Yoon T. P. Enantioselective Photocatalytic [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones. J. Am. Chem. Soc. 2016, 138, 4722–4725. 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X. T.; Gan C. C.; Liu S. Y.; Zhou F.; Wu H. H.; Zhou J. Utilization of CO2 as a C1 Building Block in a Tandem Asymmetric A3 Coupling-Carboxylative Cyclization Sequence to 2-Oxazolidinones. ACS Catal. 2017, 7, 8588–8593. 10.1021/acscatal.7b03370. [DOI] [Google Scholar]
- Da B. C.; Liang Q. J.; Luo Y. C.; Ahmad T.; Xu Y. H.; Loh T. P. Copper-Catalyzed Stereo- and Enantioselective 1,4-Protosilylation of α,β-Unsaturated Ketimines to Synthesize Functionalized Allylsilanes. ACS Catal. 2018, 8, 6239–6245. 10.1021/acscatal.8b01547. [DOI] [Google Scholar]
- Ghoshal A.; Sarkar A. R.; Manickam G.; Kumaran R. S.; Jayashankaran J. Rhodium-Catalyzed Asymmetric Hydrosilylation of Ketones Employing a New Ligand Embodying the Bis(Oxazolinyl)Pyridine Moiety. Synlett 2010, 2010, 1459–1462. 10.1055/s-0029-1219949. [DOI] [Google Scholar]
- Nishiyama H.; Ito J. Bis(Oxazolinyl)Phenyl Transition-Metal Complexes: Asymmetric Catalysis and Some Reactions of the Metals. Chem. Commun. 2010, 46, 203–212. 10.1039/B918923H. [DOI] [PubMed] [Google Scholar]
- Shiomi T.; Adachi T.; Ito J. I.; Nishiyama H. Intermolecular Antiselective and Enantioselective Reductive Coupling of Enones and Aromatic Aldehydes with Chiral Rh(Phebox) Catalysts. Org. Lett. 2009, 11, 1011–1014. 10.1021/ol802939u. [DOI] [PubMed] [Google Scholar]
- Itoh K.; Tsuruta A.; Ito J. I.; Yamamoto Y.; Nishiyama H. Enantioselective Synthesis of Optically Active 3,3-Diarylpropanoates by Conjugate Hydrosilylation with Chiral Rh-Bis(Oxazolinyl)Phenyl Catalysts. J. Org. Chem. 2012, 77, 10914–10919. 10.1021/jo302357b. [DOI] [PubMed] [Google Scholar]
- Naganawa Y.; Kawagishi M.; Ito J. I.; Nishiyama H. Asymmetric Induction at Remote Quaternary Centers of Cyclohexadienones by Rhodium-Catalyzed Conjugate Hydrosilylation. Angew. Chem., Int. Ed. 2016, 55, 6873–6876. 10.1002/anie.201601636. [DOI] [PubMed] [Google Scholar]
- Shiomi T.; Adachi T.; Toribatake K.; Zhou L.; Nishiyama H. Asymmetric β-Boration of α,β-Unsaturated Carbonyl Compounds Promoted by Chiral Rhodium-Bisoxazolinylphenyl Catalysts. Chem. Commun. 2009, (40), 5987–5989. 10.1039/b915759j. [DOI] [PubMed] [Google Scholar]
- Toribatake K.; Nishiyama H. Asymmetric Diboration of Terminal Alkenes with a Rhodium Catalyst and Subsequent Oxidation: Enantioselective Synthesis of Optically Active 1,2-Diols. Angew. Chem., Int. Ed. 2013, 52, 11011–11015. 10.1002/anie.201305181. [DOI] [PubMed] [Google Scholar]
- Smith J. R.; Collins B. S. L.; Hesse M. J.; Graham M. A.; Myers E. L.; Aggarwal V. K. Enantioselective Rhodium(III)-Catalyzed Markovnikov Hydroboration of Unactivated Terminal Alkenes. J. Am. Chem. Soc. 2017, 139, 9148–9151. 10.1021/jacs.7b05149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito J. I.; Ujiie S.; Nishiyama H. New Bis (Oxazolinyl)Phenyl-Ruthenium (II) Complexes and Their Catalytic Activity for Enantioselective Hydrogenation and Transfer Hydrogenation of Ketones. Organometallics 2009, 28, 630–638. 10.1021/om800953f. [DOI] [Google Scholar]
- Ito J. I.; Ujiie S.; Nishiyama H. A New NCN Pincer Ruthenium Complex and Its Catalytic Activity for Enantioselective Hydrogenation of Ketones. Chem. Commun. 2008, (16), 1923–1925. 10.1039/b800387d. [DOI] [PubMed] [Google Scholar]
- Ito J. I.; Teshima T.; Nishiyama H. Enhancement of Enantioselectivity by Alcohol Additives in Asymmetric Hydrogenation with Bis(Oxazolinyl)Phenyl Ruthenium Catalysts. Chem. Commun. 2012, 48, 1105–1107. 10.1039/C1CC16057E. [DOI] [PubMed] [Google Scholar]
- Ito J. I.; Asai R.; Nishiyama H. Asymmetric Direct Alkynylation Catalyzed by Chiral Ru-Bis(Oxazolinyl)Phenyl Complexes. Org. Lett. 2010, 12, 3860–3862. 10.1021/ol1015338. [DOI] [PubMed] [Google Scholar]
- Ito J. I.; Fujii K.; Nishiyama H. Direct Conjugate Addition of Alkynes with α,β-Unsaturated Carbonyl Compounds Catalyzed by NCN-Pincer Ru Complexes. Chem. - Eur. J. 2013, 19, 601–605. 10.1002/chem.201203380. [DOI] [PubMed] [Google Scholar]
- Ubukata S.; Ito J. I.; Oguri R.; Nishiyama H. Asymmetric Three-Component Coupling Reaction of Alkyne, Enone, and Aldehyde Catalyzed by Chiral Phebox Ruthenium Catalysts. J. Org. Chem. 2016, 81, 3347–3355. 10.1021/acs.joc.6b00374. [DOI] [PubMed] [Google Scholar]
- Ohshima T.; Kawabata T.; Takeuchi Y.; Kakinuma T.; Iwasaki T.; Yonezawa T.; Murakami H.; Nishiyama H.; Mashima K. C1-Symmetric Rh/Phebox-Catalyzed Asymmetric Alkynylation of α-Ketoesters. Angew. Chem., Int. Ed. 2011, 50, 6296–6300. 10.1002/anie.201100252. [DOI] [PubMed] [Google Scholar]
- Morisaki K.; Sawa M.; Nomaguchi J. Y.; Morimoto H.; Takeuchi Y.; Mashima K.; Ohshima T. Rh-Catalyzed Direct Enantioselective Alkynylation of α-Ketiminoesters. Chem. - Eur. J. 2013, 19, 8417–8420. 10.1002/chem.201301237. [DOI] [PubMed] [Google Scholar]
- Morisaki K.; Sawa M.; Yonesaki R.; Morimoto H.; Mashima K.; Ohshima T. Mechanistic Studies and Expansion of the Substrate Scope of Direct Enantioselective Alkynylation of α-Ketiminoesters Catalyzed by Adaptable (Phebox)Rhodium(III) Complexes. J. Am. Chem. Soc. 2016, 138, 6194–6203. 10.1021/jacs.6b01590. [DOI] [PubMed] [Google Scholar]
- Ito J. I.; Ujiie S.; Nishiyama H. Chiral Bis(Oxazolinyl)Phenyl RuII Catalysts for Highly Enantioselective Cyclopropanation. Chem. - Eur. J. 2010, 16, 4986–4990. 10.1002/chem.200903514. [DOI] [PubMed] [Google Scholar]
- Owens C. P.; Varela-Álvarez A.; Boyarskikh V.; Musaev D. G.; Davies H. M. L.; Blakey S. B. Iridium(III)-Bis(Oxazolinyl)Phenyl Catalysts for Enantioselective C-H Functionalization. Chem. Sci. 2013, 4, 2590. 10.1039/c3sc50886b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K.; Sibi M. P. Dibenzofuran-4,6-Bis(Oxazoline) (DBFOX). A Novel Trans -Chelating Bis(Oxazoline) Ligand for Asymmetric Reactions. Org. Biomol. Chem. 2018, 16, 5551–5565. 10.1039/C8OB01010B. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Tokunaga E.; Suzuki S.; Shiro M. Enantioselective Friedel-Crafts Reaction of -Trifluoromethylated Acrylates with Pyrroles and Its. Org. Lett. 2010, 86, 2008–2010. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Suzuki S.; Liu G.; Tokunaga E.; Shiro M.; Shibata N. Asymmetric Synthesis of Chiral Trifluoromethylated Heliotridane via Highly Catalytic Asymmetric Friedel-Crafts Alkylation with β-Trifluoromethylated Acrylates and Pyrroles. New J. Chem. 2011, 35, 2614–2621. 10.1039/c1nj20550a. [DOI] [Google Scholar]
- Murarka S.; Deb I.; Zhang C.; Seidel D. Catalytic Enantioselective Intramolecular Redox Reactions: Ring-Fused Tetrahydroquinolines. J. Am. Chem. Soc. 2009, 131, 13226–13227. 10.1021/ja905213f. [DOI] [PubMed] [Google Scholar]
- Qiu J. S.; Wang Y. F.; Qi G. R.; Karmaker P. G.; Yin H. Q.; Chen F. X. Highly Enantioselective α-Cyanation with 4-Acetylphenyl Cyanate. Chem. - Eur. J. 2017, 23, 1775–1778. 10.1002/chem.201605610. [DOI] [PubMed] [Google Scholar]
- Shibata N.; Ishimaru T.; Reddy D. S.; Horikawa T.; Nakamura S.; Toru T. DBFOX-Ph/Metal Complexes: Evaluation as Catalysts for Enantioselective Fluorination of 3-(2-Arylacetyl)-2-Thiazolidinones. Beilstein J. Org. Chem. 2008, 4, 55–57. 10.3762/bjoc.4.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy D. S.; Shibata N.; Horikawa T.; Suzuki S.; Nakamura S.; Toru T.; Shiro M. A DBFOX-Ph-Based Combinatorial Catalyst for Enantioselective Fluorination of Aryl Acetyl and 3-Butenoyl Thiazolidinones. Chem. - Asian J. 2009, 4, 1411–1415. 10.1002/asia.200900164. [DOI] [PubMed] [Google Scholar]
- Reddy D. S.; Shibata N.; Nagai J.; Nakamura S.; Toru T.; Kanemasa S. Desymmetrization-like Catalytic Enantioselective Fluorination of Malonates and Its Application to Pharmaceutically Attractive Molecules. Angew. Chem., Int. Ed. 2008, 47, 164–168. 10.1002/anie.200704093. [DOI] [PubMed] [Google Scholar]
- Reddy D. S.; Shibata N.; Nagai J.; Nakamura S.; Toru T. A Dynamic Kinetic Asymmetric Transformation in the α-Hydroxylation of Racemic Malonates and Its Application to Biologically Active Molecules. Angew. Chem., Int. Ed. 2009, 48, 803–806. 10.1002/anie.200804476. [DOI] [PubMed] [Google Scholar]
- Sibi M. P.; Coulomb J.; Stanley L. M. Enantioselective Enolate Protonations: Friedel-Crafts Reactions with α-Substituted Acrylates. Angew. Chem., Int. Ed. 2008, 47, 9913–9915. 10.1002/anie.200804221. [DOI] [PubMed] [Google Scholar]
- Banerjee B.; Capps S. G.; Kang J.; Robinson J. W.; Castle S. L. Second-Generation DBFOX Ligands for the Synthesis of β-Substituted α-Amino Acids via Enantioselective Radical Conjugate Additions. J. Org. Chem. 2008, 73, 8973–8978. 10.1021/jo801721z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Cheng F.; Kou Da Y.; Pang S.; Shen Y. C.; Huang Y. Y.; Shibata N. Catalytic Asymmetric 1,3-Dipolar Cycloaddition of β-Fluoroalkylated α,β-Unsaturated 2-Pyridylsulfones with Nitrones for Chiral Fluoroalkylated Isoxazolidines and γ-Amino Alcohols. Angew. Chem., Int. Ed. 2017, 56, 1510–1514. 10.1002/anie.201610605. [DOI] [PubMed] [Google Scholar]
- Shen X.; Li Y.; Wen Z.; Cao S.; Hou X.; Gong L. A Chiral Nickel DBFOX Complex as a Bifunctional Catalyst for Visible-Light-Promoted Asymmetric Photoredox Reactions. Chem. Sci. 2018, 9, 4562–4568. 10.1039/C8SC01219A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. M.; Gao Q.; Li J.; Liu Y.; Li C. A Robust Ru-PNNP Catalyst System for the Asymmetric Hydrogenation of α,β-Unsaturated Ketones to Allylic Alcohol. Tetrahedron Lett. 2013, 54, 7013–7016. 10.1016/j.tetlet.2013.10.051. [DOI] [Google Scholar]
- Dai W.; Li G.; Chen B.; Wang L.; Gao S. A Porphyrin-Inspired Iron Catalyst for Asymmetric Epoxidation of Electron-Deficient Olefins. Org. Lett. 2015, 17, 904–907. 10.1021/acs.orglett.5b00018. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Suzuki T.; Sakakura A.; Ishihara K. Catalytic Enantioselective Inverse Electron Demand Hetero-Diels-Alder Reaction with Allylsilanes. Angew. Chem., Int. Ed. 2014, 53, 6131–6134. 10.1002/anie.201402934. [DOI] [PubMed] [Google Scholar]
- Sakakura A.; Kondo R.; Matsumura Y.; Akakura M.; Ishihara K. Rational Design of Highly Effective Asymmetric Diels-Alder Catalysts Bearing 4,4-Sulfonamidomethyl Groups. J. Am. Chem. Soc. 2009, 131, 17762–17764. 10.1021/ja906098b. [DOI] [PubMed] [Google Scholar]
- Perrotta D.; Wang M. M.; Waser J. Lewis Acid Catalyzed Enantioselective Desymmetrization of Donor-Acceptor Meso-Diaminocyclopropanes. Angew. Chem., Int. Ed. 2018, 57, 5120–5123. 10.1002/anie.201800494. [DOI] [PubMed] [Google Scholar]
- Shao Q.; Wu L.; Chen J.; Gridnev I. D.; Yang G.; Xie F.; Zhang W. Copper (II)/RuPHOX-Catalyzed Enantioselective Mannich-Type Reaction of Glycine Schiff Bases with Cyclic Ketimines. Adv. Synth. Catal. 2018, 360, 4625–4633. 10.1002/adsc.201800850. [DOI] [Google Scholar]
- An Q.; Liu D.; Shen J.; Liu Y.; Zhang W. The Construction of Chiral Fused Azabicycles Using a Pd-Catalyzed Allylic Substitution Cascade and Asymmetric Desymmetrization Strategy. Org. Lett. 2017, 19, 238–241. 10.1021/acs.orglett.6b03529. [DOI] [PubMed] [Google Scholar]
- Huo X.; Yang G.; Liu D.; Liu Y.; Gridnev I. D.; Zhang W. Palladium-Catalyzed Allylic Alkylation of Simple Ketones with Allylic Alcohols and Its Mechanistic Study. Angew. Chem., Int. Ed. 2014, 53, 6776–6780. 10.1002/anie.201403410. [DOI] [PubMed] [Google Scholar]
- Huo X.; Quan M.; Yang G.; Zhao X.; Liu D.; Liu Y.; Zhang W. Hydrogen-Bond-Activated Palladium-Catalyzed Allylic Alkylation via Allylic Alkyl Ethers: Challenging Leaving Groups. Org. Lett. 2014, 16, 1570–1573. 10.1021/ol5000988. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Li J.; Ye J.; Liu D.; Zhang W. Synthesis of Chiral Chromanols: Via a RuPHOX-Ru Catalyzed Asymmetric Hydrogenation of Chromones. Chem. Commun. 2018, 54, 13571–13574. 10.1039/C8CC07787H. [DOI] [PubMed] [Google Scholar]
- Dai W.; Li J.; Li G.; Yang H.; Wang L.; Gao S. Asymmetric Epoxidation of Alkenes Catalyzed by a Porphyrin-Inspired Manganese Complex. Org. Lett. 2013, 15, 4138–4141. 10.1021/ol401812h. [DOI] [PubMed] [Google Scholar]
- Dai W.; Shang S.; Chen B.; Li G.; Wang L.; Ren L.; Gao S. Asymmetric Epoxidation of Olefins with Hydrogen Peroxide by an in Situ-Formed Manganese Complex. J. Org. Chem. 2014, 79, 6688–6694. 10.1021/jo501178k. [DOI] [PubMed] [Google Scholar]
- Dai W.; Li J.; Chen B.; Li G.; Lv Y.; Wang L.; Gao S. Asymmetric Oxidation Catalysis by a Porphyrin-Inspired Manganese Complex: Highly Enantioselective Sulfoxidation with a Wide Substrate Scope. Org. Lett. 2013, 15, 5658–5661. 10.1021/ol402612x. [DOI] [PubMed] [Google Scholar]
- Dai W.; Mi Y.; Lv Y.; Chen B.; Li G.; Chen G.; Gao S. Development of a Continuous-Flow Microreactor for Asymmetric Sulfoxidation Using a Biomimetic Manganese Catalyst. Adv. Synth. Catal. 2016, 358, 667–671. 10.1002/adsc.201501023. [DOI] [Google Scholar]
- Ficks A.; Sibbald C.; John M.; Dechert S.; Meyer F. Dinuclear Allylpalladium Complexes of C2-Symmetric Pyrazolate-Bridged Bis(Oxazoline) Ligands (Pyrbox’s): Structures, Dynamic Behavior, and Application in Asymmetric Allylic Alkylation. Organometallics 2010, 29, 1117–1126. 10.1021/om900835f. [DOI] [Google Scholar]
- Jalba A.; Régnier N.; Ollevier T. Enantioselective Aromatic Sulfide Oxidation and Tandem Kinetic Resolution Using Aqueous H 2 O 2 and Chiral Iron-Bis(Oxazolinyl)Bipyridine Catalysts. Eur. J. Org. Chem. 2017, 2017, 1628–1637. 10.1002/ejoc.201601597. [DOI] [Google Scholar]
- Zhou Y. Y.; Sun X. L.; Zhu B. H.; Zheng J. C.; Zhou J. L.; Tang Y. Modification of Pseudo-C3-Symmetric Trisoxazoline and Its Application to the Friedel-Crafts Alkylation of Indoles and Pyrrole with Alkylidene Malonates. Synlett 2011, 2011, 935–938. 10.1055/s-0030-1259721. [DOI] [Google Scholar]
- Wang P.; Tao W. J.; Sun X. L.; Liao S.; Tang Y. A Highly Efficient and Enantioselective Intramolecular Cannizzaro Reaction under TOX/Cu(II) Catalysis. J. Am. Chem. Soc. 2013, 135, 16849–16852. 10.1021/ja409859x. [DOI] [PubMed] [Google Scholar]
- Zhou Y. Y.; Li J.; Ling L.; Liao S. H.; Sun X. L.; Li Y. X.; Wang L. J.; Tang Y. Highly Enantioselective [3 + 3] Cycloaddition of Aromatic Azomethine Imines with Cyclopropanes Directed by π-π Stacking Interactions. Angew. Chem., Int. Ed. 2013, 52, 1452–1456. 10.1002/anie.201207576. [DOI] [PubMed] [Google Scholar]
- Ren H.; Song X. Y.; Wang S. R.; Wang L.; Tang Y. Highly Enantioselective Nickel-Catalyzed Oxa-[3 + 3]-Annulation of Phenols with Benzylidene Pyruvates for Chiral Chromans. Org. Lett. 2018, 20, 3858–3861. 10.1021/acs.orglett.8b01442. [DOI] [PubMed] [Google Scholar]
- Cao P.; Deng C.; Zhou Y. Y.; Sun X. L.; Zheng J. C.; Xie Z.; Tang Y. Asymmetric Nazarov Reaction Catalyzed by Chiral Tris(Oxazoline)/ Copper(II). Angew. Chem., Int. Ed. 2010, 49, 4463–4466. 10.1002/anie.200907266. [DOI] [PubMed] [Google Scholar]
- Zhou Y. Y.; Wang L. J.; Li J.; Sun X. L.; Tang Y. Side-Arm-Promoted Highly Enantioselective Ring-Opening Reactions and Kinetic Resolution of Donor-Acceptor Cyclopropanes with Amines. J. Am. Chem. Soc. 2012, 134, 9066–9069. 10.1021/ja302691r. [DOI] [PubMed] [Google Scholar]
- Kang Q. K.; Wang L.; Liu Q. J.; Li J. F.; Tang Y. Asymmetric H2O-Nucleophilic Ring Opening of D-A Cyclopropanes: Catalyst Serves as a Source of Water. J. Am. Chem. Soc. 2015, 137, 14594–14597. 10.1021/jacs.5b10310. [DOI] [PubMed] [Google Scholar]
- Xu H.; Hu J. L.; Wang L.; Liao S.; Tang Y. Asymmetric Annulation of Donor-Acceptor Cyclopropanes with Dienes. J. Am. Chem. Soc. 2015, 137, 8006–8009. 10.1021/jacs.5b04429. [DOI] [PubMed] [Google Scholar]
- Liao S.; Sun X. L.; Tang Y. Side Arm Strategy for Catalyst Design: Modifying Bisoxazolines for Remote Control of Enantioselection and Related. Acc. Chem. Res. 2014, 47, 2260–2272. 10.1021/ar800104y. [DOI] [PubMed] [Google Scholar]
- Li W. J.; Qiu S. X. A Novel D2-Symmetrical Chiral Tetraoxazoline Ligand for Highly Enantioselective Hydrosilylation of Aromatic Ketones. Adv. Synth. Catal. 2010, 352, 1119–1122. 10.1002/adsc.201000018. [DOI] [Google Scholar]
- Torres M.; Maisse-François A.; Bellemin-Laponnaz S. Highly Recyclable Self-Supported Chiral Catalysts for the Enantioselective α-Hydrazination of β-Ketoesters. ChemCatChem 2013, 5, 3078–3085. 10.1002/cctc.201300395. [DOI] [Google Scholar]