

Abstract
Purpose:
Metabotropic glutamate receptor 2 (mGluR2) has been implicated in various psychiatric and neurological disorders, such as schizophrenia and Alzheimer’s disease. We have previously developed [11C]7 as a PET radioligand for imaging mGluR2. Herein, [18F]JNJ-46356479 ([18F]8) was synthesized and characterized as the first 18F-labeled mGluR2 imaging ligand to enhance diagnostic approaches for mGluR2-related disorders.
Procedures:
JNJ-46356479 (8) was radiolabeled via the copper (I)-mediated radiofluorination of organoborane 9. In vivo PET imaging experiments with [18F]8 were conducted first in C57BL/6J mice and Sprague-Dawley rats to obtain whole-body biodistribution and brain uptake profile. Subsequent PET studies were done in a cynomolgus monkey (Macaca fascicularis) to investigate the uptake of [18F]8 in the brain, its metabolic stability as well as pharmacokinetic properties.
Results:
JNJ-46356479 (8) exhibited excellent selectivity against other mGluRs. In vivo PET imaging studies showed reversible and specific binding characteristic of [18F]8 in rodents. In the non-human primate, [18F]8 displayed good in vivo metabolic stability, excellent brain permeability, fast and reversible kinetics with moderate heterogeneity across brain regions. Pre-treatment studies with compound 7 revealed time dependent decrease of [18F]8 accumulation in mGluR2 rich regions based on SUV values with the highest decrease in the nucleus accumbens (18.7 ± 5.9%) followed by the cerebellum (18.0 ± 7.9%), the parietal cortex (16.9 ± 7.8%) and the hippocampus (16.8 ± 6.9%), similar to results obtained in the rat studies. However, the volume of distribution (VT) results derived from 2T4k model showed enhanced VT from a blocking study with compound 7. This is probably because of the potentiating effect of compound 7 as an mGluR2 PAM as well as related non-specific binding in the tissue data.
Conclusions:
[18F]8 readily crosses the blood-brain barrier and demonstrates fast and reversible kinetics both in rodents and in a non-human primate. Further investigation of [18F]8 on its binding specificity would warrant translational study in human.
Keywords: mGluR2, PET, Rodents, Nonhuman Primate, Kinetic Modeling, Neurological Diseases
Introduction
Metabotropic glutamate receptor 2 (mGluR2) along with mGluR3 are group II metabotropic glutamate receptors that are heterogeneously distributed in the brain and modulate synaptic transmission and neuroplasticity [1, 2]. Abnormal mGluR2 expression and function have been implicated in various psychiatric and neurological disorders, such as schizophrenia [3, 4], depression [5], anxiety [6, 7], pain [8] and Alzheimer’s disease [9, 10]. Determination of mGluR2 expression in normal and disease conditions could provide opportunities to diagnose disease at early stage and monitor disease progression. As a noninvasive imaging technique, positron emission tomography (PET) is advantageous in mapping and quantifying pathological and biochemical changes in vivo at the molecular level.
To date, several mGluR2 PET imaging ligands have been reported, two of which have been evaluated in human clinical trials, namely, [11C]JNJ-42491293 (1) [11, 12] and an undisclosed radioligand from Merck (Fig. 1) [13, 14]. Although these radioligands exhibited an excellent binding profile in vitro, when advanced to in vivo PET imaging studies, they suffered poor brain uptake and low mGluR2 specificity. For example, it was demonstrated that the brain retention observed with [11C]JNJ-42491293 (1) was mostly due to off-target binding which was initially noticed due to an unexpected high accumulation in the myocardium [11]. The mGluR2 negative allosteric modulators (NAMs), [11C]QCA (2) [15], [11C]MMP (3) [16] and [11C]MG2–1904 (4) [17] showed also off-target binding, though [11C]MG2–1904 had enhanced brain uptake and specificity. Other PET tracers also have been disclosed in conference abstracts and patents, albeit, with limited information on experimental results [18, 19]. Previously we have developed mGluR2/3 antagonists as PET tracers, [11C]MMMHC (5) [20] in 2003 and [11C]CMGDE (6) [21] in 2012. Our recent efforts on mGluR2 positive allosteric modulators (PAMs) has led to the mGluR2 PAM ligand [11C]7 [22], which showed promising pharmacological characteristics and reversible and specific binding toward mGluR2 in rodent studies. Despite satisfactory imaging results obtained with [11C]7, the short half-life of carbon-11 (20.4 min) limits its broader use with distribution of radioactivity and longitudinal investigations.
Fig. 1.
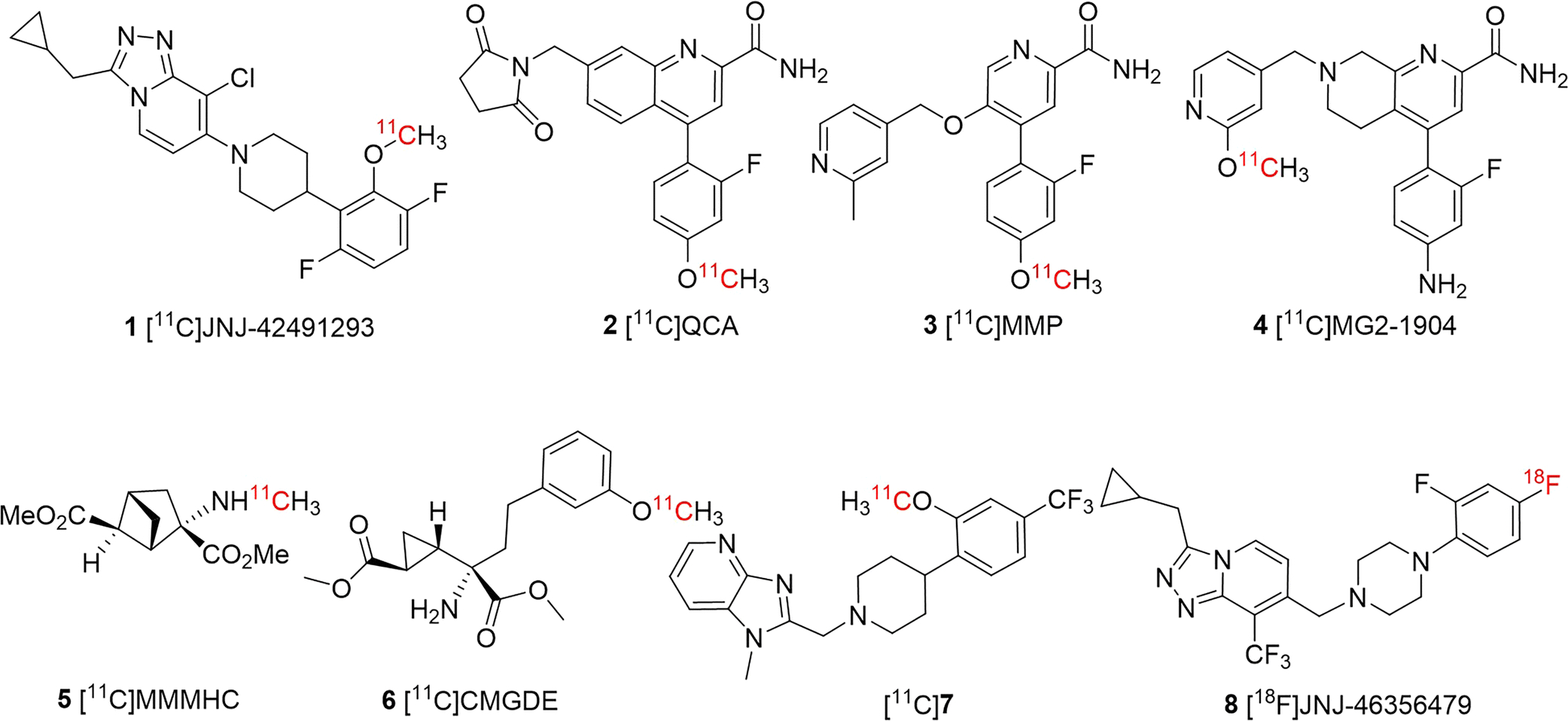
Developed PET imaging ligands for mGluR2.
Recently, Cid et al. reported the discovery of JNJ-46356479 (8) as a potent mGluR2 PAM (EC50 = 78 nM, Emax (%) = 256), with favorable physiochemical and pharmacological properties [23]. Compound JNJ-46356479 (8) also was used by Leurquin-Sterk et al. as a selective blocking reagent to investigate the off-target binding of [11C]JNJ-42491293 (3) in rats and non-human primates [11]. Furthermore in our recent work pre-administration of 8 reduced the uptake of [11C]7 by 28–37% in the rat brain [22]. The presence of an aryl-fluoride in JNJ-46356479 (8) allows incorporation of fluorine-18 isotope without modifying its chemical structure and generation of a mGluR2 PET tracer with a long half-life (109.8 min). In this work, we report the radiosynthesis of [18F]8 as well as preliminary in vivo characterization in rodent and primate studies.
Materials and Methods
Radiochemistry
The fully automated radiosynthesis of [18F]8 in GE TRACERLab™ FXFN was recently reported by our group [24]. Briefly, [18F]fluoride was produced by a GE PETtrace 16.5 MeV cyclotron (GE Healthcare, Waukesha, WI, USA) using the 18O(p, n)18F nuclear reaction with 18O-enriched water (Isoflex Isotope, San Francisco, CA). The [18F]fluoride solution was trapped onto a QMA Sep-Pak Cartridge (Sep-Pak plus light, Waters, Milford, MA) and then released by a solution of tetraethylammonium bicarbonate (TEAB, 2.7 mg, 14.1 μmol) in acetonitrile and water (1 mL, v/v 7:3) into the reactor. The [18F]fluoride solution was azeotropically dried at 80 °C for 10 min and then 100 °C for 3 min with addition of another portion of anhydrous acetonitrile (1 mL). The residue was taken up by n-BuOH (0.4 mL) followed by a solution of 9 (3.5 mg, 6.2 μmol) and [Cu(OTf)2Py4] (9.0 mg, 13.3 μmol) in anhydrous dimethylacetamide (DMA, 0.8 mL). The reaction was heated at 130 °C for 10 min and quenched with water (4.0 mL) at 50 °C. The desired [18F]8 was isolated from a semi-preparative HPLC system (Waters 4000 system equipped with an Xbridge BEH C18 OBD column: 130 Å, 5 μm, 10×250 mm) with a mobile phase of acetonitrile/0.1 M ammonium formate aqueous (55/45, v/v) at a flow rate of 6 mL/min. The fraction containing [18F]8 was diluted with 25 mL high purity water and then loaded on a C18 column (light Sep-Pak, Waters, Milford, MA). Finally, [18F]8 was eluted from the column with 0.6 mL ethanol and formulated into 10% ethanolic saline solution for injection. The molar activity (Am), radiochemical identity and purity of injected [18F]8 were determined by an analytical HPLC system using an analytical column (Waters, XBridge, C18, 3.5 μ, 4.6 × 150 mm) with a mobile phase of acetonitrile/0.1M aqueous ammonium formate solution (60/40, v/v) at a flow rate of 1 mL/min and UV absorption at λ = 254 nm.
PET Imaging Studies in Rodents
Whole Body Biodistribution Study in Mice
Quantitative biodistribution studies of [18F]8 were performed in six healthy C57BL/6J mice (male, 25–30 g). After anesthetization (2% isoflurane with oxygen flow of 1.5 L/min) the mice were administrated with the [18F]8 (5.6–10 MBq (0.15–0.27 mCi)) using tail vein injection and scanned with Triumph II Preclinical Imaging System (Trifoil Imaging, LLC, Northridge, CA) for 90 min after administration of the radioactivity. The regions of interest at different tissues were analyzed with ASIPro, including brain, lung, heart, liver, kidney and bladder. The biodistribution of [18F]8 at different tissue regions were determined and expressed as the percent of the injected radioactivity (% ID/g).
In vivo Characterization in Rats
Altogether five normal Sprague Dawley rats (male, 275–400 g) were used in eleven studies to investigate the in vivo imaging characteristics of [18F]8. Baseline studies of [18F]8 distribution were conducted in 4 rats, which were also used for the blocking experiments with compound 8. Two rats in this group as well as an additional rat from another group were employed during the blocking studies with compound 7. For the imaging studies, rats were anesthetized with isoflurane/nitrous oxide (1.0–1.5% isoflurane, with oxygen flow of 1–1.5 L/min) and the tail vein was catheterized for administration of the imaging ligand [18F]8. The rats were positioned in the scanner for imaging (Triumph II Preclinical Imaging System, Trifoil Imaging, LLC, Northridge, CA). The vital signs such as heart rate and/or breathing were monitored throughout the imaging. Data acquisition of 60 min was started from the injection of radioligand [18F]8 (20–41 MBq (0.54–1.11 mCi), i.v.).
In blocking experiments, 7 or 8 was dissolved in a saline solution with 10% ethanol and 10% Tween 20 and administered (i.v., 4 mg/kg) 10 min before radiotracer injection. CT scan was performed after PET imaging study for anatomical information and attenuation correction. The PET imaging data were corrected for uniformity, scatter, and attenuation and reconstructed using a maximum-likelihood expectation-maximization (MLEM) algorithm with 30 iterations to dynamic volumetric images with time bins of 9×20s, 7×60s, 6×300s, 2×600s. CT data were reconstructed using a modified Feldkamp algorithm using matrix volumes of 512×512×512 and pixel size of 170 μm. The ROIs, i.e., cortex, striatum, thalamus, hypothalamus, hippocampus, cerebellum, and whole brain were drawn onto coronal PET slices according to the brain outlines as derived from the rat brain atlas and corresponding TACs (time-activity curves) were created by PMOD 3.2 (PMOD Technologies Ltd., Zurich, Switzerland). Percent changes between the control and blocking studies were calculated in the selected brain areas at the 1–10 min time window after injection of [18F]8.
In vivo Studies in a Cynomolgus Monkey
Preparation for PET Imaging
Imaging studies were performed on a 5-kg female cynomolgus monkey. Prior to each study, the monkey was sedated with ketamine/xylazine (10/0.5 mg/kg IM) and intubated for maintenance anesthesia with isoflurane (1–2% in 100% O2). For PET studies, venous and arterial catheters were placed for infusion of the radiotracer and blood sampling for the arterial input function, respectively. The animal was then positioned on a heating pad on the bed of the scanner for the duration of the study.
Magnetic Resonance Imaging in Monkey
Before PET studies the animal was scanned on a 3T Biograph mMR (Siemens Medical Systems) to acquire MRI for anatomical reference. Data was acquired using a 3D structural T1-weighted multi-echo magnetization-prepared rapid gradient-echo (MEMPRAGE) sequence with repetition time (TR) = 2,530 ms, echo time (TE) = 5.82 ms, inversion time (TI) = 1,100 ms; flip angle = 7°, voxel size = 0.7×0.7×0.7 mm3, matrix size = 256×256×160, and number of averages = 1.
PET Imaging in Monkey
PET studies were done on a Discovery MI (GE Healthcare) PET/CT scanner. The animal had two baseline scans which were separated by eleven months as well as two “blocking” studies with pre-treatment of unlabeled compound 7 (1 mg/kg) 10 min prior radiotracer injection. A CT scan was acquired for attenuation correction of the PET images. Emission PET data were collected for 120 min starting from the injection of [18F]8 into the lateral saphenous vein followed by a saline flush. All injections were performed as 3-minute infusion using Medfusion 3500 syringe pumps. Radiotracer activity at the time of injection was 190.1 ± 3.4 MBq (range 188.0 – 194.4 MBq) with an averaged Am of 180 GBq/μmol. Corresponding injected mass was 476.7 ± 8.5 ng (range 471.5 – 487.5 ng). During the studies, arterial blood samples were collected as described in the next section. Dynamic images were reconstructed with a 3D ordered subset expectation maximization (OSEM) reconstruction algorithm using 3 iterations and 34 subsets. Corrections were applied for scatter, attenuation, dead-time, random coincident events, detector normalization with modeling of point spread function (PSF). List mode data were framed into time bins of 6×10 sec, 8×15 sec, 6×30 sec, 8×60 sec, 8×120 sec and 18×300 sec. Reconstructed images had matrix size of 256×256×89 with voxel sizes of 1.17×1.17×2.8 mm3.
Blood Measurements and Analyses in Monkey
First, a 3-mL blood sample was drawn immediately before radiotracer injection to determine plasma protein binding of [18F]8 by ultracentrifugation. Briefly, the sample was centrifuged to obtain 800 microliters of plasma which was spiked with 80 microliters of [18F]8 diluted in PBS to 22.2 MBq/mL. The solution was incubated for 10–15 min and 200 microliter aliquots were loaded onto Centrifree Ultrafiltration Devices (Millipore Sigma). The samples were centrifuged for 15 min at 1500 g. 20 microliters of the ultrafiltrate (Cfree) and 20 microliters of the radioactive plasma mixture (Ctotal) were measured for radioactivity concentration. The entire process was replicated using PBS instead of plasma to determine the nonspecific retention of radiotracer of the ultrafiltration device. The entire procedure was performed in triplicate and plasma free fraction fp (bioavailable fraction unbound to protein) was calculated as the ratio of Cfree to Ctotal, corrected for the nonspecific retention of the PBS standard. Then, upon radiotracer injection, arterial blood samples of 1 to 2 mL were drawn every 30 seconds during the first 5 minutes and decreased in frequency to every 15 minutes toward the end of the PET acquisition. Radioactivity concentration in whole blood was measured using a well counter and calibrated scale with weight corrected for the density of blood. Whole blood was then centrifuged, and the supernatant was collected for determination of radioactivity concentration in plasma using similar methods. Radiotracer metabolism was measured from selected plasma samples drawn at 3, 5, 10, 15, 30, 60, 90, 120 min. These samples were assayed with column-switching radio-HPLC (High Performance Liquid Chromatography) system [25, 26] to determine the fraction of total radioactivity that was attributable to intact radiotracer. Briefly, a mobile phase of 99:1 water: acetonitrile at 1.8 mL/min (Waters 515 pump) was used to trap the plasma sample on a capture column (Waters Oasis HLB 30 μm) and after 4 minutes the catch column was backflushed with 60:40 MeCN:0.1M ammonium formate in water at 1 mL/min (second Waters 515 pump) and directed onto a Waters XBridge BEH C18 (130 Å, 3.5 μm, 4.6 mm x 100 mm) analytical column. Eluent was collected in 1-minute intervals and assayed for radioactivity with a Wallac Wizard 2480 gamma counter to measure the percent parent in plasma (%PP). Measured %PP time courses were then fitted with a sum of two decaying exponentials plus a constant. The resulting model fit and the time course of total plasma radioactivity concentration were multiplied to derive the metabolite corrected arterial input function, which was used for tracer kinetic modeling as detailed below.
Image Processing and Analyses for Monkey
Data processing was performed in MATLAB and FSL [27] was used for registration purposes. First, PET images were rigidly co-registered to the MEMPRAGE images. The MEMPRAGE was then aligned into an MR rhesus template space [28] using a 12-parameter affine transformation followed by non-linear warping, and the resulting transformations were applied to the PET images. Monkey atlases derived from Paxinos et al. [29] and from McLaren et al. [30] were warped into the aforementioned monkey template space and were subsequently transformed into the original PET space for extraction of regional TACs. Regional TACs were analyzed by compartmental modeling using the metabolite-corrected arterial plasma input function. One- (1T) and two- (2T) tissue compartment model configurations were investigated with a fixed vascular contribution of the whole blood radioactivity to the PET signal set to 5%. Estimates of kinetic parameters were obtained by nonlinear weighted least-squares fitting with the weights chosen as the frame durations. Regional total volume of distributions (VT) were derived from the estimated microparameters and were calculated following the consensus nomenclature described in Innis et al. [31]. The Logan graphical method for estimation of VT was also investigated.
Results
Pharmacology & Radiochemistry
Compound 8 was re-examined for its mGluR2 PAM activity. Our results show that compound 8 has an EC50 value of 166 nM as shown in the Suppl. Fig. S1 (see ESM), which is consistent but higher than the previously reported value by Cid et al. using different functional assay [23]. Compound 8 also shows weak mGluR2 agonism (EC50 = 3.55 μM) and excellent selectivity against other mGlu receptors as shown in the Suppl. Table S1 (> 100-fold, see ESM).
[18F]8 was synthesized in a one-step manner from its boronic pinacol ester 9 via our recently described method using a modified alcohol-enhanced Cu-mediated 18F-fluorination (Scheme 1) [24, 32]. Briefly, the tetraethyl ammonium bicarbonate (TEAB) was used as a base and phase transfer agent to form SNAr nucleophile of [18F]TEAF. The cosolvent n-BuOH was added to tolerate the sensitive [1,2,4]triazolo[4,3-a]pyridine heterocycle in 9. [Cu(OTf)2py4] was used as a catalyst. The fully automated synthesis of [18F]8 was achieved in the commercially available TRACERLab™ FXF-N platform. The final [18F]8 was obtained with isolated RCY of 5 ± 3% (non-decay-corrected), and molar activity of 180 ± 102 GBq/μmol at end of synthesis (EOS, 45 min, n > 10). [18F]8 was formulated into 6.0 mL 10% ethanolic saline solution before injection with excellent chemical and radiochemical purities (> 95%).
Scheme 1.
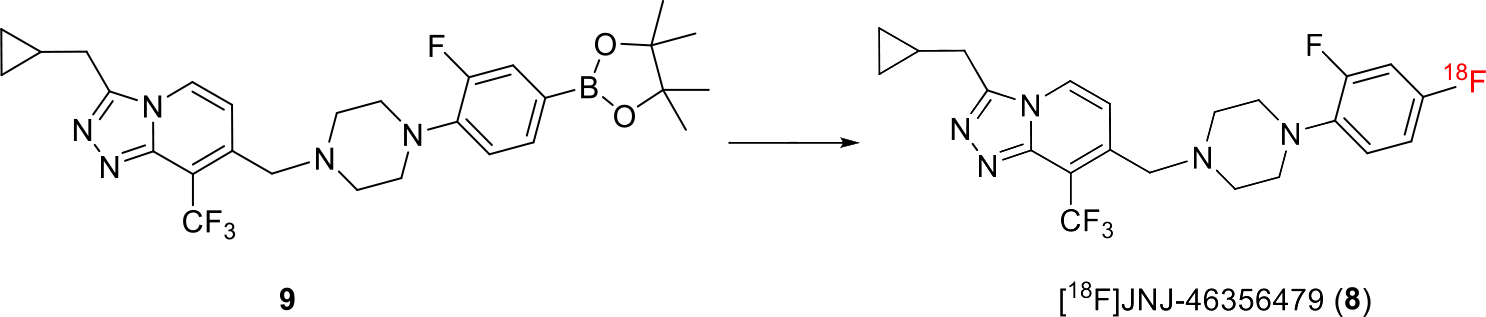
Synthesis of [18F]8. Reaction condition: 18F-, 9 (6.2 μmol), TEAB (14.1 μmol), [Cu(OTf)2py4] (13.3 μmol), DMA (0.8 mL), n-Butanol (0.4 mL), T = 130 °C, 10 min
PET Imaging Results in Rodents
Whole body PET imaging done in six C57BL/6J mice showed the highest accumulation of [18F]8 in the liver (21.3 ± 3.12% ID/g) followed by kidney (14.59 ± 1.66% ID/g), bladder (13.77 ± 6.96% ID/g), heart (7.84 ± 1.21% ID/g) and lungs (5.0 ± 0.97% ID/g). The pharmacokinetic studies support reversible accumulation of [18F]8 with the highest radioactivity 10 min after radioactivity injection, but in the bladder the radioactivity steadily increased up to 60 min and in the brain and lungs the highest activity was obtained at 5 min (Fig. 2). The high radioactivity uptake in liver and kidney suggests combined hepatobiliary elimination and renal excretion for [18F]8 clearance. The average accumulation of [18F]8 in the mouse brain at 5 min was 4.02 ± 0.73% ID/g (Fig. 2). This result indicates a rapid BBB penetration of [18F]8, which was consistent with the following in vivo brain imaging studies.
Fig. 2.
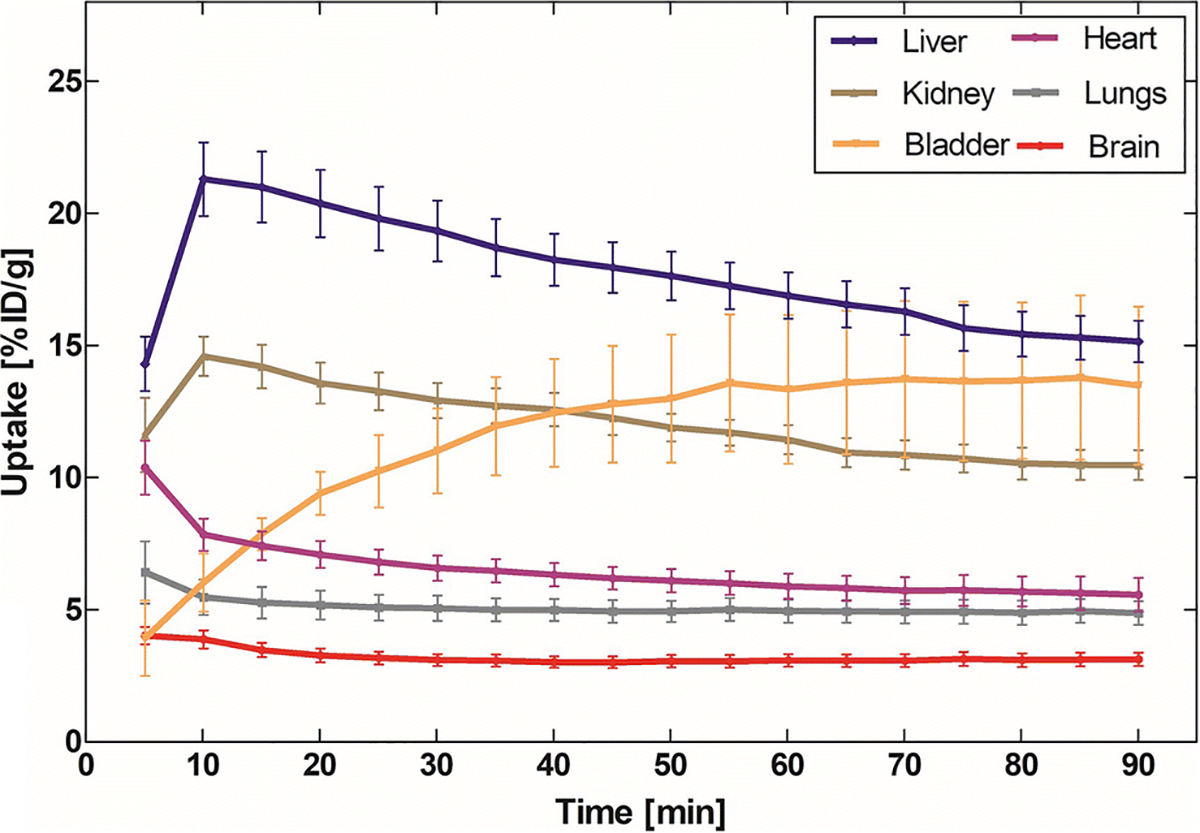
Whole body biodistribution of [18F]8 in mice.
Investigation of in vivo brain uptake for [18F]8 was first conducted using Sprague-Dawley rats. Dynamic PET data were acquired for 60 min after tail vein injection of [18F]8. Fig. 3a shows representative PET images of cumulative volumetric distribution of [18F]8 at time interval of 5–15 min on five coronal, axial and sagittal levels. [18F]8 accumulates in the mGluR2-rich regions in rat brain. Time-activity curves showed fast accumulation and progressive declining of radioactivity in all brain areas (Figs. 3b & 3c). The highest accumulation of [18F]8 was in the thalamus, followed by cerebellum, hypothalamus, hippocampus, striatum and cortex. Blocking studies were conducted to investigate selectivity of [18F]8 for mGluR2. Pretreatment with the structurally distinct in vivo active mGluR2 PAM ligand 7 (4 mg/kg i.v.) 10 min before [18F]8 injection resulted 24.1%−31.5% decrease of [18F]8 uptake in different brain areas at the 1–10 min time window (Fig. 3d and Table S2, see ESM). On the other hand, administration of unlabeled compound 8, using a dose of 4 mg/kg iv. 10 min before [18F]8 injection, resulted in a smaller decline (11.6–18.8%) of radioactivity uptake in the different brain areas at the same time window. The cerebellum had the highest blocking effect with both compounds 7 and 8, namely, 31.5% vs 18.8%, respectively. In conclusion, the in vivo rodent studies suggest that [18F]8 has specific binding to mGluR2 in rat brain.
Fig. 3.
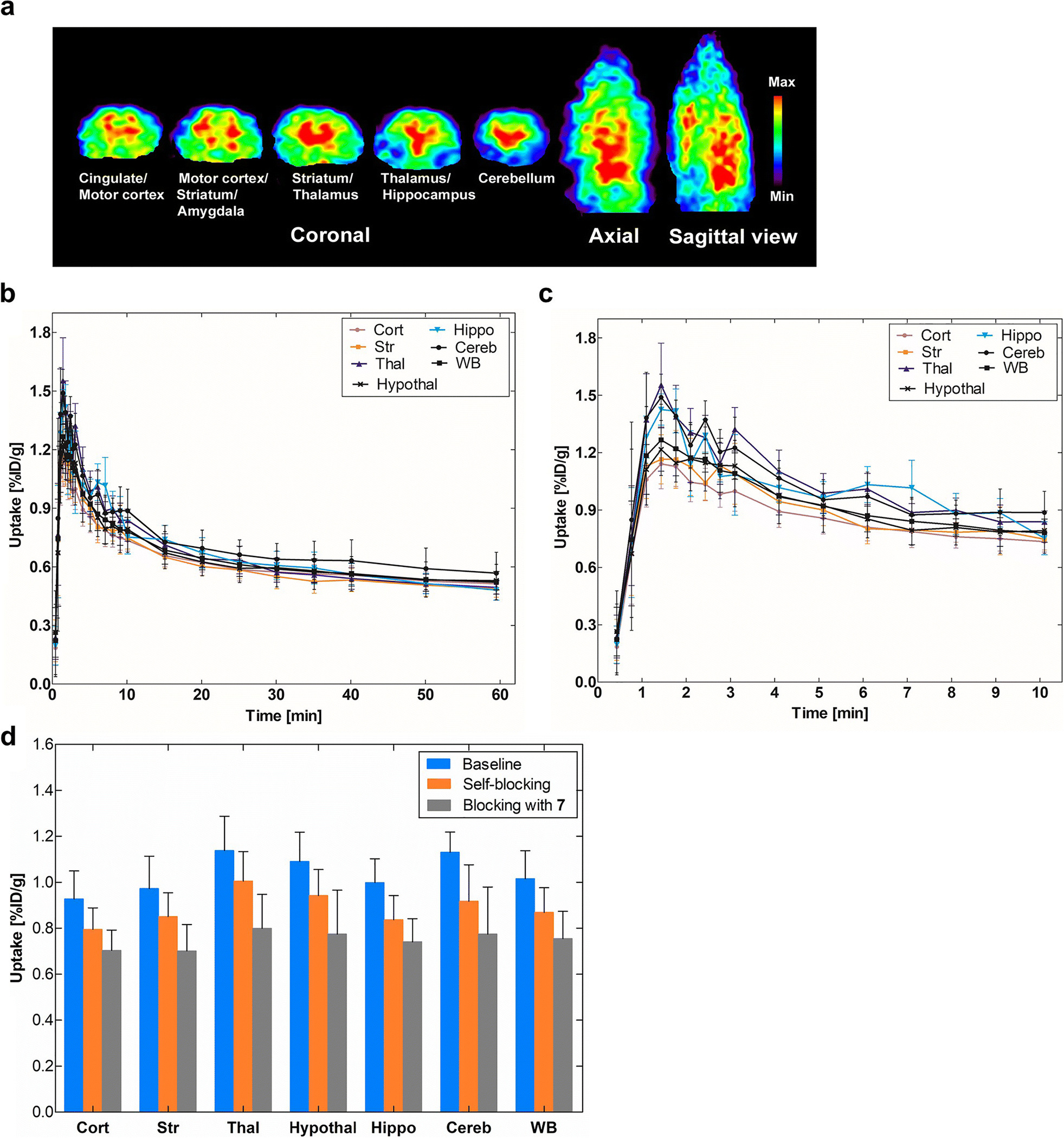
PET imaging results of [18F]8 in rat brain. a) Accumulation of [18F]8 in different brain areas at the time interval 5–15 min. Slice thickness is 1.25 mm; b) Time-activity distribution of [18F]8 for the whole 60-min dynamic scan; c) For the first 10-min dynamic scan (n = 4); d) Blocking studies of [18F]8 uptake. The blocking was investigated by administering unlabeled compounds 8 or 7 (4 mg/kg i.v.) 10 min before radioligand [18F]8. The blocking effect was calculated at the time window of 1–10 min after the administration of the radioligand. Cort = cortex, Str = striatum, Thal = thalamus, Hypothal = hypothalamus, Hippocamp = hippocampus, Cereb = cerebellum and WB = whole brain.
Results of PET Experiments in Cynomolgus Monkey
Fig. 4a. shows whole-blood radioactivity SUV time course for all studies. In one study (Pre-treatment 2) we were not able to draw arterial blood samples during the first 2.5 minutes after injection of [18F]8, possibly due to pharmacological changes in arterial blood pressure upon pre-treatment with the mGluR2 PAM ligand 7. Whole blood/plasma ratio was consistent across the studies and reached a plateau after 5 min post tracer injection with a mean value of 0.86 ± 0.02. RadioHPLC measurements indicated the presence of primarily polar and moderately polar metabolites as shown by the radiochromatograms (Fig. 4b). Percent parent in plasma measurements revealed moderate rate of metabolism with 44.4 ± 4.2% of plasma activity attributable to unmetabolized [18F]8 at 30 min and 33.7 ± 1.9% at 90 min (Fig. 4c). Fig. 4d shows the corresponding individual metabolite-corrected [18F]8 SUV time courses in plasma. Plasma free fraction was 0.15 ± 0.04 across studies (range 0.12–0.21).
Fig. 4.
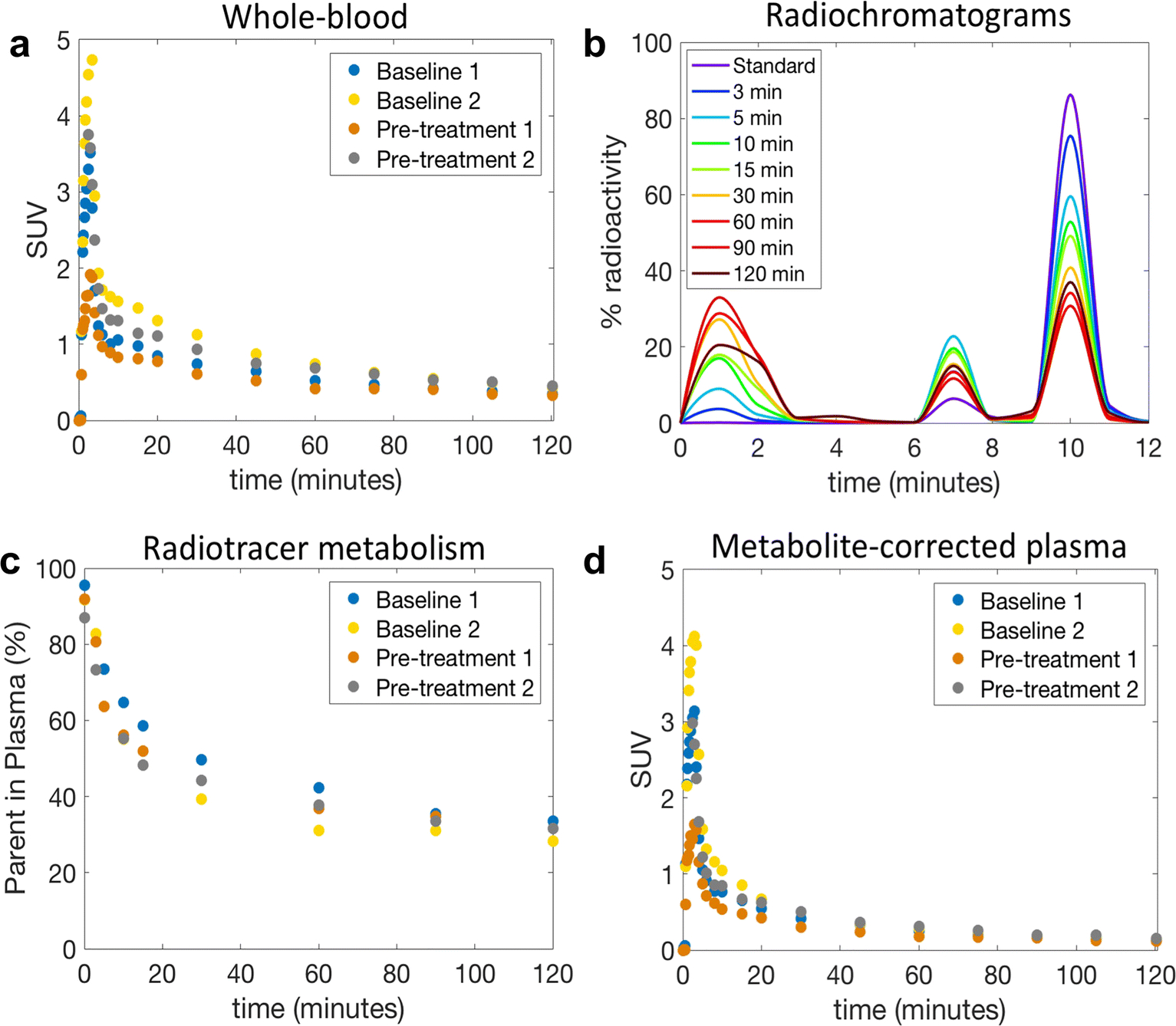
Whole-blood and plasma analysis of [18F]8 in the NHP. a) Individual whole-blood SUV time courses; b) RadioHPLC chromatogram of plasma samples from a representative study. The injection of an aliquot of [18F]8 (standard) confirmed that the parent began eluting from the analytical column at ~9 min; c) Individual time course of remaining parent compound in plasma; d) Individual metabolite-corrected [18F]8 SUV time courses in plasma.
Fig. 5a shows the relative distribution of [18F]8 in the monkey brain. Representative brain TACs along with compartmental model fits for the cortex, thalamus, cerebellum grey, hippocampus, striatum and white matter, at baseline condition (Fig. 5b) and after pre-treatment with 7 (Fig. 5c). [18F]8 peaked relatively quickly in the brain at ~5 min, followed by moderately fast washout. TACs were best described by a reversible two-tissue model with a fixed vascular contribution (2T4k) which provided stable regional total volume of distribution VT estimates. VT measurements showed relatively good reproducibility between the two baseline studies with average differences across brain regions of 12.8 ± 9.0%, calculated as %Baseline1-Baseline2 = 100*2*abs(Baseline1-Baseline2)/(Baseline1+Baseline2). VT values estimated with the Logan graphical method (t* = 40 min) were highly correlated with those obtained from the 2T4k model (VT Logan = 0.97 × VT 2T4k - 0.19; R2 = 0.93) despite a small underestimation (mean difference = −6.0 ± 3.9%). K1 values, reflecting tracer delivery, was ~0.5 mL/min/cc in the whole brain, indicating high brain penetration. Bar plots are shown for regional SUVs calculated from 30 to 60 min and from 60 to 120 min post tracer injection (Fig. 5d). SUVs showed regionally decreased accumulation after pretreatment with 7. The highest decline during 30–60 min after the injection of radioactivity was in the nucleus accumbens (18.7 ± 5.9%) followed by the cerebellum (18.0 ± 7.9%), the parietal cortex (16.9 ± 7.8%) and the hippocampus (16.8 ± 6.9%). The relative distribution of [18F]8 in the monkey brain (Fig. 5a) as well as the rank order of the semi-quantitative SUVs measured across brain regions (Fig. 5d) closely corresponded to the known distribution of mGlu2 receptors expressed throughout the cerebral cortex as well as in striatum, thalamus, cerebellum, hippocampus and amygdala [11, 23]. However, the calculated estimates for VT values were increased after the pre-treatment with an averaged enhancement of 19.4% (Fig. 5e). A relatively high white matter uptake was observed most distinctively late in the scan. The outcome measures, VT or late SUVs, showed opposite response of PET signal after pre-treatment with 1 mg/kg of compound 7; SUV was decreased by 8.6% while VT was enhanced by 19.4%.
Fig. 5.
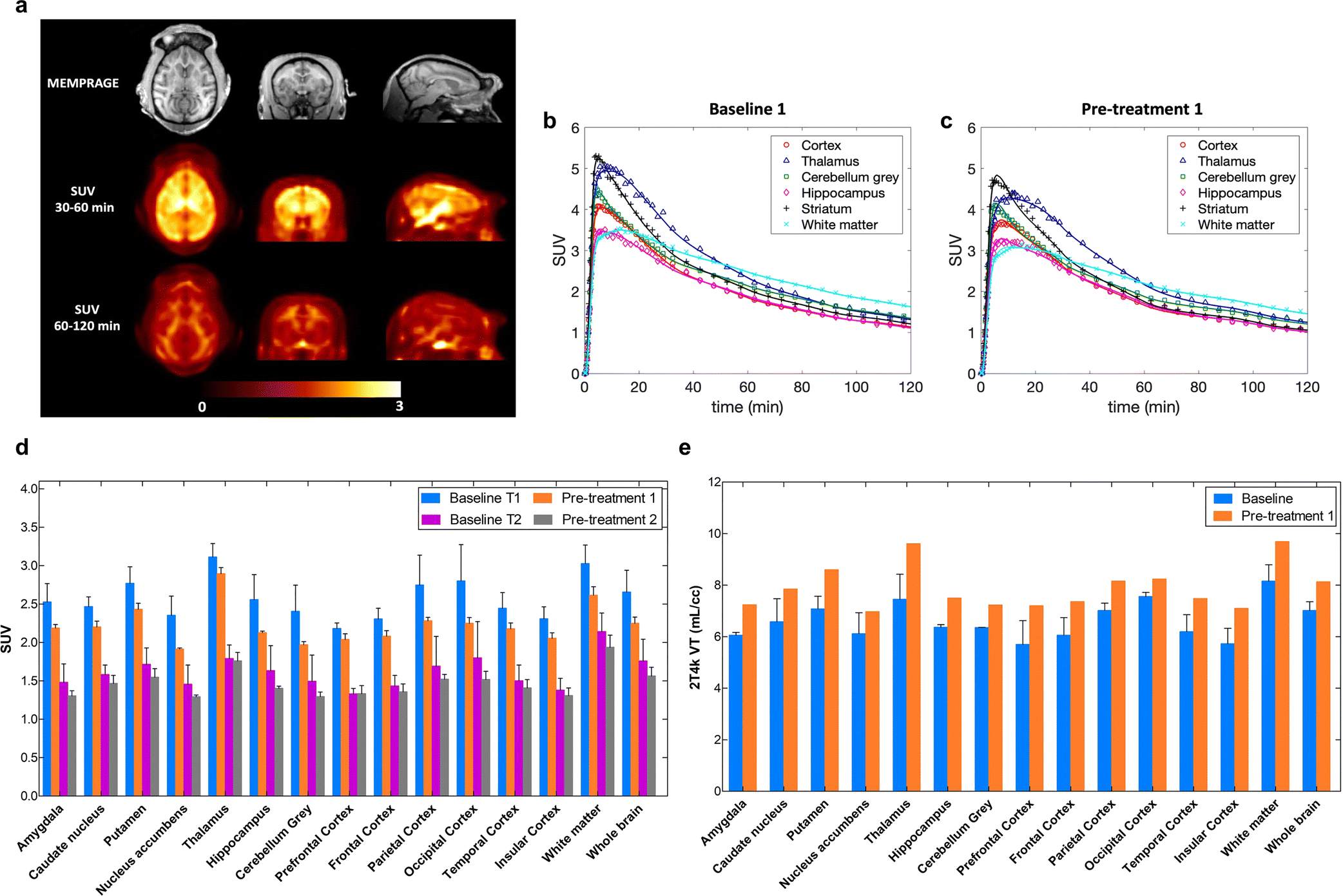
In vivo experiments in primates. a) Structural MRI (MEMPRAGE), and [18F]8 SUV images of the monkey brain. Images are presented in the NIMH Macaque Template (NMT) space [28]. Late SUV images demonstrated relatively high uptake in the white matter; b) Baseline study of [18F]8 kinetics and 2T4k model fits in different brain regions of NHP; c) TACs after pre-treatment with compound 7 (1 mg/kg, i.v.); d) SUVs averaged from 2 baseline and 2 blocking studies show moderate blocking effect in different brain areas. Unlabeled compound 7 (1 mg/kg) was administered 10 min before [18F]8. T1 refers to the time interval of 30–60 min and T2 represents time interval of 60–120 min; e) Increase of volume of distribution, VT in the different brain areas in the blocking studies of [18F]8 uptake in the monkey brain.
Discussion
We have previously developed [11C]MMMHC (5) and [11C]CMGDE (6) as orthosteric PET radioligands for imaging mGluR2/3 in the brain. Recently, we developed [11C]7 as a PAM PET radiotracer to avoid the off-target effects of mGluR3 in mGluR2/3 ligands [22]. In this study, we describe the synthesis, in vitro and in vivo characterization of the first fluorine-18 labeled mGluR2 PAM [18F]8 in the rodent and monkey studies.
The radiosynthesis of [18F]8 is achieved with a modified protocol based on the alcohol-enhanced Cu-mediated 18F-fluorination [24, 32]. In vivo whole body biodistribution experiments in mice revealed that [18F]8 was CNS penetrant and had a reversible pharmacokinetic profile suitable for brain PET radioligand. The observed relatively high uptake in the heart tissue is due to the proximity of the high accumulation in the liver. In rats, the in vivo PET imaging also confirmed the BBB permeability of [18F]8. The radioactivity accumulated in the mGluR2-rich regions with the maximum % ID/g value of 1.52 in the thalamus at 2 min post-injection. The blocking experiment with mGluR2 PAM 7 significantly reduced the radioactivity uptake in all investigated brain areas with the highest effect by 31.5% in the cerebellum, whereas lower blocking effect (18.8%) was observed in the cerebellum during self-blocking with 8. The blockade via a structurally distinct mGluR2 PAM, 7 produced significant decrease in the uptake of [18F]8 in all investigated brain areas confirming the specificity of [18F]8 as mGluR2 PET radiotracer.
In the cynomolgus monkey, [18F]8 displayed a moderate change in plasma and whole blood. The radiotracer readily crossed the BBB and accumulated in frontal cortex, thalamus, cerebellum, hippocampus, amygdala and white matter. Noteworthy, mGluR2 expression especially in axons and fiber bundles of white matter was previously reported in immunohistological studies by Ohishi et al. [33].
The 2T4k model derived TACs demonstrated fast and reversible kinetics of [18F]8 with peak uptake reached within 10 min post-injection in all brain regions. In the blocking study, pretreatment of the animal with compound 7 (1 mg/kg) reduced the binding of [18F]8 across all brain regions. The distribution volumes estimated with the two-tissue compartment model showed a good correlation with the Logan graphical analysis. However, the regional VT values in these areas showed reverse results, indicating increased uptake of [18F]8. This is probably because of the potentiating effect of the compound 7 as a mGluR2 PAM and/or non-specific binding. As reported by O’Brien et al. [34], mGluR2 PAMs, e.g., BINA, AZD8418 and JNJ-42491293 (1), exhibited both affinity and efficacy cooperativities with the glutamate in their assays. Moreover, Doornbos et al demonstrated the glutamate-dependent increase of the amount of mGluR2 binding sites (Bmax) with the mGluR2 PAM ligand [3H]JNJ-46281222 [35].
It should be also noticed that even the baseline studies in rats and primates support the similar accumulation of [18F]8 in the brain, the results from the blocking studies are slightly different. There are several aspects that might effect on it. In rodent studies, the blocking agents were administered as a single dose of 4 mg/kg i.v. 10 min before radioactivity while in the primate studies, to avoid any physiological effect on animals, a significantly lower amount (1mg/kg) of the unlabeled compound 7 was administered using i.v. infusion starting 10 min prior the radioactivity.
Conclusion
We have successfully developed the first 18F-labeled PET radioligand for imaging mGluR2 in the brain. [18F]8 was synthesized by a modified copper (I)-mediated radiofluorination method in commercially available GE TRACERLab™ FXF-N system. The in vivo PET imaging studies in rodents demonstrated the selective binding profile of [18F]8 in the brain. PET studies of [18F]8 in the cynomolgus monkey demonstrated moderate radiotracer stability in plasma and whole blood. The TACs of [18F]8 confirmed its selective binding toward mGluR2 in monkey brain. However, the VT results derived from 2T4k model showed enhanced radioactivity uptake from blocking studies with compound 7. Although the uptake of [18F]8 in white matter suggested its possible off-target binding, mGluR2 is known to express in axons and fiber bundles of the white matter. Altogether, [18F]8 is a promising PET imaging ligand for mGluR2.
Supplementary Material
Acknowledgement
mGluR1–6 and mGluR8 agonist and antagonist functional data as well as mGluR2 PAM activity were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth (email to: bryan_roth@med.unc.edu) at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll (email to: jdrisco1@mail.nih.gov) at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP web site https://pdspdb.unc.edu/pdspWeb/ (https://pdspdb.unc.edu/pdspWeb/).
Funding This project was financially supported by NIH grants [1R01EB021708 and 1R01NS100164] and the grants [1S10RR023452–01 and 1S10OD025234–01] for the imaging instrumentation and characterization of the organic compounds as well as the NIH grants [S10OD018035 and P41EB022544] to support the blood counting and metabolite analysis equipment used in the primate studies.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Compliance with Ethical Standards
Ethical approval All animal experiments were approved and done under the guidelines of the Subcommittee on Research Animals of the Massachusetts General Hospital and Harvard Medical School in accordance with the Guide of NIH for the Care and Use of Laboratory Animals.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Tanabe Y, Masu M, Ishii T, et al. (1992) A family of metabotropic glutamate receptors. Neuron 8:169–179. [DOI] [PubMed] [Google Scholar]
- 2.Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75:889–907. [DOI] [PubMed] [Google Scholar]
- 3.Chaki S (2010) Group II metabotropic glutamate receptor agonists as a potential drug for schizophrenia. Eur J Pharmacol 639:59–66. [DOI] [PubMed] [Google Scholar]
- 4.Downing AM, Kinon BJ, Millen BA, et al. (2014) A double-bind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry 14:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feyissa AM, Woolverton WL, Miguel-Hidalgo JJ, et al. (2010) Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry 34:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conn PJ, Jones CK (2009) Promise of mGluR2/3 activators in psychiatry. Neuropsychopharmacol 34:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muguruza C, Meana JJ, Callado LF (2016) Group II metabotropic glutamate receptors as targets for novel antipsychotic drugs. Front Pharmacol 7:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazzitelli M, Palazzo E, Maione S, et al. (2018) Group II Metabotropic Glutamate receptors: role in pain mechanisms and pain modulation. Front Mol Neurosci 11:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards G, Messer J, Faull RLM, et al. (2010) Altered distribution of mGlu2 receptors in β-amyloid-affected brain regions of Alzheimer cases and aged PS2APP mice. Brain Res 1363:180–190. [DOI] [PubMed] [Google Scholar]
- 10.Caraci F, Molinaro G, Battaglia G, et al. (2011) Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer’s disease: selective activation of mGlu2 receptors amplifies β-amyloid toxicity in cultured neurons, whereas dual activation of mGlu2 and mGlu3 receptors is neuroprotective. Mol Pharmacol 79:618–626. [DOI] [PubMed] [Google Scholar]
- 11.Leurquin-Sterk G, Celen S, Van Laere K, et al. (2017) What we observe in vivo is not always what we see in vitro: development and validation of 11C-JNJ-42491293, A novel radioligand for mGluR2. J Nucl Med 58:110–116. [DOI] [PubMed] [Google Scholar]
- 12.Andres JI, Alcazar J, Cid JM, et al. (2012) Synthesis, evaluation, and radiolabeling of new potent positive allosteric modulators of the metabotropic glutamate receptor 2 as potential tracers for positron emission tomography imaging. J Med Chem 55:8685–8699. [DOI] [PubMed] [Google Scholar]
- 13.Lohith T, McQuade P, Salinas C, et al. (2016) First-in-human PET imaging of mGluR2 receptors. J Nucl Med 57:213. [Google Scholar]
- 14.McQuade P, Joshi A, Miller P, et al. (2016) Discovery and preclinical evaluation of an mGluR2-NAM PET radioligand. J Nucl Med 57:290. [Google Scholar]
- 15.Zhang X, Kumata K, Yamasaki T, et al. (2017) Synthesis and preliminary studies of a novel negative allosteric modulator, 7-((2,5-dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro-4-[11C]methoxyphenyl)quinoline-2-carboxamide, for imaging of metabotropic glutamate receptor 2. ACS Chem Neurosci 8:1937–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumata K, Hatori A, Yamasaki T, et al. (2019) Synthesis and evaluation of 4-(2-fluoro-4-[11C]methoxyphenyl)-5-((2-methylpyridin-4-yl)methoxy)picolinamide for PET imaging of the metabotropic glutamate receptor 2 in the rat brain. Bioorg Med Chem 27:483–491. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Zhang Y, Chen Z, et al. (2020) Synthesis and preliminary studies of 11C-labeled tetrahydro-1,7-naphthyridine-2-carboxamides for PET imaging of metabotropic glutamate receptor 2. Theranostics 10:11178–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Krause S, Suzuki M, et al. (2016) Radiotracer compounds. WO2016033190 (A1) 1–83. [Google Scholar]
- 19.Van Gool MLM, Andres-Gil JI, Alcazar-Vaca MJ, et al. (2016) Radiolabelled mGluR2 PET ligands. WO2016087489 (A1) 1–80. [Google Scholar]
- 20.Yu M, Nagren K, Chen YI, et al. (2003) Radiolabeling and biodistribution of methyl 2-(methoxycarbonyl)-2-(methylamino) bicyclo [2.1.1]-hexane-5-carboxylate, a potent neuroprotective drug. Life Sci 73:1577–1585. [DOI] [PubMed] [Google Scholar]
- 21.Wang J-Q, Zhang Z, Kuruppu D, et al. (2012) Radiosynthesis of PET radiotracer as a prodrug for imaging group II metabotropic glutamate receptors in vivo. Bioorg Med Chem Lett 22:1958–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan G, Qu X, Zheng B, et al. (2020) Design, synthesis, and characterization of benzimidazole derivatives as positron emission tomography imaging ligands for metabotropic glutamate receptor 2. J Med Chem 63:12060–12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cid JM, Tresadern G, Vega JA, et al. (2016) Discovery of 8-trifluoromethyl-3-cyclopropylmethyl-7-[(4-(2,4-difluorophenyl)-1-piperazinyl)methyl]-1,2,4-triazolo[4,3-a]pyridine (JNJ-46356479), a selective and orally bioavailable mGlu2 receptor positive allosteric modulator (PAM). J Med Chem 59:8495–8507. [DOI] [PubMed] [Google Scholar]
- 24.Yuan G, Shoup TM, Moon SH, et al. (2020) A concise method for fully automated radiosyntheses of [18F]JNJ-46356479 and [18F]FITM via Cu-mediated 18F-fluorination of organoboranes. RSC Adv 10:25223–25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilton J, Yokoi F, Dannals RF, et al. (2000) Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol 27:627–630. [DOI] [PubMed] [Google Scholar]
- 26.Collier T, Normandin M, El Fakhri G, et al. (2013) Automation of column-switching HPLC for analysis of radiopharmaceuticals and their metabolites in plasma. J Nucl Med 54: 1133. [Google Scholar]
- 27.Jenkinson M, Beckmann CF, Behrens TE, et al. (2012). Fsl. Neuroimage 62:782–790. [DOI] [PubMed] [Google Scholar]
- 28.Seidlitz J, Sponheim C, Glen D, et al. (2018) A population MRI brain template and analysis tools for the macaque. Neuroimage 170:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paxinos G, Huang XF, Toga AW (1999) The rhesus monkey brain in stereotaxic coordinates. San Diego, CA: Academic Press. [Google Scholar]
- 30.McLaren DG, Kosmatka KJ, Oakes TR, et al. (2009) A population-average MRI-based atlas collection of the rhesus macaque. Neuroimage 45:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Innis RB, Cunningham VJ, Delforge J, et al. (2007) Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 27:1533–1539. [DOI] [PubMed] [Google Scholar]
- 32.Zischler J, Kolks N, Modemann D, et al. (2017) Alcohol-enhanced Cu-mediated radiofluorination. Chem-Eur J 23:3251–3256. [DOI] [PubMed] [Google Scholar]
- 33.Ohishi H, Neki A, Mizuno N (1998) Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res 30:65–82. [DOI] [PubMed] [Google Scholar]
- 34.O’Brien DE, Shaw DM, Cho HP, et al. (2018) Differential pharmacology and binding of mGlu2 receptor allosteric modulators. Mol Pharmacol 93:526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doornbos ML, Perez-Benito L, Tresadern G, et al. (2016) Molecular mechanism of positive allosteric modulation of the metabotropic glutamate receptor 2 by JNJ-46281222. Br J Pharmacol 173:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.