

Abstract
Immune checkpoint blockade (ICB) has become a standard therapy for several cancers; however, the response to ICB is inconsistent and a method for non-invasive assessment has not been established to date. To investigate the capability of multi-modal imaging to evaluate treatment response to ICB therapy, hyperpolarized 13C MRI using [1-13C] pyruvate and [1,4-13C2] fumarate and dynamic contrast enhanced (DCE) MRI were evaluated to detect early changes in tumor glycolysis, necrosis, and intratumor perfusion/permeability, respectively. Mouse tumor models served as platforms for high (MC38 colon adenocarcinoma) and low (B16-F10 melanoma) sensitivity to dual ICB of PD-L1 and CTLA-4. Glycolytic flux significantly decreased following treatment only in the less-sensitive B16-F10 tumors. Imaging [1,4-13C2] fumarate conversion to [1,4-13C2] malate showed a significant increase in necrotic cell death following treatment in the ICB-sensitive MC38 tumors, with essentially no change in B16-F10 tumors. DCE-MRI showed significantly increased perfusion/permeability in MC38-treated tumors, while a similar, but statistically non-significant, trend was observed in B16-F10 tumors. When tumor volume was also taken into consideration, each imaging biomarker was linearly correlated with future survival in both models. These results suggest that hyperpolarized 13C MRI and DCE MRI may serve as useful non-invasive imaging markers to detect early response to ICB therapy.
Introduction
Immunotherapy using immune checkpoint blockade (ICB) has emerged as a promising cancer treatment (1). ICB therapy removes the inhibitory signals of T cell activation by blocking co-stimulatory receptors or ligands such as PD-1, PD-L1, and CTLA-4, enabling tumor-reactive T cells to override regulatory mechanisms and exhibit antitumor activity (2). A key feature of this therapy is the highly durable tumor response, resulting in a plateau in the tail of the survival curve unlike chemotherapy or genomically targeted therapy, the benefits of which tend to diminish as time progresses (3). Despite the potential of this type of therapy, the remarkable responses to ICB are limited to a fraction of patients (4). Thus, an accurate and reliable tool for assessment of early treatment response is needed (5). While several biomarkers including PD-L1 expression (6), tumor mutation burden (7,8), mismatch repair deficiency (7,9), radiomic features (10,11), and tumor-infiltrating lymphocytes (TILs) have been proposed (12), none of them has become a gold standard to predict treatment response to date (13). The unique response to immunotherapy also complicates the search for a biomarker. Unlike cytotoxic therapy, where the effectiveness can be frequently judged by the reduction in the size of the tumor, the treatment response in immunotherapy is often delayed; tumors often transiently enlarge or new lesions will appear in the early stages only to be followed by tumor shrinkage or long-term stability of tumor size (14,15). The delayed response can pose a potential problem in designing treatment plans as the effectiveness of the therapy cannot be judged until the optimal treatment window has passed. In order to alleviate this problem, the immune-related response criteria (irRC), immune-related response criteria (irRECIST), and immune RECIST (iRECIST) standards were developed for imaging assessment of treatment response. These criteria require a consecutive scan at least 4 weeks apart for confirmation of progressive disease (15,16). When the treatment is found ineffective, patients may lose the opportunity to receive other treatments during the assessing time. Thus, developing an early prediction of the treatment response to immunotherapy is of great importance (17).
Treatment response to cancer immunotherapy is closely related to the tumor microenvironment. Immunologically “hot” tumors are typically characterized by a high degree of CD8+ cytotoxic T cells infiltration (18). A lack of CD8+ T cell infiltration and the presence of immune suppressor cells including regulatory T cells, myeloid derived suppressor cells, and type II macrophages are part of the extrinsic mechanisms of resistance to immunotherapy (19). Recently, increased metabolic competition between tumor cells and immune cells was also reported. Aerobic glycolysis (the “Warburg effect”), a metabolic effect characteristic of cancer cells (20), causes depletion of extracellular glucose and restricts glucose availability to T cells. The decrease in the availability of glucose leads to a suppression of glycolytic metabolism in T cells (21). Decreased glycolysis results in decreased effector function as phosphoenolpyruvate produced as a byproduct of glycolysis is essential in maintaining the high cytosolic Ca2+ levels necessary to support T cell activation (22,23). In this context, it is reported that PD-L1 promotes glycolysis in tumors via Akt/mTOR activation, and that PD-1 blockade can decrease glycolysis in tumors (21). The microenvironment of tumors are also a challenging environment for the immune system in other respects as it is characterized by hypoxia, low pH, high interstitial fluid pressure, and immune-inhibitory metabolites such as lactic acid (24,25). The impaired perfusion of abnormal tumor blood vessels can create a highly hypoxic microenvironment susceptible to invasion by immune suppressant cells (26). Recent studies have demonstrated that tumor hypoxia is negatively associated with the efficacy of immunotherapy and that the stimulation of immune cell functions with PD-1 and CTLA-4 blockade can also help to normalize tumor vessels (27,28). In this regard, it is potentially feasible to assess the treatment efficacy of immunotherapy by evaluating the metabolism and microenvironment of the tumors before a reduction in the size of the tumor can be detected.
A few studies have reported that 18F-FDG-PET/CT can potentially predict early response to PD-1 blockade or CTLA-4 blockade in patients with non-small cell lung cancer or melanoma (29,30). However, several studies have also demonstrated the inability of 18F-FDG-PET/CT to distinguish patients with pseudoprogression from those with progressive disease in melanoma patients (31,32). Thus, to date, non-invasive imaging approaches to clarify the early response to effective immunotherapy in vivo have not been established.
The purpose of this study is to investigate the capability of non-invasive metabolic and physiologic imaging to evaluate early response to immune checkpoint blockade therapy. New advancements in imaging using dynamic nuclear hyperpolarization (DNP) enhance the usually undetectable 13C signal by approximately four orders of magnitude in vivo (33). By using 13C pyruvate as a probe, it is possible to monitor the glycolytic profile of a tumor non-invasively, which has proven useful for monitoring the treatment response to several chemotherapeutics such as anti- angiogenic agent sunitinib and hypoxia-activated prodrug TH-302 (34,35). Similarly, the [1,4-13C2] fumarate probe has been shown to detect necrotic tumor cell death in vivo (36,37).
To test the applicability of these new imaging techniques along with conventional dynamic contrast enhanced (DCE) MRI in detecting ICB treatment response, we imaged mice bearing murine colon adenocarcinoma carcinoma (MC38) tumor or melanoma (B16-F10) tumor as ICB sensitive and less ICB sensitive models, respectively. Using the combination therapy of PD-L1/PD-1 and CTLA-4 as a treatment model, which has an improved therapeutic effect compared with either monotherapy (38-40), multimodal metabolic and physiologic imaging showed substantial differences between the sensitive and less sensitive models and correlated with survival time within each model and therefore may serve as a guide for further development of imaging biomarkers.
Methods
Mice and tumor
Female C57BL/6 (B6) mice were supplied by the Frederick Cancer Research Center, Animal Production (Frederick, MD) and housed in a specific pathogen-free environment and used at an age of 8-12 week. MC38 colon adenocarcinoma and B16-F10 melanoma were used as sensitive or less sensitive models to immune checkpoint blockade therapy. MC38 was purchased from Kerafast (Boston, MA). B16-F10 was used from the frozen stock in our lab and tested in Feb 2020 and authentificated by IDEXX RADIL (Columbia, MO) using a panel of microsatellite markers. Molecular testing of cell lines for multiple pathogens, including mycoplasma, was performed at the time of receipt and prior to in vivo studies. Both cell lines were maintained in DMEM containing 4.5 g/L glucose supplemented with 10% fetal calf serum and antibiotics.
In vivo checkpoint blockade treatment
For the in vivo treatment model, 1 x 105 MC38 tumor cells or 2 x 105 B16-F10 tumor cells were inoculated s.c. into the right leg of B6 mice. Tumor bearing mice were injected i.p. with 200 μg of αPD-L1 (10F.9G2) or αCTLA-4 (9H10) antibody or with isotype control antibodies (all from BioXcell) on days 9, 12, and 15 post tumor inoculation at the size of approximately 100-150 mm3. Imaging experiments were performed on day 13 or 14 after 2nd injection of antibodies.
In vitro treatment
For in vitro treatment assay, tumor cells were cultured with 100 U/ml of recombinant murine IFN-γ for 48h followed by 10 μg/ml αPD-L1 and αCTLA-4 antibody treatment for an additional 24h before the assay.
Flow cytometry
To generate activated tumor-draining lymph node (TDLN) cells, B6 mice were inoculated subdermally with 1 x 105 MC38 tumor cells on both flanks to stimulate TDLNs. 13 days later, TDLNs (inguinal) were harvested, and single-cell suspensions were prepared mechanically. To analyze tumor infiltrating lymphocytes, single-cell suspensions were prepared from solid tumors by digestion with a mixture of 0.1 % collagenase, 0.01 % DNase, and 2.5 U/ml hyaluronidase for 3 h at 37 deg C. FITC-conjugated mAbs against CD4 (RM4-5); PE-conjugated mAbs against CD3; Cy-chrome-conjugated mAbs against CD8 (53-6.7) were purchased from BD Biosciences. The cell surface phenotypes were determined by direct immunofluorescence staining with conjugated mAbs and analyzed using FACS Calibur (BD Biosciences, San Jose, CA). Digested cells were stained with αCD3, αCD8, and αCD4 antibody. Data were collected on FACS Calibur flow cytometer. Tumor infiltrating lymphocytes were identified and gated on a forward scatter vs side scatter plot.
Western blot
In vitro treated tumor cells were lysed on the dish with radioimmunoprecipitation assay buffer (RIPA buffer, Thermo Scientific) supplemented with protease and phosphatase inhibitors (Roche). In vivo treated tumor tissue was excised and homogenized in T-PER™ Tissue Protein Extraction Reagent (Thermo Fisher Scientific). Protein concentrations were measured by the bicinchoninic acid assay (BCA protein assay, Thermo Fisher Scientific). Glut-1, Hexokinase-2, lactate dehydrogenase A (LDHA), monocarboxylate transporter 1 (MCT1), MCT4 proteins were separated on 4 % to 20 % Tris-Glycine gel (Life Technologies) by SDS-PAGE and were transferred to nitrocellulose membrane. The membranes were blocked for 1 h in blocking buffer (3 % nonfat dry milk in 0.1 % Tween 20/TBS), which was then replaced by the primary antibody (1:500-1:1000), diluted in blocking buffer, and then incubated for 1 h at room temperature. The membranes were then washed three times in washing buffer (0.1 % Tween 20/TBS). The primary antibody was detected using the appropriate horseradish peroxidase conjugated secondary antibody and measured by the Fluor Chem HD2 chemiluminescent imaging system (Alpha Innotech Corp.). Density values for each protein were normalized to actin.
Seahorse metabolism assay
The extracellular acidification rate (ECAR) was analyzed on a XF96 Extracellular Flux Analyzer using XF Glycolysis Stress Test Kit (Agilent Technology, Santa Clara, CA) in accordance with the manufacturer's instructions. Briefly, in vitro treated cells on 96 well plates were first incubated in XF base medium without glucose or pyruvate. Glucose was injected and the ECAR is measured as the rate of glycolysis under basal conditions. Then, oligomycin, an ATP synthase inhibitor was injected and ECAR was monitored (maximum glycolytic capacity). Finally, 2-deoxy-glucose (2-DG), a glucose analog that inhibits glycolysis through competitive binding to glucose hexokinase, was injected to confirm that the ECAR produced in the experiment was due to glycolysis.
Histological assessment
Tumor tissues were excised, frozen with Tissue-Tek O.C.T. compound (Sakura Finetek USA Inc.) by ultra-cold ethanol and sectioned (10 mm) using a cryostat, with the sections being thaw-mounted on glass slides. After fixing with 4 % paraformaldehyde, sections were treated with cold acetone for 15 minutes. After blocking nonspecific-binding sites on sections with Protein Block Serum-Free reagent (Dako North America Inc.) for 30 minutes, the slides were covered by CD3 antibody (BD Biosciences; 1:250) combined with CD8 antibody (Abcam, Inc.; 1:250) overnight at 4 C. The sections were then incubated with Alexa Fluor 488 antimouse and the Alexa Fluor 546 F(ab')2 fragment of goat antirabbit IgG (HþL) (Invitrogen; 1:2,000) for 1 h at room temperature, before being mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). CD31 was stained with same procedure using CD31 antibody (BD Biosciences; 1:250) and Alexa Fluor 488 antirat (Invitrogen; 1:250). The sections were also stained with hematoxylin and eosin and mounted on permount for histological observation. The stained slides were scanned using a BZ-9000 microscope (Keyence), and the immunostain-positive area was quantified using ImageJ software (downloaded from https://imagej.nih.gov/ij/).
Hyperpolarized 13C MRI
Details of the hyperpolarization procedure were reported previously (34,35). Briefly, [1-13C] pyruvic acid (30 μL) or [1,4-13C2] fumaric acid (2.5 M in 30 μL deuterated DMSO), containing 15 mmol/L Ox063 and 2.5 mmol/L of the gadolinium chelate ProHance (BraccoDiagnostics) was polarized in Hypersense DNP polarizer (Oxford Instruments). After the polarization reached 80 % of the plateau value, the hyperpolarized sample was rapidly dissolved in 4.5 mL of a superheated alkaline buffer consisted of 40 mmol/L HEPES, NaOH, and 100 mg/L EDTA. Hyperpolarized [1-13C] pyruvate or [1,4-13C2] fumarate solution was rapidly injected intravenously through a catheter placed in the tail vein of each mouse (12 mL/g body weight). Hyperpolarized 13C MRI studies were performed on a 3 T dedicated MR Solutions animal scanner (MR SOLUTIONS Ltd., Boston, MA) using a 17 mm home-built 13C solenoid coil placed inside of a saddle coil tuned to 1H frequency. Both 1H and 13C were tuned and matched and anatomical image was taken after shimming on proton. 13C spectra was acquired every 1 s for 240 s from the whole leg including each tumor. The repetition time, spectral width, flip angle, and number of average were 1000 ms, 3300 Hz, 10°, and 1, respectively.
Fumarase activity assay
A serum sample was obtained from isotype control antibody treated mice and from αPD-L1 and αCTLA-4 antibodies treated mice one day after 2nd injection. Fumarase activity was measured with the Fumarase Assay Kit (BioAssay Systems, Hayward, CA) in accordance with the manufacturer's instruction manual.
DCE-MRI of Gd-DTPA
DCE-MRI studies were performed on a 1 T scanner (Bruker BioSpin MRI GmbH). T1-weighted fast low-angle shot (FLASH) images were obtained with TR = 156 ms; TE = 4 ms; flip angle = 45°; four slices; 0.44 x 0.44 mm resolution; 15-second acquisition time per image; and 45 repetitions. Gd-DTPA solution (4 mL/g of body weight of 50 mmol/L Gd-DTPA) was injected through a tail vein cannula 1 minutes after the start of the dynamic FLASH sequence. To determine the local concentrations of Gd-DTPA, T1 maps were calculated from three sets of Rapid Imaging with Refocused Echoes (RARE) images obtained with TR = 300, 600, 1000, and 2,000 ms, with the acquisitions being made before running the FLASH sequence.
Statistics
The significance of the differences between groups was analyzed using the Student’s t test for two groups and Tukey-Kramer test for more than two groups. Cohen’s d was calculated to assess the effect size of tumor reduction by the treatment. Cohen's d = (Mean (Treatment) – Mean (Control)) / (SD (Treatment)2 + SD (Control)2) / 2)1/2. Kaplan-Meier curves were constructed for the survival of mice; the differences between groups were identified using the log-rank test. A two-tailed p value, 0.05 was considered significant.
Study approval
The animal experiments were conducted according to a protocol approved by the Animal Research Advisory Committee of the NIH (RBB-159-3E) in accordance with the National Institutes of Health Guidelines for Animal Research.
Results
In vivo treatment effect of immune checkpoint inhibitors in a murine tumor model.
As a model for the response to immune checkpoint blockade in vivo, we evaluated the response of two xenograft models to either Anti-PD-L1 monoclonal antibody (mAb) monotherapy or Anti-PD-L1 monoclonal antibody therapy in combination with anti-CTLA-4 mAb. Mice were inoculated subcutaneously with either MC38 (colon adenocarcinoma) or B16-F10 (melanoma) cells into the right leg and treated with either or both immune checkpoint inhibitors by i.p. injection every three days for a total of three injections.
By itself, anti-PD-L1 Ab therapy inhibited tumor growth in MC38 colon adenocarcinoma tumors (Figure 1A) and extended survival times (Figure 1B and C) compared to the control group (median survival time 29.5 days compared to 24 days without treatment, p = 0.0003). Combining anti-PD-L1 with anti-CTLA-4 Ab treatment resulted in a stronger inhibition of tumor growth and a statistically significant increase in survival time relative to anti-PD-L1 Ab alone or the control group (33.5 days to 29.5 days or 24 days, p = 0.047, p < 0.0001, Figure 1B). Anti-PD-L1 Ab + anti-CTLA-4 Ab also delayed tumor growth (Figure 1D) and extended survival in B16-F10 tumors (median survival time 24 days for the combination treatment compared to 19.5 days without treatment, p = 0.0018 Figure 1E and F). However, the cytoreductive effect of anti-PD-L1 Ab + anti-CTLA-4 Ab was less in B16-F10 than in MC38 tumors (Cohen’s d; −2.88 to −3.88, Supplemental Figure 1). The survival benefit from anti-PD-L1 Ab + anti-CTLA-4 Ab was also less in B16-F10 compared to the MC38 cell line (mean survival benefit 4.2 days to 12.1 days) These results are consistent with previous reports showing that, while both cell lines respond to a statistically measurable degree to ICB treatment, MC38 displays a greater response (more sensitive) than B16-F10 (41).
Figure 1. Tumor growth and survival of immune checkpoint blockade therapy in two murine tumor models with different sensitivity.
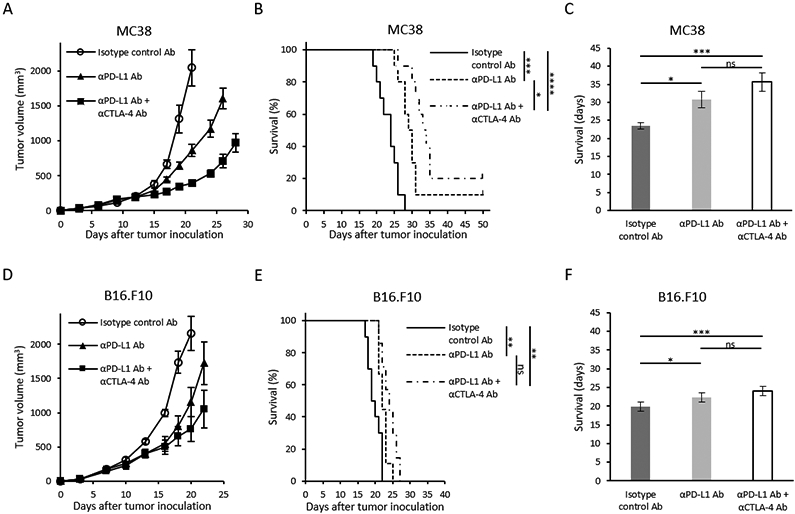
(A and D) Growth kinetics of each tumor. MC38 or B16-F10 inoculated mice were treated with either Isotype control Ab, anti-PD-L1 Ab, or combination anti-PD-L1 Ab + anti-CTLA-4 Ab on days 9, 12, and 15 post tumor inoculation (n = 5 per group). Data are shown as mean ± SE at each time point. (B and E) Kaplan-Meier survival curve for each tumor (MC38; n = 10 per group, B16-F10; n =7-10 per group). Survival refers to the time before reaching the maximally allowed tumor volume of 2,000 mm3. (C and F) Bar plot of survival from figure B and E. Data are shown as mean ± SE. Statistical significance between groups was determined by log-rank test for B and E, and by Tukey-Kramer test for C and F. *p < 0.05, **p < 0.01, ***p < 0.001 ****p < 0.0001.
This difference in sensitivity to ICB therapy was not reflected in T-cell counts (Figure 2A). Intratumor levels of CD3+CD8+ T cells increased substantially in both tumor models after 4 days of anti-PD-L1 Ab + anti-CTLA-4 Ab treatment, suggesting effective infiltration of lymphocytes into the tumors. ICB treatment raised levels of CD3+CD4+ and CD3+CD8+ T cells in the tumor draining lymph nodes in MC38 tumors, but the populations of both types of cells was already at a similar high level in the absence of treatment in B16-F10 tumors. Since no change was observed upon treatment in the B16-F10 model, the fraction of CD3+CD4+ and CD3+CD8+ cells in the tumor draining lymph nodes is not likely to serve as a strictly generalizable marker for treatment response. To more quantitatively measure total and activated T cells in situ, histological assessment was performed on tumor slices to confirm T cells proliferation. Immunofluorescence staining of CD3 and CD8 showed that infiltration of CD3+ T cells and CD8+ cytotoxic T cells significantly increased in ICB treated MC38 tumors compared to isotype control Ab treated MC38 tumors (p < 0.0001, p < 0.0001, Figure 2B). In B16-F10 tumors, the absolute number of lymphocytes was generally smaller than in MC38 tumors. The infiltration of CD3+ T cells and CD8+ cytotoxic T cells was significantly higher in both tumor models treated with ICB combination therapy compared to the corresponding tumors treated with the isotype control Ab treated (p < 0.0001, p < 0.0001, Figure 2C). These findings confirmed an active immune response as early as day 4 after the ICB treatment in both tumor models. The CD8 to CD3 ratio was similar between groups in both tumors, suggesting a similar level of activation between CD4 and CD8 T cells (Figure 2B and Figure 2C).
Figure 2. In vivo treatment effect of immune checkpoint blockade therapy.

(A) Flow cytometry analysis of tumor infiltrating lymphocytes and tumor draining lymph node cells. Tumor bearing mice were treated with each Ab on day 9 and 12. Tumors and lymph nodes were harvested on day 13. The figure on each plot shows the percentage of CD3+CD4+ T cells or CD3+CD8+ T cells. (B and C) Immunofluorescence staining of CD3 and CD8 in MC38 tumor (B) or B16-F10 tumor (C) (scale bar = 100 μm). Tumor bearing mice were treated with each Ab on day 9 and 12. Tumors were harvested on day 13. Number of cells were manually counted in randomly selected 5 fields per tumor section (MC38; n = 4 per group, B16-F10; n = 5 per group). Data are shown as mean ± SE. Statistical significance between groups was determined by Student’s t test.
Changes in tumor glycolytic metabolism induced by immune checkpoint blockade
Activation, proliferation, and differentiation of T cells all require the synthesis of numerous macromolecules. To generate synthetic intermediates to support these processes, naïve T cells often undergo metabolic reprogramming to enhance glycolytic flux (42). Decreases in glycolytic flux therefore could serve as a potential biomarker to predict ICB efficacy. To investigate whether glycolytic metabolism of tumor cells is altered directly by PD-L1 blockade, we first measured the in vitro extracellular acidification rate (ECAR) of tumor cells by the Seahorse assay to determine the extent of lactic acid fermentation (Figure 3A). Anti-CTLA4 Ab was added to anti-PD-L1 Ab so that the treatment regimen, although not the system itself, is comparable with the in vivo experimental model. The ECAR of MC38 cells in vitro was not altered after ICB treatment. In contrast, the ECAR of the less ICB sensitive B16-F10 cell line was significantly reduced after ICB treatment (Figure 3A), suggesting that the glycolytic metabolism of B16-F10 is more dependent on the PD-L1/PD-1 pathway than that of MC38. To investigate whether expression of glycolytic enzymes and transporters are affected by PD-L1 blockade, PD-L1 inhibitor treated tumor cells were grown and treated in vitro and analyzed by western blotting. No significant changes in the expression of glycolytic enzymes and transporters were found after PD-L1 blockade in either cell line (Figure 3B and Supplemental Figure 2). Since the tumor cells themselves only form part of the tumor microenvironment, this experiment was repeated in vivo with tumors treated with anti-PD-L1 Ab + anti-CTLA-4 Ab and excised from mice (Supplemental Figure 3). Mirroring the in vitro experiment, no significant changes were observed glycolytic enzyme levels in MC38 tumors. In B16-F10, the expression of specific glycolytic enzymes including lactate dehydrogenase A (LDHA), Glut1, Hexokinase 2 and Monocarboxylate transporter 4 (MCT4) was reduced after ICB treatment. However, these differences were not statistically significant at the α=0.05 level. Therefore, downregulation of glycolytic enzymes in response to PD-L1 treatment is a possible, but unproven, factor in the decrease in glycolytic flux in B16-F10.
Figure 3. Metabolic shift induced by immune checkpoint blockade can be detected in selected cancer whose metabolism relies on glycolysis.

(A) Extracellular acidification rate (ECAR) of MC38 and B16-F10 cells in vitro. Tumor cells pre-treated with IFN-γ for 48 h followed by anti-PD-L1 Ab + anti-CTLA-4 Ab or isotype control Ab for 24 h were measured (n = 5 per group). For each time point, mean ± SE is plotted. Bar plots show the parameters (non-glycolytic acidification and glycolysis) calculated from ECAR kinetics. Data are shown as mean ± SE. (B) Western blot analysis for LDHA on in vivo ICB treated tumor. MC38 or B16-F10 tumor bearing mice were treated with isotype control Ab or anti-PD-L1 Ab + anti-CTLA-4 Ab on day 9 and 12. Tumors were harvested and processed on day 13. Bar plot shows the quantification of protein bands. Data are shown as mean ± SE. The mean of the control group is set as 1 (n = 5 per group). (C-G) MRI of hyperpolarized 13C pyruvate metabolism in two murine tumor models. Tumor bearing mice treated with isotype control Ab or anti-PD-L1 Ab + anti-CTLA-4 Ab were scanned after the 2nd injection of antibodies (MC38; n = 23, n = 16 each group, B16-F10; n = 17, n = 16 each group). (C) Representative dynamic 13C spectra of MC38 and B16.F10 tumor and T2-weighted 1H anatomical image. (D and F) Lactate to Pyruvate (Lac/Pyr) ratio of MC38 (D) or B16.F10 (F) sorted by treatment. Data are shown as box-and-whisker plot (median, maximum, minimum, first quartile, and third quartile); individual values are shown. (E and G) Correlation between Lac/Pyr ratio and tumor size in MC38 (E) and B16.F10 (G). Statistical significance between groups was determined by Student’s t test for A, B, D, and F.
To evaluate changes in glycolysis upon ICB treatment as a potential imaging biomarker for ICB response, we measured the flux through the lactate dehydrogenase pathway in vivo by hyperpolarized 13C MRI using [1-13C] pyruvate. Non-localized 1D spectra encompassing the entire tumor implanted leg were acquired continuously for 240 s after the injection of hyperpolarized [1-13C] pyruvate and the conversion of pyruvate to lactate was quantitatively evaluated (Figure 3C). The lactate/pyruvate ratio is a direct measure of flux through the lactate dehydrogenase pathway (43) and an indirect measure of glycolysis. In the ICB sensitive MC38 tumors, ICB treated tumors showed lower Lac/Pyr ratios compared to control tumors, although the difference was not statistically significant (median Lac/Pyr ratio 0.80 for the ICB treatment compared to 0.90 for the control, p = 0.075, Figure 3D). A cluster of large tumors with high Lac/Pyr ratios likely undergoing anaerobic fermentation is evident in the control group (Figure 3E). No correlation of the Lac/Pyr ratio with the treatment group was found when controlled for tumor size to remove this effect (p = 0.13, Supplemental Table 1A). The decrease in glycolytic flux after ICB treatment was more marked in B16-F10 tumors. The Lac/Pyr ratio was significantly lower in the ICB treatment group relative to the control group (median Lac/Pyr ratio 0.61 for the ICB treatment compared to 0.82 for the control, p = 0.0073, Figure 3F). Even when controlled for tumor size, the Lac/Pyr ratio remained negatively correlated with the treatment with statistical significance (p = 0.018, Supplemental Table 1B and Figure 3G). The Bicarbonate/Pyruvate ratio was similar between treated and untreated control groups in both tumors (Supplemental Figure 4). Taken together, although these results suggested that Lac/Pyr tended to be lower in anti-PD-L1 Ab + anti-CTLA-4 Ab treated tumor compared to control tumors in both tumor models, the Lac/Pyr ratio did not reflect the effect size of ICB therapy (larger in MC38 compared to B16-F10).
Necrotic change induced by immune checkpoint blockade
Tumor cell necrosis is potentially a more direct biomarker for ICB efficacy. To evaluate necrotic cell death in tumors, hyperpolarized 13C MRI using [1,4-13C2] fumarate was performed on the same sets of tumors. Fumarate is not actively transported into the cell and the plasma membrane is impermeable to fumarate when intact. A loss of membrane integrity during necrosis allows entry of fumarate and the conversion to malate by cytosolic fumarate hydratase, which can be detected as a doublet distinct from fumarate at 182-183 ppm. Figure 4A shows the representative dynamic 13C spectra of ICB sensitive MC38.. Figure 4B shows the malate to fumarate (Mal/Fum) ratio of MC38 tumor bearing mice sorted by treatment group. Mal/Fum ratios were significantly higher in ICB treated MC38 tumors relative to the control group (median Fum/Mal ratio 0.091 for the ICB treatment compared to 0.055 for the control, p = 0.0006, Figure 4B) . The Mal/Fum ratio remained a significant variable even when tumor size was controlled (p = 0.013, Supplemental Table 1C and Figure 4C), suggesting that tumor cell necrosis is induced as a treatment effect of ICB therapy in vivo. Mal/Fum was slightly higher in the ICB treated B16-F10 tumors (Figure 4D), but the difference was not significant (median Fum/Mal ratio 0.096 for the ICB treatment compared to 0.081 for the control, p = 0.21, Figure 4E). Multiple regression analysis confirmed that Mal/Fum ratio was not a significant predictor of the treatment group (p = 0.42, Supplemental Table 1D and Figure 4F). No difference in serum fumarase activity was found between ICB treated MC38 mice and the control (p = 0.72, Figure 4G), suggesting that serum fumarase activity cannot be used as a surrogate marker to monitor ICB efficacy.
Figure 4. Tumor cell death can be detected by 13C fumarate MRI as treatment effect of immune checkpoint blockade.

(A-G) MRI of hyperpolarized 13C fumarate metabolism in two murine tumor models. Tumor bearing mice treated with isotype control Ab or anti-PD-L1 Ab + anti-CTLA-4 Ab were scanned after 2nd injection of antibodies (MC38; n = 16, n = 14 each group, B16.F10; n = 11, n = 14 each group). Representative dynamic 13C spectra of MC38 (A) or B16.F10 (D) tumor and T2-weighted 1H anatomical image. Signal of Malate is quintupled in the bottom plot. (B and E) The Malate to Fumarate (Mal/Fum) ratio of MC38 (B) or B16.F10 (E) sorted by treatment. Data are shown as box-and-whisker plot (median, maximum, minimum, first quartile, and third quartile); individual values are shown. (C and F) Correlation between Mal/Fum ratio and tumor size in MC38 (A) and B16.F10 (C). (G) Fumarase activity assay of mice serum. MC38 tumor bearing mice were treated with isotype control Ab or anti-PD-L1 Ab + anti-CTLA-4 Ab on day 9 and 12. Blood were collected on day 13. Data are shown as mean ± SE (n = 5, n = 7 each group). Statistical significance between groups was determined by Student’s t test for B, E, and G.
To confirm the necrotic change by histology, tumor sections were stained with hematoxylin and eosin according to standard procedures. In MC38 tumors, infiltration of lymphocytes was confirmed by focal areas of hematoxylin negative spaces (dot-line area, Figure 5A). Nucleic enlargement in tumor cells (white arrow) indicating necrotic cell damage was also observed (Figure 5A). The overall decrease in hematoxylin positive area, indicating the replacement with interstitial tissue after tumor cell death, was quantified using ImageJ software, and found to be significantly increased in ICB treated MC38 tumors compared to the control (p = 0.027, Figure 5B) (Hematoxylin and eosin staining of all the samples in supplemental Figure 5). These histologic findings support the necrotic cell death as a consequence of antitumor effect of lymphocytes induced by ICB therapy. The increase in necrotic area and lymphocyte infiltration was also evident in the less ICB sensitive B16-F10 tumors. Such changes were observed more diffusely in B16-F10 tumors. However, the decrease in hematoxin positive level was milder in B16-F10 tumors, and the difference between treatment and control groups did not meet the test for statistical significance (p = 0.13, Figure 5B). The histological diffrerence in the level of tumor cell death by the cell line is consistent with the difference in Mal/Fum ratio detected with hyperpolarized 13C MRI using [1,4-13C2] fumarate.
Figure 5. Histological assessment of HE staining.

(A) Representative hematoxylin and eosin staining (HE) of MC38 and B16-F10 tumors (scale bar = 100 μm). MC38 or B16-F10 tumor bearing mice were treated with each Ab on day 9 and 12. Tumors were harvested on day 13. (B) Quantification of hematoxylin stained area. Data are shown as mean ± SE (MC38; n = 4 per group, B16-F10; n = 5 per group). Statistical significance between groups was determined by Student’s t test.
Changes in Intratumor blood perfusion induced by immune checkpoint inhibitors
Tumor perfusion/permeability changes in response to ICB treatments were investigated by DCE-MRI using the Toft model. In the Toft model, permeability changes are characterized by the influx forward volume transfer constant (Ktrans) from plasma into the extravascular-extracellular space (EES), which reflects the sum of all processes (predominantly blood flow and capillary leakage) that determine the rate of gadolinium influx from plasma into the EES. We examined isotype control Ab treated and anti-PD-L1 Ab + anti-CTLA-4 Ab treated tumor bearing mice after the 2nd Ab injection. Relative to the control, Gd-DTPA uptake was increased (Figure 6A and B) and Ktrans was significantly higher in ICB treated MC38 tumors (p = 0.0025, Figure 6C). The separate effects of perfusion and an increase in the volume of the extravascular extracellular space through tissue clearance can be distinguished by the AUC of the calculated Gd concentration at short (1 min) and long (10 min), respectively. Significant increases in both perfusion and permeability were observed (p = 0.0037 , p = 0.0029, Figure 6D). Changes in the ICB less sensitive B16-F10 tumors were relatively less (Figure 6A). ICB treated tumors showed higher Ktrans but did not meet the strict criterion for statistical significance (p = 0.052, Figure 6E). Perfusion (AUC 1min) was significantly higher in ICB treated B16-F10 tumors, while no difference in the equilibrium (AUC 10 min) was detected, suggesting a change in the perfusion without a significant volume increase in the extravascular extracellular space (p = 0.021, p = 0.25, respectively, Figure 6F). These findings suggest that ICB treatment improved blood perfusion in MC38 tumors, but caused caused only limited changes in B16-F10 tumors.
Figure 6. Tumor perfusion and permeability are improved by immune checkpoint blockade.

(A-F) Dynamic contrast enhanced MRI in two murine tumor model. Tumor bearing mice treated with isotype control Ab or anti-PD-L1 Ab + anti-CTLA-4 Ab were scanned after treatment (MC38; n = 14, n = 12 each group, B16-F10; n = 15, n = 17 each group). (A) Representative time-intensity kinetic curve of Gd-DTPA in MC38 tumor and B16-F10. (B) Gd-DTPA intensity with T2-weighted anatomical image of MC38 and B16-F10 tumor. (C and E) Ktrans and Ve value of MC38 (C) or B16-F10 (E) sorted by treatment group. Data are shown as a box-and-whisker plot (median, maximum, minimum, first quartile, and third quartile); individual values are shown. (D and F) Area under curve (AUC) 1 min and AUC 10 min of Gd-DTPA concentration in MC38 (D) and B16-F10 (F) tumor. Data are shown as box-and-whisker plot and individual values. Statistical significance between groups was determined by Student’s t test for C-F.
To examine the changes in blood vessel density, IHC was performed with tumor sections. Supplemental Figure 6 shows the staining of CD31, which is a marker of vascular endothelial cells, as well as the staining of H&E in same section. Compared to the control tumor, CD31 staining was increased in ICB treated MC38 tumor (Supplemental Figure 6A). CD31 positive area increased notably in hematoxylin less staining area where tumor cells are less (solid-line area), while CD31 positive area were poor in hematoxylin dense area where tumor cells are rich (dot-line area) in both control Ab and ICB treated tumors (Supplemental Figure 6A). Quantification of CD31 positive area showed a significant increase in ICB treated tumor (p < 0.0001, Supplemental Figure 6B). In contrast, no significant difference was observed in CD31 staining for B16-F10 tumors (p = 0.81, Supplemental Figure 6C and 6D). These findings suggest a difference exists between MC38 and B16-F10 tumors in the improvement of vascular flow by ICB therapy, which supports the findings of tumor perfusion/permeability differences in DCE MRI.
Correlation between imaging biomarkers and clinical outcome
The previous experiments have the implicit assumption that after treatment a valid biomarker will change significantly in the ICB sensitive model (MC38) but not in the less sensitive model and control groups. An invalid biomarker for ICB, by contrast, either not change significantly after treatment in model known to be ICB sensitive model or the magnitude of the change will be similar in the non-treated control groups and less sensitive models. While useful for probing the underlying metabolic and physiologic changes that occur during treatment, this paradigm does not strictly correspond to the clinical situation where it is usually the ability of biomarkers to predict progression within similar tumor types that is primarily of interest.To investigate each imaging biomarker’s prognostic capability, a part of the mice scanned in hyperpolarized 13C MRI or DCE-MRI experiments in Figure 3C-G, Figure 4A-F, or Figure 6 were followed until their clinical endpoint. As expected from Figure 1, there was strong separation for both B16-F10 and MC38 from the control groups (Figure 7A). No correlation of survival was observed for any biomarker with the control group. Correlations were relatively strong but with significant scatter (Supplemental Table 2B). Single regression analysis showed that the Lac/Pyr ratio had a significant negative correlation with survival in B16-F10 tumors (R2=0.59±0.06 and p = 0.02, Supplemental Table 2B). Ktrans was also significantly positively correlated with survival in MC38 tumor and B16-F10 tumors (Figure 7A, R2=0.76±0.01 and p = 0.02, Supplemental Table 2B).
Figure 7. Correlation between imaging markers and clinical outcome.
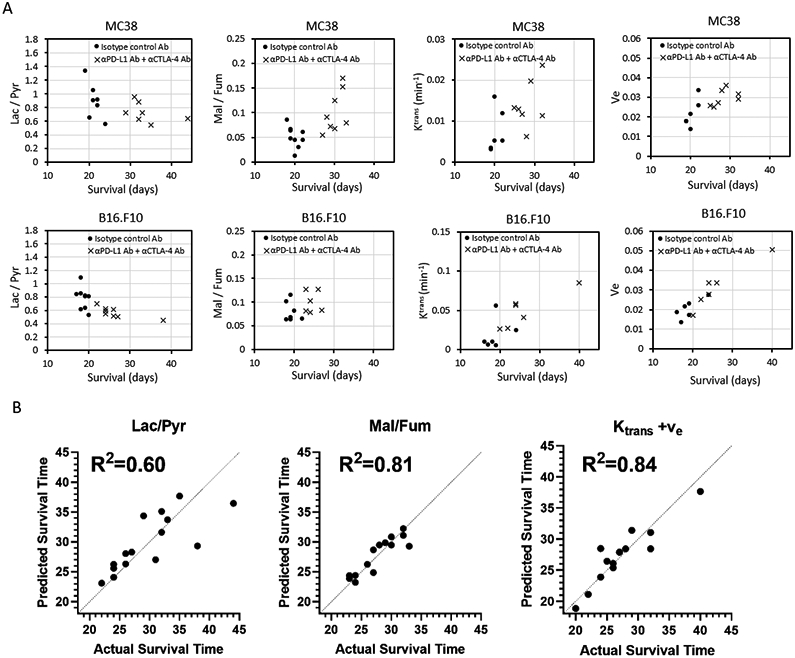
(A-C) Some of experimental mice scanned by 13C Pyruvate MRI, 13C Fumarate MRI, or DCE-MRI in Figure 3, Figure 4, or Figure 6 were followed until the end of their clinical endpoint to determine their survival. The survival data of these mice (MC38 and B16-F10) treated with both PD-L1 and CTLA-4 dual blockade was examined for the prognostic capability of each imaging biomarker. Survival refers to the time before reaching the maximally allowed tumor volume of 2,000 mm3. (A) Correlation between Lac/Pyr ratio and survival in MC38 and B16-F10 (MC38; n = 7 per group, B16-F10; n = 9, n = 8 each group). (B) Correlation between Mal/Fum ratio and survival in MC38 and B16-F10 (MC38; n = 9, n = 8 each group, B16-F10; n = 7, n = 6 each group). (C) Correlation between Ktrans and survival in MC38 and B16-F10 (MC38; n = 6, n = 7 each group, B16-F10; n = 6 per group). (D) Correlation between Ve and survival in MC38 and B16-F10 (MC38; n = 6, n = 7 each group, B16-F10; n = 6 per group). (E) Correlation between the Lac/Pyr ratio, Mal/Fum and Ktrans/ve in a multilinear regression model using tumor volume and each imaging biomarker with cell type as a categorical variable.
Some of the single time point studies suggested tumor volume may be a complicating factor in the relationship between metabolic markers and ICB response (Supplemental Table 1B). Since tumor volume is easily obtainable from an anatomical MRI, we constructed multilinear regression prediction models for survival using the imaging biomarker,the tumor volume, and the cell line as a categorical variable. Both Ktrans and ve were included in the DCE model (Figure 7B). Correlation significantly improved in each case. The least accurate predictior, the Lac/Pyr ratio, still obtained an R2 of 0.6, although the Lac/Pyr variable itself was not statistically significant (p=0.13, β=−18.6). The Mal/Fum ratio (R2=0.81) and the DCE Ktrans/ve (R2=0.84) appear to be better predictors of survival than the Lac/Pyr ratio in this model.
Discussion
In this study, we investigated the capability of non-invasive metabolic and physiologic imaging to evaluate the early response to ICB therapy in tumor bearing animal models. To our knowledge, this is the first report of metabolic imaging assessment to detect the early response to immune checkpoint blockade therapy using hyperpolarized 13C MRI.
Expression of PD-L1 on cancer cells promotes glucose uptake and production of lactate as part of the metabolic programming that enhances the survival of cells in hypoxic conditions and provides fuel for further growth. The exact mechanism by which this occurs is not fully understood. Previous reports have shown that blocking PD-L1 may directly dampen tumor glycolysis by inhibiting mTOR activity (24). In vivo tumor metabolism when evaluated by 13C pyruvate MRI showed a significant decrease in glycolysis in B16-F10 tumors after ICB treatment yet had little independent effect on MC38 after adjustment for tumor size, despite the greater impact of ICB on tumor volumes and survival time on this cell line (Figure 3C-G). Within the same cell line, Lac/Pyr emerged as the least efficient of the imaging biomarkers chosen for predicting treatment outcome (Figure 7B). The reasons for the relative insensitivity of Lac/Pyr as biomarker for treatment are not entirely clear. The in vivo tumor microenvironment consists of tumor cells with substantial contributions from the tumor stroma and immune cells. The expression of PD-1 on T cells is reported to inhibit glycolysis or amino acid metabolism and up-regulate fatty acid oxidation leading to impaired energy generation, which compromises proliferation and effector functions (44). CTLA-4 can also cause decreased expression of GLUT1, increased mitochondrial oxidation and fatty acid uptake, and decreased biosynthesis on T cells (45,46). Therefore, inhibition of PD-1/PD-L1 and CTLA-4 would preferentially promote T-cell function. ICB treatment might restore the metabolic balance in the competiton between tumor and immune cells in favor of T cells, leading to a less glycolytic profile when the tumor is considered as a whole. In our study, glycolytic flux in MC38 tumors was not affected by ICB when only tumor cells are considered whereas the glycolytic flux in B16-F10 tumors was significantly altered (Figure 3A), in line with previous reports that show the higher glycolytic activity in B16-F10 tumors results in high acidification of the tumor microenvironment when compared to MC38 tumors (47). This suggests that B16-F10 is more dependent on glycolytic metabolism for energy production than MC38. Moreover, a greater decrease in lactate production (Figure 3F) and in the expression levels of glycolytic enzymes and transporters in tumors after ICB treatment (Supplemental Figure 3) suggest that the glycolytic metabolism in B16-F10 may be more susceptible to ICB therapy. In this respect, 13C pyruvate MRI may detect the metabolic shift by ICB therapy in selected cancers whose metabolism strongly relies on glycolysis.
13C fumarate MRI showed significantly increased Mal/Fum in ICB treated tumors relative to control tumors, suggesting necrotic tumor cell damage was induced in response to ICB therapy (Figure 4). The Mal/Fum ratio also emerged as a strong predictor of survival time within the same cell line when combined with the tumor volume (Figure 7B). The utility of the malate production from hyperpolarized [1,4-13C2] fumarate probe as a sensitive indicator of tumor cell death has been previously shown in several tumor types treated with either a cytotoxic agent, anti-VEGF drug, or multi-kinase inhibitor (36,48,49). In contrast to chemotherapy which directly shows cytotoxicity on tumor cells, the effect of ICB is induced via activation of immune system. Blockade of immune checkpoints such as PD-L1, PD-1, CTLA-4, whose signals help keep T cells from killing cancer cells, release the “brakes” on the immune system and enable T cells to activate and show cytotoxicity. There are two main mechanisms involved in cytotoxic T cells-mediated tumor cell death: one elicited by granule exocytosis (perforin and granzymes) and the other via the death ligand/death receptor system like Fas ligand (FasL) and TNF-related apoptosis inducing ligand (TRAIL). It has been assumed that the final consequence of granule exocytosis is the induction of cell death by apoptosis. However, recent experimental evidence indicating that perforin and granzymes of cytotoxic lymphocytes can activate non-apoptotic pathways of cell death (50). Recently, it was reported that death receptor can induce a mixed mechanism of cell death called “necroapoptosis” (51). Necroapoptosis is a programmed form of necrosis, or inflammatory cell death, whereas necrosis is associated with unprogrammed cell death resulting from cellular damage. In necroapoptosis, rupture of the cell membrane and excretion of intracellular substrates occur as in necrosis. Thus, we assume that mixed mechanisms of tumor cell death including a mechanism similar to necrosis allows 13C fumarate MRI to detect tumor cell death in response to ICB therapy in our study. Mal/Fum showed a significant increase in the treated group of ICB sensitive MC38 model (Figure 4B), and a much smaller change in the less ICB sensitive B16-F10 model (Figure 4E). The results suggest that 13C fumarate MRI detects treatment effect of ICB therapy more directly and accurately than 13C pyruvate MRI.
Treatment effects were also detectable by measurements of intratumor perfusion/permeability, by DCE MRI (Figure 6). Previous studies have shown that abnormal tumor vasculature contributes to immune suppression through multiple mechanisms. Hypoxia promoted by impaired vessel perfusion not only increases the recruitment of immunosuppressive cells through the secretion of cytokines such as TGF-β, VEGF, and IL-10, but also upregulates the expression of CTLA-4 or LAG3 on regulatory T cells and the expression of PD-L1 on myeloid-derived suppressor cells, tumor-associated macrophages, and tumor cells (26,52,53). Treatment with dual anti-CTLA-4 and anti-PD1 therapy induces tumor vessel normalization through a dynamic process that involves various immune populations at different stages of tumor development (26,28). Although the precise mechanism remains unknown, it is reported that type 1 helper T cells play a crucial role in vascular normalization by immune checkpoint blockade (28). An increase in tumor vascularity during immune rejection has previously been detected by DCE-MRI in a E.G7-OVA tumor-bearing mice model which undergoes spontaneous regression (54). As for ICB therapy, there was one report showing that DCE-MRI showed no alteration in the relative blood distribution volume after anti-PD-L1 and anti-CTLA-4 AB treatment in a CT26 tumor (55). By contrast, another report gave evidence of vascular normalization after ICB therapy as they showed the number of functional vessels covered by pericytes increased after dual blockade of PD-1 and CTLA-4 (28). In the current study, Ktrans increased in treated MC38 and B16-F10 tumors relative to control tumors and the increase for each cell line was parallel in magnitude to the corresponding treatment effect (Figure 6C and E).
Quantitative analyses of DCE-MRI has been used in previous clinical studies and found to be useful in the assessment of anticancer therapies as well as in the prediction of eventual response in a variety of cancers (56,57). Routine clinical use of DCE is widespread (58,59). However, many factors determine the accuracy and precision of the parameter values obtained from DCE MRI including the estimation of the arterial input function, spatial and temporal resolutions, pharmacokinetic models, and curve fitting strategies (60). Thus, further investigation is warranted for determining the efficacy of ICB therapy in a clinical setting.
Several limitations of this study should also be acknowledged. First, we could not monitor changes in individual mice over time due to the potential invasiveness of repeated probe injections. The reaction to ICB is not instantaneous and it takes at least a few days for immune system to be activated. During this period, the tumor is still growing which may complicate the interpretation of metabolic or physiologic changes. Next, in the current study, we used vehicle treated mice as a negative control for ICB treatment. The use of vehicle treated mice allows the comparison of immune-activated models with non-activated ones without considering other confounding factors. However, by not using a completely refractory tumor model as a control, treatment effects that cause changes in imaging biomarkers may be overlooked if they are caused by mechanisms unrelated to immune activation. Further study on ICB refractory models would be needed to examine the possibility. Finally, the subcutaneous tumors used are not ideal because the unnatural tumor microenvironment may result in different metabolic profiles in comparison to more orthotopic models. Further investigation using orthotopic xenograft with multiple cell lines or a spontaneous tumor model is needed to further validate the biomarkers proposed here.
Supplementary Material
Significance.
Hyperpolarized 13C MRI and dynamic contrast enhanced MRI in murine tumor models provide useful insight into evaluating early response to immune checkpoint blockade therapy.
Acknowledgements
This research was supported by the Intramural Research Program, Center for Cancer Research, NCI, NIH (grant number Z01BC010477). Y.S. is a JSPS (Japan Society for the Promotion of Science) -NIH Research Fellow.
Footnotes
Conflict of interest:
The authors have declared that no conflict of interest exists.
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer discovery 2018;8:1069–86 [DOI] [PubMed] [Google Scholar]
- 3.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ventola CL. Cancer Immunotherapy, Part 3: Challenges and Future Trends. P T 2017;42:514–21 [PMC free article] [PubMed] [Google Scholar]
- 5.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer 2019;7:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol 2019;12:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019;30:44–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trebeschi S, Drago SG, Birkbak NJ, Kurilova I, Calin AM, Delli Pizzi A, et al. Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann Oncol 2019;30:998–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tunali I, Gray JE, Qi J, Abdalah M, Jeong DK, Guvenis A, et al. Novel clinical and radiomic predictors of rapid disease progression phenotypes among lung cancer patients treated with immunotherapy: An early report. Lung Cancer 2019;129:75–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JY, Yang GF, Chen FF, Peng CW. Evaluating the prognostic significance of tumor-infiltrating lymphocytes in solid tumor: practice of a standardized method from the International Immuno-Oncology Biomarkers Working Group. Cancer Manag Res 2019;11:6815–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nature reviews Clinical oncology 2017;14:655–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwak JJ, Tirumani SH, Van den Abbeele AD, Koo PJ, Jacene HA. Cancer immunotherapy: imaging assessment of novel treatment response patterns and immune-related adverse events. Radiographics : a review publication of the Radiological Society of North America, Inc 2015;35:424–37 [DOI] [PubMed] [Google Scholar]
- 15.Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clinical cancer research : an official journal of the American Association for Cancer Research 2009;15:7412–20 [DOI] [PubMed] [Google Scholar]
- 16.Weiss J, Notohamiprodjo M, Bedke J, Nikolaou K, Kaufmann S. Imaging response assessment of immunotherapy in patients with renal cell and urothelial carcinoma. Current opinion in urology 2018;28:35–41 [DOI] [PubMed] [Google Scholar]
- 17.Butterfield LH, Palucka AK, Britten CM, Dhodapkar MV, Hakansson L, Janetzki S, et al. Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17:3064–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature reviews Drug discovery 2019;18:197–218 [DOI] [PubMed] [Google Scholar]
- 19.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annual review of immunology 2013;31:259–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017;168:657–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015;162:1229–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sukumar M, Roychoudhuri R, Restifo NP. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015;162:1206–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015;162:1217–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hope HC, Salmond RJ. Targeting the tumor microenvironment and T cell metabolism for effective cancer immunotherapy. European journal of immunology 2019;49:1147–52 [DOI] [PubMed] [Google Scholar]
- 25.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell metabolism 2016;24:657–71 [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nature reviews Immunology 2018;18:195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, Stylianopoulos T, Duda DG, Fukumura D, Jain RK. Benefits of vascular normalization are dose and time dependent--letter. Cancer research 2013;73:7144–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017;544:250–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaira K, Higuchi T, Naruse I, Arisaka Y, Tokue A, Altan B, et al. Metabolic activity by (18)F-FDG-PET/CT is predictive of early response after nivolumab in previously treated NSCLC. European journal of nuclear medicine and molecular imaging 2018;45:56–66 [DOI] [PubMed] [Google Scholar]
- 30.Cho SY, Lipson EJ, Im HJ, Rowe SP, Gonzalez EM, Blackford A, et al. Prediction of Response to Immune Checkpoint Inhibitor Therapy Using Early-Time-Point (18)F-FDG PET/CT Imaging in Patients with Advanced Melanoma. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2017;58:1421–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bier G, Hoffmann V, Kloth C, Othman AE, Eigentler T, Garbe C, et al. CT imaging of bone and bone marrow infiltration in malignant melanoma--Challenges and limitations for clinical staging in comparison to 18FDG-PET/CT. European journal of radiology 2016;85:732–8 [DOI] [PubMed] [Google Scholar]
- 32.Kong BY, Menzies AM, Saunders CA, Liniker E, Ramanujam S, Guminski A, et al. Residual FDG-PET metabolic activity in metastatic melanoma patients with prolonged response to anti-PD-1 therapy. Pigment cell & melanoma research 2016;29:572–7 [DOI] [PubMed] [Google Scholar]
- 33.Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, et al. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc Natl Acad Sci U S A 2003;100:10158–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto S, Saito K, Takakusagi Y, Matsuo M, Munasinghe JP, Morris HD, et al. In vivo imaging of tumor physiological, metabolic, and redox changes in response to the anti-angiogenic agent sunitinib: longitudinal assessment to identify transient vascular renormalization. Antioxid Redox Signal 2014;21:1145–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto S, Kishimoto S, Saito K, Takakusagi Y, Munasinghe JP, Devasahayam N, et al. Metabolic and Physiologic Imaging Biomarkers of the Tumor Microenvironment Predict Treatment Outcome with Radiation or a Hypoxia-Activated Prodrug in Mice. Cancer Res 2018;78:3783–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallagher FA, Kettunen MI, Hu DE, Jensen PR, Zandt RI, Karlsson M, et al. Production of hyperpolarized [1,4-13C2]malate from [1,4-13C2]fumarate is a marker of cell necrosis and treatment response in tumors. Proceedings of the National Academy of Sciences of the United States of America 2009;106:19801–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kishimoto S, Oshima N, Yamamoto K, Munasinghe J, Ardenkjaer-Larsen JH, Mitchell JB, et al. Molecular imaging of tumor photoimmunotherapy: Evidence of photosensitized tumor necrosis and hemodynamic changes. Free radical biology & medicine 2018;116:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. The New England journal of medicine 2015;372:2006–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine 2015;373:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. The Lancet Oncology 2018;19:1480–92 [DOI] [PubMed] [Google Scholar]
- 41.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. The Journal of experimental medicine 2017;214:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grist JT, Jarvis LB, Georgieva Z, Thompson S, Kaur Sandhu H, Burling K, et al. Extracellular Lactate: A Novel Measure of T Cell Proliferation. J Immunol 2018;200:1220–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill DK, Orton MR, Mariotti E, Boult JK, Panek R, Jafar M, et al. Model free approach to kinetic analysis of real-time hyperpolarized 13C magnetic resonance spectroscopy data. PLoS One 2013;8:e71996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature communications 2015;6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allison KE, Coomber BL, Bridle BW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology 2017;152:175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends in immunology 2015;36:257–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nature immunology 2018;19:1319–29 [DOI] [PubMed] [Google Scholar]
- 48.Bohndiek SE, Kettunen MI, Hu DE, Brindle KM. Hyperpolarized (13)C spectroscopy detects early changes in tumor vasculature and metabolism after VEGF neutralization. Cancer research 2012;72:854–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mignion L, Dutta P, Martinez GV, Foroutan P, Gillies RJ, Jordan BF. Monitoring chemotherapeutic response by hyperpolarized 13C-fumarate MRS and diffusion MRI. Cancer research 2014;74:686–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21:5047–56 [DOI] [PubMed] [Google Scholar]
- 51.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature reviews Molecular cell biology 2010;11:700–14 [DOI] [PubMed] [Google Scholar]
- 52.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer research 2014;74:665–74 [DOI] [PubMed] [Google Scholar]
- 53.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nature immunology 2013;14:1173–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu DE, Beauregard DA, Bearchell MC, Thomsen LL, Brindle KM. Early detection of tumour immune-rejection using magnetic resonance imaging. British journal of cancer 2003;88:1135–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fiegle E, Doleschel D, Koletnik S, Rix A, Weiskirchen R, Borkham-Kamphorst E, et al. Dual CTLA-4 and PD-L1 Blockade Inhibits Tumor Growth and Liver Metastasis in a Highly Aggressive Orthotopic Mouse Model of Colon Cancer. Neoplasia (New York, NY) 2019;21:932–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hahn OM, Yang C, Medved M, Karczmar G, Kistner E, Karrison T, et al. Dynamic contrast-enhanced magnetic resonance imaging pharmacodynamic biomarker study of sorafenib in metastatic renal carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2008;26:4572–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim KA, Park MS, Ji HJ, Park JY, Han KH, Kim MJ, et al. Diffusion and perfusion MRI prediction of progression-free survival in patients with hepatocellular carcinoma treated with concurrent chemoradiotherapy. Journal of magnetic resonance imaging : JMRI 2014;39:286–92 [DOI] [PubMed] [Google Scholar]
- 58.Barnes SL, Whisenant JG, Loveless ME, Ayers GD, Yankeelov TE. Assessing the reproducibility of dynamic contrast enhanced magnetic resonance imaging in a murine model of breast cancer. Magnetic resonance in medicine 2013;69:1721–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galbraith SM, Lodge MA, Taylor NJ, Rustin GJ, Bentzen S, Stirling JJ, et al. Reproducibility of dynamic contrast-enhanced MRI in human muscle and tumours: comparison of quantitative and semi-quantitative analysis. NMR in biomedicine 2002;15:132–42 [DOI] [PubMed] [Google Scholar]
- 60.Abramson RG, Arlinghaus LR, Dula AN, Quarles CC, Stokes AM, Weis JA, et al. MR Imaging Biomarkers in Oncology Clinical Trials. Magnetic resonance imaging clinics of North America 2016;24:11–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.